

ABSTRACT
Epithelial ovarian cancer (EOC) is a leading cause of cancer-related death in women. EOC is often diagnosed at late stages, with peritoneal metastases and ascites production. Current surgery and platinum-based chemotherapy regimes fail to prevent recurrence in most patients. High levels of Transforming growth factor-β (TGF-β) within ascites has been linked to poor prognosis. TGF-β signaling promotes epithelial-mesenchymal transition (EMT) in EOC tumor cells, and immune suppression within the tumor microenvironment, with both contributing to chemotherapy resistance and metastasis. The goal of this study was to develop specific synthetic inhibitory antibodies to the Type II TGF-β receptor (TGFBR2), and test these antibodies in EOC cell and tumor models. Following screening of a phage-displayed synthetic antigen-binding fragment (Fab) library with the extracellular domain of TGFBR2, we identified a lead inhibitory Fab that suppressed TGF-β signaling in mouse and human EOC cell lines. Affinity maturation of the lead inhibitory Fab resulted in several derivative Fabs with increased affinity for TGFBR2 and efficacy as suppressors of TGF-β signaling, EMT and EOC cell invasion. In EOC xenograft and syngeneic tumor models, blockade of TGFBR2 with our lead antibodies led to improved chemotherapy response. This correlated with reversal of EMT and immune exclusion in these tumor models with TGFBR2 blockade. Together, these results describe new inhibitors of the TGF-β pathway that improve antitumor immunity, and response to chemotherapy in preclinical EOC models.
KEYWORDS: Ovarian cancer, TGF-β, EMT, immune exclusion, synthetic antibodies, combination therapies
Epithelial ovarian cancer (EOC) is the leading cause of gynecological cancer-associated death in women.1 EOC is typically diagnosed at late stages with metastases already present within the peritoneal cavity.2 Although most EOC patients show initial responses to platinum-based chemotherapy, recurrence is frequent, leading to an overall survival rate of only 30%.3 Within the EOC tumor microenvironment, TGF-β signaling promotes survival of cancer stem cells, epithelial-to-mesenchymal transition (EMT), and chemoresistance.4–6 The observed impact of TGF-β signaling in EOC suggests that inhibition of this pathway is a reasonable therapeutic complement to the existing platinum-based chemotherapies.
Targeting TGF-β signaling to treat cancer is a contentious issue due to the paradoxical role of TGF-β in tumor suppression and progression.7 In early stages of tumor development, TGF-β signaling acts as a tumor suppressor by inducing apoptosis, promoting senescence, and maintaining tissue homeostasis.8 However, in late stage cancers, TGF-β signaling promotes cell invasion, tumor angiogenesis, immune suppression, and survival of cancer stem cells.9 The variable effects of TGF-β signaling on tumor growth is dependent on the tumor microenvironment, with paracrine signaling between tumor and stroma dictating the impact of TGF-β signaling.10 Inhibiting TGF-β signaling in a tumor microenvironment may therefore have profound effects on the tumor, as evident from several recent studies implicating TGF-β signaling as an antagonist of immunotherapy responses in multiple cancer types.11–14 However, appropriate use of this potential therapeutic strategy requires a robust understanding of TGF-β signaling, and its role in specific cancer types and microenvironments.
TGF-β signaling is initiated when latent pools of ligands TGF-β1, TGF-β2 or TGF-β3 within the extracellular matrix are released by proteases that allow ligand binding to Type II receptor (TGFΒR2) on the cell surface.15 TGFΒR2 is a constitutively active serine/threonine/tyrosine kinase that forms a heterotetramer with Type I receptor (TGFBR1/ALK5) upon TGF-β binding. TGFΒR1 activation leads to phosphorylation of receptor-regulated SMAD proteins (SMAD2/SMAD3) that promotes interaction with SMAD4 and regulation of TGF-β target genes in the nucleus.15 Non-canonical TGF-β signaling to mitogen-activated protein kinases (MAPKs) and Rho GTPases contributes to the potent effects of TGF-β in promoting cancer cell motility and invasion.7 A number of small molecules, a ligand trap, and antibodies have been developed to inhibit the TGF-β pathway.16 However, these inhibitors have not translated effectively from preclinical studies to clinical use.17 Thus, improvements are needed in our understanding and means of targeting the TGF-β pathway to realize the potential advantages of limiting aberrant TGF-β signaling in the tumor microenvironment.
In this study, we report on new tools for TGF-β pathway blockade and their effects on preclinical EOC tumor models. A single framework, phage-displayed synthetic antibody library produced a panel of anti-TGFBR2 Fabs when screened against the Fc-tagged extracellular domain (ECD) of TGFBR2 (TGFBR2-Fc). The Fabs were stratified based on their binding affinities and TGF-β signaling blockade. A lead inhibitory Fab (5775) was selected for affinity maturation and the resulting clones were formatted and purified as human IgG1 antibodies, which showed improved avidity and efficacy in blocking TGF-β-induced EOC cell invasion and mesenchymal gene expression. Two affinity matured IgGs (8311 and 8322) were also capable of reversing EMT and immune exclusion in metastatic EOC tumor xenograft and syngeneic models, and thus facilitating an improved response to chemotherapy.
Results
Novel synthetic antibodies for TGF-β pathway blockade identified by phage display
To identify novel inhibitory antibodies for the blockade of TGF-β signaling, we prepared an expression construct composed of the complete TGFBR2 extracellular domain (ECD) expressed as a fusion protein with IL-2 signal sequence (IL2ss) and the human IgG1 Fc domain (TGFBR2-Fc, Figure 1(a)). This construct, and Fc domain alone, were expressed in FreeStyle™ 293-F cells by transient transfection and were readily purified from conditioned media (Figure 1(b)). Purified TGFBR2-Fc was then used as antigen to screen a Fab-phage library for selective human monoclonal antibodies.18 After 4 rounds of positive and negative selection with TGFBR2-Fc and Fc respectively, we identified 30 unique TGFBR2-specific Fab-phage clones from clonal ELISA (Fig. S1A). Based on unique paratope sequence properties, 18 clones were selected for Fab protein production and validated for binding to the cognate antigen by protein ELISA (Fig. S1B). Binding kinetics of antigen specific Fabs were measured by surface plasmon resonance (SPR) revealing several Fabs with high affinity (KD < 10 nM) for the immobilized human TGFΒR2-Fc protein (Figure 1(c)). Among the panel of Abs, Fab 5775 (F5775) showed the highest Fab affinity (4.08 x1010 M) and no cross-reactivity with TGFBR1 at saturating concentrations (Fig. S1C/D).
Figure 1.
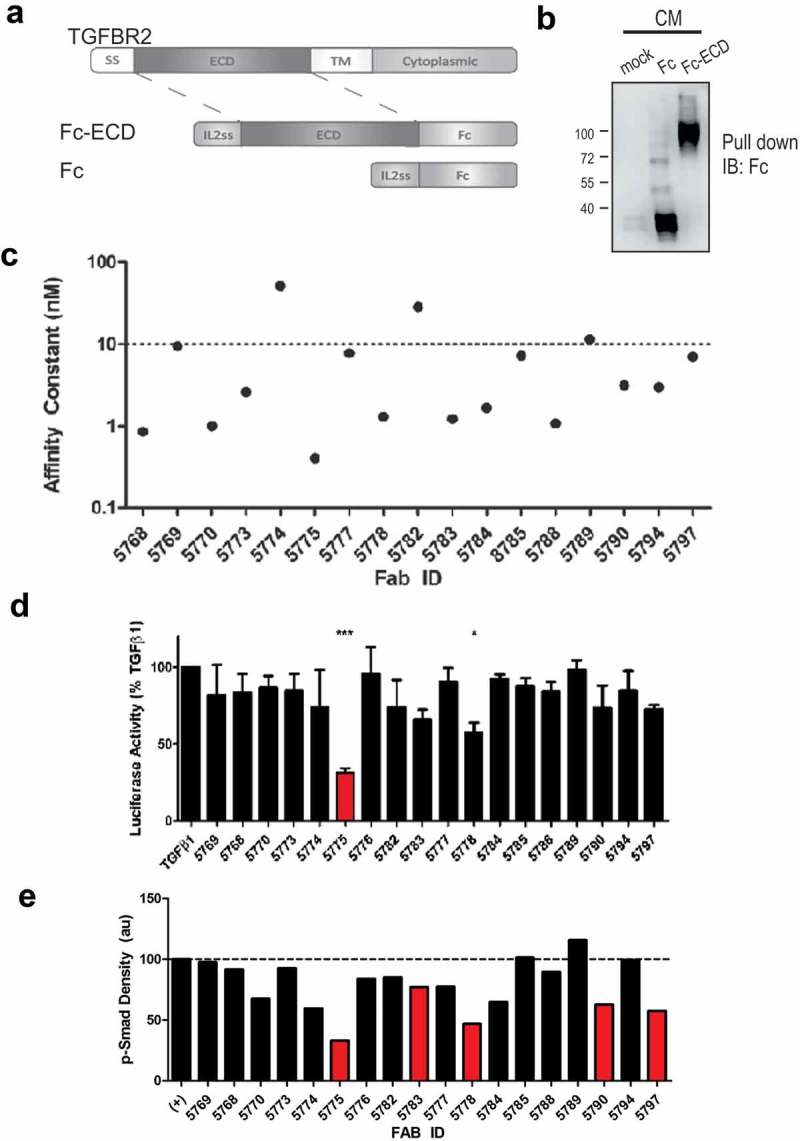
Blockade of TGF-β signaling with synthetic Fabs identified by phage display screening.
a) The extracellular domain of TGFBR2 was inserted between an IL2 signal sequence (N-terminus) and the human Fc IgG1 domain (C-terminus). b) Expression and capture of the Fc and Fc-ECD proteins from transfected HEK293F cell conditioned media (CM) using Protein A Sepharose beads and immunoblot (IB) with anti-human Fc (mock represents no transfection, positions of molecular mass markers indicated on the left). c) Affinity constants (KD) represent the interactions between unique Fabs and the TGFBR2-Fc protein, as measured by SPR. d) A Dual luciferase assay was used to measure effects of Fab treatments on Smad2/3 activity in HEK293T cells treated with TGFβ1 (10 ng/ml) for the final 10 hours of a 48 hour period post transfection (* p < 0.05, *** p < 0.001). e) Graph depicts densitometry analysis of immunoblot results for the relative SMAD2/3 phosphorylation levels in TGF-β-treated MDA-MB-231 cells with or without pretreatment with the indicated Fab’s (500 nM).
To screen for effects of these Fabs on TGF-β signaling, we transfected HEK293T cells with a TGF-β/SMAD2/3-responsive firefly luciferase reporter, along with a constitutively expressed renilla luciferase reporter as a control. The following day, cells were seeded in 12 well plates, serum starved overnight, and treated with TGF-β1 (10 ng/ml) alone, or with Fabs (500 nM) for 10 hours. Dual luciferase assays revealed the relative firefly:renilla luciferase ratio for TGF-β1 treatment alone (set at 100%) compared to co-treatments with each Fab (Figure 1(d)). Results from three independent experiments revealed significant but incomplete suppression of TGF-β-induced SMAD2/3 activation by F5775 (Figure 1(d)). To directly assess effects of Fabs on SMAD2/3 phosphorylation, we pretreated MDA-MB-231 cells for 1 hour with Fabs (500 nM), followed by treatment with TGF-β1 (10 ng/ml) for 1 hour. The relative phosphorylation of SMAD2/3 was measured by immunoblotting and densitometry revealed similar results with strong suppression by several Fabs including F5775 (Figure 1(e)). Together, these results identify F5775 as a novel synthetic Fab that inhibits TGF-β signaling via TGFBR2.
Fab 5775 suppresses TGF-β signaling and cell invasion in mouse and human EOC cells
Since F5775 was the most effective inhibitor of TGF-β signaling, we selected this Ab as our lead candidate for further testing in EOC models. We selected human SKOV3 and mouse ID8 cell lines for testing, as these widely used EOC models are responsive to TGF-β treatment.19–21 Indeed, pre-treatment of SKOV3 and ID8 cells with increasing doses of F5775 (0.5–4 μM) led to dose-dependent suppression of TGF-β1-induced SMAD2/3 phosphorylation in both cell models (Figure 2(a)). The higher doses required for F5775 to suppress pSMAD in ID8 cells expressing mouse TGFBR2, is consistent with F5775 being cross-reactive with mouse TGFBR2, but with lower affinity than the human ortholog.
Figure 2.
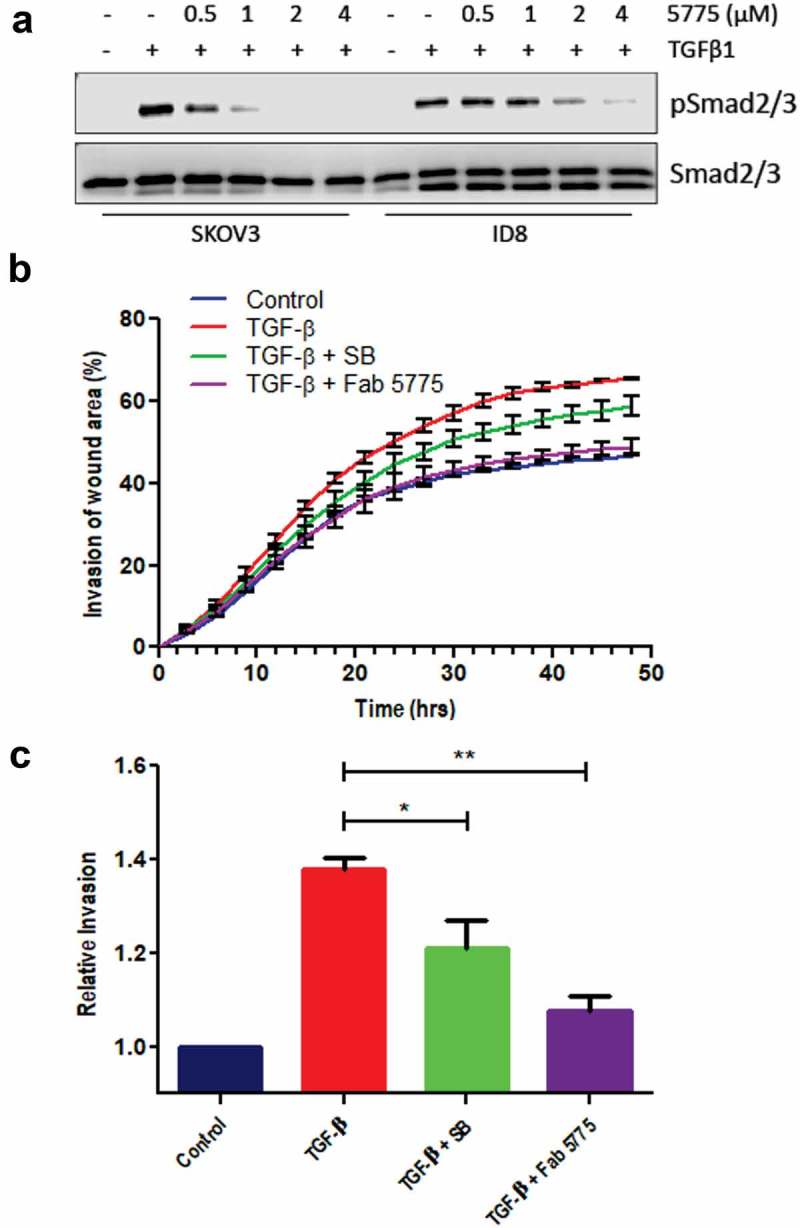
Inhibition of TGF-β signaling and EOC cell invasion by Fab 5775.
a) The effects of Fab 5775 on TGFβ1-induced SMAD2/3 phosphorylation (pSMAD2/3) was tested at the indicated doses in human SKOV3 and mouse ID8/Trp53−/- cells by IB with pSMAD2/3 and pan reactive SMAD2/3 antibodies. b) Graph depicts the kinetics of SKOV3 cell invasion over 48 hours in media with or without TGFβ1 (10 ng/ml), TGFβ1 + SB431542 (SB, 1 µM) or TGFβ1 + Fab 5775 (1 µM) using an IncuCyte ZOOM system. c) Graph depicts quantification of relative cell invasion for each treatment group described above at the assay endpoint (* p < 0.05, ** p < 0.01).
To further characterize the effects of our lead inhibitor on phenotypic changes induced by TGF-β in EOC cells, we analyzed the effects of treatments with TGF-β and F5775 on the kinetics and extent of SKOV3 cell invasion. SKOV3 cells were grown to confluence in a 96 well plate, and a wound area was generated prior to overlay with Matrigel, we then measured invasion of the wound area by SKOV3 cells treated with or without TGF-β alone, or together with small molecule TGFBR1 inhibitor SB431542 (SB, 1 µM), or F5775 (1 µM). As expected, TGF-β treatment increased the rate and extent of SKOV3 cell invasion, and this was suppressed by SB or F5775 (Figure 2(b)). At endpoint, the suppression of SKOV3 cell invasion by SB and F5775 were statistically significant, with F5775 showing the largest effect (Figure 2(c)). Together, these results identify F5775 as a novel inhibitor of TGF-β signaling in human and mouse EOC cells that can limit cell invasion.
Improved blockade of TGF-β signaling following affinity maturation screening
To further optimize the cellular potency of Ab 5775, we aimed to further improve the affinity of F5775 to hTGFBR2. To achieve this, we attempted to affinity mature F5775 by reselecting antibody sequences from a phage-displayed sublibrary based on the paratope sequence of F5775. For the sublibrary, we ‘soft-randomized’ CDRs L3, H1, H2 and H3 of F5775 to create a repertoire of combinatorially mutated clones with a bias for the F5775 paratope sequence.22 The resulting library was screened against TGFBR2-Fc with increased stringency to bias the phage pool towards higher affinity clones. Through this process we identified 45 unique TGFBR2-specific Fab-phage clones from enriched phage pools, of which 20 clones were prioritized for Fab expression based on increased affinity compared to F5775 in a single-point competitive phage ELISA (Fig. S2). The resulting 20 Fab clones were purified and compared to F5775 in the dual luciferase assays described above. Results from three independent experiments identified 8 Fabs that were significantly improved inhibitors of the TGF-β-responsive luciferase reporter compared to F5775 (Figure 3(a)). Further testing of these Fabs was performed by measuring their effects on TGF-β-induced pSMAD2/3 in SKOV3 or ID8 cells pretreated with SB, F5775 or the 11 inhibitory Fabs for 1 hour. In SKOV3 cells, treatment with 10 of these Fabs showed complete loss of pSMAD2/3 at 200 nM (Fig. S3). The effects in ID8 cells were more variable and allowed us to select several promising inhibitors (F8311, F8322, F8326, F8293) that suppressed TGF-β-induced pSMAD2/3 in mouse EOC cells (Fig. S3). Comparative titration of the matured Fabs in the dual luciferase assay revealed improved potency for these affinity matured Fab clones compared to F5775 in HEK293T cells (Figure 3(b)). All 4 of these lead inhibitory Fabs were effective in limiting TGF-β-induced pSMAD2/3 in SKOV3 cells compared to F5775 or SB (Figure 3(c)). In mouse ID8 cells, F8322 was the most potent inhibitor (Figure 3(c)). These lead inhibitory Fabs were converted to the human IgG1 format, and screened for homogeneity by size exclusion chromatography (Fig. S4). We also evaluated their kinetic binding properties towards both human and mouse TGFBR2 (Fig. S5). These assays revealed that IgG-8311 had the highest avidity for both human and mouse TGFBR2 (Table 1), and a candidate for lead inhibitor of TGFBR2 in our EOC cancer models.
Figure 3.
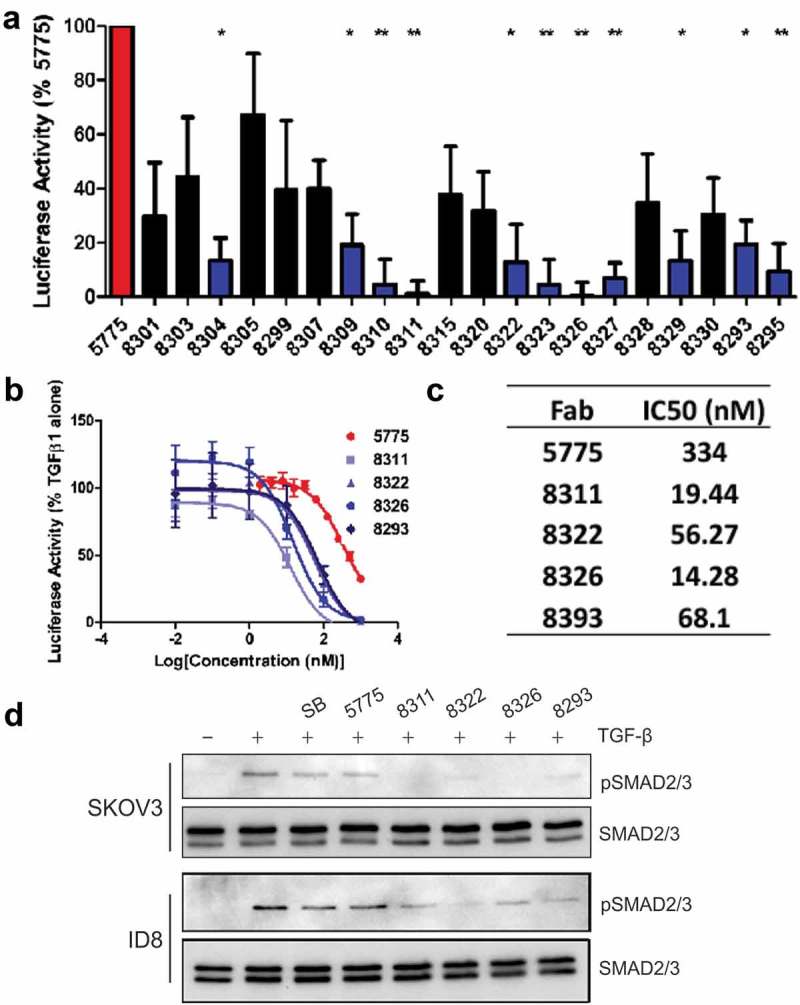
Identification of lead Fabs after affinity maturation.
a) Dual luciferase assay measuring augmented inhibition of affinity matured Fabs relative to Fab 5775 (statistical significance compared to Fab 5775 are indicated by * p < 0.05, ** p < 0.01, *** p < 0.001). b) The IC50 values for lead Fabs was determined in dual luciferase assays over a range of doses (1000–0.01 nM). c) The IC50 values are provided for each Fab based on values obtained from 3 experiments. d) The effects of affinity matured Fabs on TGFβ signaling relative to Fab 5775 and SB431542 (SB) was analyzed by IB with pSMAD2/3 and pan SMAD2/3 antibodies in SKOV3 cells (all inhibitors used at 200 nM) and ID8/Trp53−/- cells (all inhibitors used at 1 µM).
Table 1.
Affinities of lead inhibitory antibodies for human and mouse TGFBR2.
| IgG clones | KD (M) for human TGFBR2-Fc* | KD (M) for mouse TGFBR2-Fc* |
|---|---|---|
| 5775 | 1.68 x10−10 | 7.30 x10−10 |
| 8311 | 5.30 x10−12 | 1.18 x10−10 |
| 8322 | 8.34 x10−11 | 4.95 x10−10 |
| 8326 | 3.95 x10−11 | 2.12 x10−10 |
| 8293 | 6.34 x10−12 | 1.38 x10−10 |
Affinity constants were calculated from SPR experiments testing binding of the indicated IgG clones to human or mouse TGFBR2 ECD constructs fused to hIgG1 Fc domain.
Optimized TGFBR2 antibodies suppress TGF-β-induced EMT and EOC cell invasion
Next, we tested the optimized inhibitory Abs for their effects on TGF-β-induced EMT and EOC cell invasion. In SKOV3 cell invasion assays using the IncuCyte ZOOM system, the increase in cell invasion mediated by treatment with TGF-β was abolished by co-treatment with F8311, F8322 or F8326 at 250 nM (Figure 4(a)). These results were comparable to the small molecule TGFBR1 inhibitor SB. These antibodies were further tested in a 3D cell motility assay, with SKOV3 cells grown as spheroids in round bottom wells prior to transfer to flat bottom wells compatible with outward migration in the presence of TGF-β. Treatments with SB or affinity matured Fabs (1 µM) led to significant suppression of SKOV3 cell motility (Figure 4(b), Fig. S6). Considering the importance of EMT for EOC cell invasion, we tested the effects of F8311 (500 nM) on the expression of selected EMT genes that are positively regulated by TGF-β using quantitative RT-PCR. The results of three independent assays revealed that F8311 significantly attenuated the effects of TGF-β treatment of SKOV3 cells on the expression of mesenchymal markers Slug, Snail and Fibronectin (Figure 4(c)). The TGF-β-induced switch from epithelial to mesenchymal morphology, and increased N-cadherin expression was also reversed by F8311 treatment in ID8/Trp53−/ – cells (Figure 4(d)). To define the mode of action of F8311, we tested the localization of F8311 in EOC cells by immunoflourescence (IF) staining relative to markers of endosomes (EEA1) and lysosomes (Lamp1). We observed diffuse localization of F8311 in small puncta throughout the cells at early times, followed by concentration in EEA1+ endosomes after 45 minutes, and Lamp1+ lysosomes at 75 minutes of treatment (Fig. S7). This likely reflects the targeting of F8311/TGF-β receptor complexes for endocytosis and degradation in EOC cells, leading to desensitization of these cells to extracellular TGF-β. Together, these results provide evidence of the effects of TGF-β blockade with our lead inhibitory antibodies in EOC cell models, allowing for prioritization of particular antibodies for testing in EOC tumor models.
Figure 4.
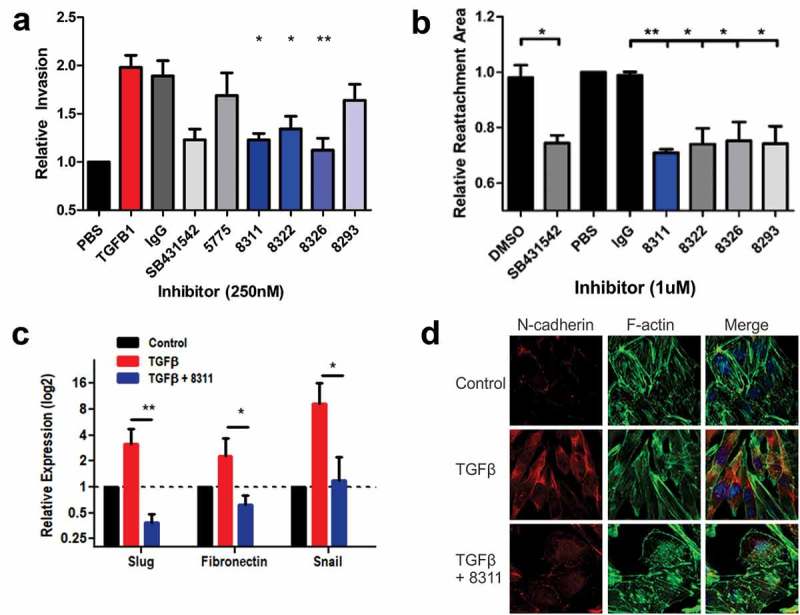
Affinity matured Fabs inhibit TGFβ-induced EMT in EOC cells.
a) Graph depicts the relative invasion of SKOV3 cells treated with or without TGFβ1 (10 ng/ml), or TGFβ1 + control IgG, SB431542, Fab 5775, or affinity matured Fabs (all inhibitors added at 250 nM). b) Spheroid reattachment motility assays were performed using SKOV3 cells treated with vehicle (DMSO), SB431542, or the indicated Fabs (1 µM) for 72 hours. c) Expression of EMT genes (Slug, Fibronectin, Snail) in SKOV3 cells were analyzed by quantitative RT-PCR to test the effects of TGFβ1 (10 ng/ml) treatment alone, or together with Fab 8311 (500 nM) for 48 hours (statistically significant differences from TGFβ1 alone are indicated by * p < 0.05, ** p < 0.01). d) Immunofluorescence staining of N-cadherin, Phalloidin staining of F-actin and DAPI staining of nuclei in ID8/Trp53−/- cells treated with or without TGFβ1 (10 ng/ml) and Fab 8311 (1 µM) for 48 hours prior to fixation.
TGFBR2 blockade improves chemotherapy response in a metastatic EOC tumor xenograft model
To test our lead inhibitor IgG-8311 in a model of metastatic EOC, we performed intraperitoneal (i.p.) injections of SKOV3-Luc cells in female Rag2−/-:IL2Rɣc−/ – mice (lacking B, T and NK cells; Figure 5(a)). After 3 weeks, mice were randomized (5 per group) between treatments with control IgG (Ctl IgG) or IgG-8311 alone (1 mg/kg, twice weekly), or in combination with carboplatin (Carbo; 15 mg/kg, weekly). After 5 weeks of treatment, mice were injected with D-luciferin to measure total metastatic tumor burden, which trended lower for mice treated with IgG-8311 alone or in combination with Carbo (Figure 5(b)). Mice were sacrificed and tumor nodules were isolated throughout the peritoneum, and a significant reduction in total tumor mass in the IgG-8311/Carbo treatment group compared to control was observed (Figure 5(c)). To investigate the effects of IgG-8311 treatment on gene expression within the SKOV3 tumors, we isolated total RNA from 3 tumors from separate mice in the Ctl IgG and IgG-8311 treatment groups. The expression of human cancer progression genes were analyzed using Nanostring technology, and differentially expressed genes with TGFBR2 blockade were visualized in a volcano plot (Figure 5(d)). The genes that were significantly upregulated in tumors treated with IgG-8311 included those that are negatively regulated by TGF-β (e.g. CDH1, SOX9, SMURF1), and target genes for SMAD or Wnt/β-catenin (Table 2, Fig. S8A). Of the genes that were downregulated in IgG-8311-treated tumors, several are known targets of the TGF-β pathway (e.g. IL-6, CXCR4, MET; Table 3). Interestingly, the expression of SMAD3 was also significantly reduced, along with other genes regulated by Wnt/β-catenin, p300/CBP, Myc and Fos/Jun (Fig. S8B). Together, these results demonstrate that TGFBR2 blockade can reverse mesenchymalization of EOC tumors, and this is known to increase sensitivity to chemotherapy drugs such as carboplatin.
Figure 5.
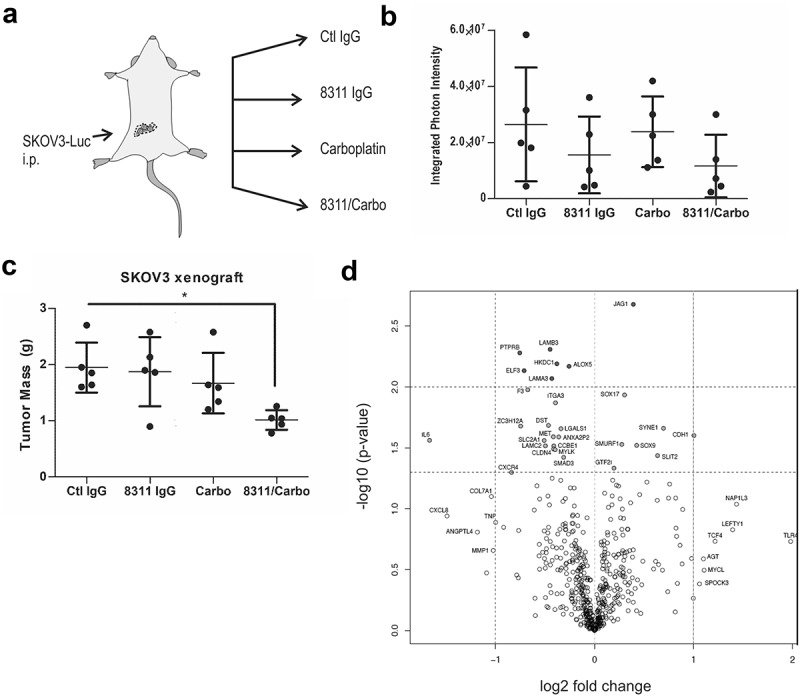
Effects of TGFBR2 blockade in metastatic EOC xenograft mouse model.
a) Schematic of metastatic EOC xenograft model involving intraperitoneal (i.p.) injection of SKOV3-Luc cells in female Rag2−/-:IL2Rɣc−/- mice. After 3 weeks, mice were randomized between control IgG or 8311 IgG (1 mg/kg, i.p., twice weekly) treatments alone, or in combination with carboplatin (Carbo, 15 mg/kg, i.p., weekly) and treated for 5 weeks. b) Biophotonic imaging of D-luciferin-injected mice was performed, with graph depicting the total photon flux for each mouse and treatment group. c) The tumor nodules were collected and total mass recorded for each mouse and treatment group (* p < 0.05). D) Volcano plot depicts differentially expressed genes in tumors from the 8311 IgG treatment compared to control (n = 3/group) using Nanostring.
Table 2.
Genes upregulated in SKOV3 tumors treated with anti-TGFBR2 IgG-8311.
| Name |
p value* |
fold change* |
TGF-β pathway* |
| CDH1 | 0.025 | 2.01 | yes |
| SYNE1 | 0.021 | 1.62 | yes |
| SLIT2 | 0.036 | 1.55 | yes |
| SOX9 | 0.03 | 1.34 | yes |
| JAG1 | 0.002 | 1.31 | yes |
| SOX17 | 0.011 | 1.23 | |
| SMURF1 | 0.029 | 1.21 | yes |
*based on Nanostring analysis of tumor RNA for IgG-8311 treatment group relative to control IgG
Table 3.
Genes downregulated in SKOV3 tumors treated with anti-TGFBR2 IgG-8311.
| Name | p value* | fold change* | TGF-β pathway* |
|---|---|---|---|
| IL6 | 0.027 | −3.176 | yes |
| CXCR4 | 0.05 | −1.788 | yes |
| PTPRB | 0.005 | −1.688 | |
| ZC3H12A | 0.021 | −1.678 | |
| ELF3 | 0.007 | −1.637 | |
| F3 | 0.01 | −1.596 | yes |
| SLC2A1 | 0.027 | −1.423 | |
| LAMC2 | 0.03 | −1.411 | |
| DST | 0.02 | −1.381 | |
| LAMB3 | 0.004 | −1.365 | yes |
| LAMA3 | 0.008 | −1.348 | |
| MET | 0.025 | −1.334 | |
| CLDN4 | 0.032 | −1.332 | |
| CCBE1 | 0.03 | −1.33 | |
| MYLK | 0.033 | −1.317 | yes |
| ITGA3 | 0.013 | −1.315 | yes |
| HKDC1 | 0.006 | −1.302 | |
| ANXA2P2 | 0.026 | −1.284 | |
| LGALS1 | 0.022 | −1.265 | |
| SMAD3 | 0.038 | −1.242 | yes |
| ALOX5 | 0.006 | −1.195 |
*based on Nanostring analysis of tumor RNA for IgG-8311 treatment group relative to control IgG
TGFBR2 blockade limits exclusion of cytotoxic immune cells in a syngeneic metastatic EOC model
To test the effects of TGFBR2 blockade in an immune competent model of metastatic EOC, we used mouse ID8 cells with CRISPR/Cas9-mediated deletion of Trp53 (ID8/Trp53−/-),23 for i.p. injections in female C57BL/6 mice. After 2 weeks, mice were randomized (5 per group) between control IgG or IgG-8322 treatments alone (1 mg/kg, i.p., twice weekly), or in combination with Carbo (15 mg/kg, i.p., weekly) treatment (Figure 6(a)). After 5 weeks of treatment, mice were sacrificed and tumor nodules were collected and weighed. Although the total tumor burden trended lower with IgG-8322 treatment, only Carbo treatment alone or in combination with IgG-8322 showed a significant reduction (Figure 6(b)). To investigate the immune modulatory effects of TGFBR2 blockade in this model, we isolated total RNA from EOC tumors from control IgG and IgG-8322 treatment groups. We then quantified RNA transcript levels for immune markers, including Cd45 (pan leukocyte), and Cd8a (CD8 T cells) by RT-qPCR. Compared to tumors from IgG control group, tumors from the IgG-8322 treatment group had significantly increased Cd8a and Cd45 levels (Figure 6(c)). This is consistent with TGF-β signaling promoting immune exclusion in EOC tumors.
Figure 6.
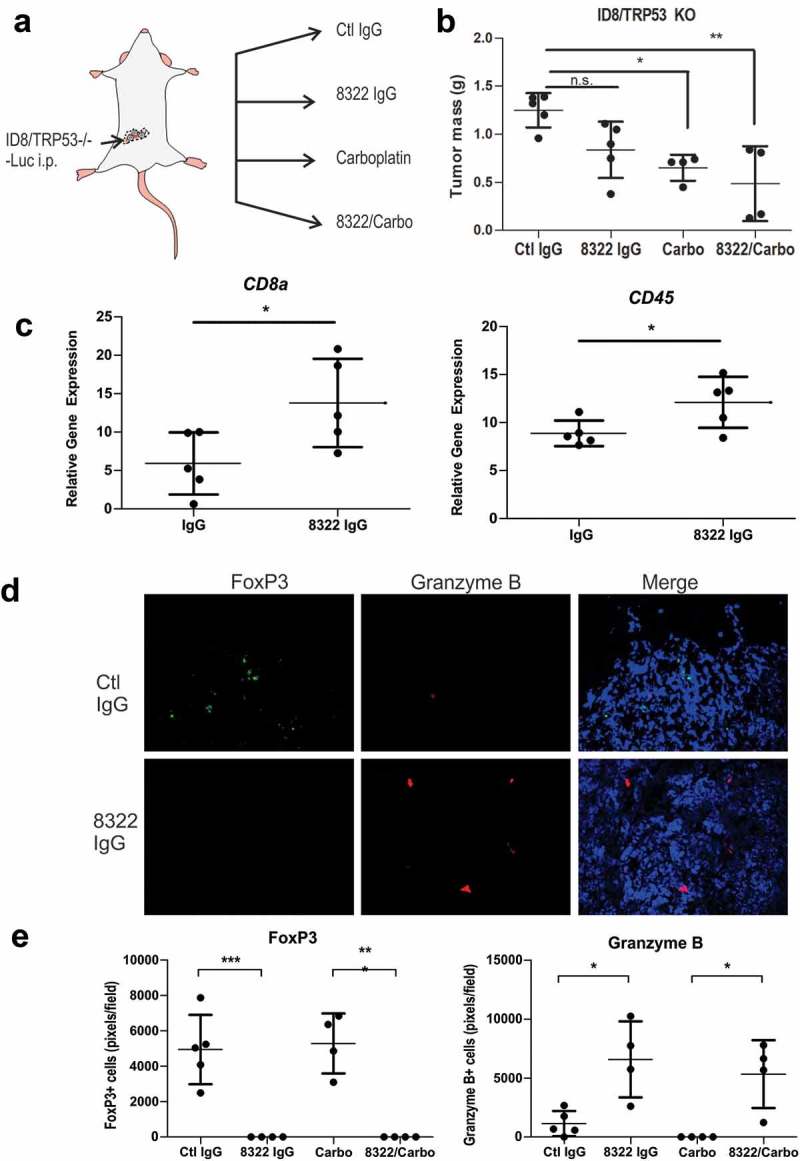
Highly cross reactive Fab 8322 modifies the immune cell landscape in a syngeneic model of EOC.
A) Schematic of sygeneic model of metastatic EOC involving i.p. injections of ID8/Trp53−/- cells in female C57BL/6 mice. After 2 weeks, mice were randomized between control IgG (Ctl) or 8322 IgG (1 mg/kg, i.p., twice weekly) treatments alone, or in combination with Carboplatin (Carbo, 15 mg/kg, i.p., weekly; n = 5/group). B) Graph depicts the total mass of tumor nodules for each mouse and treatment group (* p < 0.05, ** p < 0.01). C) Total RNA was extracted from Ctl IgG and 8322 IgG treated tumors and subjected to RT-qPCR with primers for Cd8a and CD45, with normalization to GAPDH. Graph depicts the mean values for each mouse in the treatment group (* p < 0.05). D) Tumor cryosections were subjected to IF staining of FoxP3+ (green) and Granzyme B+ (red) immune cell populations in mice treated with control IgG or 8322 (total area marked by DAPI+ nuclei). E) Graph depicts scoring for each mouse and treatment group (* p < 0.05, ** p < 0.01, *** p < 0.001).
To further investigate the immune state of the EOC tumors, we prepared cryosections for immunofluorescence staining of markers for regulatory T cells (Tregs, FoxP3) and cytotoxic CD8 T cells or NK cells (Granzyme B). We observed a significant skewing of these immune populations, with fewer Tregs and increased numbers of cytotoxic immune cells when introducing the TGFBR2 blockade alone or in combination with chemotherapy (Figure 6(c), (d)). Our results are consistent with the immune suppressive and immune exclusion effects of TGF-β signaling reported in other cancers,11–14 and the potential benefit of TGF-β blockade to sensitize EOC tumors to chemotherapy and antitumor immune responses.
Discussion
Treatments for EOC patients have remained largely unchanged for decades, and although chemotherapy response rates are initially high, recurrence is inevitable and results in low survival rates.3 In this paper, we developed novel synthetic antibodies to inhibit TGF-β signaling, and tested their effects on EMT, immune suppression, and chemotherapy responses in EOC using both cell and tumor models (Figure 7). We identified several promising synthetic antibodies that were effective inhibitors of TGF-β signaling in vitro. By deploying these antibodies in EOC tumor models, we demonstrated the sensitizing effects of TGF-β signaling blockade for effective chemotherapy responses through reversal of EMT and immune exclusion. In future, there may also be strong potential for TGFBR2 blockade to synergize with other emerging forms of cancer therapies for EOC, including immunotherapies.
Figure 7.
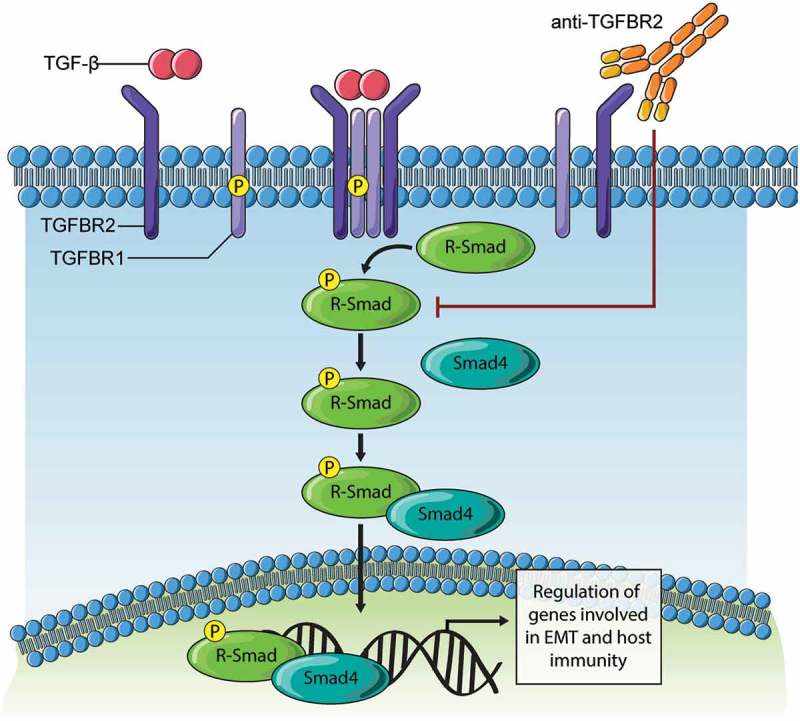
Model of TGFBR2 blockade effects on TGF-β signaling.
Binding of TGF-β ligand to TGFBR2 subunits results in clustering in a complex with TGFBR1/ALK5 that is competent for downstream signaling. The subsequent phosphorylation of receptor-activated SMAD (R-Smad) proteins (Smad2, Smad3) triggers interaction with Smad4 and nuclear entry. In tumor cells, this Smad complex promotes expression of genes driving EMT. In immune cells, this Smad complex promotes immune suppressive gene expression. The development of synthetic antibodies targeting TGFBR2 reported in this study, and their deployment in cancer models results in loss of TGF-β binding and downstream signaling. Thus, TGFBR2 blockade can evoke improved responses to chemotherapy in ovarian cancer models, and possibly other forms of therapy that rely on immune activation within the tumor microenvironment.
In this study, we report on a panel of novel synthetic antibodies that were engineered to selectively bind TGFBR2 and mediate the blockade of TGF-β signaling in both mouse and human cancer cells and tumor models. The primary screen led to identification of a lead inhibitory antibody clone (F5775) with low nanomolar affinity that was most effective in suppressing TGF-β1-induced expression of a SMAD2/3-responsive luciferase reporter. From this clone, a number of derivatives were obtained by affinity maturation screening, and four of these clones functioned as optimized inhibitors of TGF-β1 signaling. To boost the avidity and stability of these lead inhibitors, we converted the Fabs to full-length human IgG1 antibodies for further testing in EOC cell and tumor models. In human EOC cells treated with TGF-β, F8311 and IgG-8311 showed potent suppression of mesenchymal gene expression and cell invasion. This correlated with rapid association rates (ka) and slow off rates (kd) for this antibody in SPR assays with human TGFBR2-ECD. In EOC cells treated with F8311, we observed co-localization of the F8311 with markers of early endosomses and lysosomes. This is consistent with these inhibitory antibodies promoting internalization and degradation of TGFBR2, causing reduced sensitivity to TGF-β in the EOC cell models. This is similar to at least one mode of action of the clinical grade HER2 inhibitory antibody Trastuzumab, that promotes internalization of HER2 in HER2-positive EOC cancer cells.24 However, further studies are needed to map the TGFBR2 epitopes of these lead inhibitory antibodies, and fully dissect their potential modes of action.
We used a xenograft model of advanced metastatic ovarian cancer to further characterize the effects of our lead inhibitory antibody. In mice with peritoneal SKOV3 tumors, treatment with IgG-8311 led to a partial reversal of EMT in these tumors, but no significant reduction in tumor burden. However, combination treatments with IgG-8311 and carboplatin led to significant reduction in tumor burden for this EOC model with chemoresistant properties. This is consistent with a previous study showing that treatment with small molecule inhibitors of TGF-β signaling in EOC spheroid cells led to increased sensitivity and response to platinum-based chemotherapies.5 This likely relates to evidence that EMT and chemoresistance, with mesenchymal or undifferentiated cells being more resistant to chemotherapy.25 In our study, treatment of SKOV3 cells in vivo with IgG-8311 led to the upregulation of genes that characterize an epithelial phenotype (CDH1, SLIT2), while reducing the expression of genes associated with a mesenchymal phenotype (ITGA3, ELF3).26–28 Moreover, evidence is mounting in favor of cancer stem cells (CSCs) activating EMT as a mechanism of intrinsic drug resistance.29 During EOC progression, SMAD3-dependent TGF-β signaling promotes EMT and the proportion of cells expressing stemness markers (CD44+/CD117+) via transglutaminase 2 (TG2).4 Interestingly, inhibition of TGF-β signaling by IgG-8311 resulted in the downregulation of SMAD3, an important regulator of the stem cell phenotype in multiple cancers, and a potential route through which the undifferentiated state of CSCs is perturbed.30–33 Considering the critical role of EMT in EOC tumor progression, and the potential contribution of CSCs to chemotherapy resistance, new strategies are needed that target CSCs and undifferentiated EOC cells. In addition to this study showing benefit of TGFBR2 blockade, another recent study using the TGFBR1 small molecule inhibitor LY2157299/Galunisertib showed promise in several EOC xenograft models.34 They showed that LY215729 treatments prior to EOC cell injection, and throughout tumor outgrowth, led to reduced ascites development and tumor growth.34 Given these recent findings, it will be important to test whether pretreatment of mice with our inhibitory TGFBR2 antibodies leads to improved efficacy as a monotherapy. It will also be important to extend further testing of LY215729 to immune competent models of EOC in combination with chemotherapy and immunotherapy regimes.
The impact of TGF-β signaling on developing a permissive EOC tumor immune microenvironment is profound, and provides rationale for TGF-β blockade in metastatic EOC. Treg cells (CD4+/CD25+/FoxP3+) are key immune suppressors in EOC, with high levels in malignant ascites linked to poor survival outcomes in patients with ovarian carcinoma.35 Tregs are thymus – or peripherally – derived, and release factors such as TGF-β that suppress the cytolytic functions of NK cells and CD8 T cells.36 Tumor-derived TGF-β can promote the conversion of CD4 T cells to Tregs and thereby promote immune evasion.37 Interestingly, our treatments with IgG-8322 in the syngeneic ID8/Trp53−/- engraftment model of metastatic EOC resulted in a significant reduction in FOXP3+ cells within the tumors. In contrast, the numbers of Granzyme B+ cells were significantly elevated in the IgG-8322-treated tumors, thus demonstrating that TGFBR2 blockade can limit exclusion of cytotoxic CD8 T cells or NK cells in this EOC model. Moreover, IgG-8322 treated tumours exhibited higher expression levels of CD45 and CD8a (Figure 6(c)) but not CD49b (data not shown). This suggests that TGFBR2 blockade was linked to enhanced infiltration of CD8 T cells, compared to promoting NK cell recruitment. The net opposing effects of TGFBR2 blockade on FOXP3+ and Granzyme B+ cells is consistent with known roles of TGF-β promoting immune suppression in EOC cancer models.36,38 In human ovarian cancer, the density of CD8 T cells in tumors has been linked to favorable response to chemotherapy.38,39 This may also explain the benefits of combining TGFBR2 blockade with Carboplatin in the syngeneic EOC tumor model studies, with this treatment group having minimal tumor burden at endpoint. It will be important to extend these combination treatments in future survival studies, and directly compare with survival benefits with Carboplatin alone. Given the skewed immune profile of the EOC tumors with TGFBR2 blockade in our study, it will also be interesting to pursue additional combination therapies with immune checkpoint inhibitors. Previous studies with PD-1 or PD-L1 blockade revealed improved survival for 25–40% of mice in the ID8 syngeneic model.40 Future studies will be needed to test the potential benefit of combination therapies with these agents and TGFBR2 blockade. These studies in EOC tumor models will build on recent studies implicating TGF-β signaling as a limiting factor for effective immune checkpoint inhibitor treatments in several other cancer types.11,13,14 In EOC tumors, we predict that TGF-β signaling blockade will limit immune exclusion, and provoke a robust response to immune checkpoint inhibitors. Given the clinical approval of several immune checkpoint inhibitors, this may lead to rapid translation of this knowledge into new combination clinical trials with TGF-β pathway inhibitors.
In conclusion, new therapies capable of eliminating metastatic ovarian cancers are urgently needed to improve overall survival in these patients. We predict that disruption of immune suppressive paracrine signaling networks in the peritoneal tumor microenvironment will sensitize these tumors to platinum-based chemotherapy treatment regimes. Specifically, the blockade of TGF-β signaling represents a promising avenue for improving the sensitivity of EOC tumors to chemotherapy and to bolster anti-tumor immunity.
Materials and methods
TGFBR2 extracellular domain cloning, expression and phage selections
A mammalian expression construct was designed for expression of amino acids 23–534 of the human TGFBR2 ECD in frame with an IL-2 signal sequence at the N-terminus and human IgG1 Fc domain at the C-terminus using the pFuse2 vector (InvivoGen, San Diego, CA, USA). The construct was expressed and affinity purified from conditioned media of FreeStyle™ 293-F cells (InvivoGen) following transient transfection using XtremeGene HP (Roche). The mouse TGFBR2-Fc (catalog number 532-R2) and human TGFBR1-Fc (catalog number 3025-BR) were purchased from R&D Systems. The ECD-Fc fusion protein was used as the antigen to screen a phage displayed synthetic humanized Fab library.18 Binding selections, phage ELISAs and Fab protein purification were performed as described.41,42 Briefly, 1013 phage from a synthetic Fab library were cycled through rounds of binding selection with the antigen immobilized on 96-well Maxisorp Immunoplates (Fisher Scientific, Nepean, ON, Canada). After four rounds of positive selection, preceded by negative selection using human IgG1 Fc domain alone, phages from round 4 were produced as individual Fab-phage clones in 96-well format and Fab-phage ELISAs were performed to detect specific binding clones. Clones with positive binding to TGFBR2-Fc compared to Fc domain were subjected to DNA sequencing. The genes encoding for variable heavy- and light-chain domains of unique positive clones were sub-cloned into vectors designed for production of Fab or hIgG1 proteins in Escherichia coli or transfected FreeStyle™ 293-F cells, respectively, and proteins were affinity-purified on Protein A columns (GE Healthcare, Mississauga, ON, Canada). Affinity maturation phagemid libraries were created using optimized procedures that allow for the rapid construction of very large phage-displayed antibody repertoires (> 1010 unique clones).41,42 A dut−/ung− E. coli CJ236 host was transformed with template plasmid (5775) and helper phage. The result of transforming a dut−/ung− host was the propagation of phage encapsulating uracil-containing ssDNA (dU-ssDNA) which was then purified by precipitation with polyethelyne glycol. Oligonucleotides containing the mutagenic sequences for soft-randomizing CDRs of 5775, flanked by fifteen base pairs of sequence complementary to the annealing region, were annealed to the dU-ssDNA template of 5775. After annealing, the mutagenic oligonucleotides served to prime synthesis by T7 DNA polymerase of a DNA strand complementary to the uracil-containing template strand, forming a synthetic daughter strand lacking uracil. T4 DNA ligase ligated the synthesized DNA fragments, forming covalently closed circular double-stranded heteroduplex DNA (CCC-dsDNA). CCC-dsDNA was desalted and concentrated in preparation for electroporation. Selections using the affinity matured library were performed as earlier with increased stringency of washes and incorporating a final incubation of TGFBR2-Fc-bound phage with solution-phase TGFBR2-Fc (200 μg/mL) in round 4 to enrich for clones with slow off-rates. Estimation of affinity for selected matured phage clones was performed using high-throughput competitive phage ELISA.43
Surface plasmon resonance
SPR measurements were performed using a Bio-Rad ProteOn XPR instrument.
Human TGFBR2-Fc protein (180 RUs) was captured on a GLC sensor chip with an immobilized anti-Fc antibody (Jackson ImmunoResearch cat # 109–005-098) for Fab affinity measurement while human and mouse TGFBR2-Fc proteins were immobilized using amine-coupling for IgG avidity measurements. Four three-fold serial dilutions of Fab (200 nM) or IgG (100 nM) protein in PBS, 0.05% Tween20 were injected for 120 seconds to monitor association, followed by injection of PBS, 0.05% Tween-20 for 900 seconds to monitor dissociation. Experiments were performed at 25 °C. Sensorgrams were fit globally to a 1:1 binding Langmuir model using Bio-Rad ProteOn Manager fitting software.
Size-exclusion chromatography analyses of TGFBR2 IgGs
Size exclusion chromatography analysis was performed on a BioRad NGC chromatography system fitted with a C96 autosampler. Fifty micrograms of an IgG sample (and Trastuzumab control) at a concentration of 1 g/l was injected using a onto a Tosoh Bioassist G3SWxL column at a flow rate of 0.5 ml/min in a phosphate-buffered saline mobile phase (pH 7.5). Protein elution was monitored by measuring absorbance at 215 and 280 nm.
Smad-dependent luciferase reporter assay
The inhibition of SMAD activation by unique Fab clones was measured using a SMAD-dependent firefly luciferase reporter construct (Caga(12)-Luc) containing 12 repeated Smad transcriptional response elements (TRE) directly upstream of firefly luciferase (provided by Dr. JJ LeBrun, McGill University). HEK293T cells were transfected with the Smad-dependent firefly reporter vector (2.5 ug) and a separate control vector, pLightSwitch_3UTR, enabling constitutive expression of Renilla luciferase (0.25 ug). After 24 hours, cells were washed and distributed across 24-well plates coated with poly-L-lysine in serum free media overnight. Cells were then washed and incubated with serum free media containing PBS, 10 ng/mL TGF-β1 (PeproTech) or 500 nM Fab for 10 hours prior to lysis and quantification of luminescence using the Dual-Luciferase Reporter Assay System (Promega). The Firefly luciferase signal was normalized to the Renilla luciferase signal for each condition and percent inhibition was measured relative to the TGFβ1 treatment alone. Titration curves were generated using a log(inhibitor) vs. response non-linear fit model using GraphPad Prism 6.0 software.
Immunoblotting and quantitative RT-PCR
For immunoblot screening of Fabs identified by phage display, cells were distributed in 24-well plates at a density of 1.25 × 105 cells/well. After adherence, cells were washed with PBS and provided serum free media for 24 hours prior to the addition of control IgG or Fab for 1 hour in fresh serum free media. Cells were then incubated with TGF-β1 (10 ng/mL) for 1 hour prior to lysis in RIPA lysis buffer (50 mM Tris, 5 mM EDTA, 150 mM NaCl, 1% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS, 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM Na3VO4, 100 μM phenylmethylsulfonyl fluoride, 50 mM NaF). Anti-SMAD2/3 (CST) and anti-phospho-SMAD2 (CST) primary antibodies were used (1:1000) during the initial screens in conjunction with a horseradish peroxidase-linked anti-rabbit secondary antibody (CST, 1:5000). ECL Western Blotting Substrate (Thermo Scientific) was used for detection. For quantitative RT-PCR, cells were incubated with media, TGF-β1 alone (10 ng/ml) or in combination with F8311 (500 nM) for 48 hours prior to RNA isolation using TRIZOL (Sigma-Aldrich). Total RNA was quantified and served as template for cDNA synthesis using iScript Select cDNA Synthesis Kit (Bio-Rad). Quantitative PCR was performed using the iO SYBR Green Supermix Kit (Bio-Rad). The comparative CT method was used to generate relative gene expression using GAPDH as the control for input. All conditions were analyzed relative to the media control.
Cell invasion assays
SKOV3 cells were distributed in 96 well plates (Sarstedt) at 20, 000 cells/well. After 24 hours, cells were pretreated overnight with the indicated Fab or IgG and controls in media supplemented with 0.2% FBS. A single wound area was made in each well using a woundmaker device (Essen Bioscience), wells were rinsed in sterile PBS, and each well was overlayed in media (with 0.2% FBS) supplemented with 10% Matrigel (BD Biosciences). We continued treatments with or without the indicated Fab or IgG and TGF-β1 (10 ng/ml) for 48 hours with phase contrast images acquired for each well at 2 hour intervals using an IncuCyte ZOOM system (Essen BioScience). The cell densities in the wound areas for triplicate wells were calculated using IncuCyte software. For spheroid invasion assays, SKOV3 cells were grown as spheroids by seeding 1,000 cells/well across a 96-well round bottom plate coated with poly-HEMA (Sigma Aldrich). After 72 hours of growth in 10% FBS, individual spheroids were retrieved and were distributed across a 24-well plate with treatment media with or without TGF-β1 (10 ng/ml) and indicated Fab or IgG (1 µM) for 24 hours. Images were acquired immediately following spheroid attachment and after 24 hours in treatment media. Differences in the area of cell dissemination compared to spheroid area at time 0 hours was quantified using Image-J software.
Fab 8311 internalization assay
SKOV3 cells were seeded on fibronectin-coated coverslips, and the following day were incubated with or without F8311 (1 µM) for 45 or 75 minutes in growth media supplemented with Leupeptin (100 µg/ml) to limit lysosomal degradation. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton-X100, and subjected to immunofluorescence staining with mouse anti-EEA1 or rabbit anti-Lamp1 overnight at 4 °C. Following extensive rinsing in PBS/0.1% Triton-X100, detection antibodies were added: Alexa Fluor 488-anti-human Fab together with either Alexa Fluor 568-conjugated goat anti-mouse IgG or Alexa Fluor 568 goat anti-rabbit IgG (DAPI was included to visualize cell nuclei). The coverslips were rinsed and mounted in Mowiol prior to spinning disk confocal imaging (Queen’s University Biomedical Imaging Centre, WAVE FX confocal microscope, Quorom Tech.). Images from each channel were merged and analyzed using Metamorph software.
EOC tumor xenograft and syngeneic models
To evaluate effects of TGFBR2 blockade on metastatic EOC tumor progression, SKOV3-pWPI-Luc cells (5 x106) were injected in the peritoneum of female Rag2−/-:IL2ɣc−/- mice. After 3 weeks, mice were randomized (5/group) between control IgG (gammaglobulin) or 8311 IgG (1 mg/kg, i.p., twice weekly) treatments alone, or in combination with carboplatin (15 mg/kg, i.p., weekly). Treatments continued for 5 weeks, until any of the animal protocol-defined endpoints were reached in control cohort. At endpoint, biophotonic imaging was conducted on mice that were injected with D-luciferin (10 µL/g of a 7.5 mg/ml solution i.p.) and anesthetized with isoflurane (Queen’s University Biomedical Imaging Centre). Following this, all mice were sacrificed and visible tumor nodules were collected within the peritoneum, and the total tumor mass recorded for each animal. Total RNA was isolated from tumor nodules from control IgG and IgG-8311 treatment groups by homogenization in TRIZOL (Sigma-Aldrich). RNA (100 ng) was hybridized to specific capture and barcoded reporter probes from the PanCancer Progression codeset (Nanostring Technologies, Seattle, WA) prior to being immobilized on a cartridge. An nCounter Digital Analyzer was used to count the fluorescent barcoded probes to quantify each target RNA molecule (Queen’s Laboratory for Molecular Pathology, Queen’s University). Processing and normalization of the data was performed using nSolver 4 software (Nanostring) to identify and rank differentially expressed genes that were statistically significant between treatment groups. The differentially expressed genes were mapped to the TGF-β pathway (Nanostring Advanced Analysis) and transcription factor networks (Enrichr). For syngeneic EOC tumor studies, the ID8/Trp53−/- cell model was used23 (kindly provided by Dr McNeish, University of Glasgow). Cells (5 x106) were injected i.p. in female C57BL/6 mice (6–8 weeks old, Charles River, Canada). After 2 weeks, mice were randomized between treatments with control IgG or IgG-8322 (1 mg/kg, i.p., twice weekly) alone, or in combination with carboplatin (15 mg/kg, i.p., weekly). At endpoint, visible tumor nodules were collected within the peritoneum, and the total tumor mass recorded for each animal.
Tumor nodules were prepared for cryosectioning, and frozen 20-μm sections were post-fixed in acetone and blocked for 1 hour with 3% bovine serum albumin prior to addition of primary antibodies overnight at 4°C. Sources and dilutions of primary antibodies were as follows: Alexa Fluor 488 rat anti-mouse FOXP3 (BioLegend, MF-14; 1:100) and rabbit anti-Granzyme B (Abcam, ab4059, 1:200). The detection antibody was Alexa Fluor 568-conjugated goat anti-rabbit secondary antibody (Molecular Probes, 1:200), and was incubated for 60 minutes at room temperature (DAPI was also included to detect cell nuclei, 1:400). Images were acquired by epifluorescence microscopy and analysis of FOXP3+ and Granzyme B+ cell density performed in Image J. Intra-tumoural gene expression by qRT-PCR was performed following tumour RNA extraction in TRIZOL according to the previous describe method. Primers (5ʹ-3ʹ) for CD8a: (Forward:CCGTTGACCCGCTTTCTGT, Reverse:TTCGGCGTCCATTTTCTTTGG) and CD45: (Forward:AAGAGTTGTGAGGCTGGCAC, Reverse:GCTCAAACTTCTGGCCTTTG). All animals were housed in a specific pathogen-free facility, and procedures were approved by the Queen’s University Animal Care Committee in accordance with the Canadian Council on Animal Care guidelines.
Statistical analysis
Data were analyzed using Graphpad Prism (GraphPad Software, La Jolla, CA) using ANOVA with Tukey’s multiple comparison test, and p < 0.05 was considered significant.
Funding Statement
This research was supported by an Innovation grant [703045] from the Canadian Cancer Society to SSS and AWC.
Acknowledgments
The authors acknowledge contributions of Stephanie Young for help with tumor xenograft and syngeneic models, and Haiming Huang at Toronto Recombinant Antibody Centre for help with SPR assays, IgG cloning and Fab production.
Disclosure of potential conflicts of interest
The authors have declared that no conflict of interest exists.
Author contributions
DN and AWC designed research studies. DN conducted experiments, and analyzed data. KW performed analysis of Nanostring data. SN and PT contributed to development cell-based assays. SB, LLB, and LC contributed to Fab-phage screening, subcloning, expression, characterization and SPR assays. JJA and SSS managed the antibody screening and engineering portions of the project. DN, SB, JJA, SSS and AWC helped to write and edit the manuscript.
References
- 1.Kohn EC, Ivy SP.. Whence High-Grade Serous Ovarian Cancer. American Society of Clinical Oncology Educational Book American Society of Clinical Oncology Meeting 2017; 37:443–448. doi: 10.14694/EDBK_174718. [DOI] [PubMed] [Google Scholar]
- 2.Singh N, McCluggage WG, Gilks CB. High-grade serous carcinoma of tubo-ovarian origin: recent developments. Histopathol 2017; 71:339–356. doi: 10.1111/his.13248. [DOI] [PubMed] [Google Scholar]
- 3.Marcus CS, Maxwell GL, Darcy KM, Hamilton CA, McGuire WP. Current approaches and challenges in managing and monitoring treatment response in ovarian cancer. J Cancer 2014; 5:25–30. doi: 10.7150/jca.7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao L, Shao M, Schilder J, Guise T, Mohammad KS, Matei D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012; 31:2521–2534. doi: 10.1038/onc.2011.429. [DOI] [PubMed] [Google Scholar]
- 5.Rafehi S, Ramos Valdes Y, Bertrand M, McGee J, Prefontaine M, Sugimoto A, DiMattia GE, Shepherd TG. TGFbeta signaling regulates epithelial-mesenchymal plasticity in ovarian cancer ascites-derived spheroids. Endocrine Cancer 2016; 23:147–159. doi: 10.1530/ERC-15-0383 [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez GC, Haisley C, Hurteau J, Moser TL, Whitaker R, Bast RC Jr., Stack MS. Regulation of invasion of epithelial ovarian cancer by transforming growth factor-beta. Gynecol Oncol 2001; 80:245–253. doi: 10.1006/gyno.2000.6042 [DOI] [PubMed] [Google Scholar]
- 7.Massague J. TGFbeta signalling in context. Nature Rev 2012; 13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colak S, Ten Dijke P. Targeting TGF-beta Signaling in Cancer. Trends Cancer 2017; 3:56–71. doi: 10.1016/j.trecan.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Santibanez JF, Obradovic H, Kukolj T, Krstic J. Transforming growth factor-beta, matrix metalloproteinases, and urokinase-type plasminogen activator interaction in the cancer epithelial to mesenchymal transition. Dev Dyn 2018; 247:382–395. doi: 10.1002/dvdy.24554. [DOI] [PubMed] [Google Scholar]
- 10.Heneberg P. Paracrine tumor signaling induces transdifferentiation of surrounding fibroblasts. Crit Rev Oncol Hematol 2016; 97:303–311. doi: 10.1016/j.critrevonc.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Canellas A, Hernando-Momblona X, et al. TGFbeta drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018; 554:538–543. doi: 10.1038/nature25492 [DOI] [PubMed] [Google Scholar]
- 12.Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, Yu H, Qin G, Sircar A, Hernandez VM, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-beta. Sci Transl Med 2018; 10 (424).DOI: 10.1126/scitranslmed.aan5488 [DOI] [PubMed] [Google Scholar]
- 13.Ravi R, Noonan KA, Pham V, Bedi R, Zhavoronkov A, Ozerov IV, Makarev E, Artemov A, Wysocki PT, Mehra R, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFbeta enhance the efficacy of cancer immunotherapy. Nature Comm 2018; 9:741. doi: 10.1038/s41467-017-02696-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018; 554:544–548. doi: 10.1038/nature25501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Katz LH, Li Y, Chen JS, Munoz NM, Majumdar A, Chen J, Mishra L. Targeting TGF-beta signaling in cancer. Exp Op Ther Targets 2013; 17:743–760. doi: 10.1517/14728222.2013.782287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Gramont A, Faivre S, Raymond E. Novel TGF-beta inhibitors ready for prime time in onco-immunology. Oncoimmunol 2017; 6:e1257453. doi: 10.1080/2162402X.2016.1257453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST, Cleverly AL, Desaiah D, Guba SC, Benhadji KA, et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Dev Ther 2015; 9:4479–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Persson H, Ye W, Wernimont A, Adams JJ, Koide A, Koide S, Lam R, Sidhu SS. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol 2013; 425:803–811. doi: 10.1016/j.jmb.2012.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin SW, Lee MT, Ke FC, Lee PP, Huang CJ, Ip MM, Chen L, Hwang JJ. TGFbeta1 stimulates the secretion of matrix metalloproteinase 2 (MMP2) and the invasive behavior in human ovarian cancer cells, which is suppressed by MMP inhibitor BB3103. Clin Exp Met 2000; 18:493–499. doi: 10.1023/A:1011888126865 [DOI] [PubMed] [Google Scholar]
- 20.Qiu X, Cheng JC, Zhao J, Chang HM, Leung PC. Transforming growth factor-beta stimulates human ovarian cancer cell migration by up-regulating connexin43 expression via Smad2/3 signaling. Cell Signal 2015; 27:1956–1962. doi: 10.1016/j.cellsig.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 21.Cho MS, Bottsford-Miller J, Vasquez HG, Stone R, Zand B, Kroll MH, Sood AK, Afshar-Kharghan V. Platelets increase the proliferation of ovarian cancer cells. Blood 2012; 120:4869–4872. doi: 10.1182/blood-2012-06-438598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gallop MA, Barrett RW, Dower WJ, Fodor SP, Gordon EM. Applications of combinatorial technologies to drug discovery. 1 Background Peptide Combinatorial Libraries J Med Chem 1994;37:1233–1251. [DOI] [PubMed] [Google Scholar]
- 23.Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, Tookman LA, Orange C, Athineos D, Mason S, et al. CRISPR/Cas9-mediated Trp53 and Brca2 knockout to generate improved murine models of ovarian high grade serous carcinoma. Cancer Res 2016. doi: 10.1158/0008-5472.CAN-16-1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baldassarre T, Truesdell P, Craig AW. Endophilin A2 promotes HER2 internalization and sensitivity to trastuzumab-based therapy in HER2-positive breast cancers. Breast Cancer Res 2017; 19:110. doi: 10.1186/s13058-017-0900-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiwari N, Gheldof A, Tatari M, Christofori G. EMT as the ultimate survival mechanism of cancer cells. Sem Cancer Biol 2012; 22:194–207. doi: 10.1016/j.semcancer.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 26.Chen WF, Gao WD, Li QL, Zhou PH, Xu MD, Yao LQ. SLIT2 inhibits cell migration in colorectal cancer through the AKT-GSK3beta signaling pathway. Intl J Colorectal Dis 2013; 28:933–940. doi: 10.1007/s00384-013-1641-9. [DOI] [PubMed] [Google Scholar]
- 27.Zheng L, Xu M, Xu J, Wu K, Fang Q, Liang Y, Zhou S, Cen D, Ji L, Han W, et al. ELF3 promotes epithelial-mesenchymal transition by protecting ZEB1 from miR-141-3p-mediated silencing in hepatocellular carcinoma. Cell Death Dis 2018; 9:387. doi: 10.1038/s41419-018-0399-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shirakihara T, Kawasaki T, Fukagawa A, Semba K, Sakai R, Miyazono K, Miyazawa K, Saitoh M. Identification of integrin alpha3 as a molecular marker of cells undergoing epithelial-mesenchymal transition and of cancer cells with aggressive phenotypes. Cancer Sci 2013; 104:1189–1197. doi: 10.1111/cas.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nature Rev Clin Oncol 2017; 14:611–629. doi: 10.1038/nrclinonc.2017.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tian J, Hachim MY, Hachim IY, Dai M, Lo C, Raffa FA, Ali S, Lebrun JJ. Cyclooxygenase-2 regulates TGFbeta-induced cancer stemness in triple-negative breast cancer. Sci Rep 2017; 7:40258. doi: 10.1038/srep40258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rao S, Zaidi S, Banerjee J, Jogunoori W, Sebastian R, Mishra B, Nguyen BN, Wu RC, White J, Deng C, et al. Transforming growth factor-beta in liver cancer stem cells and regeneration. Hepatol Comm 2017; 1:477–493. doi: 10.1002/hep4.1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chiu WT, Huang YF, Tsai HY, Chen CC, Chang CH, Huang SC, Hsu KF, Chou CY. FOXM1 confers to epithelial-mesenchymal transition, stemness and chemoresistance in epithelial ovarian carcinoma cells. Oncotarget 2015; 6:2349–2365. doi: 10.18632/oncotarget.2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bellomo C, Caja L, Moustakas A. Transforming growth factor beta as regulator of cancer stemness and metastasis. Br J Cancer 2016; 115:761–769. doi: 10.1038/bjc.2016.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Q, Hou X, Evans BJ, VanBlaricom JL, Weroha SJ, Cliby WA. LY2157299 Monohydrate, a TGF-betaR1 Inhibitor, Suppresses Tumor Growth and Ascites Development in Ovarian Cancer Cancers 2018; 10(8). doi: 10.3390/cancers10080260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Carcia JR, Zhang L, Burow M, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10:942–949. doi: 10.1038/nm1093 [DOI] [PubMed] [Google Scholar]
- 36.Yang L, Pang Y, Moses HL. TGF-beta and immune cells: an important regulatory axis in the tumor microenvironment and progression. Trends Immunol 2010; 31:220–227. doi: 10.1016/j.it.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu VC, Wong LY, Jang T, Shah AH, Park I, Yang X, Zhang Q, Lonning S, Teicher BA, Lee C. Tumor evasion of the immune system by converting CD4+CD25- T cells into CD4+CD25+ T regulatory cells: role of tumor-derived TGF-beta. J Immunol 2007; 178:2883–2892. [DOI] [PubMed] [Google Scholar]
- 38.Milne K, Kobel M, Kalloger SE, Barnes RO, Gao D, Gilks CB, Watson PH, Nelson BH. Systematic analysis of immune infiltrates in high-grade serous ovarian cancer reveals CD20, FoxP3 and TIA-1 as positive prognostic factors. PloS One 2009; 4:e6412. doi: 10.1371/journal.pone.0006412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preston CC, Maurer MJ, Oberg AL, Visscher DW, Kalli KR, Hartmann LC, Goode EL, Knutson KL. The ratios of CD8+ T cells to CD4+CD25+ FOXP3+ and FOXP3- T cells correlate with poor clinical outcome in human serous ovarian cancer. PloS One 2013; 8:e80063. doi: 10.1371/journal.pone.0080063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Duraiswamy J, Freeman GJ, Coukos G. Therapeutic PD-1 pathway blockade augments with other modalities of immunotherapy T-cell function to prevent immune decline in ovarian cancer. Cancer Res 2013; 73:6900–6912. doi: 10.1158/0008-5472.CAN-13-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fellouse FA, Esaki K, Birtalan S, Raptis D, Cancasci VJ, Koide A, Jhurani P, Vasser M, Wiesmann C, Kossiakoff AA, et al. High-throughput generation of synthetic antibodies from highly functional minimalist phage-displayed libraries. J Mol Biol 2007; 373:924–940. doi: 10.1016/j.jmb.2007.08.005 [DOI] [PubMed] [Google Scholar]
- 42.Rajan S, Sidhu SS. Simplified synthetic antibody libraries. Meth Enzymol 2012; 502:3–23. doi: 10.1016/B978-0-12-416039-2.00001-X. [DOI] [PubMed] [Google Scholar]
- 43.Sidhu SS, Li B, Chen Y, Fellouse FA, Eigenbrot C, Fuh G. Phage-displayed antibody libraries of synthetic heavy chain complementarity determining regions. J Mol Biol 2004; 338:299–310. doi: 10.1016/j.jmb.2004.02.050. [DOI] [PubMed] [Google Scholar]