

Abstract
Over the past few years, numerous nanotechnology-based drug delivery systems have been developed in an effort to maximize therapeutic effectiveness of conventional drug delivery, while limiting undesirable side effects. Among these, carbon nanotubes (CNTs) are of special interest as potential drug delivery agents due to their numerous unique and advantageous physical and chemical properties. Here, we show in vivo favorable biodistribution and enhanced therapeutic efficacy of cisplatin (CDDP) encapsulated within ultra-short single-walled carbon nanotube capsules (CDDP@US-tubes) using three different human breast cancer xenograft models. In general, the CDDP@US-tubes demonstrated greater efficacy in suppressing tumor growth than free CDDP in both MCF-7 cell line xenograft and BCM-4272 patient-derived xenograft (PDX) models. The CDDP@US-tubes also demonstrated a prolonged circulation time compared to free CDDP which enhanced permeability and retention (EPR) effects resulting in significantly more CDDP accumulation in tumors, as determined by platinum (Pt) analysis via inductively-coupled plasma mass spectrometry (ICP-MS).
Keywords: Cisplatin, single-walled carbon nanotube, drug delivery, breast cancer, nanotechnology
Graphical abstract
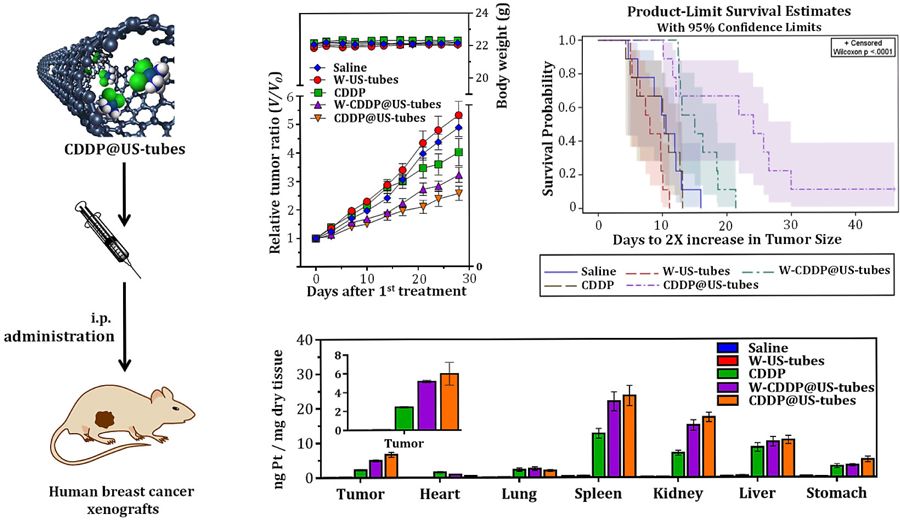
1. Introduction
Despite some advancement over the past few decades in conventional chemotherapy, the efficacy of the vast majority of cancer therapeutics is often limited for reasons such as low water solubility, rapid elimination, nonspecific biodistribution, rapid breakdown in vivo, insufficient accumulation in tumor tissue, undesirable side effects, and development of resistance. These limitations necessitate the development of more efficient ways to administer anticancer drugs systemically to more selectively target tumor tissue, thereby improving efficacy while minimizing undesirable side effects. Over the last decade, with advances in nanotechnology and nanomedicine, numerous nanoparticle-based drug delivery systems have been developed to enhance tumor-specific delivery. One of the major advantages of using nanoparticles as drug delivery vehicle for cancer therapy is that nanoparticles, upon systemic injection, can preferentially accumulate in tumor tissue by taking advantage of irregular tumor vasculature or vessel leakiness. This phenomenon has been termed the enhanced permeability and retention (EPR) effect [1]. Among diverse classes of nanomaterials, carbon nanotubes (CNTs) are of special interest in the area of drug delivery due to their numerous unique physical and chemical properties such as high surface area, high aspect ratio, high thermal conductivity, and remarkable optical and electronic properties [2–5]. Owing to their high surface area, CNTs enable the engineering of surface modification for a variety of therapeutics molecules either by specific adsorption or by covalent bioconjugation [6–11]. Due to their hollow cylindrical structure, CNTs also provide internal cavities which are capable of accommodating small molecules or ions [12– 14]. While the toxicological effects of CNTs themselves have been widely debated in the literature [15–22], it has been recently shown that appropriately functionalized and highly-purified single-wall CNTs (SWCNTs) can be nontoxic and well-tolerated in vivo [23]. Several reports have demonstrated that CNTs readily cross cell membranes due to their intrinsic lipophilic character and high aspect ratio (needle-like structure), and thus are able to transport biological molecules including drug molecules, proteins, plasmid DNA and siRNA into cells [11,24–29].
Over the years, a number of CNT-based drug delivery systems have been explored. In these systems, drug molecules have been mainly attached onto the surface or sidewalls of the nanotubes either by specific adsorption or by covalent attachment [6,11,30,31]. In addition to surface attachment, it has been shown that such drug molecules can be encapsulated within the interior cavity of CNTs [12,13,32–34] to provide an insulating environment for drug molecules. This feature prevents degradation and leakage and other unwanted interaction in vivo before the drug reaches its target sites. Recent theoretical studies based on molecular dynamics simulation have also demonstrated that drug molecules can stay within nanotubes for a long period of time during circulation due to the organization of water molecules outside the nanotubes, while the release of drug molecules from nanotubes can be favored near the cell membrane because of advantageous electrostatic interactions of nanotubes with hydrophilic parts of the cell [33,35]. This drug encapsulation approach also preserves the external surface of CNTs for further chemical modification for desired cancer cell targeting with peptide and/or antibodies. Although the ideal length of CNTs for biomedical application is uncertain, ultra-short single-walled carbon nanotubes (US-tubes, ~1.4 nm × 20 – 80 nm) which are produced from full-length SWCNTs via a fluorination and pyrolysis procedure (Figure S1) [36], may be especially well suited for bioactive agent delivery due to their short and relatively uniform-lengths (ca. 95% • 5500 nnmm)) which could help them avoid the reticuloendotheial system (RES), while enhancing their cellular uptake properties and eventual elimination profiles. US-tubes, with sidewall defects from the chemical cutting process (Figure S1) [36], have proven a convenient platform as nanocapsules for the loading and containment of ions, molecules, and drugs whose cytotoxicity may be sequestered within the US-tubes [13,37–40]. Additionally, it has been demonstrated that the exterior surface of US-tubes can be modified with chemical moieties for enhanced solubilization [41], with peptides for biological targeting purposes [42], and with monoclonal antibodies (MAbs) for the specific targeting of cancer cells. Moreover, a medical imaging agent derived from US-tubes (Gadonanotubes) has been shown to translocate into the cells without exhibiting significant toxicity [43]. Finally, one recent study [23] has shown that highly-purified US-tubes are well-tolerated in vivo by Swiss mice, even at very high doses (~ 0.5 g kg-1 b.w.).
We recently developed a new CNT-based drug delivery platform for the treatment of cancer that is comprised of US-tubes loaded with the chemotherapeutic drug cisplatin (CDDP) or (CDDP@US-tubes) [13]. In these previous studies, the encapsulation of CDDP within US-tubes was achieved by a loading procedure that is reproducible as detailed in the Materials & Methods section and as outlined in Figure S2. The resulting CDDP@US-tube material was characterized extensively by several microscopic and spectroscopic methods (Figure S3). Moreover, it was also demonstrated that CDDP@US-tubes release CDDP upon dialysis in PBS at 37 °C much more slowly when CDDP@US-tubes are wrapped with Pluronic®-F108 surfactant (W-CDDP@US-tube), which is likely due to the Pluronic wrapping around the US-tubes covering the sidewall defects sites and ends of the US-tubes as a sheath to help prevent premature drug release (Figure S4). It was shown that the CDDP@US-tube exhibited greater efficacy against MCF-7 and MDA-MB-231 breast cancer cell lines in vitro, when compared to free CDDP. Finally, it was also shown that the US-tube platform assists the delivery of encapsulated CDDP into drug-resistant cells, which suggests that CDDP@US-tubes help to overcome CDDP resistance by increasing drug accumulation in otherwise resistant cell. In this study, we report in vivo biodistribution and therapeutic efficacy of cisplatin encapsulated within US-tubes (as CDDP@US-tubes) using three different human breast cancer xenograft models.
2. Materials And Methods
2.1. Sample Preparation
US-tubes, CDDP@US-tubes and pluoronic-F108-wrapped CDDP@US-tubes (W-CDDP@US-tubes) were prepared as previously reported [13]. Briefly, full-length SWCNTs (Carbon Solution Inc.), produced by the electric-arc discharge method, were cut into US-tubes by fluorination followed by pyrolysis at 1000 °C under an inert atmosphere.[36] US-tubes were first purified in concentrated HCl for 1h by bath-sonication to remove amorphous carbon and metal catalyst impurities (nickel and yttrium), then chemically reduced by a metallic Na°/THF reduction procedure to produce debundled US-tubes [44]. Next, the US-tubes were refluxed for 5 min in 6N HNO3 and then repeatedly washed with deionized water. The debundled US-tubes were dispersed in deionized water via bath-sonication for 60 min, and then CDDP was added to the US-tube suspension and vigorously stirred for 24 h, which was then left undisturbed overnight whereupon CDDP-loaded US-tubes (CDDP@US-tubes) flocculated from solution. The CDDP@US-tubes were collected by filtration on a glass filter, washed excessively with deionized water to remove all exterior CDDP from outer surface of the US-tubes, as judged from platinum analysis on the filtrate aliquots by inductive-coupled plasma optical emission spectroscopy (ICP-OES) and then dried at 80 °C. The concentration of CDDP within US-tubes was calculated by quantifying the Pt concentration of CDDP@US-tube samples via ICP-OES. As an average weight percentage, CDDP@US-tubes contained 9.80 % wt (± 0.88) of CDDP. Extensive characterization of this CDDP@US-tubes material has been also reported previously [13]. W-US-tubes and W-CDDP@US-tubes samples were prepared by suspending either dry US-tubes or CDDP@US-tube samples in a 0.17% (w/v) Pluronic®-F108 surfactant solution via probe sonication for 2 min, followed by centrifugation at 3200 rpm for 10 min (3x) to remove unsuspended CDDP@US-tubes. Pluronic®-F108 is a neutrally-charged, non-cytotoxic surfactant that is commonly used to administer CNT materials for in vitro and in vivo testing.
2. 2. Establishment of Xenografts and Treatment
All animal experiments were performed under a protocol approved by Baylor College of Medicine Institutional Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Experimental Animals. MCF-7 and MDA-MB-231 human breast cell line xenografts were generated by inoculation of 1×106 of either MCF-7 or MDA-MB-231 cells suspended in 20 µL Matrigel-PBS into the epithelium-free “cleared” fat pads of SCID/Beige mice [45]. Human, patient-derived breast cancer xenografts (PDX) (BCM-4272 transplant) were established as described previously [46]. For each xenograft experiment, a total of forty human breast-tumor-bearing mice were used. The tumor volumes were monitored weekly by caliper measurement. When the average tumor volume reached at least 100 mm3, mice were randomly divided into five groups of eight mice each. Animals in Group 1(G1) received saline solution (0.9 % w/v NaCl) as a control. Group 2 (G2) was treated with empty Pluronic-wrapped US-tubes as a vehicle control. Group 3 (G3) was treated with dissolved CDDP and diluted with saline solution. Group 4 (G4) was treated with Pluronic-wrapped CDDP@US-tubes. Finally, Group 5 (G5) was treated with CDDP@US-tubes dispersed in saline solution via bath-sonication. The final concentration of CDDP in G3, G4, and G5 was normalized to be 2.5 mg/kg mouse body weight. The quantity of US-tubes was the same for G2, G4, G5, with the concentration being calculated from their weight% values obtained by ICP-OES for Ni and Y, which remains within US-tubes as remnant metal catalysts from the growth of the original full-lengths SWCNTs from which the US-tubes are derived. For treatment, 100 µL of different formulations of CDDP, CDDP@US-tubes, W-US-tubes or W-CDDP@US-tubes in saline was administered via i.p. once a week. Treatment was performed weekly for a period of 4 weeks. The administration dose was normalized to be 2.5 mg CDDP per kg (2.5 mg/kg). Tumor size was measured by a caliper two times a week and calculated as the volume = [(tumor length) × (tumor width) 2] / 2. At the end of the experiment, the animals were sacrificed and their tissues collected to obtain a quantitative measurement of the biodistribution of CDDP and US-tube samples in each tissue by quantifying the platinum (Pt) and yttrium (Y) concentration in each sample via inductively-coupled plasma mass spectrometry (ICP-MS). Pieces of tissue (heart, liver, lung, spleen, kidney, and tumor) were taken from animals of each group, formalin fixed, paraffin embedded, frozen, and processed to 4-µm slides by a tissue-processing core laboratory at BCM. One slide from each tissue sample was stained with hematoxylin and eosin.
2. 3. Biodistribution and Blood Circulation Studies
For biodistribution studies, a total of 45 MDA-MB-231 tumor-bearing mice (tumor size, ~200 mm3) were randomly divided into three groups (CDDP, CDDP@US-tubes, and W-CDDP@US-tubes) of 15 mice each. Three mice from each treatment group were sacrificed at 4, 24, 48, 72, and 168 h after receiving 100 µL of either CDDP, CDDP@US-tube or W-CDDP@US-tube samples in saline at a dose of 2.5 mg CDDP per kg mouse via an i.p injection. The blood samples were collected by cardiac puncture for the blood circulation studies. After harvest, the organs/tissues were immediately frozen in liquid nitrogen, lyophilized to dryness and weighted. Blood and the dried tissue samples were digested by first heating with concentrated HNO3 and then by 26% HClO3. The concentration of Pt and Y was then determined by ICP-MS for blood circulation and biodistribution analysis.
2. 4. Inductively-Coupled Plasma Emission Mass Spectrometry (ICP-MS)
ICP-MS analyses were carried out using a Perkin Elmer Elan Optima 9000 DV instrument with a CDD detector. The quantity of CDDP in each sample was determined by measuring the platinum concentration, which was detected at 265.95 nm. For all ICP-MS measurements, the collected samples were transferred into glass vials and digested first using 10 ml of concentrated HNO3 at elevated temperature (~ 230 °C), and then with 2 ml 26% HClO3. Following digestion, the samples were diluted with 2% v/v trace metal grade HNO3. The concentrations were determined from an average of five scans for each sample. Germanium (371.029 nm) was used as the internal standard.
2. 5. Histology Staining
For histology analysis, a piece of tissue (tumor, heart, liver, lung, spleen, and kidney) was taken from the animals in each group, formalin fixed, paraffin embedded, frozen, and processed to 4-µm slides by the tissue-processing core laboratory at BCM. To understand the effects of treatment, one slide from each tissue sample was stained with hematoxylin and eosin (H&E), standard fluorescent terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) (apoptosis assay), and Ki67 (proliferation assay) staining procedures to assess histological alterations by microscopy.
2. 6. Statistical Analysis
All the data are expressed as mean values ± standard error (SE). Graphs were constructed and all statistical analyses were performed using GraphPad© Prism 6.0 (GraphPad Software, Inc.). Statistical comparison between two experimental groups was done using a t test (unpaired, two-sided). Comparison of multiple groups was made with two-way ANOVA using Tukey’s multiple comparison test. Time to tumor doubling (tripling for MDA-MB-231) was computed for each animal. The data were summarized with Kaplan-Meier curves and 95% confidence regions (log-log transform) and compared using the generalized Wilcoxon test. Pairwise comparisons with Sidak p-value adjustment were used to determine which groups differed from each other. A value of less than 0.05 (p < 0.05) was used for statistical significance.
3. Results
3.1. Tumor Growth Suppressing Effect of CDDP@US-tubes on MCF-7 Xenografts in SCID/Beige Mice
In previous in vitro studies [13], we determined the efficacy of CDDP@US-tubes against two different breast cancer cell lines (MCF-7 and MDA-MB-231), which are known to be CDDP resistant and showed that US-tubes helped increase the accumulation of the drug in cells compared to free CDDP. To examine the ability of US-tubes to deliver anticancer drugs and to determine the efficacy of CDDP@US-tubes on tumor growth suppression in mice in vivo, a series of treatment studies were conducted using three different breast cancer xenograft models (MCF-7, MDA-MB-231, and BCM-4272).
To determine the in vivo efficacy of the CDDP@US-tubes, we first tested MCF-7 xenografts established in SCID/Beige mice (as detailed in the Materials and Methods Section). Figure 1 displays the comparative in vivo therapeutics efficacy of the given treatment groups. As shown in Figure 1A, a time-related increase in tumor volume was observed in all treatment groups except for the CDDP@US-tube treated group. Compared to the control injection, W-US-tube injected mice showed a similar tumor growth rate, although it was not as effective as CDDP alone or the W-CDDP@US-tubes. As showed in Figure 1A, there was also no significant difference between the mean body weight changes in all treatment groups over the course of the experiment. The mean fold increase in tumor volume (from day 0 to day 28) of CDDP@US-tube treated mice was significantly lower compared to CDDP alone or W-CDDP@US-tubes (Figure 1B). Five out of eight tumors from mice treated with CDDP@US-tubes failed to grow at all, while all control groups got larger. Determination of the tumor growth doubling time (Figure 1C) revealed that CDDP@US-tubes significantly improved mouse survival.
Figure 1.
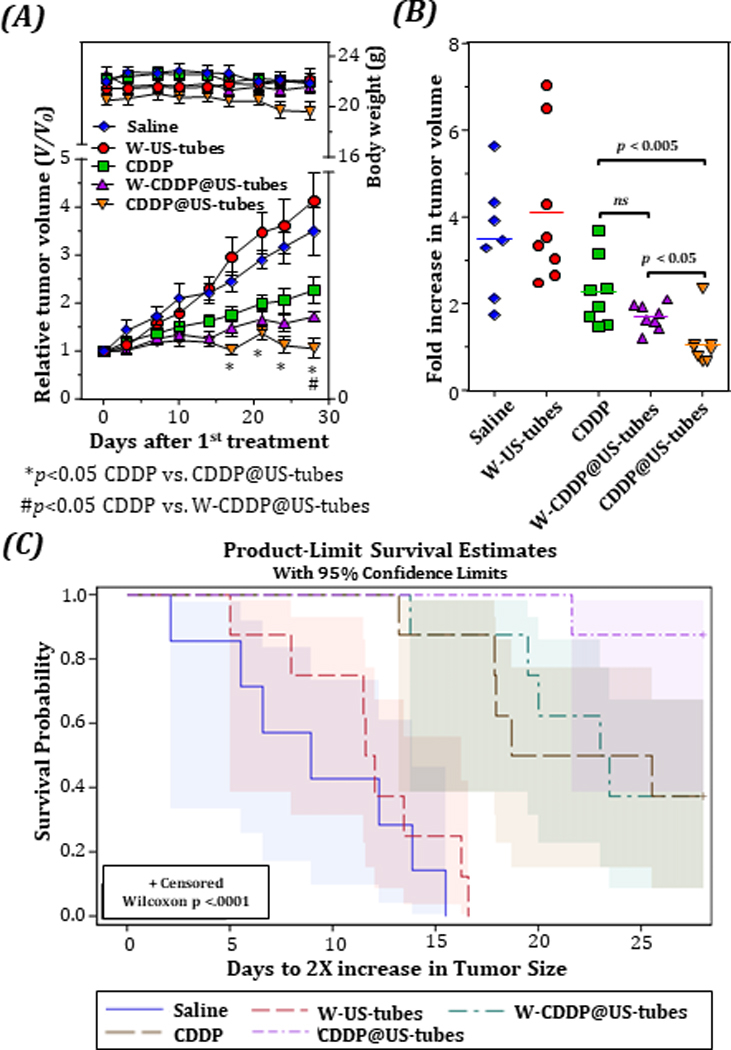
Suppression of tumor growth by intraperitoneal injection of CDDP@US-tubes to an MCF-7 breast cancer xenograft. (A), Tumor growth of MCF-7 tumor-bearing mice after receiving different treatments as indicated. (* and # indicates p < 0.05, unpaired, two-sided t-test, n=8; error bars= s.e.) The mean body weight changes is also plotted in A. Fold increase in tumor volume (B) and Kaplan-Meier survival curves (C) depicting the tumor doubling times for different treatment groups.
At the end of the experiment, animals from each treatment group were sacrificed and tissues were collected to obtain a quantitative measurement of the accumulation of CDDP and US-tubes samples in each tissue by quantifying Pt (for CDDP) and Y (for US-tubes) in each tissue samples via inductively coupled mass spectrometry (ICP-MS) analysis. ICP-MS analysis revealed that the CDDP@US-tube treated mice had significantly more Pt in their tumor, spleen and stomach compare to other treatment groups (Figure 2A). By analyzing the Y concentration in tissue, we also determined that the level of Y detected in spleen, liver, kidneys, and stomach was found to be greatest, while the amount found in other organs was much lower (Figure 2B). The analysis showed that CDDP@US-tube treated mice had significantly more Y in their spleen and stomach compare to mice treated with empty W-US-tube, and W-CDDP@US-tubes. To understand the effects of treatment, a piece of tissue including tumor, liver, lungs, spleen, and kidneys that was taken from the animals in each treatment group was stained by H&E staining to assess histological alterations. From the microscopy examination, no obvious damage was noticed in the tissues (Figure S5).
Figure 2.
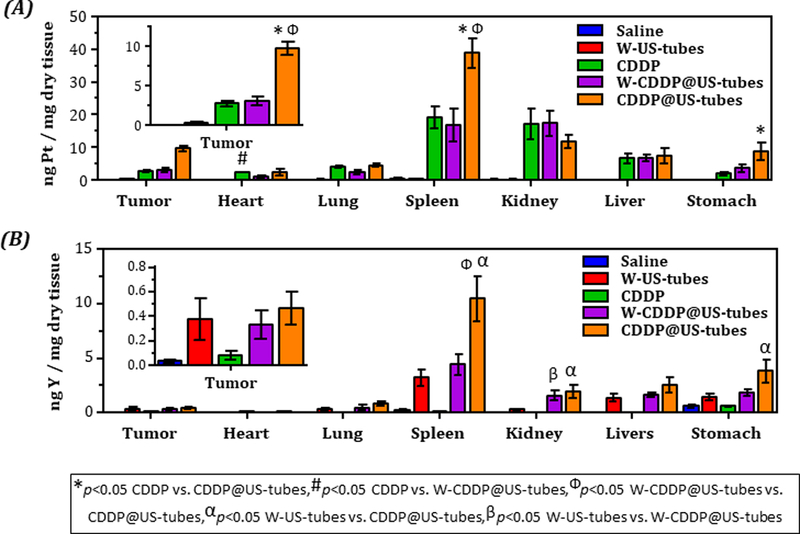
Platinum and yttrium concentration in tissue from MCF-7 tumor-bearing mice treated over a period of ~ 45 d with weekly saline, W-US-tubes, CDDP, CDDP@US-tubes, and W-CDDP@US-tubes administration via i.p. injection. (A) Concentration of platinum (from cisplatin) and (B) yttrium (from US-tubes) in tissue (mean ± s.e., n = 8). Statistically significant different (p < 0.05) are marked with symbols as indicated above (unpaired, two-sided t-test).
3. 2. Tumor Growth Suppressing Effect of CDDP@US-tubes on MDA-MB-231 Xenografts in SCID/Beige Mice
In the second experiment, the in vivo anticancer efficacy of the CDDP@US-tubes material was tested in mice bearing MDA-MB-231 human breast cancer xenograft in a manner similar to that described above for MCF-7 xenograft. The tumors were established as described in Materials & Method Section and i.p. injections were started when the average tumor volume reached ~100 mm3. Changes in relative tumor volume and body weight are shown in Figure 3A. As plotted in Figure 3A, the mean body weights of mice in all treatment groups showed a similar increase and there was no significant difference between the treatment groups over the course of the experiment. As also seen in Figure 3A, the control and the empty W-US-tubes treated group showed a fast tumor growth pattern, resulting in an approximately 12-fold increase in their tumor volume (from day 0 to day 28). No significant difference in the relative tumor growth ratio (V/V0) of these two treatment groups confirmed that the empty W-US-tubes themselves do not affect the tumor growth in mice. The relative tumor growth ratios (V/V0) of CDDP, W-CDDP@US-tubes, and CDDP@US-tubes were similar (Figure 3A) and there was no statistically significant difference between the treatment groups. On the other hand, the mean fold increase in tumor volume (from day 0 to day 28) of CDDP@US-tube (7.45 ± 0.77) treated mice was significantly lower than mice in the control treatment group (13.03 ± 2.02, p = 0.03) (Figure 3B), while the fold increase in mice treated with free CDDP (9.09 ± 1.73) and W-CDDP@US-tube (9.57 ± 1.88) was not significant different (p = 0.16, and p = 0.24, respectively). The determination of tumor growth tripling time shown that the median survival of the CDDP@US-tube and W-CDDP@US-tube treatment groups, (10 d and 14 d, respectively) was longer than that of mice treated with free CDDP, 7 d (Figure 3C).
Figure 3.
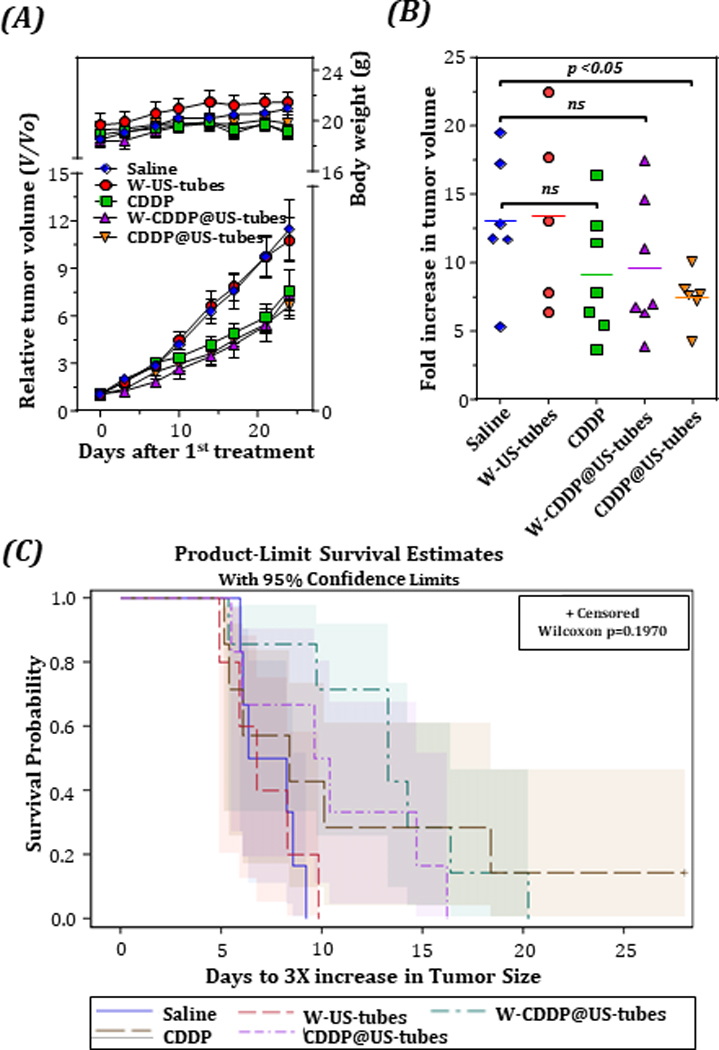
Suppression of tumor growth by intraperitoneal injection of CDDP@US-tubes to an MDA-MB-231 breast cancer xenograft. (A), Tumor growth of MDA-MB-231 tumor-bearing mice after receiving different treatments as indicated. ( p < 0.05, unpaired, two-sided t-test, n=8; error bars= s.e.) The mean body weight changes is also plotted in A. Fold increase in tumor volume (B) and Kaplan-Meier survival curves (C) depicting the tumor tribling (3X) times for different treatment groups.
Although the efficacy of free CDDP, W-CDDP@US-tube, and CDDP@US-tube on the MDA-MB-231 xenograft was found to be similar (Figure 3A), the ICP-MS analysis of tissue collected from the animals at the end of the experiment revealed that the mice treated with CDDP@US-tubes (4.28 ± 1.17 ng Pt/mg dry tissue, p = 0.02) and W-CDDP@US-tubes (2.16 ± 0.16 ng Pt/mg dry tissue, p = 0.01) had significantly more Pt in their tumor than mice treated with free CDDP (1.17 ± 0.18 ng Pt/mg dry tissue) (Figure 4A). Consistent with the MCF-7 xenograft study, the level of Y in mice treated with W-US-tubes, W-CDDP@US-tubes, and CDDP@US-tubes was found highest in spleen followed by stomach, liver, and kidney, confirming the higher retention of these materials in these organs.
Figure 4.
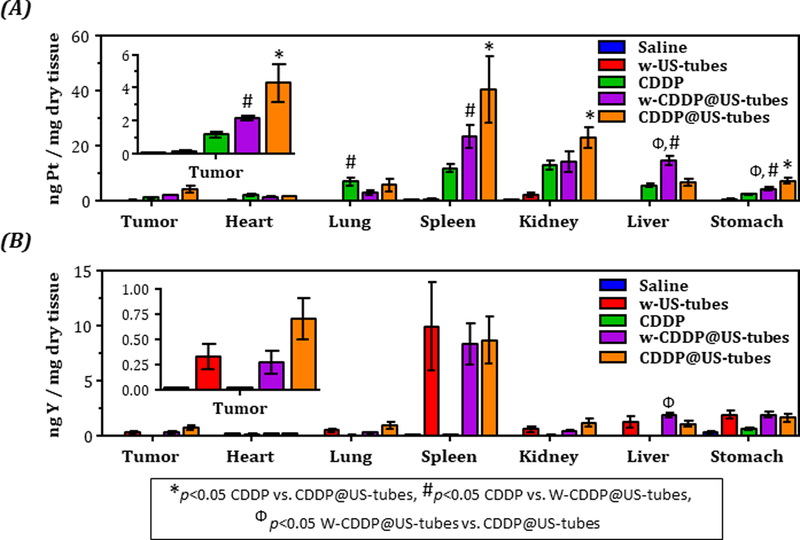
Platinum and yttrium concentration in tissue from MDA-MB-231 tumor-bearing mice treated over a period of ~ 45 d with weekly saline, W-US-tubes, CDDP, CDDP@US-tubes, and W-CDDP@US-tubes administration via i.p. injection. (A) Concentration of platinum (from cisplatin) and (B) yttrium (from US-tubes) in tissue (mean ± s.e., n = 8). Statistically significant different (p<0.05) are marked with symbols as indicated above (unpaired, two-sided t-test).
3. 3. Tumor Growth Suppressing Effect of CDDP@US-tubes on BCM-4272 PDX Xenografts in SCID/Beige Mice
To extend our studies beyond cell line xenografts, we evaluated the efficacy of CDDP@US-tubes material on a human breast cancer patient-derived xenograft (PDX) model, where human tumor tissue is surgically resected and transplanted directly into immune-deficient mice. Because traditional cell lines have been cultured for many years and have accumulated many genetic/epigenetic alterations, there is some argument as to whether their response to treatment accurately reflects patient responses. We have shown that PDX models show a high degree of similarity in response as the tumor of origin in the patient when challenged with a comparable agent [46]. Because of their ability to predict clinical tumor response, PDXs are increasingly being considered to be more relevant in vivo preclinical models, and thus, are expected to increase the success of identifying new active antitumor agents for clinical trials [47]. BCM-4272 xenografts, were established as described previously [46]. As shown in Figure 5A, the tumors in the control and the empty W-US-tubes groups showed a slow but stable growth. Compared with the control group, the CDDP treatment group slowed down tumor growth, but it was not as effective as the W-CDDP@US-tubes or CDDP@US-tubes treatment groups with both of these treatment groups showing a considerable tumor suppression (regression) (p < 0.005) compared to CDDP treatment. There were no significant body weight changes between treatment groups until the end of the experiment. The mean fold increase in tumor volume (from day 0 to day 28) of control (4.88 ± 0.32) and W-US-tubes treated group (5.32 ± 0.49) were similar, however the CDDP@US-tubes (2.57 ± 0.25) treated group displayed a significantly lower fold increase than those of free CDDP treated mice (4.02 ± 0.48, p = 0.01) which was also not significantly different than the control group (4.88 ± 0.32, p = 0.15). The fold increase in tumor volume for the W-CDDP@US-tubes (3.21 ± 0.26) was found to be significantly lower than the control group (p = 0.001), while it was not statistically different than the free CDDP treatment group (Figure 5B). As a result, an increased survival of animals was observed in mice treated with CDDP@US-tubes or W-CDDP@US-tubes compared to mice treated with free CDDP (Figure 5C).
Figure 5.
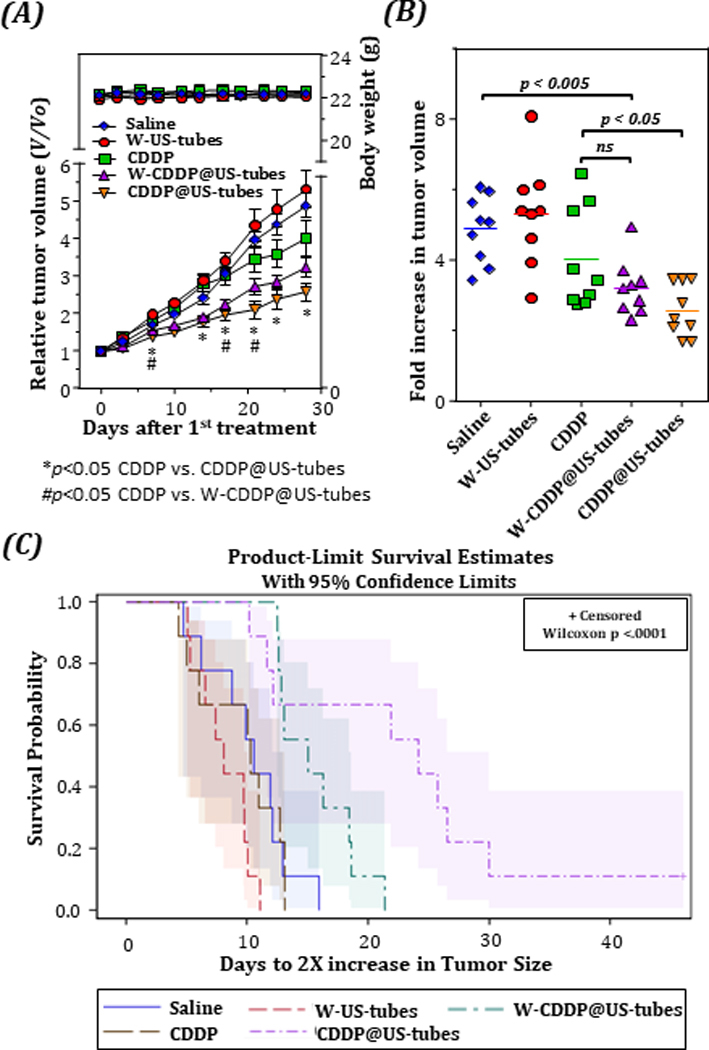
Suppression of tumor growth by intraperitoneal injection of CDDP@US-tubes to an Patient-derived, 4272 transplant, xenograft. (A), Tumor growth of Patient-derived tumor-bearing mice after receiving different treatments as indicated. (*p < 0.05 CDDP@US-tubes vs. CDDP, #p < 0.05 W-CDDP@US-tubes vs. CDDP, unpaired, two-sided t-test, n=9; error bars= s.e.) The mean body weight changes is also plotted in A. Fold increase in tumor volume (B) and Kaplan-Meier survival curves (C) depicting the tumor doubling times for different treatment groups.
ICP-MS analysis of the tissues showed that CDDP@US-tube and W-CDDP@US-tube treated mice had significantly more Pt in their tumor, spleen and kidney than the mice treated with free CDDP, while the free CDDP treated mice displayed higher Pt accumulation in heart compare to other treatment groups (Figure 6A). Consistent with measurement from MCF-7, and MDA-MB-231 xenograft studies, the Y concentration was found highest in the spleen followed by kidney, liver and stomach, indicating higher retention of US-tube materials in these organs (Figure 6B). Compared with W-CDDP@US-tubes and W-US-tubes treated mice, the level of Y in the spleen was found significantly higher in mice treated with CDDP@US-tubes. To understand the effects of treatment, a piece of tissue including tumor, liver, lungs, spleen, and kidneys that was taken from the animals in each treatment group was stained by H&E staining to assess histological alterations. From the microscopy examination, no obvious damage was noticed in the tissues (Figure S6).
Figure 6.
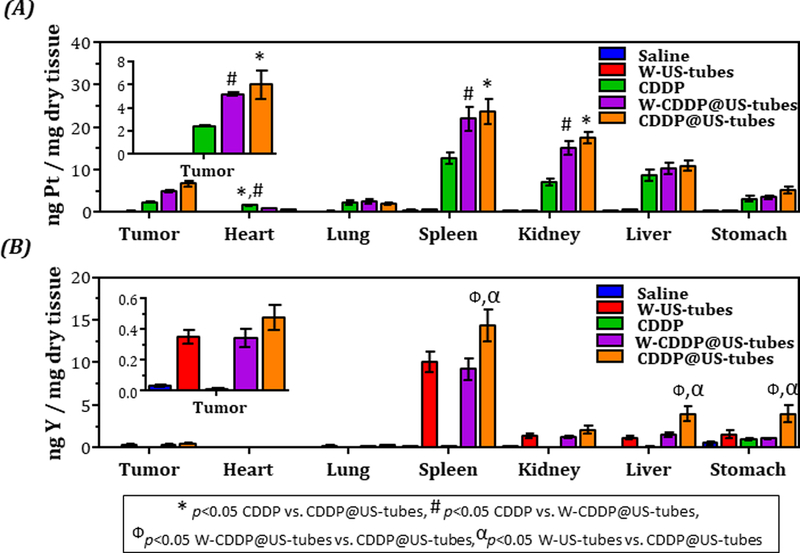
Platinum and yttrium concentration in tissue from Patient-derived tumor-bearing mice treated over a period of ~ 45 d with weekly saline, w-US-tubes, CDDP, CDDP@US-tubes, and w-CDDP@US-tubes administration via i.p. injection. (A) Concentration of platinum (from cisplatin) and (B) yttrium (from US-tubes) in tissue (mean ± s.e., n = 8). Statistically significant different (p < 0.05) are marked with symbols as indicated above (unpaired, two-sided t-test).
3. 4. Blood Circulation and the Biodistribution Studies
To understand the treatment efficacy of CDDP@US-tube materials, it was important to investigate the biodistribution of the CDDP@US-tubes in a time-dependent manner. The biodistribution studies have been conducted after a single i.p. administration of CDDP, W-CDDP@US-tubes, and CDDP@US-tubes (CDDP concentration 2.5 mg/kg) for MDA-MB-231 tumor-bearing mice by determining the Pt (from CDDP) and the Y (from US-tubes) concentrations in plasma and in main organs/tissues (tumor, liver, spleen, kidney, heart, lungs, and stomach) via ICP-MS analysis at different time points (4, 24, 48, 72, and 168 h). The mice from each treatment groups were sacrificed and harvested for blood circulation and biodistribution analysis (see Materials and Methods Section). Figure 7A illustrates the comparative blood circulation data of free CDDP, W-CDDP@US-tubes and CDDP@US-tubes for MDA-MB-231 tumor-bearing mice as a function of time. ICP-MS analysis of the blood samples revealed that CDDP@US-tubes and W-CDDP@US-tubes exhibited a much longer blood circulation time than CDDP, which was quickly cleared from the blood. At 4 h, the Pt concentration in blood for mice treated with CDDP, W-CDDP@US-tubes, and CDDP@US-tubes was 271.55 ± 21.51 ng/mL, 250.40 ± 57.03 ng/mL, and 456.00 ± 71.40 ng/mL, respectively. In comparison, CDDP-treated mice showed a significantly lower Pt concentration in the plasma at 24 h (102.78 ± 9.92 ng/mL) which gradually declined until the end of the experiment, while the mice treated with W-CDDP@US-tubes and CDDP@US-tubes maintained relatively higher Pt concentrations (221.11 ± 33.26 ng/mL and 327.03 ± 80.01 ng/mL, respectively) at 24 h, and continued to be relatively higher until the end of the study. The concentration of Pt in blood at 168 h p.i., the last point of evaluation, in mice treated with W-CDDP@US-tubes and CDDP@US-tubes was 2.3-fold (48.27 ± 7.41 ng/mL) and 3.8-fold (78.62 ± 12.83 ng/mL), respectively, higher than in mice treated with free CDDP (20.88 ± 5.14 ng/mL, p<0.05). One phase exponential decay analysis of the Pt concentration in blood over time for each treatment group showed that the plasma half-lifes are markedly extended for W-CDDP@US-tubes and CDDP@US-tubes (80 h and 130 h, respectively) compare to free CDDP (24.5 h) (Figure S7).
Figure 7.
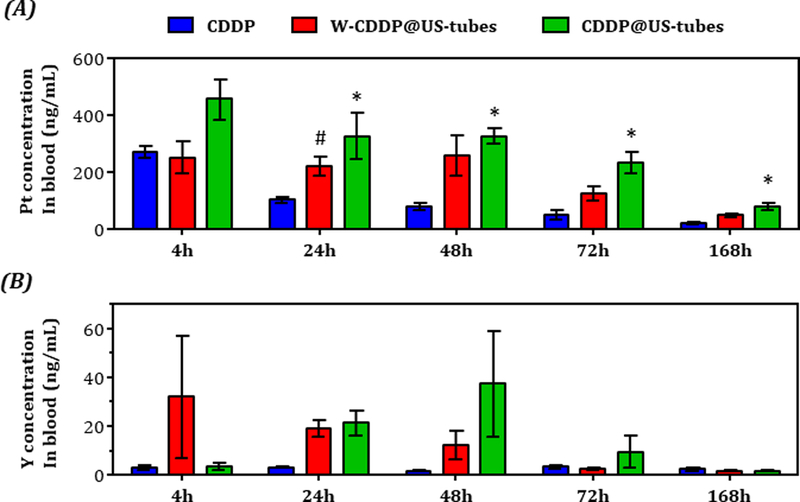
Blood circulation data of CDDP, W-CDDP@US-tubes, and CDDP@US-tubes in MDA-MB-231 tumor-bearing mice were determined by measuring (A) Pt (from CDDP) and (B) Y (from US-tubes) concentration in blood via ICP-MS at different time points post injection. (*p < 0.05 CDDP vs. CDDP@US-tubes, #p < 0.05 CDDP vs. W-CDDP@US-tubes, unpaired, two-sided, t-test, n=3, error bars = s.e)
ICP-MS analysis showed that the Pt concentration of CDDP-treated mice in each tissue gradually declined over time. The Pt levels detected in liver, spleen, and kidney was highest, while the amount measured for other organs was much lower. On the contrary, the mice treated with W-CDDP@US-tubes and CDDP@US-tubes displayed a significantly different biodistribution than mice treated with free CDDP (Figure 8), with much higher Pt concentrations in tumor, spleen, and kidney, while being similar in lung, stomach, heart, and liver (Figure 8A).
Figure 8.
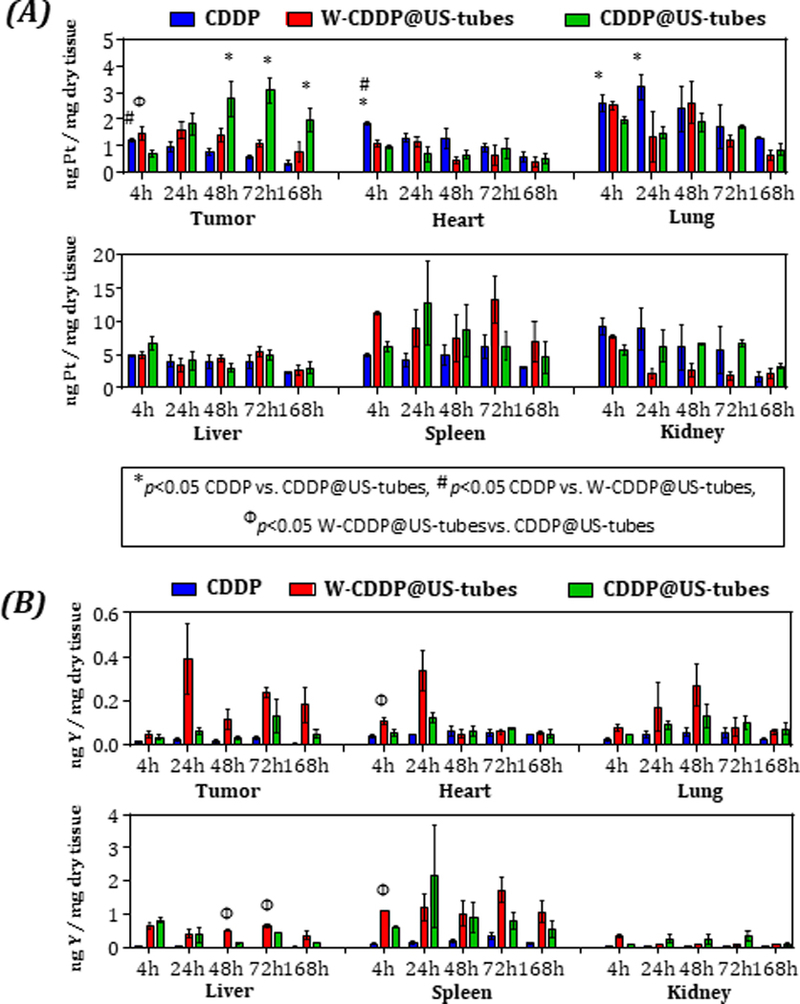
Time-dependent in vivo biodistribution of intraperitoneally injected CDDP, W-CDDP@US-tubes, and CDDP@US-tubes in MDA-MB-231 tumor bearing mice. (A) Concentration of platinum (from CDDP), (B) Concentration of yttrium (from US-tubes) in tissue (mean ± s.e., n = 3). Statistically significant different (p < 0.05) are marked with symbols as indicated above (unpaired, two-sided t-test).
As shown in Figure 8A, the concentration of Pt in tumors of all treatment groups were similar at 4 h, and then started to increase, and became significantly higher at 48 h and peaked at 72 h in CDDP@US-tubes-treated mice (3.09 ± 0.48 ng/mg dry tissue), which was more than 5-fold higher than in mice treated with free CDDP (0.56 ± 0.08 ng/mg dry tissue) and approximately 3-fold higher than W-CDDP@US-tube-treated mice (1.08 ± 0.12 ng/mg dry tissue). There was also a significant difference between the CDDP and W-CDDP@US-tube treatment groups at 72 h (p = 0.024). At 168 h, the Pt concentration levels in tumors of all treatment groups decreased; however, the accumulation of Pt for the CDDP@US-tubes (1.96 ± 0.44 ng/mg dry tissue) was still significantly higher than that in the CDDP treated groups (0.34 ± 0.1 ng/mg dry tissue, p = 0.022).
The Pt concentrations detected in kidney, liver, and spleen after free CDDP administration was found to be highest, while the amount found in other organs was much lower. The Pt concentration in kidney of mice treated with free CDDP (9.27 ± 1.26 ng/mg dry tissue), and W-CDDP@US-tubes (7.64 ± 0.18 ng/mg dry tissue) were the highest and found to be significantly higher than mice treated with CDDP@US-tubes (4.87 ± 0.05 ng/mg dry tissue) at 4 h and then gradually declined until the end of the study. In contrast, the CDDP@US-tubes treated mice displayed a relatively constant Pt concentration in kidney up to 48 h, followed by a decreased and significantly higher Pt concentration at 72 h (4.14 ± 0.3 ng/mg dry tissue) compare with both the free CDDP (1.38 ± 0.49 ng/mg dry tissue, p = 0.005) and W-CDDP@US-tube (2.16 ± 0.82 ng/mg dry tissue, p = 0.02) treatment groups. Although the Pt levels detected in the liver were lower than those measured in the kidneys in the first 24 h, in the long-term (at 168 h), the Pt levels were greater in the liver than in the kidneys. The liver Pt levels in all treatment groups displayed a similar trend at all evaluated time points except at 4 h, with a greater Pt concentration for CDDP@US-tubes (6.05 ± 0.32 ng/mg dry tissue) compared to free CDDP (4.87 ± 0.11 ng/mg dry tissue, p = 002).
Figure 8A shows that the CDDP@US-tubes and W-CDDP@US-tubes afforded considerably higher Pt accumulation in the spleen compare to free CDDP at 48 h and 72 h, while the mice treated with free CDDP exhibited higher Pt accumulation only at 4 h, and then gradually decreased over time. As shown in Figure 8B, ICP-MS analysis confirmed the high retention of the CDDP@US-tube and W-CDDP@US-tube materials in the spleen, where the Y concentration was higher than for all other organs.
4. Discussion
Although CDDP is one of the most potent anticancer drugs, its use in cancer therapy is hampered due to severe side effects caused by the lack of selectivity and resistance issues which is mainly attributed to reduced drug accumulation in tumor cells [48–50]. Because of its reactive nature, CDDP has limited bioavailability. Upon administration, CDDP rapidly binds to albumin and other plasma proteins and spontaneously degrades in the bloodstream, leading to irreversible deactivation of nearly 90% of the injected dose, and it is rapidly cleared from the blood by glomerular excretion [51]. Because this form of CDDP is inactive, it has no therapeutic effect [52]. It is important to note here that during current clinical administration of CDDP, only ~1% of administered dose reaches its cellular target by the EPR effect [53]. Thus, an ideal drug delivery systems is expected to not only increase the concentration of CDDP in tumor cells, but also protect it from plasma deactivation.
Recently, we developed a new CNT-based drug delivery system that is comprised of CDDP encapsulated within the US-tubes (CDDP@US-tubes) that showed enhanced in vitro antitumoral efficacy against two different breast cancer cell lines, MCF-7 and MDA-MB-231, which are known to be CDDP resistant against free CDDP [13]. Although the in vivo biodistribution of platinum-based drugs (CDDP and an inert Pt(IV) complex) trapped within the interior of multi-walled CNTs (MWCNTs) has been previously reported [32], the present study is the first to explore both the biodistribution and in vivo efficacy of CDDP encapsulated within a SWCNT (US-tube) material in mice for three different breast cancer xenograft models.
The in vivo biodistribution and pharmacokinetic studies of CNTs have been extensively studied by a number of research groups using different CNT materials, different surface functionalization, different tracking techniques, and after different administration methods (i.p. vs. i.v.) resulting in varying and sometimes controversial results [54]. Previous in vivo toxicity assessment studies have demonstrated that high doses of US-tubes (up to 1000 mg/kg b.w.) after i.p. administration are well tolerated by Swiss mice. The study also showed that small aggregates and well-individualized US-tubes reached the systemic circulation and subsequently accumulated in a variety of tissues and cells, by way of the lymphatic system and that these US-tubes eventually escaped the reticuloendothelial system to be excreted though the kidney and bile ducts, as evidenced by US-tubes in the urine and feces [23]. It was also observed that the US-tubes had a strong interaction with organ surfaces, and that contrary to full-length SWCNTs, they rapidly diffused inside the organs. Although small aggregates of US-tubes did not induce granuloma formation, they persisted inside cells for up to 5 months after administration. From this study, it was reasoned that i.p. administration of CDDP@US-tube materials would also avoid possible complications associated with i.v. injection such as mechanical blockage of the vasculature system. In addition, i.p. administration of free CDDP has been also studied extensively, and the dose of free CDDP that can be given safely by i.p. is substantially higher than by i.v. administration. Moreover, the concentration of active cisplatin in the plasma is not decreased after i.p. administration compared to i.v. injection. To determine the pharmacokinetics and tissue specific distribution of CDDP vs. US-tubes, we quantified the Pt (from CDDP) and Y (in the US-tubes as remnant metal catalyst) concentrations in samples via ICP-MS analysis which allowed the determination of precise Pt and Y concentrations in tissue samples because of its high sensitivity (1 ppb) and specificity due to the absence of background Pt and Y in biological samples. To our knowledge, this is the first study that has measured remnant metal catalyst concentration (Y in our study) to trace and quantitatively determine blood clearance and biodistribution behavior of a CNTs material in vivo. To date, most of the in vivo biodistribution and pharmacokinetic studies of CNT materials reported in the literature have been determined using radiolabels or spectroscopic tags for indirect detection of CNTs [55–58]. In other studies, rather insensitive CNT Raman scattering intensities have been sometimes employed as a tracer [59].
The results of the present study demonstrate that CDDP@US-tubes have greater tumor suppression efficacy than free CDDP in both MCF-7 and BCM-4272 PDX breast tumor models, while it was found to have a similar effect in a MDA-MB-231 xenograft. In all three xenograft studies, the CDDP@US-tubes have afforded higher CDDP uptake in tumors compared to free CDDP. The similar tumor suppression effects of CDDP@US-tubes and CDDP on the MDA-MB-231 xenograft is possibly due to more aggressive tumor growth of the MDA-MB-231 xenograft compared to MCF-7 and BCM-4272 PDX xenografts, which resulted in considerable lower Pt concentration in the tumor per mg of dry tissue. In fact, it was observed that the mean fold increase (from day 0 to day 28) in tumor volume of the control (saline) treated group for the MDA-MB-231 xenograft (11.415 ± 1.906) was notably higher compared to the MCF-7 (3.486 ± 0.494) and BCM-4272 PDX (4.88 ± 0.319) xenografts.
The reason for the higher CDDP uptake in tumors for CDDP@US-tubes and W-CDDP@US-tubes is likely due to their prolonged blood circulation time compare to free CDDP which facilitates substantial tumor targeting by the EPR effect. Consistent with previous reports, we observed a short blood circulation time for free CDDP [60,61]. However, the CDDP@US-tubes and W-CDDP@US-tubes displayed significantly higher and an almost constant Pt concentration in blood with time. This observation can be explained by the long-term retention of CDDP within US-tubes throughout the circulation time. As previously observed and reported[62], blood proteins coat the US-tubes in vivo which apparently limits the release of CDDP from both the CDDP@US-tube and the W-CDDP@US-tube samples until this coating is likely stripped away during transit through the cell membrane. This view agrees well with previously observed extensive aggregation of US-tubes within stem cells [43] and some recent theoretical reports [33,35]. The adsorption of free CDDP by various organs occurred very rapidly, especially for kidney, liver and spleen. However, the mice treated with W-CDDP@US-tubes and CDDP@US-tubes displayed a significantly different biodistribution than mice treated with free CDDP, with much higher Pt accumulation in tumor, spleen, and kidney, while being similar in lung, stomach, heart, and liver. By also analyzing the Y concentration in various tissues, we determined a higher retention of US-tubes in spleen, liver, kidneys, and stomach, while the amount found in other organs was much less.
From the above encouraging results, we believe that it might be possible to control CDDP release from CDDP@US-tubes by wrapping them with covalently-attached activatable cancer-specific peptide sequences instead of surfactant molecules. Recently, a catalytic method to functionalize US-tubes with various amino acids and peptides was reported which improved solubility, biocompatibility, and biological targeting capabilities [42]. In future work, using this functionalization protocol, a peptide chain, containing an enzyme-specific substrate sequence for a cancer cell antigen, such as the well-known prostate specific antigen (PSA) and/or matrix metalloproteinase-2 (MMP-2) might be synthesized and covalently-attached to CDDP@US-tubes. This peptide chain should cover the sidewall defect sites and ends of the US-tubes as a sheath and help prevent premature drug release until the peptide is cleaved upon PSA and/or MMP-2 activation either within or in the extracellular environment of cancer cells, as shown schematically in Figure S8. Inserting different cancer specific peptide sequences into the peptide chain could also allow for the treatment of a wide variety of cancers. Using this strategy, peptide-wrapped CDDP@US-tube could become a smart drug delivery platform that will not only activate CDDP drug release within or in the extracellular environment of cancer cells but could also allow administration of a higher dose of CDDP without producing undesirable side effects.
5. Conclusion
In this study, in vivo biodistribution and therapeutic efficacy of CDDP@US-tube materials have been evaluated against three different breast cancer xenograft mouse (SCID/Beige) models, and found to exhibit greater efficacy in suppressing tumor growth than free CDDP for both a MCF-7 cell line xenograft model and a BCM-4272 patient-derived xenograft (PDX) model. The studies have shown the potential of the US-tube platform to assist delivery of encapsulated CDDP by increasing the accumulation of drug in cancer resistance cells. The CDDP@US-tube materials also demonstrated a prolonged circulation time compared to free CDDP, which enhanced permeability and retention (EPR) effects to produce significantly more CDDP accumulation in tumors. Together, these studies have suggested that tumor specific accumulation of CDDP might also be further enhanced by covalently functionalizing the CDDP@US-tube platform with activatable cancer-specific peptide sequences.
Supplementary Material
STATEMENT OF SIGNIFICANT.
Over the past decade, drug-loaded nanocarriers have been widely fabricated and studied to enhance tumor specific delivery. Among the diverse classes of nanomaterials, carbon nanotubes (CNTs), or more specifically ultra-short single-walled carbon nanocapsules (US-tubes), have been shown to be a popular, new platform for the delivery of various medical agents for both imaging and therapeutic purposes. Here, for the first time, we have shown that US-tubes can be utilized as a drug delivery platform in vivo to deliver the chemotherapeutic drug, cisplatin (CDDP) as CDDP@US-tubes. The studies have demonstrated the ability of the US-tube platform to promote the delivery of encapsulated CDDP by increasing the accumulation of drug in breast cancer resistance cells, which reveals how CDDP@US-tubes help overcome CDDP resistance.
Acknowledgment
A.G. gratefully acknowledges the Turkish Ministry of Education and the Scientific and Technological Research Council of Turkey for financial support.
Funding Sources
This work was supported by the Welch Foundation (Grant C-0627) (L.J.W.), NIH/NCI Specialized Program for Research Excellence (SPORE) grant P50-CA50183 (M.T.L.), NIH/NCI U54 Grant CA149196 (M.T.L.), NIH/NCI grant RO1-CA127857 (M.T.L.), and NIH/NCI Baylor
College of Medicine Cancer Center Grant P30-CA125123.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure of Potential Conflicts of Interest
M.T.L is a Founder and Limited Partner in StemMed Ltd, and a Manager in StemMed Holdings LP, but is not compensated by the company.
REFERENCES
- [1].Matsumura Y, Maeda H, A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs, Cancer Res 46 (1986) 6387–6392. [PubMed] [Google Scholar]
- [2].Baughman RH, Zakhidov AA, de Heer WA, Carbon nanotubes--the route toward applications, Science 297 (2002) 787–792. doi: 10.1126/science.1060928. [DOI] [PubMed] [Google Scholar]
- [3].Bianco A, Kostarelos K, Prato M, Applications of carbon nanotubes in drug delivery, Current Opinion in Chemical Biology 9 (2005) 674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- [4].Yang W, Thordarson P, Gooding JJ, Ringer SP, Braet F, Carbon nanotubes for biological and biomedical applications, Nanotechnology 18 (2007) 412001. doi: 10.1088/0957-4484/18/41/412001. [DOI] [Google Scholar]
- [5].Bianco A, Kostarelos K, Partidos CD, Prato M, Biomedical applications of functionalised carbon nanotubes, Chem. Commun (2005) 571–577. doi: 10.1039/B410943K. [DOI] [PubMed] [Google Scholar]
- [6].Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ, Soluble Single-Walled Carbon Nanotubes as Longboat Delivery Systems for Platinum(IV) Anticancer Drug Design, J. Am. Chem. Soc 129 (2007) 8438–8439. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dhar S, Liu Z, Thomale J, Dai H, Lippard SJ, Targeted Single-Wall Carbon Nanotube-Mediated Pt(IV) Prodrug Delivery Using Folate as a Homing Device, J. Am. Chem. Soc 130 (2008) 11467–11476. doi: 10.1021/ja803036e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H, Drug delivery with carbon nanotubes for in vivo cancer treatment, Cancer Res 68 (2008) 6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kam NWS, Liu Z, Dai H, Carbon Nanotubes as Intracellular Transporters for Proteins and DNA: An Investigation of the Uptake Mechanism and Pathway, Angewandte Chemie International Edition 45 (2006) 577–581. doi: 10.1002/anie.200503389. [DOI] [PubMed] [Google Scholar]
- [10].Kam NWS, O’Connell M, Wisdom JA, Dai H, Carbon nanotubes as multifunctional biological transporters and near-infrared agents for selective cancer cell destruction, PNAS 102 (2005) 11600–11605. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Liu Z, Sun X, Nakayama-Ratchford N, Dai H, Supramolecular Chemistry on Water-Soluble Carbon Nanotubes for Drug Loading and Delivery, ACS Nano 1 (2007) 50–56. doi: 10.1021/nn700040t. [DOI] [PubMed] [Google Scholar]
- [12].Ren Y, Pastorin G, Incorporation of Hexamethylmelamine inside Capped Carbon Nanotubes, Advanced Materials 20 (2008) 2031–2036. doi: 10.1002/adma.200702292. [DOI] [Google Scholar]
- [13].Guven A, Rusakova IA, Lewis MT, Wilson LJ, Cisplatin@US-tube carbon nanocapsules for enhanced chemotherapeutic delivery, Biomaterials 33 (2012) 1455–1461. doi: 10.1016/j.biomaterials.2011.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hong SY, Tobias G, Al-Jamal KT, Ballesteros B, Ali-Boucetta H, Lozano-Perez S, Nellist PD, Sim RB, Finucane C, Mather SJ, Green MLH, Kostarelos K, Davis BG, Filled and glycosylated carbon nanotubes for in vivo radioemitter localization and imaging, Nat Mater 9 (2010) 485–490. doi: 10.1038/nmat2766. [DOI] [PubMed] [Google Scholar]
- [15].Kayat J, Gajbhiye V, Tekade RK, Jain NK, Pulmonary toxicity of carbon nanotubes: a systematic report, Nanomedicine 7 (2011) 40–49. doi: 10.1016/j.nano.2010.06.008. [DOI] [PubMed] [Google Scholar]
- [16].Morimoto Y, Hirohashi M, Ogami A, Oyabu T, Myojo T, Todoroki M, Yamamoto M, Hashiba M, Mizuguchi Y, Lee BW, Kuroda E, Shimada M, Wang W-N, Yamamoto K, Fujita K, Endoh S, Uchida K, Kobayashi N, Mizuno K, Inada M, Tao H, Nakazato T, Nakanishi J, Tanaka I, Pulmonary toxicity of well-dispersed multi-wall carbon nanotubes following inhalation and intratracheal instillation, Nanotoxicology 6 (2012) 587–599. doi: 10.3109/17435390.2011.594912. [DOI] [PubMed] [Google Scholar]
- [17].Kagan VE, Konduru NV, Feng W, Allen BL, Conroy J, Volkov Y, Vlasova II, Belikova NA, Yanamala N, Kapralov A, Tyurina YY, Shi J, Kisin ER, Murray AR, Franks J, Stolz D, Gou P, Klein-Seetharaman J, Fadeel B, Star A, Shvedova AA, Carbon nanotubes degraded by neutrophil myeloperoxidase induce less pulmonary inflammation, Nat Nano 5 (2010) 354–359. doi: 10.1038/nnano.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Takagi A, Hirose A, Nishimura T, Fukumori N, Ogata A, Ohashi N, Kitajima S, Kanno J, Induction of mesothelioma in p53+/− mouse by intraperitoneal application of multiwall carbon nanotube, J Toxicol Sci 33 (2008) 105–116. [DOI] [PubMed] [Google Scholar]
- [19].Muller J, Delos M, Panin N, Rabolli V, Huaux F, Lison D, Absence of carcinogenic response to multiwall carbon nanotubes in a 2-year bioassay in the peritoneal cavity of the rat, Toxicol. Sci 110 (2009) 442–448. doi: 10.1093/toxsci/kfp100. [DOI] [PubMed] [Google Scholar]
- [20].Dumortier H, Lacotte S, Pastorin G, Marega R, Wu W, Bonifazi D, Briand J-P, Prato M, Muller S, Bianco A, Functionalized Carbon Nanotubes Are Non-Cytotoxic and Preserve the Functionality of Primary Immune Cells, Nano Lett 6 (2006) 1522–1528. doi: 10.1021/nl061160x. [DOI] [PubMed] [Google Scholar]
- [21].Sayes CM, Liang F, Hudson JL, Mendez J, Guo W, Beach JM, Moore VC, Doyle CD, West JL, Billups WE, Ausman KD, Colvin VL, Functionalization density dependence of single-walled carbon nanotubes cytotoxicity in vitro, Toxicology Letters 161 (2006) 135–142. doi: 10.1016/j.toxlet.2005.08.011. [DOI] [PubMed] [Google Scholar]
- [22].Saha D, Heldt CL, Gencoglu MF, Vijayaragavan KS, Chen J, Saksule A, A study on the cytotoxicity of carbon-based materials, Materials Science and Engineering: C 68 (2016) 101–108. doi: 10.1016/j.msec.2016.05.094. [DOI] [PubMed] [Google Scholar]
- [23].Kolosnjaj-Tabi J, Hartman KB, Boudjemaa S, Ananta JS, Morgant G, Szwarc H, Wilson LJ, Moussa F, In Vivo Behavior of Large Doses of Ultrashort and Full-Length Single-Walled Carbon Nanotubes after Oral and Intraperitoneal Administration to Swiss Mice, ACS Nano 4 (2010) 1481–1492. doi: 10.1021/nn901573w. [DOI] [PubMed] [Google Scholar]
- [24].Pantarotto D, Briand J-P, Prato M, Bianco A, Translocation of bioactive peptides across cell membranes by carbon nanotubes, Chem. Commun. (Camb.) (2004) 16–17. doi: 10.1039/b311254c. [DOI] [PubMed] [Google Scholar]
- [25].Liu Y, Wu D-C, Zhang W-D, Jiang X, He C-B, Chung TS, Goh SH, Leong KW, Polyethylenimine-grafted multiwalled carbon nanotubes for secure noncovalent immobilization and efficient delivery of DNA, Angew. Chem. Int. Ed. Engl 44 (2005) 4782–4785. doi: 10.1002/anie.200500042. [DOI] [PubMed] [Google Scholar]
- [26].Kam NWS, Liu Z, Dai H, Functionalization of Carbon Nanotubes via Cleavable Disulfide Bonds for Efficient Intracellular Delivery of siRNA and Potent Gene Silencing, J. Am. Chem. Soc 127 (2005) 12492–12493. doi: 10.1021/ja053962k. [DOI] [PubMed] [Google Scholar]
- [27].Liu Z, Winters M, Holodniy M, Dai H, siRNA delivery into human T cells and primary cells with carbon-nanotube transporters, Angew. Chem. Int. Ed. Engl 46 (2007) 2023–2027. doi: 10.1002/anie.200604295. [DOI] [PubMed] [Google Scholar]
- [28].Yaron PN, Holt BD, Short PA, Lösche M, Islam MF, Dahl KN, Single wall carbon nanotubes enter cells by endocytosis and not membrane penetration, Journal of Nanobiotechnology 9 (2011) 45. doi: 10.1186/1477-3155-9-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Boyer PD, Ganesh S, Qin Z, Holt BD, Buehler MJ, Islam MF, Dahl KN, Delivering Single-Walled Carbon Nanotubes to the Nucleus Using Engineered Nuclear Protein Domains, ACS Appl. Mater. Interfaces 8 (2016) 3524–3534. doi: 10.1021/acsami.5b12602. [DOI] [PubMed] [Google Scholar]
- [30].Wu W, Wieckowski S, Pastorin G, Benincasa M, Klumpp C, Briand J-P, Gennaro R, Prato M, Bianco A, Targeted Delivery of Amphotericin B to Cells by Using Functionalized Carbon Nanotubes, Angewandte Chemie International Edition 44 (2005) 6358–6362. doi: 10.1002/anie.200501613. [DOI] [PubMed] [Google Scholar]
- [31].Boyer PD, Shams H, Baker SL, Mofrad MRK, Islam MF, Dahl KN, Enhanced intracellular delivery of small molecules and drugs via non-covalent ternary dispersions of single-wall carbon nanotubes, J. Mater. Chem. B 4 (2016) 1324–1330. doi: 10.1039/C5TB02016F. [DOI] [PubMed] [Google Scholar]
- [32].Li J, Pant A, Chin CF, Ang WH, Ménard-Moyon C, Nayak TR, Gibson D, Ramaprabhu S, Panczyk T, Bianco A, Pastorin G, In vivo biodistribution of platinum-based drugs encapsulated into multi-walled carbon nanotubes, Nanomedicine: Nanotechnology, Biology and Medicine 10 (2014) 1465–1475. doi: 10.1016/j.nano.2014.01.004. [DOI] [PubMed] [Google Scholar]
- [33].Mejri A, Vardanega D, Tangour B, Gharbi T, Picaud F, Encapsulation into Carbon Nanotubes and Release of Anticancer Cisplatin Drug Molecule, J. Phys. Chem. B 119 (2015) 604–611. doi: 10.1021/jp5102384. [DOI] [PubMed] [Google Scholar]
- [34].Hilder TA, Hill JM, Modeling the Loading and Unloading of Drugs into Nanotubes, Small 5 (2009) 300–308. doi: 10.1002/smll.200800321. [DOI] [PubMed] [Google Scholar]
- [35].Khalifi ME, Bentin J, Duverger E, Gharbi T, Boulahdour H, Picaud F, Encapsulation capacity and natural payload delivery of an anticancer drug from boron nitride nanotube, Phys. Chem. Chem. Phys 18 (2016) 24994–25001. doi: 10.1039/C6CP01387B. [DOI] [PubMed] [Google Scholar]
- [36].Gu Z, Peng H, Hauge RH, Smalley RE, Margrave JL, Cutting Single-Wall Carbon Nanotubes through Fluorination, Nano Lett 2 (2002) 1009–1013. doi: 10.1021/nl025675+. [DOI] [Google Scholar]
- [37].Sitharaman B, Kissell KR, Hartman KB, Tran LA, Baikalov A, Rusakova I, Sun Y, Khant HA, Ludtke SJ, Chiu W, Laus S, Tóth E, Helm L, Merbach AE, Wilson LJ, Superparamagnetic gadonanotubes are high-performance MRI contrast agents, Chem. Commun. (Camb.) (2005) 3915–3917. doi: 10.1039/b504435a. [DOI] [PubMed] [Google Scholar]
- [38].Hartman KB, Hamlin DK, Wilbur DS, Wilson LJ, 211AtCl@US-tube nanocapsules: a new concept in radiotherapeutic-agent design, Small 3 (2007) 1496–1499. doi: 10.1002/smll.200700153. [DOI] [PubMed] [Google Scholar]
- [39].Rivera EJ, Sethi R, Qu F, Krishnamurthy R, Muthupillai R, Alford M, Swanson MA, Eaton SS, Eaton GR, Wilson LJ, Nitroxide Radicals@US-Tubes: New Spin Labels for Biomedical Applications, Advanced Functional Materials 22 (2012) 3691–3698. doi: 10.1002/adfm.201102826. [DOI] [Google Scholar]
- [40].Rivera EJ, Tran LA, Hernández-Rivera M, Yoon D, Mikos AG, Rusakova IA, Cheong BY, Cabreira-Hansen M. da G., Willerson JT, Perin EC, Wilson LJ, Bismuth@US-tubes as a potential contrast agent for X-ray imaging applications, J. Mater. Chem. B 1 (2013) 4792–4800. doi: 10.1039/C3TB20742K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gizzatov A, Dimiev A, Mackeyev Y, Tour JM, Wilson LJ, Highly water soluble multilayer graphene nanoribbons and related honey-comb carbon nanostructures, Chem. Commun 48 (2012) 5602–5604. doi: 10.1039/C2CC31407J. [DOI] [PubMed] [Google Scholar]
- [42].Mackeyev Y, Hartman KB, Ananta JS, Lee AV, Wilson LJ, Catalytic Synthesis of Amino Acid and Peptide Derivatized Gadonanotubes, J. Am. Chem. Soc 131 (2009) 8342–8343. doi: 10.1021/ja900918x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tran LA, Krishnamurthy R, Muthupillai R, da Graça Cabreira-Hansen M, Willerson JT, Perin EC, Wilson LJ, Gadonanotubes as Magnetic Nanolabels for Stem Cell Detection, Biomaterials 31 (2010) 9482–9491. doi: 10.1016/j.biomaterials.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Ashcroft JM, Hartman KB, Mackeyev Y, Hofmann C, Pheasant S, Alemany LB, Wilson LJ, Functionalization of individual ultra-short single-walled carbon nanotubes, Nanotechnology 17 (2006) 5033. doi: 10.1088/0957-4484/17/20/001. [DOI] [Google Scholar]
- [45].Deome KB, Faulkin LJ, Bern HA, Blair PB, Development of mammary tumors from hyperplastic alveolar nodules transplanted into gland-free mammary fat pads of female C3H mice, Cancer Res 19 (1959) 515–520. [PubMed] [Google Scholar]
- [46].Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M, Wong H, Fuqua SW, Contreras A, Gutierrez C, Huang J, Mao S, Pavlick AC, Froehlich AM, Wu M-F, Tsimelzon A, Hilsenbeck SG, Chen ES, Zuloaga P, Shaw CA, Rimawi MF, Perou CM, Mills GB, Chang JC, Lewis MT, A Renewable Tissue Resource of Phenotypically Stable, Biologically and Ethnically Diverse, Patient-Derived Human Breast Cancer Xenograft Models, Cancer Res 73 (2013) 4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zhang X, Lewis MT, Establishment of Patient-Derived Xenograft (PDX) Models of Human Breast Cancer, in: Auwerx J, Brown SD, Justice M, Moore DD, Ackerman SL, Nadeau J (Eds.), Current Protocols in Mouse Biology, John Wiley & Sons, Inc, Hoboken, NJ, USA, 2013. http://www.currentprotocols.com/WileyCDA/CPUnit/refId-mo120140.html (accessed December 3, 2013). [DOI] [PubMed] [Google Scholar]
- [48].Kelland LR, Preclinical Perspectives on Platinum Resistance, Drugs 59 (2000) 1–8. doi: 10.2165/00003495-200059004-00001. [DOI] [PubMed] [Google Scholar]
- [49].Siddik ZH, Cisplatin: mode of cytotoxic action and molecular basis of resistance, Oncogene 22 (2003) 7265–7279. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- [50].Fuertes MA, Alonso C, Pérez JM, Biochemical Modulation of Cisplatin Mechanisms of Action: Enhancement of Antitumor Activity and Circumvention of Drug Resistance, Chem. Rev 103 (2003) 645–662. doi: 10.1021/cr020010d. [DOI] [PubMed] [Google Scholar]
- [51].C. H, J. I, van der V. Wj, Platinum complexes in cancer medicine: pharmacokinetics and pharmacodynamics in relation to toxicity and therapeutic activity., Cancer Surv 17 (1992) 189–217. [PubMed] [Google Scholar]
- [52].Comenge J, Sotelo C, Romero F, Gallego O, Barnadas A, Parada TG-C, Domínguez F, Puntes VF, Detoxifying Antitumoral Drugs via Nanoconjugation: The Case of Gold Nanoparticles and Cisplatin, PLoS ONE 7 (2012) e47562. doi: 10.1371/journal.pone.0047562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Cepeda V, Fuertes M, Castilla J, Alonso C, Quevedo C, Perez J, Biochemical Mechanisms of Cisplatin Cytotoxicity, Anti-Cancer Agents in Medicinal Chemistry 7 (2007) 3–18. doi: 10.2174/187152007779314044. [DOI] [PubMed] [Google Scholar]
- [54].Liu Z, Tabakman S, Welsher K, Dai H, Carbon Nanotubes in Biology and Medicine: In vitro and in vivo Detection, Imaging and Drug Delivery, Nano Res 2 (2009) 85–120. doi: 10.1007/s12274-009-9009-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Wang H, Wang J, Deng X, Sun H, Shi Z, Gu Z, Liu Y, Zhaoc Y, Biodistribution of Carbon Single-Wall Carbon Nanotubes in Mice, Journal of Nanoscience and Nanotechnology 4 (2004) 1019–1024. doi: 10.1166/jnn.2004.146. [DOI] [PubMed] [Google Scholar]
- [56].Singh R, Pantarotto D, Lacerda L, Pastorin G, Klumpp C, Prato M, Bianco A, Kostarelos K, Tissue biodistribution and blood clearance rates of intravenously administered carbon nanotube radiotracers, PNAS 103 (2006) 3357–3362. doi: 10.1073/pnas.0509009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].McDevitt MR, Chattopadhyay D, Kappel BJ, Jaggi JS, Schiffman SR, Antczak C, Njardarson JT, Brentjens R, Scheinberg DA, Tumor targeting with antibody-functionalized, radiolabeled carbon nanotubes, J. Nucl. Med 48 (2007) 1180–1189. doi: 10.2967/jnumed.106.039131. [DOI] [PubMed] [Google Scholar]
- [58].Al Faraj A, Fauvelle F, Luciani N, Lacroix G, Levy M, Cremillieux Y, Canet-Soulas E, In vivo biodistribution and biological impact of injected carbon nanotubes using magnetic resonance techniques, Int J Nanomedicine 6 (2011) 351–361. doi: 10.2147/IJN.S16653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu Z, Davis C, Cai W, He L, Chen X, Dai H, Circulation and long-term fate of functionalized, biocompatible single-walled carbon nanotubes in mice probed by Raman spectroscopy, PNAS 105 (2008) 1410–1415. doi: 10.1073/pnas.0707654105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Newman MS, Colbern GT, Working PK, Engbers C, Amantea MA, Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice, Cancer Chemother Pharmacol 43 (1999) 1–7. doi: 10.1007/s002800050855. [DOI] [PubMed] [Google Scholar]
- [61].Johnsson A, Olsson C, Nygren O, Nilsson M, Seiving B, Cavallin-Stahl E, Pharmacokinetics and tissue distribution of cisplatin in nude mice: platinum levels and cisplatin-DNA adducts, Cancer Chemother. Pharmacol 37 (1995) 23–31. doi: 10.1007/BF00685625. [DOI] [PubMed] [Google Scholar]
- [62].Ge C, Du J, Zhao L, Wang L, Liu Y, Li D, Yang Y, Zhou R, Zhao Y, Chai Z, Chen C, Binding of blood proteins to carbon nanotubes reduces cytotoxicity, PNAS 108 (2011) 16968–16973. doi: 10.1073/pnas.1105270108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.