

ABSTRACT
The pharmaceutical industry’s interest in monoclonal antibodies (mAbs) and their derivatives has spurred rapid growth in the commercial and clinical pipeline of these effective therapeutics. The complex micro-heterogeneity of mAbs requires in-depth structural characterization for critical quality attribute assessment and quality assurance. Currently, mass spectrometry (MS)-based methods are the gold standard in mAb analysis, primarily with a bottom-up approach in which immunoglobulins G (IgGs) and their variants are digested into peptides to facilitate the analysis. Comprehensive characterization of IgGs and the micro-variants remains challenging at the proteoform level. Here, we used both top-down and middle-down MS for in-depth characterization of a human IgG1 using ultra-high resolution Fourier transform MS. Our top-down MS analysis provided characteristic fingerprinting of the IgG1 proteoforms at unit mass resolution. Subsequently, the tandem MS analysis of intact IgG1 enabled the detailed sequence characterization of a representative IgG1 proteoform at the intact protein level. Moreover, we used the middle-down MS analysis to characterize the primary glycoforms and micro-variants. Micro-variants such as low-abundance glycoforms, C-terminal glycine clipping, and C-terminal proline amidation were characterized with bond cleavages higher than 44% at the subunit level. By combining top-down and middle-down analysis, 76% of bond cleavage (509/666 amino acid bond cleaved) of IgG1 was achieved. Taken together, we demonstrated the combination of top-down and middle-down MS as powerful tools in the comprehensive characterization of mAbs.
Keywords: monoclonal antibodies, top-down mass spectrometry, middle-down mass spectrometry, proteoforms, post-translational modifications
Introduction
Therapeutic monoclonal antibodies (mAbs) are a class of biotherapeutics in the fast-growing new drug development areas of the pharmaceutical industry.1 Since the commercialization of the first therapeutic mAb over thirty years ago, more than 60 antibody drugs have been approved.1,2 Currently, over 550 mAb candidates and their derivatives are in the clinical development stage.1 The high selectivity and specificity towards given targets led to the success of antibody-based drugs in the treatment of a variety of diseases including cardiovascular, infectious, immunological diseases and cancer.3–5
Therapeutic mAbs are predominantly developed from immunoglobins gamma (IgGs) and their derivatives.6 These IgGs are “Y”-shaped homodimers with a molecular mass of ~ 150 kDa, consisting of two identical heavy chains (Hcs, ~ 50 kDa) and two identical light chains (Lcs, ~ 25 kDa) joined together by disulfide bonds. The integration of primary structures and various sequence modifications on Lcs and Hcs regulates the interactions between mAbs and the targets.7 Moreover, the molecular heterogeneity resulting from post-translational modifications (PTMs), C-terminal processing on Hc, and sequence mutations, may affect the quality and pharmacological properties of mAbs to different extents.8–10 Thus, a deep characterization of mAb structural variants is critical for the assessment of structure-function relationships and effects on therapeutic safety, stability, and efficacy.
Mass spectrometry (MS) has been applied to the analysis of therapeutic mAbs over the past decades and has become the predominant tool in the characterization of their structures.11–13 Currently, top-down, middle-down and bottom-up MS are three main approaches applied in the characterization of PTMs, localization of disulfide bonds, and verification of protein sequences in analyzing therapeutic mAbs.14–16 Among these three approaches, a bottom-up approach combining liquid chromatography (LC) and tandem MS (MS/MS) is routinely used in the pharmaceutical industry owing to its standardized workflow and availability of instrument choice and software. Nevertheless, bottom-up MS may suffer from the introduction of artifacts from lengthy sample preparation, limited protease choice for full protein sequence coverage, and loss of proteoform information in convoluted peptide pools.17,18 In contrast to the bottom-up approach, top-down MS analyzes intact proteins directly with minimum sample preparation, which provides an overview of major proteoforms19 and minimizes sample artifacts.20 However, the large size of mAbs (~ 150 kDa) together with intra- and inter-chain disulfide bonds restrict efficient fragmentation of intact proteins, and therefore only ~ 15–35% bond cleavage can be achieved in top-down MS/MS analysis.21–24
In addition to bottom-up and top-down approaches, middle-down MS is also commonly used in the characterization of mAbs by analyzing subunits derived from limited protein proteolysis or disulfide bond reduction. For example, Fornelli et al. used high-resolution Orbitrap Fourier transform MS (FTMS) coupled with electron transfer dissociation (ETD) to perform a middle-down analysis on therapeutic mAbs and their oxidized forms.25 Recently, Cotham et al. investigated the performance of 193 nm ultraviolet photodissociation (UVPD) in the middle-down characterization of mAb subunits and made a comparison on fragmentation efficiency between UVPD and ETD.26 Fornelli et al. also showed that up to 90% sequence information of mAb subunits could be achieved in middle-down MS by combining three different fragmentation techniques, UVPD, ETD, and electron transfer/higher-energy collision dissociation (EThcD), in six LC runs.27 Compared with the bottom-up approach, middle-down MS reduces the chance of proteoform information loss by analyzing larger polypeptides (> 3 kDa) that may contain multiple PTMs and unique isoform sequences.28–30 Moreover, as the typical bond cleavage of mAbs in a middle-down experiment is 50%~ 70%, this middle-down approach improves the bond cleavage coverage in protein characterization that is limited in top-down MS.25,26,31
Although a variety of MS approaches have been established for the characterization of therapeutic mAbs,32 most of them focused on the high-abundance N-linked glycoforms.33,34 High mass accuracy from isotopically resolved high-resolution MS data could be used to increase the confidence level of protein identification with detection of PTMs and variations in sequence.35 The glycosylation pattern of an intact mAb including other low-abundance glycoforms has never been isotopically resolved for confident identification based on highly accurate molecular mass. In addition, a comprehensive characterization of mAbs including micro-variants, such as C-terminal glycine clipping, C-terminal proline amidation and low-abundance N-linked glycoforms, is still lacking. Here, we used a 12 T Fourier Transform Ion Cyclotron Resonance (FTICR) mass spectrometer coupled with electron capture dissociation (ECD) and collisionally activated dissociation (CAD) to comprehensively characterize a human mAb IgG1 by combining top-down and middle-down approaches. Accurate molecular mass measurements of six different IgG1 glycoforms at unit mass resolution were achieved in top-down MS analysis. The following MS/MS analysis of intact IgG1 allowed characterization of a major IgG1 proteoform at the intact protein level. In middle-down analysis, in addition to the primary structures of Lcs and Hcs, we also characterized micro-variants including G0 glycoform, G2F glycoform and C-terminal processing of Hc with confident bond cleavage higher than 44% at the subunit level. In total, 76% bond cleavage (509/666 amino acid bond cleaved) of IgG1 was obtained by combining top-down and middle-down analysis.
Results
Unit mass resolution and isotopic distribution simulation of intact mAb
The FTICR mass spectrum of intact IgG1 at the charge state of 54+ from 2710 to 2730 m/z is shown in Figure 1A. A unit mass resolution was achieved by averaging 496 transients with 9.2 s each transient (16 M transient size) and 0.008 s ion accumulation time. The most abundant peak at 2720 m/z with an experimental mass of 146,730.37 Da was identified as IgG1 with one G0F and one G1F oligosaccharide chains (G0F/G1F glycoform), matching the calculated mass 146,730.12 Da with 1.7 ppm mass error considering two N-terminal pyroglutamates, sixteen disulfide bonds, one G0F glycan and one G1F glycan. The mass difference of 162 Da from two neighboring peaks indicates the addition of a hexose to the glycan, thus the other two major peaks at 2717 m/z and 2723 m/z were identified as IgG1 with G0F/G0F, and G1F/G1F or G0F/G2F glycoforms, respectively. The peak to the left of G0F/G0F was identified as G0/G0F glycoform based on the mass shift of −146 Da from the G0F/G0F glycoform. In addition to these four glycoforms, the other two peaks at 2726 m/z and 2729 m/z were considered as G1F/G2F and G2F/G2F glycoforms based on consecutive mass shift of 162 Da from the G1F/G1F glycoform. The observed glycoform population contains ~ 6% G0/G0F, ~ 22% G0F/G0F, ~ 33% G0F/G1F, ~ 24% G1F/G1F or G0F/G2F, ~ 11% G1F/G2F, and ~ 4% G2F/G2F based on their signal intensity on the mass spectrum at 54+ (Figure 1A). Figure 1B shows the experimental isotopic distribution of G0F/G0F proteoforms and their simulated isotopic distribution. The primary G0F/G0F glycoform at 2717 m/z has an experimental mass of 146,568.02 Da, matching well with the calculated mass of 146,568.08 Da with 0.4 ppm error. Besides the primary G0F/G0F glycoform, the isotopic distribution simulations showed the potential presence of proteoforms of −58 Da, loss of water, varying levels of oxidation, Na+ adduct, K+ adduct and phosphate adduct of the major glycoform in the range of 2715 to 2720 m/z. The simulated isotopic distribution peaks match well with the experimental results, confirming the identification of various proteoforms.
Figure 1.
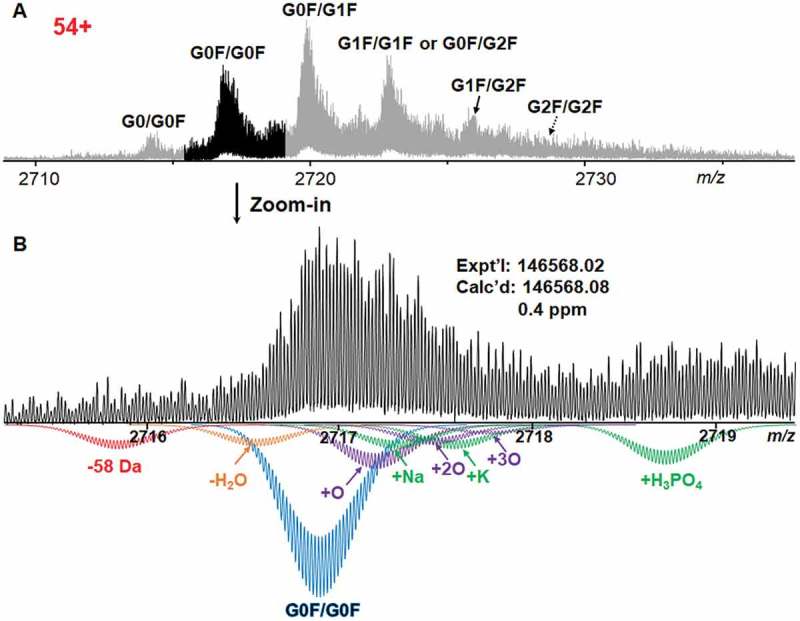
Mass spectrum of intact IgG1 at the charge state of 54 + . A Mass spectrum showing six different glycoforms of IgG1, including G0/G0F, G0F/G0F, G0F/G1F, G1F/G1F or G0F/G2F, G1F/G2F, and G2F/G2F. B Zoomed-in experimental mass spectrum of G0F/G0F proteoforms at isotopic distribution with simulated isotopic distribution of G0F/G0F proteoforms. In addition to the primary G0F/G0F glycoform, loss of 58 Da, loss of water, multiple oxidized proteoforms, Na+ adduct, K+ adduct and phosphate adduct of the major glycoform were also simulated to match the experimental mass spectrum. GxF indicates N-oligosaccharides carrying x number of terminal galactoses.
Top-down MS/MS analysis of intact IgG1
Unlike most top-down analysis of mAbs relying on the combination of multiple charge states to enhance fragment signal,22,24 here we isolated single charge state of IgG1 for MS/MS analysis to minimize the introduction of fragment ions from adjacent species in a broad isolation window. The charge state of 51+ at 2883 m/z was isolated with a window of 50 m/z for both ECD and CAD fragmentations. Figure 2 shows the fragmentation map by combining two ECD and one CAD analysis. In total, 172 fragment ions were identified, including 33 c ions and 21 b ions from the Lc and 60 c ions, 43 z● ions, and 15 b ions from the Hc. A total of 23% bond cleavage of IgG1 sequence was achieved. The fragment ions also help assign the connectivity of intramolecular disulfide bonds based on the mass shift of 2 or 4 Da.
Figure 2.

Protein sequence map of intact IgG1 showing bond cleavages and disulfide bonds. Intact IgG1 at 51+ was isolated in a 50 m/z window for MS/MS analysis. The bond cleavages shown in protein sequence map are the results from combining two ECD and one CAD analysis. Intermolecular and intramolecular disulfide bonds are shown as purple lines. Cysteines connected by disulfide bonds are highlighted in green. The pyroglutamate on Lc and glycosylation site Asn301 on Hc are highlighted in red.
Although the tandem MS analysis of intact IgG1 at a single charge state provides valuable information on the protein sequence, all the proteoforms at this charge state were fragmented and analyzed at the same time. To specifically characterize one single proteoform of IgG1, we performed tandem MS analysis on the G0F/G1F glycoform with an isolation window of 5 m/z. The fragmentation map in Figure S1 shows 12 c ions, and 13 b ions from the Lc and 32 c ions, 32 z● ions, and 12 b ions from the Hc with a total bond cleavage of 14% (90/666 amino acid bond cleaved).
Online LC/MS analysis of IgG1 subunits by middle-down mass spectrometry
Even though top-down MS analysis of intact IgG1 provides a “bird’s eye view” of all the proteoforms, the PTM sites remain difficult to completely characterize in intact protein analysis owing to the presence of disulfide bond linkage between/within Hc and Lc, the large protein size of IgG1, and the limited fragmentation ability on large proteins of the instrument. Here, to fully characterize the glycosylation sites and micro-variants of IgG1, IdeS enzyme and reducing agent TCEP were applied to digest and then reduce the intact IgG1 mAb into three subunits with molecular weights around 25 kDa, Fc/2, Fd, and Lc (Figure S2).
The total ion current (TIC) chromatogram from online LC/MS shows three proteolytic subunits, Fc/2, Lc, and Fd in Figure 3A. The small peak before Fc/2 was identified as a truncated form of Fc/2 from Asp-Pro hydrolysis (DP hydrolysis) based on MS/MS analysis (Figure S3), which is a main protein degradation product under acidic and heating condition. Experimental and deconvoluted mass spectra of the three proteolytic subunits were shown in Figure 3B. Deconvoluted mass of Lc and Fd were 22,928.23 Da and 25,201.74 Da, respectively, matching well with the theoretical mass calculated based on amino acid sequences within 10 ppm error. Consistent with the results from top-down MS analysis, two major glycoforms, G0F and G1F, and two minor glycoforms, G0 and G2F, were localized on Fc/2 subunit based on the accurate experimental mass of these four glycoforms, 25,042.57 Da (G0), 25,188.64 Da (G0F), 25,350.69 Da (G1F), and 25,512.74 Da (G2F), considering the N-glycosylation, cysteines in reduced forms and the absence of C-terminal lysine on Fc/2. The observed glycoform population contains ~ 4% G0, ~ 48% G0F, ~ 42% G1F, and ~ 6% G2F based on their signal intensity on deconvoluted mass spectrum, which is consistent with our results in top-down MS. The highly accurate mass measurement also showed oxidation, neutral loss of water, and −58 Da proteoforms next to G0F and G1F glycoforms (Table S1), leading to the subsequent MS/MS characterization of these micro-variants.
Figure 3.
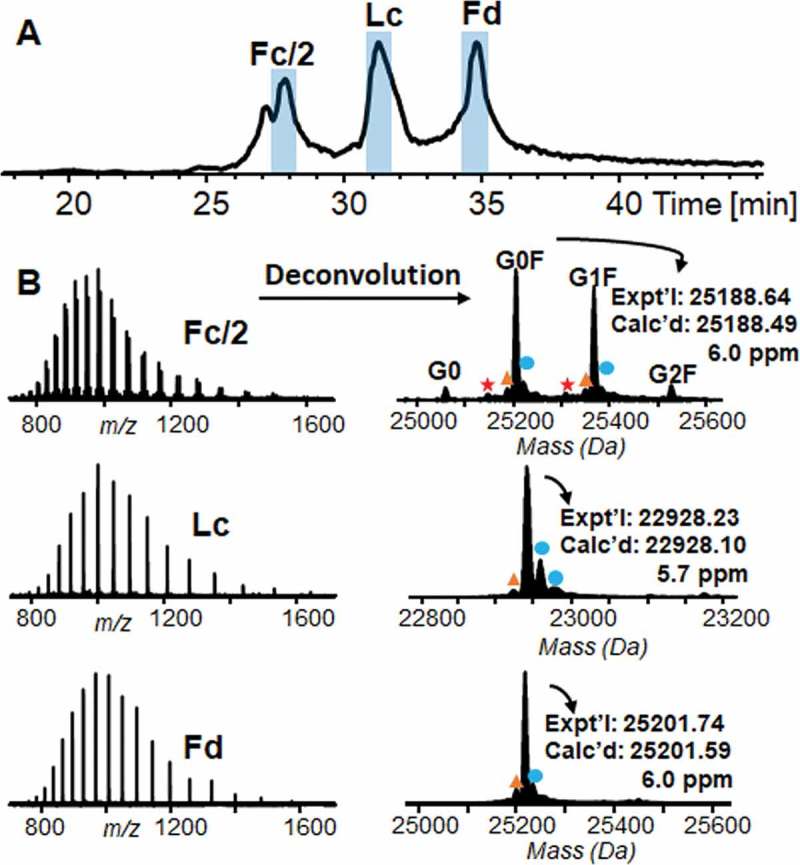
Chromatogram and mass spectra of proteolytic subunits from IdeS digestion. A TIC chromatogram of three proteolytic subunits, Fc/2, Lc and Fd, from online LC/MS analysis. B Left, experimental mass spectra of Fc/2, Lc, and Fd. Right, deconvoluted mass spectra of Fc/2, Lc, and Fd. Asteroid, −58 Da from C-terminal glycine clipping and proline amidation; triangle, −18 Da from neutral loss of water; dot, + 16 Da from oxidation. Expt’l, experimental monoisotopic mass based on data from MS experiments. Calc’d, calculated monoisotopic mass based on amino acid sequences.
Offline characterization of IgG1 by middle-down mass spectrometry
To better characterize the modification sites and to maximize the bond cleavage, IdeS-derived subunits were separated and fractionated by reverse phase chromatography (RPC) and were then subject to offline high-resolution MS/MS analysis. The CAD mass spectrum, representative fragment ions from ECD and CAD, and sequence map of one major glycoform Fc/2-G0F were shown in Figure 4. In addition to fragment ions from amino acid backbone, Figure 4A also shows the ions from labile glycan fragmentation in the low m/z range from 200 to 700. Four fragment ions with m/z of 204.09, 366.14, 528.19, and 690.24 were detected, corresponding to one GlcNAc, one GlcNAc and one hexose, one GlcNAc and two hexoses, and one GlcNAc and three hexoses, respectively. Compared to the signal of ions from peptide backbone cleavage, the signal of fragment ions from the G0F glycan is extremely low, indicating that the majority of glycosylation structures was preserved in CAD condition applied to Fc/2 subunit. In Figure 4B, four fragmentation ions, c60, c61, z●149, and y167, narrow down the glycosylation site to Asn61 in Fc/2 sequence. A bond cleavage of 66% was obtained by counting all the cleavage sites resulted from 70 c ions, 78 z● ions, 38 b ions, and 23 y ions in one ECD and one CAD (Figure 4C). The MS/MS analysis of Fc/2-G1F also gives a high bond cleavage of ~ 60% with the confirmation of glycosylation site at Asn61 by combining only one ECD and one CAD (Figure S4). In addition, 75% bond cleavage of Lc and 71% bond cleavage of Fd were obtained in one ECD and one CAD analysis (Figure S5). A pyroglutamate modification was also characterized at N-terminus of Lc based on a variety of b and c ions with mass shift of −17 Da.
Figure 4.
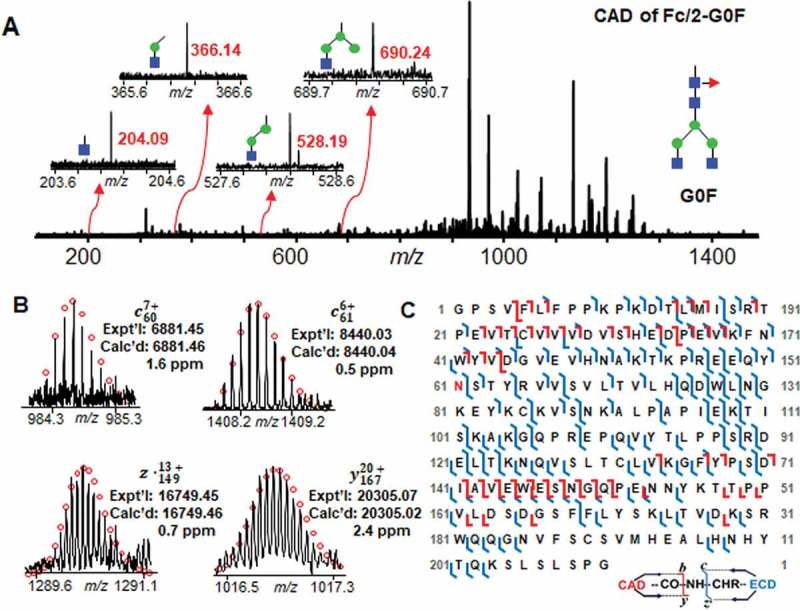
Mass spectrum, fragment ions, and sequence map of Fc/2-G0F glycoform fragmentation. A Mass spectrum of CAD fragmentation of Fc/2-G0F. Inset, representative fragment ions from labile glycan fragmentation. B Representative fragment ions from ECD and CAD fragmentations localizing the glycosylation site at Asn61. C Fc/2 subunit sequence map showing bond cleavages and glycosylation site. The glycosylation site at Asn61 is highlighted in red. The amino acid number here is based on the Fc/2 subunit sequence.
Besides the highly abundant Fc/2-G0F, Fc/2-G1F, Fd and Lc, we also characterized the micro proteoforms including Fc/2-G0, Fc/2-G2F, and the −58 Da proteoform next to Fc/2-G0F and Fc/2-G1F. Figure 5 shows the representative fragment ions and sequence maps of two micro glycoforms, Fc/2-G0 and Fc/2-G2F. Even though the signal of these two glycoforms is much lower than the highly abundant G0F and G1F forms, the fragment ions we got were sufficient to localize the glycosylation site at Asn61 and map the protein sequence. By combining one ECD and one CAD analysis, 58% and 44% bond cleavage were obtained for Fc/2-G0 and Fc/2-G2F, respectively. The micro proteoforms labeled as red asteroid next to G0F and G1F with a mass loss of 58 Da (Figure 3B) also attracted our attention. However, the intensities of these small peaks are too low to get enough fragment ions for sequence identification. To characterize this low-abundance proteoform, we deglycosylated the intact IgG1 mAb using the endoglycosidase IgGZERO so that the intensity of the −58 Da proteoform could be increased by converting all the −58 Da proteoforms to the same deglycosylated Fc/2 form. Based on the fragment ions from CAD and ECD fragmentation shown in Figure 6, the −58 Da proteoform is identified as the C-terminal glycine clipping with proline amidation.
Figure 5.
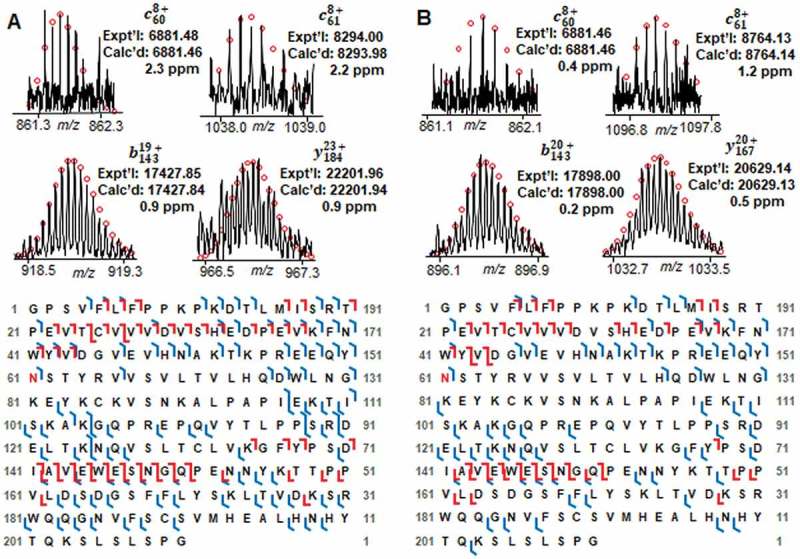
Fragment ions and sequence maps of A Fc/2-G0 and B Fc/2-G2F. Top, representative fragment ions from ECD and CAD fragmentations identifying the glycosylation site at Asn61. Bottom, Fc/2 subunit sequence map showing bond cleavages and glycosylation site. The glycosylation site Asn61 is highlighted in red. The amino acid number here is based on the Fc/2 subunit sequence.
Figure 6.
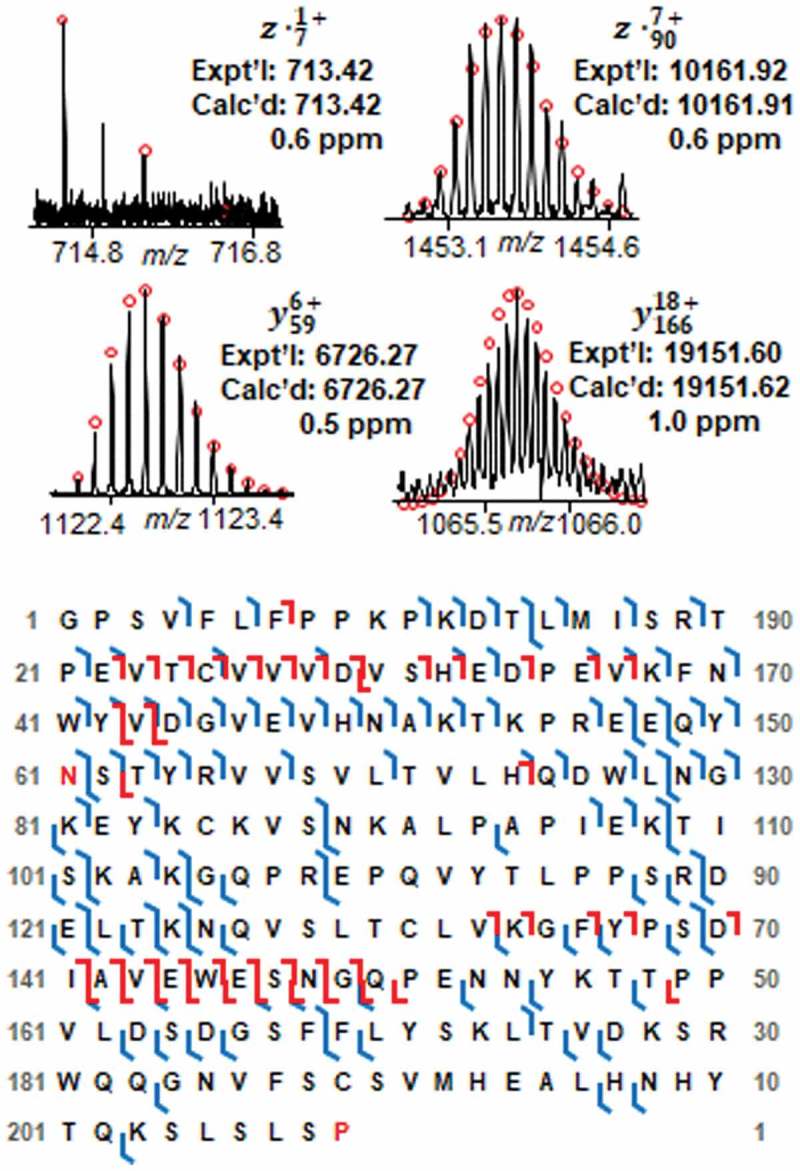
Fragment ions and sequence map of the −58 Da proteoform of Fc/2. Top, representative fragment ions. Bottom, Fc/2 subunit sequence map showing bond cleavages, glycosylation site, loss of C-terminal glycine, and C-terminal proline amidation. The glycosylation site Asn61 and amidated Pro209 are highlighted in red. The amino acid number here is based on the Fc/2 subunit sequence.
Discussion
Here, we developed an integrated strategy by combining top-down and middle-down MS to carry out a comprehensive characterization of a human mAb IgG1. Intact IgG1 was isotopically resolved using 12 T FTICR mass spectrometer, allowing accurate mass measurements, and thus, the confident identification of intact IgG1 proteoforms. MS/MS analysis performed on the single proteoform of intact IgG1 demonstrated the feasibility of characterizing a single mAb proteoform in top-down MS approach, which could be further applied to the proteoform characterization of large proteins. Moreover, all the glycoforms and other PTMs were characterized in middle-down MS with 44–75% bond cleavages at the subunit level by simply combining one ECD and one CAD analysis.
A unit mass resolution was achieved in our top-down MS analysis of intact IgG1, allowing the identification of six different IgG1 glycoforms at one specific charge state and clearly shows the glycosylation pattern of IgG1, which is consistent with the reported information in previous studies.36 The comprehensive analysis of intact mAb proteoforms at unit mass resolution is still challenging due to broad charge state distribution, addition of adducts, neutral losses, and wide isotopic distribution range resulting from large molecular weight of intact mAbs.36 These drawbacks led to the decrease in MS signal. Even though the unit mass resolution of intact mAb was achieved by Valeja et al.37 and Nicolardi et al.23 previously in FTICR analysis, only one or two major glycoforms were isolated and resolved isotopically. Here, we demonstrated the isotopic resolution of all the IgG1 proteoforms at a single charge state using higher magnetic field (12 T), higher resolving power (~ 350,000) and more averaged acquisitions (496 acquisitions) compared to 9.4 T, ~ 290,000 resolving power and 125–235 averaged acquisitions in a previous study by Valeja et al..37 In addition, a larger number of data points (16 M) was acquired for our unit mass resolution data, as compared to 1 M or 2 M data points in the previous study by Nicolardi et al.,23 enabling identification of the six different glycoforms based on accurate mass measurements. Our in-house developed software MASH Suite Pro further guarantees accurate molecular mass measurements by allowing manually validation of the peak assignments.38
The subsequent top-down MS/MS analysis on intact IgG1 at a single charge state provides 23% bond cleavage, which is comparable to the bond cleavage of 25% reported by Mao et al.21 in their ECD analysis of an IgG1κ. The presence of 16 disulfide bonds limits the fragmentation efficiency of ECD and CAD in S-S linked regions, thus only 23% peptide bonds were cleaved. To increase the percentage of bond cleavage, disulfide bonds could be reduced and the resulting reduced Hc and Lc could be analyzed separately. Besides the characterization of intact IgG1 at a single charge state, we also performed tandem MS analysis on the G0F/G1F glycoform to characterize a single IgG1 proteoform. Different from previous tandem MS analysis of intact mAb in a wide m/z range by online LC/MS/MS22,24 or offline MS/MS with 9.4 T FTICR,21 here we used a high-resolution 12 T FTICR to enable the offline MS/MS analysis of a single mAb proteoform with 5 m/z isolation window. Even though the glycosylation site could not be characterized due to the presence of disulfide bonds, we obtained 14% bond cleavage, and the results here show the feasibility of fragmenting a single proteoform of intact proteins with molecular mass as high as 150 kDa, which could be further applied to the detailed characterization of large intact proteins.
Besides top-down MS analysis, we also carried out middle-down MS analysis to completely characterize the PTM sites of IgG1. In online LC/MS analysis, all the proteoforms of three IgG1 subunits could be isotopically resolved in limited online LC elution time window with 1 M transient size and 0.04 s accumulation time, providing accurate molecular mass for proteoform identification. The baseline resolution in a limited LC time window also allows further targeted online MS/MS analysis for protein characterization. The following offline MS/MS characterization gives 66% bond cleavage of Fc/2-G0F, 75% bond cleavage of Lc and 71% bond cleavage of Fd by combining only one ECD and one CAD analysis. Previously, the online ETD analysis by Fornelli et al. 25 gave a bond cleavage of ~ 70% for Fc/2 and Lc, and ~ 60% for Fd, by combining data from 4–10 LC runs. Here, similar degree of bond cleavage could be obtained by simply collecting proteolytic subunits from one RPC separation and combining one offline ECD and CAD analysis, which greatly reduces the time of experimental period.
Previous middle-down MS analysis of mAb glycosylation focused on highly abundant Fc/2 glycoforms, such as G0F and G1F.26,33 In this study, we not only analyzed the high-abundance glycoforms, but also characterized the micro glycoforms with confident bond cleavages. We obtained 58% bond cleavage of Fc/2-G0 and 44% bond cleavage of Fc/2-G2F in offline MS/MS analysis with the localization of glycosylation site at Asn61. Moreover, we characterized a micro-variant with C-terminal glycine clipping and proline amidation. Previously, Johnson et al.39 used weak cation exchange-high performance LC separation and bottom-up MS to detect the C-terminal amidation in an IgG1 heavy chain. Here we demonstrated the characterization of C-terminal amidation by middle-down MS, which avoids the lengthy sample preparation and improves the confidence of identification by analyzing large polypeptide fragment of ~ 25 kDa. It was reported that C-terminal amidation is a general PTM of therapeutic mAbs and it could be catalyzed by peptidylglycine alpha-amidating monooxygenase.40–42 However, no effect of C-terminal amidation on activity of therapeutic mAbs was observed so far.43
Overall, an integrated strategy combining top-down and middle-down MS was developed for comprehensive characterization of a human mAb IgG1. In total, we achieved 76% bond cleavage (509/666 amino acid bond cleaved) of IgG1 by combining middle-down and top-down analysis. This integrated strategy affords an overview of intact IgG1 proteoforms and at the same time allows a detailed proteoform characterization with simple sample preparation and data analysis method. We anticipate the application of this integrated strategy in comprehensive analysis of more mAbs and other biotherapeutics such as antibody-drug conjugates.44 More bond cleavages can be achieved by combining other MS/MS techniques such as UVPD.27 Multiple-attribute method (MAM), which combines high-end MS and dedicated software to provide highly specific and quantitative information for monitoring different attributes of biotherapeutics, could also be used in characterization and quality control of biotherapeutics, as FTICR might not be readily accessible in some cases.45–47
Materials and methods
Chemicals
All chemicals were obtained from Millipore Sigma Inc. (St Louis, MO, USA) unless noted otherwise. The SILu Lite SigmaMAb universal antibody standard human IgG1 (MSQC4) was purchased from Millipore Sigma Inc. (St Louis, MO, USA). The modified IdeS protease FabRICATOR and IgG-specific endoglycosidase IgGZERO were obtained from Genovis (Cambridge, MA, USA). All solutions were prepared using HPLC grade water (Fisher Scientific, Fair Lawn, NJ, USA).
Top-down mass spectrometry
The SILu human IgG1 mAb was reconstituted in water to a final concentration of 2 µg/µL. ~ 50 µg of IgG1 was desalted using 100 kDa molecular weight cut-off filters with 0.1% formic acid (FA) in water. The desalted IgG1 was diluted using equal volume of acetonitrile (ACN) with 0.1% FA and used for direct infusion. Intact IgG1 was analyzed by 12 T solariX FTICR mass spectrometer (Bruker Daltonics, Bremen, Germany) equipped with an automated chip-based nano-electrospray ionization (nanoESI) source (Triversa NanoMate; Advion Bioscience, Ithaca, NY, USA). The spray voltage of the NanoMate was set to 1.4 kV and the gas pressure was 0.3 psi. MS spectra were acquired over 200–3000 m/z range with 16 M transient size. MS/MS spectra were acquired over 100–4000 m/z with 4 M transient size. Isolation window of ECD and CAD was 5 or 50 m/z. The ECD bias was set at 0.5–0.8 V and the collision DC bias for CAD was 20–24 V. 1000–3000 transients were acquired for MS/MS spectra.
Middle-down mass spectrometry
In IdeS digestion, 25 units of IdeS was added to ~ 25 µg IgG1 in phosphate-buffered saline. The resulting solution was incubated at 37 °C for 30 min. Then, the proteolytic product was reduced by 30 mM TCEP at pH = 3 at 65 °C for 1 h.
IdeS-derived subunits were further separated by RPC using home-packed PLRP column (PLRP-S, 200 mm length x 500 μm id, 10 μm particle size, 1,000 Å pore size, Agilent). 6 μL proteolytic product (~ 0.2 μg/μL) was injected into a nanoACQUITY UPLC system (Waters, Milford, MA, USA) with a flowrate of 20 μL/min for RPC separation. The separation was performed in a 60 min gradient with mobile phase B from 20% to 95% (Mobile phase A: 0.1% FA in water, Mobile phase B: 0.1% FA in 1:1 isopropanol:ACN). The nanoACQUITY UPLC system was coupled to the 12 T solariX FTICR mass spectrometer for online LC/MS analysis.
For fractionation of IgG1 subunits, 10 μL proteolytic product (~ 0.4 μg/μL) was injected into the UPLC system for RPC separation using the same home-packed PLRP column. The fractions were collected with approximated concentration of 0.1 μg/μL for offline high-resolution MS/MS analysis by 12 T solariX FTICR. Samples were introduced into the mass spectrometer via NanoMate using 1.3–1.5 kV spray voltage and 0.3 psi gas pressure. All the MS/MS spectra were collected in 2 M transient size over 100–3000 m/z range. The isolation window of ECD and CAD was 1.5–3 m/z. The ECD bias was set from 0.2 to 0.8 V and the collision DC bias for CAD was set from 6 V to 10 V. MS/MS spectra were averaged for 200–800 transients.
Data analysis
In top-down MS, MS spectra of intact IgG1 mAb were analyzed by DataAnalysis software from Bruker Daltonics. The Simulate Pattern in DataAnalysis was used for peak simulations. MS/MS data were analyzed by the in-house developed software MASH Suite Pro. A minimum fit of 60% and signal-to-noise (S/N) threshold of 3 were set for peak picking. Fragment ion lists from MS/MS analysis were generated for manually validation and localization of PTM sites. Fragment ions including c, c-1,z●, and z●+ 1 ions in ECD and b, y ions in CAD were validated with a mass error tolerance of 10 ppm. All the reported mass is monoisotopic mass.
In middle-down MS approach, online LC/MS data was processed and analyzed by DataAnalysis software. The Maximum Entropy algorithm incorporated in DataAnalysis software was used for the deconvolution of IgG1 subunit profiling. MS/MS data of IdeS-derived subunits were analyzed by MASH Suite Pro as mentioned above.
Abbreviations
- ACN
acetonitrile
- CAD
collisionally activated dissociation
- ECD
electron capture dissociation
- ETD
electron transfer dissociation
- EThcD
electron transfer/higher-energy collision dissociation
- FA
formic acid
- FTICR
Fourier Transform Ion Cyclotron Resonance
- FTMS
Fourier Transform Mass Spectrometry
- Hc
heavy chain
- IgG
immunoglobin gamma
- Lc
light chain
- LC
liquid chromatography
- mAb
monoclonal antibody
- MAM
multi-attribute method
- MS
mass spectrometry
- MS/MS
tandem mass spectrometry
- PTM
post-translational modification
- qTOF
quadrupole time-of-flight
- RPC
reverse phase chromatography
- TIC
total ion current
- UVPD
ultraviolet photodissociation
Funding Statement
This work was supported by the AbbVie Inc.; National Institutes of Health [1R01GM117058-01]; National Institutes of Health [1S10OD018475-01].
Acknowledgments
We would like to thank Yanlong Zhu, Zhijie Wu and Bifan Chen for their discussion in manuscript preparation. Financial support was kindly provided by AbbVie. We also would like to acknowledge the high-end instrument grant S10OD018475 and NIH R01 grants, GM117058, HL109810, and HL096971 (to Y.G.).
Disclosure of potential conflicts of interest
The authors declare the following competing financial interest(s): Dr. Zhaorui Zhang, Dr. Qunying Zhang, and Dr. Wayne Pritts are employees of AbbVie Inc.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Carter PJ, Lazar GA.. 2017. Next generation antibody drugs: pursuit of the ‘high-hanging fruit’. Nat Rev Drug Discov. 17:197–223. doi: 10.1038/nrd.2017.227. [DOI] [PubMed] [Google Scholar]
- 2.Ecker DM, Jones SD, Levine HL.. 2015. The therapeutic monoclonal antibody market. Mabs-Austin. 7:9–14. doi: 10.4161/19420862.2015.989042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chames P, Van Regenmortel M, Weiss E, Baty D. 2009. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 157:220–233. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weiner GJ. 2015. Building better monoclonal antibody-based therapeutics. Nat Rev Cancer. 15:361–370. doi: 10.1038/nrc3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaplon H, Reichert JM. 2018. Antibodies to watch in 2018. Mabs-Austin. 10:183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reichert JM. 2012. Marketed therapeutic antibodies compendium. Mabs-Austin. 4:413–415. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breedveld FC. Therapeutic monoclonal antibodies. Lancet. 355;2000:735–740. [DOI] [PubMed] [Google Scholar]
- 8.Liu HC, Gaza-Bulseco G, Faldu D, Chumsae C, Sun J. 2008. Heterogeneity of monoclonal antibodies. J Pharm Sci-Us. 97:2426–2447. doi: 10.1002/jps.21180. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Singh S, Zeng DL, King K, Nema S. 2007. Antibody structure, instability, and formulation. J Pharm Sci-Us. 96:1–26. doi: 10.1002/jps.20727. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee R, Adhikary L, Khedkar A, Iyer H. 2010. Probing deamidation in therapeutic immunoglobulin gamma (IgG1) by ‘bottom-up’ mass spectrometry with electron transfer dissociation. Rapid Commun Mass Sp. 24:879–884. doi: 10.1002/rcm.4464. [DOI] [PubMed] [Google Scholar]
- 11.Ladwig PM, Barnidge DR, Willrich MAV. Mass spectrometry approaches for identification and quantitation of therapeutic monoclonal antibodies in the clinical laboratory. Clin Vaccine Immunol. 2017;24. doi: 10.1128/CVI.00545-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang ZQ, Pan H, Chen XY. 2009. Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom Rev. 28:147–176. doi: 10.1002/mas.20190. [DOI] [PubMed] [Google Scholar]
- 13.Rogstad S, Faustino A, Ruth A, Keire D, Boyne M, Park J. 2017. A retrospective evaluation of the use of mass spectrometry in FDA biologics license applications. J Am Soc Mass Spectr. 28:786–794. doi: 10.1007/s13361-016-1531-9. [DOI] [PubMed] [Google Scholar]
- 14.Beck A, Sanglier-Cianferani S, Van Dorsselaer A. 2012. Biosimilar, biobetter, and next generation antibody characterization by mass spectrometry. Anal Chem. 84:4637–4646. doi: 10.1021/ac3002885. [DOI] [PubMed] [Google Scholar]
- 15.Mazur MT, Seipert RS, Mahon D, Zhou QW, Liu T. 2012. A platform for characterizing therapeutic monoclonal antibody breakdown products by 2D chromatography and top-down mass spectrometry. Aaps J. 14:530–541. doi: 10.1208/s12248-012-9361-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beck A, Debaene F, Diemer H, Wagner-Rousset E, Colas O, Van Dorsselaer A, Cianferani S. Cutting-edge mass spectrometry characterization of originator, biosimilar and biobetter antibodies. J Mass Spectrom. 2015;50:285–297. doi: 10.1002/jms.3554. [DOI] [PubMed] [Google Scholar]
- 17.Ren D, Pipes GD, Liu DJ, Shih LY, Nichols AC, Treuheit MJ, Brems DN, Bondarenko PV. An improved trypsin digestion method minimizes digestion-induced modifications on proteins. Anal Biochem. 2009;392:12–21. doi: 10.1016/j.ab.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 18.Chait BT. 2006. Mass spectrometry: bottom-up or top-down? Science. 314:65–66. doi: 10.1126/science.1133987. [DOI] [PubMed] [Google Scholar]
- 19.Smith LM, Kelleher NL, Proteomics CTD. 2013. Proteoform: a single term describing protein complexity. Nat Methods. 10:186–187. doi: 10.1038/nmeth.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin YT, Peng Y, Lin ZQ, Chen YC, Wei LM, Hacker TA, Larsson L, Ge Y. Comprehensive analysis of tropomyosin isoforms in skeletal muscles by top-down proteomics. J Muscle Res Cell M. 2016;37:41–52. doi: 10.1007/s10974-016-9443-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao Y, Valeja SG, Rouse JC, Hendrickson CL, Marshall AG. 2013. Top-down structural analysis of an intact monoclonal antibody by electron capture Dissociation-Fourier Transform ion cyclotron resonance-mass spectrometry. Anal Chem. 85:4239–4246. doi: 10.1021/ac303525n. [DOI] [PubMed] [Google Scholar]
- 22.Tsybin YO, Fornelli L, Stoermer C, Luebeck M, Parra J, Nallet S, Wurm FM, Hartmer R. Structural analysis of intact monoclonal antibodies by electron transfer dissociation mass spectrometry. Anal Chem. 2011;83:8919–8927. doi: 10.1021/ac201293m. [DOI] [PubMed] [Google Scholar]
- 23.Nicolardi S, Deelder AM, Palmblad M, van der Burgt YEM. 2014. Structural analysis of an intact monoclonal antibody by online electrochemical reduction of disulfide bonds and Fourier Transform ion cyclotron resonance mass spectrometry. Anal Chem. 86:5376–5382. doi: 10.1021/ac500383c. [DOI] [PubMed] [Google Scholar]
- 24.Fornelli L, Damoc E, Thomas PM, Kelleher NL, Aizikov K, Denisov E, Makarov A, Tsybin YO. Analysis of intact monoclonal antibody IgG1 by electron transfer dissociation orbitrap FTMS. Mol Cell Proteomics. 2012;11:1758–1767. doi: 10.1074/mcp.M112.019620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO. 2014. Middle-down analysis of monoclonal antibodies with electron transfer dissociation orbitrap fourier transform mass spectrometry. Anal Chem. 86:3005–3012. doi: 10.1021/ac4036857. [DOI] [PubMed] [Google Scholar]
- 26.Cotham VC, Brodbelt JS. 2016. Characterization of therapeutic monoclonal antibodies at the subunit-level using middle-down 193 nm ultraviolet photodissociation. Anal Chem. 88:4004–4013. doi: 10.1021/acs.analchem.6b00302. [DOI] [PubMed] [Google Scholar]
- 27.Fornelli L, Srzentic K, Huguet R, Mullen C, Sharma S, Zabrouskov V, Fellers RT, Durbin KR, Compton PD, Kelleher NL. Accurate sequence analysis of a monoclonal antibody by top-down and middle-down orbitrap mass spectrometry applying multiple ion activation techniques. Anal Chem. 2018;90:8421–8429. doi: 10.1021/acs.analchem.8b00984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin YT, Wei LM, Cai WX, Lin ZQ, Wu ZJ, Peng Y, Kohmoto T, Moss RL, Ge Y. Complete characterization of cardiac myosin heavy chain (223 kDa) enabled by size-exclusion chromatography and middle-down mass spectrometry. Anal Chem. 2017;89:4922–4930. doi: 10.1021/acs.analchem.7b00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cannon J, Lohnes K, Wynne C, Wang Y, Edwards N, Fenselau C. 2010. High-throughput middle-down analysis using an orbitrap. J Proteome Res. 9:3886–3890. doi: 10.1021/pr1000994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siuti N, Kelleher NL. 2007. Decoding protein modifications using top-down mass spectrometry. Nat Methods. 4:817–821. doi: 10.1038/nmeth1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He LD, Anderson LC, Barnidge DR, Murray DL, Hendrickson CL, Marshall AG. 2017. Analysis of monoclonal antibodies in human serum as a model for clinical monoclonal gammopathy by use of 21 Tesla FT-ICR top-down and middle-down MS/MS (vol 28, pg 827, 2017). J Am Soc Mass Spectr. 28:839. doi: 10.1007/s13361-017-1652-9. [DOI] [PubMed] [Google Scholar]
- 32.Kaltashov IA, Bobst CE, Abzalimov RR, Wang GB, Baykal B, Wang SH. 2012. Advances and challenges in analytical characterization of biotechnology products: mass spectrometry-based approaches to study properties and behavior of protein therapeutics. Biotechnol Adv. 30:210–222. doi: 10.1016/j.biotechadv.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tran BQ, Barton C, Feng JH, Sandjong A, Yoon SH, Awasthi S, Liang T, Khan MM, Kilgour DPA, Goodlett DR, et al. Comprehensive glycosylation profiling of IgG and IgG-fusion proteins by top-down MS with multiple fragmentation techniques. J Proteomics. 2016;134:93–101. doi: 10.1016/j.jprot.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, Cui WD, Gross ML. 2014. Mass spectrometry for the biophysical characterization of therapeutic monoclonal antibodies. Febs Lett. 588:308–317. doi: 10.1016/j.febslet.2013.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mann M, Kelleher NL. 2008. Precision proteomics: the case for high resolution and high mass accuracy. Proc Natl Acad Sci U S A. 105:18132–18138. doi: 10.1073/pnas.0800788105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S. 2013. Characterization of therapeutic antibodies and related products. Anal Chem. 85:715–736. doi: 10.1021/ac3032355. [DOI] [PubMed] [Google Scholar]
- 37.Valeja SG, Kaiser NK, Xian F, Hendrickson CL, Rouse JC, Marshall AG. 2011. Unit mass baseline resolution for an intact 148 kDa therapeutic monoclonal antibody by Fourier Transform ion cyclotron resonance mass spectrometry. Anal Chem. 83:8391–8395. doi: 10.1021/ac202429c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai WX, Guner H, Gregorich ZR, Chen AJ, Ayaz-Guner S, Peng Y, Valeja SG, Liu XW, Ge Y. MASH Suite Pro: A comprehensive software tool for top-down proteomics. Mol Cell Proteomics. 2016;15:703–714. doi: 10.1074/mcp.O115.054387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson KA, Paisley-Flango K, Tangarone BS, Porter TJ, Rouse JC. 2007. Cation exchange-HPLC and mass spectrometry reveal C-terminal amidation of an IgG1 heavy chain. Anal Biochem. 360:75–83. doi: 10.1016/j.ab.2006.10.012. [DOI] [PubMed] [Google Scholar]
- 40.Luo J, Zhang J, Ren DY, Tsai WL, Li F, Amanullah A, Hudson T. Probing of C-terminal lysine variation in a recombinant monoclonal antibody production using Chinese hamster ovary cells with chemically defined media. Biotechnol Bioeng. 2012;109:2306–2315. doi: 10.1002/bit.24510. [DOI] [PubMed] [Google Scholar]
- 41.Kaschak T, Boyd D, Lu F, Derfus G, Kluck B, Nogal B, Emery C, Summers C, Zheng K, Bayer R, et al. Characterization of the basic charge variants of a human IgG1 Effect of copper concentration in cell culture media. Mabs-Austin. 2011;3:577–583. doi: 10.4161/mabs.3.6.17959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsubaki M, Terashima I, Kamata K, Koga A. 2013. C-terminal modification of monoclonal antibody drugs: amidated species as a general product-related substance. Int J Biol Macromol. 52:139–147. doi: 10.1016/j.ijbiomac.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 43.Dada OO, Jaya N, Valliere-Douglass J, Salas-Solano O. 2015. Characterization of acidic and basic variants of IgG1 therapeutic monoclonal antibodies based on non-denaturing IEF fractionation. Electrophoresis. 36:2695–2702. doi: 10.1002/elps.201500219. [DOI] [PubMed] [Google Scholar]
- 44.Campuzano IDG, Netirojjanakul C, Nshanian M, Lippens JL, Kilgour DPA, Van Orden S, Loo JA. Native-MS analysis of monoclonal antibody conjugates by Fourier Transform ion cyclotron resonance mass spectrometry. Anal Chem. 2018;90:745–751. doi: 10.1021/acs.analchem.7b03021. [DOI] [PubMed] [Google Scholar]
- 45.Xu W, Jimenez RB, Mowery R, Luo H, Cao M, Agarwal N, Ramos I, Wang X, Wang J. A Quadrupole Dalton-based multi-attribute method for product characterization, process development, and quality control of therapeutic proteins. Mabs-Austin. 2017;9:1186–1196. doi: 10.1080/19420862.2017.1364326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rogers RS, Abernathy M, Richardson DD, Rouse JC, Sperry JB, Swann P, Wypych J, Yu C, Zang L, Deshpande R. A View on the Importance of “Multi-Attribute Method” for measuring purity of biopharmaceuticals and improving overall control strategy. Aaps J. 2017;20:7. doi: 10.1208/s12248-017-0168-3. [DOI] [PubMed] [Google Scholar]
- 47.Rogers RS, Nightlinger NS, Livingston B, Campbell P, Bailey R, Balland A. 2015. Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. Mabs-Austin. 7:881–890. doi: 10.1080/19420862.2015.1069454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.