

ABSTRACT
Regulatory T cells (Tregs) facilitate primary and metastatic tumour growth through the suppression of anti-tumour immunity. Emerging evidence suggests a distinct role for Tregs in mediating tissue repair and barrier integrity in the lungs by IL-33 mediated production of the growth factor amphiregulin (AREG). Dependent on the type of cancer and local microenvironment, AREG may induce tumour cell proliferation, invasion, migration or resistance to apoptosis by signaling through the epidermal growth factor receptor (EGFR). We have found that IL-33 is dramatically increased in and around metastatic tumour foci in the lungs of mice bearing orthotopic murine mammary tumours. We observed that Tregs express significantly more of the IL-33 receptor, ST2, relative to conventional T cells, that ST2+ Tregs accumulate in the lungs of metastatic tumour-bearing mice, and that ST2+ Tregs produce significantly more AREG than ST2− Tregs. The intranasal administration of recombinant IL-33 increased the proportion of AREG producing ST2+ Tregs and enhanced the level of phosphorylated EGFR in the metastatic lungs. While recombinant AREG did not impact mammary tumour cell proliferation in vitro despite inducing a dose-dependent increase in phosphorylated EGFR, intranasal administration of AREG resulted in a ten-fold increase in pulmonary metastatic tumour burden in vivo. Further, the intranasal administration of recombinant IL-33 significantly increased metastatic tumour burden in the lungs in an amphiregulin-dependent manner. These data identify ST2+ Tregs as a microenvironmental source of AREG in the lungs of mice with orthotopic metastatic mammary tumours and highlight an important role for AREG in promoting metastatic tumour growth in the lungs.
KEYWORDS: Regulatory T cells, ST2, IL-33, amphiregulin, metastasis, breast cancer
Introduction
Tregs are an immune suppressive T cell population essential for mediating peripheral tolerance toward innocuous antigens, preventing autoimmunity,1 and limiting inflammation-induced tissue damage.2 Tregs may suppress the activity of effector T cells through contact-dependent mechanisms such as expression of the cell surface receptors cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death protein or ligand 1 (PD-1/PD-L1), through the sequestration of IL-2, or by production of soluble suppressive molecules such as interleukin-10 (IL-10), TGF-β, and adenosine.3 The immune suppressive function of Tregs may be co-opted by tumours as a mechanism to evade immune detection and Tregs may actively contribute to cancer progression by suppressing anti-tumour immunity.4 Elevated proportions of tumour-infiltrating CD4+CD25+Foxp3+ Tregs are associated with the growth and metastasis of many types of solid tumours including those of the lung5 and breast.6,7 While the function of Tregs in promoting primary tumour growth has been well characterized across a variety of malignancies, the role of Tregs in promoting metastatic development is an area of active investigation.4 Innovative therapeutic strategies are needed for the treatment of metastases, which account for more than 90% of cancer-associated deaths.8
There is currently great interest in targeting Tregs to promote the activity of anti-tumour effector cells. Indeed, we and others have shown that inhibition of Treg homing,9 targeting of Treg function,10 or systemic Treg ablation11 can reduce primary and/or metastatic tumour growth. In addition to promoting tumour growth by immune suppression, Tregs may exert a tissue protective effect in an “innate” manner by producing the growth factor amphiregulin (AREG), perhaps in response to stimulation with the IL-1 family cytokine, IL-33.12 IL-33 demonstrates pleiotropic activity as a damage-associated molecular pattern (DAMP), a regulator of pro-inflammatory gene expression, and a cytokine that drives Th2 immunity in the context of allergy,13 fibrosis,14 maintenance of tissue barrier integrity, and wound repair.15 Under homeostatic conditions, IL-33 is principally expressed in the nucleus of epithelial and endothelial cells bound to chromatin. In response to cell stress or necrosis-associated tissue damage, full length IL-33 is released extracellularly to serve as an alarm signal that recruits immune modulatory cells such as neutrophils, which produce proteases (cathepsin G and elastase) that cleave IL-33 to an enhanced bioactive form to subsequently promote Th2 cytokine production (IL-4, IL-5, IL-13).16 Activated myeloid cells may also produce IL-33 as a danger signal in response to toll-like receptor (TLR) stimulation during infection.17 The pro-inflammatory function of IL-33 may promote immune suppression by recruitment of suppressive immune cell populations expressing the cognate receptor ST2 (IL1RL1).18 The membrane-bound variant of ST2 functions as the only known receptor for IL-33 and initiates MyD88/NF-κB signaling upon ligand binding, whereas the soluble form of ST2 (sST2) acts as a decoy to bind extracellular IL-33 and inhibit downstream signaling.19,20 ST2 can be expressed on mast cells, Th2 cells, Tregs, type 2 innate lymphoid cells (ILC2s), and is weakly expressed by several myeloid cell subsets including M2 macrophages.21
AREG is a downstream product of IL-33/ST2 signaling in ILC2 cells in the gut and lungs,22,23 and AREG can be produced by epithelial cells, endothelial cells, and several leukocyte populations including Tregs.24,25 AREG production can also be stimulated by interaction of IL-18 with the IL-18R expressed by Tregs and several other cell types.12 Secretion of AREG by Tregs is important for mediating skeletal muscle repair26 and for the repair of infection-induced tissue damage in the lungs,12 with Treg specific deficiency in AREG associated with lung tissue damage and decreased oxygenation during challenge with influenza virus.12 AREG is a ligand of the epidermal growth factor receptor (EGFR) and, in the context of epithelial malignancies, AREG may promote tumour cell proliferation, invasion, migration or resistance to apoptosis depending on the type of cancer and local microenvironment.24 AREG functions as a growth factor for mammary epithelial cells by signaling through EGFR,27 and in breast cancer patients AREG expression has been associated with the presence of invasive disease.28 It was recently shown that AREG expression is increased by intra-tumoural Tregs after intravenous injection of EO771 mammary tumour cells or Lewis lung carcinoma (LLC) cells, and that CD4+ T cell-specific knockout of AREG reduced the development of EO771 and LLC foci in the lungs after i.v. injection.29 However, the role of AREG in IL-33-stimulated growth of spontaneously metastasizing mammary tumours in the lungs and the viability of AREG as a therapeutic target to treat mammary tumour metastases are unknown.
Using orthotopically implanted models of metastatic murine mammary carcinoma, we found robust IL-33 staining in and around metastatic tumour foci that develop in the lungs. We identified that Tregs express significantly more ST2 relative to conventional T cells in the lungs and that AREG-producing ST2+ and IL-18R+ Tregs were more abundant in the lungs of mice bearing metastatic mammary tumours compared to tumour-free mice. Although exogenous IL-33 did not stimulate the production of AREG from ST2+ Tregs ex vivo, intranasal administration of IL-33 increased the proportion of AREG producing ST2+ Tregs, EGFR activation, and metastatic tumour growth in the lungs. While we observed that AREG is dispensable for the growth and viability of 4T1 and 4T07 mammary tumour cells in vitro, we found that intranasal administration of recombinant AREG induced a ten-fold increase in lung metastatic burden. Further, using a combination of IL-33 administration and immunological inhibition of AREG, we found that IL-33 increases metastatic tumour burden in the lungs in an AREG dependent manner. These data identify ST2+ Tregs as a microenvironmental source of AREG in the lungs of mice bearing orthotopic metastatic mammary tumours and indicate active roles for IL-33 and AREG in promoting metastatic tumour growth in the lungs.
Results
IL-33 is increased in the lungs of mice bearing metastatic mammary carcinomas
We have previously shown that MDSCs and immune suppressive macrophages accumulate in the lungs of mice bearing metastatic mammary tumours,30 and that inflammatory myeloid cells in the lungs of tumour-bearing mice can induce lung tissue damage.31 In our hands, 4T1 tumour-bearing mice have ~ 1.6x107 CD11b+Gr1+ cells30,31 and several thousand metastatic tumour cells9,30,31 in the lungs 3 weeks after orthotopic implant of 4T1 tumour cells (with primary tumours of 700-750mg). We postulated that IL-33 may be produced by inflammatory myeloid cells and/or tumour cells in the lungs during metastatic tumour growth and observed a 5-fold increase in IL-33 in the lungs of mice three weeks after orthotopic implant of 4T1 tumours relative to the lungs of naïve mice (Figure 1a). We also observed significantly increased IL-33 in the lungs of mice bearing orthotopic mammary tumours such as 4T07, EO771, and the EO771-LMB lung metastatic variant of the parental EO771 cells. Analysis of IL-33 production by IHC showed a prominent increase in cytoplasmic IL-33 staining in lung metastases formed from 4T1 or LMB tumour-bearing mice relative to the predominantly nuclear localization of IL-33 in the naïve lung tissue of BALB/c and C57BL/6 mice (Figure 1b). Lung tissue from naïve mice intranasally administered the protease allergen papain and lungs from IL-33 knockout mice were used as positive and negative controls, respectively.32 Additional controls for the assessment of IL-33 by IHC are shown in Supplemental Figure 1. The morphology of the IL-33+ cells detected by IHC was consistent with neutrophils, macrophages, and tumour cells. Indeed, we observed IL-33 expression by CD11b+ myeloid cells in the lungs by immunofluorescence (Figure 1c), and both neutrophils and macrophages are known to express CD11b. Taken together, these data show a marked increase of IL-33 expression in the lungs of both BALB/c and C57BL/6 mice bearing orthotopic mammary tumours and suggest that CD11b+ myeloid cells and tumour cells are sources of IL-33 in the metastatic lungs.
Figure 1.
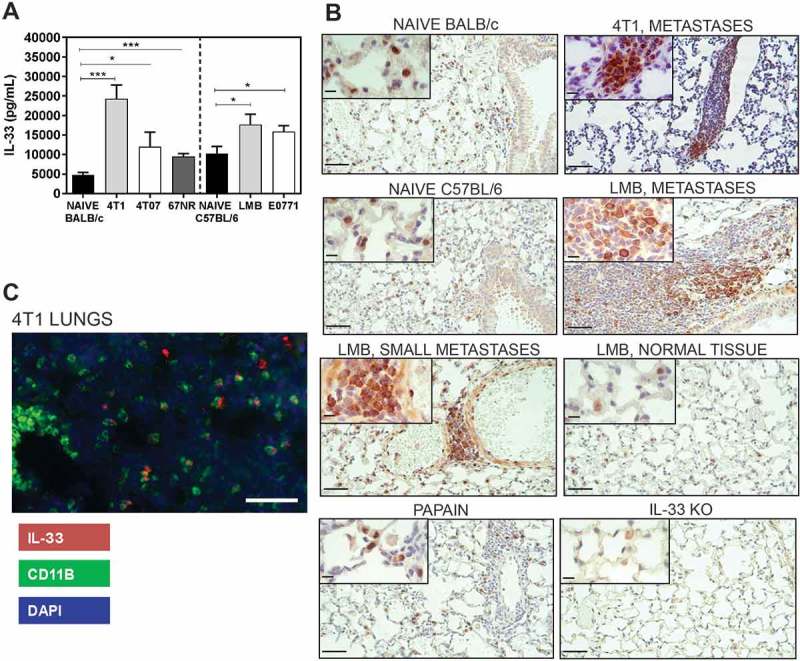
IL-33 levels are increased in the lungs of mice bearing mammary carcinomas 3 weeks after orthotopic implant. (a) IL-33 levels were quantified by ELISA of lung lysates from naïve or tumour-bearing BALB/c or C57BL/6 mice. Data are n = 5–6 mice per group from two independent experiments analyzed using a Student’s two-tailed t-test *p < 0.05, ***p ≤ 0.001. (b) IL-33 is located in the nucleus of naïve lung tissue cells from BALB/c and C57BL/6 mice in comparison to the strong cytoplasmic staining observed in the lung metastases of tumour-bearing mice. Lungs from mice given intranasal papain or lungs from IL-33 knockout mice were used as positive and negative controls, respectively. Scale bars = 50µm; inset = 10µm. (c) CD11b+ cells express IL-33 in the lungs of 4T1 tumour-bearing mice (scale bar = 125µm).
ST2+ Tregs accumulate in the metastatic lungs
To determine whether Tregs express the IL-33 receptor ST2, we performed flow cytometry analyses of lung, primary tumour, and spleen tissue from tumour-bearing mice 3 weeks after orthotopic implant of the indicated tumour cell lines. Representative flow cytometry plots of Tregs from the lungs of a 4T07 tumour-bearing mouse are shown in Figure 2a. We found that Balb/c mice bearing metastatic 4T1 or 4T07 tumours had increased ST2+ Tregs relative to naïve lung tissue or mice bearing non-metastatic 67NR tumours (Figure 2b). In contrast, we did not observe a significant change in ST2+ Tregs in the lungs of C57BL/6 mice bearing orthotopic EO771-LMB mammary tumours or subcutaneous LLC tumours (Figure 2b). Pulmonary Tregs from all naïve or tumour-bearing mice had a greater proportion of ST2+ cells relative to ST2 expression by conventional CD4+ T cells (Tconvs; CD4+CD25lo/-Foxp3−) or by CD4− cells representing the overall lung tissue (Figure 2c). The proportions of Tregs expressing ST2 in the lungs of mice with 4T1 or 4T07 tumours were particularly elevated relative to the lungs of naïve mice. We previously reported that Treg recruitment to the lungs of 4T1 or 4T07 tumour-bearing mice is regulated by CCR5,9 and we found that ST2 was highly co-expressed with CCR5 in the lungs of 4T1 or 4T07 bearing mice (Supplemental Figure 2). The percentage of ST2+ Tregs was also increased in the primary tumours (Figure 2d) and spleens (Figure 2e) of tumour-bearing mice. Collectively, these data indicate that ST2+ Tregs accumulate in mice bearing metastatic primary mammary tumours and that Tregs preferentially express ST2 relative to CD4+ Tconvs.
Figure 2.
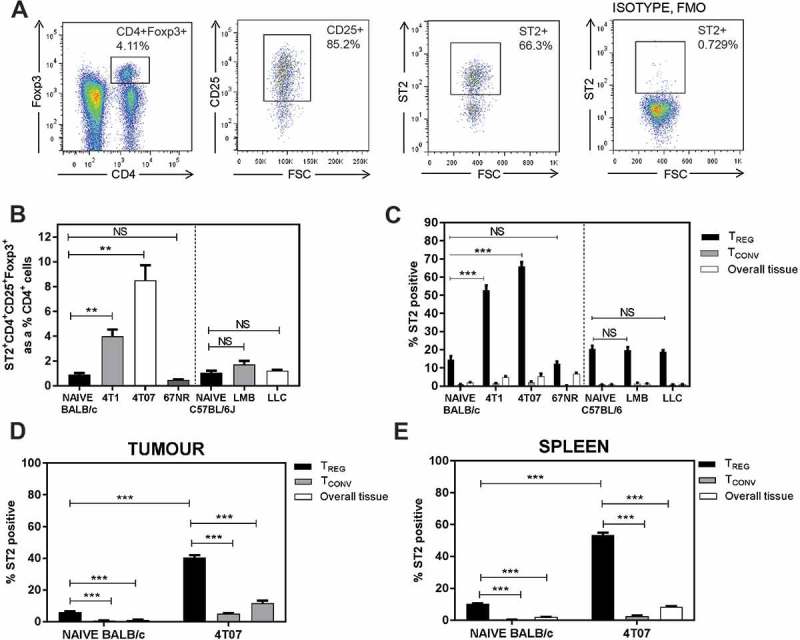
ST2+ Tregs accumulate in the lungs of mice bearing metastatic tumours 3 weeks post orthotopic implant. (a) Representative flow plots of lungs from a 4T07-bearing mouse; ST2 isotype fluorescence minus one (FMO) used for ST2 gating. (b) ST2+ Tregs, represented as a percentage of CD4+ T cells, are elevated in BALB/c mice bearing orthotopic 4T1 or 4T07 mammary carcinomas. Pulmonary ST2+ Tregs were not different in naïve vs EO771-LMB or LLC tumour-bearing C57BL/6 mice. (c) Elevated ST2+ Tregs in the lungs of 4T1 and 4T07 tumour-bearing mice. The percentage of Tregs expressing ST2 in the lungs was increased relative to ST2 expressing conventional CD4+ T cells (Tconv, CD4+CD25low/-Foxp3−). ST2+ Tregs were also increased in (d) primary tumour and (e) spleen of 4T07 tumour-bearing mice relative to naïve control tissue (mammary fat pads for tumours). Data are n = 4–6 mice per group from two independent experiments analyzed using Student’s two-tailed t-test, ** p ≤ 0.01, *** p ≤ 0.001.
Tregs and Tconvs in the metastatic lungs express IL-18R
The expression of IL-18R by Tregs may promote the release of AREG in response to IL-18.12 We therefore assessed if Tregs isolated from the lungs of tumour-bearing mice 3 weeks post orthotopic implant express IL-18R. Representative flow cytometry plots of Tregs from the lungs of a 4T07 tumour-bearing mouse are shown in Figure 3a. We observed a significant increase in IL-18R+ Tregs in the lungs and spleens of mice with 4T07 (Figure 3b) or EO771-LMB (Figure 3c) tumours. In contrast to the high proportion of ST2+ Tregs relative to ST2+ Tconvs in Figure 2, IL-18R+ Tconvs were significantly greater than IL-18R+ Tregs in the lungs and spleens of 4T07 tumour-bearing BALB/c (Figure 3d–e) and EO771-LMB-bearing C57BL/6 (Figure 3f–g) mice. The overall lung tissue of 4T07 tumour-bearing mice had increased IL-18R+ cells relative to naïve mice, suggesting that other cell population(s) express IL-18R and accumulate in the lungs of BALB/c mice in the presence of a metastatic primary tumour (Figure 3d). Thus, although both ST2+ and IL-18R+ Tregs may contribute to the production of AREG, ST2 is predominantly expressed by Tregs while IL-18R is highly expressed by CD4+ Tconvs and CD4− cells in the lungs of mammary tumour-bearing BALB/c and C57BL/6 mice.
Figure 3.
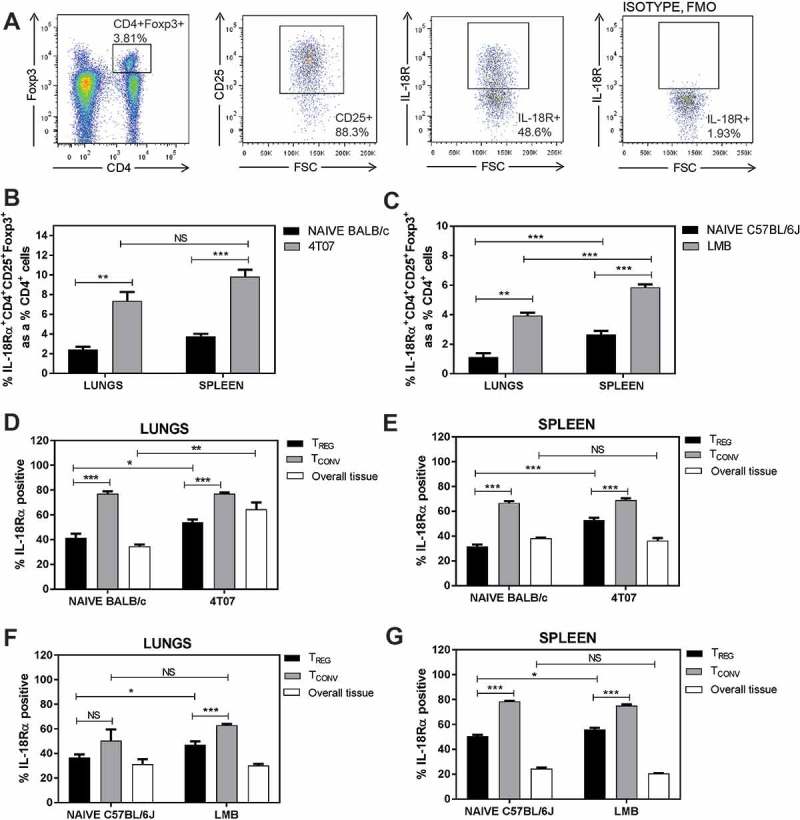
IL-18R expression is abundant in the lungs of naïve and tumour-bearing mice. (a) Representative flow cytometry plots for gating. (b-c) IL-18R+ Tregs are increased in the lungs and spleens of 4T07 and EO771-LMB bearing mice relative to naïve control tissues. IL-18R+CD4+ Tconvs are more abundant than IL-18R+ Tregs in the lungs and spleens of naïve and tumour-bearing 4T07 (d-e) or LMB (f-g) mice. Data are n = 5–6 mice per group from two independent experiments analyzed using Student’s two-tailed t-test * p < 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Tregs produce high levels of AREG in the metastatic lungs
AREG derived from Tregs expressing ST2 and IL-18R were recently found to mediate lung tissue repair following influenza infection,12 and CD4+ T cell-derived AREG can increase lung colonization by i.v. injected tumour cells.29 We found an increase in the percentage of Tregs expressing AREG in the metastatic lungs of mice bearing 4T1 tumours, but not 4T07 tumours, relative to naïve control lungs (Figure 4a). AREG expressing Tregs were approximately 2.5-fold more abundant than AREG expressing Tconvs in the lungs of 4T1-bearing mice, and there was no significant difference evident in the proportion of AREG+ Tconvs between naïve mice and mice with 4T1 or 4T07 tumours (Figure 4a). Tregs isolated from tumour-bearing mice secreted significantly more AREG ex vivo on a per cell basis than Tregs isolated from naïve control mice (Figure 4b). AREG secretion was dependant on TCR stimulation of the Tregs and, somewhat surprisingly, the addition of recombinant murine IL-33 (rmIL-33) did not increase AREG production by Tregs ex vivo (Figure 4b). By sorting Tregs based on ST2 expression prior to ex vivo culture, we found that AREG secretion was significantly higher in ST2+ Tregs relative to ST2− Tregs, but we again did not observe elevated AREG production with addition of rmIL-33 (Figure 4c). Interestingly, the level of phosphorylated EGFR and phosphorylated Akt were modestly increased in Tregs cultured with IL-33 relative to untreated Tregs (Figure 4d), and ST2+ Tregs had higher total EGFR, AKT, and ERK levels compared to ST2− Tregs. These data indicate that ST2+ Tregs are enriched for AREG production, that Tregs from the lungs of 4T1 tumour-bearing mice express significantly more AREG than CD4+ Tconvs, and that IL-33 slightly increases EGFR signaling in Tregs without increasing AREG production ex vivo.
Figure 4.
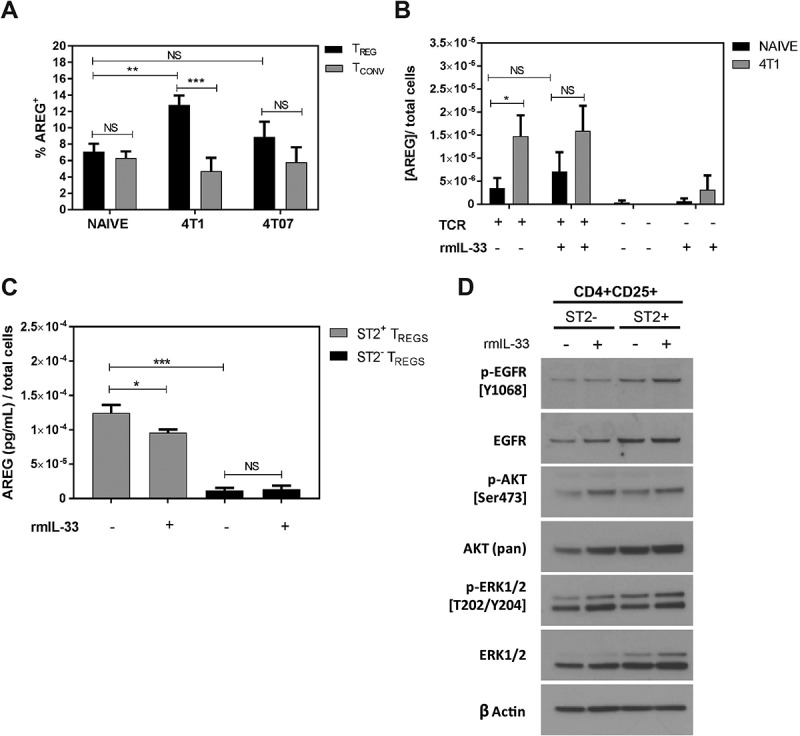
AREG expressing Tregs are increased in the lungs of 4T1 tumour-bearing mice relative to naïve mice. (a) Flow cytometry-based quantification of AREG+ Tregs and Tconvs from naïve and tumour-bearing mice show an increased percentage of AREG+ Tregs in the lungs of 4T1-bearing mice. Data are n = 7–10 mice per group. (b) Tregs were isolated from 4T1 tumour-bearing mice and stimulated with 10µg/mL αCD3, 2.5µg/mL αCD28 and 100U/mL rIL-2 (TCR) in the presence or absence of rmIL-33 (100ng/mL). AREG secretion was quantified by ELISA and shows the influence of TCR stimulation on AREG production. Data are from n = 6 mice per group. (c) Measurement of AREG secreted by ST2+ or ST2− Tregs cultured ± rmIL-33. Data are from three independent experiments performed in duplicate. All data analyzed using Student’s two-tailed t-test * p < 0.05, ** p ≤ 0.01, *** p ≤ 0.001. (d) Western blots showing the levels of total and phosphorylated EGFR, AKT, and ERK in ST2+ and ST2− Tregs cultured in the presence or absence of rmIL-33.
Recombinant IL-33 increases AREG producing ST2+ Tregs
After finding that IL-33 is increased in lung metastases and that Tregs express a high level of AREG in the metastatic lungs, we tested whether administration of rmIL-33 could increase AREG production by ST2+ Tregs in vivo. Tumour-bearing mice were given intranasal rmIL-33 (0.25µg/40μL) or PBS for 3 days according to a previously published protocol to assess pulmonary IL-33 activity in naïve mice.33 4T1 tumour cells colonize the lungs as early as 9 days after orthotopic implantation in our hands (data not shown), and therefore rmIL-33 treatment was initiated after metastatic foci had developed (Figure 5a). IL-33 levels were maintained in the lungs of rmIL-33 treated mice at the experimental endpoint (Figure 5b). We found that rmIL-33 increased total Treg levels in the lungs (Figure 5c) by expanding the proportion of ST2+IL-18R− Tregs (Figure 5d). The total number of CD4+ T cells did not increase with IL-33 treatment (data not shown), suggesting that rather than inducing an influx of Tregs to the lungs, IL-33 may promote the differentiation of CD4+ T cells into ST2+IL-18R− and ST2+IL-18R+ Tregs. We found that IL-33 increased the number of AREG-expressing Tregs in the lungs of tumour-bearing mice (Figure 5e), and that nearly 50% of the AREG+ Tregs were derived from the ST2+IL-18R− Treg subpopulation (Figure 5f). We also found that rmIL-33 treatment induced a marked increase in phospho-EGFR levels in the lungs relative to PBS treated tumour-bearing mice (Figure 5g, Supplemental Figure 3). Positive control lung tissue for phospho-EGFR staining (Supplemental Figure 3) was obtained from CCSP-rtTA;TetO-EGFRL858R transgenic mice with doxycycline-inducible expression of the activated EGFRL858R mutant in type II airway epithelial cells.34 Taken together, our data indicate that in the lungs of mice bearing metastatic 4T1 tumours IL-33 can increase the proportion of ST2+IL-18R− Tregs, which are a major source of AREG under these conditions, and that IL-33 can increase phospho-EGFR levels in and around metastatic tumour foci.
Figure 5.
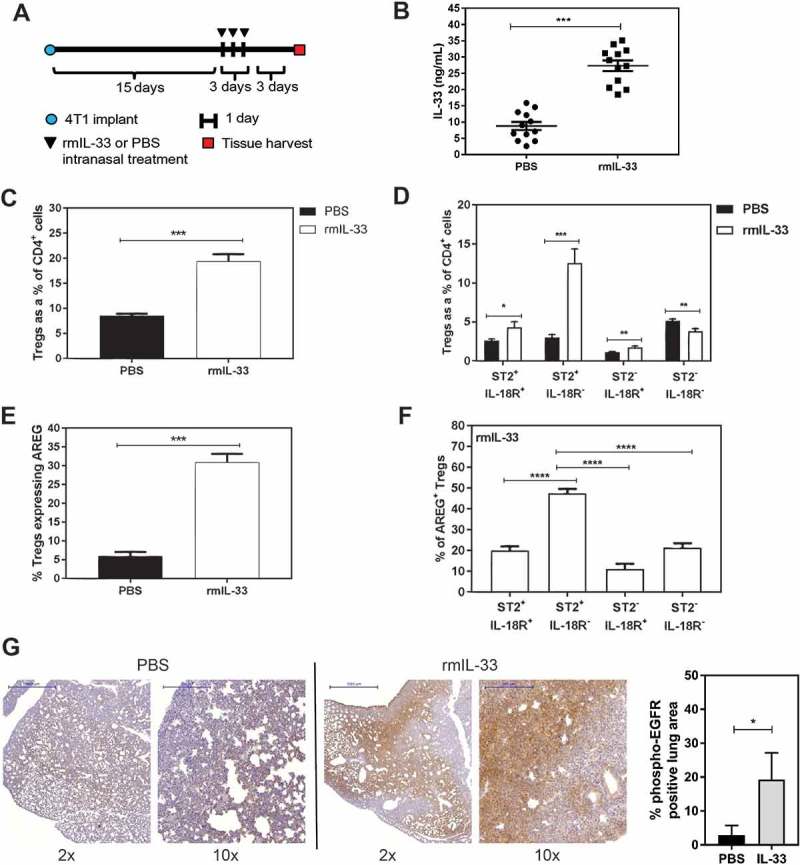
Recombinant IL-33 increases ST2+ AREG-expressing Tregs in the lungs of mice bearing 4T1 mammary tumours. (a) Schematic depicting the treatment time course for intranasal administration of rmIL-33 (0.25µg/40μL) or PBS (40μL) to 4T1 tumour-bearing mice. (b) Pulmonary IL-33 levels are elevated at experimental endpoint. (c) rmIL-33 increases total Tregs in the lungs, and (d) the majority of these Tregs are ST2+IL-18R+ Tregs. (e) The percentage of total Tregs expressing AREG is increased with rmIL-33 treatment. (f) The majority of AREG-expressing Tregs in the lungs of mice bearing 4T1 mammary tumours and treated with IL-33 is derived from ST2+IL-18R− Tregs. Data are from three independent experiments (n = 12) analyzed using a Student’s two-tailed t-test, * p < 0.05, ** p ≤ 0.01, *** p ≤ 0.001. (g) pEGFR staining of lungs from 4T1 tumour-bearing mice treated with intranasal PBS (left) or rmIL-33 (middle) with quantification of pEGFR+ lung area from n = 3 mice per group. Scale bars: 2x = 1000µm; 10x = 200µm.
Murine mammary carcinomas secrete AREG in vitro but are not sensitive to EGFR or AREG inhibition
We next examined if the tumour cell lines used in this study express AREG or EGFR, and are sensitive to AREG or EGFR inhibition in vitro. AREG was produced by all cell lines except 67NR cells (Figure 6a), although none of the five cell lines expressed detectable levels of ST2 or IL-18R (Supplemental Figure 4). All five cell lines expressed EGFR, although only 4T1 cells had detectable levels of phosphorylated EGFR in culture (Figure 6b). The second-generation tyrosine kinase inhibitor afatinib inhibits autophosphorylation of human EGFR, HER2 (ErbB2), and HER4 (ErbB4).35 We found that the half maximal inhibitory concentration (IC50) of afatinib over a 72 hour treatment timeframe was > 10µM for 4T1 and 4T07 murine cell lines, which is over 1,000-fold higher than the afatinib sensitive H1975 (EGFRL858R/T790) and 4-fold higher than the afatinib resistant H23 (KRASG12C) human lung adenocarcinoma cell lines (Supplemental Figure 5). These data suggest 4T1 and 4T07 cells are resistant to EGFR inhibition, although afatinib is an inhibitor of human EGFR and is not ideal for inhibiting murine EGFR (an antagonist of murine EGFR is not commercially available). Treatment of 4T1 cells with afatinib at 50% of the IC50 in reduced serum conditions decreased phospho-EGFR and phospho-ERK levels, although immunological inhibition of AREG did not affect EGFR or phospo-ERK (Figure 6c). Recombinant murine AREG (rmAREG) induced a dose-dependent increase in pEGFR in 4T1 cells, but pEGFR was not detected in 4T07 cells with AREG treatment (Figure 6d). The survival of 4T1 and 4T07 cells after 8 days in culture was decreased with increasing concentrations of afatinib (Figure 6e–f). However, the proliferation and survival of 4T1 and 4T07 cells in vitro was unaffected by AREG inhibition (Figure 6g–h) or by addition of rmAREG under low serum conditions (Figure 6i–j) or serum free conditions (data not shown). Collectively, our findings indicate that while rmAREG can stimulate phosphorylation of EGFR in 4T1 cells, AREG does not promote the proliferation or survival of 4T1 or 4T07 tumour cells under low serum conditions in vitro.
Figure 6.
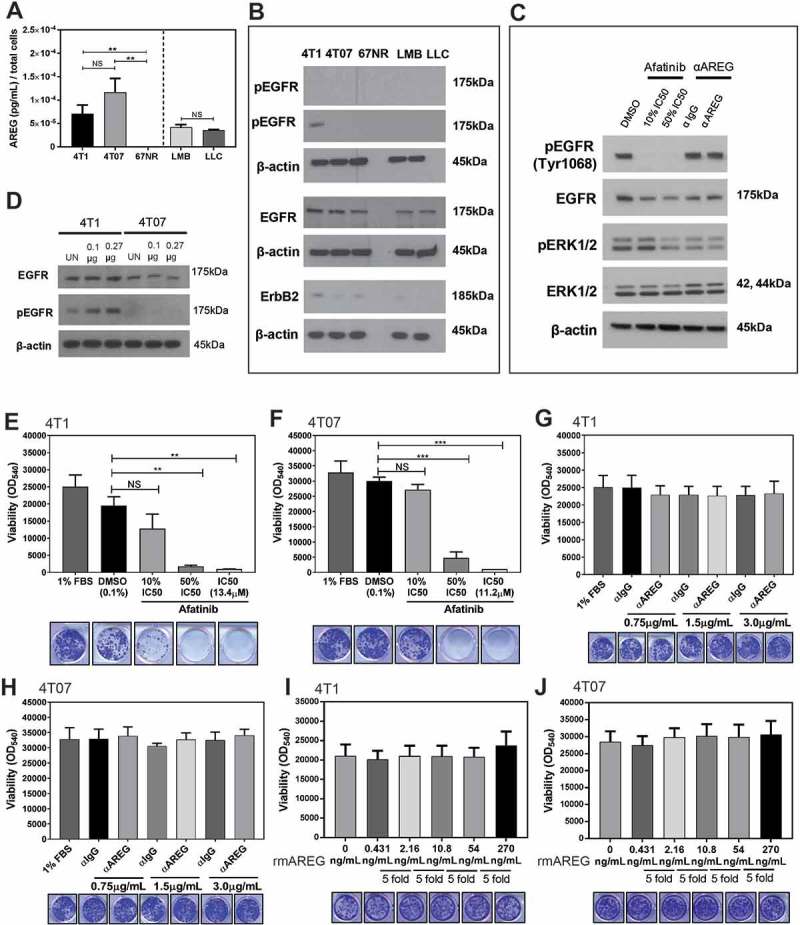
Murine mammary carcinoma cells secrete AREG in vitro but are not sensitive to EGFR or AREG inhibition. (a) AREG is secreted by several murine tumour cell lines in vitro. Data are from three independent experiments performed in triplicate analyzed using a Student’s two-tailed t-test ** p ≤ 0.01. (b) Expression of EGFR, phospho-EGFR, and ErbB2 by cell lines in vitro. (c) EGFR signaling is reduced in 4T1 cells (1% FBS) treated with high doses of afatinib, but not in cells treated with 0.75µg/mL αAREG. (d) pEGFR expression is increased by 4T1 cells in a dose dependent manner after 2 hours of culture with 0.1µg or 0.27µg rmAREG. The survival of 4T1 (d) and 4T07 (e) cells was decreased in a dose dependent manner by treatment with afatinib, but use of a neutralizing antibody against AREG did not impact 4T1 (f) or 4T07 (g) viability. Culture of 4T1 (h) and 4T07 (i) cells with 5-fold increasing concentrations of rmAREG did not impact cell proliferation in vitro. The lowest concentration of rmAREG (0.431ng/mL) used in culture was selected as an average of the AREG produced by the 4T1 and 4T07 cell lines, as measured by ELISA. Clonogenic data are from three independent experiments analyzed using Student’s two-tailed t-test * p < 0.05, ** p ≤ 0.01, *** p ≤ 0.001.
Recombinant AREG increases lung metastases and AREG inhibition blocks the pro-metastatic activity of IL-33
While AREG did not influence tumour cell survival or proliferation in vitro, AREG can exert a variety of pro-tumourigenic functions depending on the tumour type and local microenvironment. We intranasally administered recombinant murine AREG (rmAREG) twice per week starting four days after orthotopic 4T1 tumour implantation (Figure 7a) and despite a reduction in primary tumour volume (Figure 7b), rmAREG induced a nearly 10-fold increase in the number of 4T1 tumour cells in the lungs (Figure 7c). We then tested whether the increase in AREG expression in the lungs induced by intranasal rmIL-33 (Figure 5) affected lung metastases. We found that intranasal rmIL-33 given 15–17 days after 4T1 tumour implant (Figure 7d) increased the growth of 4T1 lung metastases, and that the pro-metastatic effect of rmIL-33 could be blocked by immunological inhibition of AREG (Figure 7e). Neither rmIL-33 nor anti-AREG affected primary tumour growth (Figure 7f). Taken together, these data indicate that AREG in the lungs promotes 4T1 metastases, and that IL-33 induces 4T1 metastatic growth in the lungs in an AREG dependent manner.
Figure 7.
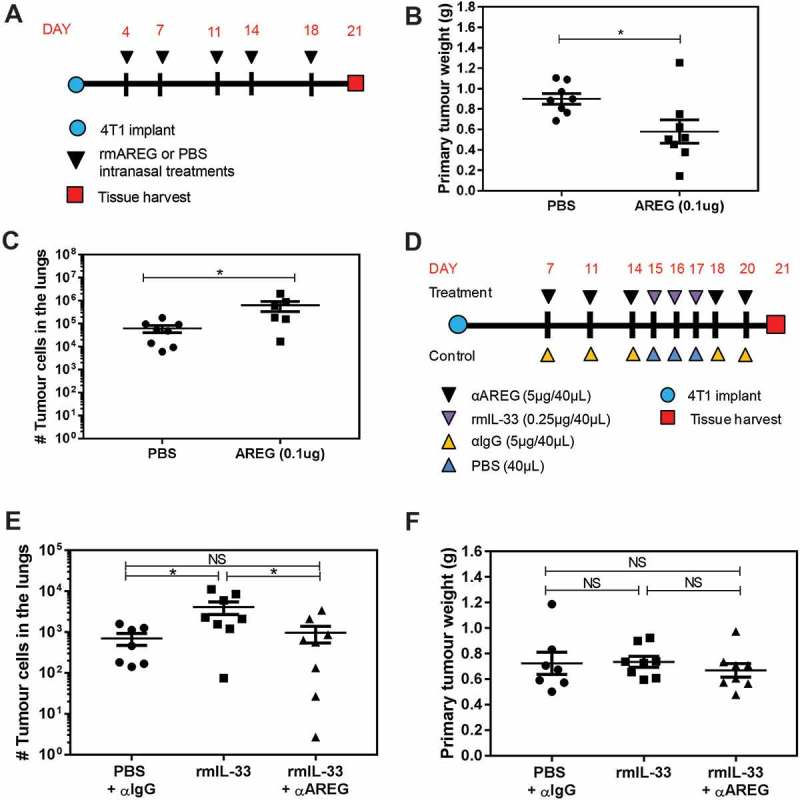
AREG and IL-33 increase 4T1 metastatic burden in the lungs, and the pro-metastatic effect of IL-33 is AREG dependent. (a) Experimental schematic depicting the timing of rmAREG (0.1µg/40µL) or PBS (40µL) administration after orthotopic 4T1 tumour implantation. (b) Intranasal rmAREG decreased primary mammary tumour growth and (c) increased 4T1 metastatic growth in the lungs. (d) Experimental schematic depicting the treatment time course for administration of rmIL-33 (0.25µg) or PBS alone or in combination with αAREG (5µg) or IgG (5µg). (e) Intranasal rmIL-33 increased 4T1 metastatic growth that was inhibited by αAREG. (f) Primary mammary tumour volume was not impacted by rmIL-33 in the presence or absence of αAREG. Data are from two independent experiments with n = 7–8 mice per group analyzed using a Student’s two-tailed t-test * p < 0.05.
Discussion
The immune suppressive activity of Tregs is thought to play a central role in promoting the growth of a variety of malignancies, including cancers of the breast, lung, head and neck, ovary, and colon.4 Increased intra-tumoural infiltration of Tregs is associated with poor prognosis and reduced disease-free survival, particularly when coupled with reduced levels of anti-tumour effector T cells. As such, the systemic inhibition of Tregs, or targeting of their intra-tumoural homing or suppressive function, has become an active area of investigation.36 In addition to immune suppression, Tregs may also facilitate tumour progression through the production of RANKL to suppress the EMT inhibitor maspin,7 or by secretion of pro-angiogenic VEGFA.37 Emerging literature indicates Tregs also promote tissue repair in the lungs by producing the growth factor AREG in response to IL-33 or IL-18.12 AREG is associated with a variety of tumour promoting effects by autocrine or paracrine signaling through the cognate receptor EGFR.24 We show herein that IL-33 is increased in the lungs of mice bearing primary orthotopic mammary tumours, and that IL-33 increases AREG within the metastatic lungs at least partially through increased numbers of ST2+ Tregs. Further, we demonstrate that IL-33 increases metastatic tumour burden in the lungs in an AREG dependent manner, highlighting an active role for AREG in the development and growth of mammary tumour metastases.
IL-33 is an alarmin that is principally expressed by epithelial and endothelial cells, as well as activated innate immune cells.38 Under homeostatic conditions, IL-33 localizes to the nucleus and is exported to the cytoplasm for subsequent release following cell damage induced by infection or tissue injury.39 To propagate the inflammatory response and facilitate tissue repair, innate immune cells may produce IL-33 to drive Th2 immunity.40 We have found increased IL-33 in the lungs of mice bearing five different orthotopic mammary tumours relative to naïve C57BL/6 or BALB/c control mice (Figure 1). Our histological data indicate prominent cytoplasmic and extracellular IL-33 staining in and around metastatic tumour foci in the lungs of mice with 4T1 or EO771-LMB tumours derived from inflammatory myeloid cells and tumour cells. IL-33 content in the lungs correlated with the metastatic propensity of the primary tumours;41 mice with macrometastatic 4T1 tumours had the most pulmonary IL-33, followed by mice with micrometastatic 4T07 tumours, and then mice with non-metastatic 67NR tumours. We also found IL-33 expression in CD11b+ myeloid cells, which is consistent with the accumulation of CD11b+Gr1+ myeloid-derived suppressor cells and CD11b+F4/80+ macrophages in the lungs of mice bearing 4T1 or 4T07 tumours.30 Our data suggest IL-33 levels in the lungs of mice with metastatic mammary tumours are associated with myeloid cell infiltration in the lungs and metastatic tumour burden, although the proportion of pulmonary IL-33 derived from metastatic tumour cells or myeloid cells is the subject of further study.
Ligation of IL-33 with the heterodimeric ST2/IL-1RAP receptor complex in Tregs has been reported to increase Foxp3 and Gata3 expression while expanding Treg numbers and enhancing Treg function through a TFG-β dependent mechanism.42 While IL-33 can directly promote the expansion of Tregs, expression of IL-2 by IL-33 stimulated ST2+ dendritic cells (DCs) may also augment this process.43 The lungs of mice with 4T1 or 4T07 mammary tumours contained increased numbers of Tregs expressing the IL-33 receptor ST2 (Figure 2). The majority of these ST2+ Tregs also expressed CCR5 (Supplemental Figure 2), consistent with our previous work showing CCL8-mediated recruitment of CCR5+ Tregs to the lungs of mice with 4T1 or 4T07 tumours.9 The proportion of ST2+ Tregs in the lungs of mice bearing 67NR, EO771, or EO771-LMB tumours was not significantly different from naïve mice despite increased IL-33 in the lungs (Figure 1), suggesting IL-33 does not influence ST2+ Treg numbers in these three models. IL-33 has been reported to induce AREG production by lung-resident Tregs during periods of cell stress due to infection,12 and we observed increased AREG-producing Tregs in the lungs of mice with 4T1 tumours relative to Tregs in naïve lungs (Figure 4). However, while ST2+ Tregs secreted significantly more AREG than ST2− Tregs, we did not observe increased AREG secretion by Tregs after IL-33 treatment ex vivo. This discrepancy may result from ex vivo Treg culture and activation conditions, the origin of the isolated cells, or the context of infection compared to metastasis. We found that ST2+ Tregs expressed higher levels of EGFR, AKT, and ERK when compared to ST2− Tregs, and that the addition of IL-33 increased phosphorylated AKT levels in the Tregs. Consistent with the role of AKT in promoting cell survival and proliferation, the intranasal administration of recombinant IL-33 significantly increased the proportion of AREG producing ST2+IL-18R− Tregs in the lungs of 4T1 tumour-bearing mice (Figure 5). While ~ 50% of AREG+ Tregs were ST2+IL-18R−, we also observed AREG production by Tregs after rmIL-33 treatment regardless of ST2 or IL-18 receptor status, suggesting intranasal IL-33 can promote AREG production in Tregs through direct or indirect mechanisms. Taken together, our data support a role for IL-33 in promoting expansion of AREG-producing ST2+ Tregs and increasing AREG production by Tregs in the lungs.
Previous studies have shown that administration of recombinant IL-33 to 4T1 tumour-bearing mice led to the recruitment of immune suppressive cells to the primary tumour, a corresponding reduction in natural killer (NK) cell cytotoxicity and increased metastasis.44 Correspondingly, ablation of ST2 abrogated the tumour-promoting effects of IL-33 and enhanced anti-tumour immunity reflected by an activated phenotype of CD4+ and CD8+ T cells and increased NK cell cytotoxicity.45 We found that in addition to increasing the proportion of AREG-producing ST2+ Tregs in the lungs, IL-33 increased phosphorylated EGFR in and around metastatic foci in the lungs of 4T1 tumour-bearing mice (Figure 5g). Activation of EGFR signaling by AREG may induce a variety of biological effects including cell proliferation, survival, migration, invasion, angiogenesis or resistance to apoptosis.24 In addition to AREG derived from ST2+ Tregs, we found that 4T1, 4T07, EO771-LMB, and LLC tumour cells produce AREG in vitro (Figure 6), suggesting tumour cells present in the lungs may represent an additional source of pulmonary AREG. It is worth noting that the level of AREG produced by ST2+ Tregs (Figure 4c) was 2-fold greater than the level of AREG produced by 4T1 tumour cells in vitro. Interestingly, while all tumour cell lines expressed EGFR in vitro, only 4T1 cells had detectable levels of pEGFR in vitro which could be increased by treatment with AREG. Neither 4T1 nor 4T07 cells were sensitive to EGFR inhibition in low serum conditions, and AREG was dispensable for the survival and proliferation of both cell lines in vitro. However, intranasal administration of AREG to 4T1 tumour-bearing mice increased metastatic burden in the lungs (Figure 7). The difference between in vitro and in vivo effects of AREG on 4T1 metastatic growth may be due to activity of the transmembrane metalloproteinase ADAM-17 and/or effects of AREG on the activity of other cell types in the lungs. Enzymatic cleavage of AREG by ADAM-17 is necessary to process AREG to an active form for binding and downstream phosphorylation of EGFR in the context of mammary ductal development.46 The expression of ADAM-17 in the local tissue microenvironment may therefore dictate the pro-tumourigenic role of AREG, and the expression of both AREG and ADAM-17 may vary dependent on the tumour type and local microenvironment. While AREG may directly induce tumour cell survival and proliferation in some contexts,47 it may also indirectly promote tumour growth by acting on normal cells in the lungs. AREG derived from CD4+ T cells was recently shown to affect lung colonization of i.v. injected EO771 cells independent of EGFR signaling on tumour cells or endothelial cells.29 AREG has also been reported to increase the suppressive function of EGFR+ Tregs mediated by EGFR/GSK3β signaling through the post-translational regulation of Foxp3 expression.48
We found that in addition to increasing ST2+ Tregs and AREG in the lungs, IL-33 also promotes metastatic 4T1 tumour growth in the lungs. Importantly, the IL-33-induced increase in metastatic growth could be prevented by immunological inhibition of AREG (Figure 7), identifying AREG as a mediator of the metastasis-promoting effects of IL-33. While we have identified ST2+ Tregs as a source of AREG production, AREG can be produced by several other cell types including mast cells, basophils, and ILC2 cells.25 More work is needed to evaluate the relative contribution of AREG derived from different immune cells and/or tumour cells to metastatic tumour growth in the lungs. Nevertheless, our data suggest that targeting AREG either as a monotherapy or in conjunction with EGFR tyrosine kinase inhibitors may represent viable therapeutic strategies to inhibit the development and growth of tumour metastases in the lungs.
While principally thought to promote tumour development through the suppression of anti-tumour immunity, a new role has emerged for Tregs in mediating tumour growth through AREG expression. Collectively, our findings indicate that IL-33 increases AREG-producing ST2+ Tregs in the lungs and emphasize the significance of IL-33 and AREG in promoting metastatic tumour growth in the lungs. Improved understanding of the crosstalk between the local immune microenvironment and tumour cells is essential to the generation of innovative therapeutic strategies for the treatment of metastatic disease.
Materials and methods
Mouse and tumour models
Female BALB/c mice (12–16 weeks of age; Simonsen Laboratories) were housed under specific-pathogen free conditions in the Animal Resource Centre at the BC Cancer Agency Research Centre in micro-isolator cages with ventilated racks. Animal experiments were performed in accordance with the Canadian Council on Animal Care and the University of British Columbia Committee on Animal Ethics.
Murine mammary carcinoma cell lines syngeneic to BALB/c mice, including 4T1, 4T07, and 67NR, were gifted by Dr. Fred Miller (Karmanos Cancer Institute, Detroit, MI). These cell lines originated from a spontaneous mammary tumour in BALB/cfC3H and are heterogeneous in their metastatic potential as previously described,41 with 4T1 and 4T07 forming macro- and micro-metastases in the lungs, respectively, and 67NR unable to disseminate from the primary tumour. Cell lines syngeneic to C57BL/6 mice include LLC (ATCC® CRL1642), EO771 (CH3 Biosystems, Amherst, NY, USA), and EO771-LMB (an enhanced pulmonary metastatic variant of the parental EO771 line49) gifted by Dr. Robin Anderson (Peter MacCallum Cancer Centre, Melbourne, Australia).
Mammary tumour cell lines were implanted orthotopically into the mammary fat pad as previously described9 at 105, 106, and 2 × 105 cells for 4T1, 4T07, and 67NR cell lines, respectively, and 5 × 105 cells for EO771 or EO771-LMB in 50µL PBS. The primary lung cancer line LLC, was injected intravenously at 2 × 105 cells in 200µL. The implanted cell numbers were optimized to generate tumour volumes that approach ethical restrictions 3–3.5 weeks after implantation. Cell lines were maintained for no more than 20 passages in Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with D-glucose, sodium pyruvate, HEPES and 10% fetal bovine serum as previously described.9
Tissue processing
Lung, tumour, and mammary fat pad tissue were mechanically disaggregated following excision and then agitated for 40mins at 37°C with 1mg/mL type II collagenase (Gibco® Life Technologies) in PBS. Spleen tissue was homogenized and lysed with ammonium chloride (NH4Cl, 1:9 ratio, 9mins on ice according to manufacturer’s recommendation, StemCell Technologies, Vancouver, BC, Canada) prior to flow cytometry as previously described.9
Flow cytometry
Single-cell suspensions from the lungs, tumour, mammary fat pad, and spleen were resuspended in PBS and stained with viability dye (eFluor® 780, eBioscience, San Diego, CA, USA) according to the manufacturer’s instructions. Cells were washed and resuspended in Hank’s balanced salt solution supplemented with 10mM HEPES (StemCell Technologies), 2% FBS, and 0.05% NaN3 and stained for 1 hour at 4°C in the dark with surface antibodies to CD4 (BV605, Biolegend, San Diego, CA, USA), CD25 (BV421, Biolegend), ST2 (PE, eBioscience) or IL-18Rα (PE, eBioscience), and CCR5 (APC, Biolegend). Intracellular staining was performed as described9 using Foxp3 (PE Cy7, eBioscience), primary AREG (biotin conjugated, R&D systems, Minneapolis, MN, USA), secondary streptavidin (FITC, eBioscience), or IL-33 (FITC, R&D systems). Myeloid cell expression of IL-33 was assessed using CD11b (FITC, eBioscience). Anti-murine CD16/32 (0.5μg/100μL, clone 2.4G2) was used to block non-specific Fc receptor binding by antibodies. Samples were assayed using an LSR Fortessa and analyzed with FACSDiva software (BD, Mississauga, ON, Canada). Treg proportions are expressed as a percentage of total CD4+ cells. Sorting of ST2+ Tregs from a CD4 enriched population isolated from 4T1 tumour-bearing mice was performed using a BD FACSAria III or BD FACSAria Fusion using the fluorochrome combinations listed above for CD4, CD25, and ST2.
Treg isolation and culture
For ex vivo analysis of AREG secretion, Tregs were isolated from single cell suspensions by magnetic bead separation for CD25+ cells following the manufacturer’s instructions (Mouse Regulatory T Cell Positive Selection kit; Stemcell Technologies). The Foxp3 expression of isolated cells was > 90% as validated by flow cytometry analysis (Supplemental Figure 6). For analysis of AREG secretion by Tregs stimulated with recombinant IL-33, a CD4 negative selection kit (EasySep mouse CD4+ T cell isolation kit, Stemcell Technologies) was used to enrich for lymphocytes prior to sorting for ST2+ or ST2− Tregs by FACS. Immediately post isolation, cells were resuspended to 106 cells/mL in expansion buffer (RPMI + 10% FBS, 50μM 2-ME (Gibco® Thermo Scientific, Waltham, MA, USA)), 2.5μg/mL αCD28 (eBioscience), 100U/mL IL-2 (eBioscience), plated into αCD3 (eBioscience) coated wells (10μg/mL), and cultured for 40 hours at 37°C with 5% CO2. Recombinant IL-33 (eBioscience) was added to ex vivo Treg cultures as indicated at a concentration of 100ng/mL as previously reported.12
ELISA
IL-33 and AREG concentrations were assessed in the lung tissue, Treg supernates, and tumour cell line supernates using murine DuoSet® ELISA development systems (R&D Systems). Lung tissue was homogenized in PBS containing protease inhibitor cocktail (1X, Sigma Aldrich, Oakville, Ontario, Canada) and centrifuged at 330xg to pellet cells. The pelleted cell fraction was processed in lysis buffer as reported previously.9 Blood was collected by cardiac puncture in heparinized syringes, centrifuged at 3,000rpm for 10mins, and the plasma stored at −80°C. Supernates were collected from tumour cell lines at 80% confluency and from Tregs following 40 hours of ex vivo culture as described above. Protease inhibitor cocktail (1X, Sigma Aldrich) was added to supernates immediately following collection and samples were frozen in aliquots at −80°C. ELISA samples were assayed in duplicate according to the manufacturer’s protocol and the results analyzed using ELISA Analysis software (ElisaAnalysis.com; Leading Technology Group, Australia).
Immunohistochemistry and immunofluorescence
For immunofluorescence staining of IL-33, lung tissue was harvested from BALB/c and C57BL/6 tumour-bearing mice 3 weeks post orthotopic implant and frozen in Optimal Cutting Temperature (OCT) medium (Sakura Finetek, Torrance, CA, USA). Serial sections of 8-10μm were cut and stained in PBS + 4% FBS with CD11b-biotin (eBioscience) and IL-33 (R&D Systems) using streptavidin-FITC (eBioscience) and Alexa Fluor® 594 (Thermo Scientific) as secondary antibodies, respectively. Images were captured with a Zeiss Imager Z1 using a cooled, monochrome CCD camera (Retiga 4000R, QImaging) and Northern Eclipse software.
Lung tissue for immunohistochemistry was harvested 3 weeks post orthotopic implant of BALB/c and C57BL/6 mice and inflated with 10% neutral buffered formalin. Samples were fixed for 24 hours in formalin and then paraffin embedded and sectioned (4-5μm thickness) by the CTAG histopathology core (BC Cancer, Vancouver, BC, Canada). Following overnight incubation at 55°C, sections were deparaffinised with xylene, followed by a succession of ethanol washes (100%, 95%, and 70%). Antigen retrieval was performed in boiling citrate solution and cells were permeabilized with Triton X-100 in 1x TBS. Blocking and chromogenic detection were performed using a VECTASTAIN Elite ABC HRP kit (Vector Laboratories) according to the manufacturer’s instructions. Primary IL-33 was purchased from R&D systems and substrate was purchased from Vector Laboratories (ImmPACT DAB Peroxidase). Detection of pEGFR by IHC was performed with a Discovery XT automated slide staining system (Ventana Medical Systems Inc.) and extended antigen retrieval using Cell Conditioning 1 (CC1, Ventana). Phospho-EGFR (Tyr1068, Cell Signaling) was diluted 1:10 in SignalStain® Antibody Diluent (New England Biolabs) and incubated for 2hrs. Following IHC, sections were dehydrated and mounted (Permount, Fisher Scientific) for subsequent imaging at 20x magnification using a 3D Histotech Pannoramic MIDI whole slide scanner (3DHISTECH, Budapest, Hungary) and Pannoramic Viewer for visualization.
Clonogenic assay
In vitro clonogenic assays were performed by seeding 4T1 and 4T07 cells into 48 well plates in RPMI + 1% FBS + 1x pen/strep (Gibco, Thermo Scientific) at 1,000 cells/mL and incubating overnight at 37°C with 5% CO2. The following day, media was exchanged for RPMI with 1% or 10% FBS (as indicated) containing DMSO (0.1%), afatinib (10%, 50%, or IC50, Selleck Chemicals, Houston, TX, USA) anti-AREG (0.75-3µg/mL) or control IgG (0.75-3µg/mL). For recombinant AREG experiments, rmAREG (R&D Systems) was added five-fold above 431pg/mL (the average secreted concentration by 4T1 and 4T07 cell lines) to a maximum concentration of 270ng/mL. Cells were incubated for 8 days (37°C, 5% CO2) and media was changed at day 4. To determine cell viability post culture, cells were incubated with AlamarBlue® (1x, Thermo Fisher) for 2.5 hours at 37°C prior to reading fluorescence with a BioTek™ Cytation™ 3 Cell Imaging Multi-Mode Reader (BioTek™, Winooski, VT, USA, 540nm excitation, emission 585nm) using Gen5™ Version 2.06 Microplate Reader software (BioTek™, Winooski, VT, USA). Cells were then washed with PBS and stained with crystal violet for imaging. For the analysis of pEGFR following administration of recombinant AREG, 4T1 and 4T07 cells were stimulated with 100ng/mL or 270ng/mL of rmAREG for 2hrs followed by western blotting, as described below.
To assess metastatic tumour burden in vivo, lung tissue was processed and erythrocytes lysed with NH4Cl as previously described.9 Cells were plated in aliquots of 104, 105 and 106 in triplicate for clonogenic assays with medium containing 60µM 6-thioguanine to allow for selective growth of 4T1 cells. Following incubation for 10 days (37°C, 5% CO2), colonies were stained with malachite green for enumeration. The total number of clonogenic tumour cells in the lungs was calculated as described.9
Western blotting
Cultured tumour cells were lysed for 30mins on ice in RIPA Lysis and Extraction buffer (Thermo Fisher Scientific) containing 1x Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). Cultured Tregs were lysed in 0.1% Triton-X (Sigma-Aldrich) in PBS containing 1x Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) and by freeze-thaw at −80°C. Lysed cells were centrifuged for 20 minutes at 14000xg at 4°C, and then the protein fraction was removed and quantified using DCTM Protein Assay (Bio-Rad Laboratories). Samples were denatured by boiling for 5 minutes with 3x loading buffer (Bio-Rad Laboratories). For all samples, 20-25µg of protein was loaded into precast NuPAGE™ 4–12% Bis-Tris Gel (Invitrogen™ Thermo Fisher Scientific). Gels were run at 200V for 50mins in NuPAGE™ MOPS SDS Running Buffer (Thermo Fisher Scientific) and subsequently transferred at 100V for 1 hour onto Immobilon-P PVDF membranes (Millipore). Membranes were blocked for 1 hour at room temperature in 5% skim milk in TBST or 5% BSA (Sigma-Aldrich) in TBST. The following primary antibodies (Cell Signalling Technology, Danvers, MA, USA) were incubated overnight at 4°C in 5% BSA in TBST: anti-EGFR (1:1000), anti-phospho-EGFR [Tyr1068] (1:500), anti-AKT (1:1000), anti-phospho-AKT [Ser473] (1:1000), anti-ERK 1/2 (1:1000), anti-phospho-ERK 1/2 [Thr202/Tyr204] (1:1000) and anti-β-actin (1:5000) or anti-β-actin HRP conjugate (1:10000). Secondary goat anti-rabbit IgG HRP conjugate (1:2000) (Bio-Rad Laboratories) was added for 1 hour at room temperature in 5% skim milk or 5% BSA in TBST. Bands were visualized using Western LightningTM chemiluminescent substrate (Perkin Elmer) or SuperSignalTM West Pico or Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) at a 1/10 dilution (phosphor-EGFR) and developed on X-ray film (Mandel Scientific).
Recombinant protein and antibody treatment in vivo
Mice implanted orthotopically with 4T1 tumour cells were anesthetized with isofluorane and intranasally administered rmIL-33 (0.25μg, eBioscience), rmAREG (0.1µg, R&D Systems), PBS, or a combination of rmIL-33 and IgG or αAREG (5ug, R&D Systems) in 40μL of PBS. Intranasal administration of rmIL-33 at the indicated dose was performed once daily for three days as reported previously in naïve mice,33 three days prior to tissue harvest. Mice were sacrificed three weeks after tumour implant with tissues collected as described above.
Statistics
Student’s t-tests were used for comparison of data with p < 0.05 designated by (*), p < 0.01 as (**) and p < 0.001 as (***). One-tailed and two-tailed t-tests were used as indicated. GraphPad Prism software version 6 was used to analyze data and perform statistical comparisons. Unless otherwise stated, all data are reported as mean ± standard error of the mean (SEM).
Funding Statement
This research was funded by the CURE Foundation and the Cancer Research Society [21441; KLB] and the Canadian Institutes of Health Research [MOP-126138 to KLB and MOP-142313 to WWL/KLB]. ECH is funded by a Four Year Doctoral Fellowship and by a Li Tze Fong Memorial Fellowship from the University of British Columbia. BJW is funded by a Canadian Institutes of Health Research (CIHR) Doctoral Research Award. RAC is funded by a Frederick Banting and Charles Best Canada Graduate Scholarship from CIHR. KLB and WWL are Michael Smith Foundation for Health Research Biomedical Research Scholars.
Acknowledgments
The authors would like to thank Dr. Meegan Larsen, DVM, DVSc from MBed Pathology for expert pathological analysis of mouse lung tissue samples, and Anita Carraro for assistance with slide imaging. Additionally, the authors would like to thank Avery Lam and Dr. Megan Levings for protocols and helpful discussions.
Disclosure statement
The authors declare no conflict of interest or financial disclosures.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M.. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol. 1995;155(3): 1151–1164. [PubMed] [Google Scholar]
- 2.Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Reviews Immunol. 2007;7(11):875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- 3.Schmidt A, Oberle N, Ph K. Molecular mechanisms of treg-mediated T cell suppression. Front Immunol. 2012;3:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Halvorsen EC, Mahmoud SM, Kl B. Emerging roles of regulatory T cells in tumour progression and metastasis. Cancer Metastasis Rev. 2014;33(4):1025–1041. doi: 10.1007/s10555-014-9529-x. [DOI] [PubMed] [Google Scholar]
- 5.Kotsakis A, Koinis F, Katsarou A, Gioulbasani M, Aggouraki D, Kentepozidis N, Georgoulias V, Ek V. Prognostic value of circulating regulatory T cell subsets in untreated non-small cell lung cancer patients. Sci Rep. 2016;6:39247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Ah B. Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clinical Oncology: Official Journal Am Soc Clin Oncol. 2006;24(34):5373–5380. doi: 10.1200/JCO.2006.05.9584. [DOI] [PubMed] [Google Scholar]
- 7.Tan W, Zhang W, Strasner A, Grivennikov S, Cheng JQ, Hoffman RM, Karin M. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470(7335):548–553. doi: 10.1038/nature09707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 9.Halvorsen EC, Hamilton MJ, Young A, Wadsworth BJ, LePard NE, Lee HN, Firmino N, Collier JL, Kl B. Maraviroc decreases CCL8-mediated migration of CCR5+ regulatory T cells and reduces metastatic tumor growth in the lungs. Oncoimmunology. 2016;5(6):e1150398. doi: 10.1080/2162402X.2016.1150398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng G, Guo Z, Kiniwa Y, Voo KS, Peng W, Fu T, Wang DY, Li Y, Wang HY, Rf W. Toll-like receptor 8-mediated reversal of CD4+ regulatory T cell function. Science (80-). 2005;309(5739):1380–1384. doi: 10.1126/science.1113401. [DOI] [PubMed] [Google Scholar]
- 11.Bos PD, Plitas G, Rudra D, Lee SY, Ay R. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210(11):2435–2466. doi: 10.1084/jem.20130762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, Ay R. A distinct function of regulatory t cells in tissue protection. Cell. 2015;162(5):1078–1089. doi: 10.1016/j.cell.2015.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Christianson CA, Goplen NP, Zafar I, Irvin C, Good JT Jr., Rollins DR, Gorentla B, Liu W, Gorska MM, Chu H, et al. Persistence of asthma requires multiple feedback circuits involving type 2 innate lymphoid cells and IL-33. J Allergy Clin Immunol. 2015;136(1):59–68. e14. doi: 10.1016/j.jaci.2014.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li D, Guabiraba R, Besnard AG, Komai-Koma M, Jabir MS, Zhang L, Graham GJ, Kurowska-Stolarska M, Liew FY, McSharry C, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134(6):1422–1432. e1411. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuswanto W, Burzyn D, Panduro M, Wang KK, Jang YC, Wagers AJ, Benoist C, Mathis D. Poor Repair of Skeletal Muscle in Aging Mice Reflects a Defect in Local, Interleukin-33-Dependent Accumulation of Regulatory T Cells. Immunity. 2016;44(2):355–367. doi: 10.1016/j.immuni.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cayrol C, Jp G. IL-33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31::31–37. doi: 10.1016/j.coi.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 17.Talabot-Ayer D, McKee T, Gindre P, Bas S, Baeten DL, Gabay C, Palmer G. Distinct serum and synovial fluid interleukin (IL)-33 levels in rheumatoid arthritis, psoriatic arthritis and osteoarthritis. Joint, Bone, Spine: Revue Du Rhumatisme. 2012;79(1):32–37. doi: 10.1016/j.jbspin.2011.02.011. [DOI] [PubMed] [Google Scholar]
- 18.Liew FY, Girard JP, Hr T. Interleukin-33 in health and disease. Nat Reviews Immunol. 2016;16(11):676–689. doi: 10.1038/nri.2016.95. [DOI] [PubMed] [Google Scholar]
- 19.Kakkar R, Rt L. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discovery. 2008;7(10):827–840. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vasanthakumar A, Moro K, Xin A, Liao Y, Gloury R, Kawamoto S, Fagarasan S, Mielke LA, Afshar-Sterle S, Masters SL, et al. The transcriptional regulators IRF4, BATF and IL-33 orchestrate development and maintenance of adipose tissue-resident regulatory T cells. Nat Immunol. 2015;16(3):276–285. doi: 10.1038/ni.3085. [DOI] [PubMed] [Google Scholar]
- 21.Griesenauer B. Paczesny S: the ST2/IL-33 axis in immune cells during inflammatory diseases. Front Immunol. 2017;8:475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monticelli LA, Osborne LC, Noti M, Tran SV, Zaiss DM, Artis D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc Natl Acad Sci U S A. 2015;112(34):10762–10767. doi: 10.1073/pnas.1509070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12(11):1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Busser B, Sancey L, Brambilla E, Coll JL, Hurbin A. The multiple roles of amphiregulin in human cancer. Biochim Biophys Acta. 2011;1816(2):119–131. doi: 10.1016/j.bbcan.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Zaiss DM, Gause WC, Osborne LC, Artis D. Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity. 2015;42(2):216–226. doi: 10.1016/j.immuni.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, Sefik E, Tan TG, Wagers AJ, Benoist C, et al. A special population of regulatory T cells potentiates muscle repair. Cell. 2013;155(6):1282–1295. doi: 10.1016/j.cell.2013.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kariagina A, Xie J, Leipprandt JR, Sz H. Amphiregulin mediates estrogen, progesterone, and EGFR signaling in the normal rat mammary gland and in hormone-dependent rat mammary cancers. Horm Cancer. 2010;1(5):229–244. doi: 10.1007/s12672-010-0048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma L, de Roquancourt A, Bertheau P, Chevret S, Millot G, Sastre-Garau X, Espie M, Marty M, Janin A, Calvo F. Expression of amphiregulin and epidermal growth factor receptor in human breast cancer: analysis of autocriny and stromal-epithelial interactions. J Pathol. 2001;194(4): 413–419. [DOI] [PubMed] [Google Scholar]
- 29.Green JA, Arpaia N, Schizas M, Dobrin A, Ay R. A nonimmune function of T cells in promoting lung tumor progression. J Exp Med. 2017;214(12):3565–3575. doi: 10.1084/jem.20170356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamilton MJ, Bosiljcic M, Lepard NE, Halvorsen EC, Ho VW, Banath JP, Krystal G, Kl B. Macrophages are more potent immune suppressors ex vivo than immature myeloid-derived suppressor cells induced by metastatic murine mammary carcinomas. J Immunol. 2014;192(1):512–522. doi: 10.4049/jimmunol.1300096. [DOI] [PubMed] [Google Scholar]
- 31.Hamilton MJ, Halvorsen EC, LePard NE, Bosiljcic M, Ho VW, Lam V, Banath J, Bennewith KL, Krystal G. SHIP represses lung inflammation and inhibits mammary tumor metastasis in BALB/c mice. Oncotarget. 2016;7(4):3677–3691. doi: 10.18632/oncotarget.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halim TY, Steer CA, Matha L, Gold MJ, Martinez-Gonzalez I, McNagny KM, McKenzie AN, Takei F. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–435. doi: 10.1016/j.immuni.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Gonzalez I, Matha L, Steer CA, Ghaedi M, Poon GF, Takei F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. 2016;45(1):198–208. doi: 10.1016/j.immuni.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 34.Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, He V. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20(11):1496–1510. doi: 10.1101/gad.1417406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keating GM. Afatinib: a review of its use in the treatment of advanced non-small cell lung cancer. Drugs. 2014;74(2):207–221. doi: 10.1007/s40265-013-0170-8. [DOI] [PubMed] [Google Scholar]
- 36.Tanaka A. Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27(1):109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Facciabene A, Peng X, Hagemann IS, Balint K, Barchetti A, Wang LP, Gimotty PA, Gilks CB, Lal P, Zhang L, et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature. 2011;475(7355):226–230. doi: 10.1038/nature10169. [DOI] [PubMed] [Google Scholar]
- 38.Moussion C, Ortega N, Jp G. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’?. PLoS One. 2008;3(10):e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin NT, Mu M. Interleukin 33 is a guardian of barriers and a local alarmin. Nat Immunol. 2016;17(2):122–131. doi: 10.1038/ni.3370. [DOI] [PubMed] [Google Scholar]
- 40.Luthi AU, Cullen SP, McNeela EA, Duriez PJ, Afonina IS, Sheridan C, Brumatti G, Taylor RC, Kersse K, Vandenabeele P, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31(1):84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 41.Heppner GH, Miller FR, Pm S. Nontransgenic models of breast cancer. Breast Cancer Res. 2000;2(5): 331–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Siede J, Frohlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, Saito H, Nakae S, Lohning M. IL-33 receptor-expressing regulatory T cells are highly activated, Th2 biased and suppress CD4 T cell Proliferation through IL-10 and TGFbeta Release. PLoS One. 2016;11(8):e0161507. doi: 10.1371/journal.pone.0161507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matta BM, Lott JM, Mathews LR, Liu Q, Rosborough BR, Blazar BR, Hr T. IL-33 is an unconventional Alarmin that stimulates IL-2 secretion by dendritic cells to selectively expand IL-33R/ST2+ regulatory T cells. J Immunol. 2014;193(8):4010–4020. doi: 10.4049/jimmunol.1400481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jovanovic IP, Pejnovic NN, Radosavljevic GD, Pantic JM, Milovanovic MZ, Arsenijevic NN, Ml L. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. Int J Cancer. 2014;134(7):1669–1682. doi: 10.1002/ijc.28481. [DOI] [PubMed] [Google Scholar]
- 45.Jovanovic I, Radosavljevic G, Mitrovic M, Juranic VL, McKenzie AN, Arsenijevic N, Jonjic S, Ml L. ST2 deletion enhances innate and acquired immunity to murine mammary carcinoma. Eur J Immunol. 2011;41(7):1902–1912. doi: 10.1002/eji.201141417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sternlicht MD, Sw S. The ADAM17-amphiregulin-EGFR axis in mammary development and cancer. J Mammary Gland Biol Neoplasia. 2008;13(2):181–194. doi: 10.1007/s10911-008-9084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurbin A, Dubrez L, Coll JL, Favrot MC. Inhibition of apoptosis by amphiregulin via an insulin-like growth factor-1 receptor-dependent pathway in non-small cell lung cancer cell lines. Ann N Y Acad Sci. 2003;1010:354–357. [DOI] [PubMed] [Google Scholar]
- 48.Wang S, Zhang Y, Wang Y, Ye P, Li J, Li H, Ding Q, Xia J. Amphiregulin confers regulatory T cell suppressive function and tumor invasion via the EGFR/GSK-3beta/Foxp3 Axis. J Biol Chem. 2016;291(40):21085–21095. doi: 10.1074/jbc.M116.717892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cn J, Ye S, Cao Y, Ad B, Rs C, Ling X, Rp R, Jp D, Bl E, Al N, et al. Functional and molecular characterisation of EO771.LMB tumours, a new C57BL/6-mouse-derived model of spontaneously metastatic mammary cancer. Dis Model Mech. 2015;8(3):237–251. doi: 10.1242/dmm.017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.