

Abstract
Next Generation Sequencing allows the characterization of the Adaptive Immune Receptor Repertoire (AIRR) in exquisite detail. These large-scale AIRR-seq data sets have rapidly become critical to vaccine development, understanding the immune response in autoimmune and infectious disease, and monitoring novel therapeutics against cancer. However, at present there is no easy way to compare these AIRR-seq data sets across studies and institutions. The ability to combine and compare information for different disease conditions will greatly enhance the value of AIRR-seq data for improving biomedical research and patient care.
The iReceptor Data Integration Platform (gateway.ireceptor.org) provides one implementation of the AIRR Data Commons envisioned by the AIRR Community (airr-community.org), an initiative that is developing protocols to facilitate sharing and comparing AIRR-seq data. The iReceptor Scientific Gateway links distributed (federated) AIRR-seq repositories, allowing sequence searches or metadata queries across multiple studies at multiple institutions, returning sets of sequences fulfilling specific criteria. We present a review of the development of iReceptor, and how it fits in with the general trend toward sharing genomic and health data, and the development of standards for describing and reporting AIRR-seq data. Researchers interested in integrating their repositories of AIRR-seq data into the iReceptor Platform are invited to contact ireceptor-help@sfu.ca.
Keywords: immune repertoires, vaccines, therapeutic antibodies, cancer immunotherapy, distributed data federation, data sharing
1. INTRODUCTION
The integration of large-scale genomic data with extensive health data is revolutionizing biomedical research and holds great potential for improving patient care. However, our ability to share these large-scale data across studies and institutions is limited. Facilitating sharing these data across studies will greatly increase sample sizes, strengthening our statistical inferences, and will be vitally important to searching for the patterns that underlie personalized medicine approaches, as we try to develop specific therapies based on an individual’s genotype, personal exposure history, and clinical response. Goodhand (1) has argued that one efficient way to facilitate sharing data across studies and institutions is by establishing federated systems of data repositories. The iReceptor Data Integration Platform takes this distributed approach and applies it to the domain of next generation sequencing (NGS) of antibody/B-cell and T-cell receptor repertoires. This review covers the development of the iReceptor Data Integration Platform, an implementation of a data commons for Adaptive Immune Receptor Repertoire (AIRR)-seq data, guided by the principles set out by the AIRR Community (airr-community.org; (2)). In this debut paper, we discuss the history and philosophy of iReceptor, the present status and future goals of the iReceptor Platform, and some of the challenges to attaining these goals through a federated system of repositories. We then present the results of two use cases to show the power of data integration across studies and repositories. Finally, we invite researchers who are producing AIRR-seq data to join the iReceptor network to facilitate sharing of their data.
2. AIRR-SEQ DATA: CHALLENGES AND COMMUNITY RESPONSE
The adaptive immune system has evolved a unique molecular diversification mechanism designed to produce a highly diverse set of antigen receptors. This diverse set of antibody/B-cell and T-cell receptors is necessary to recognize and remove the vast and ever-changing array of pathogens that an individual will encounter over a lifetime, while differentiating these pathogens from self. This unique genetic mechanism, and the sheer immensity of the Antibody/B-cell and T-cell response, presents challenges for producing, storing, sharing and analyzing these data.
The unique mechanism involves recombining sets of V-, D-, and J-genes that encode these receptors, along with the introduction of variability at the joints between these recombined gene segments (3). As a result of this recombination process, the random pairing of Ig heavy and light B-cell receptor (BCR) chains (or paired T-cell receptor (TCR) chains), and somatic hypermutation (which is unique to B-cell receptors (4)), the diversity of the adaptive immune receptor repertoire greatly exceeds the coding capacity of the genome. For example, it is estimated that humans express a hundred million or more unique B-cell and T-cell receptors (5)(6) (7). It was in 2009 that NGS approaches were first used to characterize this Adaptive Immune Receptor Repertoire in exquisite detail, producing 106 or 107 sequences, for multiple time points, per sample (AIRR-seq data). These data sets have grown quickly in size and number, and exist in multiple repositories across labs, studies and institutions.
Not only do these AIRR-seq data sets often comprise many millions of sequences per sample, they also require extensive analysis or “processing” after sequencing and prior to being interpreted. Such analyses are performed in a sequential series of steps or “data analysis pipelines” that vary between investigators. A typical data analysis pipeline begins with raw reads (often in the form of FASTQ sequences) produced by NGS sequencing technology. Low-quality sequences are removed from these “base-call” data, which is often accomplished with arbitrary cut-offs. “Paired-end” reads are merged into a single sequence to obtain “full reads”, often with seemingly arbitrary rules for excluding short sequences and imprecise merges. Different algorithms are then used for assigning Variable (V-), Diversity (D-) and Joining (J-) gene segment usage and for assigning somatic mutations in the case of antibody/B-cell sequences (reviewed in (8)(9)). Furthermore, several very different approaches can be used to identify and characterize clonal lineages (each clonal lineage being the set of descendants of a given “ancestral” B- or T-cell produced during the development of an individual). For example, clones can be recognized on the basis of shared V- and J- gene usage, and CDR3 length and diversity (CDR3 is a specific region in an immune receptor, often important in binding to pathogens and other molecules) (10). Alternatively, phylogenetic, hierarchical clustering or probabilistic approaches can be used to assign sequences to clones (11)(12)(13). All this information describing the processing of the sequences, and the results of this processing, must be stored along with the sequence data.
In summary, these AIRR-seq data require unique annotation tools, specialized database models for storing annotated data, and idiosyncratic ways of defining and tracking clonal relationships, in addition to requiring massive storage. As these datasets grow in size and number, they also grow in importance for research in infectious diseases and vaccine development, development of therapies for autoimmune diseases, and in novel cancer immunotherapy approaches, among other applications (see section 2.1). These challenges haven’t been fully addressed, but in section 2.2 we describe an initiative to adopt community protocols and standards to facilitate sharing and integrating these immense AIRR-seq data sets, and starting in Section 3 we describe a federated database approach to integrating these data, as implemented in the iReceptor Data Integration Platform.
2.1. Recent applications of AIRR-seq data
Since 2009, AIRR-seq data have been applied to a broad range of biomedical questions, including autoimmune diseases (e.g., (14), infectious diseases and vaccine development, particularly HIV (e.g., (15) (16) and flu (e.g. (17)), and cancer immunotherapy ((18) (19). Here we review some of the latest applications of this type of data, emphasizing studies that would benefit from performing analyses across federated repositories covering many studies, labs and institutions.
One of the latest breakthroughs based on these highly diverse AIRR-seq data is a novel way to diagnose infectious diseases. As a proof of principle, Emerson et al. (20) showed that NGS sequencing of the T-cell receptor (TCRβ) repertoire could be used to distinguish individuals who were infected with CMV from those who were not. They employed a training set of 666 subjects and a validation cohort of 120 subjects infected with CMV, all compared to 640 CMV negative subjects. Their method depended on identifying more than 164 CMV-associated TCRβ sequences in a majority of individuals (“public” T-cell receptor sequences associated with CMV infection). Once these were identified, then by sequencing ~200K sequences per individual, and comparing the frequency of CMV-associated versus private TCRβ sequences in each individual, they were able to correctly identify CMV+ individuals with 93% accuracy. This demonstration that the TCRβ repertoire is shaped by exposure, and thus can be used to identify whether an individual is infected with a specific pathogen, will become increasingly important as a diagnostic tool for diseases more difficult to identify. However, the sample sizes needed to differentiate signal from noise for this application to other diseases will necessitate integrating large data sets across multiple institutions, labs.
AIRR-seq data are also being used in multiple ways to investigate the causes and possible treatments of autoimmune diseases. One recent example is a study of celiac disease by Gunnarsen et al. (21), showing how these data can be used to answer long-standing questions in autoimmunity. Celiac disease (CD) occurs primarily in individuals with particular alleles at the HLA loci, a set of genes that are often associated with infectious and autoimmune disease susceptibility. Individuals with these susceptibility alleles for CD at the HLA loci are characterized by a T-cell response against very specific binding motifs in the gluten molecule. Analysis of T-cell receptor repertoires sequenced from the blood of 9 celiac disease patients after gluten challenge identified 536 unique T-cell receptor clonotypes, which were further studied for the specific molecular interactions promoting the disease. Based on these results, the authors were able to show how these molecular interactions recruited a pathogenic TCR repertoire in celiac disease, leading to one explanation why TCR-biased repertoires are so frequently seen in HLA-associated diseases. Examining these patterns in more autoimmune diseases, using larger sample sizes, could determine the generality of this approach.
One of the most important applications of AIRR-seq data is to the field of anti-cancer immunotherapy. Malignant cancers are characterized by multiple mutations compared to normal tissue, and thus the adaptive immune system should recognize malignant tissue and remove it. However, many tumors release checkpoint molecules that suppress this immune response, thus allowing tumors to grow uncontrollably. Immunotherapy has become a key weapon against cancer, through the development of “anti-checkpoint therapies” that suppress these checkpoint molecules, thus releasing the immune system against the malignant tumors. This approach has seen some amazing successes; for example, anti-checkpoint immunotherapy has raised the 3-year overall survival for advanced melanoma from 12% before 2010 to ~60% in 2017(22). However, in other types of cancer, as few as 10% of patients treated respond to the immunotherapy, leading to an intense search for biomarkers that could differentiate responders from non-responders (one of the priority questions in personalized medicine). Many studies are examining whether there are AIRR-seq data signals that can make this determination, and this effort is one of the main drivers of the explosion of studies sequencing B-cell and T-cell repertoires in multiple cancers.
One recent example of using TCR repertoires to monitor and predict response to anti-checkpoint therapy is provided by Snyder et al. (23), who studied urothelial cancer patients treated with atezolizumab. These researchers concentrated on the clonality of the TCRβ repertoire, the set of sequences making up one of the two chains in a T-cell receptor. High clonality would mean a repertoire was less diverse, in that many of the sequences in the repertoire would consist of only a few dominant clones. They observed that high pretreatment peripheral blood TCRβ clonality was strongly associated with poor clinical outcomes; this suggests that clonality could be a biomarker, possibly capable of stratifying patients into potential good and poor responders in personalized cancer immunotherapy. As emphasized by the authors (23), the sample sizes in this study were small; only 19 individuals were available who had been treated with the monoclonal antibody. The authors state: “The patients under study were treated at a single institution and represent a small subset of the overall study, limiting statistical power.” This clearly shows the necessity of the iReceptor approach, which would allow these researchers to easily compare their results and patterns with other individuals being treated with anti-checkpoint therapies.
As exciting as the anti-checkpoint therapy approach is, there is at least one challenge to its general application, and that is that these anti-checkpoint drugs may also induce inflammatory responses and toxicities termed immune-related adverse events (irAEs) in a significant proportion of patients. These irAEs are often low grade and manageable, but severe irAEs may lead to prolonged hospitalizations or fatalities. It is only very recently that patterns in the AIRR-seq repertoire data have been examined in an attempt to predict who may or may not exhibit such responses. For example, Subudhi et al. (24) studied metastatic prostate cancer patients undergoing androgen deprivation therapy plus ipilimumab, an anti-checkpoint monoclonal antibody. They showed that the number of expanded clones (leading to lower diversity) in the CD8 T-cell repertoire strongly correlated with the probability of suffering adverse effects. Unfortunately, these results are nearly the opposite of those of Oh et al. (25) who also studied metastatic prostate cancer patients treated with ipilimumab. In the Oh et al. study, irAE patients exhibited a greater diversity of sequences in the T-cell repertoire. This points out that it is essential that the results of such studies be compared across diseases, treatments, and institutions, so that we can look for consistent biomarker patterns in the T-cell (and B-cell) repertoires, predicting clinical outcomes such as immune adverse effects.
2.2. The AIRR Community
The AIRR Community (airr-community.org) was established in May 2015 to address the challenges of effectively sharing AIRR-seq data (2). The first AIRR Meeting was organized by Felix Breden, Jamie Scott, and Thomas Kepler in Vancouver, BC, Canada. Membership in the AIRR Community is open and is intended to cover all aspects of AIRR-seq technology and its uses. AIRR-Community members include researchers expert in the generation of AIRR-seq data; statisticians and bioinformaticians versed in their analysis; informaticians and data security experts experienced in their management; basic scientists and physicians who turn to such data for critical insights; and experts in the ethical, legal, and policy implications of sharing AIRR-seq data. The AIRR Community meets as a community annually; the most recent meeting convened in December 2017 at the NIH’s Fishers Lane facility.
At the 2015 AIRR Community Meeting, Working Groups were formed to address three priority areas: (1) Minimal Standards for the publication and sharing of AIRR-seq datasets (26); (2) Tools and Resources to facilitate the comparison of AIRR-seq datasets; and (3) a Common Repository to establish an AIRR Data Commons. These Working Groups are dynamic and often collaborate with each other, as methods evolve and applications of standards in one area (for example, metadata standards) impact other areas (data repository requirements).
At the 2017 AIRR Community Meeting, these working groups were expanded, with the following groups established and actively meeting on a regular basis:
Minimal Standards - developing a set of metadata standards for the publication and sharing of AIRR-seq datasets
Common Repository - establishing the requirements for repositories that will store AIRR-seq data - an AIRR Data Commons
Data Representation - developing standard file formats and schemas to represent annotated antibody/B-cell and T-cell receptor sequences allowing interoperability between analysis pipelines from different developers
Germline Database - promoting the comprehensive and accurate identification, description, classification, annotation, curation, and consistent use of germline IG (immunoglobulin) and TR (T-cell receptor) genes and alleles across species, strains, and populations
Software Standards - developing a list of standard datasets with which software tools can be tested and compared. This will include both real and simulated data with a variety of characteristics matched to potential applications
Biological Standards - recommending a set of biological standards that can be used for normalization of data sets allowing more direct comparison of data generated by different library preparation methods
The iReceptor team is very involved in the AIRR Community, with iReceptor PI Breden the current Chair of the AIRR Community Executive. The broader iReceptor technical team helps drive several of the AIRR Working Groups. Of particular relevance to iReceptor is the work of the AIRR Common Repository Working Group and the Community’s goal of establishing an AIRR-seq data commons.
3. IRECEPTOR - TOWARDS AN AIRR-SEQ DATA COMMONS
3.1. What is iReceptor?
iReceptor is a distributed data management system and scientific gateway for mining “Next Generation” sequence data from immune responses. The main goal of iReceptor is to provide a web-based platform that will lower the barrier to immune genetics researchers who need to federate large, distributed, AIRR-seq data repositories to answer complex questions about the immune response. At the most basic level, iReceptor consists of two key components: a distributed network of data repositories (an AIRR-seq data commons) and a web based Scientific Gateway that allows researchers to discover, federate, explore, and analyze AIRR-seq data of interest across that network of data repositories (Figure 1).
Figure 1:
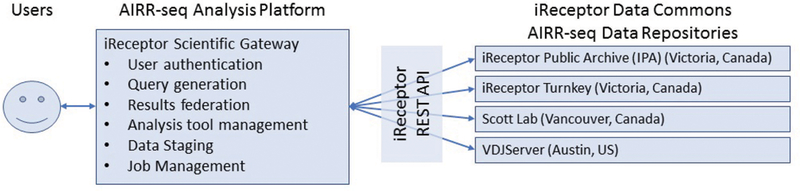
The iReceptor architecture showing a user interacting with the Scientific Gateway, which directs queries to the AIRR-seq data repositories
3.2. Why an AIRR Data Commons and why be FAIR?
Both the iReceptor Data Integration Platform and the AIRR Community are dedicated to following open access principles. The modern Open Science/Open Access movement (27) has been gaining momentum over the past 15 years as researchers embrace the opportunities presented by broad digital access to both research results and research data. In its 2007 report “OECD Principles and Guidelines for Access to Research Data from Public Funding” (28) the OECD states that “Databases are rapidly becoming an essential part of the infrastructure of the global science system” and cites the following benefits of open access to data:
Reinforces open scientific inquiry;
Encourages diversity of analysis and opinion;
Promotes new research;
Makes possible the testing of new or alternative hypotheses and methods of analysis;
Supports studies on data collection methods and measurement;
Facilitates the education of new researchers;
Enables the exploration of topics not envisioned by the initial investigators; Permits the creation of new data sets when data from multiple sources are combined.
In 2014, a group of stakeholders identified the need to improve the infrastructure for supporting the sharing and reuse of research data, with an emphasis on enhancing the ability to use technology, in particular through third parties, to find and reuse those data. Over the next year, this group defined what are known as the “FAIR” data management principles (29). Today, it is widely accepted that for research outputs to be truly globally useful, it is necessary for data to be Findable, Accessible, Interoperable, and Reusable (FAIR).
The importance of data repositories that are collectively owned and managed by a community of users (a data commons), and in particular data sharing across such repositories, is demonstrated through the funding that agencies are dedicating to this area, especially in the life sciences and biomedical research. In 2013 the US National Institutes of Health (NIH) established the Big Data to Knowledge (BD2K) program “... to support the research and development of innovative and transformative approaches and tools to maximize and accelerate the integration of big data and data science into biomedical research” (30). The first phase of the program ($200M USD 2014–2017) focused on fostering use, developing analysis methods and tools, and establishing Centers of Excellence. The second phase (expected to be $95.5M USD 2017–2020) (31) will focus on making the products from Phase 1 usable, discoverable, and disseminated to the user community through the creation of a set of data common pilot projects. The NIH defines a data commons as “... a shared virtual space where scientists can work with the digital objects of biomedical research such as data and analytical tools.” (31). This definition is important, because it stresses not only being able to find and federate data, but also the ability to apply advanced analytical tools to those federated data.
In parallel with the NIH Big Data to Knowledge initiative, the Global Alliance for Genomics and Health (GA4GH) (32) emerged as an international organization that is taking on an important role in setting policy and technical standards to enable the effective sharing of genomic data. Established in 2013, GA4GH has an established set of work streams focusing on developing best practices and technical standards around areas such as data security, regulatory and ethics, data discovery, genomics knowledge standards, and large-scale genomics. These work streams are driven by GA4GH Driver Projects, which are active genomics data initiatives that drive the policy and standards work of the organization. The GA4GH projects span a wide range of data initiatives, many of which can be considered data commons.
Although the technologies with which groups build data commons continues to evolve, and therefore different groups and communities take different approaches to implementing data commons, four basic structural models have emerged, varying in their level of centralization (33) These structural models are:
Fully centralized, where all data are integrated into a single repository (e.g. The Cancer Data Commons - (34))
Intermediate distributed, where repositories are distributed and separately maintained but are connected by a central resource and/or share technical service components such as a registry. Such systems have been emerging for some time, in particular in the Life Sciences, with systems like the GA4GH’s Beacon system for sharing genetic variant data and the Match Maker Exchange (1) as well as the BioMart platform for data federation (35) and the tools built on top of it such as the International Cancer Genome Consortium (ICGC) and its ICGC “franchise” data sets (36). This category encompasses iReceptor and the iReceptor Data Commons
Fully distributed, where repositories are distributed, not technically integrated, but share a common legal or policy framework
Noncommons, where repositories lack technical and legal interoperability and at most, might share a common index
3.3. Why distributed data repositories for AIRR-seq data?
Given the uptake, and the success, of projects that use the intermediate distributed data model (as discussed above), in particular by organizations such as GA4GH and the ICGC, iReceptor has chosen to use a distributed data model for its AIRR-seq data commons. A distributed data model, although difficult to support, is we believe critical to the success of research in this area for two main reasons.
Firstly, next generation sequencing has caused an explosion in the data available to labs that are carrying out research on AIRR-seq data. To answer complex research questions, these labs need to collaborate in a variety of ways. Although large-scale repositories for sequence data exist, it is our belief that it is not practical to provide a central repository at the scale that will be required by the AIRR research community. We know this from our own experience where we have seen the challenges of trying to effectively query even a small lab’s data. We also know that this is only the first challenge, as a central repository would need to provide similar resources for hundreds of groups nationally and internationally. Alternatively, a distributed data model, although it doesn’t exclude some large, public repositories in the network, means that it is possible for each lab to store and manage its own data if they so choose. This provides a mechanism for groups to “scale-up” individual repositories to handle large amounts of data while at the same time providing the ability for the community to “scale-out” by federating tens, if not hundreds of repositories. If interfaces to these repositories can be defined consistently and adhering to community standards, then tools can access the distributed repositories to perform complex analyses which in turn allow complex research questions (through queries that federate data from distributed repositories) to be answered.
Secondly, AIRR-seq data are mostly patient data, and therefore need to be treated with confidentiality and security. Data use typically goes through institutional ethics boards, requiring data stewards at given institutions to be confident that data are treated securely. A distributed data model enables a data steward to store, monitor, and share data as appropriate to the study’s ethics and sharing agreements while at the same time having explicit and direct control over who has access to those data.
The goal of iReceptor is to hide the technical complexities of the above problems, while at the same time empowering AIRR-seq researchers to perform very sophisticated (and in many cases, computationally expensive) analyses on federated data from multiple, distributed repositories.
3.4. The Past - iReceptor v1.0
The evolution of iReceptor followed a very similar timeline to that of both the NIH Big Data to Knowledge and the GA4GH Initiative. The original concept of iReceptor emerged from discussions at Simon Fraser University between the iReceptor Principal Investigators Felix Breden, Jamie Scott, and Brian Corrie. These discussions focused on the challenges researchers faced in sharing and analyzing AIRR-seq data between research labs, at first, when determining the characteristics of broadly-neutralizing anti-HIV antibodies (37). Based on finding solutions to these challenges, the original iReceptor proposal was funded in 2014 by the CANARIE Network Enabled Platform (NEP) program. CANARIE is Canada’s national advanced network provider and has been providing Research Software funding since 2007. The CANARIE NEP program’s goal is to foster “... the development of software tools that accelerate discovery by simplifying access to digital infrastructure” (38). The CANARIE NEP program funded the development of version 1.0 (v1.0), including the initial version of the iReceptor Scientific Gateway, as well as a small number of AIRR-seq data repositories.
One of the key challenges in sharing data is having a mechanism for exchanging data in a rigorous and reproducible way. In May 2014, the iReceptor team collaborated with the international ImMunoGeneTics information system (IMGT) team, led by Marie-Paule Lefranc at the Centre National de la Recherche Scientifique/Universite de Montpellier in Montpellier, France to define a set of baseline metadata and sequence annotation data that were considered fundamental for federated data analysis. Based on this initial metadata definition, the iReceptor team created a relational data repository model (using MySQL) that represented key data elements (study, subject, sample, sequence, and sequence annotation, etc.) and their relationships. In iReceptor v1.0, three data repositories were created: one a central, open repository (the iReceptor Public Archive or IPA), one a lab-specific data repository curated and managed by the Scott Lab at Simon Fraser University, and a second, open repository that was tightly coupled to the Advanced Research Computing platforms provided by Compute Canada (the national Advanced Research Computing infrastructure provider in Canada). These three repositories comprised the original iReceptor AIRR-seq data repository network.
To create an abstraction layer between the data repository implementation (the actual database technology used) and the tools that would utilize and query those repositories, iReceptor v1.0 also implemented a web-based application programing interface (REST API) for querying and returning data from these AIRR-seq data repositories. iReceptor v1.0 also created an iReceptor Repository service that translated queries/requests received through the REST API into queries on the actual repository that return the correct data. Assuming all data repositories in the network have iReceptor compliant data in them, the combination of the iReceptor API and the iReceptor Repository Service provides a mechanism for external tools to query the network of AIRR-seq repositories using a single, consistent API, federate the results of those queries, and perform complex analyses on those federated data to answer complex research questions.
The iReceptor REST API defines a set of queries, and as a result, each iReceptor Repository Service must implement those queries. In iReceptor v1.0, three query levels were supported: queries at the metadata summary level (e.g. return summaries of the types of data your repository contains), the biological sample level (e.g. return all samples that satisfy certain criteria, such as all data that are from female subjects associated with cancer studies), and the annotated sequence level (e.g. return all sequences that contain the annotated V-gene allele IGLV3–1*01). Each query type is implemented through a web service through a web query of the form:
The iReceptor Scientific Gateway provides a web based graphical user interface (GUI) that allows researchers to pose queries of this form and makes use of the iReceptor REST API to query and federate data from the iReceptor repository network. The iReceptor Gateway then either presents the user with summary statistics of federated results of the query, allows the user to download the federated data, or pass the data to an analysis application for federated data analysis.
One of the key goals of iReceptor is to make it easy for researchers to manage, publish, and share their data. In 2014, to seed the iReceptor Data Commons, the team began to populate the iReceptor IPA repository with curated data sets that were judged to have value to the general AIRR-seq community. At the end of 2015 when the CANARIE funding ended, the IPA repository had approximately 137 Million annotated sequences from ~290 biological samples and 11 studies across the three repositories available to the general AIRR-seq research community. Although in iReceptor v1.0 it was possible to interactively explore the approximately 290 biological samples and their relevant study and subject metadata (search across characteristics of the study, subject, and sample), due to the scale of the sequence data (with between 3,000 and 13,000,000 sequences/sample) it was not possible to interactively explore the sequence and sequence annotation data in iReceptor v1.0.
With the focus of iReceptor on sharing of data, it is critical that we don’t overlook the fact that AIRR-seq data are often human health data, and therefore data security and data provenance need to be considered. Recall from above that one of the key benefits of a distributed data model is that a local data repository allows a data steward to store, monitor, and share data in accordance with the associated ethics and sharing agreements. Although only implemented at a very basic level, the iReceptor Repository Service and iReceptor API provide a security layer that can either be turned on (for repositories that require security) or off (for repositories that provide open access to public data). It is the responsibility of the data steward operating the repository to ensure that the data in the repository are protected with the correct level of data security according to the study’s ethics and sharing constraints.
The iReceptor v1.0 security layer relies on a trust relationship between the iReceptor Repository Service and the iReceptor Gateway. When a Repository Service registers with the iReceptor Gateway, it provides a shared secret to the Gateway (a password for authentication to the service). The Repository Service allows access to the repository based on user level authorization. On every connection to the Repository Service, the Gateway provides the Repository Service with the shared secret and the Gateway user name of the user requesting access. The Repository Service authenticates the Gateway using the shared secret, ensuring the client is trusted. Once authenticated, the service then maps the gateway user requesting access to a local user and determines if that user is authorized to access the repository. If so, the Repository Service responds to the request. If not, the service responds with an unauthorized access response. All data are communicated over an SSL encrypted communication channel.
It is important to point out the limitations of this security model. The only authentication that occurs is between the iReceptor Gateway and the iReceptor Repository Service. This establishes the trust relationship. The iReceptor Repository Service must manage the mapping of iReceptor Gateway users to users that are authorized to access the local repository. There is currently no locally supported authentication at the repository level, requiring the repository to trust that when the Gateway says that John Doe wants access that the Gateway is really requesting that access on behalf of John Doe. In addition, there is currently no fine-grained authorization providing different levels of user access to different parts of the repository. Although these are on the current roadmap for implementation within the iReceptor framework, only the authentication and authorization described above are currently supported.
Although one of the deliverables of the iReceptor v1.0 proposal was to provide the ability for research groups to download and install the software that implemented the iReceptor data repository (MySQL data model), the iReceptor Repository Service, and the iReceptor REST API, it was recognized early in development that many research groups will have invested in their own data repository technologies and their own data collections. In these instances, it is not practical for such a lab to change repository technologies. Instead, the iReceptor model provides a mechanism for a lab with an existing repository to join the iReceptor AIRR-seq network. For a repository to “join” the iReceptor AIRR-seq network, the repository owner needs to 1) translate their internal data representation of the repository into the iReceptor standard metadata fields, 2) provide an implementation of a data service that implements the iReceptor API queries, and 3) return the iReceptor data fields in an iReceptor API compliant format.
As a repository maintainer, taking the above steps to integrate your repository into the iReceptor Repository Network may seem like a daunting task. To demonstrate the feasibility of such an integration, the iReceptor team collaborated with Dr. Lindsay Cowell and the VDJServer team at the University of Texas Southwestern Medical School to integrate VDJServer’s repository into the iReceptor Repository Network. VDJServer (39) is a web based platform that allows users to upload and process AIRR-seq data using a range of analysis pipelines. As part of iReceptor v1.0, the iReceptor and VDJServer teams developed an iReceptor Repository Service that acted as a proxy for the VDJServer repository and produced iReceptor API compliant data. In this fashion, the iReceptor Gateway was able to not only query the three iReceptor repositories but was also able to query the VDJServer repository (through this proxy). Although just a proof of concept, the ability to hide the data repository implementation (by implementing a service that accepted standard iReceptor queries) while at the same time searching and returning the relevant data in a standard compliant manner (through the iReceptor API) demonstrated the power of the distributed, federated, approach that iReceptor takes to linking disparate AIRR-seq data repositories into a single iReceptor Data Commons.
3.5. The Present - iReceptor v2.0
In 2016, iReceptor received funding to continue the development of the iReceptor Data Integration Platform from the Canada Foundation for Innovation (CFI) Cyberinfrastructure Challenge 1 program. The goal of the iReceptor CFI project, entitled “Immune Receptor Repertoire Data Commons for Personalized Immunotherapy”, is the creation of an international AIRR-seq data commons, a platform that integrates AIRR-seq data sets by combining: 1) a large, scalable, controlled access repository of AIRR-seq data (the iReceptor IPA); 2) an international network of AIRR-seq data repositories; 3) the ability to federate AIRR-seq data across these distributed repositories; 4) the ability to perform advanced analyses on these federated AIRR-seq data; and 5) a scientific gateway, or web portal, that hides the complexity of performing research queries and analyses using this advanced research data infrastructure.
The emergence of the iReceptor CFI project coincides with the inception and development of the AIRR Community. The goal of the iReceptor project is to leverage the developments made by the AIRR Community to implement components (from a repository perspective) and tools (from the Scientific Gateway perspective) that are part of an AIRR-seq data commons. Since the AIRR Community’s inception in May 2015, the iReceptor team has been active within the community to help establish and implement standards and protocols that will enable the creation of such an AIRR-seq data commons, evolving components of the iReceptor architecture to become “AIRR Compliant” as AIRR standards and protocols are defined.
One of the most recent outcomes of the AIRR Community and its working groups has been the publication of the Minimal Information for AIRR-seq data (MiAIRR) standard (26). The MiAIRR standard has been approved by the AIRR Community and is considered the set of metadata that is minimally necessary to interpret and compare AIRR-seq experiments and data sets. It consists of six high high-level classes of metadata that provide information about different aspects of an AIRR-seq based study:
Study (e.g. study title, study type, lab name, and funding), subject (e.g. subject ID, organism, sex, age) and diagnosis (e.g. diagnosis, disease stage)
Sample collection (e.g. tissue type, collection time)
Sample processing and sequencing (e.g. cell subset phenotype, library generation protocol)
Raw sequences
Data processing (e.g. software used, version numbers, quality thresholds)
Processed sequences with annotations (e.g. germline reference database, V gene, J gene)
The establishment of such a standard is a critical step in the process of making AIRR-seq data Accessible, Interoperable, and Reusable (adhering to the FAIR principles). The MiAIRR standard is published and available for developers through a GitHub repository (40). This repository includes formal specifications of the standard fields (including a Swagger (41) definition of the MiAIRR fields for implementation in a REST API), tools and resources to process data in this format, as well as resources to help researchers upload data in a MiAIRR compliant form to national repositories such as NCBI.
iReceptor has made extensive use of the MiAIRR standardization efforts to direct its development.
The iReceptor data curation methodology that is used to load data into the iReceptor Public Archive (IPA) has been extended to include the use of MiAIRR terms. Although the iReceptor data curators have little control over the data that authors publish, when loading data into IPA any MiAIRR compliant data that are available from a paper is included in the repository.
All iReceptor repositories have been extended to be able to include all MiAIRR data fields. If a study includes a MiAIRR field then the iReceptor repositories will be able to store it.
The iReceptor Repository Services have been extended to utilize the MiAIRR fields in the repositories for both searching and returning data in MiAIRR compliant forms. The iReceptor Repository Service can search on any MiAIRR term.
The iReceptor REST API is also MiAIRR compliant. The iReceptor API definition uses the MiAIRR Swagger definitions from the AIRR Community Git Repository to ensure that the iReceptor REST API can query MiAIRR terms and return fields based on the MiAIRR definition in its API response. The iReceptor REST API also returns the AIRR Community defined file format in response to those API calls that download large amounts of data.
The iReceptor Scientific Gateway web portal uses MiAIRR terminology to ensure that a common language is used to describe and explore AIRR-seq data. In this manner, the same terms and ontologies are used to both upload and explore AIRR-seq data. Of course, the iReceptor Scientific Gateway also utilizes the MiAIRR compliant iReceptor REST API to query the iReceptor repository network.
In iReceptor v1.0, the iReceptor REST API implemented three query levels. In iReceptor v2.0, we have simplified the interface to include only two types of queries, those at the sample level and those at the annotated sequence level. The functionality of the metadata query capability in v1.0 has been replaced by having the samples and sequences REST APIs return summary statistics as well as the data that results from the queries. The iReceptor REST API is defined through a Swagger API definition (which includes the MiAIRR Swagger definition) and is available for download through GitHub (42).
One of the key active areas of development within the AIRR Community is through the AIRR Common Repository Working Group (CRWG). The CRWG is working to define the components of an AIRR Data Commons, and therefore iReceptor works closely with CRWG to further develop both the iReceptor REST API and the query interfaces supported by the iReceptor Repository Services. Just as iReceptor adapted the MiAIRR standard, we will implement the CRWG REST APIs and query interfaces in the next generation iReceptor REST API and iReceptor Repository Services. Eventually, it is expected that the iReceptor Data Commons and the AIRR Data Commons will converge on a single data commons architecture.
Another critical component in an AIRR-seq data commons is the ability to discover AIRR-seq data repositories (the “Find” from the FAIR principles). In both iReceptor v1.0 and v2.0, the iReceptor Gateway maintains an internal registry of AIRR-seq data repositories. Recall that in the iReceptor v1.0 (and v2.0) security model, data repositories that require secure authentication from their clients must register themselves with the iReceptor Scientific Gateway and provide a “Shared Secret” (a password for the gateway to authenticate to the repository service). This registration allows iReceptor to manage an internal registry of repositories. Even if a repository provides completely open data (does not require authentication from the client), it is still necessary for the repository to register with the iReceptor Gateway. It is anticipated that over the next year, the AIRR CRWG will define a protocol and API for an AIRR-seq data repository registry. Once this registry is defined and implemented, iReceptor will transition to using the AIRR Data Commons repository registry rather than its internal registry.
Given that all iReceptor data repositories support the MiAIRR standard, these repositories form the basis of what can be thought of as an AIRR-seq data commons. Although all components of the iReceptor Data Commons are not yet defined by the AIRR Community, the fundamental components to provide an AIRR-seq data commons currently exist within the iReceptor Data Integration System. In addition, our collaboration with the VDJServer team has extended the iReceptor v1.0 VDJServer proof of concept into a production resource that has been integrated into the iReceptor Data Commons in iReceptor v2.0. VDJServer now provides a publication mechanism where researchers can publish data they store on VDJServer into a public repository. The VDJServer team has developed an iReceptor Repository Service that directly queries the VDJServer repository and responds with iReceptor REST API compliant (and therefore MiAIRR compliant) responses. As a result, the iReceptor Scientific Gateway can now seamlessly query all iReceptor repositories (including the VDJServer repository), federate data across those repositories, and perform analyses on those federated data (see the Use Cases below). This capability is currently in production on the iReceptor Gateway and demonstrates the feasibility of federating AIRR-seq data across disparate data repositories.
Of course, a network of repositories is only as useful as the data that are in those repositories, and one of the key goals of iReceptor is to make it easy for researchers to manage, publish, and share their data. To this end, the iReceptor platform operates the iReceptor Public Archive (IPA), a public repository that contains data from studies that the iReceptor Data Curation team has identified as being significant and relevant to the general community. The security model on the iReceptor v2.0 Repository is the same as that of the v1.0 repository and is not suitable for storing protected health information. Currently, the IPA contains only anonymized, public data that has been downloaded and curated from other public repositories such as the NCBI SRA.
One of the key challenges of any data repository is to be able to perform searches on the data in the repository as the repository grows. iReceptor and its Scientific Gateway support two types of data repository operations:
Synchronous operations are those that can be performed on a data repository in “user interactive” time frames. By “user interactive” we mean time frames that a user would be willing to wait in front of a computer screen to get a response, i.e. one to two minutes per operation. Our goal in iReceptor is to be able to support interactive data exploration at the sequence level, and therefore perform simple search operations on key data elements across millions of sequences. For example, we want to be able to support substring searches on key MiAIRR sequence annotation fields such as V-, D-, J-gene annotation, CDR3/Junction amino acid sequences as well as searches on CDR3/Junction lengths.
Asynchronous operations are operations that require longer than 2 minutes to perform. Asynchronous operations are typically either operations where the user wants to perform a complex query at the sequence level and/or perform some sort of detailed analysis applied to a federated data set. Asynchronous operations can take minutes to days to complete and are managed as “jobs”. In these cases, the iReceptor Gateway manages federating data from the repositories, staging those data to a computational resource, running the analysis application, staging the results back to the gateway, and notifying the user when the analysis is complete using the Science-as-a-Service AGAVE platform (43).
Since iReceptor’s inception in 2014, we have constantly struggled with data repository scalability. Metadata at the study, subject, and sample level (MiAIRR Level 1, Level 2, Level 3, and Level 5) are manageable by most data repository technologies. For example, typical studies will have anywhere from 10 to 100 biological samples (along with appropriate metadata - see the MiAIRR standard for details). Even with thousands of studies in a repository, the number of sample records in a repository would be less than one million, and data at such a scale can be interactively queried relatively easily. This clearly does not hold true at the sequence level. For each sample, the number of sequences could range from the thousands to 10s of millions. Searching synchronously at this scale (e.g. return the sequences in your repository that contain the following CDR3/Junction AA sequence: CASSQVGTGVYEQYF) is extremely challenging.
In iReceptor v1.0, we utilized MySQL as our repository technology. As we explored the types of interactive queries that we wanted to perform with our user community, we rapidly realized that MySQL would not scale to the level that we desired. Over the past year, the iReceptor team has been exploring the use of MongoDB (44) a widely used NoSQL data repository. MongoDB is able to scale up through its “sharding” capability. Sharding essentially divides the repository across multiple servers (data are distributed based on a shard key), parallelizing queries across the shards through load balancing. As long as the data are divided equally across the shards, parallel performance can be achieved. In particular, in repositories such as the iReceptor AIRR-seq repositories, for which the primary operations are searches (i.e., data are read, but not written) and write consistency is not time critical, MongoDB shards can be very effective in accelerating searches. In particular, MongoDB is known to perform very well if indexes on its data fields are carefully selected and can be kept in memory on the sharded repository servers.
For the iReceptor repositories, we have spent an extensive amount of time carefully optimizing both the indexes required to support fast searchers at the sequence level as well as optimizing the hardware configuration for the IPA repository to ensure that indexes fit into memory across all shards. As a result, we are currently able to run interactive substring queries (on the order of 10s of seconds) for targets such as V-, D-, and J-gene annotations and CDR3/Junction amino acid strings across 100M sequences. Although our performance analyses are preliminary and continue as we add more data, we believe that it will be possible to continue to scale a single MongoDB repository to 100s of Millions of sequences and their annotations.
In addition, it is important to note that once we find a limit to MongoDB’s scalability for AIRR-seq data, as we inevitably will, the distributed data model on which iReceptor is built makes it simple to add another repository (or indeed many more repositories) of the same scale. This gives iReceptor the ability to both “scale up” a single repository as well as “scale out” to multiple large and/or many small repositories as required.
To facilitate the uptake in sharing AIRR-seq data, iReceptor has built on its knowledge of using MongoDB for AIRR-seq data, to create what we call the iReceptor Turnkey Repository. The iReceptor Turnkey Repository is an easy to install package that enables research labs to install and manage their own iReceptor compliant repositories. The Turnkey platform includes an iReceptor MongoDB Repository (with appropriate indexes on critical fields), an iReceptor Repository Service, and a data import pipeline for MiAIRR metadata and V-, D-, and J-gene annotation from a variety of tools, including IMGT VQuest (45), igBlast (46), and MiXCR (11). The iReceptor Turnkey Repository is available for download from GitHub (47).
Last, but certainly not least, we recognize that many labs have already invested significantly in their own repository technologies and their own data collections. Through the definition of the iReceptor REST API and the implementation of data services that implement that API (in particular through its reliance on the MiAIRR standard), it is possible for data repositories to expose and share their data. This has been demonstrated in practice through iReceptor’s collaboration with VDJServer, showing that is feasible for a lab to extend their own data repository such that it can participate as a node in the iReceptor Data Commons and ultimately the AIRR-seq Data Commons.
In summary, as of the writing of this paper, the iReceptor Data Commons consists of four data repositories:
The iReceptor Public Archive (IPA)(Victoria, Canada): The main iReceptor public repository, with ~145M sequences from 761 biological samples, 17 studies, and 13 research labs (Figure 2). As discussed above, we anticipate that this repository will grow to the order of 100s of Millions of sequences.
The iReceptor Turnkey Archive (Victoria, Canada): An example lab scale repository, based on the iReceptor Turnkey Repository, with ~1.2M sequences from one study and one lab.
The Scott Lab Repository (Vancouver, Canada): A lab-scale repository operated by the iReceptor team on behalf of iReceptor co-PI Jamie Scott, with ~12M sequences from 155 biological samples and 3 studies from the Scott Lab. Note this repository is a private repository with access provided only to authorized users.
The VDJServer Repository (Austin, Texas): The VDJServer public data repository, with ~1.7M sequences from one study and one lab. We anticipate the VDJServer repository will grow to 10s or 100s of Millions of sequences.
Figure 2:
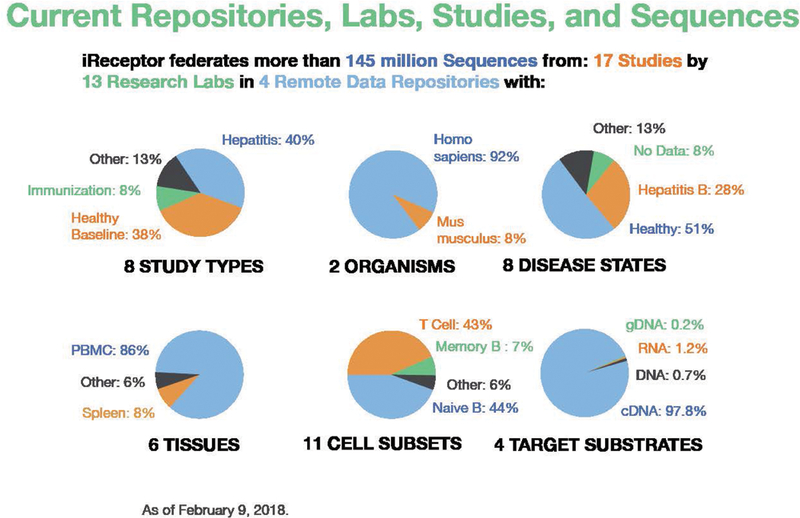
AIRR-seq repositories and data available in iReceptor as of February 2018
As the AIRR Community converges on a definition of an AIRR Data Commons, we anticipate convergence of the iReceptor and AIRR Data Commons. This will result in all iReceptor repositories being compliant with, and participating in, the AIRR Data Commons. We anticipate that the AIRR Data Commons will soon consist of 10s, if not 100s, of repositories that span the scales listed above.
iReceptor v2.0 will be released in the Spring of 2018, with the iReceptor Scientific Gateway (gateway.ireceptor.org) available for general use by the AIRR-seq research community. Researchers who are interested in using the iReceptor Gateway are encouraged to contact the iReceptor team at support@ireceptor.org.
3.6. The Future - iReceptor v3.0
The iReceptor v3.0 timeline is targeted for mid-2019 and will entail the further development of each of the iReceptor v2.0 components listed above. One of the key development milestones will be the convergence of the iReceptor Data Commons with the emerging definition on an AIRR Data Commons. This will include tracking developments with the MiAIRR Standard and work emerging from the AIRR Common Repository Working Group around refinements to ontological definitions of MiAIRR terms, the establishment of relationships between groupings of the MiAIRR terms, the definition of a set of queries to which AIRR-seq repositories should be able to respond, and the establishment of an AIRR repository registry. It is expected that for the iReceptor v3.0 release, the iReceptor Scientific Gateway will be querying a network of AIRR Data Commons repositories as defined by the criteria above.
Components that will be impacted by the evolution of the AIRR Community and its standards are:
The iReceptor repositories, in particular the iReceptor Public Archive and the iReceptor Turnkey Archive, will evolve to meet the requirements established by the AIRR CRWG
The iReceptor Repository Service will evolve to ensure that iReceptor repositories can implement the queries defined by the CRWG
The iReceptor REST API, will evolve to implement a richer query interface based on the queries defined by the AIRR CRWG
The iReceptor Scientific Gateway will evolve to incorporate changes such that it can perform the analyses required for the scientific use cases that the AIRR community requires
One of the critical components of iReceptor development for v3.0 is data scalability: both scaling up the iReceptor Public Archive (having single repositories contain more data) as well as scaling out the network of repositories in the iReceptor Data Commons (supporting new repositories). Over the next twelve months, we will continue to work towards scaling up the iReceptor IPA as our data curation team continues to work with collaborators to import their data. At the same time, we will be working with a number of partner organizations and collaborators, both in research and in industry, to scale out the iReceptor repository network to include the installation of iReceptor Turnkey repositories as well as integrating other AIRR-seq repositories into the iReceptor network through the use of the iReceptor REST API.
Two other areas of significant development remain on the roadmap for iReceptor v3.0. Firstly, data security remains of critical importance. The iReceptor architecture at the iReceptor Repository Service and iReceptor REST API layers provide a basic security model in v2.0 with the implementation of a layered security model planned for iReceptor v3.0. The goal of this implementation is to enable data stewards to securely share data that requires protection under privacy or ethics constraints among collaborators who have permission to access that data.
Secondly, although iReceptor v1.0 and v2.0 provide a basic level of analysis on federated data, through the use of AGAVE’s Science-as-a-Service capabilities (43), there has been little work on integrating advanced analysis applications into the iReceptor Scientific Gateway to date. Given that both VDJServer and iReceptor make use of AGAVE as a middleware platform, it is anticipated that we will be able to leverage each other’s work in this area. VDJServer’s current capabilities in terms of providing analysis pipelines (39) is far more advanced than iReceptor’s, but it is anticipated that our ongoing collaboration will result in shared developments around integrating these analysis tools on federated AIRR-seq data.
4. IRECEPTOR USE CASES
As previously stated, the primary purpose of the iReceptor resource is to lower the barrier for researchers to share, reuse, explore, and analyze AIRR-seq data to answer questions about immunogenetics and the immune response. The usefulness of the iReceptor platform is highlighted by the following two simple use cases. Use Case 1 demonstrates a search for partial CDR3 sequences across datasets from different labs and repositories. Use Case 2 demonstrates the interactive search of sample metadata across different repositories to identify candidate AIRR-seq data sets whose analysis might help validate a research hypothesis. Interviews with iReceptor users and general discussions within the AIRR Community suggest that these two types of searchers, one searching for a specific sequence and one over metadata, will encompass most of the searches that researchers will perform on iReceptor and other AIRR-seq resources.
4.1. Use Case 1 - looking for a needle in a haystack!
The CDR3 region is the most diverse part of the full adaptive immune receptor gene. Given this diversity, we wouldn’t expect to find common CDR3 sequences among unrelated individuals. However, specific B-cell and T-cell receptors are found in multiple individuals for various immune responses, and are referred to as public clonotypes (48). The role of these public BCRs and TCRs in human health is still being studied, but one role would be fighting common pathogens within a population. One such common pathogen in humans is the Epstein-Barr virus (EBV), which can cause life-threatening infections and cancers in immunocompromised individuals. Nguyen et al. (49) characterized TCRs from EBV-specific CD8+ T cells in post lung transplant patients before clinical detection of the EBV. Thus, searching for these TCRs based on CDR3 sequence features would be a simple and useful exercise for researchers interested in exploring the relative abundance/presence of such public CDR3 sequences in larger population samples of immunocompetent (healthy) and immunocompromised individuals (such as cancer patients).
4.1.1. The challenge
This use case in some ways presents a worst-case scenario for any data repository. It is essentially searching for an entity at the finest granularity of detail (the needle – in this case an annotated sequence feature such as a partial CDR3 sequence) across the entire data set (the haystack - in the distributed repository case a search across all data in all repositories). It is a brute force search without any data refinement or data reduction before the detailed search takes place.
In a world in which there were no AIRR-seq data commons, individual researchers and labs would store their own processed data, associated metadata, and in some cases (but not all) publications uploaded to central repositories like GenBank and SRA. In this case, performing the above search would be extremely labor intensive and time consuming, with a researcher having to perform the following steps:
Finding: Identify a set of papers and/or labs that would have relevant study data.
Federating: Find the appropriate data sets from those studies and downloading them to local storage.
Curating: Reconstruct the appropriate MiAIRR compliant metadata from those studies so the downloaded data can be compared. If studies do not archive the metadata required for comparisons with the sequence data, it would be necessary to reconstruct the metadata from the text of the papers.
Annotating: If a study has open, published data, they are most likely to be the “raw” sequence data (FASTA files) without the sequence annotations (e.g., without V-, D-, and J-gene and CDR3 identification). In these cases, it is necessary to reproduce the annotation pipeline used in the paper and/or develop your own annotation pipeline to process the “raw” sequence data to attain the gene and CDR3 sequence annotation.
Analyzing: Search the annotated sequences for the CDR3 sequence feature of interest.
It is easy to see why such a process is challenging and often beyond the resources of many research groups.
Within the iReceptor Data Commons, this process becomes relatively straightforward:
Finding: Data repositories with MiAIRR compliant data are registered with the Data Commons.
Federating: The iReceptor Scientific Gateway performs queries across the federated repositories on behalf of the user as well as federating the results of the queries into a single manageable data set.
Curating: Data in the repositories is MiAIRR compliant to the degree that it can be, and therefore no curation is required during the exploration process. This does not mean that curation does not need to happen, but the curation happens only once when the data are added to a repository.
Annotating: Curated MiAIRR data are annotated as part of the curation process.
Analyzing: The iReceptor Scientific Gateway can perform simple analyses such as searching for a specific gene annotation or a CDR3 sequence interactively, while more complex analyses can be managed through asynchronous jobs.
The iReceptor Data Commons (and indeed any data commons) puts a much larger burden on the curation process, but once curated these data can be shared seamlessly for the broad AIRR-seq research community to answer a wide range of research questions.
4.1.2. Using iReceptor to find the needle!
Using the iReceptor Scientific Gateway in Use Case 1 is quite simple. The researcher would:
Log in to the iReceptor Scientific Gateway
Select all the data in the repository (a simple select all button)
Enter the CDR3 sequence or partial sequence of interest
Ask the Gateway to perform the search.
In fact, the iReceptor Gateway implements a shortcut for this use case, providing a Quick Search function that assumes Step 2 above and simply allows the user to choose a sequence annotation feature to search for V-, D-, or J-gene or CDR3 sequences. The Gateway would search all the datasets across all repositories in the iReceptor Data Commons on behalf of the researcher, without the researcher being aware that a federated query was being executed. A researcher can then explore the summary statistics around the metadata of the studies, subjects, and samples whose repertoires contained TCRs with the queried CDR3 sequence feature to get insights on the CDR3 sequence and/or TCR of interest. This also allows the researcher to identify the study and contact the researcher who generated the dataset if other information is necessary about the data or study (Figure 3).
Figure 3:
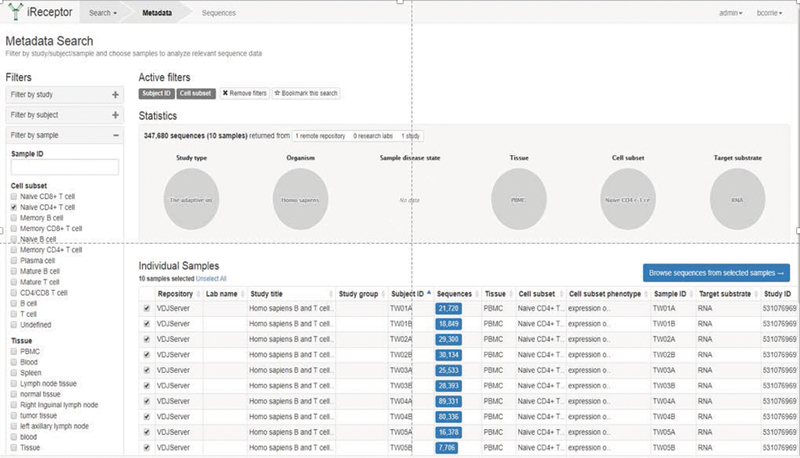
Metadata search on iReceptor Gateway restricted to naïve CD4+ T- cell for use case 2.
4.1.3. Use Case 1 results
In Use Case 1, we compared results from a study on EBV-specific public TCR clonotypes found in 3 healthy patients and 3 lung transplant patients from Nguyen et al. (49) to data curated in the IPA repository on Compute Canada resources and VDJServer at UTSW, both of which are nodes on the iReceptor network of repositories. The authors sorted EBV-specific T cells that bound to a highly immunogenetic target on the surface of EBV, and sequenced the TCRα and TCRβ chains from these single cells. Thus, they were able to capture the exact pairing of the TCRα and TCRβ chains in the TCR repertoire. Based on this small number of samples (6 individuals), the authors identified many new public clonotypes (paired CDR3 sequences occurring in 3–6 of the individuals). In addition, they classified a high proportion of the dominant sequences in these repertoires into 5 consensus clonotypes. A consensus clonotype consists of multiple clonotypes that exhibit similar binding motifs and gene pairings. Of the 5 consensus clonotypes, one was well-known in EBV research, but four were newly discovered. Since these public and consensus clonotypes were discovered from only 3 healthy and 3 lung transplant individuals, an obvious question is how common these sequences would be among a broader range of individuals.
In order to examine how common these EBV-associated sequences are in a larger sample of individuals, we extracted all the TCRβ chain binding motifs from the paper (N = 63). Of these 63 motifs observed in the healthy and immunocompromised individuals, 24 were classified as belonging to one of the 5 consensus clonotypes, while the remaining 37 clonotypes did not cluster with these consensus clonotypes. Our analysis looked for the presence of these TCRβ motifs in a total of 362,022,383 functional TCRβ sequences (50,720,436 TCRβ CDR3 sequences from 19 healthy individuals and the rest from 16 cancer patients (Table 1)) from the IPA resource curated by the iReceptor team, and the repository curated by VDJServer.
Table 1.
Sequence data sets used in Use Cases 1 and 2
| Study | Disease | Data Source |
No. of Subjects |
No. of Samples |
No. of functional reads |
No. of unique CDR3s |
|---|---|---|---|---|---|---|
| Wang et al. 2017 |
Breast Cancer |
IPA | 16 | 48 | 311,301,947 | 8,962,685 |
| Putinsteva et al. 2013 |
Healthy | IPA | 9 | 9 | 4,9999,999 | 12,391,299 |
| Rubelt et al. 2016 |
Healthy | VDJServer | 20 | 60 | 720,437 | 486,118 |
| Total | 35 | 117 | 362,022,383 | 21,840,102 |
Table 2 shows the number of times these motifs were observed among the 117 samples sequenced from these 35 individuals (only 40 of the 63 motifs were observed in the repertoires from these new individuals and are listed in Table 2). Some of these motifs were very common in the new data sets, with one of them found in 44 of the 117 samples. Table 2 also identifies motifs corresponding to the 5 consensus clonotypes identified in the EBV-specific sequences, signified by the same 5 colors used in the Nguyen et al. study. This analysis shows that a larger proportion of the motifs identified in the broader set of individuals come from the consensus clonotypes (19/26 or 73%) than from the non-consensus clonotypes (21/37, or 57%). In summary, many of the public clonotypes observed in the study of EBV-specific T-cell receptor repertoires were found in a larger survey of individuals, and the consensus sequences observed in the EBV-specific repertoires were possibly enriched in these broader samples.
Table 2.
Occurrence of sequence motifs analysed in Use Case 1
| IPA | VDJServer | |||
|---|---|---|---|---|
| Motifs | Number of samples with reads containing the motif |
Total number of reads containing the motif |
Number of samples with reads containing the motif |
Total number of reads containing the motif |
| GGLSSYNEQ | 44 | 6,606 | 7 | 12 |
| GTGGTNEKL | 42 | 116,700 | 0 | 0 |
| RDRVGNTI | 28 | 2,691 | 0 | 0 |
| RGQGDEQ | 26 | 2,350 | 2 | 2 |
| SGGTSSYNEQ | 24 | 1,116 | 0 | 0 |
| GAGGTNEKL | 24 | 721 | 0 | 0 |
| RDRTGNGY | 22 | 21,534 | 0 | 0 |
| GSGGTNEKL | 21 | 10,748 | 0 | 0 |
| RTGVGDTQ | 21 | 3,683 | 1 | 1 |
| PGTLNTEA | 20 | 399 | 2 | 2 |
| SVVGETQ | 19 | 1,278 | 1 | 1 |
| SVVGGNEQ | 17 | 582 | 0 | 0 |
| SVAGGDEQ | 16 | 353 | 0 | 0 |
| GSAGTNEKL | 16 | 2,915 | 1 | 2 |
| SSTTEQ | 14 | 140 | 0 | 0 |
| SQSPGGTQ | 13 | 1,401 | 0 | 0 |
| RDGTGNGY | 9 | 662 | 0 | 0 |
| RDRVGNGY | 9 | 192 | 0 | 0 |
| SVGGEAYEQ | 9 | 40 | 1 | 1 |
| GGSSYQETQ | 8 | 51 | 0 | 0 |
| RDSTGNGY | 8 | 398 | 0 | 0 |
| SFSSGTTDTQ | 8 | 302 | 0 | 0 |
| SLSGGINEQ | 8 | 50 | 0 | 0 |
| SQSPGGEQ | 7 | 202 | 0 | 0 |
| RDQTGNGY | 6 | 984 | 0 | 0 |
| SISGDYGY | 5 | 187 | 1 | 1 |
| REDSTNEKL | 5 | 351 | 0 | 0 |
| SQAGLAAYNEQ | 5 | 45 | 0 | 0 |
| RDRGIGNTI | 5 | 19 | 0 | 0 |
| SFGTFETQ | 4 | 1,964 | 0 | 0 |
| SPVSGSSYEQ | 4 | 11 | 0 | 0 |
| PGLAVPGEL | 4 | 5 | 0 | 0 |
| RSETGNTI | 3 | 249 | 0 | 0 |
| GSDGTNEKL | 3 | 11 | 0 | 0 |
| RDTTGNGY | 2 | 8 | 0 | 0 |
| PITVQETQ | 1 | 2 | 0 | 0 |
| KEGSGNEKL | 1 | 2 | 0 | 0 |
| SMAGGGRNEQ | 1 | 2 | 0 | 0 |
| RDSRIGNTI | 1 | 1 | 0 | 0 |
| GTQGTNEKL | 1 | 1 | 0 | 0 |
4.2. Use Case 2 – comparing stacks of different types of needles
In many cases, researchers would like to have access to more data that are relevant to their research hypothesis. Use Case 2 demonstrates the use of iReceptor in such a situation. Consider the use case where a researcher is interested in exploring the relationship between heredity and CDR3 diversity, hypothesizing that the diversity in CDR3 sequences vary from most diverse between unrelated individuals to least diverse for monozygotic (MZ) twins, with related siblings having a diversity between these two extremes. One could test this hypothesis by measuring the overlap of CDR3 sequences between repertoires sequenced from pairs of individuals, and determining if there is a relationship between the overlap and how closely the individuals are related.
4.2.1. The challenge
In this use case, the challenges are less about scale (e.g., number of samples) and more about the fidelity of the comparisons that the researcher wants to explore. All the challenges from the Use Case 1 still apply, but the challenge around data curation, data comparison, and data analysis are paramount for Use Case 2.
Whenever it is necessary to find and compare data with different characteristics, the quality of the metadata that describes those characteristics are critical. That is, we want to partition all the data across all the distributed repositories (our haystack) into subsets (the different types of needles) that we can then analyze. For example, in this use case, the researcher is interested in sibling relationships, both twin and non-twin. For the iReceptor Data Commons to support such a use case, it is necessary for the researcher to be able to find studies that fit these criteria. In the MiAIRR standard, there is a piece of metadata that describes the relationship between subjects that would work for this purpose, but this still relies on the data curation process to capture these relationships. In general, it is difficult if not impossible for a standard such as MiAIRR to capture all metadata and relationships that a researcher might need. As a result, it is critical that the data federation tools allow for flexible and powerful searchers. As in this case, if the relationship of interest (relationship of subjects) is not captured as part of the curation process then the researcher is left with the problem: “How do I find studies that involve sibling relationships”. In such cases, and indeed in this case, it is necessary to revert to searching for keywords (sibling, child, twin) across metadata fields such as the study title or a study description. The power of the combination of the iReceptor Scientific Gateway and the iReceptor Data Commons is that such searches are not only possible, but relatively straightforward.
Both exploratory analysis and detailed analysis are a challenge in Use Case 2. Initially, the researcher wants to explore studies at a high level, looking for studies that not only involve siblings, but possibly only involve specific characteristics such as disease (e.g. cancer) and/or cell type (B Cell vs. T Cell). Once a set of studies are found that meet the researcher’s criteria, detailed analyses need to be performed on the resulting federated repertoire data sets. iReceptor as a platform excels at performing the iterative exploration of metadata and specific annotation characteristics such as V-, D-, and J-Genes or CDR3 sequences, as well as federating those data so the researcher can perform detailed analyses. As discussed above, iReceptor’s capabilities around supporting and coordinating those detailed, asynchronous analyses are still under development. How these two types of searches are used in this use case are discussed in more detail below.
4.2.2. Using iReceptor to find (and analyze) the needles
The search performed in Use Case 2 is much more complex than Use Case 1. In this analysis, the researcher is starting with an exploratory analysis, looking for studies that might be relevant to their research question around heredity and CDR3 diversity. Using the iReceptor Scientific Gateway to perform this exploratory analysis is relatively easy – the researcher would:
Log in to the iReceptor Scientific Gateway
Perform a metadata search looking for studies of interest
As a start, the most promising search criteria would be to search for keywords such as “heredity”, “child”, “parent”, “mother”, “father”, and “twin” in fields where you would expect these keywords to be used (e.g. Study Title). Performing such a search on the iReceptor Scientific Gateway performs a query across all repositories in the iReceptor Data Commons and discovers two studies of relevance.
Both the keyword “mother” and “child” discover a deep profiling study on mother and child T-Cell repertoires by Putinseva et al. (48). This study compared the V segment usage and the relative overlap of CDR3 sequence features between TCR repertoires of mother and children for which the child siblings were non-twins. The iReceptor Gateway provides access to the metadata for this study, including the fact that it consists of samples from three mothers and three pairs of siblings (one per mother) for a total of 9 biological samples. The overall study consists of over 55 million annotated sequences and was found in the iReceptor Public Archive repository. The iReceptor Gateway links back to other relevant resources for this study, such as its NCBI BioProject information.
Searching for the keyword “twin” also finds a relevant study of the repertoires from 5 pairs of monozygotic twins (50). In this study, Rubelt et al. characterized the impact of heritable factors on both V(D)J recombination and on thymic selection in the TCR repertoires of these monozygotic twins. The study also showed chromosomal bias in the usage of V- and J-gene segments based on the analysis of B- and T-cell repertoires from the 5 pairs of twins, and that V gene usage was most similar between twins than unrelated non-twins. Again, the iReceptor Gateway provides a summary of the metadata for this study, including that it consists of 60 repertoires from 5 pairs of twins (10 individuals), with cell sorting carried out to identify six different cell phenotypes for each of the 10 individuals. The study consists of over 1.7 million annotated sequences and was found in the VDJServer repository.
Once data sets of interest have been identified, the researcher can perform more interactive exploration of each study and its metadata, through the iReceptor Scientific Gateway. For example, the researchers might choose to explore the characteristics of a single cell phenotype (e.g., Naïve CD4+ T- cell) across all the twins from the Rubelt et al. study (Figure 3).
Eventually, it is likely that a federated data analysis step would need to be carried out on these data to assess, for example, CDR3 diversity across the two data sets. Such an analysis is beyond the scope of the interactive explorations provided by the iReceptor Scientific Gateway’s web portal. Although at the current time the iReceptor Gateway supports the management of asynchronous (long running) analyses, it does not have a significant number of analysis applications integrated into the platform (recall that this is on the iReceptor v3.0 roadmap). As a result, it is necessary for a researcher to download the federated data and perform such an analysis offline.
Fortunately, the iReceptor Gateway can still be of assistance, as it is possible for the researcher to request the download of both data sets from each repository in a single consistent file format that makes comparative analyses straightforward. The iReceptor Scientific Gateway uses the AIRR Community’s emerging AIRR TSV file format as its data interchange format. As a result, the annotated data from both the Rubelt et al. and the Putintseva et al. studies can be downloaded by the researcher in a format that makes comparative analyses relatively straightforward.
To demonstrate iReceptor’s functionality, the comparative analysis for Use Case 2 was performed as an offline analysis once the federated data were downloaded from the iReceptor Gateway. The current analysis was limited to a comparison of CDR3 from sequence features of T-cell receptors, but this is easily generalizable to compare other parameters such as V segment usage between the repertoires of the related non-twins and twins as needed.
The Rubelt et al. study assessed the impact of heredity on the repertoires generated among monozygotic twins while the Putintseva et al. assessed and showed the effects of heredity on repertoires generated among non-twin siblings and their maternal parent. While the Rubelt et al. study showed evidence for shared bias in V-gene usage among monozygotic twins, the Putintseva et al. study did not find strong evidence of biased overlap between repertoires of related and unrelated mother-child pairs.
The Rubelt et al. study could have benefited by access to AIRR-seq data from non-monozygotic siblings, while the Putintseva et al. study could have extended their observations of low overlap of TCR repertoires among more closely related siblings by accessing data from monozygotic twins. In this use case, the expectation or non-expectation of relative increase in overlap between repertoires of unrelated individuals (sampled from different geographical locations) to most closely related individuals (data from monozygotic twins) is tested in an explorative manner by comparing the overlap of unique CDR3s across the repertoires of individuals from both the Rubelt et al. and the Putinseva et al. studies.
4.2.3. Use Case 2 results
When using monozygotic twin pairs to estimate the effect of heredity on any phenotype the most critical comparison is similarity between monozygotic twins compared to related sibling pairs. The expectation is that monozygotic twins, by having the same genotype, will exhibit higher similarity compared to non-monozygotic siblings. We explored this expectation by comparing monozygotic twin pairs from Rubelt et al. and siblings from the Putinsteva et al. studies. At first we were surprised that there was not a strong difference between these types of pairs, for various measures of overlap between repertoires. The amount of data per repertoire was very different, with the read depth from Putinseva et al. ranging from 17,000 to 66,000 sequences, while read depths per repertoire in Rubelt et al. were typically several million. We attempted to adjust for these differences, but finally resorted to randomly drawing 50,000 reads per sample from the Rubelt et al. study to make read depths similar between studies.
Repertoire overlap for these pairs of individuals are presented in Table 3. In this case, overlap was calculated as follows. First the unique CDR3 sequences were determined for each member of a pair, say A and B, and the set of shared CDR3 sequences was determined. Then the number of sequence reads for individual A that matched any of the shared sequences, divided by the total reads in the repertoire of A, is the repertoire overlap with A as the reference repertoire and B as the target repertoire (reported in Table 3). As shown explicitly in Table 3, the average for 5 pairs of monozygotic twins from Rubelt et al. was compared to the average overlap for 3 pairs of full sibling pairs from Putinseva et al. The average overlap for all monozygotic twins was 6.31, which was not much higher than the average value of 4.81 for full sibling pairs. This result could be confounded by differences between these studies, such as sequencing methods and ethnicity of sampled individuals. These differences are potential problems when combining data from multiple studies. Even so, this comparison shows a surprising result, which could motivate more controlled comparisons. This data exploration potential is one of the strengths of the data commons approach, as exemplified by iReceptor, and this will only be stronger when the data commons includes more repositories to be mined and more controlled descriptions of the metadata associated with each study in the set of repositories to facilitate better matched comparisons.
Table 3.
Repertoire overlap calculated for monozygotic twins and full siblings analyzed in Use Case 2
| Sibling Pair | Reference Repertoire |
Overlap | Reference Repertoire |
Overlap |
|---|---|---|---|---|
| Monozygotic twins TW01 |
TW01A | 13.65 | TW01B | 6.02 |
| Monozygotic twins TW02 |
TW02A | 5.55 | TW02B | 2.70 |
| Monozygotic twins TW03 |
TW03A | 3.75 | TW03B | 12.37 |
| Monozygotic twins TW04 |
TW04A | 4.48 | TW04B | 4.24 |
| Monozygotic twins TW05 |
TW05A | 2.70 | TW05B | 7.64 |
| Average overlap monozyotic twin pairs = 6.31 | ||||
| Full siblings Child A1 & A2 |
Child A1 | 2.85 | Child A2 | 4.39 |
| Full siblings Child B1 & B2 |
Child B1 | 4.30 | Child B2 | 6.84 |
| Full siblings Child C1 & C2 |
Child C1 | 3.01 | Child C3 | 7.48 |
| Average overlap full sibling pairs = 4.81 | ||||
5 |. INVITATION TO PARTICIPATE IN IRECEPTOR NETWORK AND AIRR COMMUNITY INITIATIVE
The AIRR Community has worked since 2015 to encourage sharing of AIRR-seq data, with the goal of improving biomedical research and patient care. The success of such initiatives depends on openness and community spirit, to which the community has been dedicated. Please see airr-community.org or contact join@airr-community.org to join this growing group of researchers.
The iReceptor team is dedicated to implementing our vision of the AIRR Community’s Data Commons (gateway.ireceptor.org). Please contact us at help@ireceptor.org to see how your lab can become a member of this growing network.
Acknowledgements
This work utilizes software provided by the Agave Platform (NSF OCI #1450459) and infrastructure hosting provided by the Texas Advanced Computing Center and Compute Canada
Funding Information
CANARIE NEP-131; Canada Foundation for Innovation Cyberinfrastructure Grant; Canada Research Chairs; Natural Sciences and Engineering Research Grant; National Institute of Allergy & Infectious Diseases (AI097403); Burroughs Wellcome Fund.
Footnotes
CONFLICT OF INTERESTS
None.
REFERENCES |
- 1.Global Alliance for Genomics and Health TGA for G and. A federated ecosystem for sharing genomic, clinical data. Science [Internet] 2016. June 10 [cited 2018 Feb 3];352(6291):1278–80. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27284183 [DOI] [PubMed] [Google Scholar]
- 2.Breden F, Luning Prak ET, Peters B, et al. Reproducibility and Reuse of Adaptive Immune Receptor Repertoire Data. Front Immunol [Internet] 2017. November 1 [cited 2018 Feb 3];8:1418 Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2017.01418/full [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakano H, Maki R, Kurosawa Y, Roeder W, Tonegawa S. Two types of somatic recombination are necessary for the generation of complete immunoglobulin heavy-chain genes. Nature 1980;286(5774):676–83. [DOI] [PubMed] [Google Scholar]
- 4.Weigert MG, Cesari IM, Yonkovich SJ, Cohn M. Variability in the lambda light chain sequences of mouse antibody. Nature 1970. [DOI] [PubMed]
- 5.Glanville J, Zhai W, Berka J, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci [Internet] 2009;106(48):20216–21. Available from: http://www.pnas.org/content/106/48/20216%5Cnhttp://www.ncbi.nlm.nih.gov/pubmed/19875695%5Cnhttp://www.pnas.org/content/106/48/20216.full.pdf%5Cnhttp://www.pnas.org/content/106/48/20216.long [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freeman JD, Warren RL, Webb JR, Warren L, Nelson BH, Holt R Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing Profiling the T-cell receptor beta-chain repertoire by massively parallel sequencing. 2009;(604):1817–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robins HS, Campregher PV, Srivastava SK, et al. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009; [DOI] [PMC free article] [PubMed]
- 8.Yaari G, Kleinstein SH. Practical guidelines for B-cell receptor repertoire sequencing analysis. Genome Med 2015. November;7:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi SA, Boyd SD. High-Throughput DNA Sequencing Analysis of Antibody Repertoires. Microbiol Spectr [Internet] 2014. October 3 [cited 2018 Feb 10];2(5). Available from: http://www.ncbi.nlm.nih.gov/pubmed/26104353 [DOI] [PubMed] [Google Scholar]
- 10.Hershberg U, Luning Prak ET. The analysis of clonal expansions in normal and autoimmune B cell repertoires. Philos Trans R Soc B Biol Sci [Internet] 2015. September 5 [cited 2018 Feb 10];370(1676):20140239 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26194753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolotin DA, Poslavsky S, Mitrophanov I, et al. MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods [Internet] 2015. May 1 [cited 2018 Feb 3];12(5):380–1. Available from: http://www.nature.com/articles/nmeth.3364 [DOI] [PubMed] [Google Scholar]
- 12.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. Change-O: A toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 2015; [DOI] [PMC free article] [PubMed]
- 13.Ralph DK, Matsen FA. Likelihood-Based Inference of B Cell Clonal Families. Peters B, editor. PLOS Comput Biol [Internet] 2016. October 17 [cited 2018 Feb 10];12(10):e1005086 Available from: http://dx.plos.org/10.1371/journal.pcbi.1005086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stern JNH, Yaari G, Vander Heiden JA, et al. B cells populating the multiple sclerosis brain mature in the draining cervical lymph nodes. Sci Transl Med 2014;6(248). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao H-X, Lynch R, Zhou T, et al. Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature [Internet] 2013. April 3 [cited 2018 Feb 10];496(7446):469–76. Available from: http://www.nature.com/doifinder/10.1038/nature12053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou T, Lynch RM, Chen L, et al. Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell 2015; [DOI] [PMC free article] [PubMed]
- 17.Avnir Y, Watson CT, Glanville J, et al. IGHV1–69 polymorphism modulates anti-influenza antibody repertoires, correlates with IGHV utilization shifts and varies by ethnicity. Sci Rep 2016;(6):20842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kirsch I, Vignali M, Robins H. T-cell receptor profiling in cancer. Mol Oncol [Internet] 2015. December [cited 2018 Feb 10];9(10):2063–70. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26404496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharma P, Allison JP. The future of immune checkpoint therapy. Science (80- ) [Internet] 2015. April 3 [cited 2018 Feb 10];348(6230):56–61. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25838373 [DOI] [PubMed] [Google Scholar]
- 20.Emerson RO, DeWitt WS, Vignali M, et al. Immunosequencing identifies signatures of cytomegalovirus exposure history and HLA-mediated effects on the T cell repertoire. Nat Genet [Internet] 2017. April 3 [cited 2018 Feb 3];49(5):659–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28369038 [DOI] [PubMed] [Google Scholar]
- 21.Gunnarsen KS, Høydahl LS, Risnes LF, et al. A TCRα framework–centered codon shapes a biased T cell repertoire through direct MHC and CDR3β interactions. JCI Insight [Internet] 2017. September 7 [cited 2018 Feb 3];2(17). Available from: https://insight.jci.org/articles/view/95193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kvistborg P, Yewdell JW. Enhancing responses to cancer immunotherapy. Science (80- ) [Internet] 2018. February 2 [cited 2018 Feb 10];359(6375):516–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29420276 [DOI] [PubMed] [Google Scholar]
- 23.Snyder A, Nathanson T, Funt SA, et al. Contribution of systemic and somatic factors to clinical response and resistance to PD-L1 blockade in urothelial cancer: An exploratory multi-omic analysis. Minna JD, editor. PLOS Med [Internet] 2017. May 26 [cited 2018 Feb 3];14(5):e1002309 Available from: http://dx.plos.org/10.1371/journal.pmed.1002309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Subudhi SK, Aparicio A, Gao J, et al. Clonal expansion of CD8 T cells in the systemic circulation precedes development of ipilimumab-induced toxicities. Proc Natl Acad Sci U S A [Internet] 2016. October 18 [cited 2018 Feb 3];113(42):11919–24. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27698113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh DY, Cham J, Zhang L, et al. Immune toxicities elicted by CTLA-4 blockade in cancer patients are associated with early diversification of the T-cell repertoire. Cancer Res 2017;77(6):1322–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rubelt F, Busse CE, Bukhari SAC, et al. Adaptive Immune Receptor Repertoire Community recommendations for sharing immune-repertoire sequencing data. Nat Immunol [Internet] 2017. November 16 [cited 2018 Feb 3];18(12):1274–8. Available from: http://www.nature.com/doifinder/10.1038/ni.3873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan L, Cuplinskas D, Eisen M, et al. Budapest Open Access Initiative [Internet] 2002. [cited 2018 Feb 7] Available from: http://www.budapestopenaccessinitiative.org/read
- 28.OECD. OECD Principles and Guidelines for Access to Research Data from Public Funding [Internet]. 2007. [cited 2018 Feb 7] p. 22 Available from: http://www.oecd.org/sti/sci-tech/38500813.pdf
- 29.Wilkinson MD, Dumontier M, Aalbersberg IjJ, et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci Data [Internet] 2016. March 15 [cited 2018 Feb 3];3:160018 Available from: http://www.nature.com/articles/sdata201618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.NIH. Big Data to Knowledge [Internet] 2012. [cited 2018 Feb 7]. Available from: https://commonfund.nih.gov/bd2k
- 31.NIH. Data Commons: NIH Common Fund [Internet]. [cited 2018 Feb 7]. Available from: https://commonfund.nih.gov/bd2k/commons
- 32.Global Alliance for Genomics and Health. Global Alliance for Genomics and Health [Internet] 2013. [cited 2018 Feb 7] Available from: https://www.ga4gh.org/
- 33.Contreras JL, Reichman JH. DATA ACCESS. Sharing by design: Data and decentralized commons. Science [Internet] 2015. December 11 [cited 2018 Feb 3];350(6266):1312–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26659039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grossman RL, Heath AP, Ferretti V, et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med [Internet] 2016. September 22 [cited 2018 Feb 3];375(12):1109–12. Available from: http://www.nejm.org/doi/10.1056/NEJMp1607591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang J, Haider S, Baran J, et al. BioMart: a data federation framework for large collaborative projects. Database [Internet] 2011. September 19 [cited 2018 Feb 3];2011(0):bar038-bar038. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21930506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudson TJ, Anderson W, Aretz A, et al. International network of cancer genome projects. Nature [Internet] 2010. April 15 [cited 2018 Feb 3];464(7291):993–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20393554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breden F, Lepik C, Longo NS, Montero M, Lipsky PE, Scott JK. Comparison of antibody repertoires produced by HIV-1 infection, other chronic and acute infections, and systemic autoimmune disease. PLoS One 2011;6(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.CANARIE. CANARIE Research Software [Internet]. [cited 2018 Feb 7]. Available from: https://www.canarie.ca/software/
- 39.Cowell L, Fonner J, Jordan C, et al. VDJServer: a web-accessible analysis portal for immune repertoire sequence data (HUM1P.317). J Immunol 2015;194(1 Supplement). [Google Scholar]
- 40.AIRR Community. AIRR Community Data Standards GitHub Repository [Internet] [cited 2018 Feb 7]. Available from: https://github.com/airr-community/airr-standards
- 41.Swagger. Swagger [Internet]. [cited 2018 Feb 7]. Available from: https://swagger.io/
- 42.iReceptor. iReceptor API Definition [Internet] [cited 2018 Feb 7]. Available from: https://github.com/sfu-ireceptor/api
- 43.Dooley R, Vaughn M, Stanzione D, Terry S, Skidmore E. Software-as-a-Service: The iPlant Foundation API. In: 5th IEEE Workshop on Many-Task Computing on Grids and Supercomputers [Internet]. 2012. Available from: http://datasys.cs.iit.edu/events/MTAGS12/p07.pdf [Google Scholar]
- 44.Chodorow K, Bradshaw S. MongoDB: The Definitive Guide, 3rd Edition.
- 45.Giudicelli V, Chaume D, Lefranc M-P. IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucleic Acids Res [Internet] 2004. July 1 [cited 2018 Feb 3];32(Web Server):W435–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15215425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye J, Ma N, Madden TL, Ostell JM. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res [Internet] 2013. July 1 [cited 2018 Feb 3];41(W1):W34–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23671333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.iReceptor. iReceptor Turnkey GitHub Repository [Internet] [cited 2018 Feb 7]. Available from: https://github.com/sfu-ireceptor/turnkey-service
- 48.Putintseva EV, Britanova OV, Staroverov DB, et al. Mother and child T cell receptor repertoires: Deep profiling study. Front Immunol 2013;4(December). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nguyen TH, Bird NL, Grant EJ, et al. Maintenance of the EBV-specific CD8 + TCRαβ repertoire in immunosuppressed lung transplant recipients. Immunol Cell Biol [Internet] 2017. Jan 1 [cited 2018 Feb 13];95(1):77–86. Available from: http://doi.wiley.com/10.1038/icb.2016.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubelt F, Bolen CR, McGuire HM, et al. Individual heritable differences result in unique cell lymphocyte receptor repertoires of naïve and antigen-experienced cells. Nat Commun [Internet] 2016;7:11112 Available from: http://www.ncbi.nlm.nih.gov/pubmed/27005435 [DOI] [PMC free article] [PubMed] [Google Scholar]