

Abstract
Rationale:
The gene deletion (5)(q22q35) is reported in 10–20% of myelodysplastic syndrome (MDS) cases and is associated with response to lenalidomide and favorable prognosis. The authors report here a clinical case of MDS transformation to B-cell acute lymphocytic leukemia (B-ALL) with an associated accrual of an additional mutation following treatment with lenalidomide.
Patient Concerns:
A 69-year-old man presented with progressive anemia, normal white blood cell count, and thrombocytopenia consistent with MDS. He was administered lenalidomide for 27 months, then developed acute B-cell lymphocytic leukemia and acquired a previously unreported mutation in the gene enhancer of zeste homolog 2 (EZH2).
Diagnoses:
After 27 months of therapy with lenalidomide, a surveillance bone marrow aspiration (BMA) revealed 90% cellularity with persistent multilineage dysplasia and a population of blasts comprising 54% of all bone marrow elements by morphology, consistent with B-ALL, even though the patient was asymptomatic. Conventional karyotype showed no signs of del(5)(q22q35) MDS, however bone marrow next–generation sequencing (NGS) demonstrated the accrual of a nonsense mutation (c.211del pL71∗) in exon 3 of EZH2. A confirmatory BMA yielded 70% blasts and clinical features indicative of B-ALL.
Interventions:
Mini-hyper-CVD (cyclophosphamide and dexamethasone at 50% dose reduction, no anthracycline, methotrexate at 75% dose reduction, cytarabine at 0.5 g/m2 × 4 doses) was administered for 21 days.
Outcomes:
A follow-up BMA was performed 2 months after mini-hyper-CVD therapy, showing dysplastic features with 25% ring sideroblasts, but no evidence of B-ALL. The patient is currently receiving monthly-low dose decitabine, ofatumumab, and dexamethasone, and is transfusion independent and asymptomatic after 7 cycles.
Lessons:
The present study shows an extremely rare progression of del(5)(q22q35) MDS to B-ALL with accompanying NGS data and a newly described acquisition of an EZH2 frameshift mutation. This case highlights the importance of NGS as a diagnostic and surveillance tool for MDS.
Keywords: acute lymphocytic leukemia, lenalidomide, myelodysplastic syndromes
1. Introduction
Myelodysplastic syndromes (MDS) are clonal hematopoietic disorders characterized by inefficient hematopoiesis, peripheral blood cytopenias, and increased risk of transformation to acute myeloid leukemia (AML).[1] Isolated gene deletion (5)(q22q35), reported in 10% to 20% of de novo MDS, is associated with favorable prognosis and response to lenalidomide.[2] Although transformation to AML can occur in up to 10% of patients with MDS with isolated del(5)(q22q35) mutations (del5q-MDS),[3] transformation to, or development of acute lymphocytic leukemia is exceedingly rare. Only 2 cases of del5q-MDS transforming to secondary B-cell acute lymphocytic leukemia (B-ALL) have been reported.[4]
EZH2, enhancer of zeste homolog 2, the catalytic subunit of Polycomb Repressive Complex 2, regulates gene expression and hematopoietic stem cell maintenance and differentiation through methylation on lysine 27 of histone 3.[5] Loss-of-function mutations in EZH2 are present in 10% of patients with MDS and are associated with worse prognosis.[5,6] Germline mutations in EZH2, including the variant c.553 G > C p. D185H, have been described in Weaver syndrome (WS), a rare condition that is associated with an increased risk of myeloid malignancies.[7] Herein, we present a patient diagnosed with del5q-MDS, with an initial EZH2 variant (c.553 G > C p. D185H), who received lenalidomide for 27 months, and then developed B-ALL and a new mutation in EZH2 (c.211del p.L71), which has not previously been reported.
Informed consent to write and publish this case report was obtained in writing from the patient. All of the work detailed here was conducted according to the ethical and medical guidelines of the University of Texas MD Anderson Cancer Center.
2. Patient information, clinical findings, and timeline
A 69-year-old man presented with progressive anemia (hemoglobin 7.9 g/dL, MCV 106 fL), normal white blood cell count of 5.4 K/L (including 38% neutrophils, 46% lymphocytes, and 12% monocytes), and thrombocytopenia (platelet count 89 K/L). Initial bone marrow aspiration (BMA) revealed 40% to 50% cellularity with erythroid predominance, trilineage dysplasia with micromegakaryocytes, 3% blasts, and 25% ring sideroblasts. Conventional karyotype revealed 46 XY, del(5)(q22q35) in 7 metaphases consistent with del5q-MDS. Amplicon-based next-generation sequencing (NGS) targeting the entire coding regions of a panel of 28 genes associated with hematological malignancies was performed on whole bone marrow DNA using a MiSeq platform (Illumina, San Diego, CA). An EZH2 c.553 G>C p. D185H polymorphism at a variant allelic frequency (VAF) of 48% was identified. He was administered lenalidomide at 10 mg daily, achieving transfusion independence and cytogenetic response 3 months later. At that time, BMA revealed 45% cellularity with 1% blasts, multilineage dysplasia, 18% ring sideroblasts, and diploid cytogenetics. Immunohistochemical staining for TP53 mutations was negative. The patient required occasional interruptions of therapy and dose reduction to 5 mg daily due to gastrointestinal toxicity.
After 27 months of therapy, a surveillance BMA revealed 90% cellularity with persistent multilineage dysplasia and a population of blasts comprising 54% of all bone marrow elements by morphology. The patient was asymptomatic. Peripheral blood at that time showed a hemoglobin count of 9.9 g/dL, white blood cell count of 2.4 K/L (0.72 K/L neutrophils, 1.44 K/L lymphocytes), platelet count of 64 K/L, and no peripheral blasts. Myeloperoxidase staining was negative in the blast population and multiparametric flow cytometry revealed 17.2% events which were negative for CD2, CD3, cyCD3, CD5, CD7, CD13, CD14, CD15, CD20 (2.1%), CD36, CD41, CD56, CD64, CD117 (0.9%), and MPO; and positive for CD4, CD19 (99%), CD22 (89.9%), CD25, CD33 (42.6%), CD34 (17.2%), CD38, CD123, TDT, HLA-DR, and cyCD79a, consistent with precursor B-ALL. Conventional karyotype showed no evidence of del5q-MDS. Fluorescence in situ hybridization (FISH) using EGR1/D5S23, D5S721, and BCR/ABL1 dual color probes were negative for del(5q31) or t(9;22). Bone marrow NGS identified the same EZH2c.553 G>C p.D185H variant (VAF 50.7%) present at diagnosis as well as a nonsense mutation (c.211del pL71∗) in exon 3 of EZH2 with a VAF of 13.6%. In order to confirm the diagnosis, a repeat BMA was performed showing 70% blasts, with identical immunophenotypic and cytogenetic features to those of the initial B-ALL diagnosis.
3. Therapeutic intervention, follow-up, and outcomes
On the basis of these findings, therapy with mini-hyper-CVD (cyclophosphamide and dexamethasone at 50% dose reduction, no anthracycline, methotrexate at 75% dose reduction, cytarabine at 0.5 g/m2 × 4 doses) was started.[8] BMA performed on day 21 of therapy revealed 70% to 80% cellularity with trilineage dysplasia, and 2% blasts by morphology. Multiparametric flow cytometry identified 0.19% events similar to those of the B-ALL diagnosis, consistent with positive minimal residual disease. FISH and karyotype analysis were negative for del5q-MDS. Peripheral cytopenias were present with hemoglobin of 9.1 g/dL, white blood cell count of 3.6 K/L (2.6 K/L neutrophils, 0.72 K/L lymphocytes), platelet count of 49 K/L, and no peripheral blasts. At this point, bone marrow DNA sequencing was not performed. Two months later, the last follow-up BMA showed persistent dysplastic features with 25% ring sideroblasts, but no evidence of acute leukemia. Minimum residual disease was negative for B-ALL and karyotype was diploid. Also, no monoclonal IgH rearrangements were observed by polymerase chain reaction analysis. A detailed evolution of leukemic burden by morphology and flow cytometry, EZH2 VAF, and cytogenetic abnormalities is shown in Fig. 1. The patient is currently being treated with monthly low-dose decitabine, ofatumumab, and dexamethasone, and is transfusion independent with no symptoms. He has received 7 cycles of therapy.
Figure 1.
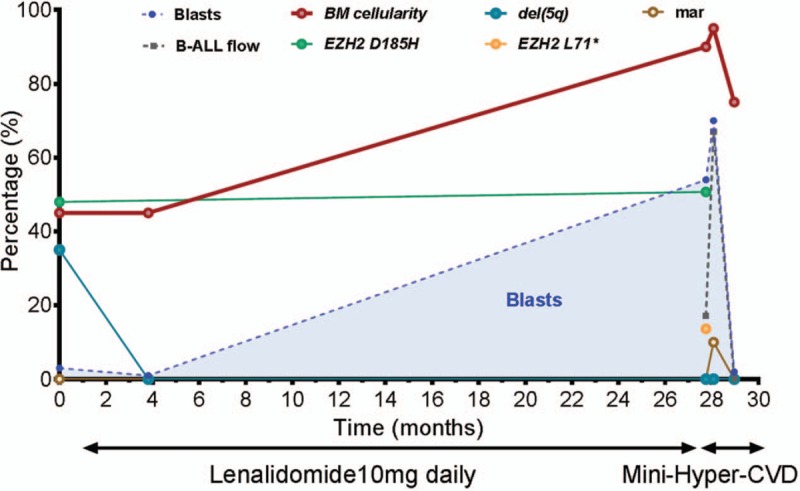
Dynamics of blast percentage, cytogenetic abnormalities, and mutational burdens through the course of therapy. Blast percentages observed by bone marrow morphologic evaluation are represented over time as blue shaded areas with blast populations defined by flow cytometry represented as dotted grey lines (B-ALL flow). Variant allele frequencies (VAFs) of somatic mutations are depicted over time for the EZH2 D185H variant in green and the EZH2 L71∗ in yellow. Frequency of cytogenetic abnormalities are represented as percentages based on the number of metaphases with the abnormality within the total number of evaluated metaphases. The current therapy received by the patient at a given time is specified under the X axis. BM cellularity = bone marrow cellularity, del(5q) = chromosome 5q deletion, mar = marker chromosome.
4. Discussion
Transformation from del5q-MDS to B-ALL is extremely uncommon, with only 2 cases reported in the literature.[4] Among 2978 patients with a diagnosis of B-ALL treated at MD Anderson Cancer Center since 1980, only 1 additional patient with synchronous del5q-MDS and B-ALL has been identified.[8] In both cases, the patients presented with additional cytogenetic abnormalities and complex karyotypes at the time of transformation, suggesting clonal evolution.[4] No mutational analysis was available in these cases. In contrast to what has been previously reported, our patient had normal cytogenetics at the time of B-ALL diagnosis. Interestingly, the initial EZH2 variant (c.553 G>C p. D185H) persisted at transformation, and an additional EZH2 missense mutation (c.211del p.L71) was acquired. Because EZH2 p. D185H is observed in up to 8% of the general population, therefore, it cannot by itself be causative of WS.[7,9]
Lenalidomide is currently the standard of care for del5q-MDS.[3] As secondary malignancies have been described with the use of lenalidomide,[10] there is a potential role for it in B-ALL transformation. Palumbo et al[10] reported secondary hematologic neoplasms after a median of 29 months of treatment with lenalidomide in myeloma patients, with 4 cases of ALL reported. Consistent with this time period, our patient developed B-ALL after 27 months of lenalidomide. In general, B-ALL as a second malignancy is infrequent, with a reported incidence between 2.3% and 3.7%.[11]
An intriguing factor in this particular case is the acquisition of novel EZH2 c.211del p.L71 mutation, at the moment of B-ALL development. Although it has not been previously reported and other studies would be required to confirm its leukemogenic potential, a frameshift mutation leading to a deletion such as this could be expected to lead to a dysfunctional protein with oncogenic activity.
As EZH2 works in a cell-context dependent manner,[12,13] it might act as a tumor suppressor in MDS and as an oncogene in leukemia and lymphoma.[14] In addition, it is likely that emergence of these mutations in different stem-cell or hematopoietic progenitor compartments may lead to different phenotypes based on the cell of origin driving the disease. Also, evidence suggests that EZH2 is a checkpoint in transitioning from pro-B to pre-B cells, perhaps by blocking differentiation, as observed in AML models.[13,15]EZH2 c.553 G>C p. D185H, presumed to be a germline mutation in this case, is reported in the Single Nucleotide Polymorphism Database (rs2302427C>G). Cohen et al[7] have shown this, and other EZH2 variants to be in vitro loss-of-function mutations. Tatton-Brown et al[9] suggest germline mutations in EZH2 c.553 G>C p. D185H represents a risk factor for developing hematologic and/or other neoplasms.[9]
Due to the absence of other phenotypic abnormalities suggestive of WS, a skin biopsy to confirm the germline nature of this SNP was not performed. WS also could not be confirmed in peripheral lymphocyte culture due to persistent lymphopenia. However, previous reports of this variant and the high VAF (50%) support its germline origin in the patient.
5. Conclusion
Although del5q-MDS can transform to AML, particularly in the presence of TP53 mutations, emergence of another lineage leukemia such as B-ALL is exceedingly rare, with only 2 cases reported in the literature. The present case provides additional evidence of such an event, and is the first to provide sequencing data at the time of leukemic evolution. By doing so, we identified a novel EZH2 frameshift mutation that we associated with the emergence of the ALL clone.
Acknowledgments
SBC would like to thank Colegio Mexicano para la Investigacion del Cancer, proyecto CONACYT Redes 280148, and Patronato del Instituto Nacional de Cancerologia Mexico for providing his observership at MD Anderson Cancer Center. Also, MJMO would like to thank FUNDALEU---Fundación para combatir la Leucemia---for granting her the Uberman scholarship to carry out her rotation at MD Anderson Cancer Center.
Author contributions
Conceptualization: Sebastian Burgos, Guillermo Montalban-Bravo, Rashmi Kanagal-Shamanna, Guillermo Garcia-Manero, Maria J Mela-Osorio.
Data curation: Guillermo Montalban-Bravo, Rashmi Kanagal-Shamanna.
Formal analysis: Guillermo Montalban-Bravo, Rashmi Kanagal-Shamanna.
Funding acquisition: Guillermo Garcia-Manero.
Investigation: Sebastian Burgos, Guillermo Montalban-Bravo, Lucia Fuente, Elias J Jabbour, Rashmi Kanagal-Shamanna, Guillermo Garcia-Manero, Maria J Mela-Osorio.
Methodology: Sebastian Burgos, Guillermo Montalban-Bravo, Lucia Fuente, Elias J Jabbour, Rashmi Kanagal-Shamanna, Guillermo Garcia-Manero, Maria J Mela-Osorio.
Project administration: Kelly A Soltysiak, Guillermo Garcia-Manero, Maria J Mela-Osorio.
Resources: Elias J Jabbour, Rashmi Kanagal-Shamanna, Maria J Mela-Osorio.
Supervision: Elias J Jabbour.
Writing – original draft: Sebastian Burgos, Guillermo Montalban-Bravo, Lucia Fuente, Elias J Jabbour, Rashmi Kanagal-Shamanna, Kelly A Soltysiak, Guillermo Garcia-Manero, Maria J Mela-Osorio.
Writing – review & editing: Sebastian Burgos, Guillermo Montalban-Bravo, Elias J Jabbour, Rashmi Kanagal-Shamanna, Kelly A Soltysiak, Guillermo Garcia-Manero, Maria J Mela-Osorio.
Footnotes
Abbreviations: AML = acute myeloid leukemia, B-ALL = B-cell acute lymphocytic anemia, BMA = bone marrow aspiration, del5q-MDS = myelodysplastic syndrome with del(5)(q22q35) mutations, EZH2 = enhancer of Zeste homolog 2, FISH = fluorescence in situ hybridization, MDS = myelodysplastic syndrome, Mini-hyper-CVD = cyclophosphamide and dexamethasone at 50% dose reduction, no anthracycline, methotrexate at 75% dose reduction, cytarabine at 0.5 g/m2 × 4 doses, NGS = next-generation sequencing, VAF = variant allelic frequency, WS = Weaver syndrome.
Funding/support: GG-M declares research support and an advisory role for Celgene Corporation and Acceleron Pharma. EJJ declares research support from Pfizer, Takeda, Amgen, AbbVie, Spectrum, and Bristol-Myers Squibb.
This work was supported in part by the University of Texas MD Anderson Cancer Center Support Grant CA016672 and by generous donations to the University of Texas MD Anderson MDS/AML Moon Shot Program.
The remaining authors have no declarations of interest.
References
- [1].Garcia-Manero G. Myelodysplastic syndromes: 2015 Update on diagnosis, risk-stratification and management. Am J Hematol 2015;90:831–41. [DOI] [PubMed] [Google Scholar]
- [2].List A, Dewald G, Bennett J, et al. Lenalidomide in the myelodysplastic syndrome with chromosome 5q deletion. N Engl J Med 2006;355:1456–65. [DOI] [PubMed] [Google Scholar]
- [3].Pellagatti A, Boultwood J. Recent advances in the 5q- syndrome. Mediterr J Hematol Infect Dis 2015;7:e2015037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Agostino NM, Ahmed B, Popescu D, et al. Transformation of the 5q- syndrome to acute lymphoblastic leukemia: a report of two cases and review of the literature. Int J Clin Exp Pathol 2011;4:322–6. [PMC free article] [PubMed] [Google Scholar]
- [5].Herviou L, Cavalli G, Cartron G, et al. EZH2 in normal hematopoiesis and hematological malignancies. Oncotarget 2016;7:2284–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 2011;364:2496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cohen AS, Yap DB, Lewis ME, et al. Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat 2016;37:301–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jain P, Tang G, Konoplev SN, et al. Synchronous del5q myelodysplastic syndrome (del5qMDS) and adult B-cell acute lymphoblastic leukemia (B-ALL) with TET2 and TP53 mutations. Am J Hematol 2016;91:354–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tatton-Brown K, Hanks S, Ruark E, et al. Germline mutations in the oncogene EZH2 cause Weaver syndrome and increased human height. Oncotarget 2011;2:1127–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Palumbo A, Bringhen S, Kumar SK, et al. Second primary malignancies with lenalidomide therapy for newly diagnosed myeloma: a meta-analysis of individual patient data. Lancet Oncol 2014;15:333–42. [DOI] [PubMed] [Google Scholar]
- [11].Pagano L, Pulsoni A, Tosti ME, et al. Acute lymphoblastic leukaemia occurring as second malignancy: report of the GIMEMA archive of adult acute leukaemia. Gruppo Italiano Malattie Ematologiche Maligne dell’Adulto. Br J Haematol 1999;106:1037–40. [DOI] [PubMed] [Google Scholar]
- [12].Sashida G, Harada H, Matsui H, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun 2014;5:4177. [DOI] [PubMed] [Google Scholar]
- [13].Tanaka S, Miyagi S, Sashida G, et al. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood 2012;120:1107–17. [DOI] [PubMed] [Google Scholar]
- [14].Xu F, Liu L, Chang CK, et al. Genomic loss of EZH2 leads to epigenetic modifications and overexpression of the HOX gene clusters in myelodysplastic syndrome. Oncotarget 2016;7:8119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Su IH, Basavaraj A, Krutchinsky AN, et al. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol 2003;4:124–31. [DOI] [PubMed] [Google Scholar]