

ABSTRACT
Monoclonal anti-programmed cell death 1 (PD1) antibodies are successful cancer therapeutics, but it is not well understood why individual antibodies should have idiosyncratic side-effects. As the humanized antibody SHR-1210 causes capillary hemangioma in patients, a unique toxicity amongst anti-PD1 antibodies, we performed human receptor proteome screening to identify nonspecific interactions that might drive angiogenesis. This screen identified that SHR-1210 mediated aberrant, but highly selective, low affinity binding to human receptors such as vascular endothelial growth factor receptor 2 (VEGFR2), frizzled class receptor 5 and UL16 binding protein 2 (ULBP2). SHR-1210 was found to be a potent agonist of human VEGFR2, which may thereby drive hemangioma development via vascular endothelial cell activation. The v-domains of SHR-1210’s progenitor murine monoclonal antibody ‘Mab005ʹ also exhibited off-target binding and agonism of VEGFR2, proving that the polyspecificity was mediated by the original mouse complementarity-determining regions (CDRs), and had survived the humanization process. Molecular remodelling of SHR-1210 by combinatorial CDR mutagenesis led to deimmunization, normalization of binding affinity to human and cynomolgus PD1, and increased potency in PD1/PD-L1 blockade. Importantly, CDR optimization also ablated all off-target binding, rendering the resulting antibodies fully PD1-specific. As the majority of changes to the paratope were found in the light chain CDRs, the germlining of this domain drove the ablation of off-target binding. The combination of receptor proteome screening and optimization of the antibody binding interface therefore succeeded in generating novel, higher-potency, specificity-enhanced therapeutic IgGs from a single, clinically sub-optimal progenitor. This study showed that highly-specific off-target binding events might be an under-appreciated phenomenon in therapeutic antibody development, but that these unwanted properties can be fully ameliorated by paratope refinement.
Keywords: antibody, therapeutic, paratope, immunogenicity, humanization, specificity, toxicity, polyreactivity, hemangioma
Introduction
Monoclonal antibodies that target programmed cell death 1 (PD1) and antagonize its function can be of substantial value in treating cancer, by boosting the immune response against malignant cells with high PD-L1 expression and/or which exhibit high levels of mutation.1 The broad utility of anti-PD1s and their potential to be productively combined in the clinic with many old and new therapeutic modalities has led to the rapid proliferation of different molecules in development.1 This is an unprecedented situation, where there are many more clinical trials of individual antibodies, with superficially-overlapping characteristics, than would be typical for any given drug target in the past.2 As many clinical trials for the first wave of immunotherapeutic antibodies are now reading out, this offers the unique opportunity to observe idiosyncratic side-effects of individual anti-PD1 antibodies, and to then identify the underlying molecular mechanisms.3
Early clinical trial reports have shown that the anti-PD1 antibody SHR-1210 (also known as camrelizumab) demonstrated the expected biological activity, but also had the unusual toxicity profile of causing capillary hemangioma.4 This highly specific side-effect has not been reported for other anti-PD1 antibodies. Heretofore, it has not been known why such an unusual and specific skin toxicity should develop when treating patients with a mouse-derived monoclonal antibody molecule which, following the theory of clonal selection,5 are commonly assumed to be inherently monospecific.
In the study reported here, we successfully identified the off-target binding specificities of SHR-1210, and thereby identified that the likely mechanism by which it stimulates vascular neogenesis (leading to hemangioma) is by modulating the vascular receptors VEGFR2, and possibly frizzled class receptor 5 (FZD5). These findings suggested that toxicity mediated by highly specific off-target binding events might be an under-appreciated phenomenon in therapeutic antibody development. Indeed, chimeric Mab005 (containing the v-domains of the progenitor mouse monoclonal antibody, before humanization) and SHR-1210 both exhibited the same off-target reactivities and potent VEGFR2 agonism, highlighting that even antibodies derived from wild-type strains of mice can be inherently polyspecific. Importantly however, through comprehensive molecular engineering we refined the antibody paratope and successfully generated a panel of novel antibodies with exquisite specificity for PD1 and optimal potency.
Results
Antibody binding specificity analyses
SHR-1210, a humanized IgG4 antibody, was derived from murine hybridoma Mab005.4 To minimize aggregation risk in the recombinant IgGs produced, and therefore reduce the risk of aberrant binding signals and crystalizable fragment (Fc) receptor interactions, the v-domains of SHR-1210 and Mab005 were both cloned and expressed in IgG1 effector null format (referred to hereafter as SHR-1210-IgG1 and Mab005-IgG1, respectively), producing high purity, monomeric IgGs. A human receptor proteome array-based binding screen on live HEK-293 cells identified that SHR-1210-IgG1 exhibited strong binding to membrane-expressed PD1, but additionally had three potential off-target binding specificities (out of 4,975 receptors screened). The three off-target specificities were identified as FZD5, UL16 binding protein 2 (ULBP2), and KDR (also known as VEGFR2). To confirm the off-target binding events, the plasmids encoding for these receptors and controls were verified by DNA sequencing. The sequence-confirmed plasmid samples for control and potential target receptors were then re-arrayed onto new chips for repeat analyses in duplicate. The effective induction of expression from all re-arrayed plasmids was confirmed via scanning the chips for ZS green, which is co-encoded on all expression plasmids as an internal control marker. This analysis showed clearly detectable ZS expression in all positions where plasmids were spotted (Figure 1A). Further identically-spotted slides were then used to re-probe transfected cells with SHR-1210-IgG1 (Figure 1B), isotype IgG1 (negative control, Figure 1C), rituximab (IgG1 positive control, Figure 1D), and a chip where only secondary antibody probe was applied (Figure 1E). These analyses showed that SHR-1210-IgG1 again demonstrated measurable binding over background (on both chips) on cells transfected with PD1, FZD5, ULBP2 and VEGFR2, but no binding to FcγR1a (due to IgG1null isotype) or any other spots (Figure 1B). Rituximab demonstrated binding to CD20 and FcγR1a (due to effector positive IgG1 isotype) as expected, with no observable binding to PD1, FZD5, ULBP2 and VEGFR2 (Figure 1D). In the chips probed with isotype control IgG1 antibody (Figure 1C) and no primary antibody (Fig, 1E), only the expected control proteins showed any signal (the mouse monoclonal secondary antibody only very weakly binds to human FcγR1a). This clean performance of the control chips suggested that SHR-1210-IgG1 binding signals on PD1, FZD5, ULBP2 and VEGFR2 were specific.
Figure 1.
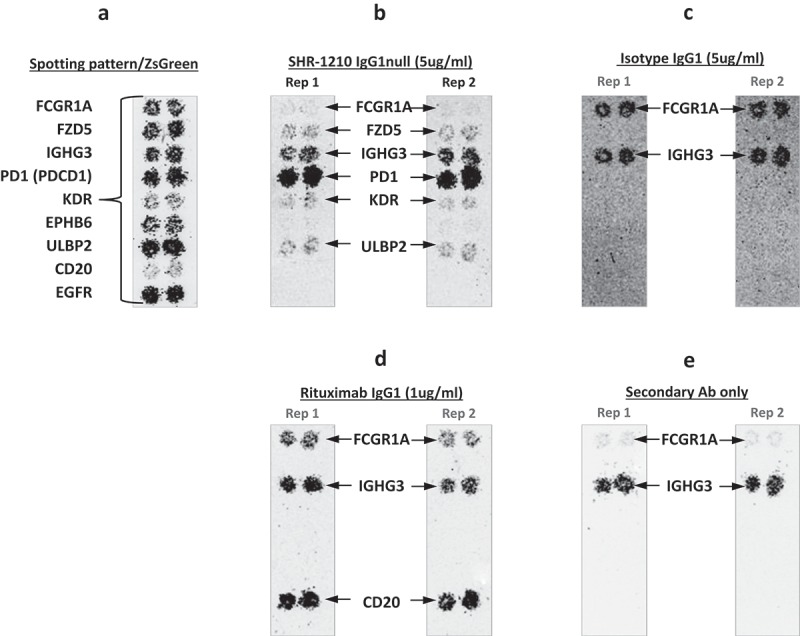
Off-target binding analysis chip re-array assay.
After performing an array-based binding screen on 4,975 human receptors for SHR-1210-IgG1, confirmatory analyses of binding specificity were performed on chips in which plasmids encoding PD1 and putative SHR-1210 off-target binding proteins were arrayed and used to transfect HEK-293 cells. Effective transfection of all plasmids was confirmed by screening for the co-encoded marker ZS green (A). Separate chips were then probed in duplicate using SHR-1210-IgG1 (B), Isotype IgG1 (C), Rituximab (D), and no primary antibody (E). These analyses confirmed that only SHR-1210-IgG1 exhibited binding to PD1, but also exhibited unexpected off-target binding to VEGFR2, FZD5 and ULBP2 proteins.
To secondarily confirm these findings with an orthogonal, high-sensitivity assay, the sequence-verified plasmids for PD1, VEGFR2, ULBP2, FZD5, and ZS green-only (negative control) were used to perform transient transfection of the human cell line HEK-293. Transfected cells were then stained using Mab005-IgG1, SHR-1210-IgG1, pembrolizumab IgG1null analog and isotype IgG1. Each antibody was used in repeat staining at high (5 μg/ml, Figure 2A, 2B) and moderate (1 μg/ml, Supplemental Figure 1A, 1B) concentration against both receptor-transfected and ZS green-transfected cells (to measure background binding). In staining of PD1-transfected cells at both 5 and 1 μg/ml (Figure 2A, Supplemental Figure 1A), all tested antibodies other than the isotype control showed the expected strong staining of PD1-transfected cells, but not ZS green-transfected cells. In contrast, staining of VEGFR2 (Figure 2A), FZD5 (Figure 2B), and ULBP2-transfected cells (Figure 2B) fully correlated with the chip data shown in Figure 1, with only Mab005-IgG1 and SHR-1210-IgG1 exhibiting strong binding signal on all 3 targets. This finding confirmed that the off-target binding reactivity of SHR-1210 derived directly from the v-domains of the chimeric Mab005 and was not solely a result of polyreactivity being induced during the humanization process, which is a known risk of v-gene engineering6. The off-target binding activity of SHR-1210 was therefore deduced to be housed directly in the complementarity-determining regions (CDRs), as it was retained when the murine CDRs of Mab005 were grafted onto human germline frameworks.
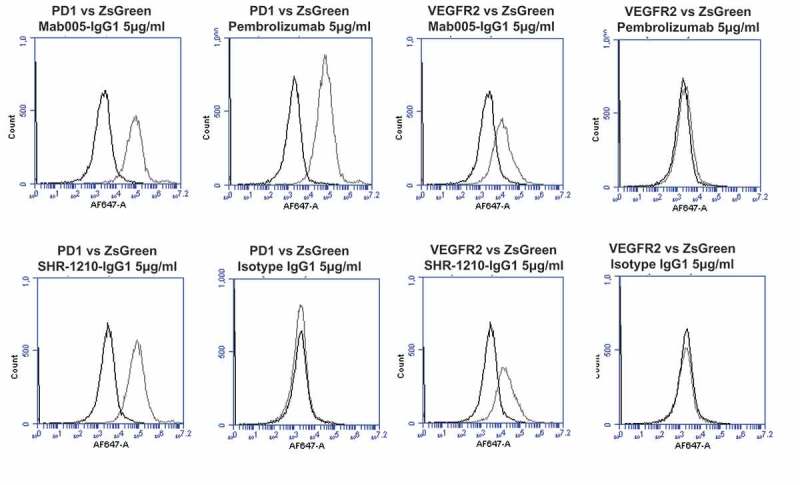
Figure 2.
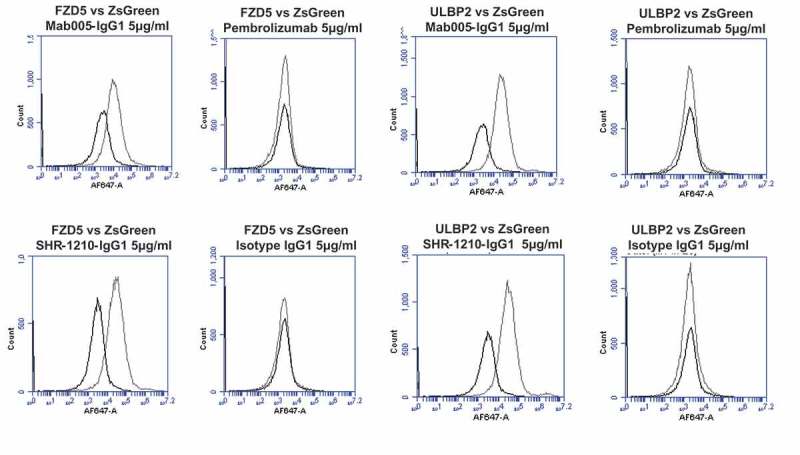
Specificity analyses by flow cytometry using transiently-transfected HEK-293 cells.
Analyses of binding specificity were performed on HEK-293 cells transiently transfected with plasmids encoding either (A) human PD1, human VEGFR2, (B) human FZD5, human ULBP2. All plots show the target of interest transfected (grey line) versus ZS green marker-only transfected cells (black line). Transfected cells were stained using Mab005-IgG1, SHR-1210-IgG1, Pembrolizumab IgG1 null analog, and isotype IgG1. Each antibody was used in repeat staining at 5 μg/ml. These analyses confirmed that all antibodies (other than the isotype control IgG1) exhibited binding to PD1, but no antibody exhibited measurable signal on ZS-green transfected cells. Both Mab005-IgG1 and SHR-1210-IgG1 also exhibited strong binding to all targets, while the isotype IgG1 and Pembrolizumab analog did not.
SHR-1210-IgG1 v-domain paratope refinement
As the off-target reactivity of Mab005 was derived from the CDRs of the antibody, we engineered the progenitor SHR-1210-IgG1 to generate new, fully novel antibodies. To bias our engineering efforts towards final lead therapeutic IgG compounds with optimal drug-like properties, we chose to graft the CDRs of the parental antibody onto germline v-domain scaffolds IGHV3-7 and IGKV1-39 (Table 1), which are known to have good solubility and drug development qualities, and are used at high frequency in the expressed human antibody repertoire.7 The CDR-grafted IGHV3-7/IGKV1-39 v-domain sequences were combined into a human antigen-binding fragment (Fab) phage display format and a mutagenesis library cassette was generated, with diversity limited to the CDRs. This library sampled human germline residues at high frequency at all positions,8 but also added non-germline mutagenesis in multiple CDRs. The final Fab library was ligated into a phage display vector and transformed into E. coli via electroporation to generate 1.3 × 109 independent clones. Library build quality was verified by sequencing 96 clones. This sequencing data showed that the positions encoding either the murine or human germline residue at each position of variance had been effectively sampled at a frequency of approximately 50%, other than in select positions in the CDRs where full amino acid diversity was sampled. Library selections were performed on biotinylated human and cynomolgus monkey PD1-Fc proteins in multiple separate branches. Post-selection screening and DNA sequencing revealed the presence of 64 unique Fab clones that exhibited strong binding to human and cynomolgus (cyno) PD1. Amongst these 64 clones, the framework sequences remained fully germline while mutations were also observed in all CDRs. Lead clones were ranked based on the level of CDR germlining versus enzyme-linked immunosorbent assay (ELISA) signals for binding to both human and cyno PD1-Fc. The v-domains of the 12 top clones from this ranking were then sub-cloned into IgG expression vectors for further testing as below.
Table 1.
Amino acid sequence of MAB005 murine anti-PD1 v-domains (mVH/mVL) and SHR-1210 CDR grafts.
| V DOMAIN | Human germline1 | Amino acid sequence2 |
|---|---|---|
| mMab005 VH | n/a | EVMLVESGGGLVKPGGSLKLSCAASGFTFSSYMMSWVRQTPEKRLEWVATISGGGANTYYPDSVKGRFTISRDNAKNTLYLQMSSLRSEDTALYYCARQLYYFDYWGQGTTLTVSS |
| SHR-1210-IgG1 VH | IGHV3-7 | EVQLVESGGGLVQPGGSLRLSCAASGFTFSSYMMSWVRQAPGKGLEWVATISGGGANTYYPDSVKGRFTISRDNAKNSLYLQMNSLRAEDTAVYYCARQLYYFDYWGQGTTVTVSS |
| mMab005 VL | n/a | DIQMTQSPASQSASLGEGVTITCLASQTIGTWLTWYQQKPGKSPQLLIYTATSLADGVPSRFSGSGSGTKFSFKISSLQAEDFVTYYCQQVYSIPWTFGGGTKLEIK |
| SHR-1210-IgG1 VL | IGKV1-39 | DIQMTQSPSSLSASVGDRVTITCLASQTIGTWLTWYQQKPGKAPKLLIYTATSLADGVPSRFSGSGSGTDFTLTISSLQPEDFATYYCQQVYSIPWTFGGGTKVEIK |
1Human germline definitions used for grafting, based on the international ImMunoGeneTics information system® system. 2CDR residues are in bold and underlined. Each sequence above shows the framework regions (FRs) and the CDRs in the following order: FR1-CDR1-FR2-CDR2-FR3-CDR3-FR4.
While germlining mutations were observed in all CDRs for the lead clones derived directly from library selections, it remained possible that sequence analyses might allow further clones to be designed to have maximal humanization. The 64 sequence-unique hits with binding signals against human and cyno protein were therefore used to analyze the retention frequency for murine amino acids in the CDRs. Positional amino acid retention frequency was expressed as a percentage found in the VL and VH domains (Figure 3A and B, respectively). Murine residues with RF < 75% (median value between the 50% encoding at key positions of murine residues, and full murine amino acid retention at 100%) were regarded as positions that were possibly not essential to the target-binding paratope and were likely to be open to mutagenesis, in a series of combinatorial designs. In the VH domain (excluding the CDR-H3), only four of nine murine residues in the CDR-H1 and H2 exhibited retention frequency above 75% and the sole murine residue (methionine, M) found in the CDR-H1 was not found in any functional clones, giving it a frequency value of 0% (Figure 3B).
Figure 3.
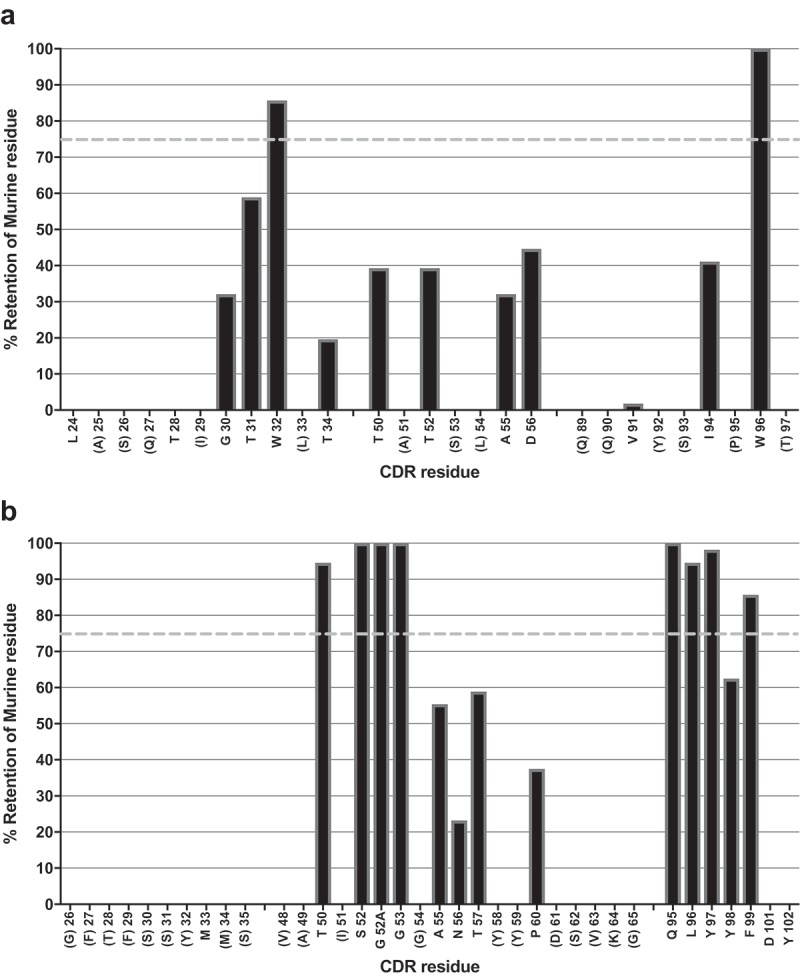
Analysis of CDR residue tolerance for mutation to germline.
A plot of murine amino acid retention frequencies in the CDRs of the ELISA-positive population of 64 unique Fab fragment clones is shown for VL (A) and VH (B) domains, respectively. Only those residues targeted for human/murine residue mutagenesis are plotted, other than in the HCDR3. CDR residues noted in parentheses on the X-axes were identical to those found in the human germlines used for grafting (IGKV1-39 and IGHV3-7). In both plots the dashed line in grey at 75% represents the cut-off for tolerance of murine residue replacement by human germline.
In the VL domain, only two of 13 murine CDR residues derived from the Mab005 sequence were retained with frequencies >75% (Figure 3A). This analysis strongly suggested that almost the entire CDR-L2 sequence could be changed to the germline IGKV1-39 sequence. Importantly, the tryptophan (W) residue found in the CDR-L3 of Mab005 was retained at almost 100% (Figure 3A), was in a position donated by the J segment during light chain splicing and was only regarded as non-human due to the use of the human JK4 sequence in the starting library. This observation allowed a redesign of the light chain in a series of designer clones to include the human JK1 sequence instead of JK4, as human JK1 naturally contains a W residue at that position, rendering the resulting light chain sequence fully germline in both the frameworks and CDR-L2 and L3. Designs containing only those murine residues with RF > 75% were cloned in IgG1 null format and co-transfected in a matrixed fashion to create 15 final designer IgGs in total. All IgGs were readily expressed and purified from transient transfections of mammalian cells.
Lead IgG specificity and potency characteristics
The purified IgGs described above were then tested for binding to human and cyno PD1-Fc in direct titration ELISA format. This analysis demonstrated that several library-derived and designer clones had human and cyno PD1 binding profiles similar to (within 2-fold), or improved over, SHR-1210-IgG1 (Figure 4A-F). As the ELISA signals for directly binding IgGs are strongly influenced by avidity, rather than true 1:1 binding affinity, we then proceeded to perform higher-sensitivity, solution-phase Alphascreen epitope competition and Biacore binding affinity determinations. An Alphascreen assay was established to allow the testing of IgGs for epitope competition with SHR-1210-IgG1 binding to biotinylated monomeric human PD1. In this assay, the top-performing library-derived and designer IgGs exhibited full inhibition of signal, showing that both the potency and epitope specificity on PD1 of SHR-1210 had been maintained (Figure 5A, B). Biacore analyses of binding affinity were performed for all IgGs to solution-phase, monomeric human and cyno PD1 ectodomain proteins. In all cases, accurate 1:1 binding affinities with low Chi2 values were obtained (Table 2). These analyses showed that library-derived clones that consistently gave the highest EC50 and IC50 values in Fab and IgG ELISA and Alphascreen assays also showed highest affinity binding to human and cyno PD1. Importantly, library-derived clones IgG1-06D02, IgG1-12B07, IgG1-12H04 and IgG1-16H10 and designer clones IgG1-04, IgG1-05, IgG1-08, IgG1-10, IgG1-11, IgG1-13 and IgG1-14 all exhibited improved binding affinities for both human and cyno PD1 in comparison to SHR-1210-IgG1. These improvements unexpectedly normalized the human/cyno affinities for these clones to within 3-fold (all KD values < 4.9 nM), as opposed to SHR-1210-IgG1, which exhibited an 8-fold differential (human KD – 4.0 nM, cyno KD – 32.0 nM).
Figure 4.
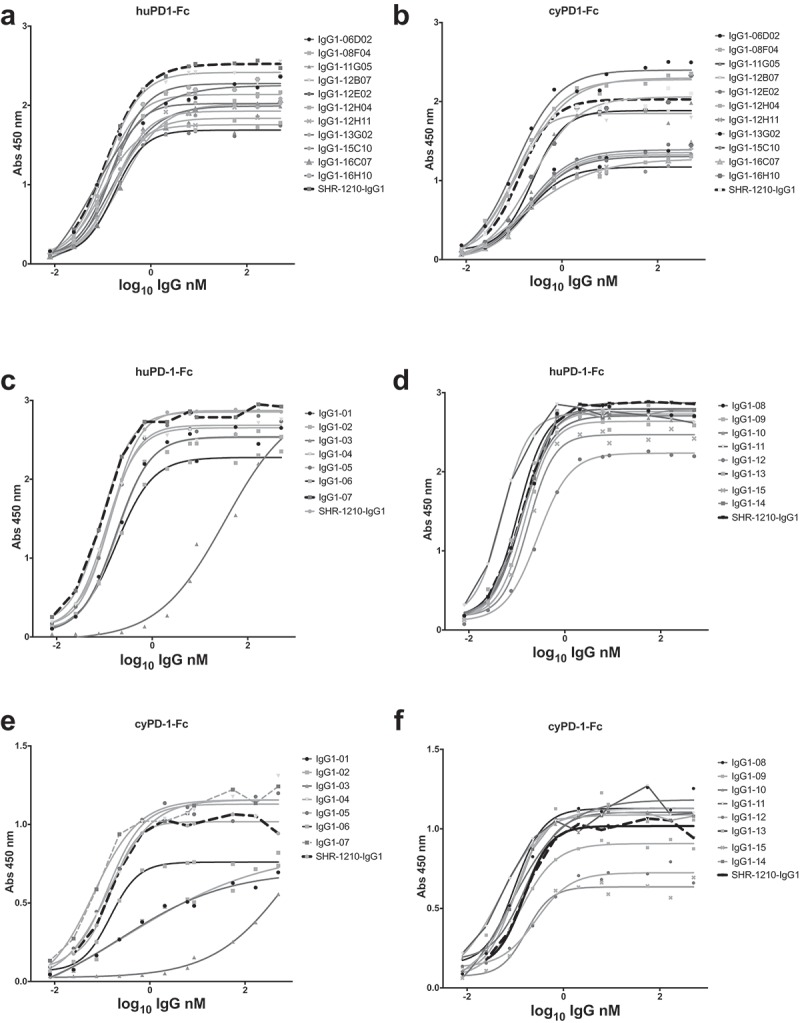
Direct titration ELISA for library-derived IgG1null clones binding to human and cyno PD1-Fc proteins.
SHR-1210-IgG1, library-derived clones (A, B) and designer clones (C-F) in human IgG1null format were titrated (in nM) in a direct binding ELISA against human and cyno PD1-Fc proteins.
Figure 5.
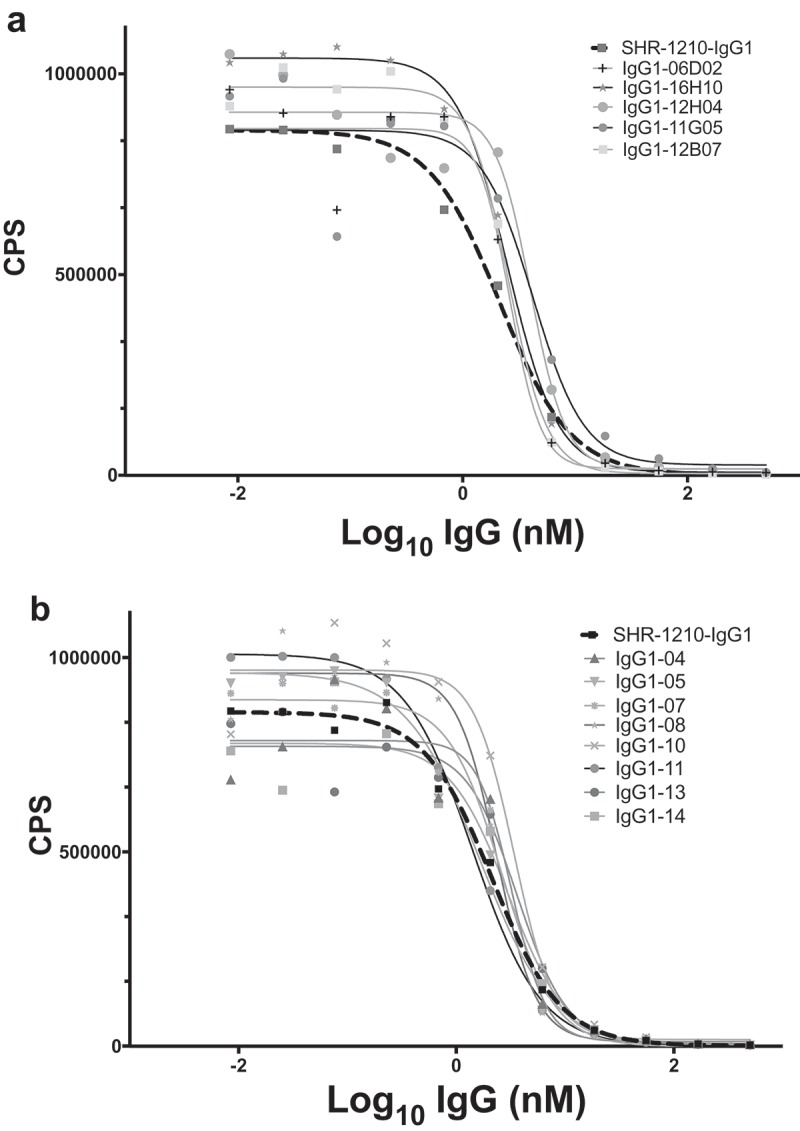
SHR-1210 epitope competition analysis of IgG1null proteins in Alphascreen.
Anti-PD1 IgG1null clones were applied in an epitope competition assay using Alphascreen technology. In this assay, library-derived (A) and designer (B) IgGs were analysed for their relative affinities and retention of the parental SHR-1210 epitope by competing for SHR-1210-IgG1 binding to human PD1 protein, in solution. All clones analysed showed strong, concentration-dependent neutralization of SHR-1210-IgG1 binding to PD1, indicating maintenance of the same binding epitope.
Table 2.
Biacore affinity values for IgG1null binding to human and cyno monomeric PD1.
| Human PD1 |
Cyno PD1 |
|||||||
|---|---|---|---|---|---|---|---|---|
| Clone name | ka (1/Ms) | kd (1/s) | Chi2 | KD (nM) | ka (1/Ms) | kd (1/s) | Chi2 | KD (nM) |
| IgG1-Pembro* | 4.9E+05 | 2.4E-03 | 1.3 | 4.8 | 3.9E+05 | 6.8E-04 | 1.7 | 1.8 |
| IgG1-Mab005 | 1.2E+05 | 4.7E-04 | 1.3 | 4.0 | 8.8E+04 | 2.8E-03 | 0.4 | 32.0 |
| IgG1-16H10 | 1.6E+05 | 3.4E-04 | 1.2 | 2.1 | 9.3E+04 | 4.2E-04 | 0.3 | 4.5 |
| IgG1-12H04 | 2.0E+05 | 3.2E-04 | 1.1 | 1.6 | 1.4E+05 | 6.4E-04 | 0.8 | 4.7 |
| IgG1-06D02 | 2.0E+05 | 2.5E-04 | 3.0 | 1.3 | 1.3E+05 | 6.2E-04 | 0.4 | 4.9 |
| IgG1-12B07 | 1.7E+05 | 2.5E-04 | 2.2 | 1.5 | 1.1E+05 | 1.6E-03 | 0.5 | 14.2 |
| IgG1-11G05 | 1.2E+05 | 5.8E-04 | 0.4 | 4.7 | 5.5E+04 | 1.3E-03 | 0.3 | 24.0 |
| IgG1-13G02 | 1.5E+05 | 2.0E-03 | 0.4 | 13.4 | 9.7E+04 | 6.4E-03 | 0.4 | 65.5 |
| IgG1-16C07 | 1.5E+05 | 1.8E-03 | 0.3 | 12.0 | 9.6E+04 | 7.5E-03 | 0.4 | 77.7 |
| IgG1-12H11 | 1.5E+05 | 2.8E-03 | 0.3 | 18.1 | 9.9E+04 | 1.0E-02 | 0.4 | 103.0 |
| IgG1-15C10 | 1.4E+05 | 1.8E-03 | 1.3 | 12.3 | 9.6E+04 | 1.4E-02 | 0.4 | 144.0 |
| IgG1-08F04 | 1.3E+05 | 2.5E-03 | 1.1 | 19.5 | 1.5E+05 | 2.6E-02 | 0.7 | 174.0 |
| IgG1-08 | 1.9E+05 | 2.8E-04 | 3.4 | 1.47 | 1.5E+05 | 5.2E-04 | 1.6 | 3.4 |
| IgG1-06 | 1.8E+05 | 2.9E-04 | 2.9 | 1.59 | 1.5E+05 | 5.2E-04 | 1.6 | 3.4 |
| IgG1-04 | 1.5E+05 | 2.7E-04 | 1.6 | 1.87 | 1.2E+05 | 3.2E-04 | 0.8 | 2.7 |
| IgG1-05 | 1.5E+05 | 2.9E-04 | 1.1 | 1.91 | 1.2E+05 | 3.3E-04 | 1.3 | 2.8 |
| IgG1-13 | 1.8E+05 | 4.4E-04 | 1.9 | 2.45 | 1.4E+05 | 9.8E-04 | 1.2 | 6.8 |
| IgG1-14 | 1.8E+05 | 4.6E-04 | 2.2 | 2.57 | 1.4E+05 | 9.7E-04 | 1.2 | 6.9 |
| IgG1-10 | 1.5E+05 | 4.0E-04 | 1.2 | 2.7 | 1.2E+05 | 7.1E-04 | 1.4 | 6.1 |
| IgG1-11 | 1.3E+05 | 4.0E-04 | 1.4 | 3.01 | 1.0E+05 | 7.3E-04 | 0.9 | 7.1 |
| IgG1-015 | 1.6E+05 | 3.5E-03 | 1.6 | 21.5 | 1.2E+05 | 8.6E-03 | 0.9 | 69.9 |
*Pembrolizumab analog
Key lead antibodies were analyzed for concentration-dependent binding at the cell surface via flow cytometry on Chinese hamster ovary (CHO) cells stably-transfected with human or cyno PD1 (Figure 6A-D). These analyses showed that lead library-derived and designer clones exhibit concentration-dependent binding to membrane-presented human and cyno PD1 with potencies equivalent to or improved over SHR-1210-IgG1 (Table 3). In particular, analyses performed on cyno PD1 CHO cells further confirmed that several library-derived and designer leads exhibited significantly improved binding for cyno PD1 in comparison to SHR-1210-IgG1 (Table 3).
Figure 6.
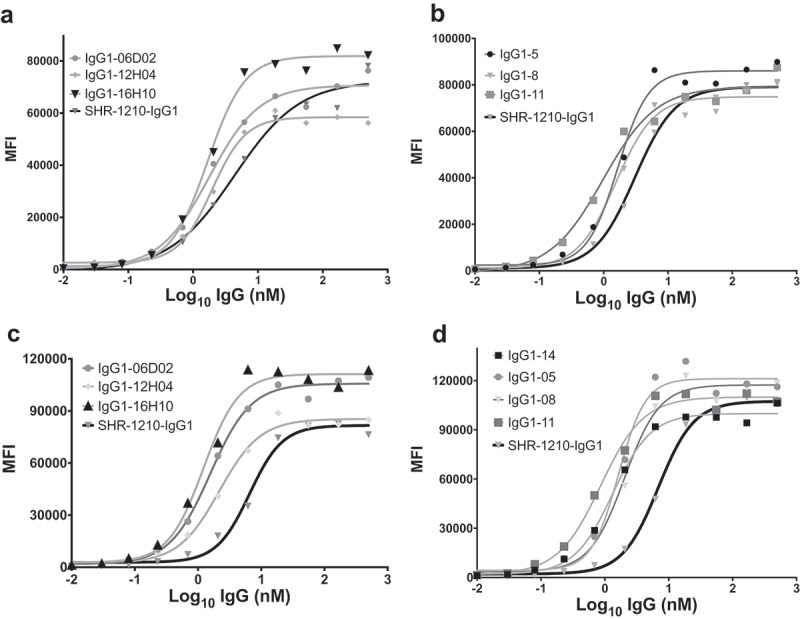
Flow cytometric binding to human and cyno PD1+ CHO cells.
SHR-1210-IgG1, lead library-derived (A) and designer (B) IgGs were examined for specific binding on CHO-K1 cells expressing human PD1. SHR-1210-IgG1, lead library-derived (C) and designer (D) IgGs were also examined for specific binding on CHO-K1 cells expressing cyno PD1. Concentration-dependent binding was observed against human and cyno PD1 for all clones, with weaker binding being observed for SHR-1210-IgG1 in each experiment.
Table 3.
EC50 values for IgG1null binding to human and cyno PD1-CHO cells.
| Clone name | huPD-1-Fc EC50 (nM) | rhPD-1-Fc EC50 (nM) |
|---|---|---|
| SHR-1210-IgG1 | 2.319 | 6.532 |
| IgG1-06D02 | 1.779 | 1.561 |
| IgG1-12H04 | 1.952 | 2.172 |
| IgG1-16H10 | 1.681 | 1.221 |
| IgG1-05 | 1.656 | 2.521 |
| IgG1-08 | 1.551 | 2.031 |
| IgG1-11 | 0.978 | 0.861 |
| IgG1-14 | 1.866 | 1.326 |
| IgG1-15 | 2.133 | ND |
| ND = Not Determined | ||
In a cell-based PD1/PD-L1 blockade reporter assay, all clones tested exhibited concentration-dependent antagonism of PD1. Indeed, multiple clones (including IgG1-06D02, IgG1-12H04, IgG1-16H10, IgG1-05, IgG1-08, IgG1-11 and IgG1-14) exhibited improved potency in PD1 blockade in comparison to SHR-1210-IgG1 (Table 4). In addition, clones IgG1-06D02 and IgG1-16H10 exhibited increased maximal signal in the assay, over SHR-1210-IgG1 and an IgG4 nivolumab analog (Figure 7).
Table 4.
EC50 values for IgG1null blockade of human PD1/PD-L1.
| Clone name | EC50 (ng/ml) |
|---|---|
| SHR-1210-IgG1 | 285 |
| IgG1-16H10 | 252 |
| IgG1-12H04 | 188 |
| IgG1-06D02 | 224 |
| IgG1-08 | 127 |
| IgG1-05 | 156 |
| IgG1-14 | 212 |
| IgG1-11 | 172 |
Figure 7.
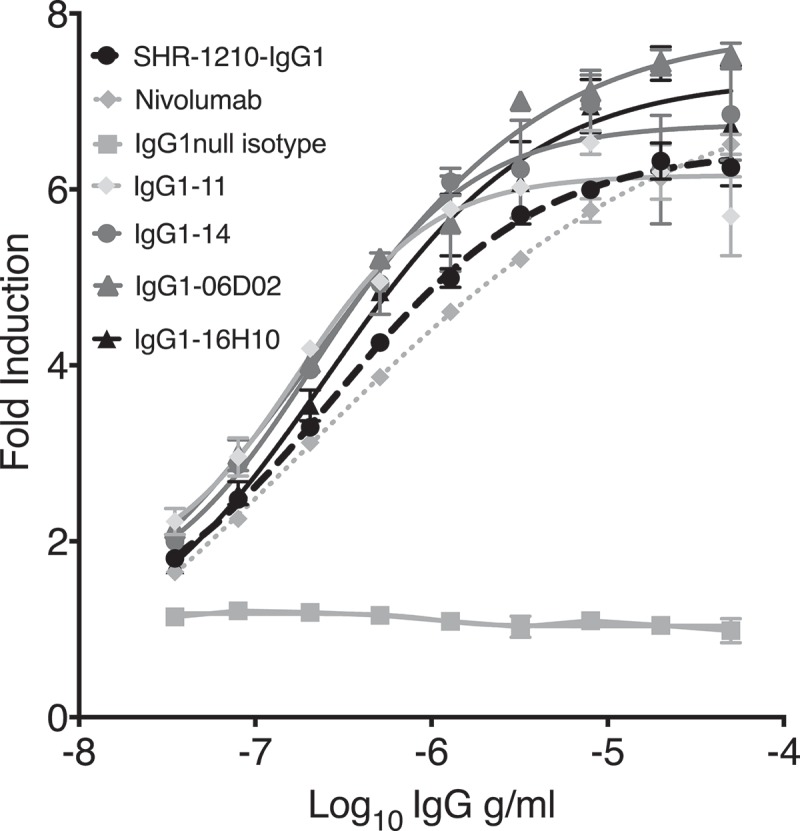
Cell-based PD1/PD-L1 antagonism assay.
Analyses of antagonism of human PD1 function at the cell surface, for example lead clones IgG1-11, IgG1-14, IgG1-06D02 and IgG1-16H10 in human IgG1null format, showed that all novel clones exhibited concentration-dependent antagonistic activity, with higher relative potency in comparison to both SHR-1210-IgG1 and IgG4 nivolumab analog.
Antibody v-domain T cell epitope analyses
In silico technologies (Abzena, Ltd.), which are based on identifying the location of T cell receptor (TCR) epitopes in therapeutic antibodies and proteins, were used for assessing the immunogenicity of both the Mab005 and lead antibody v-domains, as outlined in the Materials and Methods section. Identified potential TCR epitope peptides were grouped into four classes: High Affinity Foreign (’HAF’ – high immunogenicity risk), Low Affinity Foreign (‘LAF’ – lower immunogenicity risk), T Cell Epitope DatabaseTM (TCED)+ (previously identified epitope in TCED database), and Germline Epitope (‘GE’ – human germline peptide sequence with high major histocompatibility complex (MHC) Class II binding affinity). GE 9mer peptides are unlikely to have immunogenic potential due to T cell tolerance, as validated by previous studies with a wide range of germline peptides and with germline constant and variable domains.9,10 Importantly, such high affinity germline v-domain epitopes (aided further by similar sequences in the human antibody constant regions) may also compete for MHC Class II occupancy at the membrane of antigen-presenting cells, reducing the risk of foreign peptide presentation being sufficient to achieve the ‘activation threshold’ required for T cell stimulation.11 High GE content is therefore a potentially beneficial quality in clinical development of an antibody therapeutic. As shown in Table 5, key lead v-domains exhibited significant beneficial changes in peptide epitope content in comparison to SHR-1210-IgG1. As the v-domain framework regions (i.e., outside the CDR sequences) of SHR-1210-IgG1 and all leads were germline in sequence (Table 1), all improvements in predicted immunogenicity came about as a result of the germlining of CDR residues. Indeed, in several clones, the VL domains were found to be fully deimmunized. GE epitope content was also found to be significantly increased in the VL regions of lead clones (from 1 to ≥ 3 in all leads), and TCED+ epitopes were eliminated from the VL domains in all leads (Table 5).
Table 5.
Human T cell epitope content in v-domains predicted by iTOPETM and TCEDTM.
| Clone Name | Germline epitopes | Low Affinity Foreign | High Affinity Foreign | TCED+ |
|---|---|---|---|---|
| SHR-1210 VL | 1 | 4 | 2 | 1 |
| SHR-1210 VH | 9 | 3 | 1 | 2 |
| 06D02 VL | 5 | 0 | 0 | 0 |
| 06D02 VH | 9 | 3 | 1 | 2 |
| 12H04 VL | 4 | 1 | 0 | 0 |
| 12H04 VH | 9 | 3 | 1 | 2 |
| 16H10 VL | 3 | 0 | 2 | 0 |
| 16H10 VH | 9 | 3 | 1 | 2 |
| IgG1-05 VL | 5 | 0 | 0 | 0 |
| IgG1-05 VH | 9 | 3 | 1 | 2 |
| IgG1-08 VL | 5 | 0 | 0 | 0 |
| IgG1-08 VH | 9 | 3 | 1 | 2 |
| IgG1-11 VL | 5 | 0 | 0 | 0 |
| IgG1-11 VH | 9 | 3 | 1 | 2 |
| IgG1-14 VL | 5 | 0 | 0 | 0 |
| IgG1-14 VH | 9 | 2 | 1 | 2 |
Binding specificity analyses for lead IgGs
To investigate whether off-target reactivity was retained in key lead IgGs, further on-chip binding analyses were performed (Figure 8A-L). The re-arrayed plasmids were again checked for transfection quality and the induction of ZS green expression was confirmed (Figure 8A). Control antibodies bound only their cognate targets and the isotype control IgG1 showed no binding (Figure 8B-D). Analyses using SHR-1210-IgG1 showed that the off-target binding signals previously identified were recapitulated (Figure 8E). Lead antibodies were then also used to probe this same chip set. Surprisingly, none of the library-derived or designer antibodies (Figure 8F-L) showed any measurable binding to any other protein than PD1. To confirm these findings, transfected cells were again used in fluorescence-activated cell sorting (FACS) analyses as in Figure 2. In staining of PD1-transfected cells at both 5 (Figure 9A) and 1 μg/ml (Supplemental Figure 2A, 2B), all tested antibodies showed the expected strong, specific staining of PD1-transfected, but not ZS green-transfected, cells. In contrast, staining of VEGFR2 (Figure 9A), FZD5 (Figure 9B) and ULBP2-transfected cells (Figure 9B) showed that none of the antibodies, IgG1-06D02, IgG1-12HO4, IgG1-05, or IgG1-08, exhibited any measurable binding above background.
Figure 8.
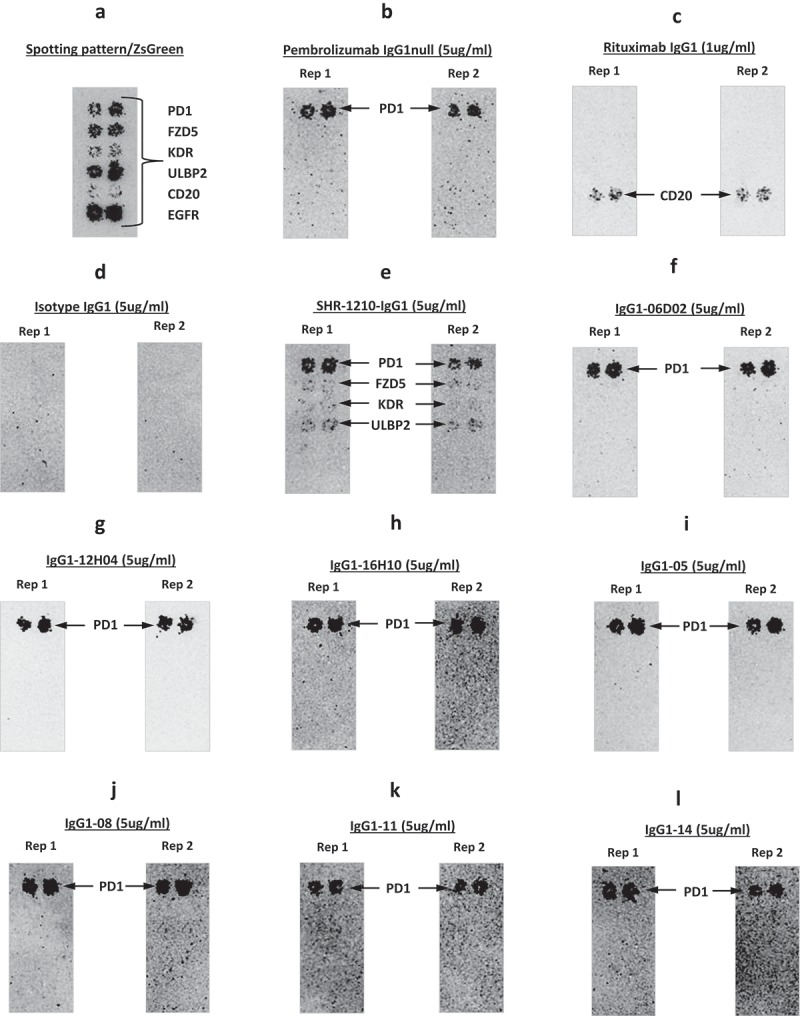
Off-target binding analysis re-array assay.
Analyses of binding specificity were performed on chips in which plasmids encoding relevant targets were arrayed and used to transfect HEK-293 cells. Transfection of all plasmids was confirmed by screening for the co-encoded marker ZS green (A). Separate chips were then probed in duplicate using pembrolizumab analog (B), rituximab (C), Isotype IgG1 (D), SHR-1210-IgG1 (E), and example library-derived and designer lead IgGs (F-L). These analyses confirmed that all anti-PD1 antibodies exhibited binding to PD1, but only SHR-1210-IgG1 also exhibited unexpected off-target binding to VEGFR2, FZD5 and ULBP2 proteins.
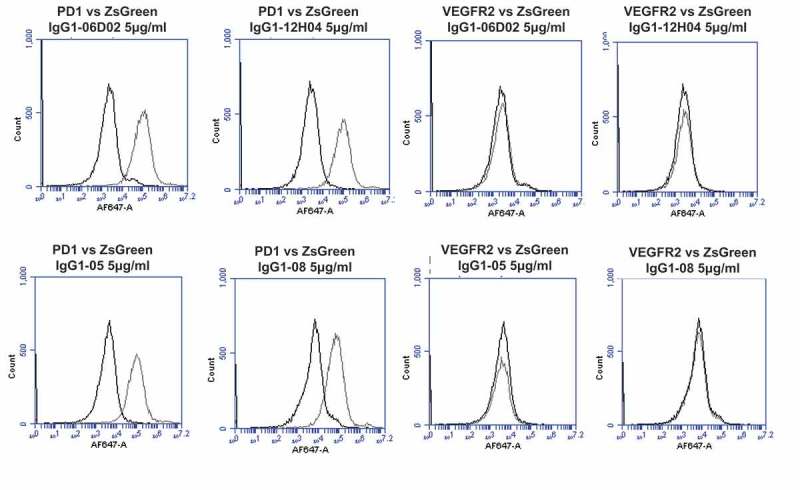
Figure 9.
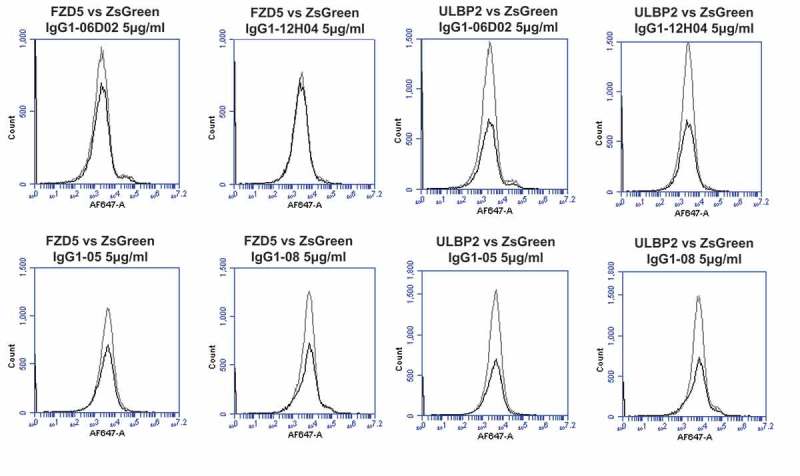
Specificity analyses for library-derived and designer lead IgGs by flow cytometry using transiently-transfected HEK-293 cells.
Analyses of binding specificity were performed on HEK-293 cells transiently transfected with plasmids encoding either (A) human PD1 and human VEGFR2, (B) human FZD5 and human ULBP2. All plots show the target of interest transfected cells (grey line) versus ZS green marker-only transfected cells (black line). Each antibody was used in repeat staining at 5 μg/ml. These analyses confirmed that all antibodies (other than the isotype control IgG1) exhibited binding to PD1, but no antibody exhibited measurable signal on ZS-green, VEGFR2, FZD5 or ULBP2 transfected cells.
To further examine whether or not the antibodies IgG1-06D02, IgG1-12HO4, IgG1-05 and IgG1-08 might still retain some minor affinity for the off-target proteins, we performed an ELISA assay where recombinant forms of each target protein were directly coated to allow low affinity, but potentially avid, interactions to be measured. SHR-1210-IgG1, Mab005-IgG1, library-derived clones and designer clones in human IgG1null format were titrated (in μg/ml) against human and cyno PD1, human and rhesus VEGFR, human FZD5, human ULBP2 and bovine serum albumin (BSA) (control) proteins. These analyses confirmed that all anti-PD1 antibodies exhibited strong binding to PD1, but only Mab005-IgG1 and SHR-1210-IgG1 exhibited measurable off-target binding to VEGFR2 and FZD5 proteins (Figure 10A-F). No binding was observed to the ULBP2 protein (data not shown) or BSA negative control (Figure 10F). In addition, assays designed to detect non-specific binding12 showed that neither SHR-1210-IgG1, nor any of several library-derived clones exhibited ‘sticky’ general polyreactivity characteristics (Supplemental Figure 3). These findings fully confirmed that the CDR sequences derived in the mutagenesis and reselection process had unexpectedly ablated the off-target binding of receptors VEGFR2, FZD5 and ULBP2 that may be the primary drivers of the clinical toxicities associated with SHR-1210, without adding pharmacokinetics (PK)-lowering polyreactivity.
Figure 10.
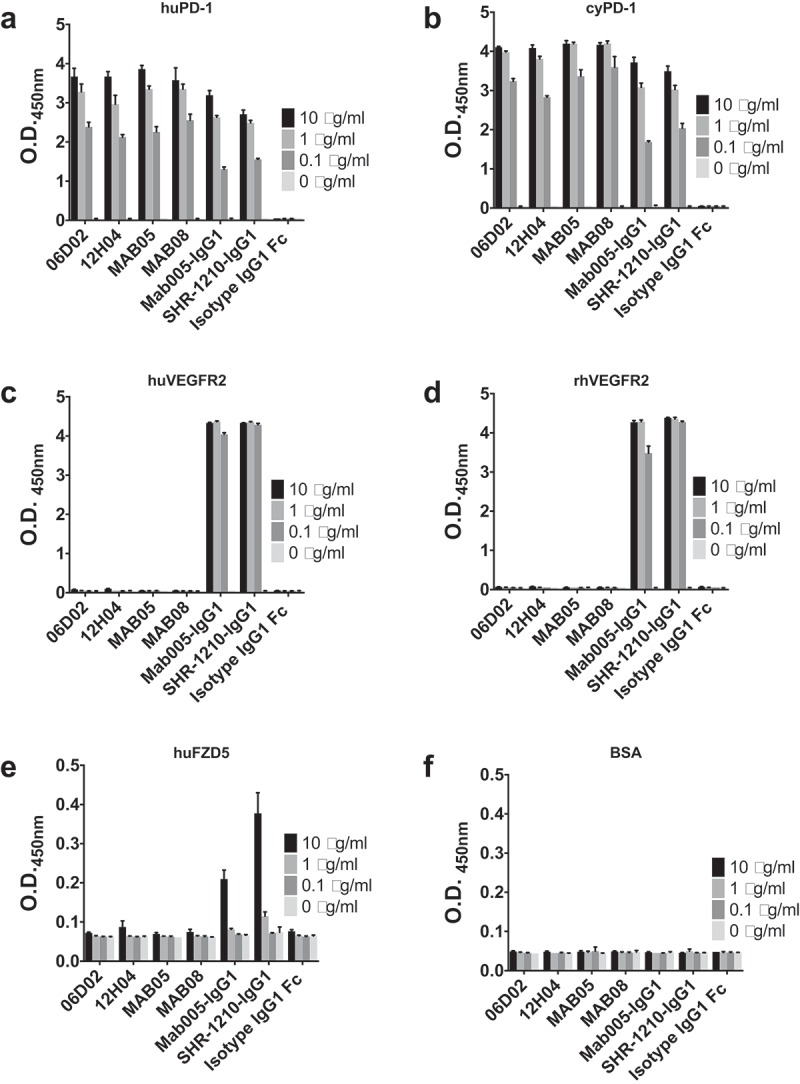
Off-target binding analysis ELISA assay.
SHR-1210-IgG1, Mab005-IgG1, library-derived clones and designer clones in human IgG1null format were titrated (in μg/ml) in a direct binding ELISA against human PD1 (A) and cyno PD1 (B), human VEGFR2 (C) and rhesus VEGFR2 (D), human FZD5 (E) and BSA (F) proteins. These analyses confirmed that all anti-PD1 antibodies exhibited binding to PD1, but only Mab005-IgG1 and SHR-1210-IgG1 exhibited measurable off-target binding to VEGFR2 and FZD5 proteins.
Finally, an attempt was made to estimate the affinity of Mab005-IgG1 and SHR-1210-IgG1 for VEGFR2 via Biacore, using the conditions described above for PD1 affinity evaluation. Human and rhesus monomeric VEGFR2-his proteins were titrated against SHR-1210-IgG1 and Mab005-IgG1. Both antibodies again exhibited binding to VEGFR2 recombinant protein, but binding signal was only observed at very high concentrations of soluble analyte (Supplemental Figure 4). As a result, complex curves with poor fit values (Chi2 > 6.6) were all that could be generated and reliable KD values could not be accurately derived. This indicated that the affinity of both antibodies for either human or rhesus VEGFR2 was probably low, likely in the μM range.
VEGFR2 activation analyses
To investigate whether the VEGFR2 reactivity in SHR-1210 was capable of activating the receptor, a human VEGFR2 reporter assay was used to examine induction of luciferase expression under control of the natural VEGF response element NFAT (Promega). In this assay, both SHR-1210-IgG1 and Mab005-IgG1 exhibited strong, concentration-dependent activation of VEGFR2 signalling (in the nM range), with SHR-1210 being more potent than Mab005 (Figure 11). The activation potency of SHR-1210-IgG1 was, however, significantly lower than that of recombinant human VEGF-163 (Figure 11). Importantly, each of clones MAB04, MAB08, 06D02 and 12H04 were also analyzed in this assay and demonstrated no observable activation signal, even at concentrations as high as 1 μM. Indeed, the signals at maximum concentration for the lead IgG clones were lower than the signals observed for the isotype control IgG1 (Figure 11).
Figure 11.
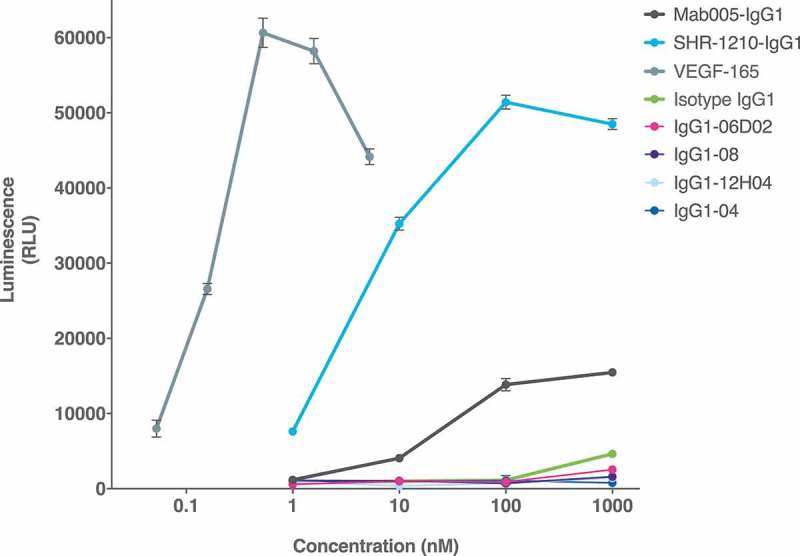
Cell-based VEGFR2 agonism assay.
SHR-1210-IgG1, Mab005-IgG1, library-derived clones and designer clones in human IgG1null format were titrated (in nM) in a human VEGFR2 signalling assay. SHR-1210-IgG1, Mab005-IgG1 and the positive control protein (human VEGF-165) all induced strong, concentration-dependent VEGFR2 agonism. Lead clones IgG1-04, IgG1-08, IgG1-06D02, and IgG1-12H04 showed no measurable agonism, even at concentrations as high as 1 μM.
Discussion
In the development of immunotherapeutics, monoclonal antibodies have been the preferred modality because they have the potential for high potency, long half-life in the body, and highly specific modulation of single drug targets.13 While well-designed therapeutic antibodies will often fulfil these criteria, not all antibodies are truly specific.14 Indeed, in molecular recognition of any target by the receptors of the immune system, specificity is a relative term. Antibodies intended for therapeutic use, derived from both humanized and fully human backgrounds, can exhibit unwanted tissue interactions via general polyreactive binding chemistry. Polyreactivity can affect PK and bioavailability, but is not associated with specific binding to non-target proteins (termed ‘polyspecificity’). Polyreactivity is rather caused, for example, by excess CDR positive charge causing low affinity interactions with negatively charged polymers such as peptidoglycans and DNA, or by excess hydrophobicity in the v-domains that may cause nonspecific membrane interactions.12,15–18 The basic chemistry issues that drive polyreactivity are now better understood and a series of assays have been developed that can identify clones with such issues early in the drug discovery process.12,17,19 Due to a lack of clear examples, however, the same cannot yet be said of the more complex issue of whether polyspecificity, via e.g., molecular mimicry in antibody epitopes, might cause off-target reactivity to a limited number of highly disparate targets. Equally, there is a paucity of understanding in how this phenomenon could drive unexpected antibody toxicities in man.
In early clinical trials, SHR-1210 has been reported to induce hemangiomas in human patients, a toxicity not observed in other anti-PD1 or anti-PD-L1 therapeutics.4 Hemangioma is a benign tumor formed by a collection of excess blood vessels, often developing in the skin, but also potentially in the liver and other organs.20 Polyspecificity has recently been recognized as an issue causing the cross-binding of similar epitopes in functionally distinct, but structurally related proteins targeted by diagnostic monoclonal antibodies.21 We hypothesized that the high level of structural redundancy in receptors could mean that SHR-1210 might similarly bind to unidentified and unpredictable receptors associated with vascular development or tissue differentiation. To examine this possibility, in vitro technologies, which are based on high-density arrays of cells expressing 4,975 unique human membrane receptors, were used to screen for off-target binding specificities.22 This screen identified that SHR-1210 exhibits polyspecificity and mediates off-target binding to FZD5, ULBP2 and, most importantly, VEGFR2. Indeed, we showed direct evidence that both Mab005 and SHR-1210 mediate potent agonism of human VEGFR2 and that the agonistic potency of Mab005 was made worse during the humanization process to generate SHR-1210.
Importantly, angiomas are known to develop spontaneously in patients with mutations in vascular biology-associated receptors such as VEGFR2 and are a reported side effect in the use of anti-VEGFR2 antagonist antibodies for cancer therapy.23,24 VEGFR2 is also known to be constitutively active in capillary haemangioma.20 In addition, FZD5 is associated with vascular neogenesis as it is a receptor for SFRP2, potentially mediating SFRP2-induced angiogenesis via calcineurin/NFATc3 pathway in endothelial cells.25 Modulation of the function of either of VEGFR2 or FZD5 is therefore likely to influence vascular biology and potentially drive the development of hemangioma. ULBP2, an MHC class 1-related protein, is a known ligand for the natural killer cell activating receptor NKG2D.26,27 so binding to this protein during cancer therapy with an anti-PD1 is of unknown consequence. Mab005 and SHR-1210 exhibited strong binding signals for ULBP2 in chip analyses and flow cytometry, but no binding in ELISA analyses. This may reflect the disruption or burial of the binding epitope on ULBP2 when the recombinant ectodomain is placed on a plate surface.
The polyspecificity observed for SHR-1210 may be reflective of molecular mimicry allowing functional epitopes to be accessed across unrelated targets.21 For SHR-1210, this polyspecificity has not resulted in a toxicity that is severe enough to halt the clinical studies being performed with the molecule, but it has nevertheless led to development and commercialization issues.28 Angiogenesis is a known driver of solid tumor growth and multiple targeted therapies exist that are designed to specifically inhibit vascular development in cancers.29 It is therefore plausible that pro-angiogenic activity in SHR-1210 may be counterproductive in solid tumor treatment, unless counteracted by the addition of an antiangiogenic therapy.
Critically, the frequency of polyspecificity issues in the myriad antibodies derived from disparate technologies that are currently intended for clinical development is unknown. It has been speculated in the past that naturally-occurring antibodies can be grouped into monoreactive, oligoreactive (i.e., polyspecific) or polyreactive subclasses.30 If polyspecificity that mediates highly specific binding to multiple off-target proteins (oligoreactivity) is more common than is currently recognized in the field, it could drive unexpected and severe toxicities in newly-developed antibody-based immunotherapy modalities with extremely potent cell-killing mechanisms of action, such as antibody-drug conjugates,31 CD3-targeting bispecifics32 or CAR-T cells.33 Indeed, despite the fact that SHR-1210 was shown in this study to cross-react to both human and monkey orthologs of VEGFR2, it has been shown that polyspecificity can be species-specific.6 In that case, human off-target reactivity might not be identified during classical pre-clinical toxicology in animals, meaning that the issue may be uncovered only after a lengthy and costly clinical development campaign.
Having proven that SHR-1210 exhibits relevant off-target binding, we estimated that this would be a complex, multifactorial problem to fix. Multiple functional properties of the molecule should preferably be simultaneously optimized, including target binding specificity, potency of PD1/PD-L1 signalling antagonism, affinity to PD1 from both human and animal test species (e.g., cynomolgus monkey, Macaca fascicularis, to facilitate accurate preclinical safety testing), v-domain biophysical stability and/or IgG expression yield. Indeed, antibody engineering studies have shown that even minor changes in key CDR sequences can have dramatic negative effects on all of these desired molecular properties.6,34 To address these issues in a single pass, we employed a rapid, in-depth screen of the CDR amino acid tolerance of SHR-1210 to both germline and non-germline mutations. This process identified multiple, globally-improved novel antibodies with improved pharmacological properties, including fully ablated binding to VEGFR2, FZD5 and ULBP2. Indeed, these clones were also shown to fully lack the ability to agonize VEGFR2 signalling, even at extremely high concentration. This paratope refinement effect was mediated principally by the near-germlining of the antibody light chain, coupled with minor alterations in amino acid content in the VH domain. While it remains possible (as for any antibody) that our paratope-refined clones might exhibit another as yet unknown off-target reactivity, the final clones appear to be highly specific for PD1 in all analyses performed and exhibit no engineering-induced polyreactivity signals.
Lead antibodies also exhibited improved immunogenicity profiles, improved potency and reduced affinity differentials between human and cyno PD1 affinities. This relative normalization of the binding to both PD1 orthologs rendered these lead clones highly similar in binding to an IgG1-pembrolizumab analog. The lower affinity binding to cyno PD1 observed for SHR-1210 was surprising because human and cyno PD1 share 96% amino acid identity, suggesting that the structural differences in human and cyno paratopes on PD1 for SHR-1210 are likely minor. The normalization of this affinity therefore further illustrates that paratope refinement can not only improve specificity, but the fitness of epitope-reactivity across orthologs. Clinically-relevant polyspecificity was therefore a phenomenon that could be identified and rapidly engineered out of the final therapeutic protein via paratope refinement.
Materials and methods
Antibody v-domain specificity testing: human receptor array analyses
Human cell membrane receptor proteome arrays were performed at Retrogenix Ltd. Primary screens: 5 µg/ml of IgG1-Mab005 antibody was screened for binding against fixed HEK-293 cells/slides expressing 4,975 human plasma membrane proteins individually (14 slide sets, n = 2 slides per slide set). All transfection efficiencies exceeded the minimum threshold. Antibody binding was detected using AF647 fluorescent secondary anti-human IgG1 antibody (Biolegend, catalog number 409,319). Primary hits (duplicate spots) were identified by analyzing fluorescence (AF647 and ZsGreen1) on ImageQuant. Vectors encoding all hits were sequenced to confirm their correct identities. Confirmation/specificity screens: Vectors encoding all hits, plus control vectors encoding MS4A1 (CD20) and epidermal growth factor receptor, were spotted in duplicate on new slides and used to reverse transfect human HEK-293 cells as before. All transfection efficiencies exceeded the minimum threshold. Identical fixed slides were treated with 5 µg/ml of each test antibody, 5 µg/ml of the negative control antibody, 1 µg/ml rituximab biosimilar (positive control), Isotype IgG1 (Ab00102 human IgG1 anti-fluorescein), or no test molecule (secondary only; negative control) (n = 2 slides per treatment). Slides were analyzed as above. Flow cytometry confirmation screen: Expression vectors encoding ZsGreen1 only, or ZsGreen1 and PD1, FZD5, VEGFR2 or ULBP2, were transfected into human HEK-293 cells. Each live transfectant was incubated with 1 and 5 mg/ml of each of the test antibodies and the Isotype control antibody. Cells were washed and incubated with the same AF647 anti-human IgG Fc detection antibody as used in the cell microarray screens. Cells were again washed and analyzed by flow cytometry using an Accuri flow cytometer (BD). A 7AAD live/dead dye was used to exclude dead cells, and ZsGreen1-positive cells (i.e., transfected cells) were selected for analyses.
PD1 library generation and selection
The PD1 Fab mutagenesis repertoire was assembled by mass oligo synthesis and PCR. The amplified Fab repertoire was then cloned via restriction-ligation into a phagemid vector, transformed into E.coli TG-1 cells, and the phage repertoire rescued essentially as previously described in detail.35 Phage selections were performed by coating streptavidin magnetic microbeads with biotinylated PD1 target protein (either human or cyno), washing the beads thrice with PBS and resuspending in PBS pH7.4 plus 5% skim milk protein. These beads were coated at 100 nM target protein in round 1 of selection, followed by reduced antigen concentrations in three successive rounds. In each round, phage were eluted using trypsin before re-infection into TG1 cells.
Periplasmic extracts production
Production of soluble Fabs in individual E. coli clones was performed. E. coli TG1 cells in logarithmic growth phase were induced with isopropyl 1-thio-β-D-galactopyranoside. Periplasmic extracts containing soluble Fab were generated by a freeze/thaw cycle: Bacterial cell pellets were frozen at −20 °C for overnight and then thawed at room temperature and resuspended in PBS pH 7.4. The supernatants containing the soluble Fab were collected after shaking at room temperature and centrifugation.
IgG expression and purification
Mammalian codon-optimized synthetic genes encoding the heavy and light chain variable domains of the lead panel anti-PD1 antibodies plus the Mab005, SHR-1210-IgG1, and a pembrolizumab analog were cloned into mammalian expression vectors comprising effector function null human IgG1 (‘IgG1null’; human IgG1 containing L234A, L235A, G237A mutations in the lower hinge that abrogate normal immunoglobulin antibody-dependent cell-mediated cytotoxicity, antibody dependent-cellular phagocytosis and complement-dependent cytotoxicity functions) and human Cκ domains, respectively. Co-transfection of heavy and light chain containing vectors in mammalian expression system was performed, followed by protein A-based purification of the IgG, quantification and QC on denaturing and non-denaturing SDS-PAGE, and monomeric status verified by size-exclusion chromatography. Control IgG1, IgG1null and IgG4 nivolumab analog antibodies were sourced commercially (Absolute Antibody Ltd., UK, catalog numbers Ab00102-10.0, Ab00102-10.3 and Ab00791-13.12, respectively).
Direct binding ELISA for IgG
Binding and cross-reactivity of the lead panel to the recombinant proteins was initially assessed by binding ELISA. The human PD1 human Fc tagged recombinant protein and the cynomolgus monkey PD1 human Fc tagged recombinant protein were coated to the surface of MaxiSorp™ flat-bottom 96-well plate at 1 µg/ml. The purified IgG samples were titrated in two-fold serial dilutions starting from 500 nM to 0.98 nM, allowed to bind to the coated antigens, and then detected using mouse anti-human IgG conjugated to horseradish peroxidase. Binding signals were visualized with 3,3ʹ,5,5ʹ-tetramethylbenzidine substrate solution (TMB) and the absorbance measured at 450 nM.
Alphascreen epitope competition assay for IgG1null antibodies
The AlphaScreen assay (Perkin Elmer) was performed in a 25 µl final volume in 384-well white microtiter plates (Greiner). The reaction buffer contained 1xPBS pH 7.3 (Oxoid, Cat. nr. BR0014G) and 0.05 % (v/v) Tween® 20 (Sigma, Cat. nr. P9416). Purified IgG samples were titrated in three-fold serial dilutions starting at 500 nM final concentration and incubated with biotinylated human PD1-His/AviTag at 0.6 nM final concentration for 20 minutes at room temperature. The parental IgG at 0.3 nM and the anti-human IgG1 Acceptor beads at 20 µg/ml (final concentrations) were added and the mix was incubated for 1 hour at room temperature. Followed by addition of the streptavidin donor beads at 20 µg/ml (final concentration) and incubation for 30 minutes at room temperature. The emission of light was measured in the EnVision multilabel plate reader (Perkin Elmer) and analysed using the EnVision manager software. Values were reported as counts per second (CPS) and corrected for crosstalk. The EC50 values were calculated using the mean fluorescence intensity (MFI) values in GraphPad Prism software (GraphPad Software, La Jolla, CA) and 4 parameters.
Biacore analyses of IgG affinity for monomeric human and cyno PD1 in solution
Affinity (KD) of purified IgGs was determined via surface plasmon resonance with antigen in solution on a Biacore 3000 (GE). A mouse anti-human antibody (BD Pharmingen, catalog number 555,784) was immobilized on a CM5 Sensor Chip to a level of 2000 RU in acetate buffer at pH 4.5 using amine coupling following the Wizard instructions for two channels. One channel was used for background signal correction. The standard running buffer HBS-EP pH 7.4 was used. Regeneration was performed with a single injection of 10 µl of 10 mM glycine at pH 1.5 at 20 µl/minute. IgG samples were injected for 2 minutes at 50 nM at 30 µl/min followed by an off-rate of 60 seconds. The monomeric antigen (His-tagged human or cynomolgus monkey PD1) was injected in two-fold serial dilutions from 100 nM to 3.1 nM, for 2 minutes at 30 µl/min followed by an off-rate of 300 seconds. The obtained sensorgrams were analysed using the Biacore 3000 evaluation (BIAevaluation) software. The KD was calculated by simultaneous fitting of the association and dissociation phases to a 1:1 Langmuir binding model.
Flow cytometry of iggs on PD1 CHO cells
Purified IgGs were tested in FACs for binding to human and cyno PD1 expressed on CHO-K1 stable cell lines and CHO-K1 wild-type cells. The IgG samples were titrated in three-fold serial dilutions starting at 500 nM to 0.98 nM. Binding of IgGs was detected with a mouse anti-human IgG conjugated to FITC (Sigma, catalog number F9512-1ML). Results were analyzed by examining the MFI of 10,000 cells per sample in the BL-1 channel detector of a flow cytometer (AttuneTM NxT Acoustic Focusing Cytometer, Invitrogen/ThermoFisher Scientific).
PD1/PD-L1 cell-based antagonism assay
The PD1/PD-L1 blockade cell-based bioassay (Promega), was used to measure the potency of antibodies in blocking the PD1/PD-L1 interaction. On the day before the assay, PD-L1 aAPC/CHO-K1 cells were thawed and transferred into cell recovery medium (90% Ham’s F12/10% fetal bovine serum (FBS)). The cell suspension was dispensed to each of the inner 60 wells of two 96-well, white, flat-bottom assay plates, at 100 µl per well. Cell recovery medium was added to each of the outside wells and the assay plates and incubated overnight at 37 °C/5% CO2. On the day of the assay the sample IgGs were diluted 4-fold in assay buffer (99% RPMI 1640/1% FBS) from 300 nM to 0.04 nM and 40 µl per dilution added to the assay plates containing the PD-L1 aAPC/CHO-K1 cells. Positive inhibition controls included the human PD1 Antibody AF1086 (R&D systems), mAb005 in IgG1null form and a pembrolizumab analog in IgG1null form. As a negative inhibition control, an irrelevant IgG was included. PD1 effector cells (Promega) were then thawed in assay buffer (99% RPMI 1640/1% FBS) and the cell suspension added to the wells of the assay plates containing the PD-L1 aAPC/CHO-K1 cells and the IgG titration samples. The assay plates were incubated for six hours in a 37 °C/5 % CO2 incubator, allowed to equilibrate to ambient temperature for 5–10 minutes, then 80 µl of Bio-Glo™ Reagent (Promega) was added. Assay plates were incubated at ambient temperature for a further 5–30 minutes and luminescence signals subsequently measured at 10, 20, and 30 minutes.
Antibody v-domain T cell epitope content: in silico analyses
In silico technologies (Abzena, Ltd.) based on identifying the location of T cell epitopes in therapeutic antibodies and proteins were used for assessing potential immunogenicity in antibody v-domains. iTopeTM was used to analyse the VL and a VH sequences of key leads for peptides with promiscuous high affinity binding to human MHC class II. Promiscuous high affinity MHC class II-binding peptides are thought to correlate with the presence of T cell epitopes that are high risk indicators for clinical immunogenicity of drug proteins. The iTopeTM software predicts favourable interactions between amino acid side chains of a peptide and specific binding pockets (in particular pocket positions; p1, p4, p6, p7, and p9) within the open-ended binding grooves of 34 human MHC class II alleles. These alleles represent the most common HLA-DR alleles found world-wide with no weighting attributed to those found most prevalently in any particular ethnic population. Twenty of the alleles contain the ‘open’ p1 configuration and 14 contain the ‘closed’ configuration where glycine at position 83 is replaced by valine. The location of key binding residues is achieved by the in silico generation of 9mer peptides that overlap by eight amino acids spanning the test protein sequence. This process successfully discriminates with high accuracy between peptides that either bind or do not bind MHC class II molecules. Analysis of the v-domain sequences was performed with overlapping 9mer peptides (with each overlapping the last peptide by 8 residues), which were tested against each of the 34 MHC class II allotypes. Each 9mer was scored based on the potential ‘fit’ and interactions with the MHC class II molecules. The peptide scores calculated by the software lie between 0 and 1. Peptides that produced a high mean binding score (>0.55 in the iTopeTM scoring function) were highlighted and, if >50% of the MHC class II binding peptides (i.e., 17 of 34 alleles) had a high binding affinity (score >0.6), such peptides were defined as ‘high affinity’ MHC class II-binding peptides, which are considered a high risk for containing CD4 + T cell epitopes. Low affinity MHC class II-binding peptides bind a high number of alleles (>50%) with a binding score >0.55 (but without a majority >0.6).
In addition, the sequences were analysed using TCEDTM searching for matches to T cell epitopes previously identified by in vitro human T-cell epitope mapping analyses of other protein sequences. The TCEDTM is used to search any test sequence against a large (>10,000 peptides) database of peptides derived from unrelated protein and antibody sequences. The sequences were used to interrogate the TCEDTM by BLAST search in order to identify any high sequence homology between peptides (T-cell epitopes) from unrelated proteins/antibodies that stimulated T cell responses in previous in vitro human T-cell epitope mapping studies performed at Abzena Ltd.
VEGFR2 cell-based agonism assay
A VEGFR2 agonism cell-based bioassay (Promega), was used to measure the potency of antibodies in inducing VEGFR2 signalling, according to the manufacturer’s protocol. Briefly, the cell suspension was dispensed to 96-well, white, flat-bottom assay plates, at 100 µl per well. IgGs were diluted in assay buffer (99% RPMI 1640/1% FBS) and added to the assay plates containing the VEGFR2/NFAT luciferase/HEK-293 cells. Positive control protein was human VEGF-165 (R&D systems). Mab005, SHR-1210, MAB04, MAB08, 06D02 and 12H04 in IgG1null form were also tested across a broad concentration range. As a negative inhibition control, an isotype IgG1 null was included. The assay plates were incubated for six hours in a 37 °C/5 % CO2 incubator, allowed to equilibrate to ambient temperature for 5–10 minutes, then Bio-Glo™ Reagent (Promega) was added. Assay plates were incubated at ambient temperature and luminescence signals subsequently measured.
Acknowledgments
All authors are Directors of UltraHuman Ltd. The authors would like to sincerely thank the teams at RxCelerate Ltd, Fairjourney Biologics and Retrogenix Ltd for their support during the performance of this study.
Abbreviations
- BSA
bovine serum albumin
- CHO
Chinese hamster ovary
- ELISA
Enzyme-Linked Immunosorbent Assay
- Fab
antigen-binding fragment
- Fc
crystalizable fragment
- FZD5
frizzled class receptor 5
- GE
Germline Epitope
- MHC
Major Histocompatibility Complex
- PD-1
Programmed Cell Death 1
- PK
pharmacokinetics
- ULBP2
UL16 binding protein 2
- VEGFR2
Vascular Endothelial Growth Factor Receptor 2.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK.. PD-1 and PD-L1 checkpoint signaling inhibition for cancer immunotherapy: mechanism, combinations, and clinical outcome. Front Pharmacol. 2017;8:561. doi: 10.3389/fphar.2017.00561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–774. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 3.Kaplon H, Reichert JM. Antibodies to watch in 2018. MAbs. 2018;10:183–203. doi: 10.1080/19420862.2018.1415671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mo H, Huang J, Xu J, Chen X, Wu D, Qu D, Wang X, Lan B, Wang X, Xu J, et al. Safety, anti-tumour activity, and pharmacokinetics of fixed-dose SHR-1210, an anti-PD-1 antibody in advanced solid tumours: a dose-escalation, phase 1 study. Br J Cancer. 2018. doi: 10.1038/s41416-018-0100-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burnet FM. A modification of Jerne’s theory of antibody production using the concept of clonal selection. CA Cancer J Clin. 1976;26:119–121. [DOI] [PubMed] [Google Scholar]
- 6.Bumbaca D, Wong A, Drake E, Reyes Ii AE, Lin BC, Stephan J-P, Desnoyers L, Shen B-Q, Dennis MS. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs. 2014;3:376–386. doi: 10.4161/mabs.3.4.15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tiller T, Schuster I, Deppe D, Siegers K, Strohner R, Herrmann T, Berenguer M, Poujol D, Stehle J, Stark Y, et al. A fully synthetic human Fab antibody library based on fixed VH/VL framework pairings with favorable biophysical properties. MAbs. 2013;5:445–470. doi: 10.4161/mabs.24218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Townsend S, Fennell BJ, Apgar JR, Lambert M, McDonnell B, Grant J, Wade J, Franklin E, Foy N, Ní Shúilleabháin D, et al. Augmented binary substitution: single-pass CDR germ-lining and stabilization of therapeutic antibodies. Proc Natl Acad Sci U.S.A. 2015;112:15354–15359. doi: 10.1073/pnas.1510944112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bogen B, Ruffini P. Review: to what extent are T cells tolerant to immunoglobulin variable regions? Scand J Immunol. 2009;70:526–530. doi: 10.1111/j.1365-3083.2009.02340.x. [DOI] [PubMed] [Google Scholar]
- 10.Eyerman MC, Zhang X, Wysocki LJ. T cell recognition and tolerance of antibody diversity. J Immunol. 1996;157:1037–1046. [PubMed] [Google Scholar]
- 11.Manz BN, Jackson BL, Petit RS, Dustin ML, Groves J. T-cell triggering thresholds are modulated by the number of antigen within individual T-cell receptor clusters. Proc Natl Acad Sci U.S.A. 2011;108:9089–9094. doi: 10.1073/pnas.1018771108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Avery LB, Wade J, Wang M, Tam A, King A, Piche-Nicholas N, Kavosi MS, Penn S, Cirelli D, Kurz JC, et al. Establishing in vitro in vivo correlations to screen monoclonal antibodies for physicochemical properties related to favorable human pharmacokinetics. MAbs. 2018;10:244–255. doi: 10.1080/19420862.2017.1417718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Granier C, De Guillebon E, Blanc C, Roussel H, Badoual C, Colin E, Saldmann A, Gey A, Oudard S, Tartour E. Mechanisms of action and rationale for the use of checkpoint inhibitors in cancer. ESMO Open. 2017;2:e000213. doi: 10.1136/esmoopen-2017-000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finton KA, Larimore K, Larman HB, Friend D, Correnti C, Rupert PB, Elledge SJ, Greenberg PD, Strong RK, Trkola A. Autoreactivity and exceptional CDR plasticity (but not unusual polyspecificity) hinder elicitation of the anti-HIV antibody 4E10. PLoS Pathog. 2013;9:e1003639. doi: 10.1371/journal.ppat.1003639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Betts A, Keunecke A, van Steeg TJ, van der Graaf PH, Avery LB, Jones H, Berkhout J. Linear pharmacokinetic parameters for monoclonal antibodies are similar within a species and across different pharmacological targets: A comparison between human, cynomolgus monkey and hFcRn Tg32 transgenic mouse using a population-modeling approach. MAbs. 2018;10:751–764. doi: 10.1080/19420862.2018.1462429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piche-Nicholas NM, Avery LB, King AC, Kavosi M, Wang M, O’Hara DM, Tchistiakova L, Katragadda M. Changes in complementarity-determining regions significantly alter IgG binding to the neonatal Fc receptor (FcRn) and pharmacokinetics. MAbs. 2018;10:81–94. doi: 10.1080/19420862.2017.1389355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohli N, Jain N, Geddie ML, Razlog M, Xu L, Lugovskoy AA. A novel screening method to assess developability of antibody-like molecules. MAbs. 2015;7:752–758. doi: 10.1080/19420862.2015.1048410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hotzel I, Theil FP, Bernstein LJ, Prabhu S, Deng R, Quintana L, Lutman J, Sibia R, Chan P, Bumbaca D, et al. A strategy for risk mitigation of antibodies with fast clearance. MAbs. 2012;4:753–760. doi: 10.4161/mabs.22189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jain T, Sun T, Durand S, Hall A, Houston NR, Nett JH, Sharkey B, Bobrowicz B, Caffry I, Yu Y, et al. Biophysical properties of the clinical-stage antibody landscape. Proc Natl Acad Sci U.S.A. 2017;114:944–949. doi: 10.1073/pnas.1616408114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boye E, Olsen BR. Signaling mechanisms in infantile hemangioma. Curr Opin Hematol. 2009;16:202–208. doi: 10.1097/MOH.0b013e32832a07ff. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma D, Baruch D, Shu Y, Yuan K, Sun Z, Ma K, Hoang T, Fu W, Min L, Lan Z-S, et al. Using protein microarray technology to screen anti-ERCC1 monoclonal antibodies for specificity and applications in pathology. BMC Biotechnol. 2012;12:88. doi: 10.1186/1472-6750-12-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Turner L, Lavstsen T, Berger SS, Wang CW, Petersen JE, Avril M, Brazier AJ, Freeth J, Jespersen JS, Nielsen MA, et al. Severe malaria is associated with parasite binding to endothelial protein C receptor. Nature. 2013;498:502–505. doi: 10.1038/nature12216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Espinosa Lara P, Medina-Puente C, Riquelme Oliveira A, Jimenez-Reyes J. Eruptive cherry angiomas developing in a patient treated with ramucirumab. Acta Oncol. 2018;57:709–711. doi: 10.1080/0284186X.2017.1410287. [DOI] [PubMed] [Google Scholar]
- 24.Lim YH, Odell ID, Ko CJ, Choate KA. Somatic p.T771R KDR (VEGFR2) mutation arising in a sporadic angioma during ramucirumab therapy. JAMA Dermatol. 2015;151:1240–1243. doi: 10.1001/jamadermatol.2015.1925. [DOI] [PubMed] [Google Scholar]
- 25.Peterson YK, Nasarre P, Bonilla IV, Hilliard E, Samples J, Morinelli TA, Hill EG, Klauber-DeMore N. Frizzled-5: a high affinity receptor for secreted frizzled-related protein-2 activation of nuclear factor of activated T-cells c3 signaling to promote angiogenesis. Angiogenesis. 2017;20:615–628. doi: 10.1007/s10456-017-9574-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Molfetta R, Quatrini L, Santoni A, Paolini R. Regulation of NKG2D-dependent NK cell functions: the yin and the yang of receptor endocytosis. In J Mol Sci. 2017;18:1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutherland CL, Chalupny NJ, Schooley K, VandenBos T, Kubin M, Cosman D. UL16-binding proteins, novel MHC class I-related proteins, bind to NKG2D and activate multiple signaling pathways in primary NK cells. J Immunol. 2002;168:671–679. [DOI] [PubMed] [Google Scholar]
- 28.Endpoints. Incyte grabs a new PD-1 checkpoint drug in $900M deal with MacroGenics In Carroll J, editor. 2017. https://endpts.com/incyte-grabs-a-new-pd-1-checkpoint-drug-in-900m-deal-with-macrogenics/ [Google Scholar]
- 29.Jayson GC, Kerbel R, Ellis LM, Harris AL. Antiangiogenic therapy in oncology: current status and future directions. Lancet. 2016;388:518–529. doi: 10.1016/S0140-6736(15)01088-0. [DOI] [PubMed] [Google Scholar]
- 30.Notkins AL. Polyreactivity of antibody molecules. Trends Immunol. 2004;25:174–179. doi: 10.1016/j.it.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 31.Rios-Doria J, Harper J, Rothstein R, Wetzel L, Chesebrough J, Marrero AM, Chen C, Strout P, Mulgrew K, McGlinchey K, et al. Antibody-drug conjugates bearing pyrrolobenzodiazepine or tubulysin payloads are immunomodulatory and synergize with multiple immunotherapies. Cancer Res. 2017. doi: 10.1158/0008-5472.CAN-16-2854. [DOI] [PubMed] [Google Scholar]
- 32.Root A, Cao W, Li B, LaPan P, Meade C, Sanford J, Jin M, O’Sullivan C, Cummins E, Lambert M, et al. Development of PF-06671008, a highly potent anti-P-cadherin/Anti-CD3 bispecific DART molecule with extended half-life for the treatment of cancer. Antibodies. 2016;5:6. doi: 10.3390/antib5010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T-cell therapy. Mol Ther Oncolytics. 2016;3:16011. doi: 10.1038/mto.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vugmeyster Y, Guay H, Szklut P, Qian MD, Jin M, Widom A, Spaulding V, Bennett F, Lowe L, Andreyeva T, et al. In vitro potency, pharmacokinetic profiles, and pharmacological activity of optimized anti-IL-21R antibodies in a mouse model of lupus. MAbs. 2010;2:335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Finlay WJ, Bloom L, Cunningham O. Optimized generation of high-affinity, high-specificity single-chain Fv antibodies from multiantigen immunized chickens. Methods Mol Biol. 2011;681:383–401. doi: 10.1007/978-1-60761-913-0_21. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.