

Supplemental Digital Content is available in the text
Keywords: β-ureidopropionase deficiency, genetic analysis, mutation, UPB1
Abstract
β-Ureidopropionase (βUP) deficiency is an autosomal recessive disease caused by abnormal changes in the pyrimidine-degradation pathway. This study aimed to investigate the mutation of β-ureidopropionase gene (UPB1) gene and clinical features of 7 Chinese patients with βUP deficiency.
We reported 7 Chinese patients with βUP deficiency who were admitted at Tianjin Children's Hospital. Urine metabolomics was detected by gas chromatography–mass spectrometry (GC–MS). Then genetic testing of UPB1 was conducted by polymerase chain reaction (PCR) method.
The patients presented with developmental delay, seizures, autism, abnormal magnetic resonance imaging, and significantly elevated levels of N-carbamyl-β-alanine and N-carbamyl-β-aminoisobutyric acid in urine. Subsequent analysis of UPB1 mutation revealed 2 novel missense mutations (c.851G>T and c.853G>A), 3 previously reported mutations including 2 missense mutations (c.977G>A and c.91G>A) and 1 splice site mutation (c.917-1 G>A).
The results suggested that the UPB1 mutation may contribute to βUP deficiency. The c.977G>A is the most common mutation in Chinese population.
1. Introduction
Pyrimidine nucleotides are essential for various biological processes such as the synthesis of RNA, DNA, phospholipids, and glycogen, as well as the sialylation and glycosylation of proteins.[1] Meanwhile, pyrimidines play an important role in the regulation of the central nervous system. Metabolic changes affecting pyrimidine levels may lead to abnormal neurological activity.[2] The pyrimidine bases uracil and thymine are degraded in 3 enzymatic steps. Dihydropyrimidine dehydrogenase is an initial and rate-limiting enzyme which catalyzes uracil and thymine reduction to 5,6-dihydrouracil and 5,6- dihydrothymine, respectively. The second enzyme, dihydropyrimidinase, catalyzes the hydrolytic ring opening of the pyrimidines. The third step results in the conversion of N-carbamy1-β-alanine and N-carbamyl-β-aminoisobutyric into β-alanine and β-aminoisobutyric acid, ammonia, as well as carbon dioxide by β-ureidopropionase (βUP).[3] Related studies have demonstrated that patients with βUP deficiency often present with elevated concentrations of N-carbamyl-β-alanine and N-carbamyl-aminoisobutyric acid, as well as moderately elevated levels of dihydrouracil and dihydrothymine in plasma, cerebrospinal fluid (CSF), and urine.[4–7] In Japan, gas chromatography–mass spectrometry (GC–MS) is used to detect these abnormalities.[6,8,9]
βUP deficiency is a rare autosomal recessive disease caused by mutations in the βUP gene, that is, β-ureidopropionase gene (UPB1). The gene is located in chromosome 22q11.2 and consists of 10 exons that encode a 384 amino acid protein. To date, approximately 30 patients worldwide have been reported to have this disease.[10–13] Moreover, the sequence of UPB1 has enabled the detection of the defect at the molecular level.[14,15] Meanwhile, only 15 mutations have been reported, including 11 missense mutations and 4 splice site mutations.[9,13,14,16]
In this study, we performed genetic and biochemical analyses in 7 Chinese patients with βUP deficiency as identified by GC–MS. Then we also analyzed the follow-up clinical characteristics of these patients.
2. Subjects and methods
2.1. Subjects
The subjects were 7 Chinese families from the Department of Neurosurgery of Tianjin Children's Hospital in China. The 7 familial cases were diagnosed independently by 2 experienced doctors according to the clinical manifestation and GC-MS results. The study protocols were planned according to the guidelines of the 2000 Declaration of Helsinki and the Declaration of Istanbul 2008 and approved by the Ethical Committee of Tianjin Children's Hospital in China. Written informed consent for the use of clinical data and blood samples was signed by the patients’ guardians.
2.2. Pyrimidine bases and degradation products in body fluids
The concentrations of uracil, thymine, dihydrouracil, dihydrothymine, N-carbamyl-β-alanine, and N-carbamy-β-aminoisobutyric acid in urine were determined using GC–MS.
2.3. Genomic DNA extraction and polymerase chain reaction (PCR) amplification of UPB1
Genomic DNA was extracted from the peripheral blood of patients and their parents using the QiAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). All exons and flanking intron regions of UPB1 were amplified using the primers designed by GenBank accession No: NC_000022.10 (Table S1). Amplification was carried out in a 25 μL volume containing 50∼100 ng of DNA, 50 mM KCl, 10 Mm Tris-HCl (pH 8.3), 1.5 Mm MgCl2, 200 μM of each dNTP, 0.5 mΜ of primer, and 1 U of Taq polymerase (TaKaRa Biotechnology Co., Dalian, China). After initial denaturation for 5 minutes at 94 °C, amplification was carried out for 35 cycles (94 °C for 30 seconds, 58 °C for 30 seconds, and 72 °C for 40 seconds) and a final extension step of 5 minutes at 72 °C. The PCR products were separated by 1.5% agarose gel electrophoresis and then sequenced on an ABI 3730 XL Automated DNA Sequencer (Applied Biosystems, Foster City, CA) with the ABI BigDye Terminator v3.1 cycle sequencing kit.
2.4. Bioinformatics analysis
The amino acid sequence of UPB1 in humans was compared with those in other vertebrates by using DNAMAN software. To analyze the perniciousness of the missense mutation, we used 2 online bioinformatic software packages, namely, Polyphen (http://genetics.bwh.harvard.edu/pph2/) and SIFT (http://sift.jcvi.org/), to predict the potential functions of these pathogenic mutations. A damaging tendency is indicated by a Polyphen score asymptotically approaching 1 and a SIFT score asymptotically approaching zero.
3. Results
3.1. Clinical details
Seven patients with βUP deficiency from 7 different Chinese families were investigated to determine the specific etiology. The clinical characteristics of all patients were as follows.
Patient 1 was a 4-year-old girl who was the first child born to nonconsanguineous parents after 39 weeks gestation through cesarean section without prenatal or neonatal complications. She was hospitalized with intermittent vomiting, fever, and diarrhea. No growth and neuropsychiatric deficits were observed. Deep-tendon reflexes were enhanced and Achilles clonus was present. No pathological reflex was observed. No evidence of causative-agent infection in blood, CSF, urine, and stool was found. X-ray, electrocardiogram and abdominal ultrasound were normal, although electroencephalogram (EEG) showed mild abnormality. Late data were unavailable due to lost follow-up results.
Patient 2 was a 2-month-old boy who was the second child born to nonconsanguineous parents. He was born after 33 gestation weeks through normal delivery with a birth weight of 1150 g. He had an elder sister who was diagnosed with cerebral palsy. He had a history of neonatal asphyxiation that improved after treatment. He was hospitalized with jaundice for nearly 2 months and progressive aggravation with occasional clay-like stool. At 2 months, his height was 57 cm, weight was 1750 g, and head circumference was 36 cm. No positive signs were found in neurological examination. Ultrasound showed poor filling of gallbladder and bile-duct dilatation. Other routine tests were normal. He showed no abnormal developmental milestones at follow-up evaluation.
Patient 3 was a 3-day-old boy who was the first child born to nonconsanguineous parents. He was born at full term through normal spontaneous vaginal delivery, but he was hospitalized because of a temporary convulsion. No obvious abnormality was found in neurological examination, and muscle tension was normal. No family history of note or exposure to infectious agent was reported. Blood-gas analysis indicated metabolic acidosis combined with respiratory alkalosis. Elevated pyrimidines and metabolites in urine were detected by GC–MS. His father also suffered from βUP deficiency, but detailed clinical data were unavailable per his own nondisclosure preference. Follow-up investigation indicated that the child had normal growth and development.
Patient 4 was a 5-month-old boy who was the first child born to nonconsanguineous parents at full term through cesarean section. He was hospitalized because of mental retardation. His mother had taken antiabortion drugs in the early stage of pregnancy. No history of mental retardation and convulsion was reported. At the time of presentation (5 months), the child cannot raise his head and turn over. Furthermore, he presented with unique features, including wide eye distance, high palatal arch, polydactyly of right little finger, and polydactyly and syndactyly of the bilateral little toe. According to the parents, his grandfather had polydactyly of right hand. Routine chromosome analysis showed a normal male 46, XY karyotype. Brainstem auditory evoked potential (BAEP) test indicated moderate hearing loss of his left ear. Visual-evoked potential test indicated abnormal bilateral visual pathway. Brain magnetic resonance imaging (MRI) revealed a high signal in the bilateral parietal white matter area about T2-weighted, in addition to a widened ventricle and being outside the brain interval. Increased pyrimidine and metabolite levels in urine were detected by GC–MS. He has developed an inability to look after himself at the follow-up age of 4 years.
Patient 5 was a 6-month-old boy and the first child of nonconsanguineous parents. He was born after 32 gestation weeks through cesarean section because the mother was in a comatose state resulting from pregnancy-induced hypertension syndrome. His birth weight was 1800 g. A history of asphyxia at birth was reported. He presented with developmental delay and muscular hypertonia. At the time of presentation (6 months), he cannot sit by himself and suck his thumb. He was thus initially diagnosed with mental retardation. GC-MS results showed increased pyrimidine and metabolite levels in urine. Growth and development of the child caught up to the normal range at the follow-up age of 4 years.
Patient 6 was a 3-month-old boy who was hospitalized because of fever and cough. He had normal growth, development, and neurological examination, as well as no hereditary-disease history. A CT scan of the throat showed no apparent abnormalities. Lung X-ray examination showed scattered inflammatory solid changes in the right lung, increased lung markings, and thickened right pleural, which suggested pneumonia of the right lung. At 3 months, the patient cannot raise his head so developmental delay was not clinically excluded. He developed an inability to look after himself at the follow-up age of 4 years.
Patient 7 was a 2-year-old boy and the first child born to nonconsanguineous patients at full term through cesarean section. The boy weighed 3900 g and was 53 cm in length. No history of postnatal asphyxiation was reported. He can sit without support at 7 months, crawl at 10 months, and walk independently at 14 months. At 2 years old, he can speak a few simple words. He had poor language comprehension and communicated only with his mother. The psychological assessment scale indicated that he suffered from autism. No family history of convulsion, seizure, and mental retardation existed. Physical and neurological examinations yielded normal results. BAEP test was normal. Brain MRI demonstrated developmental delay of myelin sheath, in addition to thickened mucosa of bilateral maxillary sinus and ethmoid sinus. Increased pyrimidine and metabolite levels in urine were detected by GC-MS. During follow-up period, the child was diagnosed with autism, but his growth and development caught up to the normal range except for language comprehension.
3.2. Pyrimidine bases and metabolite degradation in urine
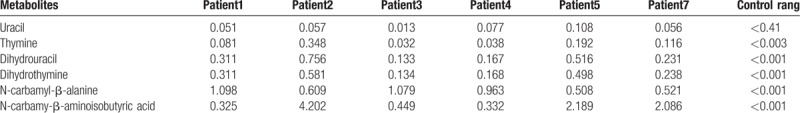
Quantitative analyses of relevant pyrimidines and metabolites in urine was performed by GC–MS. Urine samples from all subjects showed significantly elevated concentrations of N-carbamyl-β-alanine and N-carbamyl-aminoisobutyric acid, as well as moderately elevated levels of dihydrouracil and dihydrothymine (Table 1).
Table 1.
Pyrimidine degradation metabolites of the patients with βUP deficiency.
3.3. Mutation analysis of UPB1
The patient's parents did not permit the collection of blood sample of patient 6. Thus, a total of 6 families with βUP deficiency were included. Genetic testing of UPB1 confirmed 4 missense mutations and 1 splice site mutation from 6 Chinese patients with βUP deficiency. Among them, c.977 G>A and c.91 G>A have been previously reported by our research group[11] and c.917-1 G>A has been reported by another group,[17] whereas 851G>T and c.853G>A in exon 7 were novel missense mutations. Genetic analysis showed the 2 above-mentioned novel mutations comprising 2 compound heterozygous mutations in patients 3 and 7, respectively. Patient 3 carried the compound heterozygous UPB1 mutations of 851G>T and c.977G>A in exons 7 and 9, respectively. Analysis of his parents showed that the father was homozygous for c.977G>A mutation and the mother was heterozygous for 851G>T mutation (Fig. 1). Patient 7 also carried the compound heterozygous UPB1 mutations of c.853G>A and c.917-1 G>A in exon 7 and intron 8, and analysis of the parents’ UPB1 indicated heterozygosity of the father for c.853G>A mutation and the mother for c.917-1 G>A mutation (Fig. 2). Therefore, the mutations of the six patients were from their parents in accordance with autosomal recessive inheritance.
Figure 1.
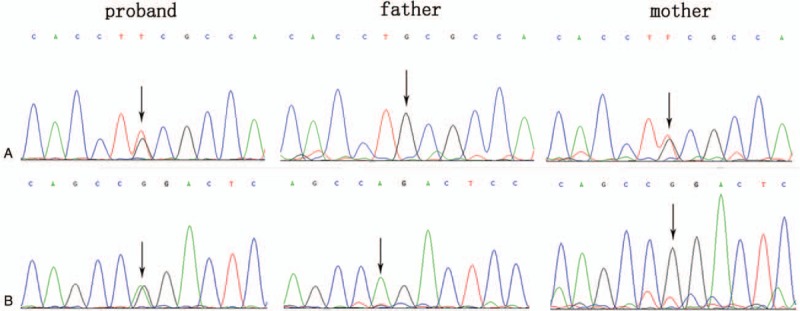
Identification of the compound heterozygous mutation of c.851G>T and c.977G>A in UPB1. A: patient with heterozygous mutation of c.851G>T, father with normal genotype, mother with heterozygous mutation of c.851G>T. B: patient with heterozygous mutation of c.977G>A, father with homozygous mutation of c.977G>A, mother with normal genotype. UPB = β-ureidopropionase gene.
Figure 2.
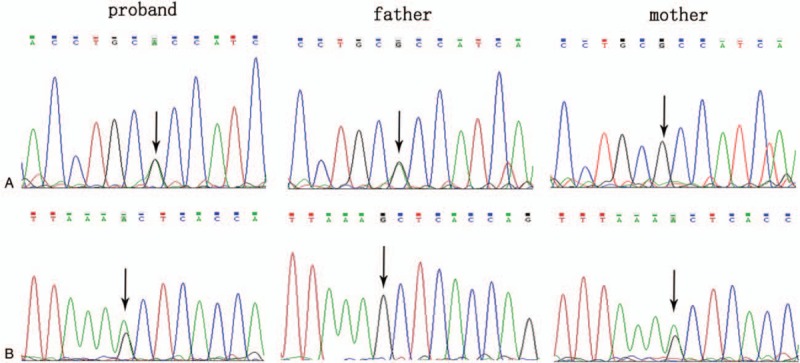
Identification of the compound heterozygous mutation of c.853G>A and c.917-1G>A in UPB1. A: patient with heterozygous mutation of c.853G>A, father with heterozygous mutation of c.853G>A, mother with normal genotype. B: patient with heterozygous mutation of c.917-1G>A, father with normal genotype, mother with homozygous mutation of c.917-1G>A. UPB = β-ureidopropionase gene.
3.4. Bioinformatics analysis
The amino acids of 285 Alanine and 284 Cysteine were highly conserved among various species and may be involved in important biological reactions (Fig. 3). The Polyphen 2 software predicted that the mutation scores of p.A285T and p.C284F were 0.635 and 0.533, respectively, indicating “damaging” tendency. SIFT predicted that the mutation scores of these sites were 0.03 and 0.01, respectively, also suggesting the “possibly damaging” tendency.
Figure 3.

Aligned amino acid sequences revealed that 284C and 285A was highly conserved among several species.
4. Discussion
βUP deficiency is a rare inherited metabolic disease. Patients with βUP deficiency usually present with strongly elevated levels of the N-carbamyl-β-alanine and N-carbamyl-β-aminoisobutyric acid in urine and plasma. Related studies have indicated that N-carbamyl-β-alanine and N-carbamyl-β-aminoisobutyric acid were weak inhibitors of dihydropyrimidinase.[18] This may explain the moderately increased levels of dihydrouracil and dihydrothymine in these patients.
To date, about 38 patients (33 families) with 18 homozygous or compound heterozygous mutations of UPB1 have been reported. In the present work, 30 reported patients had 15 mutations and 8 patients had 2 novel mutations (Table 2). The phenotypes were highly variable, that is, ranging from severe neurological involvement to asymptomatic.[10] Despite large variations in clinical presentation, the most frequently observed clinical manifestations were mainly neurological, namely, MRI abnormalities, seizures, intellectual disabilities, dystonia, and other neurological symptoms.[10] Neurological symptoms seem to be the most common in this disease. However, asymptomatic cases have also been described.[10,13] In this study, only 3 of 7 patients presented with developmental delay, muscular hypertonia, autism, or abnormal MRI. Furthermore, patient 4 suffered from polydactyly of the right little finger, as well as polydactyly and syndactyly of the bilateral little toe. No similar phenotype has been reported on congenital pyrimidine metabolic disorders. We further found that his grandfather had only polydactyly of the right hand, leading to the speculation that polydactyly was a phenomenon of intergenerational inheritance in this family, as well as this phenotype of polydactyly, was heavier than that of his grandfather. However, more studies are needed to determine any possible relation to abnormal pyrimidine metabolism. Moreover, patient 7 suffered from autism in addition to mild developmental delay. Related research has indicated that autism has obvious genetic susceptibility, and genes associated with this disease are located in 1q21.1, 3q29, 7q11.23, 16p11.2, 15q11.2-13, and 22q11.2.[23,24] Interestingly, human UPB1 is also located in 22q11.2. However, large sample validation and molecular mechanism studies need to be conducted to verify whether this gene is a new candidate gene for autism.
Table 2.
genetic and phenotypic finding of patients with βUP deficiency.
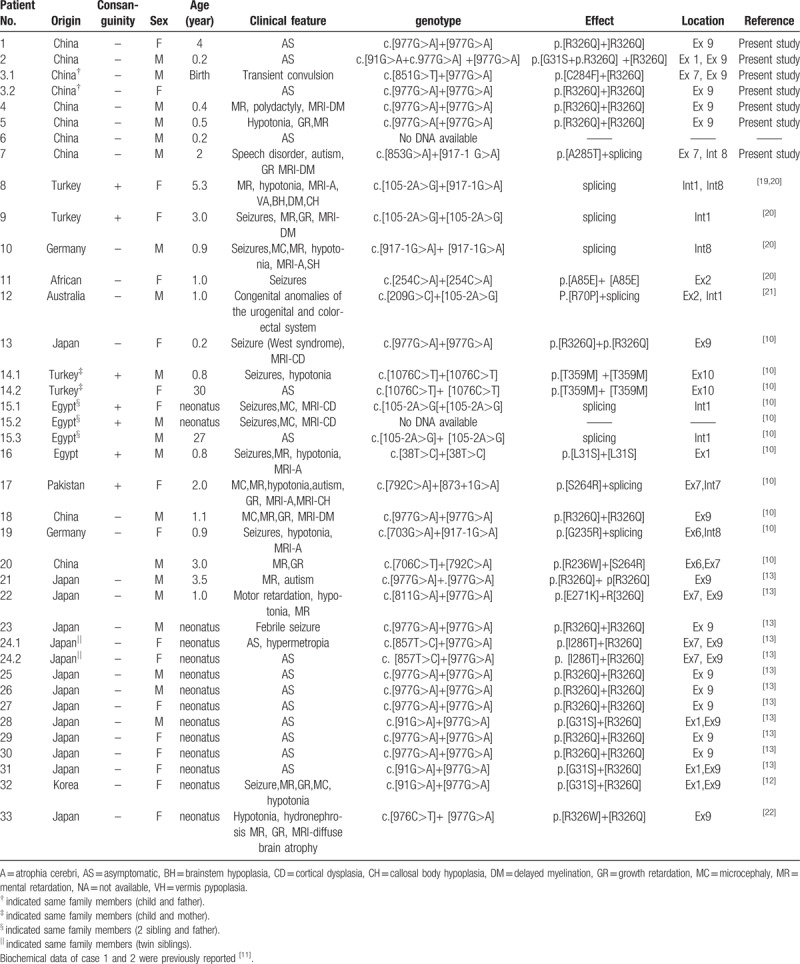
UPB1 is mapped to chromosome 22q11.2 and contains 10 coding exons.[14] The open reading frame codes a protein consisting of 384 amino acids with a molecular weight of 43.1 kDa. To date, only a total of 15 mutations have been reported worldwide, and most of them are missense mutations except for 4 splice site mutations in UPB1. In the present work, we identified 2 novel missense mutations and 3 recently reported mutations (2 missense mutations and 1 splice site mutation) in 6 Chinese patients from 6 unrelated Chinese families. Notably, the homozygous mutation c.977G>A was observed in 67% (4/6). Five patients carried this mutation on 1 or both alleles, resulting in an allele frequency of 75% (9/12). Thus, c.977G>A mutation was the common mutation in this study, which is consistent with previous results.[11] Bioinformatics analysis showed that amino acids of 285 Alanine and 284 Cysteine were highly conserved between species. The mutaions of c.853G>A and c.851G>T were predicted as damaging tendency. The splice site mutation c.917-1 G>A has been observed in many patients.[17,25] In the current work, genetic testing of UPB1 showed that cases 3 and 7 were compound heterozygous mutations (c.851G>T/c.977G>A and c.853G>A /c.917-1 G>A) originating from their parents, in accordance with autosomal recessive inheritance. Therefore, these mutations may be the cause of the disease.
In fact, there were a few limitations in this study. First, present study only involved gene mutation and bioinformatics analysis. Functional verification in vivo is not available in the present study due to the limitations of experimental condition, which is useful in the further study of the disease mechanism. Second, the patients with c.977G>A homozygous mutation presented without any clinical abnormalities in our study. The result indicated that additional factors may be involved in the clinical outcome, such as other genes and environmental factors. So, further study is needed to validate this hypothesis. Last, the follow-up period seemed to be short for assessment of growth and development.
5. Conclusions
Our study showed that c.977G>A mutation was the most common mutation of UPB1 in the Chinese population. Moreover, 2 novel missense mutations were found, which further enriched the gene-mutation spectrum of the disease and provided reliable basis for genetic consultation and prenatal diagnosis.
Acknowledgments
We are grateful to all the families enrolled in this study. We also extend our thanks to the staff of Tianjin Children's Hospital for their cooperation and support in the collection of samples.
Author contributions
Data curation: Yulian Fang, Chunquan Cai, Jianbo Shu.
Methodology: Yulian Fang, Bei Sun, Jianbo Shu, Wenchao Hu.
Resources: Yingtao Meng, Yuqin Zhang, Jianbo Shu, Wenxuan Fan.
Software: Chao Wang, Xinjie Zhang.
Supervision: Shuxiang Lin, Chunhua Zhang.
Writing – original draft: Yulian Fang, Chunquan Cai.
Writing – review & editing: Yulian Fang, Chunquan Cai, Yuqin Zhang, Jianbo Shu.
Yulian Fang orcid: 0000-0003-0136-0078.
Supplementary Material
Footnotes
Abbreviations: βUP = β-ureidopropionase, BAEP = Brainstem auditory evoked potential, CSF = cerebrospinal fluid, GC–MS = gas chromatography–mass spectrometry, MRI = magnetic resonance imaging, PCR = polymerase chain reaction, UPB1 = β-ureidopropionase gene.
YLF and CQC contributed equally to this study.
This work was supported by the Natural Science Foundation of Tianjin City (Grant No.16JCQNJC11900), the National Natural Science Foundation of China (Grant No.81771589), the Doctoral Program of Natural Science Foundation of Shandong Province (Grant No. ZR2016HB34), and the Program of Tianjin Science and Technology Plan (Grant No. 18ZXDBSY00170).
The study was conducted in accordance with the principles of the Declaration of Helsinki and guidelines of Tianjin Children's Hospital.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Website (www.md-journal.com).
References
- [1].Huang M, Graves LM. De novo synthesis of pyrimidine nucleotides; emerging interfaces with signal transduction pathways. Cell Mol Life Sci 2003;60:321–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Connolly GP, Simmonds HA, Duley JA. Pyrimidines and CNS regulation. Trends Pharmacol Sci 1996;17:106–7. [DOI] [PubMed] [Google Scholar]
- [3].Van Kuilenburg AB, Stroomer AE, Van Lenthe H, et al. New insights in dihydropyrimidine dehydrogenase deficiency: a pivotal role for beta-aminoisobutyric acid. Biochem J 2004;379:119–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Van Kuilenburg AB, Van Lenthe H, Assmann B, et al. Detection of beta-ureidopropionase deficiency with HPLC-electrospray tandem mass spectrometry and confirmation of the defect at the enzyme level. J Inherit Metab Dis 2001;24:725–32. [DOI] [PubMed] [Google Scholar]
- [5].Hartmann S, Okun JG, Schmidt C, et al. Comprehensive detection of disorders of purine and pyrimidine metabolism by HPLC with electrospray ionization tandem mass spectrometry. Clin Chem 2006;52:1127–37. [DOI] [PubMed] [Google Scholar]
- [6].Ohse M, Matsuo M, Ishida A, et al. Screening and diagnosis of beta-ureidopropionase deficiency by gas chromatographic/mass spectrometric analysis of urine. J Mass Spectrom 2002;37:954–62. [DOI] [PubMed] [Google Scholar]
- [7].Moolenaar SH, Gohlich-Ratmann G, Engelke UF, et al. beta-Ureidopropionase deficiency: a novel inborn error of metabolism discovered using NMR spectroscopy on urine. Magn Reson Med 2001;46:1014–7. [DOI] [PubMed] [Google Scholar]
- [8].Kuhara T. Noninvasive human metabolome analysis for differential diagnosis of inborn errors of metabolism. J Chromatogr B Analyt Technol Biomed Life Sci 2007;855:42–50. [DOI] [PubMed] [Google Scholar]
- [9].Kuhara T, Ohse M, Inoue Y, et al. Five cases of beta-ureidopropionase deficiency detected by GC/MS analysis of urine metabolome. J Mass Spectrom 2009;44:214–21. [DOI] [PubMed] [Google Scholar]
- [10].van Kuilenburg AB, Dobritzsch D, Meijer J, et al. β-ureidopropionase deficiency: phenotype, genotype and protein structural consequences in 16 patients. Biochim Biophys Acta 2012;1822:1096–108. [DOI] [PubMed] [Google Scholar]
- [11].Shu J, Lv X, Jiang S, et al. Genetic analysis of the UPB1 gene in two new Chinese families with beta-ureidopropionase deficiency and the carrier frequency of the mutation c.977G>A in Northern China. Childs Nerv Syst 2014;30:2109–14. [DOI] [PubMed] [Google Scholar]
- [12].Lee JH, van Kuilenburg AB, Abeling NG, et al. A Korean case of beta-ureidopropionase deficiency presenting with intractable seizure, global developmental delay, and microcephaly. JIMD Rep 2015;19:117–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nakajima Y, Meijer J, Dobritzsch D, et al. Clinical, biochemical and molecular analysis of 13 Japanese patients with beta-ureidopropionase deficiency demonstrates high prevalence of the c.977G > A (p.R326Q) mutation [corrected]. J Inherit Metab Dis 2014;37:801–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Vreken P, van Kuilenburg AB, Hamajima N, et al. cDNA cloning, genomic structure and chromosomal localization of the human BUP-1 gene encoding beta-ureidopropionase. Biochim Biophys Acta 1999;1447:251–7. [DOI] [PubMed] [Google Scholar]
- [15].van Gennip AH, van Kuilenburg AB. Defects of pyrimidine degradation: clinical, molecular and diagnostic aspects. Adv Exp Med Biol 2000;486:233–41. [DOI] [PubMed] [Google Scholar]
- [16].Thomas HR, Ezzeldin HH, Guarcello V, et al. Genetic regulation of dihydropyrimidinase and its possible implication in altered uracil catabolism. Pharmacogenet Genomics 2007;17:973–87. [DOI] [PubMed] [Google Scholar]
- [17].van Kuilenburg AB, Meinsma R, Beke E, et al. beta-Ureidopropionase deficiency: an inborn error of pyrimidine degradation associated with neurological abnormalities. Hum Mol Genet 2004;13:2793–801. [DOI] [PubMed] [Google Scholar]
- [18].Kikugawa M, Kaneko M, Fujimoto-Sakata S, et al. Purification, characterization and inhibition of dihydropyrimidinase from 841 rat liver. Eur J Biochem 1994;219:393–9. [DOI] [PubMed] [Google Scholar]
- [19].Assmann B, Gohlich-Ratmann G, Wagner L, et al. Presumptive ureidopropionase deficiency as a new defect in pyrimidine catabolism found with in vitro H-NMR spectroscopy. J Inherit Metab Dis 1998;21Suppl 2: [Google Scholar]
- [20].van Kuilenburg ABP. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer 2004;40:939–50. [DOI] [PubMed] [Google Scholar]
- [21].Yaplito-Lee J, Pitt J, Meijer J, et al. Beta-ureidopropionase deficiency presenting with congenital anomalies of the urogenital and colorectal systems. Mol Genet Metab 2008;93:190–4. [DOI] [PubMed] [Google Scholar]
- [22].Akiyama T, Shibata T, Yoshinaga H, et al. A Japanese case of (-ureidopropionase deficiency with dysmorphic features. Brain Dev 2017;39:58–61. [DOI] [PubMed] [Google Scholar]
- [23].Sanders SJ, He X, Willsey AJ, et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 2015;87:1215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Varghese, Keshav N, Jacot-Descombes S, et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol 2017;134:537–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Assmann B, Gohlich G, Baethmann M, et al. Clinical findings and a therapeutic trial in the first patient with beta-ureidopropionase deficiency. Neuropediatrics 2006;37:20–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.