

ABSTRACT
Due to the increase in the number of infliximab products, the need for global harmonization of the bioactivity of this monoclonal antibody was recognized by the World Health Organization (WHO). In response, the National Institute for Biological Standards and Control (NIBSC) developed the first international standard (IS) for infliximab, which targets tumour necrosis factor (TNF). Each ampoule is assigned values of 500 IU of TNF neutralizing activity and 500 IU of binding activity. Two preparations of infliximab were formulated and lyophilized at NIBSC prior to evaluation in a collaborative study for their suitability to serve as an IS for the in vitro biological activity of infliximab. The study involved participants using in vitro cell-based bioassays (TNF neutralization, antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity) and binding assays. The results of this study showed that the candidate preparation, coded 16/170, is suitable as an IS for infliximab bioactivity. This infliximab IS from NIBSC, is intended to support in vitro bioassay calibration and validation by defining international units of bioactivity. The proposed unitages, however, are not intended to revise product labelling or dosing requirements, as any decisions regarding this relies solely with the regulatory authorities. Furthermore, the infliximab IS is not intended for determining the specific activity of products, nor to serve any regulatory role in defining biosimilarity. We briefly discuss the future use of WHO international standards in supporting the global harmonisation of biosimilar infliximab products.
KEYWORDS: monoclonal antibodies, biosimilar, international standard, infliximab, TNF Neutralisation, ADCC, CDC, global harmonisation, bioactivity, bioassay
Introduction
A core remit in the constitution of the World Health Organization (WHO) is to “develop, establish and promote international standards (IS) with respect to food, biological, pharmaceutical and similar products” as well as “to standardize diagnostic procedures as necessary”. The provision of international biological reference standards makes a critically important contribution to high standards of efficacy, quality, purity and safety of many biological medicines used worldwide in the prevention, treatment or diagnosis of disease or conditions. WHO has a large portfolio of IS for many different products, including vaccines, biopharmaceuticals, in vitro diagnostics and has recently recognized the need for provision of international reference standards for monoclonal antibodies (mAbs) through a consultation with major stakeholders from all sectors of mAb manufacturing and regulation. It was agreed that publicly available reference standards for mAbs would be valuable in several ways: 1) providing a benchmark for biological activity; 2) method development and assessment of system suitability; 3) the calibration of national, pharmacopoieal or in-house reference standards; 4) assessing the potency of multisource products; 5) facilitating product surveillance and life-cycle management; and 6) supporting the development of novel methods.1 The recommendations explicitly state that an IS is an independent entity to reference medicinal products (RMPs) used to define biosimilarity, and the two entities have different roles and are not interchangeable. In particular, the WHO IS is intended for the in vitro calibration of bioassays, which use complex biological systems to test activity and can be variable from test to test. By using a WHO IS of known activity or potency, bioassay results can be compared and calibrated to give a consistent result, no matter when or where the bioassay is performed. WHO IS are not intended to serve any role in defining biosimilarity, specific activity, product labelling or therapeutic dosage. The key differences between the reference standards have been discussed in detail elsewhere2,3 and are summarised in Table 1.
Table 1.
A comparison of the distinct roles of the reference medicinal product and the WHO International Standard.
| Characteristic | Reference Medicinal Product | WHO International Standard |
|---|---|---|
| Authorisation | Approved by the relevant competent authorities for defined clinical indications | Established by the WHO Expert Committee for Biological Standardization |
| Product characteristics | Clinical product for human use.
|
Not a product for clinical use in humans Manufactured as per WHO specifications for use as a primary reference standard for bioactivity |
| Regulatory role | Serves as a ‘comparator’ product for biosimilarity assessments as per biosimilar guidance | No formal regulatory rolea |
| Form/Presentation | Lyophilised/liquid in product specific formulation Vials or prefilled syringes, pen cartridge etc |
Small amounts lyophilised in product specific formulation Glass ampoules |
| Labelling | Labelled and dosed in ‘mass’ | International Units (IU) per ampoule with no stated ‘mass’ |
| Bioactivity | Expressed in proprietary U/ml ± acceptance limits | Arbitrary IU/ampoule |
| Specific Activity | U/mg | Not applicable |
| Stability and Expiry date | Stable within expiration date (~ 2 years) if stored as per manufacturer’s recommended conditions | As defined in the ‘Instructions for Use’. Usually stable over many decades as predicted by accelerated degradation studies |
| Role in assay calibration, standardisation and data harmonization | None | As the highest order (primary) standard
|
| Availability | Restricted | Publicly available from a WHO custodian laboratory, e.g., NIBSC |
aProduct bioactivity expressed in units traceable to the IS can support regulatory decisions
The development of IS follows a stepwise process described in the flowchart shown in Figure 1. Consistent with this, the active substance for WHO IS for biopharmaceuticals is sourced from a single batch of bulk drug substance (usually obtained from a donation from a product manufacturer) and is reformulated and lyophilized to WHO guidelines4 to produce a standard that is stable for many decades. The suitability of the preparations to serve as a WHO IS is assessed in a multi-centre international collaborative study in which the participants are relevant stakeholders who perform relevant bioassays in order to characterise the standard. The biological activities of the standard are expressed in terms of arbitrary International Units (IU).
Figure 1.
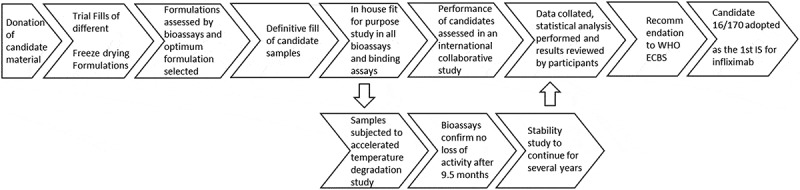
Flow chart summarising the steps in establishing the first WHO International Standard for infliximab.
mAb therapeutics are large complex bioengineered proteins. The most common therapeutic isotype is IgG1, which has a mass of ~ 150kDa and is composed of human IgG1 heavy chains and kappa light chains, with a single glycosylation site in the heavy chain. They are produced in mammalian cell lines using recombinant DNA technology, and, as a result, minor differences, i.e., micro-heterogeneity, will be present between batches of mAb. Until recently there has only been one marketed product for each mAb, therefore the bioactivity of the batches of product has been determined using the manufacturers own in-house reference standards. However, the patents of several mAbs have either expired or will imminently expire, resulting in the development of many biosimilar products, which are versions of marketed innovator products referred to as reference medicinal products (RMPs), with demonstrated similarity in physicochemical characteristics, efficacy and safety, based on comprehensive comparability assessments.5-7 Over 150 biosimilar mAb products were known to be in development as of 2016,8 and once a biosimilar is approved it then becomes a stand-alone product in its own right. It can be subjected to manufacturing and process changes independent of the originator product with no regulatory requirement to demonstrate biosimilarity against the RMP. However, evaluation of the effect of such changes in the manufacturing process of the biosimilar should be conducted, just as for the originator product, as outlined in the International Conference on Harmonisation Q5E document.9 With multiple products on the market, each with their own life-cycle, there remains the potential for divergence of critical quality attributes (CQAs) and biological activities among the different biosimilars and also relative to the originator product. Drifts and shifts in CQAs of products can occur through unintended or unexpected process changes or through planned process changes.10 Furthermore, the different activities of a mAb can vary independently of each other, e.g., antigen binding, Fc receptor function.11,12 Whilst predicting the effect of these changes remains difficult, demonstration of comparability at each manufacturing change ensures a consistent product such that the clinical safety and efficacy is maintained. 13,14 Once licensed, all the different products, i.e., biosimilars as well as the originator product, should be monitored; therefore, robust quality and pharmacovigilance systems need to be in place.15 Taking all of this into account represents a challenge for manufacturers and regulatory bodies because each manufacturer develops a different quality system and their own ‘in-house’ standards for monitoring CQAs, including the calibration of in vitro bioassays. Previously, for biological medicines derived from naturally occurring products such as erythropoietin and insulin, WHO IS preparations for bioactivity assessments were already available when recombinant biosimilar products were developed. This simplified the global harmonization of biological potency across many different products. In contrast, mAbs have no naturally occurring counterpart, and so mAb products have been developed in the absence of publicly available standards.
The National Institute for Biological Standards and Control (NIBSC) is the UK’s official medicines control laboratory for biological medicines and is the world’s major producer and distributor of WHO IS and reference materials (supplying over 95% of WHO standards worldwide).16 With support from the WHO, we launched a program to develop WHO IS for mAbs after they endorsed the development of IS for anti-TNF mAbs.17
Soluble TNF plays a role in many debilitating diseases such as rheumatoid arthritis (RA), Crohn’s disease (CD) and ulcerative colitis (UC). CD and UC are often referred to collectively as inflammatory bowel disease (IBD).18 As autoimmune diseases driven by TNF affect people of working age, they inflict huge economic burden.19,20 In the absence of a cure, substantial efforts were made over the past few decades to develop anti-TNF biotherapeutics that can control TNF-mediated diseases. Centocor’s anti-TNF mAb cA2, later known as infliximab, showed efficacy in both RA and UC, improving all aspects of the diseases.21-23 In RA, antigen binding that neutralizes TNF is the primary mechanism of action;24 however, in IBD Fc functions including antibody-dependent cell-meditated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), are also thought to be important in disease resolution.25
Infliximab (marketed as Remicade® by Johnson and Johnson, now Janssen) was the first anti-TNF mAb approved for use in humans. Licensed in the US in 1998 and in the EU in 1999, it has since become a blockbuster product with 2015 global sales in excess of $8bn.26 With patent protection already expired in the EU and due to expire in the US in September 2018, there has been intense activity to develop biosimilar anti-TNF products, including infliximab. The first two biosimilar mAbs to be licensed in Europe and the US were infliximab products, Remsima®27 and Flixabi®,28 and several others are now licenced worldwide (Table 2).
Table 2.
Anti-TNF biosimilar products that are currently authorised in the EU and US.
| Anti-TNF | Reference medicinal product | Patent Expiry EU (US) | Approved Biosimilar products |
|
|---|---|---|---|---|
| EU | US | |||
| Infliximab | Remicade® (Janssen) | 2015 (2018) | Remsima®/Inflectra® (Celltrion/Hospira2, Sep 2013), Flixabi® (Samsung Bioepis, May 2016) Zessly® (Sandoz, May 2018) | Inflectra®(Celltrion/Hospira2, Apr 2016), Renflexis® (Samsung Bioepis, May 2017), Ixifi® (Pfizer, Dec 2017) |
| Etanercept | Enbrel® (Amgen/Pfizer) | 2015 (2028) | Benepali (Samsung Bioepis, Jan 2016), Erelzi (Sandoz, June 2017) | Erelzi (Sandoz, Aug 2016) |
| Adalimumab | Humira® (Abbvie) | 2018 (2016) |
1Amgevita® (Amgen, Mar 2017), 1Solymbic® (Amgen, Mar 2017), Imraldi® (Samsung Bioepis, Aug 2017), Cyltezo® (Boehringer Ingelheim, Nov 2017) |
Amjevita® (Amgen, Sep 2016), Cyltezo® (Boehringer Ingelheim, Aug 2017) |
1Duplicate marketing authorization; 2Now acquired by Pfizer
In response to a marketplace with an expanding portfolio of infliximab products, we developed a candidate IS for the in vitro biological activity of infliximab at NIBSC. Here, we describe the results of a collaborative study designed to assess the suitability of the candidate to serve as the first WHO IS for infliximab, with an assigned arbitrary IU for infliximab bioactivities.
Results
Evaluation of lyophilization formulations for the IS
The formulation of mAb products such as Remicade® need to be compatible with in vivo human use, therefore the available excipients are limited. Moreover, the product is formulated with a high concentration of infliximab (10 mg/ml), which minimizes losses through adsorption to vials. As a result, the shelf life of Remicade® batches is limited. Conversely, the amount of mAb material in an IS is much lower (50 µg/ml), and it is important that the ampoules of IS are stable for many decades. As IS are for in vitro use only, we use a carrier protein and evaluate different formulations to minimise losses and achieve the desired stability. Prior to lyophilisation of the IS, we evaluated two freeze-drying formulations (supplementary material, Table S1) and examined their effect on the bioactivity of the freeze-dried material. Both the trial fills contained 50 µg of infliximab, which would provide enough material for assay calibration. SS-571 was formulated in a sucrose, mannitol and human serum albumin (HSA) formulation used previously in the etanercept IS29 and SS-575 was formulated in a sodium citrate and HSA formulation used previously in the rituximab IS.30 For each formulation both the freeze-dried material and the frozen formulated material was tested for TNF neutralization biological activity relative to the bulk infliximab drug substance. KLJ reporter gene assays were performed in duplicate (Figure 2) and ED50 values were determined for the samples and expressed as a percentage potency of the ED50 of unformulated bulk drug product. This revealed that, upon formulation and freeze-drying, SS-571 retained a mean biological activity of 76.5% relative to the unformulated bulk drug product, whereas SS-575 retained 90.4% mean activity. The frozen formulated but not freeze-dried samples retained almost identical mean activities (75.6% and 90.3% for SS-571 and SS-575, respectively) relative to the bulk as the freeze-dried samples. This suggests that most of the decrease in activity is a result of freezing rather than lyophilisation. As the freeze-drying formulation of SS-575 retained more activity than that of SS-571, this was taken forward as the formulation for the IS.
Figure 2.
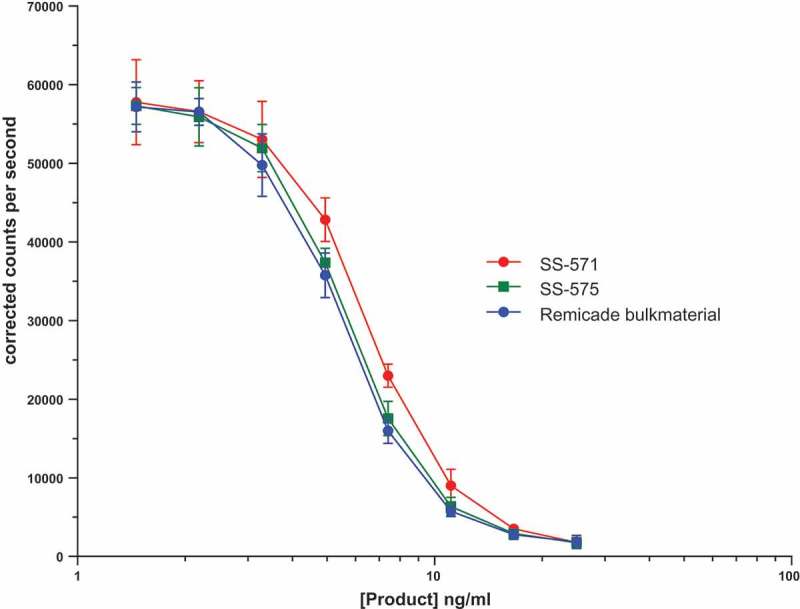
Representative TNF neutralisation assay using KLJ TNF responsive reporter gene cells showing the differential loss of biological activity of freeze-dried infliximab in the two freeze drying formulations SS-571 and SS-575. Each point is represented as a mean and standard deviation of four individual wells.
Production of the IS and comparator standard
To increase the stability and preserve the biological activity of the IS, the material is formulated and freeze-dried in flame-sealed glass ampoules that are back filled with dry nitrogen. This ensures a low oxygen and moisture content over the lifespan of the IS, something that cannot be achieved with stoppered vials commonly used for infliximab products. Infliximab bulk drug substance was donated to WHO and the IS was formulated, ampouled and freeze-dried at NIBSC using established protocols defined by the WHO.4 A second freeze-dried preparation to act as a comparator was produced in exactly the same way; however, the material sourced was from a purchased clinical batch of Remicade® drug product. To achieve the desired 50 µg/ml concentration, the starting material had to be diluted 812 and 200 fold, respectively, for the IS and the comparator. As a consequence, any differences in the formulation of the starting material (bulk drug substance vs. pharmaceutical product) was not considered an issue. Details of the products and the parameters from filling and freeze drying are summarised in the supplementary material, Table S2 and were all within specifications dictated by the WHO.
Stability of the candidate preparations
WHO IS have no shelf life or expiry date assigned; however, the stability of the IS needs to be demonstrated and any yearly loss estimated through an accelerated temperature degradation (ATD) study. Ampoules of the material are stored at elevated temperatures and bioactivities are compared relative to a sample stored at −70°C, and any predicted annual loss of bioactivity is stated in the instructions for use for the standard. The stability of the candidate preparation 16/170 is being assessed by an ongoing ATD study, and, at the time of writing, one time point (9.5 months) was interrogated; there was no detected loss of bioactivity when samples were stored at −20°C, + 4°C, + 20°C, + 37°C and + 45°C, compared to a control sample stored at −70°C (see supplementary material, Table S3). Samples in the accelerated thermal degradation studies will continue to be assessed. Similarly, no loss of bioactivity was observed when reconstituted samples were subjected to one-week storage at + 4°C or room temperature (Supplementary material, Table S4) or subjected to four freeze thaw cycles (Supplementary material, Table S5).
International collaborative study design
TNF neutralisation is the “batch-release” assay for infliximab products, but there are many other methods that are used to assess infliximab batches during product development, such as ADCC and CDC assays and binding to TNF. To determine the suitability of these preparations to serve as the first IS for infliximab, the lyophilised preparations of infliximab were subjected to an international collaborative study. This study consisted of 26 participants based in 15 countries with laboratories from regulatory agencies, official medicine control institutions, contract research organisations, academia and the pharmaceutical sector (Table 3). Participants were asked to evaluate the candidate preparations, in their laboratories, using their qualified “in-house” methods and reference standards for assaying infliximab bioactivity and binding activity.
Table 3.
Participants of the collaborative study.
| Melanie Morris, Lea-Ellen Hogie and Keith Mortimer, Biochemistry Section, Office of Laboratory & Scientific Services, Therapeutic Goods Administration,136 Narrabundah Lane, Symonston, Canberra ACT 2609, Australia. |
| Haibin Wang, Lei Li and Bingjie Hao, Zhejiang Hisun Pharmaceutical Co Ltd, 46 Waisha Rd. Jiaojiang, Taizhou, Zhejiang, China. |
| Meng Li and Lan wang, Division of Monoclonal Antibodies, NIFDC, No2.Tiantan Xili, Beijing, 100050, China. |
| Jaana Vesterinen, Finnish Medicines Agency, Mannerheimintie 166, P.O.Box 55,00300 Helsinki, Finland. |
| Sylvie Jorajuria and Marie-Emmanuelle Behr-Gross, European Directorate for the Quality of Medicines and HealthCare (EDQM) Council of Europe, 7 allée Kastner, CS 30026, F-67081 Strasbourg, France. |
| Jean-Claude Ourlin, ANSM, 635 Rue de la Garenne,CS 60007, 34740 Vendargues Cedex, France. |
| Michael Tovey and Christophe Lallemand, Biomonitor SAS, Villejuif Bio Park, 1 mail du Professor George Mathé, 94800 Villejuif, France. |
| Ulrike Herbrand and Kerstin Brack, Charles River Biopharmaceutical Services GmbH, Max-Planck-Str. 15A, 40699, Erkrath, Germany. |
| Shubrata Khedkar, Prabhavathy Munagala and Ranjan Chakrabarti, Biologics & Biotechnology Division, United States Pharmacopeia-India (P) Ltd, Plot No. D6 & D8, IKP Knowledge Park, Genome Valley, Shameerpet, Hyderabad, 500078, R.R. District, Telangana, India. |
| Ezra Mulugeta and Charlotta Mark, Medical Products Agency, P.O. Box 26, SE-751 03 Uppsala, Sweden. |
| Cornelius Fritsch and Ruzica Puljic, Biologics Process R&D, Novartis Pharma AG, Klybeckstrasse 141, CH-4052 Basel, Switzerland. |
| Chris Bird, Paula Dilger and Clive Metcalfe, Cytokines and Growth Factors Section, Biotherapeutics Group, NIBSC, Blanche Lane, South Mimms, Potters Bar, Herts, EN6 3QG, UK. |
| Stuart Dunn, Covance Laboratories Ltd, BioCMC, Otley Road, Harrogate HG3 1PY, UK. |
| Guoping Wu and Todd Geders, Bioassay, R&D Systems, Inc.614 McKinley Place NE, Minneapolis, MN 55413,USA. |
| Yong Suk Yang and Gye Mee Jang, QC C&I, Celltrion Plant 2, 20, Academy-ro 51, Yeonsugu, Incheon, 22014, Korea. |
| Jia-Ming Yang and Yucai Peng, ADQC Department, Livzon Mabpharm Inc., No.38 Chuangye North Road, Jinwan District, Zhuhai City, Guangdong Province, 519045, China. |
| Hiroko Shibata, Masato Kiyoshi, Akiko Ishii-Watabe, Division of Biological Chemistry and Biologicals, National Institute of Health Sciences, 1–18-1, Kamiyoga, Setagaya-ku, Tokyo 158–8501, Japan. |
| Yeonjoo Hong, Bioassay Group 1, QE, Samsung Bioepis, 107, Cheomdan-daero, Yeonsu-gu, Incheon, 406–840, Korea. |
| Omer Dushek, Marcus Bridge and John Nguyen, Molecular Immunology Group, Sir William Dunn School of Pathology, South Parks Road, Oxford, OX1 3RE, UK. |
| Chaomei Lin, Joseph Albanese and Ton Geurts, Janssen Biologics BV, Einsteinweg 101, 2333 CB Leiden, The Netherlands. |
| James Kessels and Raf Berghmans, apDia bvba, Raadsherenstraat 3, 2300 Turnhout, Belgium. |
| Annick de Vries and Tom Lourens, Biologicals Laboratory, Sanquin Diagnostic Services, Plesmanlaan 125, 1066CX Amsterdam, The Netherlands. |
| Daniel Nagore and Susana Catarino, Progenika Biopharma S.A., Parque Tecnológico Bizkaia, Ed. 504, 48160 Derio, Spain. |
| Melissa Snyder and Maria Willrich, Division of Clinical Biochemistry and Immunology, College of Medicine, Mayo Clinic, 200 First St SW, Rochester, MN 55905, USA. |
| Zehra Arkir, Viapath, Reference Chemistry, Biochemical Sciences, 5th Floor, North Wing, St Thomas' Hospital, SE1 7EH, UK. |
| Ermis Parussini and Simon Daviere, Theradiag, 14 Rue Ambroise Croizat, 77183 Croissy Beaubourg, France. |
Participants in the collaborative study were sent candidate 16/170 (designated sample A), the comparator 16/160 (designated sample B) and a coded duplicate of 16/170 (designated sample C) along with the third WHO TNF IS (coded 12/154). This common source of TNF was included to reduce assay variability arising from use of human TNF from different suppliers.
Assays performed in the collaborative study
The TNF neutralisation bioassays performed in the study are summarised in Table 4. These have been described previously29 and consisted of three different assay strategies, TNF-induced cytotoxicity assays, TNF-induced apoptosis assays and reporter gene assays of TNF binding (supplementary material, Table S6a). The majority performed cytotoxicity assays, measuring the cytotoxic effect of TNF on either murine fibroblast, L92931 or fibrosarcoma, WEHI-16432 cell-lines. Apoptosis assays were performed using the human histiocytic lymphoma cell-line, U93733 Reporter gene assays utilised the engineered cell line K56234 or HEK-293 cells transfected with the TNF responsive NFκB regulated Firefly luciferase reporter-gene construct. Only three participants performed ADCC assays (supplementary material, Table S6b) all using engineered cells expressing a non-cleavable mutant of membrane-bound TNF as targets, while effectors were either the natural killer cell line NK92, which lyses target cells upon activation,35 or reporter gene containing cells that luminesce in response to crosslinking of CD16 (FcγIII receptor) by infliximab in the presence of cells presenting surface-bound TNF antigen.36 Two participants performed CDC assays (supplementary material, Table S6b) in which infliximab induces lysis of Jurkat cells expressing membrane-bound TNF in the presence of complement.35 Assays evaluating the relative TNF binding of the candidates were also performed and are summarised in Table 5. The majority of binding assays were variations of enzyme-linked immunosorbent assays (ELISAs); however, one participant used fluorescence resonance energy transfer (FRET) and surface plasmon resonance (SPR).
Table 4.
Summary of the bioassays performed in the collaborative study.
| Bioassay Type | Cell Line | No of Participants | TNF (IU/ml) | Assay Period (hrs) | Assay Readout | Readout Reagent |
|---|---|---|---|---|---|---|
| TNF Neutralisation Assays | ||||||
| Cytotoxicity | WEHI-164 | 9 | 2–100 | 18–24 | Absorbance | WST-8/CCK-8, MTS |
| L929 | 8 | 7.2–20 | 18–24 | Fluorescence, Absorbance, Luminescence | Resazurin/Alamar Blue, CCK-8, Cell-Titer Glo | |
| Apoptosis | U937 | 3 | 40–60 | 2.5 | Luminescence | Caspase Glo 3/7 |
| Rep-Gene | HEK 293 | 2 | 17.2–50.0 | 16–24 | Luminescence | Steady Glo |
| KLJ | 1 | 40 | 4 | Luminescence | Steady-Glo Plus | |
| Other Assays – target cell-lines express non-cleavable human TNF | ||||||
| CDC | Target: Jurkat | 2 | - | 4–22 | Absorbance, Luminescence | CCK-8, Cyto-Tox Glo |
| ADCC | Target: 3T3 cells, Effector: CD16 expressing NK92 | 1 | - | 4 | Luminescence | Cyto-Tox Glo |
| Target: CHO-K1 cells, Effector: CD16 expressing Jurkat cells linked to luciferase reporter gene | 1 | - | 16–24 | Luminescence | Bio-Glo | |
| Target: HEK-293 cells, Effector: CD16 expressing Jurkat cells linked to luciferase reporter gene | 1 | - | 6 | Luminescence | Dual Glo | |
Table 5.
Brief details of binding titrations contributing to the study.
| Lab Code | Assay Platform | Assay description | Readout | Specific for infliximab |
|---|---|---|---|---|
| 1 | FRET | Europium labelled infliximab and Cy5 labelled TNF form fluorescent complex which is competitively inhibited by unlabelled infliximab | Resonance energy transfer | Yes |
| 2 | SPR | Infliximab titrated onto chip coated with anti-Fc and fixed concentration of TNF passed over | Response units at binding saturation | N/A |
| 8, 18 | ELISA | Plates coated with TNF, infliximab captured and detected with anti-Kappa chain-HRP and TMB substrate. | Absorbance 450nm | No |
| 22, 24, 27 | ELISA | Plates coated with anti-TNF, TNF captured, infliximab captured by bound TNF and detected by anti-infliximab-HRP and TMB substrate | Absorbance 450nm | Yes |
| 23 | ELISA | Plates coated with TNF, infliximab captured and detected with anti-IgG -HRP and TMB substrate | Absorbance 450nm | No |
Statistical analysis of infliximab preparations and assay validity
Upper equivalence bounds used in the assessment of dose-response curve similarity, as described in materials and methods, and the percentage of invalid TNF neutralisation bioassays obtained using these values are summarised in the supplementary material Table S7, grouped by assay readout type. For an assay to be concluded as valid, equivalence had to be demonstrated for all three parameters (α, β and δ). The percentage of invalid assays per laboratory is shown in the supplementary material, Table S8, illustrating the wide range in relative performance of the participating laboratories using the defined equivalence bounds. In 70% of the laboratories, invalidity rates were ≤ 25% with no invalid assays found in 39% of these laboratories. The majority of invalid assays were observed in the remaining 30% of laboratories, in particular for laboratory 8, where between 46 and 100% (depending on the cell line) of their assays were invalid, suggesting that the chosen statistical model was not appropriate. Re-analysis of data from this laboratory using no response transformation was performed using CombiStats 5.0,37 after which > 90% of assays gave valid estimates of relative potency, illustrating that the best choice of assay response transformation may not be the same across all laboratories.
For ADCC assays, no valid relative potency estimates were calculated for assays performed by laboratory 14, as only four dilutions of each sample were tested and neither a sigmoid curve nor a parallel line model could be fitted (due to lack of convergence or non-linearity, respectively). For laboratory 1, there was a technical issue with the assay media, and therefore their result was excluded from the study. The majority of ADCC assays from laboratory 15 gave valid estimates for some or all of the coded samples tested. CDC assays were performed by one laboratory only and all gave valid estimates of relative potency. A majority of binding assays gave valid estimates for some or all of the coded samples tested.
In general, the data showed that the infliximab preparations were active in all the bioassays performed by the participants. The performance of the preparations including the candidate in the bioassays were comparable to in-house standards, confirming that the preparations are suitable for use in calibration of bioassays.
Estimates of relative potency to in-house reference standards and to the candidate preparation for bioassays and TNF binding assays
Currently, each manufacturer or testing laboratory establishes and validates their own in-house standards from a given batch of infliximab. Different batches of infliximab can have differing biological activity within pre-determined limits, resulting in in-house standards that may not be comparable between laboratories. This can result in inter-laboratory variability in potency estimates if an infliximab product is assayed in several different laboratories. However, if the potency estimates are determined against a common reference standard that does not vary between laboratories, nor over time, the inter-laboratory variability should be improved, resulting in more harmonised potency estimates of infliximab products.
To test this hypothesis, geometric means (GM) and associated geometric coefficients of variation (GCV) of potency estimates of the preparations, calculated relative to in-house reference standards or relative to candidate sample A, were determined. Inter-laboratory GCVs relative to in-house reference standards were calculated after exclusion of laboratories 3 (used commercial TNF rather than IS 12/154) and 16 (a non-infliximab product was used as the in-house standard).
For all the neutralisation assays combined, as shown in Table 6, the GM of the relative potencies of candidate A and comparator B were 1.02 and 0.98, respectively, when determined relative to the participants in-house standards. Furthermore, the relative potency of the coded duplicate C being 1.03 was in good agreement with candidate A. Inter-laboratory GCVs for potency estimates of the preparations were between 5.9 and 6.5% when estimates were determined relative to the in-house standards. When candidate A was used by the participants as the reference standard in TNF neutralisation bioassays, the GM of the relative potencies was similar, 0.95 for comparator B and 1.01 for the coded duplicate C, but the GCVs of the potency estimates were reduced to 2.7% and 3.9% for B and C, respectively (Figure 3). The two most popular TNF neutralisation assays, L929 and WEHI-164 cell-based cytotoxicity assays, showed similar trends to that of the overall combined neutralisation data for relative potency and GCV of the candidates relative to both participants’ in house standards and candidate A (supplementary material Table S9). Candidate A performed particularly well in the WEHI-164 assay, with inter-laboratory GCV reduced from 6.7% and 7.5% when estimated relative to in-house standards to 1.3% and 2.6% when estimated relative to comparator B and coded duplicate C, respectively (supplementary material, Figure S1).
Table 6.
Overall geometric mean relative potency estimates for TNF neutralisation and binding assays.
| Method | Sample | Potencies relative to Candidate A |
Potencies relative to participants IH reference |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| GM | LCL | UCL | GCV | n | GM | LCL | UCL | GCV | n | ||
| Neutralisation (all) | A | 1.02 | 0.99 | 1.06 | 5.9% | 16 | |||||
| B | 0.95 | 0.94 | 0.96 | 2.7% | 22 | 0.98 | 0.96 | 1.01 | 5.1% | 16 | |
| C | 1.01 | 1.00 | 1.03 | 3.9% | 21 | 1.03 | 1.00 | 1.07 | 6.5% | 17 | |
| Binding | A | 1.09 | 0.83 | 1.43 | 18.6% | 4 | |||||
| B | 0.95 | 0.86 | 1.05 | 10.4% | 6 | 1.00 | 0.78 | 1.29 | 17.1% | 4 | |
| C | 1.00 | 0.91 | 1.09 | 8.6% | 6 | 1.06 | 0.88 | 1.28 | 12.5% | 4 | |
GM: Geometric Mean; LCL and UCL: Lower and Upper 95% Confidence Limits;
GCV: Inter-laboratory Geometric Coefficient of Variation (%); n: Number of laboratories used in calculation of GM and GCV; IH: In-house reference
Figure 3.
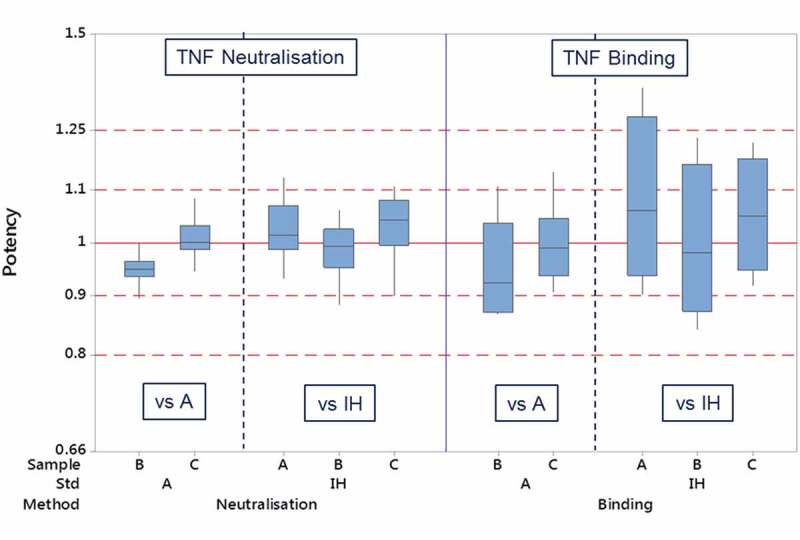
Box and whisker plot of laboratory geometric mean relative potencies of infliximab preparations using either participants in-house reference or candidate A as the assay standard.
For laboratory 8, potency estimates from neutralisation assays, summarised in supplementary material, Table S10, showed GM potencies and levels of intra-laboratory variability that were comparable to those obtained in other laboratories which were calculated using a different assay response transformation.
Individual laboratory GM relative potency estimates for the preparations are shown in the supplementary material, Table S11. Intra-laboratory GCV values ranged from 0.8% to 28.9% in neutralisation assays, with a median value of 4.3% and the majority (82%) of values less than 10%, demonstrating generally good intermediate precision in participating laboratories over the valid assays. Levels of variability were not observed to be related to the reference standard or cell line used.
For TNF binding assays, the estimates of relative potency were much more variable than those of the TNF neutralisation bioassays (Table 6, Figure 3). Overall across all the different binding assay methods, the GM potencies of candidate A and comparator B relative to in-house standards were 1.09 and 1.00, with GCVs of 18.6% and 17.1%, respectively. The coded duplicate C has a potency estimate of 1.06 and GCV of 12.5%. Potency estimates when determined relative to candidate A were identical to those seen in the TNF neutralisation bioassays (0.95 for comparator B and 1.00 for coded duplicate C). Inter-laboratory GCVs were also much improved to 10.4% and 8.6%, respectively for B and C. Intra-laboratory GCV values (supplementary material, Table S12) ranged from 3.3% to 22.1%, although they were only calculated in a small number of cases from the seven laboratories that performed this assay.
ADCC and CDC assays were each only performed by a single laboratory and relative potency estimates of the candidate A, comparator B and coded duplicate C calculated by each laboratory were in line with those seen in the other assays (supplementary material, Table S9). Intra-laboratory variability (supplementary material, Table S12) was lower in the CDC assay (2.4% to 4.0%) and higher for ADCC assay (9.0% to 26.3%).
Estimates of ED50 derived from neutralisation assays
As expected, the geometric mean ED50 values varied between different laboratories and assay methods (supplementary material, Table S13), with GCVs ranging from 3.0% to 24.4 % in L929 assays (except for laboratory 3, which used commercial TNF) and from 0.4% to 19.5% in WEHI-164 assays (except laboratories 8 and 28 where the GCVs were higher), and in U937 assays from 1.1% to 16.8%. Since infliximab inhibits TNF bioactivity, the inhibitory activity should be expressed in terms of units of TNF, in this case the 3rd IS for TNF, 12/154. Inhibitory activity can be determined for candidate A based on the ED50 values derived for both L929 and WEHI-164 cytotoxicity assays from laboratories using the same fixed amount of TNF IS in the assays, Equation 1.
For WEHI-164 and L929 assays, four laboratories used the same amount of TNF (supplementary material, Table S14). Based upon this limited data, 0.04 IU of infliximab candidate A inhibits the cytotoxic effect of 20 IU of TNF IS in an L929 cytotoxicity assay. A slightly higher amount of 0.08 IU of infliximab candidate A inhibits the cytotoxic effect of 40 IU of TNF IS in a WEHI-164 cytotoxicity assay. It is assumed the mechanism of inhibition of TNF by infliximab is identical in both WEHI-164 and L929 cytotoxicity assays, with just the TNF responsive cell line changing. This assumption is corroborated as twice as much infliximab is required in the WEHI-164 assay to neutralise twice as much TNF when compared to the L929 assay.
Assignment of international units of bioactivity for the preparation
Multiple methods are used to assign a value to the infliximab IS, as per WHO guidance.4 As a result, the definition of the IU is not related to a specific method of determination. Candidate preparation A (NIBSC code 16/170) has been assigned 500 IU of TNF neutralising activity per ampoule and 500 IU of TNF binding activity per ampoule and will serve as the first IS for infliximab. Since only a limited number of laboratories performed the ADCC and CDC assays in the current collaborative study, a unitage has not been assigned for these activities; however, the standard can be used as an assay control for ADCC and CDC.
The mass content of infliximab filled in each ampoule is approximate and no declared mass content for the ampoule of the IS is provided. Thus, the proposed unitage cannot be used to define the specific activity of the preparation, nor can it be used to derive or infer any specific activity for regulatory purposes or to revise product labelling and dosing requirements. The IS is not a substitute for the RMP and should not play a role in defining biosimilarity. It is designed to serve as a higher order reference standard for calibration of bioassays, thus facilitating harmonisation of bioactivity across infliximab products throughout their entire product lifecycle.
In-house fit for purpose assessment of the proposed IS against different infliximab products
After establishing that candidate 16/170 was fit for purpose to serve as the IS for infliximab, we conducted an in-house study to gauge its suitability to determine relative TNF neutralization potencies of different infliximab products. Remicade® (2 batches), Remsima® (2 batches) and Flixabi® (1 batch) were assayed for TNF neutralisation in duplicate L929 cell cytotoxicity assays. ED50 values were determined for the samples and expressed as a percentage potency of the ED50 of candidate IS 16/170; data from a representative assay is shown in Figure 4. Relative to the candidate IS, the two batches of Remicade® showed mean potencies of 102.4% and 105.7% (n = 2), the two batches of Remsima showed mean potencies of 112.7% and 106.4% (n = 2) and the batch of Flixabi® showed a relative potency of 94.5% (n = 2). This confirmed that the candidate IS 16/170 was suitable for use in calibrating assays involving both originator infliximab and biosimilar products.
Figure 4.
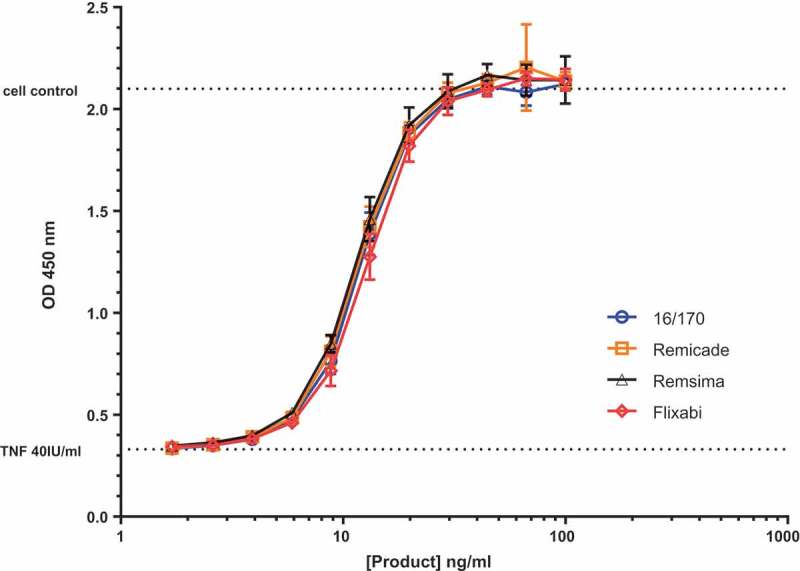
Representative TNF neutralisation assay using L929 cells showing the performance of the proposed IS 16/170 with different infliximab products on the market. The assay readout was CCK-8 and each point is represented as a mean and standard deviation of three individual wells.
Discussion
The international collaborative study described herein was undertaken to assess the performance of a lyophilised preparation of infliximab (NIBSC code 16/170) in biological assays that are routinely used to characterise infliximab products. These assays are used in all parts of a product’s lifecycle from product development, quality assurance and batch release to post marketing surveillance. As the infliximab product used to manufacture the candidate standard was a biosimilar, a commercially sourced lot of the originator product (Remicade®) was reformulated identically to the candidate and included as a comparator (NIBSC code 16/160). This was included to ensure that there was no difference in assay performance between a bulk drug substance and formulated drug product after reformulation and freeze drying.
All the lyophilised preparations were active and performed well in all the different assays and methods included in the study. For the purpose of this study, rigorous equivalence criteria were applied to the data, ensuring the subsequent analysis was carried out on high quality data. For TNF neutralisation, even with these stringent validity parameters applied, between 74 and 83% of the assays performed were valid depending on the standard and the assay readout type.
The TNF neutralisation assays proved to be very robust, even in the absence of a common standard. Inter-laboratory GCVs of estimated potencies were all below 8.5% when potencies were determined relative to in-house standards. The robustness can be attributed to the fact that, because TNF neutralisation takes place in solution without involvement of the cell, all that varies is the amount of free soluble TNF that is available to bind to the cells and instigate the cytotoxic, apoptotic or reporter response. Therefore, no one particular assay method was more variable and all reported potencies were similar, as has been seen in previous studies for the third WHO IS for TNF38 and the first WHO IS for etanercept.29 Other cell-based assays such as ADCC are highly influenced by the target, the effector cell type, the expression of FcγRIII receptors, receptor polymorphism, the assay conditions and the readout employed. The glycosylation pattern of the mAb, particularly the degree of afucosylation is also known to affect Fc-mediated ADCC activity, depending on the sensitivity of the assay system used.39 Consequently, these factors can intrinsically lead to higher levels of variability, as was recently observed in the collaborative study for the first WHO IS for rituximab, but are not attributable to the performance of the reference standard per se.30 In this study, we were unable to determine the levels of inter-laboratory variability in ADCC and CDC, or whether these were improved by introduction of a common standard because each assay was only performed by a single laboratory. However, all the candidate materials were active in the assays that were conducted and similar relative potencies were reported to those seen in the other assay types when the potencies were expressed relative to candidate A.
The most variable assays in this study were the binding assays. Although this might be due to the smaller number of assays performed compared to the TNF neutralisation assays, there could be several other contributing factors. Unlike TNF neutralisation, the methods used were diverse and included ELISA, FRET and SPR platforms. Even the five participants performing ELISA adopted different methodologies, with TNF being adsorbed directly onto the detection plate or adoption of a sandwich approach where the plate is first coated with an anti-TNF capture antibody. Detection methods were equally varied, with detection accomplished using antibodies either for the heavy or light chain isotypes (anti-kappa or anti-IgG1) or more specific anti-infliximab antibodies. Secondly, because the third WHO IS for TNF contained carrier albumin, it was unsuitable for use in ELISA experiments; therefore, each participant or kit manufacturer sourced their own TNF, which could potentially have different activity.
Overall, when infliximab products are assayed relative to in-house standards, there is significant variation in the relative potency estimates reported. However, the introduction of a common bioassay reference standard reduced inter-laboratory variability. As a result of this, the precision of the estimates was increased, comparator B was estimated to have a potency of 95% of candidate A and C when each laboratory used candidate A as a reference whereas, when each laboratory used their own in-house standard, differences in the relative potencies of candidate A and comparator B could not be resolved. Remarkably, this was completely independent of assay type, with the same relative potencies determined for the preparations over four different neutralisation assay platforms, several ELISA platforms, as well as other less common platforms such as SPR and FRET. Isotope dilution mass spectrometry revealed this small difference between the preparations was due to less material being filled in the ampoules of comparator B. The outcome of this collaborative study confirms that the infliximab candidate preparation 16/170 is suitable to harmonise biological potency estimates of infliximab products in many different assay platforms. Based on this, the WHO established the preparation, coded 16/170 as the first IS for infliximab, with each ampoule containing 500 IU of activity for TNF neutralisation and 500 IU of activity for TNF binding. The unitages have been assigned independent of each other in order to aid in replacement of the standard. As we only manufacture a single large batch of standard (~ 7000), expected to last several years, any subsequent replacement will be designated the second IS for infliximab (and not a subsequent batch of this first IS). It will be an independent standard with independent unitages for each activity, calibrated against the first IS, thus maintaining continuity and traceability of unitage relative to the current infliximab IS.
Future implications
Over the last two decades mAb therapy has revolutionised the treatment of TNF-mediated inflammatory disorders, but this has come at great cost to healthcare systems. In the UK, it is estimated that the cost of such therapy is £10,000 per year per patient, and therefore biological therapy is only usually offered as a last resort, after the failure of other treatments.40 In contrast to adopted practice, there is also growing evidence that both RA and IBD patients have a window of opportunity where early treatment can slow disease progression,41,42 so a framework for wider access to these therapies would be beneficial to healthcare systems worldwide. A recent report suggests that if physicians in the UK exclusively prescribed biosimilar anti-TNF mAbs to both newly diagnosed patients and those on maintenance doses, the cost of treatment could be halved to £5,000 per patient per annum, with a projected annual saving of £3m for the National Health Service.40 Such reductions in the price of therapy will provide many more patients access to these advanced treatments, increasing patient quality of life, enabling them to continue working and reducing the burden of disease on economies.41 However, prior to authorisation of biosimilar mAbs there were concerns about switching patients who had been receiving the originator product to a biosimilar product. So far, the concerns appear to be unfounded as the results of switching studies for Remsima®, the first infliximab biosimilar authorised have revealed its safety and efficacy to be comparable to Remicade®, both in the short term, and over extended periods of administration.43-45 However, there are currently relatively few infliximab products authorised in Europe and the US, resulting in limited data available. As more products come to market, they will all adopt their own lifecycle, and, as with the originator product, there remains the possibility of divergence during the life cycle of the different products, potentially resulting in a market littered with distinct quasi products. Therefore, it is paramount that the CQAs of products continue to be aligned post authorisation. This study, and a similar one conducted for rituximab,30 show that WHO International Standards for mAbs are tools that, on universal adoption, can play a pivotal role in globally harmonising the biological activity of antibody products for many years to come.
Materials and methods
Materials and processing
A preparation of recombinant infliximab, from a single batch of bulk drug substance was kindly donated to WHO (see Acknowledgement) and a commercial batch of Remicade® (Janssen) was purchased to act as a comparator.
Trial fills were conducted using two different formulations; A) 10 mM Tris, pH 7.4, 4% D-mannitol, 1% sucrose, 0.2% HSA; and B) 25 mM sodium citrate tribasic dihydrate, pH 6.5, 150 mM sodium chloride, 1% HSA. The biological activity of the lyophilized preparations was compared with the bulk material in a cytotoxicity assay and a reporter gene assay.
Final lyophilizations of the candidate and comparator B were carried out at NIBSC using WHO guidelines.4 For this, buffers and excipients, were prepared using nonpyrogenic water and depyrogenated glassware and solutions filtered using sterile nonpyrogenic filters (0.22 µm Stericup filter system, Millipore, USA) where appropriate.
The two preparations were coded as described in the supplementary material, Table S2. The mass content of the protein in the ampoules, given as ‘predicted µg’ is calculated from the dilution of the bulk material of known protein mass content as provided by the manufacturer.
For both preparations, a solution of infliximab at a theoretical protein concentration predicted to be 50 µg/ml was distributed in 1 ml aliquots into 5 ml ampoules and lyophilised under optimised and controlled conditions. The glass ampoules were sealed under dry nitrogen by heat fusion and stored at −20°C in the dark until shipment at room temperature.
For each fill, a percentage of ampoules were weighed, residual moisture of each preparation was measured by the coulometric Karl-Fischer method (Mitsubishi CA100) and the headspace oxygen content was determined by frequency modulated spectroscopy using the Lighthouse FMS-760 Instrument (Lighthouse Instruments, LLC). The mean fill weights, moisture content and headspace oxygen content, which is a measure of ampoule integrity, are reported in the supplementary material, Table S2. Testing for microbial contamination using the total viable count method did not show any evidence of microbial contamination.
Study design
Participating laboratories, listed in Table 3, were provided with a sample pack, which consisted of 5 ampoules each of the study samples A-C, for each different type of assay they were undertaking, along with 5 ampoules of the third WHO TNF IS (coded 12/154) for the bioassays. A study protocol was provided that outlined the aims and objectives of the study, suggested assay layouts and contained spreadsheets for reporting of results. Instructions for use for each of the candidate preparations and the TNF IS were also provided.
Prior to performing the assays for the study, participants were advised to perform a pilot assay using the study samples for each of the assay types they intended to undertake to ensure appropriate assay conditions and optimal dose response curves. For TNF neutralisation bioassays, participants were also advised to select a suitable dose of TNF.
Following establishment of suitable conditions, participants were asked to assay all samples concurrently on a minimum of three separate occasions using their own routine methods, within a specified layout that allocated the samples across 3 plates and allowed testing of replicates as per the study protocol. It was requested that participants perform at least 8 dilutions of each preparation using freshly reconstituted ampoules for each assay and include their own in-house standard where available on each plate. Participants were requested to return their raw assay data, using spreadsheet templates provided, and also their own calculations of potency of the study samples relative to preparation A or their own in-house standard.
For binding assays, participants were requested to use their proprietary assay kits or in-house assays to assess the binding to human TNF of the three candidate preparations and their in-house standard using serial dilutions. Participants were requested to perform three independent assays on three separate occasions and return raw data in a format that was appropriate for the assay technique used.
Statistical analysis
Analysis of dose-response curve data was performed using a four-parameter logistic (sigmoid curve) model, Equation 2. Assay responses (e.g., absorbance, luminescence) were log10 transformed for all neutralisation assays. For binding, ADCC and CDC assays, no transformation of assay response was used.
For neutralisation assays, models were fitted using the R package ‘drc’. Parallelism (similarity) for a pair of dose-response curves was concluded by demonstrating equivalence of the parameters α, β and δ. For this approach, differences in these parameters for the two samples under consideration were calculated and approximate 90% confidence limits for these differences (dL, dU) were determined using the delta method. Extreme values, defined as max(|dL|,|dU|) were calculated and equivalence concluded in cases where these were below pre-defined upper equivalence bounds. The calculated upper equivalence bound values are shown in the supplementary material Table S7 and were based on the data obtained for the coded duplicate samples that are expected to have parallel dose-response curves, i.e., equivalent values of all model parameters.
As the binding, ADCC and CDC assays were performed by fewer laboratories, analysis was performed using CombiStats v5.037 and the validity of the assays was concluded when no significant non-parallelism (p < 0.01) was found by analysis of variance. In cases where significant non-parallelism appeared to result from low or underestimated residual variability, correlation coefficients were confirmed to be > 0.985 (R2 > 0.97) and slope ratios confirmed to be within the range [0.90, 1.11] before potency estimates were accepted as valid.
All relative potency estimates were combined to generate unweighted geometric mean potencies for each laboratory and these laboratory means were used to calculate overall unweighted geometric mean potencies. Variability between assays and laboratories has been expressed using geometric coefficients of variation (GCV = {10s-1} × 100% where s is the standard deviation of the log10 transformed potencies).
Stability studies
Accelerated degradation studies were performed to predict the long-term stability of the candidate standard. Ampoules of the lyophilised preparation were stored at different temperatures, namely 45°C, 37°C, 20°C and 4°C and tested at indicated time points in L929 cell cytotoxicity assays, together with ampoules stored at the recommended temperature of −20°C and −70°C as baseline reference temperature. There was no observed loss in potency after 9.5 months, so no attempt to predict degradation rates has been undertaken. Real-time monitoring of stability is ongoing.
where y denotes the assay response, x is the concentration, α is the upper asymptote, δ is the difference between upper and lower asymptotes, β is the slope factor and γ is the ED50
Funding Statement
This work was supported by the UK Department of Health [044/069].
Acknowledgments
We are grateful to Celltrion Healthcare for donation of the material to develop and manufacture the standard and to the participants of the collaborative study for performing bioassays and providing data. We thank the different official medicines control laboratories for their support of the study. We thank Paula Dilger for technical assistance, Paul Matejtschuk, Kiran Malik and Chinwe Duru for their help in the formulation of the pilot fills and the staff of the Standards Processing Division for the preparation of the candidate materials and dispatching the study preparations. Finally, we thank Chris Burns for critical review of the manuscript, Anne Cook (Licensing division, MHRA) and Sandra Prior for helpful discussions. This work was funded, in part, by the UK Department of Health’s Policy Research programme, Grant Number 044/0069
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed on the publisher's website..
Abbreviations
- ADCC
antibody-dependent cell-meditated cytotoxicity
- CDC
complement dependent cytotoxicity
- CQA
critical quality attributes
- CD
Crohn’s disease
- ELISA
enzyme linked immunosorbent assay
- FRET
fluorescent resonance energy transfer
- GCV
geometric coefficient of variance
- GM
geometric mean
- IH
in-house
- IBD
irritable bowel disease
- IS
international standard
- IU
international units
- mAb
monoclonal antibody
- RMP
reference medicinal product
- RA
rheumatoid arthritis
- SPR
surface plasmon resonance
- TNF
tumour necrosis factor alpha
- UC
ulcerative colitis
- WHO
World Health Organisation
References
- 1.WHO Expert Committee on Biological Standarisation Sixty sixth Report. Report of a WHO informal consulation on international standards for biotherapeutic products; 2016. WHO Technical Report Series 999, p. 13–15.
- 2.WHO Expert Committee on Biological Standardization Sixtieth report. Guidelines on evaluation of similar biotherapeutic products (SBPs). WHO Technical Report Series, No.977, Annex 2; 2013. http://www.who.int/biologicals/expert_committee/TR.
- 3.Thorpe R, Wadhwa M.. Intended use of Reference Products & WHO International Standards/Reference Reagents in the development of Similar Biological Products (Biosimilars). Biologicals. 2011;39:262–265. doi: 10.1016/j.biologicals.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 4.WHO Expert Committee on Biological Standarisation Fifty fifth Report. Recommendations for the preparation, characterization and establishmentof international and other biological reference standards. WHO Technical Report Series 932; 2006, p. 73–130.
- 5.Weise M, Bielsky MC, De Smet K, Ehmann F, Ekman N, Narayanan G, Heim HK, Heinonen E, Ho K, Thorpe R, et al. Biosimilars-why terminology matters. Nat Biotechnol. 2011;29:690–693. doi: 10.1038/nbt.1867. [DOI] [PubMed] [Google Scholar]
- 6.Weise M, Kurki P, Wolff-Holz E, Bielsky M-C, Schneider CK.. Biosimilars: the science of extrapolation. Blood. 2014;124:3191–3196. doi: 10.1182/blood-2014-06-583617. [DOI] [PubMed] [Google Scholar]
- 7.McCamish M, Woollett G.. The state of the art in the development of biosimilars. Clin Pharmacol Ther. 2012;91:405–417. doi: 10.1038/clpt.2011.343. [DOI] [PubMed] [Google Scholar]
- 8.Udpa N, Million RP.. Monoclonal antibody biosimilars. Nat Rev Drug Discov. 2016;15:13–14. doi: 10.1038/nrd.2015.12. [DOI] [PubMed] [Google Scholar]
- 9.Guideline on similar biological medicinal products. Committee for Medicinal Products for Human Use. European Medicines Agency; 2014;44:1–7. [Google Scholar]
- 10.Lamanna WC, Holzmann J, Cohen HP, Guo X, Schweigler M, Stangler T, Seidl A, Schiestl M. Maintaining consistent quality and clinical performance of biopharmaceuticals. Expert Opin Biol Ther. 2018;18:369–379. doi: 10.1080/14712598.2018.1421169. [DOI] [PubMed] [Google Scholar]
- 11.Kim S, Song J, Park S, Ham S, Paek K, Kang M, Chae Y, Seo H, Kim HC, Flores M. Drifts in ADCC-related quality attributes of Herceptin®: impact on development of a trastuzumab biosimilar. MAbs. 2017;9:704–714. doi: 10.1080/19420862.2017.1305530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29:310–312. doi: 10.1038/nbt.1867. [DOI] [PubMed] [Google Scholar]
- 13.Schneider KC. Biosimilars in rheumatology: the wind of change. Ann Rheum Dis. 2013;72:315–318. doi: 10.1136/annrheumdis-2012-202941. [DOI] [PubMed] [Google Scholar]
- 14.Tebbey PW, Varga A, Naill M, Clewell J, Venema J. Consistency of quality attributes for the glycosylated monoclonal antibody Humira® (adalimumab). MAbs. 2015;7:805–811. doi: 10.1080/19420862.2015.1073429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ramanan S, Grampp G. Drift, Evolution, and Divergence in Biologics and Biosimilars Manufacturing. BioDrugs. 2014;28:363–372. doi: 10.1007/s40259-014-0088-z. [DOI] [PubMed] [Google Scholar]
- 16.NIBSC - International standards http://www.nibsc.org/standardisation/international_standards.aspx.
- 17.WHO Expert Committee on Biological Standarisation Sixty third Report. Proposed projects for endorsement. WHO Technical Report Series 980; 2014, p. 40–41. doi: 10.1177/1753193413487470. [DOI]
- 18.Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62. doi: 10.1038/nrrheum.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Services for people with rheumatoid arthritis. Report by the comptroller and auditor general. London: National Audit Office; 2009. [Google Scholar]
- 20.The Economic Burden of Rheumatoid Arthritis. Maidenhead: National Rheumatoid Arthritis Society; 2010. [Google Scholar]
- 21.van Dullemen HM, van Deventer SJH, Hommes DW, Bijl HA, Jansen J, Tytgat GNJ, Woody J. Treatment of Crohn’s disease with anti-tumor necrosis factor chimeric monoclonal antibody (cA2). Gastroenterology. 1995;109:129–135. doi: 10.1016/0016-5085(95)90277-5. [DOI] [PubMed] [Google Scholar]
- 22.Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti-TNF therapy: past, present and future. Int Immunol. 2015;27:55–62. doi: 10.1093/intimm/dxv003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Côté-Daigneault J, Bouin M, Lahaie R, Colombel J-F, Poitras P. Biologics in inflammatory bowel disease: what are the data? United Eur Gastroenterol J. 2015;3:419–428. doi: 10.1177/2050640615590302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taylor PC, Michael Peters A, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN. Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor α blockade in patients with rheumatoid arthritis. Arthritis Rheum. 2000;43:38–47. doi:. [DOI] [PubMed] [Google Scholar]
- 25.Danese S. Mechanisms of action of infliximab in inflammatory bowel disease: an anti-inflammatory multitasker. Dig Liver Dis. 2008;40:225–228. doi: 10.1016/S1590-8658(08)60530-7. [DOI] [PubMed] [Google Scholar]
- 26.PharmaCompass Top drugs by sales revenue in 2015: who sold the biggest blockbuster drugs?; 2016. https://www.pharmacompass.com/radio-compass-blog/top-drugs-by-sales-revenue-in-2015-who-sold-the-biggest-blockbuster-drugs.
- 27.Jung SK, Lee KH, Jeon JW, Lee JW, Kwon BO, Kim YJ, Soo JB, Kim DII, Lee SY, Chang SJ. Physicochemical characterization of Remsima®. MAbs. 2014;6:1163–1177. doi: 10.4161/mabs.32221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong J, Lee Y, Lee C, Eo S, Kim S, Lee N, Park J, Park S, Seo D, Jeong M, et al. Physicochemical and biological characterization of SB2, a biosimilar of Remicade® (infliximab). MAbs. 2017;9:364–382. doi: 10.1080/19420862.2016.1264550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wadhwa M, Bird C, Dilger P, Rigsby P, Jia H, Gross BME, Participants of the study. Establishment of the first WHO International Standard for etanercept, a TNF receptor II Fc fusion protein: report of an international collaborative study. J Immunol Methods. 2017;447:14–22. doi: 10.1016/j.jim.2017.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Prior S, Hufton SE, Fox B, Dougall T, Rigsby P, Bristow A. Participants of the study. International standards for monoclonal antibodies to support pre- and post-marketing product consistency: evaluation of a candidate international standard for the bioactivities of rituximab. MAbs. 2018;10:129–142. doi: 10.1080/19420862.2017.1386824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meager A, Leung H, Woolley J. Assays for tumour necrosis factor and related cytokines. J Immunol Methods. 1989;116:1–17. doi: 10.1016/0022-1759(89)90306-2. [DOI] [PubMed] [Google Scholar]
- 32.Khabar SK, Siddiqui S, Armstrong AJ. WEHI-13VAR: a stable and sensitive variant of WEHI 164 clone 13 fibrosarcoma for tumor necrosis factor bioassay. Immunol Lett. 1995;46:107–110. doi: 10.1016/0165-2478(95)00026-2. [DOI] [PubMed] [Google Scholar]
- 33.Minafra L, Di Cara G, Albanese NN, Cancemi P. Proteomic differentiation pattern in the U937 cell line. Leuk Res. 2011;35:226–236. doi: 10.1016/j.leukres.2010.07.040. [DOI] [PubMed] [Google Scholar]
- 34.Lallemand C, Kavrochorianou N, Steenholdt C, Bendtzen K, Ainsworth MA, Meritet JF, Blanchard B, Lebon P, Taylor P, Charles P, et al. Reporter gene assay for the quantification of the activity and neutralizing antibody response to TNFα antagonists. J Immunol Methods. 2011;373:229–239. doi: 10.1016/j.jim.2011.08.015. [DOI] [PubMed] [Google Scholar]
- 35.Mitoma H, Horiuchi T, Tsukamoto H, Tamimoto Y, Kimoto Y, Uchino A, To K, Harashima S, Hatta N, Harada M. Mechanisms for cytotoxic effects of anti-tumor necrosis factor agents on transmembrane tumor necrosis factor alpha-expressing cells: comparison among infliximab, etanercept, and adalimumab. Arthritis Rheum. 2008;58:1248–1257. doi: 10.1002/art.23794. [DOI] [PubMed] [Google Scholar]
- 36.Cheng JZ, Garvin D, Paguio A, Moravec R, Engel L, Fan F, Surowy T. Development of a robust reporter-based ADCC assay with frozen, thaw-and-use cells to measure Fc effector function of therapeutic antibodies. J Immunol Methods. 2014;414:69–81. doi: 10.1016/j.jim.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 37.CombiStats v5.0 EDQM- Council of Europe. www.combistats.eu.
- 38.Wadhwa M, Bird C, Dilger P, Hockley J, Rigsby P. Report on a collaborative study for proposed 3rd international standard for tumor necrosis factor -alpha (TNF-α). Geneva: World Health Organization; 2013. [Google Scholar]
- 39.Lee C, Jeong M, Lee JAJ, Seo S, Cho SC, Zhang W, Jaquez O. Glycosylation profile and biological activity of Remicade® compared with Flixabi® and Remsima®. MAbs. 2017;9:968–977. doi: 10.1080/19420862.2017.1337620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.National clinical audit of biological therapies - Annual report 2016. London: Royal College of Physicians; 2016. [Google Scholar]
- 41.Breedveld F. The value of early intervention in RA-a window of opportunity. Clin Rheumatol. 2011;30:33–39. doi: 10.1007/s10067-010-1638-5. [DOI] [PubMed] [Google Scholar]
- 42.Danese S, Fiorino G, Fernandes C, Peyrin-Biroulet L. Catching the therapeutic window of opportunity in early Crohn’s disease. Curr Drug Targets. 2014;15:1056–1063. doi: 10.2174/1389450115666140908125738. [DOI] [PubMed] [Google Scholar]
- 43.Jørgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, Lundin KEA, Mørk C, Jahnsen J, Kvien TK, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389:2304–2316. doi: 10.1016/S0140-6736(17)30068-5. [DOI] [PubMed] [Google Scholar]
- 44.Smits LJT, Grelack A, Derikx LAAP, de Jong DJ, van Esch AAJ, Boshuizen RS, Drenth JPH, Hoentjen F. Long-term clinical outcomes after switching from Remicade® to biosimilar CT-P13 in inflammatory bowel disease. Dig Dis Sci. 2017;62:3117–3122. doi: 10.1007/s10620-017-4448-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smits LJT, Derikx LAAP, de Jong DJ, Boshuizen RS, van Esch AAJ, Drenth JPH, Hoentjen F. Clinical outcomes following a switch from Remicade® to the biosimilar CT-P13 in inflammatory bowel disease patients: A prospective observational cohort study. J Crohn’s Colitis. 2016;10:1287–1293. doi: 10.1093/ecco-jcc/jjw087. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.