

Abstract
Purpose of review
microRNAs (miRNAs) are short noncoding RNAs that function as sequence-specific inhibitors of gene expression. Autosomal dominant polycystic kidney disease (ADPKD) is the most frequent genetic cause of end-stage kidney failure with limited treatment options. The realization that miRNA upregulation, and thus its gain-of-function, can drive the progression of ADPKD has raised the possibility that anti-miRs represent a novel drug class for this disorder.
Recent findings
A common set of miRNAs are aberrantly expressed in various murine models of polycystic kidney disease. In particular two miRNAs, miR-17 family and miR-21, are both upregulated in kidney cysts and promote ADPKD progression in mouse models. miR-17 rewires cyst epithelial metabolism to enhance cyst proliferation. On the other hand, miR-21 represses proapoptotic genes and thus inhibits cyst apoptosis. Importantly, an anti-miR-17 drug has advanced through preclinical ADPKD studies, whereas an anti-miR-21 drug has already cleared phase I clinical trial.
Summary
miRNAs have emerged as new regulators of ADPKD pathogenesis. Anti-miRs represent a feasible and an entirely new class of drugs for the treatment of ADPKD.
Keywords: anti-miRs, microRNAs, miR-17, miR-21, polycystic kidney disease
INTRODUCTION
microRNAs (miRNAs) were first discovered in the early 1990s in the model organism Caenorhabditis Elegans as short, noncoding RNAs (~22-nucleotide in length) that inhibit posttranscriptional gene expression [1]. It wasn’t until the year 2000 that miRNAs were found to be evolutionarily conserved in higher organisms including humans. Since then, a groundswell of miRNA-related research has linked these noncoding RNAs to virtually all aspects of mammalian biology [2] and more importantly, aberrant miRNA function is now known to regulate pathogenesis of common human disorders, including cancer, heart disease, and many forms of kidney diseases [3–5]. The improved understanding of basic miRNA biology and its role in disease pathogenesis has led to the development of an entirely new class of drugs that either inhibit or mimic miRNA function.
Over 1000 different miRNAs are encoded by the human genome, a majority of which are conserved in other species. Within the genome, miRNAs are either present in intergenic DNA regions as independent transcriptional units or located inside introns or even exons of protein-coding genes. Initially transcribed as a primary-miRNA (pri-miRNA) transcript that is several kilo-bases in length, the mature miRNA is eventually produced by sequential processing by RNAse III enzymes DROSHA and DICER (Fig. 1) [6,7]. The mature miRNA then physically associates with a protein complex called miRNA-induced silencing complex, in which it functions as a sequence-specific inhibitor of mRNA translation. The specificity of miRNA–mRNA interaction is conferred by a stretch of nucleotides at position 2 through 8 at the 5’ end of the mature miRNA called the ‘seed sequence’. Watson–Crick base-pairing between the seed sequence and complementary sequences located primarily in the 3’-untranslated region (3’-UTR) of target mRNA results in its destabilization and thus prevents its translation into proteins.
FIGURE 1.
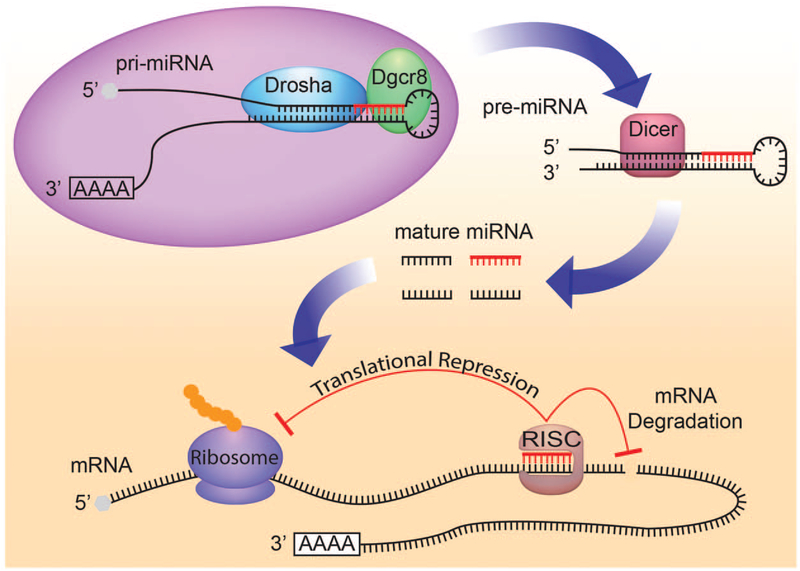
microRNA biogenesis and function. microRNAs are transcribed as long transcripts called primary microRNA. These transcripts are sequentially processed by two RNAse III enzymes DROSHA and DICER to eventually produce small, approximately 22-nucelotide, mature microRNAs. Mature microRNAs physically associate with the microRNA-induced silencing complex, where they bind to target mRNAs and function as sequence-specific inhibitors of protein translation.
miRNAs are essential for ureteric bud branching, renal vesicle formation, renal tubule elongation, and proper patterning of the renal stroma [8–11]. Thus, miRNAs are required for multiple aspects of normal kidney development. Conversely, dysregulated miRNA expression is thought to promote common chronic kidney diseases such as autosomal dominant polycystic kidney disease (ADPKD) [3,4]. In this review, we provide a brief overview of the role of miRNAs in the pathogenesis of ADPKD. In particular, we have highlighted two pathogenic miRNAs – miR-17 family and miR-21 – and discussed the potential of a miRNA-based therapeutic approach for ADPKD.
microRNAs THAT PROMOTE AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE PROGRESSION
ADPKD is amongst the most common monogenetic disorders known to humans. With an incidence of approximately 1 : 800 live births, an estimated 600 000 people with ADPKD live in the United States, and there are over 12.4 million ADPKD patients worldwide. The clinical hallmark of this disorder is massive bilateral kidney enlargement due to the presence of innumerable, fluid-filled, renal tubule-derived cysts. Over the span of decades, the cysts become more numerous and enlarge causing kidney failure. Nearly 50% of ADPKD patients develop end-stage renal disease requiring dialysis or kidney transplantation [12].
ADPKD is caused by mutation of either polycystic kidney disease (PKD)1 or PKD2. Over the years, dysregulation of multiple cellular processes such as enhanced cyst epithelial proliferation, low levels of apoptosis, excessive fluid secretion, rewiring of mitochondrial metabolism and inflammatory response, and aberrant polarity have been shown to underlie kidney cyst growth. Considering that dys-regulation of so many critical cellular processes is observed, it is not surprising that many well studied signaling pathways including mammalian Target of Rapamycin signaling, c-Myc, Janus kinase/signal transducer and activator of transcription, and cyclic adenosine monophosphate (cAMP)/cAMP response element-binding protein (CREB) are aberrantly activated in ADPKD [13]. Recently, miRNA-regulated signaling has emerged as a new mechanism to explain imbalance in proliferation and apoptosis of cyst epithelial cells. Specifically, two miRNAs – miR-17~92 cluster and miR-21 – have been shown to have proproliferative and antiapoptotic effects, respectively, on cyst epithelial cells.
miR-17 rewires metabolism and promotes cyst proliferation
miR-17~92, also called ‘oncomir-1’ for its role in cancer, is a conserved miRNA cluster located on human chromosome 13 within the third intron of the nonprotein coding gene MIR17HG. This cluster is transcribed as a large pri-miR-17~92 transcript, which in turn is processed into smaller fragments to eventually produce six mature miRNAs: miR-17, miR-18a, miR-19a, miR-20a, miR-19b-1, and miR-92–1. Functionally, these miRNAs can be classified into four families: miR-17 family (miR-17 and miR-20a), miR-18 family, miR-19 family (miR-19a and miR-19b-1), and miR-25 family (miR-92a-1). Members of each family harbor an identical seed sequence and thus are predicted to inhibit same mRNA targets. Interestingly, evolutionarily ancient MIR17HG gene duplication events have given rise to two paralogous miRNA clusters, miR-106a~363 (located on human chromosomeX) and miR-106b~25 (located on human chromo-some 7), both of which generate miRNAs that are homologous to miRNAs derived from the miR-17~92 cluster. Although miR-17~92 is abundantly expressed across most tissues, especially during embryogenesis, its paralogous clusters exhibit very low levels of expression. Accordingly, germline deletion of miR-17~92 leads to perinatal lethality in mice due to lung, heart, skeletal, and hematopoietic development defects, whereas germline deletion of miR-106a~363 or miR-106b~25 does not produce any obvious phenotypes [14]. Thus, amongst the three clusters, miR-17~92 appears to be indispensable, and its physiological function is to aid in normal embryogenesis. Further supporting this notion, microdeletions of miR-17~92 has been shown to cause a human developmental disorder called Feingold syndrome, which is characterized by mental retardation, skeletal, heart, kidney, and gastrointestinal developmental defects [15].
Similar to many other developmental genes, miR-17 expression is activated in adult injured tissues, for example after acute kidney injury, and its proproliferative function is often repurposed to promote diseases such as ADPKD. In fact, several lines of evidence have now conclusively linked miR-17 92 to ADPKD pathogenesis [16,17▀ ▀]. We have shown that miR-17~92 transcription is elevated in multiple orthologous ADPKD mouse models. The oncogene c-Myc binds to the miR-17~92 promoter in kidney cells and activates miR-17~92 expression in cystic kidneys. Transgenic overexpression of either c-Myc or miR-17~92 in normal mice produces kidney cysts proving direct pathogenicity of the c-Myc-miR-17~92 axis in ADPKD. In an opposite approach, we have demonstrated that inactivation of miR-17~92 in a variety of ADPKD mouse models slows cyst proliferation, reduces cyst size, improves renal function, and substantially prolongs survival. This benefit is observed irrespective of whether Pkd1 or Pkd2 were mutated, the type of Pkd1/2 mutation (i.e., inactivating vs. hypomorphic), or the dynamics of cyst growth (i.e., fast and aggressive vs. slow and long-lived). Importantly, expression of miR-17 is increased in a subset of cysts in humans with ADPKD. Moreover, miR-17 inhibition slows cyst growth in an in-vitro human ADPKD model, suggesting that the observations in mouse models may be translatable to human.
Two main themes have emerged regarding how miR-17~92 may promote ADPKD progression. In keeping with the idea that miRNAs can regulate entire signaling nodes by repressing multiple mRNA targets within the same pathway, the first theme is that the miR-17~92 cluster miRNAs inhibit a network of cystic kidney diseases genes including PKD1, PKD2, and HNF1 β [18]. How could inhibition of PKD1/2 promote ADPKD progression when these genes are already mutated? Recent studies have shown that a substantial portion of ADPKD patients harbor hypomorphic PKD1/2 mutations, which do not inactivate the gene but instead reduce the gene dosage [19–21]. In this context, further reduction in gene dosage by miR-17 would aggravate cyst growth. miR-17~92 is also known to be upregulated in other forms of cystic kidney diseases such as autosomal recessive PKD and nephronophthisis [16,17▀ ▀]. Thus, by reducing PKD1/2 and HNF1 β gene dosage, miR-17~92 could potentially aggravate these diseases as well.
The second theme involves miR-17-mediated rewiring of mitochondrial metabolism [17▀ ▀]. Rather than ATP, a more pressing need for proliferating cells is to use nutrients to produce building blocks for the formation of a new cell [22,23]. c-Myc rewires metabolism of cancer cells so that instead of primarily being used to generate ATP via mitochondrial oxidative phosphorylation (OXPHOS), carbon can be shuttled to produce DNA, proteins, and lipids for cell membranes/organelles [24]. Our recent work has shown that miR-17 functions downstream of c-Myc to inhibit OXPHOS through direct repression of PPARα, a transcription factor that activates the expression of many OXPHOS and fatty acid oxidation (FAO) genes (Fig. 2) [17▀ ▀]. Moreover, we found that genetic deletion of Pparα worsens, whereas its activation slows cyst growth in ADPKD mouse models. Whether reduced OXPHOS can independently drive ADPKD progression has not been formally studied. However, several lines of circumstantial evidence indeed support this idea. First, OXPHOS is reduced in Pkd1-null cells [25]. Second, mutations in several OXPHOS genes produce human syndromes that are characterized by kidney cysts [26–28], and finally, many PPARα OXPHOS/FAO target genes are downregulated in human ADPKD cysts [29].
FIGURE 2.
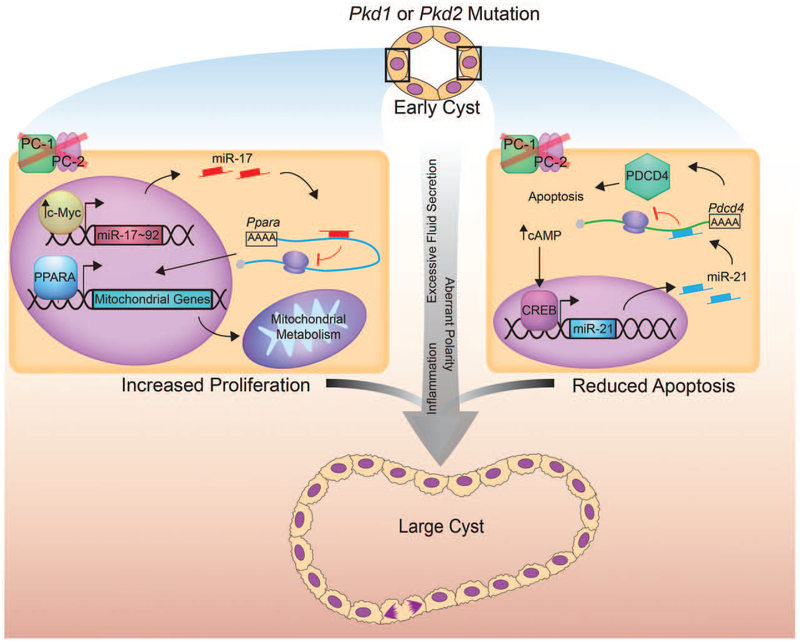
Functions of miR-17 and miR-21 in autosomal dominant polycystic kidney disease progression. PKD1 or PKD2 mutations results in cyst initiation and eventually cyst enlargement due to dysregulation of several cellular processes, such as excessive proliferation and fluid secretion, dysregulated apoptotic, metabolic and inflammatory response, and aberrant cell polarity. Recent studies have shown that microRNAs can modulate at least two of these processes. The c-Myc → miR-17 – | Pparα axis promotes proliferation of cyst epithelial cells through rewiring of mitochondrial metabolism (left). On the other hand, the cAMP/CREB → miR-21 –| Pdcd4 axis results in reduced apoptosis (right). These two mechanisms in concert, along with other dysregulated pathways, leads to cyst enlargement.
miR-21 inhibits apoptosis and promotes survival of cyst epithelial cells
miR-21 is located on the human chromosome 17 within the last exon of the protein-coding gene VMP1. Despite this intragenic location, miR-21 functions as an independent transcriptional unit and is transactivated through its own unique promoter located inside intron 10 of VMP1. Even though miR-21 is widely expressed in multiple cell types and tissues, including renal tubules, miR-21-null mice are viable and fertile indicating that miR-21 is not needed for normal development. Instead, its physiological function may be to aid in maintaining tissue homeostasis. Indeed, miR-21 is upregulated after injury in multiple tissues and where it functions as an antiapoptotic, prosurvival miRNA.
miR-21 has primarily been implicated in the pathogenesis of diseases in two broad categories of cancer and fibrosis [30,31]. With regard to the kidney, robust miR-21 upregulation is observed in the setting of persistent tubular injury and inhibiting miR-21 is effective in abrogating progressive fibrosis[32]. In contrast to these findings, at least one study so far has shown that loss of miR-21 induces podocyte apoptosis and worsens fibrosis in diabetic nephropathy mouse models [33]. We have recently demonstrated that transcriptional activation of miR-21 is a common feature of the murine form of ADPKD [34▀ ▀]. Moreover, miR-21 is also upregulated in human ADPKD cysts. Interestingly, we found that cAMP/CREB signaling, a critical pathogenic pathway in ADPKD, transactivates the miR-21 promoter. Accordingly, treatment with tolvaptan, which reduces renal cAMP levels and has been approved for ADPKD treatment outside of the United States, reduces miR-21 expression in mouse models. We found that, like tolvaptan treatment, miR-21 genetic deletion attenuates cyst growth in an ADPKD mouse model. The potential mechanism by which miR-21 aggravates cyst growth may involve direct inhibition of the proapoptotic tumor suppressor PDCD4. In support of this notion, miR-21 deletion was associated with upregulation of Pdcd4 expression and elevated rates of cyst cell apoptosis. Moreover, a subset of Pdcd4−/− mice is known to develop kidney cysts spontaneously [35]. Thus, cAMP/CREB-miR-21-PDCD4 represents a new signaling axis in ADPKD.
THE POTENTIAL OF A microRNA-BASED THERAPEUTIC APPROACH FOR AUTOSOMAL DOMINANT POLYCYSTIC KIDNEY DISEASE
The realization that miRNA upregulation, and thus its gain-of-function, can drive the disease progression has led to the development of a novel drug class called anti-miRs [36,37▀ ▀]. Anti-miRs basically are synthetic oligonucleotides that are complementary to specific miRNAs. In normal tissues, there is an optimal balance between the number of target mRNAs that undergo translation vs. the ones that are inhibited by miRNAs. However, in diseased states when a particular miRNA is overexpressed, this balance can be tipped in favor of excessive miRNA-mediated mRNA target inhibition (Fig. 3). Anti-miR binds to cognate miRNAs and displaces them from target mRNAs, thereby restoring the balance between mRNA translation and mRNA inhibition.
FIGURE 3.
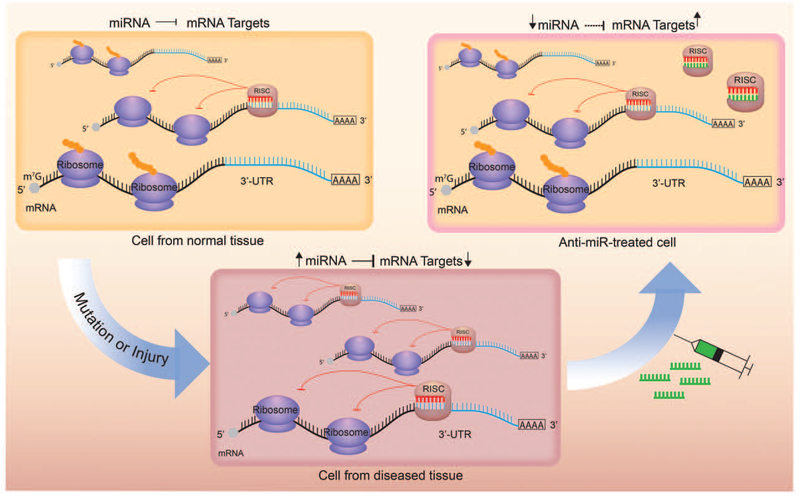
Anti-miRs are synthetic oligonucleotides that are complimentary to microRNAs. In normal tissue there is an optimal balance between microRNA-induced mRNA silencing and mRNA translation (top left). In diseased tissue, when a particular microRNA is upregulated, the balance can be shifted in favor of microRNA-induced mRNA silencing (bottom). Anti-miRs (green) bind to cognate microRNAs and displace them from target mRNAs, thereby restoring the balance between mRNA translation and microRNA-induced mRNA silencing (top right).
Even if injected systemically, anti-miRs are readily delivered to the liver and the kidney. Accordingly, the primary route of their eventual elimination is urine and bile. Anti-miR half-life is only about 8 h in blood, but they last for up to 3 weeks in kidney and liver. In the healthy kidney, anti-miRs primarily accumulate in proximal tubules likely because they are filtered through the glomerulus. Interestingly, however, we have found that anti-miRs can be delivered to cysts even if they are derived from collecting ducts perhaps due to the altered vasculature in cystic kidneys (Fig. 4). Favorable tissue distribution and a long half-life make anti-miRs extremely attractive choices for treatment of chronic kidney diseases such as ADPKD and Alport syndrome. We recently tested whether anti-miR-17 drugs are feasible therapeutic agents for ADPKD [17▀ ▀]. Functionally active miRNAs are present in high-molecular-weight (HMW) polysomes, where they prevent ribosomes from translating the target mRNAs [38]. Anti-miR-17 compound displaced miR-17 from HMW polysomes in kidney tissue signifying functional miR-17 inhibition. Moreover, anti-miR-17 was delivered to cyst epithelia, slowed cyst proliferation, and improved renal function in an aggressive, fast cyst-growth ADPKD mouse model. Importantly, anti-miR-17 demonstrated cyst-reducing effects, but no overt toxicity, in a second 6-month, preclinical trial involving a slow cyst-growth mouse model. These observations have led to the development of the drug candidate RGLS4326, a specific anti-miR-17 inhibitor. Currently, additional safety and therapeutic efficacy preclinical trials are ongoing. Filing of investigational new drug application to the Food and Drug Administration, paving the way for initial human testing, is expected in late 2017. A second miRNA drug called RG-012, a direct miR-21 inhibitor has already cleared phase I clinical trial and now is being tested in a phase II trial involving Alport syndrome patients. Similar to miR-21 genetic deletion, whether RG-012 or some other anti-miR-21 drug also slows ADPKD progression remains to be seen.
FIGURE 4.
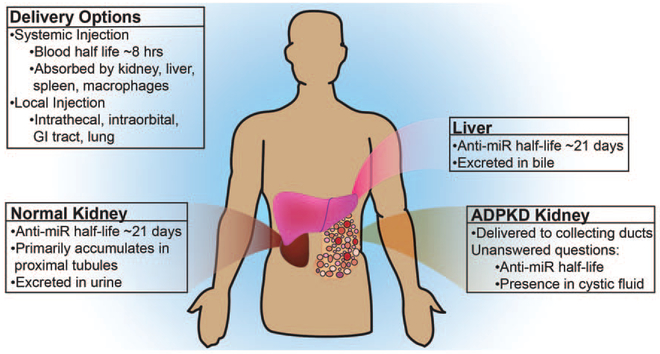
Pharmaceutical properties of anti-miRs. Anti-miRs can be delivered both locally and systemically. With systemic administration, anti-miRs are readily absorbed by the kidney and liver. They are excreted in the urine and bile. The half-life in blood is about 8 h, whereas it is approximately 3 weeks in the kidney and the liver. In a normal kidney, anti-miRs accumulate primarily in the proximal tubules. In cystic kidneys, they are also delivered to the collecting duct cysts.
Despite this early success and excitement, several unanswered questions and future challenges lie ahead. An obvious question is if anti-miR therapy can be safely tolerated over the long term. Another concern is whether there will be off-target effects of these drugs. A significant research focus moving forward should also be on understanding the unintended consequence of miR-17 or miR-21inhibition. For example, does it affect podocyte function, recovery after AKI, and others.
CONCLUSION
ADPKD is a common but incurable genetic cause for kidney failure. miRNAs have emerged as novel regulators of ADPKD progression. In particular, miR-17 and miR-21 are both upregulated in kidney cysts, and their inhibition slows disease progression in ADPKD mouse models. The proto-oncogene c-Myc transactivates miR-17~92, which in turn rewires cyst epithelial metabolism to enhance cyst proliferation. The cAMP/CREB pathway activates miR-21, which in turn promotes cyst cell survival by inhibiting proapoptotic genes. Importantly, an anti-miR-17 drug has advanced through preclinical ADPKD studies, whereas an anti-miR-21 drug has already cleared phase I clinical trial.
KEY POINTS.
A common set of dysregulated miRNAs have been identified in murine models of ADPKD.
miR-17, a c-Myc-regulated miRNA, rewires cyst epithelial cell metabolism to enhance cyst proliferation.
miR-21, a cAMP/CREB signaling responsive miRNA, inhibits proapoptotic genes and thereby promotes cyst cell survival.
Based on therapeutic efficacy in mouse models of PKD, an anti-miR-17 drug has now been advanced to investigational new drug-enabling studies.
An anti-miR-21 drug developed for Alport syndrome has already cleared phase I clinical trial. This drug could be tested for ADPKD.
Acknowledgments
Financial support and sponsorship
The work form authors’ laboratory is supported by grants from the National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases (R01DK102572 to V.P.).
Footnotes
Conflicts of interest
V.P. along with Regulus Therapeutics has applied for a patent related to the treatment of PKD with miR-17 inhibitors. V.P. serves as a consultant, and the Patel lab has a sponsored research agreement with Regulus Therapeutics.
REFERENCES AND RECOMMENDED READING
Papers of particular interest, published within the annual period of review, have been highlighted as:
▀ of special interest
▀ ▀ of outstanding interest
- 1.Ambros V The functions of animal microRNAs. Nature 2004; 431:350–355. [DOI] [PubMed] [Google Scholar]
- 2.Stefani G, Slack FJ. Small noncoding RNAs in animal development. Nat Rev Mol Cell Biol 2008; 9:219–230. [DOI] [PubMed] [Google Scholar]
- 3.Noureddine L, Hajarnis S, Patel V. MicroRNAs and polycystic kidney disease. Drug Discov Today Dis Models 2013; 10:e137–e1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patel V, Noureddine L. MicroRNAs and fibrosis. Curr Opin Nephrol Hypertens 2012; 21:410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendell JT, Olson EN. MicroRNAs in stress signaling and human disease. Cell 2012; 148:1172–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15:509–524. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004; 116:281–297. [DOI] [PubMed] [Google Scholar]
- 8.Patel V, Hajarnis S, Williams D, et al. MicroRNAs regulate renal tubule maturation through modulation of Pkd1. J Am Soc Nephrol 2012; 23:1941–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagalakshmi VK, Ren Q, Pugh MM, et al. Dicer regulates the development of nephrogenic and ureteric compartments in the mammalian kidney. Kidney Int 2011; 79:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nagalakshmi VK, Lindner V, Wessels A, Yu J. microRNA-dependent temporal gene expression in the ureteric bud epithelium during mammalian kidney development. Dev Dyn 2015; 244:444–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakagawa N, Xin C, Roach AM, et al. Dicer1 activity in the stromal compartment regulates nephron differentiation and vascular patterning during mammalian kidney organogenesis. Kidney Int 2015; 87:1125–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Patel V, Chowdhury R, Igarashi P. Advances in the pathogenesis and treatment of polycystic kidney disease. Curr Opin Nephrol Hypertens 2009; 18:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris PC, Torres VE. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J Clin Invest 2014; 124:2315–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ventura A, Young AG, Winslow MM, et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008; 132:875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Pontual L, Yao E, Callier P, et al. Germline deletion of the miR-17 approximately 92 cluster causes skeletal and growth defects in humans. Nat Genet 2011; 43:1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patel V, Williams D, Hajarnis S, et al. miR-17~92 miRNA cluster promotes kidney cyst growth in polycystic kidney disease. Proc Natl Acad Sci U S A 2013; 110:10765–10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. ▀ ▀.Hajarnis S, Lakhia R, Yheskel M, et al. microRNA-17 family promotes poly-cystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun 2017; 8:14395.This study demonstrated that miR-17 is a viable drug target for autosomal dominant polycystic kidney disease. The authors also showed that miR-17 promotes cyst proliferation by rewiring cyst epithelial proliferation.
- 18.Sun H, Li QW, Lv XY, et al. MicroRNA-17 posttranscriptionally regulates polycystic kidney disease-2 gene and promotes cell proliferation. Mol Biol Rep 2010; 37:2951–2958. [DOI] [PubMed] [Google Scholar]
- 19.Hopp K, Ward CJ, Hommerding CJ, et al. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J Clin Invest 2012; 122:4257–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heyer CM, Sundsbak JL, Abebe KZ, et al. Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2016; 27:2872–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hwang YH, Conklin J, Chan W, et al. Refining genotype-phenotype correlation in autosomal dominant polycystic kidney disease. J Am Soc Nephrol 2016; 27:1861–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell 2008; 134:703–707. [DOI] [PubMed] [Google Scholar]
- 23.Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov 2011; 10:671–684. [DOI] [PubMed] [Google Scholar]
- 24.Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 2013; 3:a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menezes LF, Lin C-C, Zhou F, Germino GG. Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 2016; 5:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kjaergaard S, Graem N, Larsen T, Skovby F. Recurrent fetal polycystic kidneys associated with glutaric aciduria type II. APMIS 1998; 106:1188–1193. [DOI] [PubMed] [Google Scholar]
- 27.Whitfield J, Hurst D, Bennett MJ, et al. Fetal polycystic kidney disease associated with glutaric aciduria type II: an inborn error of energy metabolism. Am J Perinatol 1996; 13:131–134. [DOI] [PubMed] [Google Scholar]
- 28.FitzPatrick DR. Zellweger syndrome and associated phenotypes. J Med Genet 1996; 33:863–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song X, Di Giovanni V, He N, et al. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet 2009; 18:2328–2343. [DOI] [PubMed] [Google Scholar]
- 30.Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008; 456:980–984. [DOI] [PubMed] [Google Scholar]
- 31.Medina PP, Nolde M, Slack FJ. OncomiR addiction in an in vivo model of microRNA-21-induced pre-B-cell lymphoma. Nature 2010; 467:86–90. [DOI] [PubMed] [Google Scholar]
- 32.Chau BN, Xin C, Hartner J, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med 2012; 4:121ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lai JY, Luo J, O’Connor C, et al. MicroRNA-21 in glomerular injury. J Am Soc Nephrol 2015; 26:805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. ▀ ▀.Lakhia R, Hajarnis S, Williams D, et al. MicroRNA-21 aggravates cyst growth in a model of polycystic kidney disease. J Am Soc Nephrol 2016; 27:2319–2330.This study demonstrated that miR-21 promotes cyst epithelial cell survival by inhibiting proapoptotic gene.
- 35.Hilliard A, Hilliard B, Zheng SJ, et al. Translational regulation of autoimmune inflammation and lymphoma genesis by programmed cell death 4. J Immunol 2006; 177:8095–8102. [DOI] [PubMed] [Google Scholar]
- 36.Obad S, dos Santos CO, Petri A, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 2011; 43:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. ▀ ▀.Gomez IG, MacKenna DA, Johnson BG, et al. AntimicroRNA-21 oligonucleo-tides prevent Alport nephropathy progression by stimulating metabolic pathways. J Clin Invest 2015; 125:141–156.This study describes pharmaceutical properties of an anti-miR-21 drug and demonstrates its therapeutic efficacy in Alport syndrome mouse models.
- 38.Androsavich JR, Sobczynski DJ, Liu X, et al. Polysome shift assay for direct measurement of miRNA inhibition by antimiRNA drugs. Nucleic Acids Res 2016; 44:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]