

ABSTRACT
Circulating T-cells that have passed thymic selection generally bear T-cell receptors (TCRs) with sub-optimal affinity for cancer-associated antigens, resulting in a limited ability to detect and eliminate tumor cells. Engineering TCRs to increase their affinity for cancer targets is a promising strategy for generating T-cells with enhanced potency for adoptive immunotherapy in cancer patients. However, this manipulation also risks generating cross-reactivity to antigens expressed by normal tissue, with potentially serious consequences. Testing in animal models might not detect such cross-reactivity due to species differences in the antigenic repertoire. To mitigate the risk of off-target toxicities in future clinical trials, we therefore developed an extensive in vitro testing strategy. This approach involved systematic substitution at each position of the antigenic peptide sequence using all natural amino acids to generate a profile of peptide specificity (“X-scan”). The likelihood of off-target reactivity was investigated by searching the human proteome for sequences matching this profile, and testing against a panel of primary cell lines. Starting from a diverse panel of parental TCRs, we engineered several affinity-enhanced TCRs specific for the cancer-testis antigen MAGE-A10. Two of these TCRs had affinities and specificities which appeared to be equally optimal when tested in conventional biochemical and cellular assays. The X-scan method, however, permitted us to select the most specific and potent candidate for further pre-clinical and clinical testing.
KEYWORDS: MAGE-A10, adoptive T-cell therapy, T-cell receptor, cross-reactivity, off-target, peptide scan
Introduction
Adoptive transfer of effector T-cells recognizing tumor antigens is a promising strategy for cancer immunotherapy in multiple indications.1,2 Cells expanded in vitro from naturally occurring tumor-reactive T-cells have shown promise in the clinic,3 but their broader application has been relatively limited. These cells have undergone thymic selection, removing those with T-cell receptors (TCRs) that bind strongly to self-antigens. Because the majority of tumor antigens are self-like, they may be recognized by the circulating repertoire of T-cells significantly less strongly than their pathogen-specific counterparts.4–6 The therapeutic efficacy of T-cells bearing such TCRs can be further reduced by low levels of peptide-major histocompatibility antigen complexes (pHLA) on the surface of some tumor cells.7
Engineering tumor-specific TCRs to enhance affinity to the higher end of the physiological affinity range8-10 can lead to improved tumor cell recognition and killing in vitro and in vivo.10–12 Moreover, patients receiving T-cells bearing an affinity-optimized tumor-specific TCR show evidence of improved clinical efficacy compared to those treated with T-cells expressing a native TCR.13 Clinical success with adoptive transfer of naturally occurring tumor-infiltrating lymphocytes has been largely limited to malignant melanoma,3 whereas affinity-enhanced TCRs have demonstrated objective clinical responses in a wider range of indications.14
The specificity by which T-cells recognize their targets has the potential to avoid the general toxicities observed with conventional chemotherapies. However, prevention and management of immune-mediated toxicities represents a significant challenge to the clinical success of adoptive T-cell therapies.12,13,15–19 A number of mechanisms have been implicated.9,17 One is “on-target” toxicity, where T-cells respond specifically to their intended pHLA target but the target is expressed by not only tumor but also normal healthy tissues.10 The risk of this form of toxicity can be reduced by extensive validation of expression patterns and selection of targets that are cancer-associated but absent or minimally expressed in normal adult tissues. Many cancer-testis antigens fit these criteria and therefore have attracted interest as cancer biomarkers and targets for adoptive T-cell therapies,18,19 in particular members of the well-characterized melanoma antigen gene (MAGE) family.20
In one pair of clinical studies, two patients were treated using adoptive transfer of T-cells expressing a TCR with enhanced affinity for an HLA-A*01-restricted peptide originating from MAGE-A3. Pre-clinical in vitro characterization of the specificity and efficacy of this TCR did not highlight any safety concerns. Unexpectedly, both patients suffered fatal acute cardiac toxicity.21 Apart from male germline cells, the MAGE-A3/HLA-A*01 epitope is restricted to tumor cells and absent from cardiac tissue. Moreover, in silico BLAST searches for peptides with high sequence similarity to the target peptide did not identify any candidate mimotope peptides. An alternative strategy was, therefore, employed. First, the main peptide positions recognized by the affinity-enhanced TCR were identified using a glycine/alanine amino acid scan.12 From the results, a degenerate motif was constructed and used for a directed in silico search using the ScanProsite tool. A peptide from the muscle protein Titin was identified as the off-target candidate. Its responsibility for the observed cardiac toxicity was confirmed by cytolytic activity of the MAGE-A3 TCR-transduced T-cells towards beating Titin-positive iPS-derived cardiac myocytes.12,22 By contrast, none of 38 cardiac-derived normal primary cell lines grown in 2D culture were recognized by T-cells expressing the affinity-enhanced MAGE-A3 TCR. No response was observed to mouse Titin peptide, demonstrating that improved tools were required for pre-clinical toxicity testing in addition to cell lines and transgenic mouse models. Herein, we describe the generation and systematic testing of affinity-enhanced TCRs recognizing an HLA-A*02 restricted epitope from the MAGE-A10 cancer testis antigen. We also demonstrate the ability of the peptide X-scan assay to distinguish between two affinity-optimized TCRs, which otherwise appear similarly potent and specific.
Results
Generation of multiple parental TCRs recognizing the HLA-A*0201-restricted MAGE-A10 peptide GLYDGMEHL254–262 epitope with a variety of sequence characteristics, binding affinities and functional performances
Twenty-one TCRs were characterized for recognition of the HLA-A*0201-restricted MAGE-A10 peptide GLYDGMEHL254–262 (hereafter MAGE-A10254–262) epitope. Surface plasmon resonance (SPR) showed that their affinities ranged from 1 to 50 µM, in line with values typically reported for effective engagement of pHLA.6,23 Ten TCRs, encompassing a range of affinities and TCR chain pairings, were selected for cloning into a lentiviral vector. Incubation of TCR-transduced primary human T-cells with T2 target cells pulsed with varying concentrations of MAGE-A10254–262 peptide indicated that this subset of parental TCRs recognize the MAGE-A10254–262 epitope with a range of sensitivities as determined by the numbers of IFN-γ releasing cells. Eight of the 10 parental TCRs were also screened for recognition of natively processed antigen. To this end, well-characterized MAGE-A10-positive and -negative cell lines and primary cells were used as targets (Figure 1). All parental TCRs demonstrated recognition of at least one MAGE-A10+ line, although several also exhibited evidence of cross-reactivity by recognizing MAGE-A10− lines.
Figure 1.
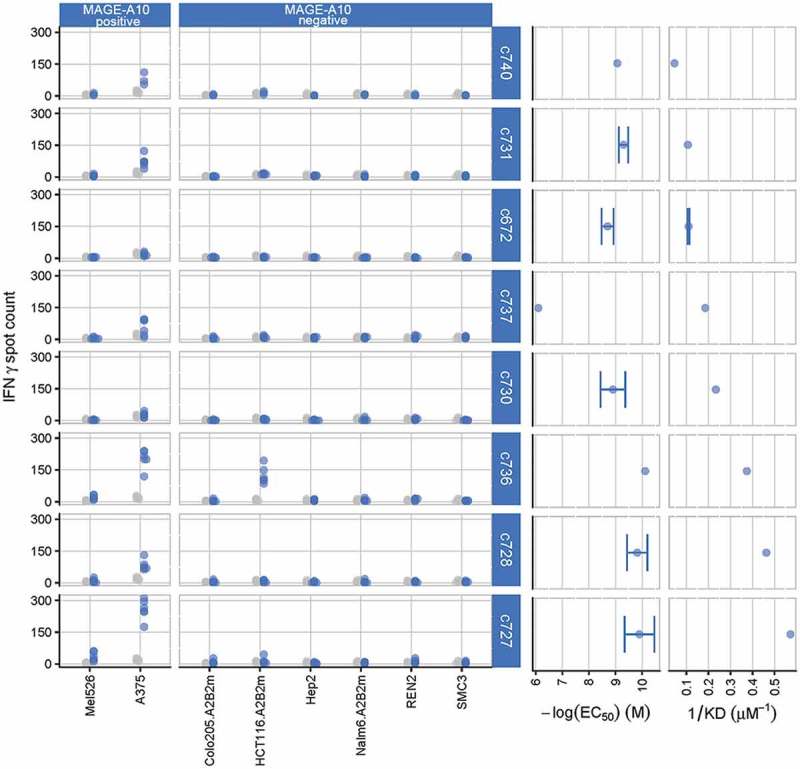
Characterization of eight parental TCRs recognizing HLA-A*0201 presenting the MAGE-A10254-262 peptide, using cellular and biochemical assays.
The response of eight of the ten parental TCRs to MAGE-A10+ and MAGE-A10− target cell lines is shown in the two left-hand panels, ordered from lowest (c740) to highest (c727) affinity. Blue points represent numbers of IFN-γ spot forming units (SFU) counted in triplicate wells for two T-cell donors, with TCR-transduced T-cells; gray points represent the responses of non-transduced T-cells (consistently < 20 SFU). The two right-hand panels illustrate the strength of interaction between the parental TCRs and the HLA-A*0201-restricted MAGE-A10 peptide GLYDGMEHL254-262, in both cell surface expression and soluble formats. EC50 values, indicating the sensitivity of TCR-engineered T-cells to antigen, were assessed by counting the numbers of T-cells releasing IFN-γ in response to T2 cells pulsed with a titration of GLYDGMEHL254-262 peptide (ELISpot assay). Points represent mean values of -log(EC50) ± SEM (error bars); control measurements were carried out in the absence of peptide. Affinities (KD values) were calculated using equilibrium analysis of SPR measurements made using a Biacore-3000TM system. Points represent mean values of KD−1 ± SEM (shown by error bars where replicate assays were performed).
Enhancing affinity for MAGE-A10254–262 through engineering parental TCRs improves sensitivity of TCR-transduced T-cells for target pHLA but can also reduce specificity
To maximize the chances of successful affinity enhancement, we selected three parental TCRs, c672, c728 and c740, which appeared to best represent the TCR α- and β-chain diversity and affinity range of the parent panel (Figure 1 and S1). TCR c672 contains a single amino acid change from the germline TRAV sequence. This mutation was reverted to the germline sequence to create c753, which then acted as the template for affinity enhancement. The CDR regions of the parental α- and β-chains were mutated, and the resulting TCRs were tested in SPR and IFN-γ enzyme-linked immunospot (ELISpot) assays for affinity, as expressed by the dissociation constant, KD, and specificity. SPR binding curves and affinity ratios of parental and mutated daughter TCRs showed that mutations conferred varying degrees of affinity enhancement for MAGE-A10254-262/HLA-A*02 (Figure 2). We achieved up to > 15-fold increases in target binding (c799, KD = 0.14 µM compared to parent c728, KD = 2.2 µM). Enhanced affinity led to greater functional potency. T-cells transduced with affinity-enhanced daughter TCRs responded to target cells pulsed at lower concentrations of peptide than their parent TCR. Transduced T-cells were further screened against a panel of MAGE-A10+ and MAGE-A10− target cell lines and primary cells (Figure 3).
Figure 2.
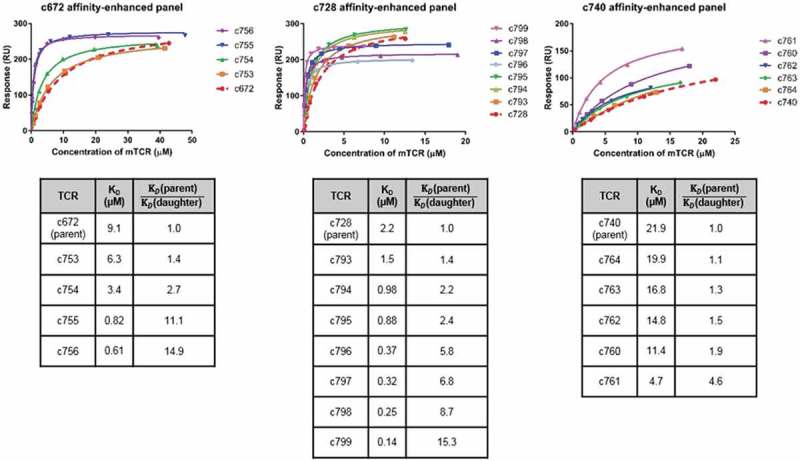
Biochemical characterization of affinity-enhanced TCR panels arising from the parental TCRs c672, c728 and c740.
Comparison of SPR data for affinity-enhanced TCR panels arising from three parental TCRs for which raw SPR data is shown in Figure S1. Note that TCR c753 is a version of c672 with the α-chain reverted fully to germline sequence, and that c753 therefore acted as the template for the daughter mutants c754-c756. Binding affinity analysis of the MAGE-A10254-262-specific mutant TCR panels is illustrated in the upper panel, which shows the binding curve fits. KD values shown in the bottom panel were obtained by equilibrium binding or kinetic analysis. From the c728 panel, c796-c799 were tested in a separate SPR experiment to c728 and c793-c795, leading to the difference in maximum binding levels.
Figure 3.
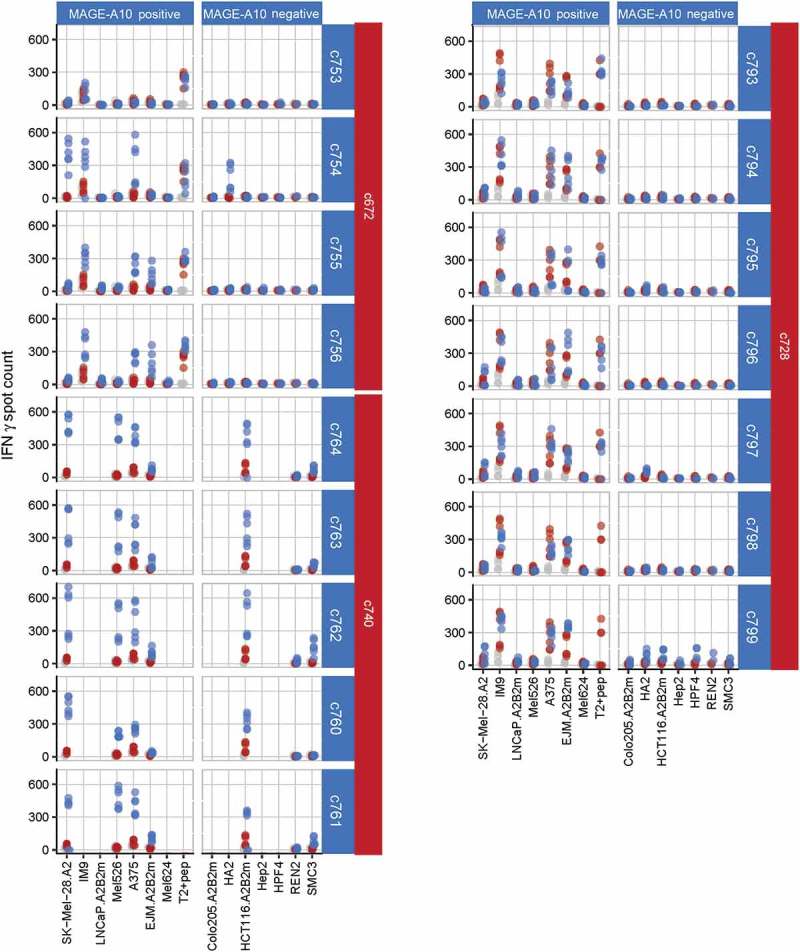
Characterization of three affinity-enhanced TCR panels arising from the c672, c740 and c728 parental TCRs, using cellular assays.
The response of each affinity-enhanced TCR panel to MAGE-A10+ and MAGE-A10− target cell lines is shown, with daughter TCRs sorted in order of increasing affinity (top to bottom) for each parent. MAGE-A10+ cell lines are shown in order of increasing MAGE-A10 expression (left to right), as assessed by RT-qPCR. Red points represent average numbers of IFN-γ spot forming units (SFU) for T-cells obtained from two donors and transduced with each parental TCR, each measured in triplicate. Blue points represent the responses of T-cells transduced with the daughter TCR, while grey points represent the responses of non-transduced cells (consistently < 50 SFU).
T-cells transduced with enhanced affinity daughter TCRs demonstrated elevated responses to MAGE-A10+ cells and/or responded to a greater number of MAGE-A10+ cell lines than T-cells transduced with their parental TCR. For example, c756-transduced cells recognized three more MAGE-A10+ cell lines than c672-transduced cells. TCRs c799 and all TCRs of the c740 panel which conferred increased cross-reactivity towards MAGE-A10-negative target cells were excluded from further testing (members of the c740 panel were also not tested against cell lines HA2 and HPF4 owing to significant responses being observed to SMC3 and REN2 primary cells).
Further analysis of daughter TCRs permits selection of two final candidates, which are functionally indistinguishable
Five TCRs were selected on the basis of strong efficacy and specificity profiles (Figure 2, 3) for more rigorous in vitro characterization. Each TCR was tested for cross-reactivity in IFN-γ release assays against an extensive panel of MAGE-A10− tumor cell lines and primary cells (Figure 4A). None demonstrated significant activation of T-cells. This observation from primary cells suggested that on-target/off-tumor activation of TCR-transduced T-cells against normal tissues was unlikely, in agreement with the well-characterized restricted expression of MAGE-A10 to male germline cells, placenta and certain cancers.24,25
Figure 4.
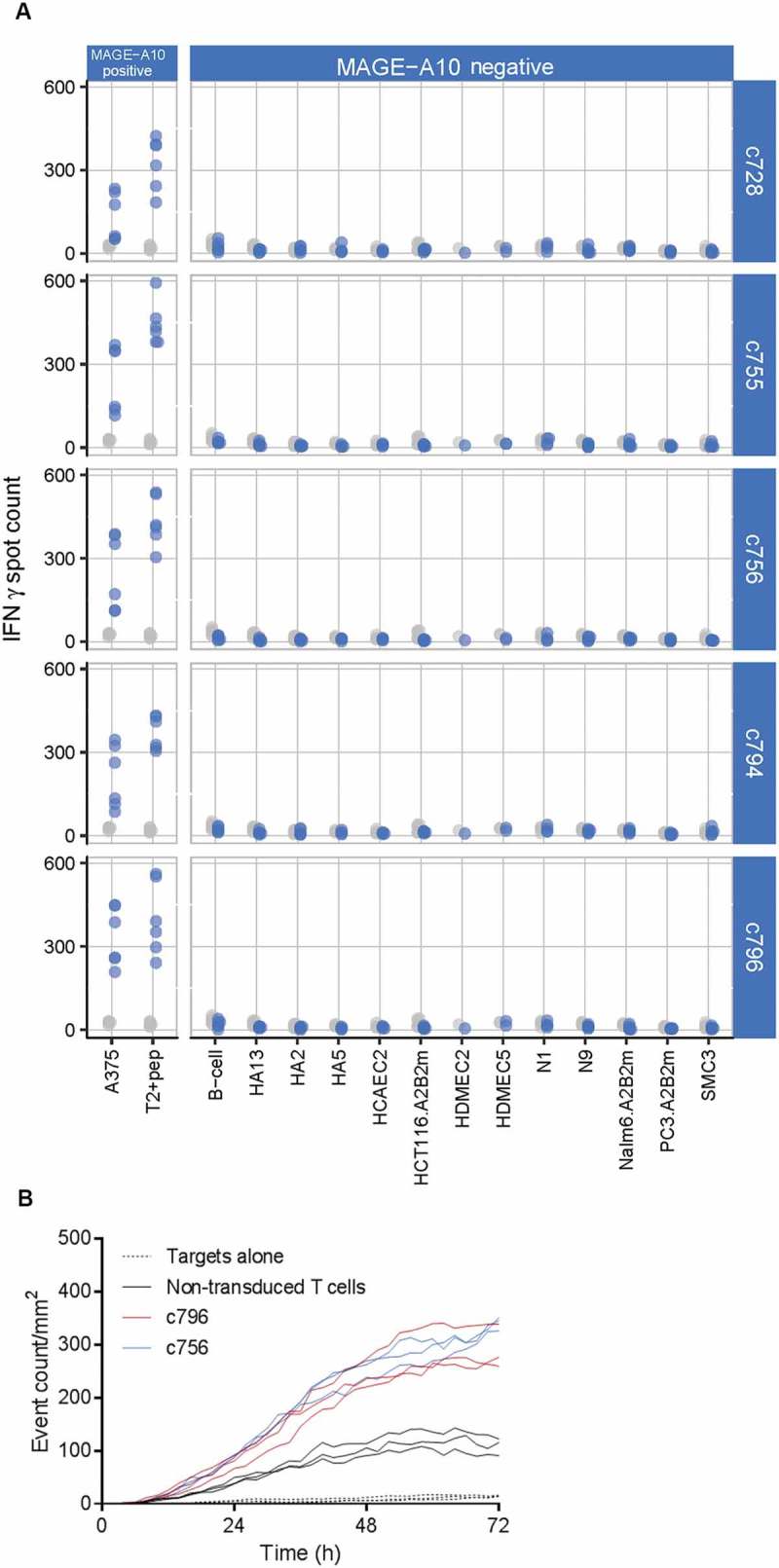
Extended specificity and functional cytotoxicity testing of final TCR candidates arising from the c672 and c728 parental TCRs.
(A) Extended tumor cell line and primary cell screen of five candidate TCRs selected on the basis of initial characterization (see Figure 3). Blue points represent numbers of IFN-γ spot forming units (SFU) counted in triplicate wells in two donors, for TCR-transduced T-cells; grey points represent the responses of non-transduced T-cells (consistently < 50 SFU). MAGE-A10− cell lines did not induce T-cell responses above the background level. (B) Potency testing of two final candidate TCRs c756 and c796 using a real-time cytotoxicity assay. Functional cytotoxicity was confirmed, and the magnitude and kinetics of the killing of MAGE-A10+ HLA-A*0201-transduced SK-Mel-28 cells were similar.
MAGE-A10+ cells stimulated high numbers of IFN-γ releasing T-cells, with the strongest responses from T-cells transduced with the highest affinity daughters from each of the c672 and c728 panels. These clones, c756 and c796, were further assessed to confirm their cytotoxic functionality by measuring killing of MAGE-A10+ target cells in real-time (Figure 4B). In these assays, both affinity-enhanced TCRs demonstrated similarly potency for target cell recognition and killing and highly similar affinities and recognition of peptide-pulsed targets: KDs of 0.37 and 0.61 µM (Figure 2), and EC50s of 10−9.5 and 10−9.3 M for c796 and c756, respectively.
A single amino acid substitution X-scan of the MAGE-A10254-262 peptide differentiates between engineered TCRs based on the potential for cross-reactivity with the normal human proteome
To determine which of the two TCRs, c756 or c796, would be the best candidate for clinical testing, we characterized their specificity and potential repertoire of recognized peptides in detail. We synthesized a set of 172 peptides in which each residue of the MAGE-A10254-262 target peptide was sequentially mutated to all 19 other naturally occurring amino acids. T2 cells were incubated with each of the substituted peptides at a concentration corresponding to the EC90 of response to the index peptide, i.e. 10−7.5 M for both c756 and c796. These cells were then used as targets in an ELISpot assay for T-cells transduced with either TCR. We considered a substitution as tolerated if the number of responding T-cells exceeded a threshold of 10% relative to the index peptide. Figure 5 summarizes the results (details are shown in Figure S2): Pie areas (Figure 5A) are proportional to the average number of tolerated substitutions at a given position, and segment areas are proportional to the residues with shared physicochemical side chain properties. For the latter, we grouped amino acids based on a substitution matrix for HLA class I binding that might have general relevance for peptide ligands.26 We differed from the scheme by clustering cysteine (monomeric Cys-SH) together with asparagine, glutamine, serine and threonine, rather than with aspartate and glutamate, according to preliminary analysis of X-scan data. The color scheme is based on Lesk,27 with modifications according to Kim et al.26
Figure 5.
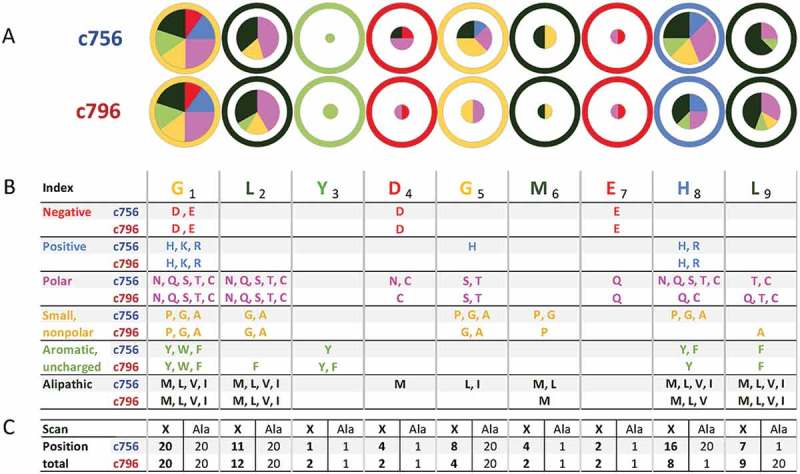
Peptide X-scan analysis to assess specificity of the c756 and c796 MAGE-A10-specific TCRs.
Number and diversity of amino acids tolerated at various peptide positions by the final candidate TCRs. This example shows search motifs for the 10% threshold (side chains in whose presence IFN-γ spot counts were at least 10% of that triggered by the index peptide).(A) Pie areas are proportional to the number of tolerated amino acids, including the index. Pie segments represent a breakdown of tolerated amino acids into groups with similar physico-chemical properties. Individual amino acids in group colors are listed in panel B, classification and color scheme are explained in the main text. The area within the outer circle corresponds to all 20 amino acids being tolerated at a 10% threshold; the color of the circle indicates the group of the index peptide. (B) Comparison of the X-scan search motifs for c756 and c796 at a response threshold of 10%. (C) Total number of tolerated residues at each peptide position, including the index. The number of residues that would be identified as potentially tolerated by an alanine scan is shown for comparison. The product of the numbers of recognized residues at each position is the motif size, or number of peptide sequences generated by all possible permutations of amino acids tolerated by a TCR.
At most peptide positions, c756 showed higher average responses and tolerated a wider range of side chain properties, embodied by more groups of similar amino acids, than c796. TCR c796 was tolerant of a greater number and diversity of amino acids at anchor positions 2 and 9. Some amino acids enhanced T-cell responses when substituted for the index position 2 residue leucine: glutamine and threonine by > 50% for c756, valine by ~ 30% for both TCRs (Figure S2). However, there was no correlation between the predicted affinity of peptides for HLA-A2 and pHLA recognition by either TCR, for any of the positions including anchors (data not shown). Tolerated residues were combined into a degenerate peptide recognition consensus motif (Figure 5B). This motif was used in a directed in silico search of the human proteome for peptides with the potential to be recognized by each of the candidate TCRs.
There is no proven way at present to extrapolate from response thresholds in vitro to clinically relevant tissue damage. Therefore, we generated recognition motifs for a range of response thresholds, and performed database searches for each of these (Figure 6A). The numbers of potentially recognized targets were fewer for TCR c796 than c756 at any response threshold, even considering experimental error in replicates of the ELISpot assay. This was largely due to the less degenerate recognition motif of c796. In addition, the c796 motif matched fewer peptides than c756, even taking into account the probability of the motifs matching a random nonameric sequence, p(Match) (Figure 6B). This observation indicates that the c796 motif space represents less common amino acid sequences in the human proteome, and further supports the finding that c796 appears to exhibit a lower chance of displaying off-target reactivity. Additionally, it demonstrates that performing proteome searches rather than just comparing the motif space sizes can reveal subtle differences in the risk for cross-reactivity.
Figure 6.
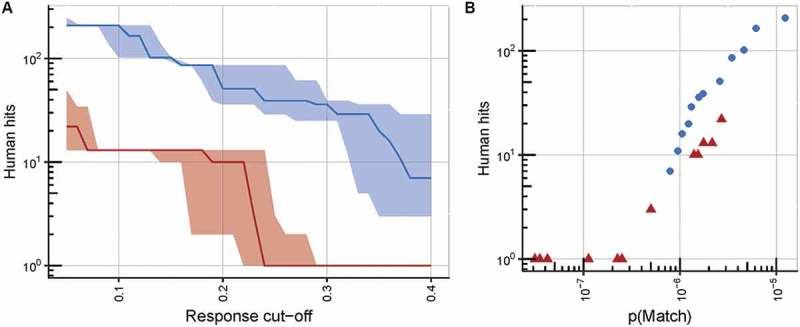
Further analysis of peptide X-scan data confirming the superior specificity of the c796 MAGE-A10-specific TCR over c756.
(A) An illustration of the superior specificity of TCR c796 over c756, at any response cut-off value (defined as the fraction of the response to the index peptide at its EC90 concentration (10−7.5 M) at which a substitution is considered to be tolerated by the TCR). Numbers of unique human peptide matches, which conform to the consensus motif based on peptide X-scan data, are plotted against the response cut-off value, with solid lines representing the median number of matches derived from data from triplicate wells, and shaded areas representing the range between the lowest and highest numbers of matches from these wells. TCR c796 is shown in red, c756 in blue. (B) Motifs derived for TCR c796 identify fewer human nonamers than those for c756, at any value of p(Match) (defined as the naïve probability of identifying matching sequences within the proteome). Points represent numbers of unique human peptide matches to which the two final TCR candidates may show cross-reactivity, plotted against p(Match) values; c796 is shown as red triangles, and c756 as blue circles.
T-cells transduced with the c756 affinity-enhanced TCR have the potential to cross-react to significantly higher numbers of off-target human peptides than c796. To confirm whether c796 is therefore the most suitable candidate for progression to pre-clinical testing, we synthesized the eight unique human peptides whose sequences matched the predicted consensus motif for c796 at a 10% threshold. HLA-A*0201-transduced T2 cells pulsed with these peptides elicited no detectable IFN-γ release by c796 transduced T-cells (manuscript in preparation).
Discussion
Engineering TCRs for enhanced affinity can generate unpredicted cross-reactivity
Although T-cells bearing affinity-enhanced TCRs have demonstrated potent anti-tumor activity both in vitro and in vivo, this strategy is not without risk.12,16,28 On-target cross-reactivity is one potential hazard common to all adoptive T-cell therapies.29 It can be mitigated by thorough investigation of normal tissues for expression of the intended target. Cancer-testis antigens often have highly restricted expression, such as that of MAGE-A10, in the immune-privileged sites of the placenta and sperm cells/testes.18,20,24 Where the same target peptide epitope is found in several closely related proteins, the expression profile of each of these needs to be considered to avoid on-target cross-reactivity. This was shown by a report of fatal on-target toxicity arising from recognition of a peptide found not only in MAGE-A3, but also MAGE-A9 and MAGE-A12.28 Such toxicity was not apparent in the HLA-transgenic mouse strain used to isolate the TCR. Of note, the MAGE-A10254-262 peptide is from a distinct and relatively divergent section of the protein. In particular, the corresponding sequence of MAGE-A12 is highly dissimilar to MAGE-A10254-262. These differences mean that peptides from other MAGE-family members should be not be recognized by our TCR, or even be presented efficiently by HLA-A2 (Table S2). Nevertheless, we subsequently assessed the reactivity of c796 TCR-transduced T-cells to peptides from homologous MAGE family members and found nothing of concern for patient safety (manuscript in preparation).
Another toxicity risk is posed by off-target TCR cross-reactivity. The underlying molecular mimicry mechanisms are incompletely understood, making it challenging to predict and test pre-clinically. One possibility is TCR recognition of non-cancer-restricted peptide epitopes sharing a degree of similarity with the intended target.30 This molecular mimicry was found to underlie unexpected, fatal toxicities following adoptive transfer of T-cells bearing an affinity-enhanced TCR towards an HLA-A*01-restricted peptide from MAGE-A3.21 This TCR was later found to recognize an HLA-A*01-restricted peptide with unrelated sequence, from Titin, which is expressed in cardiac and skeletal muscle.12 A level of degeneracy in TCR antigen recognition is established via positive thymic selection for non-peptide contacts31,32 and is essential to ensure that an individual’s circulating T-cell repertoire is capable of responding to the highly diverse range of pathogenic peptide epitopes they might encounter.33–35 In an adoptive T-cell therapy setting, this plasticity also means that an apparently cancer-specific TCR might also unexpectedly bind “off-target” to non-cancer-restricted epitopes, leading to clinical toxicity. This risk increases when TCRs are affinity-enhanced in vitro, without the safety mechanism of negative thymic selection.8,9,16 The experience with the MAGE-A3/Titin cross-reactive TCR12 has highlighted the usefulness of amino acid scanning for identifying potential cross-reactivity and clinical toxicity in an adoptive therapy setting.
Here, we have applied an extension of this approach and integrated it into our workflow for generating enhanced affinity TCRs for adoptive T-cell therapy (Figure 7). First, we generated a significant number of mutated daughter TCRs from diverse parents recognizing the MAGE-A10254-262 peptide presented by HLA-A*0201. We reasoned that, since efficacy of parental TCRs can be very variable, a larger number would increase our chances of finding a potent and highly specific candidate. Parental TCRs demonstrated a range of responses in biochemical and cellular assays, with little to no correlation between KD and EC50 values (determined by SPR and IFN-γ release, respectively), or between EC50 values and reactivity to antigen-positive cell lines (Figure 1). This result is likely due to differing modes of engagement between the diverse parental TCRs and their pHLA target in the different assays: the SPR assay measures binding of a soluble reagent, whereas in cellular assays, TCR and pHLA bind and signal in a physiologically relevant manner, membrane bound with other co-factors present. Although the relationship between biochemical affinity (SPR) and functional avidity (EC50) is complex, we and others have found an affinity of range of KD ~ 1–10 μM to be optimal for signaling of engineered TCRs while avoiding non-specific reactivity8–10,12 (Figures 2 and 3). Importantly, the reproducible and precise nature of SPR makes it more capable than cellular assays in differentiating between mutant TCRs, provided they originate from the same parent (Figures 2 and 3). Because these mutants share most of the sequence of the parental CDR loops, it is likely that they also preserve the binding geometry of the parent.
Figure 7.
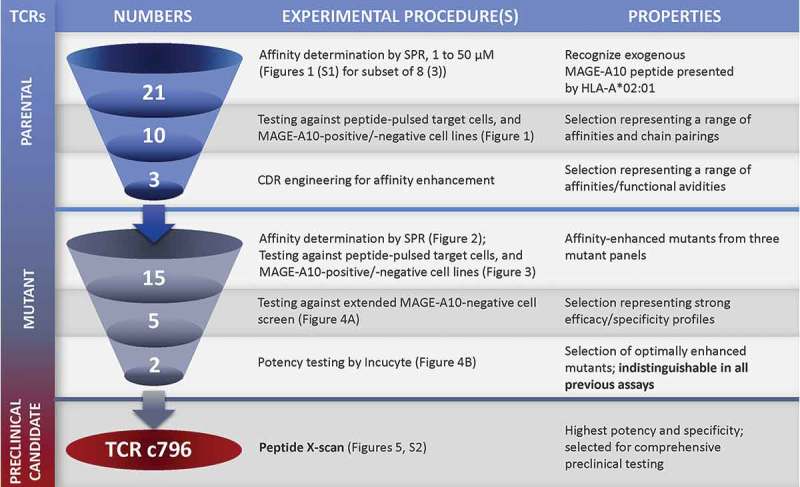
Schematic overview of the workflow for selecting an HLA-A*0201 MAGE-A10254-262-specific TCR candidate to progress to comprehensive pre-clinical testing.
The number of TCRs under investigation at each stage and the types of experimental procedures involved in progressing candidates from one stage to the next are shown. This resulted in two mutant TCRs with desirable functional properties that were indistinguishable by previously described assays. The X-scan approach identified c796 as the TCR with the highest potency and specificity.
A small subset of enhanced affinity TCR candidates demonstrated an optimal balance between functional potency and specificity when tested against a sizable panel of cell lines and primary cells (Figures 3, 4). From these TCRs, two that performed equally well in potency assays were selected. We reasoned that subtle differences might exist in their peptide recognition motifs. To define and compare these we employed the X-scan, whereby each residue of the target peptide is mutated to all possible amino acids in turn. Compared to the glycine/alanine scan,12, 16 we found that this approach resulted in significantly enhanced sensitivity and specificity for detection of tolerated substitutions. Alanine substitutions were a poor predictor of the biochemical properties and number of other tolerated amino acids, in some cases substantially overestimating the latter. On the other hand, the consensus motif for the 10 % threshold included several residues, but not alanine, at 5 out of 9 positions for both TCRs (positions 3, 4, 6, and 7 for both, and one each at positions 8 and 9). In these cases, an alanine scan would have failed to detect the corresponding peptides had they existed in the human proteome. A similar observation was recently reported, where an alanine failed to identify a cross-reactive target that was confirmed by a complete mutational scan.36
Although the specificity of the X-scan means that, overall, a smaller number of residues are identified as tolerated than with an alanine scan (Figure 5C), their distribution over a larger number of peptide positions can result in greater combinatorial diversity. For our candidate TCR c796, the size of the X-scan motif was therefore 7 times greater than that which would have resulted from alanine substitutions alone. This resulted in a moderately larger number of proteomic hits, although these did not represent truly cross-reactive peptides when tested in cellular assays (manuscript in preparation). However, we consider the additional effort reasonable in return for increased patient safety through the ability to detect non-specific recognition in cases where it does not involve alanine.
Recognition of peptide HLA complexes by TCRs c756 and c796
A closer look at the nature of tolerated changes reveals how peptides may be recognized by the two TCRs. Charged amino acids at positions 2 and 9 completely abrogated responses, independent of the TCR (Figure 5), which is compatible with the predicted role of these positions as the primary peptide anchors, buried in hydrophobic pockets of HLA-A2. Other substitutions resulted in variable and in some cases super-agonistic responses (Figure S2), supporting the inclusion of “conserved” HLA anchors in the peptide scans. Both TCRs were most sensitive to changes at positions 3, 4 and 7. Their main differences existed at positions 5 and 8, for which c796 displayed a higher stringency of recognition (Figure 5). The number and diversity of tolerated side chains suggest that the overall specificity of TCR c756 is reliant on fewer peptide positions than that of c796. It has been reported that cross-reactivity increases when binding focuses on fewer side chains, although lower specificity can also result from other mechanisms such as conformational flexibility.37,38
There are limitations to the X-scan approach. We acknowledge that nuances of the assay setup may affect the absolute outcome for a given TCR, in terms of the consensus motif and number of unique human peptide matches obtained. However, in the present study, this was not a concern, because our explicit aim was to distinguish between two TCRs of very similar affinities and EC50s for the index peptide. The outcome of this relative comparison, the conclusion that c796 has less potential for off-target reactivity than c756, appears to be robust even when alternative conditions are considered (Figure 6).
We developed an affinity-enhanced TCR with high specificity and potency against cells expressing HLA-A*0201 and the cancer antigen MAGE-A10. Starting from 21 parental receptors, three were selected, from which we generated panels of mutants with diverse germline and CDR loop sequences. As the first step of an extensive pre-clinical testing package we employed a novel in vitro specificity screen. This allowed us to select and focus resources on an optimal candidate for further pre-clinical safety and potency assessment. Details of these tests, involving hundreds of tumor, primary and allo-MHC lines to confirm potency and specificity, will be published in a second manuscript (currently in preparation). Together they describe an experimental strategy which we believe is both economic and efficient for mitigating the risk of unexpected off-target clinical toxicities, and applicable to the selection of engineered TCRs for any target.
Materials and methods
Isolation of parental MAGE-A10254-262 TCRs and preparation of affinity-enhanced TCR mutant genes
Parental TCRs recognizing the HLA-A*0201-restricted MAGE-A10254-262 peptide GLYDGMEHL were isolated either from antigen specific T-cell clones or from proprietary naïve phage display libraries. CD8+ T-cell clones specific for MAGE-A10254-262 were generated and maintained in-house from peripheral blood mononuclear cells isolated from an HLA-A*0201-positive healthy donor. Informed consent was obtained, according to Research Ethics Committee approval numbers 13SC0227, 13SC0226, 13SW0240 and 14LO1777. To activate T-cells, the cells were stimulated several times with autologous antigen-presenting cells pulsed with the MAGE-A10254-262 peptide, initially at 1 µM and thereafter at 100 nM. These T-cells were then screened for specific activation by IFN-γ ELISpot, and specific clones were sorted for further culture. TCR α- and β-chain variable domains were isolated from T-cell cDNA by rapid amplification of cDNA ends (RACE), as previously described.39 TCR α and β chain sequences from c753, c728 and c740 were used as templates for PCR mutagenesis, to construct TCRs bearing mutations in their CDR loop regions. This resulted in three panels of affinity-enhanced TCRs (one panel from each parent), with mutations in their α- and/or β-chains as confirmed by DNA sequencing (Source Bioscience).
Protein expression and purification
Parental and mutant TCR α- and β-chains (modified to promote formation of soluble disulfide-stabilized heterodimers during refolding), β2 microglobulin (residues 1–99) and the HLA-A*0201 heavy chain (residues 1–276) were inserted into the pGMT7 expression vector using PCR cloning. These constructs were expressed separately as inclusion bodies from the BL21 (DE3) Rosetta pLysS E. coli strain, and refolded and purified as described,40–41 with minor modifications. The HLA-A*0201 heavy chain was expressed with a C-terminal BirA biotinylation tag, and biotinylated pHLA monomers prepared as described.41,42 Synthetic peptides were obtained from Peptide Protein Research Limited (> 90 % pure). Refolds were dialyzed extensively against 10 mM Tris pH8 at 4°C and purified using Poros 50HQTM 10/100 (Life Technologies) and Superdex S200 HRTM 10/300 (GE Healthcare) columns. Protein concentration was determined by measurement of OD280 using an extinction coefficient calculated from the sequence, and quality assessed by SDS-PAGE and staining with Coomassie Blue.
Surface plasmon resonance
The dissociation constants (KD) of purified TCRs for their cognate pHLA ligand were determined by SPR analysis, using a Biacore3000TM instrument and flow cells (GE Healthcare) as described.43 Biotinylated pHLA monomers were immobilized onto streptavidin-coupled CM5 sensor chips (approximately 300 response units per flow cell) and the chips blocked with biotin. All experiments included a control pHLA monomer immobilized onto flow cell 2. Binding measurements for serial dilutions of each TCR were performed at 25°C in phosphate-buffered saline (PBS) (Sigma) at a flow rate of 20 μl/min, starting from the lowest analyte concentration.
SPR binding data for all parental and most daughter mutant TCRs were processed using equilibrium binding analysis; exceptions were c796 and c799, which were processed using kinetic analysis owing to longer dissociation half-lives. pHLA-specific equilibrium binding responses were determined for each concentration using BIAevaluation software. Specific equilibrium response levels were then plotted against TCR concentration using GraphPad Prism software, and KD values were calculated by fitting to the 1:1 Langmuir binding model. For kinetic analysis, chip and reagent preparation was conducted identically to equilibrium analysis, and the experiments were performed in the same way. The kon and koff values were determined separately by fitting of the association and dissociation curves, respectively, assuming 1:1 Langmuir binding. The KD could then be calculated using BIAevaluation software as the ratio koff/kon.
Cell lines, primary cells, and effector cells
Tumor cell lines A375, HCT-116, IM9, SK-Mel-28, PC3, T2, LNCaP, and colo205 were obtained from the American Type Culture Collection (ATCC), Mel526 and Mel624 were from Thymed, and NALM6 and EJM were provided by Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ). Normal primary cell lines HEP2, HDMEC2, HDMEC5, HA2, HA13, SMC3, and REN2 were obtained from ScienCell. HPF4, HA5 and N9 were obtained from Lonza, and HCAEC2 and N1 from PromoCell. All cell lines were maintained in appropriate medium, under conditions recommended by the supplier. MAGE-A10 expression was determined by RT-qPCR analysis, and HLA class I status was assessed by PCR-SSOP typing (ProImmune). Where necessary, cell lines were lentivirally transduced with HLA-A*0201. HLA-A*0201 levels were confirmed by flow cytometry or T-cell activation after peptide pulsing.
Quantitative real-time PCR
Total RNA was extracted using the RNeasy Mini kit (Qiagen, Manchester, UK) and cDNA prepared using the qScript cDNA SuperMix (Quanta Biosciences, Beverly, MA, USA) according to the manufacturer’s protocol. Subsequent quantitative PCR (qPCR) was carried out on the QuantStudio7 Real-time PCR system (ThermoScientific, Loughborough, UK) using the QuantiTect Probe and QuantiTect Multiplex Probe PCR kits (Qiagen, Manchester, UK).
Gene expression was quantified by comparing the CT value of the test samples to a standard curve with known number of copies (plasmid). Data are represented following normalization to the average of a high (RPL32) and low (HPRT1) expression housekeeping gene. The combination was chosen from 38 housekeeping genes because it generated the most stable results in a panel of 95 different samples, including immune cells. Primer and TaqMan sequences can be found in Table S1.
Cellular assays for potency and safety
The activity of TCRs in a cellular context was assessed by the production of IFN-γ from TCR-modified effector T-cells in the presence of target antigen, as measured by ELISpot assay according to the manufacturer’s instructions (BD BioSciences). Human peripheral blood mononuclear cells were isolated from fresh venous blood from healthy donors to prepare effector cells, and TCR modified T-cells were prepared by lentiviral mediated transduction as previously described.12,44 T-cell transduction efficiency was assessed by flow cytometry using antibodies specific for the Vβ domain of the introduced TCR and was > 20% for all transductants.
The MAGE-A10254-262 peptide was either naturally processed and presented by human cells or loaded on cells. For peptide-loading assays, synthetic peptides were obtained from Peptide Protein Research Limited (> 90% pure) and mixed with T2 cells at the indicated final concentrations. ELISpot assays were set up as described12,22 and evaluated on an automated ELISpot reader (ImmunoSpot Series 5 analyzer, Cellular Technology Ltd.). Non-transduced T-cells were used as controls to assess background activation. All assays were carried out in triplicate and results analyzed (including calculation of EC50 values) using GraphPad Prism.
Cytotoxicity assay
The cytolytic activity of TCR-transduced T-cells to the SK-Mel-28 tumor cell line was assessed using the IncuCyteTM FLR platform (Essen BioScience), as previously described.45 MAGE-A10+ HLA-A*0201-transduced SK-Mel-28 cells were plated out at 20,000 cells per well and incubated with TCR-transduced or non-transduced T-cells in the presence of CellPlayer™ 96-Well Kinetic Caspase-3/7 reagent. SK-Mel-28 targets incubated alone were included as a control. Images were taken every 4 hours over a duration of 72 hours, and the number of apoptotic cells (“objects”) per mm2 was quantified for each image and plotted against time. An exclusion gate of > 100 µm2 was set to remove dead T-cells from the analysis.
X-scan assay
X-scan assays were performed using protocols as for standard IFN-γ ELISpot assays (described above), with TCR-transduced T-cells assessed for reactivity towards T2 cells pulsed with a panel of 172 synthetic peptides (Peptide Protein Research Limited) (> 90% pure). This panel represents a complete positional scan of the index peptide MAGE-A10254-262 (GLYDGMEHL) comprised of the index itself as well as all possible peptides with single natural amino acid substitutions. Peptides were added directly to ELISpot plate wells at a concentration of 10−7.5 M, approximately equal to the EC90 of the index peptide for both c756 and c796. Responses to variant peptides were assessed relative to the index peptide, after subtraction of responses using non-transduced T-cells.
Substitutions were defined as tolerated by the TCR where the response was > 10% of the response to the index peptide at its EC90 concentration, and Prosite motifs were generated to express which residues were tolerated at each position. Nonameric sequences contained within the human proteome which comply with the derived motifs were identified using an in-house R script to query a local copy of the UniProtKB/Swiss-Prot database with splice variants. The naïve probability of identifying matching sequences within the proteome was defined as:
where n = sequence length and ri = number of tolerated residues at position i within the sequence.
Funding Statement
This work was funded by Adaptimmune.
Acknowledgments
The authors particularly acknowledge E.W. Carter for generating data (Figures 1 and 2), performing affinity maturation for one of the parental TCRs, and producing certain of the TCR-encoding lentiviral constructs discussed in this report. We thank J.V. Harper for generating T-cell clones from which certain parental TCRs were isolated, J.W. Robinson for performing RACE analysis, A. Volkov for isolating further parental TCRs from proprietary naïve phage display libraries, and P.T. Todorov and M.C.C. Raman for performing biochemical assays on some of these parental TCRs. We thank J. Long and W. Bennett for producing some of the lentiviral preparations used in cellular assays, and J. Senra, Z. Wolchinsky and Z. Ferjentsik for performing qRT-PCR assays. We are grateful to our blood donors, without whose contributions this work would not have been possible.
Disclosure of potential conflicts of interest
All authors are employees of Adaptimmune.
A patent has been filed on the sequence and utility of the TCRs used in this study (WO2016/055785).
Author contributions
E.C.B. conceived and prepared the manuscript, performed biochemical assays and affinity enhancement, and generated TCR constructs; J.P.S. and T.W. contributed figures and analysis of data for the manuscript, and assisted in its preparation; A.B.G. and N.J.P. contributed to the conception and design of the experiments. J.P.S. and E.C.B. contributed equally, and all authors significantly to the study.
Supplementary material
Supplementary data can be accessed here.
References
- 1.Aranda F, Buque A, Bloy N, Castoldi F, Eggermont A, Cremer I, Starmann J, Tjwa M, Plate KH, Sültmann H, et al. Trial watch: adoptive cell transfer for oncological indications. Oncoimmunology. 2015;4:e1046673. doi: 10.1080/2162402X.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang M, Yin B, Wang HY, Wang RF.. Current advances in T-cell-based cancer immunotherapy. Immunotherapy. 2014;6:1265–1278. doi: 10.2217/imt.14.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, Citrin DE, Restifo NP, Robbins PF, Wunderlich JR, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res: An Off J Am Assoc Cancer Res. 2011;17:4550–4557. doi: 10.1158/1078-0432.CCR-11-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aleksic M, Liddy N, Molloy PE, Pumphrey N, Vuidepot A, Chang KM, Jakobsen BK. Different affinity windows for virus and cancer-specific T-cell receptors: implications for therapeutic strategies. Eur J Immunol. 2012;42:3174–3179. doi: 10.1002/eji.201242606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van der Merwe PA, Davis SJ. Molecular interactions mediating T cell antigen recognition. Annu Rev Immunol. 2003;21:659–684. doi: 10.1146/annurev.immunol.21.120601.141036. [DOI] [PubMed] [Google Scholar]
- 6.Bridgeman JS, Sewell AK, Miles JJ, Price DA, Cole DK. Structural and biophysical determinants of αβ T-cell antigen recognition. Immunology. 2012;135:9–18. doi: 10.1111/j.1365-2567.2011.03515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garrido F, Cabrera T, Aptsiauri N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: implications for immunotherapy. Int J Cancer. 2010;127:249–256. doi: 10.1002/ijc.25270. [DOI] [PubMed] [Google Scholar]
- 8.Tan MP, Gerry AB, Brewer JE, Melchiori L, Bridgeman JS, Bennett AD, Pumphrey NJ, Jakobsen BK, Price DA, Ladell K, et al. T cell receptor binding affinity governs the functional profile of cancer-specific CD8+ T cells. Clin Exp Immunol. 2015;180:255–270. doi: 10.1111/cei.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hebeisen M, Oberle SG, Presotto D, Speiser DE, Zehn D, Rufer N. Molecular insights for optimizing T cell receptor specificity against cancer. Front Immunol. 2013;4:154. doi: 10.3389/fimmu.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhong S, Malecek K, Johnson LA, Yu Z, Vega-Saenz de Miera E, Darvishian F, McGary K, Huang K, Boyer J, Corse E, et al. T-cell receptor affinity and avidity defines antitumor response and autoimmunity in T-cell immunotherapy. Proc Natl Acad Sci U S A. 2013;110:6973–6978. doi: 10.1073/pnas.1221609110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol. 2015;6:582. doi: 10.3389/fimmu.2015.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cameron BJ, Gerry AB, Dukes J, Harper JV, Kannan V, Bianchi FC, Grand F, Brewer JE, Gupta M, Plesa G, et al. Identification of a Titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci Transl Med. 2013;5:197ra03. doi: 10.1126/scitranslmed.3006034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, Kammula US, Royal RE, Sherry RM, Wunderlich JR, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–546. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan DA, Feldman SA, Davis JL, Morgan RA, Merino MJ, Sherry RM, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther: J Am Soc Gene Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Feldman SA, Yang JC, Sherry RM, Phan GQ, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linette GP, Stadtmauer EA, Maus MV, Rapoport AP, Levine BL, Emery L, Litzky L, Bagg A, Carreno BM, Cimino PJ, et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood. 2013;122:863–871. doi: 10.1182/blood-2013-03-490565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amos SM, Duong CP, Westwood JA, Ritchie DS, Junghans RP, Darcy PK, Kershaw MH. Autoimmunity associated with immunotherapy of cancer. Blood. 2011;118:499–509. doi: 10.1182/blood-2011-01-325266. [DOI] [PubMed] [Google Scholar]
- 19.Kunert A, Obenaus M, Lamers CHJ, Blankenstein T, Debets R. T-cell receptors for clinical therapy: in vitro assessment of toxicity risk. Clin Cancer Res: An Off J Am Assoc Cancer Res. 2017;23:6012–6020. doi: 10.1158/1078-0432.CCR-17-1012. [DOI] [PubMed] [Google Scholar]
- 20.van Loenen MM, de Boer R, Amir AL, Hagedoorn RS, Volbeda GL, Willemze R, van Rood JJ, Falkenburg JHF, Heemskerk MHM. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proceedings of the National Academy of Sciences of the United States of America 2010; 107:10972–10977. doi: 10.1073/pnas.1005802107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caballero OL, Chen YT. Cancer/testis (CT) antigens: potential targets for immunotherapy. Cancer Sci. 2009;100:2014–2021. doi: 10.1111/j.1349-7006.2009.01303.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hickman ES, Lomax ME, Jakobsen BK. Antigen selection for enhanced affinity T-cell receptor-based cancer therapies. J Biomol Screen. 2016;21:769–785. doi: 10.1177/1087057116637837. [DOI] [PubMed] [Google Scholar]
- 23.Weon JL, Potts PR. The MAGE protein family and cancer. Curr Opin Cell Biol. 2015;37:1–8. doi: 10.1016/j.ceb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Raman MC, Rizkallah PJ, Simmons R, Donnellan Z, Dukes J, Bossi G, Le Provost GS, Todorov P, Baston E, Hickman E, et al. Direct molecular mimicry enables off-target cardiovascular toxicity by an enhanced affinity TCR designed for cancer immunotherapy. Sci Rep. 2016;6:18851. doi: 10.1038/srep18851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fremont DH, Rees WA, Kozono H. Biophysical studies of T-cell receptors and their ligands. Curr Opin Immunol. 1996;8:93–100. [DOI] [PubMed] [Google Scholar]
- 26.De Plaen E, Arden K, Traversari C, Gaforio JJ, Szikora JP, De Smet C, Brasseur F, van der Bruggen P, Lethe B, Lurquin C, et al. Structure, chromosomal localization, and expression of 12 genes of the MAGE family. Immunogenetics. 1994;40:360–369. [DOI] [PubMed] [Google Scholar]
- 27.Rimoldi D, Salvi S, Reed D, Coulie P, Jongeneel VC, De Plaen E, Brasseur F, Rodriguez AM, Boon T, Cerottini JC. cDNA and protein characterization of human MAGE-10. Int J Cancer. 1999;82:901–907. [DOI] [PubMed] [Google Scholar]
- 28.Kim Y, Sidney J, Pinilla C, Sette A, Peters B. Derivation of an amino acid similarity matrix for peptide: MHC binding and its application as a Bayesian prior. BMC Bioinformatics. 2009;10:394. doi: 10.1186/1471-2105-10-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lesk A. Introduction to Bioinformatics. Oxford, UK: Oxford University Press; 2013. . [Google Scholar]
- 30.Birnbaum ME, Mendoza JL, Sethi DK, Dong S, Glanville J, Dobbins J, Özkan E, Davis MM, Wucherpfennig KW, Garcia KC. Deconstructing the peptide-MHC specificity of T cell recognition. Cell. 2014;157:1073–1087. doi: 10.1016/j.cell.2014.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang H, Lim HS, Knapp B, Deane CM, Aleksic M, Dushek O, van der Merwe PA. The contribution of major histocompatibility complex contacts to the affinity and kinetics of T cell receptor binding. Sci Rep. 2016;6:35326. doi: 10.1038/srep35326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Attaf M, Holland SJ, Bartok I, Dyson J. alphabeta T cell receptor germline CDR regions moderate contact with MHC ligands and regulate peptide cross-reactivity. Sci Rep. 2016;6:35006. doi: 10.1038/srep35006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mason D. A very high level of crossreactivity is an essential feature of the T-cell receptor. Immunol Today. 1998;19:395–404. [DOI] [PubMed] [Google Scholar]
- 34.Sewell AK. Why must T cells be cross-reactive? Nat Rev Immunol. 2012;12:669–677. doi: 10.1038/nri3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wooldridge L, Ekeruche-Makinde J, van den Berg HA, Skowera A, Miles JJ, Tan MP, Dolton G, Clement M, Llewellyn-Lacey S, Price DA, et al. A single autoimmune T cell receptor recognizes more than a million different peptides. J Biol Chem. 2012;287:1168–1177. doi: 10.1074/jbc.M111.289488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bijen HM, van der Steen DM, Hagedoorn RS, Wouters AK, Wooldridge L, Falkenburg JHF, Heemskerk MHM. Preclinical strategies to identify off-target toxicity of high-affinity TCRs. Mol Ther: J Am Soc Gene Ther. 2018. doi: 10.1016/j.ymthe.2018.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cole DK, van den Berg HA, Lloyd A, Crowther MD, Beck K, Ekeruche-Makinde J, Miles JJ, Bulek AM, Dolton G, Schauenburg AJ, et al. Structural mechanism underpinning cross-reactivity of a CD8+ T-cell clone that recognizes a peptide derived from human telomerase reverse transcriptase. J Biol Chem. 2017;292:802–813. doi: 10.1074/jbc.M116.741603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang YH, Terabe M, Pendleton CD, Stewart Khursigara D, Bera TK, Pastan I, Berzofsky JA. Identification and enhancement of HLA-A2.1-restricted CTL epitopes in a new human cancer antigen-POTE. PLoS One. 2013;8:e64365. doi: 10.1371/journal.pone.0064365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moysey R, Vuidepot AL, Boulter JM. Amplification and one-step expression cloning of human T cell receptor genes. Anal Biochem. 2004;326:284–286. doi: 10.1016/j.ab.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Boulter JM, Glick M, Todorov PT, Baston E, Sami M, Rizkallah P, Jakobsen BK. Stable, soluble T-cell receptor molecules for crystallization and therapeutics. Protein Eng. 2003;16:707–711. [DOI] [PubMed] [Google Scholar]
- 41.Cole DK, Pumphrey NJ, Boulter JM, Sami M, Bell JI, Gostick E, Price DA, Gao GF, Sewell AK, Jakobsen BK. Human TCR-binding affinity is governed by MHC class restriction. J Immunol. 2007;178:5727–5734. [DOI] [PubMed] [Google Scholar]
- 42.Garboczi DN, Hung DT, Wiley DC. HLA-A2-peptide complexes: refolding and crystallization of molecules expressed in Escherichia coli and complexed with single antigenic peptides. Proc Natl Acad Sci U S A. 1992;89:3429–3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.C A O’Callaghan, Byford MF, Wyer JR, Willcox BE, Jakobsen BK, McMichael AJ, Bell JI. BirA enzyme: production and application in the study of membrane receptor-ligand interactions by site-specific biotinylation. Anal Biochem. 1999;266:9–15. doi: 10.1006/abio.1998.2930. [DOI] [PubMed] [Google Scholar]
- 44.Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. [DOI] [PubMed] [Google Scholar]
- 45.McCormack E, Adams KJ, Hassan NJ, Kotian A, Lissin NM, Sami M, Mujić M, Osdal T, Gjertsen BT, Baker D, et al. Bi-specific TCR-anti CD3 redirected T-cell targeting of NY-ESO-1- and LAGE-1-positive tumors. Cancer Immunol, Immunother. 2013;62:773–785. doi: 10.1007/s00262-012-1384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.