

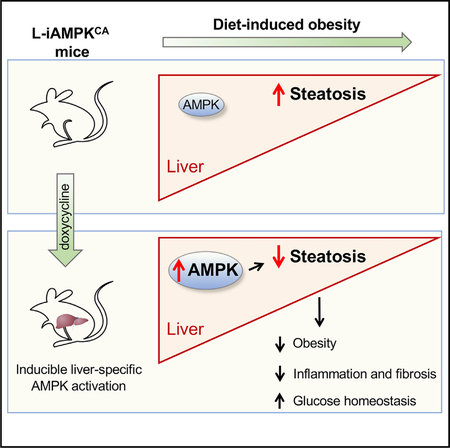
SUMMARY
The AMP-activated protein kinase (AMPK) is a highly conserved master regulator of metabolism, whose activation has been proposed to be therapeutically beneficial for the treatment of several metabolic diseases, including nonalcoholic fatty liver disease (NAFLD). NAFLD, characterized by excessive accumulation of hepatic lipids, is the most common chronic liver disease and a major risk factor for development of nonalcoholic steatohepatitis, type 2 diabetes, and other metabolic conditions. To assess the therapeutic potential of AMPK activation, we have generated a genetically engineered mouse model, termed iAMPKCA, where AMPK can be inducibly activated in vivo in mice in a spatially and temporally restricted manner. Using this model, we show that liver-specific AMPK activation reprograms lipid metabolism, reduces liver steatosis, decreases expression of inflammation and fibrosis genes, and leads to significant therapeutic benefits in the context of diet-induced obesity. These findings further support AMPK as a target for the prevention and treatment of NAFLD.
Graphical Abstract
In Brief
Garcia et al. present a GEMM, the “iAMPKCA” mouse, where AMPK is inducibly activated in vivo in a tissue-specific and temporal on and off nature. Liver-specific AMPK activation in these mice protects from diet-induced obesity and reduces liver steatosis, inflammation, and fibrosis. These data further support AMPK as a therapeutic target for NAFLD.
INTRODUCTION
Modern lifestyle, characterized by overnutrition and sedentariness, has led to increasing rates of metabolic diseases. Nonalcoholic fatty liver disease (NAFLD) is the most common chronic liver disease, with alarming prevalence around the world (Samuel and Shulman, 2018; Tilg et al., 2017; Younossi et al., 2018). NAFLD is associated with obesity and is characterized by excessive accumulation of lipids in the liver (steatosis). Moreover, NAFLD is an important risk factor for many metabolic diseases, and it commonly precedes more serious conditions, such as nonalcoholic steatohepatitis (NASH), cirrhosis, hepatocellular carcinoma (HCC), type 2 diabetes (T2D), and cardiovascular dis eases (Samuel and Shulman, 2018; Tilg et al., 2017; Younossi et al., 2018). Despite intense research, there are no currently approved drugs for the treatment of NAFLD. Therefore, identification of therapeutic targets for the treatment of NAFLD remains critically important.
AMP-activated protein kinase (AMPK) is a heterotrimeric protein kinase complex that is activated by low energy status and restores energy balance through inhibition of ATP-consuming processes and promotion of ATP-generating processes (Garcia and Shaw, 2017; Hardie et al., 2012). AMPK is thus a master regulator of multiple metabolic pathways that impact whole-body metabolism and physiology. Accordingly, AMPK activation has become an attractive potential candidate for the treatment of metabolic diseases, such as obesity and T2D (Day et al., 2017). Indeed, AMPK is indirectly activated by many clinically relevant compounds, most notably metformin, and might mediate many of their therapeutic effects (Duca et al., 2015; Fullerton et al., 2013; Hardie, 2013; Rena et al., 2017). A769662, the first described AMPK-specific small-molecule activator (that mostly targets AMPK in the liver), reduced liver steatosis and improved glucose parameters in obese mice (Boudaba et al., 2018; Cool et al., 2006). Moreover, a number of next-generation direct AMPK small-molecule activators with improved bioavailability across many tissues have been recently reported (Cokorinos et al., 2017; Myers et al., 2017; Steneberg et al., 2018). These studies demonstrated that AMPK activation efficiently lowered blood glucose levels in both healthy and diabetic animals, mainly through increased glucose uptake in muscle.
The therapeutic efficacy of AMPK activation has also been analyzed in genetically engineered mouse models (GEMMs), leading to conflicting and controversial findings (Carling, 2017). In particular, mice in which whole-body basal AMPK activity is increased, due to knockin activating mutations in the AMPKγ2 subunit, exhibited divergent phenotypes in terms of obesity (Yang et al., 2016; Yavari et al., 2016). One study reported that AMPK activation in AMPKγ2 mutant mice led to obesity due to hyperphagia and impaired insulin secretion (Yavari et al., 2016). The other study reported that AMPKγ2 mutant mice had reduced fat mass and liver steatosis in the context of diet-induced obesity, but AMPK activation also led to heart and kidney damage (Yang et al., 2016). In addition, in a recently developed mouse model of constitutive liver-specific AMPK activation, AMPK activation had surprisingly no effect in mice fed a high-fat diet (HFD) but resulted in protection against liver steatosis only when mice were fed a high-fructose diet (Woods et al., 2017). Of note, AMPK is activated during embryonic development in all of these mouse models, either in all cells or in the developing liver. The activation of AMPK in utero in these models makes it difficult to assess which of these phenotypes, either beneficial or deleterious, arise from developmental compensatory mechanisms and, therefore, which of the phenotypes reported might translate to the context of pharmacological activation of AMPK in adults. Thus, genetic activation of AMPK in previous GEMMs has not always substantiated the presumed therapeutic potential of AMPK activation.
In all, although the therapeutic potential for specific AMPK activation in metabolic diseases is increasingly evident, whether AMPK activation is a viable treatment for NAFLD requires further investigation. Here, we report the generation of a GEMM, termed the iAMPKCA mouse, which allows for spatial and temporal activation of AMPK in vivo. iAMPKCA mice exhibit robust, genetically inducible activation of AMPK that is comparable to pharmacologically relevant activation in its magnitude, thus validating the iAMPKCA GEMM as an improved system to model therapeutic activation of AMPK. We find that adult liver-specific AMPK activation in vivo leads to a reprogramming of lipid metabolism and a reduction in lipid levels in the liver due to promotion of fatty acid oxidation and inhibition of de novo lipogenesis. Importantly, liver-specific AMPK activation in iAMPKCA mice conferred substantial protection against weight gain, liver steatosis, and inflammation in the context of diet-induced obesity, which further validates AMPK as a therapeutic target for NAFLD.
RESULTS
Generation and Validation of iAMPKCA Mice
To study the effects of AMPK activation and its potential therapeutic benefits in vivo, we generated a GEMM in which we could inducibly activate AMPK. Truncated AMPKα protein (amino acids 1–312), which comprises the kinase domain, has previously been shown to exhibit high activity and is frequently used as a constitutively active allele of AMPK (Crute et al., 1998). We therefore engineered transgenic mice in which the expression of a cDNA encoding truncated AMPKα1 (AMPKα1(312)) was coupled to a tetracycline-responsive promoter (Figures S1A and S1B). Specifically, these mice have a modified Col1A1 locus, in which a doxycycline-inducible AMPKα1(312) cassette has been knocked in via recombinase-mediated cassette exchange, and a modified Rosa26 locus, in which a Cre-inducible rtTA3 cassette has been knocked in (Figure 1A) (Dow et al., 2014). We call these mice iAMPKCA mice (i, inducible; AMPKCA, constitutively active AMPK). Thus, iAMPKCA mice have three main components: (1) a Col1A1 transgene that serves as a genetic switch by which the AMPK pathway can be activated inducibly in a doxycycline-dependent manner, (2) a Rosa26 trans-gene that allows control of where the activation can take place (either whole body or tissue specific, depending on which Cre line is used), and (3) fluorescent markers that label induction of AMPKα1(312) (GFP) and expression of rtTA3 (mkate2, a red fluorescent protein). Importantly, doxycycline and Cre are both required for AMPK activation in this model.
Figure 1. Generation and Validation of iAMPKCA Mice.

(A) Diagram detailing the genetically engineered components of iAMPKCA mice at the Col1A1 and Rosa26 loci. CAG, CMV early enhancer chicken β-actin promoter; FRT, Flp recombination target; IRES, internal ribosome entry site; LSL, loxP-Stop (neomycin-resistance gene)-loxP cassette; mkate2, next-generation red-fluorescent protein; pA, polyA signal; rtTA3, reverse tetracycline-controlled transactivator 3; TRE, tet-response element.
(B) Table depicting the components required for liver-specific activation of AMPK using the iAMPKCA mouse model. Only doxycycline-treated L-iAMPKCA mice activate AMPK in liver. The other three groups serve as control mice.
(C) Western blot analysis of the expression of constitutively active AMPK and subsequent AMPK signaling activation in livers from iAMPKCA and L-iAMPKCA mice. Each lane is a separate mouse. d, day; Rev, 2 weeks doxycycline and then 1 week without doxycycline (reversed); w, weeks.
(D) Western blot analysis comparing the magnitude of AMPK activation in livers of iAMPKCA and L-iAMPKCA mice fed for 3 days with chow, plus or minus doxycycline, to AMPK activation by metformin in liver (1 hr, at indicated doses; or 3 hr where indicated). Each lane is a separate mouse. met, metformin; mpk, mg per kg.
(E) Livers of iAMPKCA and L-iAMPKCA mice, plus or minus 4 weeks doxycycline, were subjected to cellular fractionation. The subcellular localization and phosphorylation of the indicated proteins were determined by western blot. Tubulin, TOM20, and DNMT3a were used to demonstrate fraction purity. cyto, cytoplasmic fraction; iA, iAMPKCA mice; L-iA, L-AMPKCA mice; mito, mitochondrial fraction; nuc, nuclear fraction.
The liver plays a key role in the regulation of whole-body energy metabolism, and therefore, AMPK activation in the liver may confer important therapeutically beneficial effects, especially in the context of NAFLD. To test the efficiency of iAMPKCA mice to activate AMPK in the liver and the potential therapeutic effects of this activation, we crossed iAMPKCA mice to mice that express Cre recombinase from the albumin promoter (AlbCre). We call these mice L-iAMPKCA mice (for liver-specific iAMPKCA mice). Unlike iAMPKCA mice, which are functionally equivalent to wild-type mice in the absence of Cre, L-iAMPKCA mice express rtTA3 in the liver and, consequently, exhibit liver-specific activation of AMPK when treated with doxycycline (Figure 1B). All experimental iAMPKCA and L-iAMPKCA mice were bred to be homozygous for both the Col1A1- AMPKα1(312) and Rosa26-rtTA3 transgenes. To test for expression of constitutively active AMPK and subsequent activation of the AMPK pathway, we fed iAMPKCA and L-iAMPKCA mice chow with or without doxycycline (50 mg/kg) for 1 day, 3 days, or 2 weeks before collecting livers. As expected, only L-iAMPKCA mice expressed the constitutively active allele of AMPK and showed robust activation of the AMPK pathway upon administration of doxycycline, as evidenced by phosphorylation of the downstream substrates acetyl-CoA carboxylase (ACC), Raptor, and ULK1 (Figure 1C), demonstrating the inducibility of AMPK activation in L-iAMPKCA mice. Importantly, removal of doxycycline stopped expression of constitutively active AMPK and phosphorylation of downstream targets, which demonstrates the reversibility of AMPK activation in L-iAMPKCA mice (Figure 1C).
To further assess the magnitude of AMPK activation achieved in L-iAMPKCA mice, we compared mice fed doxycycline for 3 days to mice treated with one dose of metformin, a clinically relevant activator of AMPK in the liver, at doses that moderately (150 mg/kg) or potently (250 mg/kg) activate AMPK. Remarkably, L-iAMPKCA mice fed doxycycline exhibited sustained AMPK activation that was comparable to the activation achieved by metformin treatment in vivo, as evidenced by phosphorylation of the AMPK substrates Raptor, ULK1, mitochondrial fission factor (MFF), and p300 (Figure 1D). Similarly, in primary hepatocytes prepared from iAMPKCA and L-iAMPKCA mice fed chow plus or minus doxycycline for 4 weeks, we also found that AMPK activation in vitro was comparable to that of control hepatocytes treated with either metformin or the synthetic AMPK activator 991 (Figure S1C) (Xiao et al., 2013). Altogether, these experiments demonstrate that the iAMPKCA system is a GEMM that achieves robust, genetically inducible activation of AMPK in vivo that matches pharmacologically relevant activation of the AMPK pathway.
Truncated AMPKα1(312) protein does not form a complex with AMPKβ and AMPKγ subunits. To confirm proper localization of AMPKα1(312), we took iAMPKCA and L-iAMPKCA mice fed chow plus or minus doxycycline for 4 weeks and performed whole-liver fractionation. Significantly, truncated AMPKα1(312) exhibited identical localization as endogenous AMPK across cytoplasmic, mitochondrial, and nuclear fractions (Figure 1E). We also evaluated the ability of AMPKα1(312) to phosphorylate known substrates in the different subcellular compartments. Notably, AMPKα1(312) was able to efficiently phosphorylate not only cytoplasmic substrates but also MFF, a mitochondrial AMPK substrate (Toyama et al., 2016), and p300, a nuclear AMPK substrate (W. Yang et al., 2001) (Figure 1E). Moreover, AMPKα1(312) phosphorylated MFF and p300 to a similar extent compared to metformin-activated endogenous AMPK (Figure 1D). Although it is impossible to completely rule out the possibility that AMPKα1(312) might not always perfectly co-localize with endogenous AMPKα, our evidence suggests that AMPKα1(312) protein expressed in doxycycline-treated L-iAMPKCA is able to localize appropriately inside cells and phosphorylate endogenous substrates in several distinct subcellular locations.
AMPK Activation Reduces Lipid Levels in the Liver through Inhibition of De Novo Lipogenesis and Promotion of Fatty Acid Oxidation
To test for potential therapeutic effects of liver-specific activation of AMPK, we first analyzed iAMPKCA mice in chow-fed conditions, plus or minus 4 weeks of doxycycline (Figure 2A). iAMPKCA and L-iAMPKCA mice showed no differences in food consumption rate (Figure S2A) or body weight (Figure 2B), regardless of doxycycline treatment. Mice with liver-specific AMPK activation had lower levels of fasting blood glucose (Figure 2C); however, they did not perform significantly better in a glucose tolerance test (GTT) (Figures S2B and S2C) or an insulin tolerance test (ITT) (Figures S2D and S2E). Moreover, L-iAMPKCA mice treated with doxycycline did not have altered levels of plasma insulin relative to control mice (Figure S2F). We also analyzed activity and gas exchange in these mice by indirect calorimetry in metabolic cages. AMPK activation in the liver did not result in differences in oxygen consumption rates (Figures S2G and S2H) or respiratory exchange ratio (RER) (Figure S2I), indicating that there were no alterations in energy expenditure or fuel utilization. No differences were observed in the activity of these mice either (Figures S2J and S2K). In all, the effects of liver-specific AMPK activation on whole-body energetic balance and glucose homeostasis in chow-fed mice are small.
Figure 2. AMPK Activation Reduces Hepatic Lipid Levels.

(A) Schematic of experimental design to determine the effects of liver-specific AMPK activation in chow-fed iAMPKCA and L-iAMPKCA mice.
(B) Body weight of iAMPKCA and L-iAMPKCA mice treated as in (A). n = 8–10 mice per condition.
(C) Fasting (6 hr) blood glucose of iAMPKCA and L-iAMPKCA mice treated as in (A). n = 10 mice per condition.
(D) Schematic of experimental design to determine the effects of AMPK activation on de novo lipogenesis in vivo. D2O, deuterium-labeled water.
(E) Levels of newly synthesized (D2O-labeled) lipids in livers from iAMPKCA and L-iAMPKCA mice treated as in (D).
(F) Fatty acid oxidation rates traced from uniformly labeled [U-13C16]-Palmitate and its oxidation into citrate in hepatocytes harvested from iAMPKCA and L-iAMPKCA mice, fed chow with or without doxycycline for 4 weeks.
(G) Oxygen consumption rate from palmitate substrate in hepatocytes harvested from iAMPKCA and L-iAMPKCA mice fed chow with or without doxycycline for 4 weeks. Results from two independent experiments are plotted.
(H) Total levels of palmitate (C16:0), stearate (C18:0), oleate (C18:1) and linoleate (C18:2) in livers from iAMPKCA and L-iAMPKCA mice fed chow with or without doxycycline for 4 weeks. n = 4 mice per condition.
(I) Volcano plots showing lipidomics analysis of fold changes in triglycerides and diglycerides species in livers from iAMPKCA and L-iAMPKCA mice fed chow with or without doxycycline for 4 weeks. n = 4 mice per condition.
All values are expressed as means, and error bars reflect SEM. Significance was determined by ANOVA (*p < 0.05; **p < 0.01; **p < 0.001; ****p < 0.0001; ns, not significant).
AMPK is well known to regulate lipid metabolism through inhibition of de novo lipogenesis and promotion of fatty acid oxidation (Garcia and Shaw, 2017; Hardie et al., 2012). To directly assess whether sustained AMPK activation affected de novo lipogenesis in vivo, we treated iAMPKCA and L-iAMPKCA mice with deuterium-containing water (D2O) during the last week of a 4-week doxycycline trial (Figure 2D). During this period, de novo synthesized lipids become labeled with D2O, which allows us also to specifically measure the levels of newly synthe sized lipids in the liver. Notably, livers from L-iAMPKCA mice treated with doxycycline exhibited significantly lower levels of D2O-labeled palmitate (C16:0), stearate (C18:0) and oleate (C18:1) relative to control mice (Figure 2E), suggesting that de novo lipogenesis is inhibited and that newly synthesized lipids do not accumulate to the same extent when AMPK is activated. To assess whether sustained AMPK activation also affected fatty acid oxidation rates, we extracted primary hepatocytes from iAMPKCA and L-iAMPKCA mice fed chow plus or minus doxycycline for 4 weeks, incubated them with uniformly labeled palmi-tate, and then measured its oxidation (Figure S2L). Hepatocytes derived from doxycycline-treated L-iAMPKCA mice had much higher levels of palmitate oxidation relative to controls (Figure 2F), indicating that sustained AMPK activation also robustly promoted fatty acid oxidation. In addition, we also measured the ability of these cells to utilize palmitate as an energy source. Indeed, AMPK-active hepatocytes exhibited much greater oxygen consumption rates when administered palmitate as the energy source relative to controls (Figure 2G), confirming that AMPK activation led to higher rates of fatty acid oxidation. Importantly, etomoxir (an inhibitor of mitochondrial fatty acid oxidation) completely ablated oxygen consumption from palmitate in these cells, demonstrating that the observed effect is through mitochondrial β-oxidation (Figure 2G). Thus, AMPK activation resulted in both inhibition of de novo lipogenesis and promotion of fatty acid oxidation in the liver. Consistent with this, doxycycline-treated L-iAMPKCA mice had lower levels of total palmitate, stearate, oleate, and also linoleate (C18:2) in the liver (Figure 2H). Moreover, lipidomics analysis confirmed that AMPK activation resulted in a generalized reduction of hepatic lipid levels. Indeed, doxycycline-treated L-iAMPKCA mice had reduced levels of multiple species of triglycerides and diglycerides in the liver (Figure 2I). Taken together, these results indicate that sustained AMPK activation substantially remodels lipid metabolism and leads to a reduction of total hepatic lipid levels in vivo.
Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and Liver Steatosis
Since sustained AMPK activation resulted in lower lipid levels in livers from chow-fed animals, we proceeded to test whether liver-specific activation of AMPK was therapeutically beneficial in the context of diet-induced obesity and NAFLD. We first tested whether AMPK activation protected against development of fatty liver and obesity. We thus fed iAMPKCA and L-iAMPKCA mice a HFD (45% calories from fat), with or without doxycycline for 8 weeks (Figure 3A). As expected, L-iAMPKCA mice treated with doxycycline exhibited robust AMPK activation, even after extended HFD and doxycycline treatment (Figure S3A). Notably, mice with liver-specific AMPK activation were resistant to weight gain and were significantly lighter at the end of the trial compared to mice without AMPK activation (Figures 3B and 3C). Importantly, the observed effects in doxycycline treated L-iAMPKCA mice were not due to decreased food intake (Figure S3B). Body composition analysis by magnetic resonance imaging (MRI) revealed that doxycycline-treated L-iAMPKCA mice on a HFD had substantially lower levels of whole-body fat relative to controls (Figures 3D and 3E). Interestingly, fat depots outside of the liver were significantly smaller in these mice (Figure 3F), suggesting that although AMPK is being activated specifically in the liver, its effects have consequences beyond that organ. Consistent with reduced adiposity, mice with liver-specific AMPK activation had lower levels of the adipokines leptin and resistin (Figures S3C and S3D). Furthermore, livers from doxycycline-treated L-iAMPKCA mice exhibited smaller lipid droplets and overall reduced levels of lipid accumulation, as evidenced by H&E and Oil Red O staining (Figures 3G and 3H). Moreover, direct measurement of lipid content in the liver showed that AMPK activation led to decreased levels of palmitate, stearate, oleate, and linoleate (Figure 3I). Interestingly, even though AMPK activation resulted in lower lipid content in the liver and at the whole-body level, we found higher triglyceride and cholesterol levels in plasma from AMPK-activated mice (Figures S3E and S3F). In all, these experiments demonstrate that liver-specific activation of AMPK confers protection against diet-induced obesity and leads to vastly reduced liver steatosis.
Figure 3. AMPK Activation in Liver Protects against Diet-Induced Obesity.

(A) Protection study: schematic of experimental design to determine the effects of liver-specific AMPK activation on iAMPKCA and L-iAMPKCA mice fed a high-fat diet (HFD).
(B) Body weights of iAMPKCA and L-iAMPKCA mice during the 8-week HFD trial. n = 14 mice per condition.
(C) Percentage weight gain in iAMPKCA and L-iAMPKCA mice at the end of the HFD trial compared to initial weight. n = 14 mice per condition.
(D) Body composition and (E) percent fat mass of iAMPKCA and L-iAMPKCA mice treated as in (A). n = 13 mice per condition.
(F) Weight of one epididymal fat depot in iAMPKCA and L-iAMPKCA mice treated as in (A). n = 6 mice per condition.
(G) Representative images of livers from iAMPKCA and L-iAMPKCA mice treated as in (A) stained with H&E or Oil Red O, as indicated. Scale bar, 50 μm.
(H) Quantification of the area stained by Oil Red O in livers from iAMPKCA and L-iAMPKCA mice treated as in (A). n = 6 mice per condition.
(I) Total levels of palmitate (C16:0), stearate (C18:0), oleate (C18:1), and linoleate (C18:2) in livers from iAMPKCA and L-iAMPKCA mice treated as in (A). n = 6 mice per condition.
(J) Western blot analysis of the indicated proteins in iAMPKCA and L-iAMPKCA mice treated as in (A). Each lane is a separate mouse.
All values are expressed as means, and error bars reflect SEM. Significance was determined by ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant).
Autophagy, which is induced by AMPK, has been shown to promote clearance of lipids (Egan et al., 2011; Kaushik and Cuervo, 2016). We thus investigated whether autophagy was affected by HFD and AMPK activation in iAMPKCA mice. Indeed, levels of LC3b-II were substantially higher in livers from mice fed a HFD, indicating that autophagosomes were accumulating due to defective or insufficient autophagy (Figure 3J). Remarkably, AMPK activation largely restored lower LC3b-II levels, indicating that LC3b-containing vesicles may be processed more efficiently and do not accumulate when AMPK is activated, which was also supported by lower levels of p62 SQSTM1 in AMPK-activated livers (Figures 3J and S3G). These data suggest that besides inhibition of de novo lipogenesis and promotion of fatty acid oxidation, the induction of autophagy by AMPK may also contribute to alleviation of lipid accumulation in the liver.
Liver-Specific AMPK Activation Modestly Improves Glucose Homeostasis in the Context of Diet-Induced Obesity
Obesity and liver steatosis often lead to insulin resistance and deterioration of glucose homeostasis (Samuel and Shulman, 2018; Tilg et al., 2017). Therefore, we next investigated whether sustained AMPK activation in the liver improved glucose parameters in mice on a HFD. Indeed, doxycycline-treated L-iAMPKCA mice (8 weeks of a HFD) had considerably lower levels of fasting blood glucose levels compared to control mice (Figure 4A). Importantly, doxycycline-treated L-iAMPKCA mice did not exhibit higher levels of fasting plasma insulin (Figure 4B), suggesting that these mice might have higher insulin sensitivity. We thus evaluated the performance of these mice on various tolerance tests. Mice with liver-specific AMPK activation maintained lower levels of blood glucose when subjected to a GTT compared to control mice (Figures 4C and 4D). Similarly, doxycycline-treated L-iAMPKCA mice also maintained lower levels of blood glucose on an ITT relative to non-AMPK active control mice (Figures 4E and 4F). In addition, mice with liver-specific AMPK activation had lower blood glucose levels during a pyruvate tolerance test (PTT) (Figures S4A and S4B), suggesting that these mice have lower hepatic glucose production despite slight increases in the gluconeogenesis genes G6PC and PCK1 (Figure S4C). Although these results are consistent with AMPK-active mice having improved glucose homeostasis, the shape of the curves suggests that the effects are rather modest. However, metabolic cage analysis provided further evidence that mice with AMPK activation indeed retained higher insulin sensitivity. At night, respiratory exchange ratio values (RER) go up when glucose is used as energy source after feeding, but this process is impaired in mice on a HFD due to insulin resistance. Accordingly, control mice on a HFD with no AMPK activation displayed a flat respiratory exchange ratio across day and night cycles, as expected, because of an inability to use glucose as an energy source due to insulin resistance (Figure 4G). On the other hand, the respiratory exchange ratio for doxycycline-treated L-iAMPKCA mice retained modest oscillation from day to night cycles, demonstrating that mice with AMPK activation are still insulin sensitive and are able to utilize glucose during periods of feeding and insulin release in a normal, non-challenged context (i.e., without fasting or administration of a bolus of glucose or insulin) (Figure 4G). Consistent with the data above, the observed effect was nevertheless small. In addition, we did not observe differences in oxygen consumption in AMPK-active mice (Figures S4D and S4E), suggesting that there are no changes in energy expenditure after liver-specific AMPK activation or in their activity (Figures S4F and S4G).
Figure 4. AMPK Activation in Liver Modestly Improves Glucose Homeostasis in the Context of Diet-Induced Obesity.

(A) Fasting (6 hr) blood glucose in iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. n = 13 mice per condition.
(B) Fasting (6 hours) plasma insulin levels in iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. n = 6 mice per condition.
(C) Glucose tolerance test (GTT) on iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. n = 6 mice per condition.
(D) Quantification of the area under the curve in (C).
(E) Insulin tolerance test (ITT) in iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. n = 6 mice per condition.
(F) Quantification of the area under the curve in (E).
(G) Metabolic cage analysis of the respiratory exchange ratio (RER) in iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. Shaded area (light gray) delineates night (6:00 p.m. to 6:00 a.m.). n = 4 mice per condition.
(H) Measurement of diacylglycerol (DAG) in livers of iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. n = 5 mice per condition.
(I) Western blot analysis of PKCε levels in fractionated livers from iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. Tubulin and caveolin-1 were used to demonstrate fraction purity. Cyto, cytoplasmic fraction; mem, cell membrane fraction.
(J) Quantification of PKCε membrane translocation in fractionated livers from iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline for 8 weeks. The ratio of membrane-associated PKCε (normalized to caveolin-1) to cytoplasmic PKCε (normalized to tubulin) is plotted. n = 4 mice per condition.
All values are expressed as means, and error bars reflect SEM. Significance was determined by ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001; ns, not significant).
Because AMPK activation led to significant reductions in hepatic lipid levels, we hypothesized that AMPK might reduce diacylglycerol (DAG) levels in the context of diet-induced obesity. DAGs have been shown to contribute to insulin resistance by promoting activation and translocation of protein kinase C (PKC) proteins to the cellular membrane, where they can dampen insulin signaling (Fullerton et al., 2013; Petersen et al., 2016). In the liver, PKCε is the isoform most strongly associated with lipid-induced hepatic insulin resistance (Samuel and Shulman, 2018). Indeed, DAG levels were reduced in livers from doxycycline-treated L-iAMPKCA mice (Figure 4H). Furthermore, the ratio of membrane-associated to cytoplasmic PKCε was also reduced in AMPK-active livers (Figures 4I and 4J), suggesting that this may be a potential mechanism by which AMPK can promote insulin sensitivity in the liver. Interestingly, the total levels of liver PKCε go down in mice on a HFD, but AMPK activation restores its levels to those observed in chow-fed mice (Figure S4H). How AMPK can modulate PKCε levels is not clear. In all, our experiments indicate that the protection from diet-induced obesity afforded by liver-specific AMPK activation did translate into improved glucose homeostasis; however, these beneficial effects are modest.
AMPK Activation in the Liver Inhibits Both the Onset of Obesity and the Progression of Established Diet-Induced Obesity
With the iAMPKCA model, we have a unique ability to control the timing of hepatic activation of AMPK. We thus took advantage of the inducibility of the L-iAMPKCA model to test (1) whether prior liver-specific activation of AMPK affected the onset of obesity and (2) the therapeutic effect of liver-specific activation of AMPK on established obesity and liver disease.
First, to test whether prior liver-specific activation of AMPK could prevent or delay the onset of obesity, we first fed iAMPKCA and L-iAMPKCA mice chow with or without doxycycline and then switched them to a HFD, with or without doxycycline (Figure 5A). Mice with prior AMPK activation in liver exhibited slower weight gain once they were switched to HFD (Figure 5B). In fact, L-iAMPKCA mice on doxycycline diet were lighter (Figure 5C) and had reduced levels of fat mass at the end of the trial relative to mice with no AMPK activation (Figures 5D and 5E). Moreover, mice with AMPK activation prior to HFD had lower levels of fasting blood glucose (Figure 5F) and more efficiently cleared blood glucose in a GTT (Figures 5G and 5H), suggesting that unlike mice with no AMPK activation, mice with prior AMPK activation in liver exhibited better glucose homeostasis. Therefore, liver-specific AMPK activation hinders the onset of diet-induced obesity.
Figure 5. AMPK Activation in Liver Inhibits the Onset of Diet-Induced Obesity.

(A) Prevention study: schematic of experimental design to determine whether prior liver-specific AMPK activation can inhibit the onset of diet-induced obesity in L-iAMPKCA mice.
(B) Body weights of iAMPKCA and L-iAMPKCA mice during the HFD trial. n = 8 mice per condition.
(C) Weight of iAMPKCA and L-iAMPKCA mice at the end of trial described in (A). n = 8 mice per condition.
(D) Body composition and (E) percent fat mass of iAMPKCA and L-iAMPKCA mice treated as in (A). n = 8 mice per condition.
(F) Fasting (6 hr) blood glucose levels in iAMPKCA and L-iAMPKCA mice treated as in (A). n = 7 mice per condition.
(G) GTT in iAMPKCA and L-iAMPKCA mice treated as in (A). n = 7 mice per condition.
(H) Quantification of the area under the curve in (G).
All values are expressed as means, and error bars reflect SEM. Significance was determined by ANOVA (*p < 0.05; **p < 0.01).
Second, we tested the therapeutic effects of liver-specific AMPK activation as an intervention against established obesity and NAFLD. Thus, we first fed iAMPKCA and L-iAMPKCA mice a HFD without doxycycline for 8 weeks to allow mice to become obese, and we then switched half of the cohort to a HFD with doxycycline for an additional 8 weeks to determine whether AMPK activation had any therapeutic effects on established obesity (Figure 6A). Remarkably, L-iAMPKCA mice that were switched to doxycycline treatment showed a clear deceleration in further weight gain after AMPK activation and were significantly lighter at the end of the trial compared to control mice (Figures 6B and 6C). Indeed, body composition analysis showed that L-iAMPKCA mice that were switched to doxycycline had lower fat mass (Figures 6D and 6E) and smaller fat depots (Figure 6F) relative to mice with no AMPK activation. Furthermore, L-iAMPKCA mice that were switched to doxycycline treatment had lower levels of lipid accumulation in the liver, as evidenced by H&E and Oil Red O staining (Figures 6G and 6H). We next tested whether L-iAMPKCA mice that were switched to doxycycline treatment exhibited improved glucose metabolism. Doxycycline-treated L-iAMPKCA mice showed slightly reduced levels of fasting blood glucose levels and performed slightly better on a GTT, but these differences were not statistically significant across all control groups (Figures 6I–6K). Moreover, doxycycline-treated L-iAMPKCA mice did not perform better in an ITT (Figures S5A and S5B). Altogether, these experiments further demonstrate that AMPK activation in liver leads to a substantial reduction in liver steatosis, even when AMPK is activated in conditions where obesity and NAFLD were already established. On the other hand, the associated benefits to glucose homeostasis are essentially gone after the extended HFD regimen tested in these mice.
Figure 6. AMPK Activation in Liver Inhibits Progression of Established Diet-Induced Obesity.

(A) Intervention study. Schematic of experimental design to determine the effects of liver-specific AMPK activation on established obesity in iAMPKCA and L-iAMPKCA mice.
(B) Weights of iAMPKCA and L-iAMPKCA mice during the intervention study. n = 9 mice per condition.
(C) Percent weight gain in iAMPKCA and L-iAMPKCA mice at end of the intervention study relative to weight on week 8. n = 9 mice per condition.
(D and E) Body composition and (E) percent fat mass of iAMPKCA and L-iAMPKCA mice at the end of intervention study. n = 9 mice per condition.
(F) Weight of one epididymal fat depot in iAMPKCA and L-iAMPKCA mice at the end of intervention study. n = 8 mice per condition.
(G) Representative images of H&E- and Oil Red O-stained livers from iAMPKCA and L-iAMPKCA mice at the end of intervention study. Scale bar, 50 μm.
(H) Quantification of Oil Red O-stained area in livers from iAMPKCA and L-iAMPKCA mice at the end of the intervention study. n = 6 mice per condition.
(I) Fasting (6 hr) blood glucose levels in iAMPKCA and L-iAMPKCA mice at the end of the intervention study. n = 8 mice per condition.
(J) GTT on iAMPKCA and L-iAMPKCA mice at the end of intervention study. n = 7 mice per condition.
(K) Quantification of the area under the curve in (J).
All values are expressed as means. and error bars reflect SEM. Significance was determined by ANOVA (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant).
Liver-Specific AMPK Activation Inhibits Liver Inflammation and Fibrosis
To further investigate the effects of AMPK activation in the liver, we performed RNA-sequencing (RNA-seq) analysis on liver RNA from fasted iAMPKCA and L-iAMPKCA mice fed 8 weeks of a HFD, with or without doxycycline. We identified 285 genes that were significantly upregulated and 360 genes that were significantly downregulated in livers from doxycycline-treated L-iAMPKCA mice relative to controls (adjusted p < 0.05 relative to each of the three groups of control mice) (Figures 7A and 7B). Metascape pathway analysis of the upregulated genes revealed that sustained AMPK activation resulted in differences in carbohydrate and lipid metabolism in doxycycline-treated L-iAMPKCA mice (Figures 7C and S6A). Examination of the genes in the terms “Carbohydrate catabolic process” and “Monocarboxylic acid metabolic process” showed upregulation of several genes involved in the mobilization of glucose and lipids (Figure 7E). Specifically, genes upregulated in AMPK-activated mice included genes involved in glycogen metabolism, such as Ppp1r3b, Pygl (glycogen phosphorylase), and Gaa (lysosomal alpha-glucosidase), and genes involved in gluconeogenesis, such as G6pc, Fbp1, Aldob, and Mpc2. Although AMPK is known to inhibit glycogen synthesis and gluconeogenesis, the induction of these genes most probably indicates that, unlike control mice, doxycycline-treated iAMPKCA mice have properly mounted a fasting response, given their lower levels of blood glucose relative to control mice (Figure 4A).
Figure 7. AMPK Activation Downregulates Inflammation and Fibrosis-Related Transcriptional Programs.

Whole-transcriptome sequencing (RNA-seq analysis) was performed on whole-liver RNA from iAMPKCA and L-iAMPKCA mice fed a HFD with or without doxycycline 8 weeks.
(A) Heatmap representation of 283 genes upregulated after AMPK activation in liver (adjusted p < 0.05 from pairwise comparison of doxycycline-treated L-iAMPKCA samples to each of the 3 control groups). Prkaa1 (AMPKα1) is pointed out.
(B) Heatmap representation of 360 genes downregulated after AMPK activation in liver (adjusted p < 0.05 from pairwise comparison of doxycycline-treated L-iAMPKCA samples to each of the 3 control groups).
(C) Metascape pathway analysis on the genes upregulated by AMPK activation.
(D) Metascape pathway analysis on the genes downregulated by AMPK activation.
(E) Heatmaps showing upregulated genes in the terms “Carbohydrate catabolic process” and “Monocarboxylic acid metabolic process” from Metascape pathway analysis in (C).
(F) Heatmaps showing downregulated genes in the terms “Extracellular matrix organization” and “Regulation of cell migration” from Metascape pathway analysis in (D).
In terms of lipid metabolism, genes induced in AMPK-activated mice included some genes involved in triglyceride synthesis, such as Gpd1, Dgat2, and Mlxipl (ChREBP). Moreover, several genes involved in lipid transport were also upregulated, such as Apoc3, Apoa5, Apoa2, and Fabp1. Upregulation of these genes, together with upregulation of other genes involved in the control of plasma levels of triglycerides, such as Angptl8 (angiopoietin like protein 8) and Angptl3 (Zhang, 2016), might explain in part the hypertriglyceridemia and hypercholesterolemia observed in mice with liver-activated AMPK. Interestingly, induction of peroxisomal genes Hacl1 and Acnat1 might indicate an uptick in peroxisomal lipid oxidation after AMPK activation.
More importantly, analysis of the genes downregulated in doxycycline-treated L-iAMPKCA mice revealed a transcriptional profile that clearly pointed to reduced inflammation and reduced fibrosis when AMPK was activated (Figures 7D and 7F). Indeed, examination of the genes in the terms “Extracellular matrix organization” and “Regulation of cell migration” (Figure 7F) showed downregulation of many genes that are produced by liver or inflammatory cells in response to inflammation, such as Saa1 (serum amyloid A1), Saa2, Rarres2 (chemerin), Cxcl9 (C-X-C motif chemokine ligand 9), Cxcl10, Il1rn (interleukin 1 receptor antagonist), Spp1 (osteopontin), Sparc (osteonectin), and matrix metalloproteinases Mmp12, Mmp13, and Mmp2. Furthermore, motif enrichment analysis, which identifies whether specific regulatory elements are enriched in a set of genes, revealed that the top motif significantly enriched in the genes downregulated in AMPK-activated livers was the motif ISRE (IFN-stimulated response element) (Figure S6D). ISRE is a motif found in the promoters of genes induced by type I interferons (Chen et al., 2017); therefore, its enrichment in the downregulated genes is consistent with reduced inflammation in AMPK-activated livers. Moreover, inflammation and fibrosis are processes that are closely linked in liver. Remarkably, besides reduced expression of inflammatory genes, AMPK-activated mice showed reduced expression in genes involved in fibrosis. Indeed, genes that were downregulated included not only several extracellular matrix genes (most notably, several collagen genes and also Lamb1 [laminin beta 1]) but also genes involved in the processing of procollagens and collagen fibers (such as Serpine1, Loxl2 and Adamts2). Hepatic stellate cells, which store vitamin A in liver, play a major role in fibrosis when activated (Tsuchida and Friedman, 2017). Importantly, downregulation of genes in the term “Retinol metabolism” (Figure 7D and S6B) (retinol is vitamin A1) and downregulation of genes that function in cell motility (increased cell motility is a feature of stellate cell activation), such as Cdh1, Eppk1, Prex1, and Dock1, suggest reduced activation of hepatic stellate cells in doxycycline-treated L-iAMPKCA mice. Interestingly, Nox4, which was recently reported to promote liver fibrosis through hepatic stel-late cell activation (Lan et al., 2015), was also expressed at lower levels in livers in which AMPK was activated. Of note, direct assessment of fibrosis in these mice did not show any evidence of the development of liver fibrosis (Figure S6E), which is not surprising given the short time on a HFD (8 weeks). Evidently, the processes that lead to liver fibrosis initiate long before overt disease is established. Thus, the significant downregulation of pro-fibrotic transcriptional programs in AMPK-activated mice indicate that AMPK can inhibit such processes even in the absence of overt liver fibrosis. Indeed, incipient deposition of collagen fibers beyond the portal tracts could be detected in control mice fed a HFD for 16 weeks, but not in similarly treated mice in which AMPK was activated (Figure S6F). In all, the RNA-seq data strongly indicate that besides potently alleviating steatosis, AMPK activation in liver also leads to reduced inflammation and fibrosis and may therefore inhibit progression of NAFLD into NASH.
Finally, it is now clear that some liver-secreted proteins (referred to as hepatokines) can play significant roles in whole-body metabolism through their interactions with other metabolically relevant tissues, such as muscle, adipose tissue, and β cells (Meex and Watt, 2017). Notably, even though doxycycline-treated L-iAMPKCA mice activate AMPK specifically in the liver, we observed robust extra-hepatic beneficial effects in the context of diet-induced obesity in these mice, namely, reduced white adipose tissue expansion. Our RNA-seq analysis indicated that AMPK activation in liver changed expression of numerous secreted proteins, including some hepatokines (such as Angptl3 and Angptl8). We thus examined the expression profile of several well-established hepatokines in our RNA-seq dataset (Figure S6C) in an effort to assess whether some of the extra-hepatic effects might be mediated by known hepatokines. Besides the aforementioned increase in Angptl3 and Angptl8 expression, we observed decreased expression of the proinflammatory hepatokines alpha-2 macroglobulin (a marker of liver fibrosis) and Lect2 with AMPK activation. However, most of the examined hepatokines exhibited small or no AMPK-dependent changes in their transcription (Figure S6C).
DISCUSSION
Activation of AMPK has long been proposed to be therapeutically beneficial in the context of metabolic diseases (Day et al., 2017; Garcia and Shaw, 2017; Hardie, 2013). In this study, we have developed a GEMM platform, which we call iAMPKCA, in which AMPK can be robustly and inducibly activated in vivo, and we have used it to test the therapeutic effects of AMPK activation in metabolic disease. The iAMPKCA model provides important advantages over previously developed GEMMs in that it allows for both spatial and temporal control of AMPK activation and can thus bypass possible embryonic compensations and adaptations to its activation in utero. Using L-iAMPKCA mice (i.e., iAMPKCA mice that express AlbuminCre), we have specifically tested the therapeutic efficacy of AMPK activation in liver. Liver-specific AMPK activation resulted in a marked lowering of liver lipid content through multiple mechanisms, including a potent increase in fatty acid oxidation rates, inhibition of de novo lipogenesis, and activation of autophagy. Importantly, the ability of AMPK activation to lower lipids in liver was therapeutically beneficial in the context of diet-induced obesity. Indeed, mice with liver-specific AMPK activation were protected against weight gain and obesity, had modestly improved glucose metabolic parameters, exhibited substantially reduced liver steatosis, and showed reduced expression of genes involved in liver inflammation and fibrosis. Moreover, reduced liver steatosis and inhibition of obesity were still observed even when AMPK was activated in mice with established obesity. Thus, our study provides further evidence for the therapeutic potential of AMPK and specifically supports AMPK as a therapeutic target for NAFLD.
AMPK controls systemic glucose metabolism at least in part through acute stimulation of glucose uptake in muscle tissue and through inhibition of hepatic gluconeogenesis, which may itself be indirectly impacted by improved insulin sensitivity from glucose clearance in muscle and liver (Cokorinos et al., 2017; Fullerton et al., 2013; Myers et al., 2017; Steneberg et al., 2018). Recent studies have reported that AMPK activation in liver does not acutely lower blood glucose levels or improve glucose homeostasis (Cokorinos et al., 2017; Esquejo et al., 2018; Woods et al., 2017). We also observed only small to no effects on glucose homeostasis after sustained AMPK activation in liver in chow-fed mice (Figures 2C and S2B–S2F). We did observe, however, some improvements in glucose homeostasis and insulin sensitivity when AMPK was specifically activated in livers of mice fed a HFD. The substantial decrease in liver lipid content and steatosis is the most robust therapeutic effect of liver-specific AMPK activation in our L-iAMPKCA model, and thus, we presume that other therapeutic benefits might stem from this lowering of hepatic lipids. Indeed, the therapeutic effects on glycemic control in mice with liver AMPK activation may be due to reduced hepatic lipid content (such as reduced DAG) and lower body weight. Nonetheless, the beneficial effects on glucose homeostasis conferred by liver-specific AMPK activation were more obvious at early time points (Figures 5G and 5H), modest after 8 weeks of HFD (Figure 4), but largely blunted when AMPK was activated in mice with established obesity (Figures 6I–6K), consistent with previous reports (Cokorinos et al., 2017; Esquejo et al., 2018). Thus, our study suggests that liver-specific AMPK activation can delay, but not prevent, the deterioration of glycemic control elicited by diet-induced obesity. Moreover, AMPK activation in liver might not be therapeutically sufficient to restore glycemic control once obesity is established.
AMPK is increasingly recognized as a potent inhibitor of inflammation, mainly through modulation of metabolic states in inflammatory cells such as macrophages (Day et al., 2017; Fullerton et al., 2015; Mills and O’Neill, 2016; Mounier et al., 2013). Surprisingly, even though AMPK is not directly activated in inflammatory cells in L-iAMPKCA mice, transcriptional profiling of livers from mice on a HFD showed that liver-specific activation of AMPK led to reduced expression of numerous genes involved in inflammation and fibrosis (Figure 7F). As above, although it is not possible to rule out that AMPK activation in hepatocytes directly inhibits inflammatory signals stemming from those cells, the lowering of liver steatosis caused by AMPK activation probably contributes in large part to the observed reduction in pro-inflammation and pro-fibrosis transcriptional signatures. In any case, our results indicate that liver-specific AMPK activation might slow down the progression of NAFLD into NASH, thus further adding substantial clinical value to the therapeutic activation of AMPK. Additional studies would be needed to test whether AMPK activation can also alleviate established NASH.
Previous studies on AMPK activation have reported several deleterious effects stemming from whole-body AMPK activation. The best-known example of this is AMPK-induced accumulation of cardiac glycogen and cardiac hypertrophy, first identified in humans with Wolff-Parkinson-White syndrome, a condition caused by activating AMPKγ2 mutations (Blair et al., 2001). Some of these phenotypes were recently recapitulated in mice with germline mutations in AMPKγ2 (Yang et al., 2016; Yavari et al., 2016) and also, more importantly, after systemic activation of AMPK by a small-molecule activator (Myers et al., 2017). Other reported side effects of systemic AMPK activation include hyperphagia and kidney dysfunction (Yang et al., 2016; Yavari et al., 2016), although these effects were not reported after systemic AMPK activation by drugs (Cokorinos et al., 2017; Myers et al., 2017; Steneberg et al., 2018). Thus, although the beneficial effects of systemic AMPK are significant, especially in terms of improvements in glucose homeostasis, targeting AMPK in a tissue-specific manner might be a better strategy in some cases. In the case of NAFLD, our data strongly indicate that liver-specific activation of AMPK is sufficient to obtain a robust therapeutic effect in the context of diet-induced obesity, namely, a marked reduction in liver steatosis and associated improvements in inflammation and fibrosis. Moreover, liver-specific AMPK activation clearly conferred beneficial extra-hepatic effects, most notably reduced expansion of white adipose depots in response to HFD. Interestingly, the liver secretes a remarkable number of proteins (Meex et al., 2015), many of which can modulate whole-body metabolism (i.e., hepatokines). We speculate that AMPK activation may induce secretion of hepatokines that therapeutically improve insulin sensitivity and obesity. Indeed, AMPK activation in doxycycline-treated L-iAMPKCA mice led to changes in the expression of many secreted proteins (Figures 7E and 7F), although we did not observe changes in several established hepatokines (Figure S6C). Thus, investigation of AMPK-induced changes in the liver secretome would be of great interest in the future.
Apart from therapeutic effects, we unexpectedly found AMPK activation in liver led to modest hypertriglyceridemia and hyper-cholesterolemia in doxycycline-treated L-iAMPKCA mice (Figures S3E and S3F). Intriguingly, hypertriglyceridemia was also reported as a side effect in humans after pharmacological inhibition of hepatic ACC, a treatment that parallels AMPK activation (AMPK inhibits ACC), as well as in mice with liver-specific deletion of ACC1 and ACC2 (Kim et al., 2017). In these studies, increased secretion of triglycerides into the bloodstream was ascribed to the activation of SREBP1 and subsequent induction of genes that function in triglyceride synthesis, especially GPAT1, the enzyme that catalyzes the first step in triglyceride synthesis (Kim et al., 2017). A similar mechanism might be operating as well in livers with AMPK activation, since we also observed upregulation in genes involved in triglyceride synthesis, most notably Dgat2 and Gpd1, and genes that directly regulate plasma triglyceride levels, including several apolipoproteins and the lipoprotein lipase inhibitors Angptl3 and Anptl8 (Figure 7E). Interestingly, a recent study also reported hypercholesterolemia in mice after pharmacological activation of AMPK in the liver (Esquejo et al., 2018). However, plasma cholesterol levels actually decreased in rats and non-human primates after similar AMPK activation in liver, suggesting that the hypercholesterolemia induced by AMPK activation in mice might be consequence of specific peculiarities of the regulation of plasma cholesterol in that species (Esquejo et al., 2018). Nevertheless, how pharmacological activation of AMPK impacts plasma levels of triglycerides and cholesterol in human patients needs to be evaluated.
NAFLD represents a major clinical challenge, given its high levels of incidence worldwide. Several promising therapeutic approaches are being researched for treatment of NAFLD, although no drug has been approved yet (Samuel and Shulman, 2018). The results presented here, employing a mouse model for genetic AMPK activation, further extend the evidence for liver-specific AMPK activation as a therapeutic avenue for NAFLD treatment and strongly validate AMPK as a therapeutic target for NAFLD (Boudaba et al., 2018; Esquejo et al., 2018; Woods et al., 2017). Next-generation, liver-specific AMPK activators with favorable pharmacological properties have now been developed (Cokorinos et al., 2017; Esquejo et al., 2018). An extensive analysis of their therapeutic effects on NAFLD in the clinic will certainly be of great interest.
Significantly, the iAMPKCA GEMM provides a valuable system in which to assess and analyze the potential therapeutic effects of AMPK activation in any preclinical mouse model. Moreover, given its modular design, the iAMPKCA model allows for specific AMPK activation either systemically, by crossing iAMPKCA mice into a line that globally expresses Cre recombinase, or in a spatially restricted manner in any tissue that can be targeted with tissue-specific Cre. In addition, the inducibility of the iAMPKCA model allows evaluation of therapeutically valuable information from the kinetic control of AMPK activation in preclinical models, such as assessment of the effects of acute versus sustained AMPK activation, assessment of the effects of AMPK activation in either the prevention or treatment of disease, and assessment of AMPK activation in young versus old animals, just to name a few. Integration of the features of the genetic activation of AMPK by the iAMPKCA GEMM with relevant preclinical mouse models will undoubtedly shed light into many additional prospective therapeutic applications for AMPK activation, including cancer, inflammation, and aging.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Reuben J. Shaw (shaw@salk.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
iAMPKCA mice were generated as described below. To generate mice with liver-specific activation of AMPK, iAMPKCA mice were crossed to Albumin-Cre mice (Jackson Laboratories #003574). All mice were 8–10 weeks of age at the beginning of experiments. Only males were used for experiments, and were randomly assigned to experimental groups. Mice were housed with a standard 12-hour day and night cycles. All procedures using animals were approved by the Salk Institute Institutional Animal Care and Use Committee (IACUC).
Primary hepatocytes
Primary mouse hepatocytes from 12-week-old mice given food with or without doxycycline for 4 weeks were isolated. Livers were perfused with Hank’s balanced salt solution (HBSS, KCl, 5.4 mM; KH2 PO4, 0.44mM; NaCl, 138mM; NaHCO3, 4.2 mM; Na2HPO4, 0.34 mM; glucose, 5.6 mM; HEPES, 55 mM; EGTA, 0.6 mM; pH 7.4) followed by perfusion with a collagenase buffer (as above but without EGTA and supplemented with CaCl2, 5 mM; Collagenase (Sigma C5138); 0.025%)). Cells were spun, washed and cell viability was determined using Trypan blue exclusion test. Hepatocytes were plated on TPP plates (Light Lab Systems) at 2×106 cells for a 6-cm plate, 4.5×105 cells per well of a 6-well plate, or at 4,000 cells per well of a 96-well Seahorse plate, as appropriate, in DMEM (Corning, 10–017CV) containing 5% FBS and supplemented with penicillin and streptomycin (GIBCO, 15140–122), and allowed to adhere for 3h. Cells were washed twice in DMEM and incubated in serum-free DMEM supplemented with penicillin and streptomycin overnight before indicated treatments. Drug doses used were as follows: 991 (10mM), metformin (1mM), doxycycline (50 ng/ml).
Generation of iAMPKCA mice
Full method details for the generation of Col1A1-transgenic mice have been described previously (Dow et al., 2012). Briefly, a cDNA encoding human AMPKα1(1−312) was cloned into a “Flp-in” targeting vector, kindly provided by Lukas E. Dow, and co-transfected with a pCAGGS-Flpe plasmid (Addgene #20733) into D34 stem cells (Dow et al., 2014), also provided by Lukas E. Dow. Recombined stem cell clones were then selected for with hygromycin (140 μg/ml) (Thermo Fisher Scientific #10687010). Correctly targeted clones were screened by PCR, Southern blotting and western blotting. A correctly targeted clone was then expanded, injected into embryos and implanted into females to generate chimeras by the Salk Transgenic Core. After germline transmission, iAMPKCA mice were backcrossed into a C57BL/6J background (Jackson Laboratories #000664).
Animal studies
Special diets
To activate AMPK, mice were fed chow with 50 mg/kg doxycycline (Envigo, #TD.150877). For diet-induced obesity studies, mice were fed a HFD (45% calories from fat) (Envigo, #TD.06415) or the equivalent HFD with doxycycline (87 mg/kg) (Envigo, #TD.160354), as indicated. Weights were monitored every two weeks. All diets were administered ad libitum.
Metformin treatment in vivo
Non-fasted iAMPKCA mice were injected intraperitoneally with either vehicle (0.9% saline) or metformin (150 mg/kg or 250 mg/kg in saline, as indicated) and euthanized 1 or 3 hours post-injection, as indicated. Liver samples were snap frozen in liquid nitrogen.
GTT, insulin tolerance test (ITT) and pyruvate tolerance test (PTT)
Mice were fasted in paper bedding for 6 hours (8:00 am to 2:00 pm) before beginning of tolerance tests. Vehicle (0.9% saline), glucose (1.5 g/kg body weight, for GTTs), insulin (.75 U/kg body weight, for ITTs) (Humulin® R, Lilly), or sodium pyruvate (2 g/kg body weight, for PTTs) were injected intraperitoneally. Blood glucose level was measured using Aviva ACCU-CHEK® glucose meter prior to injection and after injection, at the times indicated.
Metabolic cages
Oxygen consumption (VO2), carbon dioxide expiration (VCO2), food consumption and activity were measured using PhenoMaster/LabMaster equipment following the manufacturer’s instructions (TSE systems). Mice were housed singly and were habituated for 3 days prior to 2 days of recording. Lighting and feeding conditions were kept identical to those of home cages.
Body composition
Body composition was measured in live mice using EchoMRI™-100H body composition analyzer.
Immunohistochemistry and image analysis
Liver samples were formalin-fixed overnight and then transferred into 70% ethanol. After processing, samples were paraffin embedded. For lipid staining, liver samples were frozen in O.C.T. compound (Tissue-Tek®, 4583). For histology and assessment of liver steatosis and fibrosis, 5 μm sections from paraffin-embedded tissues were prepared and stained with hematoxylin and eosin or Masson’s Trichrome, and frozen sections were prepared and stained with Oil Red O at the Salk Institute Histology Core. Oil Red O staining was quantified using ImageJ.
Fatty acid oxidation measurement (Seahorse)
Fatty acid oxidation was determined by measuring oxygen consumption rates using palmitate as substrate with a Seahorse XF 96 Flux Analyzer (Agilent Technologies). 4,000 primary hepatocytes per well were seeded in XF96 cell culture microplates, with at least 5 wells per condition, and incubated overnight in DMEM containing 25mM Glucose. On the day of the assay, cells were switched to KHB buffer (111 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 2 mM MgSO4, 1.2 mM NaH2PO4, 2.5 mM glucose, 0.5 mM carnitine, and 5 mM HEPES pH 7.4) and incubated for 60 minutes at 37 C. Immediately before starting the seahorse assay, palmitate-BSA conjugate (Agilent Technologies, #102720–100) was added to each well. Basal oxygen consumption was measured before injection. Oxygen consumption was also measured after the following compounds were injected: oligomycin (1μM), FCCP (125nM), Antimycin A/rotenone (1μM each) (Agilent Technologies, #103015–100). When indicated, etomoxir (40μM) (Sigma, E1905) was added to the well 15 min prior to adding palmitate-BSA.
Plasma biochemistry
Mice were fasted for 6 hours (8:00 am to 2:00 pm) before blood was collected via retro-orbital bleeding using EDTA microvette tubes (Sarstedt 16444100). Plasma was obtained by centrifugation. Plasma triglycerides and cholesterol were measured with Thermo Scientific Infinity™ reagents (#TR22421 and #TR13421, respectively), according to manufacturer’s instructions. Plasma levels of insulin, leptin and resistin were measured using Bio-Plex® Pro Mouse Diabetes immunoassay kit (Bio-Rad, # 171F7010M), according to manufacturer’s instructions. Blood glucose level was measured using Aviva ACCU-CHEK® glucose meter.
Liver fractionation
For isolation of cytoplasmic, mitochondrial and nuclear fractions from mouse liver, livers were homogenized using a glass dounce homogenizer in buffer (250 mM Sucrose, 25 mM KCl, 5 mM MgCl2, 10 mM Tris pH 7.4, 50 mM sodium fluoride, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 0.2 mM PMSF, and protease inhibitors). Nuclei were pelleted at 1,000 g for 10 min and purified through a 30% iodixanol solution by centrifugation (Optiprep, Sigma D1556). Nuclear proteins were then extracted with buffer (250 mM Sucrose, 1 mM EDTA, 800 mM NaCl, 10 mM Tris pH 7.4). Mitochondria were recovered from the post-nuclear supernatant by centrifugation at 9000 g for 10min and were resuspended in CST lysis buffer (recipe below). Supernatant was used as cytoplasmic fraction. For isolation of cytoplasmic and membrane fractions (for analysis of PKCε translocation), 100 mg of pulverized liver from mice fasted for 6 hours was homogenized with a dounce homogenizer in 0.6 mL of buffer A (20 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.25 mM EGTA, 250 mM sucrose, and freshly added protease (cOmplete, Roche) and phosphatase inhibitors (PhosSTOP, Roche)) and centrifuged for 60 minutes at 100,000g (4°C). Supernatant was used as cytoplasmic fraction. The pellet was washed once with buffer B (250 mM Tris-HCl, pH 7.4, 1 mM EDTA, 0.25 mM EGTA, 2% Triton X-100, and freshly added protease (cOmplete, Roche) and phosphatase inhibitors (PhosSTOP, Roche)). The pellet was then resuspended in buffer B, incubated for 45 minutes at 4°C, and then centrifuged for 60 minutes at 100,000g (4°C). The resulting supernatant was used as the membrane fraction. The collected fractions were subsequently analyzed by western blotting.
Western blotting
For biochemical analysis of liver, liver fragments were harvested, immediately snap frozen in liquid nitrogen and homogenized on ice in RIPA lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, 50 mM sodium fluoride, 2.5 mM sodium pyrophosphate, 2 mM beta-glycerophosphate, 1 mM Na3VO4, 10 nM Calyculin A) supplemented with protease inhibitors (Roche, cOmplete™, #11836170001). For biochemical analysis of hepatocytes, cells were washed with ice-cold PBS and lysed directly on plates using CST lysis buffer (20mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 50 mM sodium fluoride, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM beta-glycerophosphate, 1 mM Na3VO4, 10 nM Calyculin A) supplemented with protease inhibitors. Protein lysates were centrifuged and protein concentration was determined using Pierce™ BCA protein assay kit (Thermo Scientific, #23225). Lysates were resolved on 8%–12% SDS-PAGE gels, depending on the experiment, and transferred to PVDF membranes for immunoblotting. When appropriate, protein levels were quantified by densitometry analysis using ImageJ.
Antibodies and reagents
For western blotting, antibodies were purchased from Cell Signaling Technologies (Denvers, MA, USA) and diluted 1:1,000 in 5% BSA (Sigma, A4503): P-ACCS79 (#3661), ACC (#3662), AMPKα (#2532) P-AMPKαT172 (#2535), P-RaptorS792 (#2083), Raptor (#2280), P-ULK1S555 (#5869), ULK1 (#8054), P-MFFS146 (#49281), MFF (#84580), GFP (#2956), LC3b (#3868), PKCε (#2683), and Caveolin-1 (#3267). Also diluted 1:1,000 in 5% BSA: p62 (#GP62-C) from Progen. Diluted 1:500 in 5% BSA, unless otherwise indicated: Dnmt3a (#sc-20703), TOM20 (#sc-1145, 1:1000 dilution), P-p300S89 (#sc-130210), p300 (#sc-585), were purchased from Santa Cruz Biotechnology. From Sigma-Aldrich, anti-β-actin (#A5441) was diluted 1:10,000 and anti-Tubulin (#T5168) was diluted 1:5,000. Other reagents include: metformin (Sigma, PHR1084), glucose (Sigma, 7528), sodium pyruvate (Sigma, P4562), 991 (Glixx Laboratories, GLCX-09267), and doxycycline (Sigma, D9891).
Quantitative real-time PCR
RNA was isolated from liver tissue using RNeasy Lipid tissue mini kit (QIAGEN cat # 74804) and cDNA synthesis performed using SuperScript III First-Strand synthesis system (Invitrogen cat # 18080051). Sybr Green PCR Master mix (Applied Biosystems cat #4309155) was used in qPCR reactions with the following primers:
G6Pase Forward: 5′-ACTGTGGGCATCAATCTCCTC-3′, G6Pase Reverse: 5′-CGGGACAGACAGACGTTCAGC-3′, PEPCK Forward: 5′-CTGCATAACGGTCTGGACTTC-3′, PEPCK Reverse: 5′CAGCAACTGCCCGTACTCC-3′, Cyclophilin Forward:5′- TGGA GAGCACCAAGACAGACA, Cyclophilin Reverse: 5′- TGCCGGAGTCGACAATGAT-3′
mRNA-seq
RNA was isolated using the RNeasy Lipid Tissue Mini kit (QIAGEN), including a DNase treatment. RNA integrity (RIN) numbers were determined using the Agilent TapeStation prior to library preparation. mRNA-seq libraries were prepared using the TruSeq stranded RNA library preparation kit (version 2) according to the manufacturer’s instructions (Illumina). Libraries were then quantified, pooled, and sequenced by single-end 50 base pairs using the Illumina HiSeq 2500 platform at the Salk Next-Generation Sequencing Core. Raw sequencing data were demultiplexed and converted into FASTQ files using CASAVA (version 1.8.2). Libraries were sequenced at an average depth of 21 million reads per sample.
mRNA-seq analysis
mRNA-seq analsysis was performed as in (Eichner et al., 2018). In brief, sequenced reads were quality-tested using the FASTQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the mouse mm10 genome using the STAR aligner version 2.4.0k. Raw gene expression was quantified across all annotated mm10 exons using HOMER. Differential expression analysis was carried out using the edgeR package version 3.6.8 using triplicates to compute within-group dispersion and correcting for batch effects. Differentially expressed genes were defined as having a false discovery rate (FDR) < 0.05. The genes were then z-score normalized, clustered using a hierarchical clustering approach with Cluster 3 and visualized by heatmaps using Java TreeView (version 1.1.6r4). Functional pathway analysis of both upregulated and downregulated genes was done using Metascape. Known motif enrichment analysis was carried out using HOMER findMotifs.pl.
D2O-labeling of newly synthesized lipids in vivo
To begin the D2O (deuterium) labeling period, mice were first injected intraperitoneally with 0.9% NaCl in D2O (2H2O), at 0.035 mls/g body weight. Drinking water was then replaced with 8% D2O enriched water for one week. Mice were fasted for 6 hours prior to plasma and tissue collection and samples were immediately snap frozen in liquid nitrogen.
GC/MS analysis of fatty acids and polar metabolites
Metabolites and total fatty acids were extracted from tissues or cells using a Folch-based methanol/chloroform/saline extraction at a ratio of 1:2:1 with inclusion of [U-2H31]palmitate and norvaline as organic and aqueous internal standards respectively. Briefly, 250 μL MeOH, 500 μL CHCl3, 250 μL saline and fatty acid isotope internal standards were added to weighed pre-ground tissue or cell pellets. This mixture was vortexed for 10 minutes followed by centrifugation at 10,000 g for 5 minutes. Both phases were then isolated and dried for storage at −20°C.
Aqueous metabolites were derivatized in 2% (w/v) methoxyamine hydrochloride (Thermo Scientific) in pyridine and incubated at 37°C for 60 minutes. Samples were then silylated with N-tertbutyldimethylsilyl-N-methyltrifluoroacetamide (MTBSTFA) with 1% tertbutyldimethylchlorosilane (tBDMS) (Regis Technologies) at 37°C for 30 min. Polar derivatives were analyzed by GC-MS using a DB-35MS column (30 m × 0.25 mm i.d. × 0.25 μm, Agilent J&W Scientific) installed in an Agilent 7890A gas chromatograph (GC) interfaced with an Agilent 5975C mass spectrometer (MS). GC oven was held at 100°C for 1 min after injection, increased to 255°C at 3.5°C/min, and finally increased to 320°C at 15°C/min and held for 3 min.
Organic phase was derivatized to form fatty acid methyl esters (FAMEs) via addition of 500 μL 2% H2SO4 in methanol and incubation at 50°C for 2 hours. FAMEs were extracted via addition of 100 μL sat. salt solution and 500 μL hexane. FAMEs were then analyzed using a Select FAME column (100 m × 0.25mm i.d.) installed in an Aglient 7890A GC interfaced with an Agilent 5975C MS. GC oven was held at 80°C for 1 min after injection, increased to 170°C by 20°C/min, increased to 204°C by 1°C/min, and finally increased to 250°C by 20°C/min and held for 10 min.
The % isotopologue distribution of each fatty acid and polar metabolite was determined by integration using in-house MATLAB-based algorithm and corrected for natural abundance using method described in Fernandez et al. (Fernandez et al., 1996).
Plasma D2O enrichment analysis
The2H labeling of water from samples or standards was determined via deuterium acetone exchange (McCabe et al., 2006; Yang et al., 1998). 5 μL of sample or standard was reacted with 4 μL of 10N NaOH and 4 μL of a 5% (v/v) solution of acetone in acetonitrile for 24 hours. Acetone was extracted by the addition of 600 μL chloroform and 0.5 g Na2SO4 followed by vigorous mixing. 100 μL of the chloroform was then transferred to a GC/MS vial. Acetone was measured using an Agilent DB-35MS column (30 m × 0.25mm i.d × 0.25 mm, Agilent J&W Scientific) installed in an Agilent 7890A gas chromatograph (GC) interfaced with an Agilent 5975C mass spectrometer (MS) with the following temperature program: 60°C initial, increase by 20°C/min to 100°C, increase by 50°C/min to 220°C, and hold for 1 min. The split ratio was 40:1 with a helium flow of 1 ml/min. Acetone eluted at approximately 1.5 min. The mass spectrometer was operated in the electron impact mode (70 eV). The mass ions 58 and 59 were integrated and the % M1 (m/z 59) calculated. Known standards were used to generate a standard curve and plasma % enrichment was determined from this. All samples were analyzed in triplicate.
In vivo de novo lipogenesis calculations
Calculation of the fraction of newly synthesized fatty acids (FNS) was based on the method described by Lee et al. (Lee et al., 2000), where FNS is described by the following equation:
Where ME is the average number of deuterium atoms incorporated per molecule (ME = 1 × m1 + 2 × m2 +3 × m3.…), p is the deuterium enrichment in water and n is the maximum number of hydrogen atoms from water incorporated per molecule. N was determined using the equation:
As described by Lee et al. (Lee et al., 1994) where q is the fraction of hydrogen atoms and p + q = 1. The molar amount of newly synthesized fatty acids was determined by:
MNS = FNS x total fatty acid amount (nmoles/mg tissue).
Lipid extraction for lipidomics analysis (LC/MS)
Lipids were extracted using a modified version of the Bligh-Dyer method (Bligh and Dyer, 1959). Briefly, liver samples were manually shaken in a glass vial (VWR) with 1 mL PBS, 1 mL methanol and 2 mL chloroform containing internal standards (13C16-palmitic acid, d7-Cholesterol and SPLASH Lipidomix from Avanti Polar Lipids) for 30 s. The resulting mixture was vortexed for 15 s and centrifuged at 2400 x g for 6 min to induce phase separation. The organic (bottom) layer was retrieved using a Pasteur pipette, dried under a gentle stream of nitrogen, and reconstituted in 2:1 chloroform:methanol for LC/MS analysis.
Lipidomic analysis was performed on a Vanquish HPLC online with a Q-Exactive quadrupole-orbitrap mass spectrometer equipped with an electrospray ion source (Thermo). Data was acquired in positive and negative ionization modes. Solvent A consisted of 95:5 water:methanol, Solvent B was 60:35:5 isopropanol:methanol:water. For positive mode, solvents A and B contained 5 mM ammonium formate with 0.1% formic acid; for negative mode, solvents contained 0.028% ammonium hydroxide. A Bio-Bond (Dikma) C4 column (5 μm, 4.6 mm × 50 mm) was used. The gradient was held at 0% B between 0 and 5 min, raised to 20% B at 5.1 min, increased linearly from 20% to 100% B between 5.1 and 55 min, held at 100% B between 55 min and 63 min, returned to 0% B at 63.1 min, and held at 0% B until 70 min. Flow rate was 0.1 mL/min from 0 to 5 min, 0.4 mL/min between 5.1 min and 55 min, and 0.5 mL/min between 55 min and 70 min. Spray voltage was 3.5 kV and 2.5 kV for positive and negative ionization modes, respectively. Sheath, auxiliary, and sweep gases were 53, 14 and 3, respectively. Capillary temperature was 275°C. Data was collected in full MS/dd-MS2 (top 5). Full MS was acquired from 100–1500 m/z with resolution of 70,000, AGC target of 1×106 and a maximum injection time of 100 ms. MS2 was acquired with resolution of 17,500, a fixed first mass of 50 m/z, AGC target of 1×105 and a maximum injection time of 200 ms. Stepped normalized collision energies were 20, 30 and 40%.
Lipid identification was performed with LipidSearch (Thermo). Data alignment, peak integration and comparison between sample conditions were performed with Dilu (https://github.com/bathyg/dilu). Mass accuracy, chromatography and peak integration of all LipidSearch-identified lipids were verified with Skyline. Skyline and Dilu-generated peak areas were used in data reporting, data were normalized using internal standards.
DAG measurement
For measurement of diacylglycerols (DAG), lipids were similarly extracted from liver samples using the Bligh-Dyer method described above. DAG levels were then quantified using a DAG kinase assay, following the manufacturer’s instructions (Cell Biolabs, Inc., Cat # MET-5028).
QUANTIFICATION AND STATISTICAL ANALYSIS
One-way ANOVA with post hoc tests was used to compare doxycycline-treated L-iAMPKCA mice or samples to control groups. Where indicated in figure legend, two-way ANOVA was used. Statistical analyses are described in each figure and were calculated using GraphPad Prism 7. Data in the figures are presented as mean ± SEM, unless otherwise indicated, with the result of the statistical test, with *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical significance was concluded at p < 0.05.
DATA AND SOFTWARE AVAILABILITY
RNaseq files can be found under GEO accession number GSE122767.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| P-ACCS179 | Cell Signaling Technology | 3661; RRID: AB_330337 |
| ACC | Cell Signaling Technology | 3662; RRID: AB_2219400 |
| P-AMPKαT172 | Cell Signaling Technology | 2535; RRID: AB_331250 |
| AMPKα | Cell Signaling Technology | 2532; RRID, AB_330331 |
| P-RaptorS792 | Cell Signaling Technology | 2083; RRID: AB_2249475 |
| Raptor | Cell Signaling Technology | 2280; RRID: AB_561245 |
| P-ULK1S555 | Cell Signaling Technology | 5869; RRID: AB_10707365 |
| ULK1 | Cell Signaling Technology | 8054; RRID: AB_11178668 |
| P-MFFS146 | Cell Signaling Technology | 49281 |
| MFF | Cell Signaling Technology | 84580; RRID: AB_2728769 |
| GFP | Cell Signaling Technology | 2956; RRID: AB_1196615 |
| LC3b | Cell Signaling Technology | 3868; RRID: AB_2137707 |
| PKCε | Cell Signaling Technology | 2683 |
| Caveolin-1 | Cell Signaling Technology | 3267; RRID: AB_2275453 |
| p62 | Progen | GP62-C; RRID: AB_1542690 |
| Dnmt3a | Santa Cruz Biotechnology | sc-20703; RRID: AB_2093990 |
| TOM20 | Santa Cruz Biotechnology | sc-1145 |
| P-p300S89 | Santa Cruz Biotechnology | sc-130210 |
| p300 | Santa Cruz Biotechnology | sc-585; RRID: AB_2231120 |
| β-actin | Sigma-Aldrich | A5441; RRID: AB_476744 |
| Tubulin | Sigma-Aldrich | T5168; RRID: AB_477579 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Metformin | Sigma | PHR1084 |
| Glucose | Sigma | 7528 |
| Sodium pyruvate | Sigma | P4562 |
| 991 | Glixx Laboratories | GLCX-09267 |
| Doxycycline | Sigma | D9891 |
| Insulin | Lilly | Humulin R |
| Etomoxir | Sigma | E1905 |
| Deuterium oxide | Sigma-Aldrich | 151882 |
| Critical Commercial Assays | ||
| Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | 103015–100 |
| Seahorse XF Palmitate-BSA FAO substrate | Agilent Technologies | 102720–100 |
| Infinity Triglyceride Reagent | Thermo Fisher Scientific | TR22421 |
| Infinity Cholesterol Reagent | Thermo Fisher Scientific | TR13421 |
| Bio-Plex Pro Mouse Diabetes immunoassay kit | Bio-Rad | 171F7010M |
| RNeasy Lipid Tissue Mini Kit | QIAGEN | 74804 |
| SuperScript III First-Strand Synthesis System | Invitrogen | 18080051 |
| TruSeq stranded-mRNA RNA Library Prep Kit v2. | Illumina | RS-122–2001 |
| DAG (Diacylglycerol) Assay Kit | Cell Biolabs, Inc. | MET-5028 |
| Deposited Data | ||
| Raw RNA seq data | This paper deposited in GEO | GEO: GSE122767 |
| Experimental Models: Organisms/Strains | ||
| Mouse: iAMPKCA mice | This paper | n/a |
| Mouse: Albumin-Cre, B6.Cg-Tg(Alb-cre)21Mgn/J | The Jackson Laboratories | 003574 |
| Mouse: C57BL/6J | The Jackson Laboratories | 000664 |
| Oligonucleotides | ||
| Primer: G6Pase Forward: 5’-ACTGTGGGCATCAATCTCCTC-3’ | This paper | n/a |
| Primer: G6Pase Reverse: 5’-CGGGACAGACAGACGTTCAGC-3’ | This paper | n/a |
| Primer: PEPCK Forward: 5’-CTGCATAACGGTCTGGACTTC-3’ | This paper | n/a |
| Primer: PEPCK Reverse: 5’-CAGCAACTGCCCGTACTCC-3’ | This paper | n/a |
| Primer: Cyclophilin Forward:5’-TGGAGAGCACCAAGACAGACA-3’ | This paper | n/a |
| Primer: Cyclophilin Reverse: 5’-TGCCGGAGTCGACAATGAT-3’ | This paper | n/a |
| Recombinant DNA | ||
| pCAGGS-Flpe | Beard et al., 2006 | Addgene plasmid #20733 |
| cTGIL-AMPKα1(312) | This paper | n/a |
| Other | ||
| Chow with doxycycline (50 mg/kg) | Envigo | TD.150877 |
| High fat diet (45% kcal from fat) | Envigo | TD.06415 |
| High fat diet with doxycyclye (87 mg/kg, 45% kcal) | Envigo | TD.160354 |
Highlights.
Creation of a genetically engineered inducible activated allele of AMPKa1, iAMPKCA mice
Hepatic AMPK activation leads to reduced steatosis and inflammation in obese mice
Hepatic AMPK activation blocks white adipose tissue expansion in mice on a high-fat diet
AMPK is a therapeutic target for nonalcoholic fatty liver disease (NAFLD)
ACKNOWLEDGMENTS
This study was supported by grants to R.J.S. from the NIH (R01DK080425, R01CA172229, R35CA220538, and P01CA120964) and The Leona M. and Harry B. Helmsley Charitable Trust (grant 2012-PGMED002). D.G. was supported by a postdoctoral fellowship to the Salk Institute Glenn Center for Aging Research and a postdoctoral fellowship from the American Cancer Society (#123811-PF-13–064-01-TBG). R.J.S. holds the William R. Brody Chair and is a professor in the Molecular and Cell Biology Department at the Salk Institute of Biological Studies. This study was supported by a grant to S.P. (NIH R01 DK115214). A.C. was supported by American Heart Association Career Development Award (18CDA34110292), Philippe Foundation Inc., New York, and a Women in Science Award from the Salk Institute. We thank K. McIntyre and the Salk Institute histology core, which is also supported in part through the Salk Institute CCSG P30 CA014195, for their assistance. This work was supported by the NGS Core Facility of the Salk Institute with funding from the NIH-NCI-CCSG (grant P30 014195), the Chapman Foundation, and the Helmsley Charitable Trust. This work was supported by the Razavi Newman Integrative Genomics and Bioinformatics Core Facility of the Salk Institute with funding from the NIH-NCI-CCSG (grant P30 014195) and the Helmsley Charitable Trust. We thank Lillian J. Eichner for assistance in the analysis of the RNA-seq data.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.036.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Beard C, Hochedlinger K, Plath K, Wutz A, and Jaenisch R (2006). Efficient method to generate single-copy transgenic mice by site-specific integration in embryonic stem cells. Genesis 44, 23–28. [DOI] [PubMed] [Google Scholar]
- Blair E, Redwood C, Ashrafian H, Oliveira M, Broxholme J, Kerr B, Salmon A, Ostman-Smith I, and Watkins H (2001). Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet 10, 1215–1220. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Boudaba N, Marion A, Huet C, Pierre R, Viollet B, and Foretz M (2018). AMPK re-activation suppresses hepatic steatosis but its downregulation does not promote fatty liver development. EBioMedicine 28, 194–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carling D (2017). AMPK signalling in health and disease. Curr. Opin. Cell Biol 45, 31–37. [DOI] [PubMed] [Google Scholar]
- Chen K, Liu J, and Cao X (2017). Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J. Autoimmun 83, 1–11. [DOI] [PubMed] [Google Scholar]
- Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjøbsted R, Jørgensen NO, Tran J-L, Jatkar A, Cialdea K, Esquejo RM, et al. (2017). Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 25, 1147–1159.e10. [DOI] [PubMed] [Google Scholar]
- Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M, Dickinson R, Adler A, Gagne G, Iyengar R, et al. (2006). Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 3, 403–416. [DOI] [PubMed] [Google Scholar]
- Crute BE, Seefeld K, Gamble J, Kemp BE, and Witters LA (1998). Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem 273, 35347–35354. [DOI] [PubMed] [Google Scholar]
- Day EA, Ford RJ, and Steinberg GR (2017). AMPK as a therapeutic target for treating metabolic diseases. Trends Endocrinol. Metab 28, 545–560. [DOI] [PubMed] [Google Scholar]
- Dow LE, Premsrirut PK, Zuber J, Fellmann C, McJunkin K, Miething C, Park Y, Dickins RA, Hannon GJ, and Lowe SW (2012). A pipeline for the generation of shRNA transgenic mice. Nat. Protoc 7, 374–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow LE, Nasr Z, Saborowski M, Ebbesen SH, Manchado E, Tasdemir N, Lee T, Pelletier J, and Lowe SW (2014). Conditional reverse tet-trans-activator mouse strains for the efficient induction of TRE-regulated transgenes in mice. PLoS ONE 9, e95236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duca FA, Côté CD, Rasmussen BA, Zadeh-Tahmasebi M, Rutter GA, Filippi BM, and Lam TKT (2015). Metformin activates a duodenal Ampk-dependent pathway to lower hepatic glucose production in rats. Nat. Med 21, 506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D, Kim J, Shaw RJ, and Guan K-L (2011). The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichner LJ, Brun SN, Herzig S, Young NP, Curtis SD, Shackelford DB, Shokhirev MN, Leblanc M, Vera LI, Hutchins A, et al. (2018). Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab Published online November 5, 2018 10.1016/j.cmet.2018.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esquejo RM, Salatto CT, Delmore J, Albuquerque B, Reyes A, Shi Y, Moccia R, Cokorinos E, Peloquin M, Monetti M, et al. (2018). Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models. EBioMedicine 31, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez CA, Des Rosiers C, Previs SF, David F, and Brunengraber H (1996). Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J. Mass Spectrom. 31, 255–262. [DOI] [PubMed] [Google Scholar]
- Fullerton MD, Galic S, Marcinko K, Sikkema S, Pulinilkunnil T, Chen Z-P, O’Neill HM, Ford RJ, Palanivel R, O’Brien M, et al. (2013). Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med 19, 1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullerton MD, Ford RJ, McGregor CP, LeBlond ND, Snider SA, Stypa SA, Day EA, Lhoták Š, Schertzer JD, Austin RC, et al. (2015). Salicylate improves macrophage cholesterol homeostasis via activation of Ampk. J. Lipid Res 56, 1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia D, and Shaw RJ (2017). AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2013). AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes 62, 2164–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, and Hawley SA (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol 13, 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushik S, and Cuervo AM (2016). AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy 12, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C-W, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, Burgess SC, Li C, Ruddy M, Chakravarthy M, et al. (2017). Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 26, 394–406.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan T, Kisseleva T, and Brenner DA (2015). Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation. PLoS ONE 10, e0129743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WN, Bassilian S, Ajie HO, Schoeller DA, Edmond J, Bergner EA, and Byerley LO (1994). In vivo measurement of fatty acids and cholesterol synthesis using D2O and mass isotopomer analysis. Am. J. Physiol 266, E699–E708. [DOI] [PubMed] [Google Scholar]
- Lee WN, Bassilian S, Lim S, and Boros LG (2000). Loss of regulation of lipogenesis in the Zucker diabetic (ZDF) rat. Am. J. Physiol. Endocrinol. Metab 279, E425–E432. [DOI] [PubMed] [Google Scholar]
- McCabe BJ, Bederman IR, Croniger C, Millward C, Norment C, and Previs SF (2006). Reproducibility of gas chromatography-mass spectrometry measurements of 2H labeling of water: application for measuring body composition in mice. Anal. Biochem 350, 171–176. [DOI] [PubMed] [Google Scholar]
- Meex RCR, and Watt MJ (2017). Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat. Rev. Endocrinol 13, 509–520. [DOI] [PubMed] [Google Scholar]
- Meex RC, Hoy AJ, Morris A, Brown RD, Lo JCY, Burke M, Goode RJA, Kingwell BA, Kraakman MJ, Febbraio MA, et al. (2015). Fetuin B Is a Secreted Hepatocyte Factor Linking Steatosis to Impaired Glucose Metabolism. Cell Metab 22, 1078–1089. [DOI] [PubMed] [Google Scholar]
- Mills EL, and O’Neill LA (2016). Reprogramming mitochondrial metabolism in macrophages as an anti-inflammatory signal. Eur. J. Immunol 46, 13–21. [DOI] [PubMed] [Google Scholar]
- Mounier R, Théret M, Arnold L, Cuvellier S, Bultot L, Göransson O, Sanz N, Ferry A, Sakamoto K, Foretz M, et al. (2013). AMPKa1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 18, 251–264. [DOI] [PubMed] [Google Scholar]
- Myers RW, Guan H-P, Ehrhart J, Petrov A, Prahalada S, Tozzo E, Yang X, Kurtz MM, Trujillo M, Gonzalez Trotter D, et al. (2017). Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357, 507–511. [DOI] [PubMed] [Google Scholar]
- Petersen MC, Madiraju AK, Gassaway BM, Marcel M, Nasiri AR, Butrico G, Marcucci MJ, Zhang D, Abulizi A, Zhang XM, et al. (2016). Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J. Clin. Invest 126, 4361–4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G, Hardie DG, and Pearson ER (2017). The mechanisms of action of metformin. Diabetologia 60, 1577–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2018). Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 27, 22–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steneberg P, Lindahl E, Dahl U, Lidh E, Straseviciene J, Backlund F, Kjellkvist E, Berggren E, Lundberg I, Bergqvist I, et al. (2018). PANAMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 3, S157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilg H, Moschen AR, and Roden M (2017). NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol 14, 32–42. [DOI] [PubMed] [Google Scholar]
- Toyama EQ, Herzig S, Courchet J, Lewis TL Jr., Losón OC, Hellberg K, Young NP, Chen H, Polleux F, Chan DC, and Shaw RJ (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351, 275–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida T, and Friedman SL (2017). Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol 14, 397–411. [DOI] [PubMed] [Google Scholar]
- Woods A, Williams JR, Muckett PJ, Mayer FV, Liljevald M, Bohlooly-Y M, and Carling D (2017). Liver-specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep 18, 3043–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, et al. (2013). Structural basis of AMPK regulation by small molecule activators. Nat. Commun 4, 3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Diraison F, Beylot M, Brunengraber DZ, Samols MA, Anderson VE, and Brunengraber H (1998). Assay of low deuterium enrichment of water by isotopic exchange with [U-13C]acetone and gas chromatography-mass spectrometry. Anal. Biochem 258, 315–321. [DOI] [PubMed] [Google Scholar]
- Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, and Leff T (2001). Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J. Biol. Chem 276, 38341–38344. [DOI] [PubMed] [Google Scholar]
- Yang X, Mudgett J, Bou-About G, Champy M-F, Jacobs H, Monassier L, Pavlovic G, Sorg T, Herault Y, Petit-Demoulière B, et al. (2016). Phys iological expression of AMPKγ2RG mutation causes Wolff-Parkinson-White syndrome and induces kidney injury in mice. J. Biol. Chem 291, 23428–23439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavari A, Stocker CJ, Ghaffari S, Wargent ET, Steeples V, Czibik G, Pinter K, Bellahcene M, Woods A, Martínez de Morentin PB, et al. (2016). Chronic activation of γ2 AMPK induces obesity and reduces β cell function. Cell Metab 23, 821–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z, Anstee QM, Marietti M, Hardy T, Henry L, Eslam M, George J, and Bugianesi E (2018). Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol 15, 11–20. [DOI] [PubMed] [Google Scholar]
- Zhang R (2016). The ANGPTL3–4–8 model, a molecular mechanism for triglyceride trafficking. Open Biol. 6, 150272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNaseq files can be found under GEO accession number GSE122767.