

Abstract
Inflammasome dysregulation is a hallmark of various inflammatory diseases. Evaluating inflammasome-associated structures (ASC specks) and caspase-1 activity by microscopy is time-consuming and limited by small sample size. The current flow-cytometric method, TOFIE, cannot visualize ASC specks or caspase-1 activity, making colocalization studies of inflammasome components and enzymatic activity impossible. We describe a rapid, high-throughput, single cell, fluorescence-based image analysis method utilizing the Amnis ImageStream®X instrument that quantitatively and qualitatively characterizes the frequency, area, and cellular distribution of ASC specks and caspase-1 activity in mouse and human cells. Unlike TOFIE, this method differentiates between singular perinuclear specks and false positives. In the described study, we have shown that presence of NLRP3 reduces the size of ASC specks, which is further reduced by the presence of active caspase-1. The capacity of our approach to simultaneously detect and quantify ASC specks and caspase-1 activity, both at the population and single-cell level, renders it the most powerful tool available for visualizing and quantifying the impact of mutations on inflammasome assembly and activity.
INTRODUCTION
Inflammasomes are multi-protein complexes comprised of a sensor which directly or indirectly recruit and activate the aspartic protease caspase-1 leading to maturation of bioactive IL-1β and IL-18 (1, 2). Active caspase-1 also cleaves gasdermin-D which initiates pyroptotic cell death (3–5). Cytosolic proteins, such as the NOD-like receptors (NLR) and AIM2-like receptors (ALR), initiate inflammasome assembly in response to pathogen- and danger- associated molecular patterns (PAMPs/DAMPs) (6–9). While some NLRs, such as NLRC4 and NLRP1 can recruit and activate caspase-1 directly through homotypic CARD-domain interactions (10, 11). most NLR-mediated inflammasomes require apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC). ASC acts as an adaptor, interacting with NLR and ALR proteins through a homotypic Pyrin-domain interaction and subsequently recruiting pro-caspase-1 via a homotypic CARD-domain interaction. NLR/ALR sensing of PAMPs/DAMPs allows rapid association with ASC to initiate an active inflammasome (1, 2, 6, 7, 9). The best-studied inflammasome, that initiated by NLRP3, is activated by many structurally divergent agonists of microbial, environmental, and host origin (2, 9). Dysregulation of NLRP3 inflammasome activity is a hallmark of pathogenesis in several human diseases (12–16), indicating its highly significant clinical relevance.
Inflammasome assembly is accompanied by the formation of a typically singular, perinuclear structure called a “speck”. Speck structures were first described for ASC and Pyrin, are readily visualized by microscopy (17, 18) and frequently have a toroidal appearance with an apparent diameter of ~1μm (17). Studies using fluorescent reporter tagged ASC tagged have observed speck diameters ranging from 2–4μm (19, 20). A time-course analysis of in vivo speck formation shows that speck size increases over a period of 15 minutes and then stabilizes (21). Speck morphology has also been debated. Some studies suggest that the speck is a conglomerate of ASC filaments arranged randomly in a localized space (21, 22). In contrast, others suggest that specks have a hollow to fibrillar organized structure composed of multiple units (17, 23, 24). Since speck formation is a rapid, all-or-none event that coincides with activation of caspase-1, it is frequently used as complementary readout for inflammasome activation (25, 26). Caspase-1 activation, a direct read-out for inflammasome activation, is typically measured by immunoblotting for the active subunit of caspase-1 and its cleavage product IL-1β or by detection of binding by the fluorescent caspase-1 inhibitor FLICA (8, 27). Microscopy- and flow cytometry-based methods are currently used to quantitate speck formation. Of these methods, only microscopy-based techniques permit visualization and analysis of inflammasome specks in single-cells. However, microscopy is time-consuming, generally restricted to small sample sizes, and frequently requires subjective determination of relevant structures versus artifacts. Moreover, quantitative evaluation of features such as speck size (e.g. ASC area) and distribution of active caspase-1 (e.g. diffused vs punctate) require subjective selection of cells followed by image analysis, an approach inherently subject to human error and bias (28, 29). Recently, a rapid and high-throughput flow-cytometry-based technique, Time of Flight Inflammasome Evaluation (TOFIE) was developed to identify speck-containing cells based on the increased ratio of pulse height to area or decreased ratio of pulse width to area (25, 30). While useful, TOFIE cannot be used to examine co-localization of ASC speck with active caspase-1 or other cell structural features.
Here we describe a gating and masking strategy that we term Inflammasome and caspase-1 activity Characterization and Evaluation (ICCE), using imaging flow cytometry and computational quantitative image analysis to perform single-cell analysis which quantifies ASC speck-containing cells, and evaluates speck size. Further, ICCE eliminates non-speck like aggregates of ASC (false positive artifacts) which TOFIE fails to exclude. Further, single-cell analysis allows for simultaneous assessment of caspase-1 activity including its distribution and localization in ASC-expressing cells. Using our analysis method we studied the speck size in the presence and absence of NLRP3. We demonstrate that presence of NLRP3 reduces the size of the specks. We further establish a negative correlation between speck size and caspase-1 activity. The advantages of quantitative imaging combined with cytometry allows the performance of quantitative single-cell analyses accurately representing data at the population level, a degree of evaluation that was previously impossible.
MATERIALS AND METHODS
Cells and Reagents
Human kidney epithelial cells (HEK239T) and immortalized bone-marrow derived macrophages (iBMDMs) were cultured in Dulbecco’s Modified Eagle Medium/high glucose(DMEM) (Hyclone, Cat.#SH30243.1) supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Cat.#S11050) and 1X-Glutamax (Gibco, Cat. #35050–061). FuGENE6 was from Roche Applied Science (Cat.#11988387001). Primary human monocytes were obtained from University of Nebraska Medical Center and cultured in DMEM supplemented with 10% Human AB Serum (Corning, Cat.#35–060-Cl). THP1 cells were cultured in RPMI-1640 (Hyclone, Cat.# SH30027.01) supplemented with 10% FBS, 1X-β-mercaptoethanol (Gibco, Cat.#21985) and 1X-Glutamax (Gibco, Cat. #35050–061). Nigericin was from InvivoGen (Cat.#tlrl-nig) and reconstituted in ethanol as per the manufacturer’s instructions. The FLICA-660 caspase-1 assay kit was purchased from Immunochemistry Technologies (Cat.#9122) and FLUORO-GEL II Mounting Medium with DAPI was from EMS (Cat.#17985–51).
Expression plasmids and DNA transfection
Expression plasmids encoding human caspase-1 (31) and human NLRP3 (32) have been previously described. ASC was cloned into the pEGFP-C3 expression vector backbone using HindIII and KpnI digestion to generate GFP-ASC. HEK293T cells (2×105) were seeded in 12-well plates (Corning) in 1ml of DMEM. Transfections were carried out at a constant DNA concentration of 1μg/well. Empty vector, pcDNA3, was used to adjust the final amount of DNA to 1μg. Transfections were performed using 2.5μl of FuGENE6 per μg of DNA.
Inflammasome reconstitution and activation
HEK293T cells (2×105) were seeded in 12 well plates in 1ml DMEM and incubated overnight at 37°C with 5% CO2. For microscopy, cells were seeded on 18mm coverslips in a 12 well plate. Individual wells were transfected with plasmids encoding pro-caspase-1 (20 or 50ng) and GFP-ASC (50ng) with or without NLRP3 (100ng) and incubated at 37°C with 5% CO2. 18 hours post-transfection, cells were treated with 5μM nigericin or vehicle for 2hr. Following treatment, media from each well was aspirated leaving 150μl and volume was made up to 200μl with media containing FLICA660 (1:45 final dilution) and incubated for 30 min at 37°C with 5% CO2. Cells were washed twice with 1X wash buffer (supplied with the FLICA kit). For ImageStream®X Mark II (ISXII) acquisition, cells were treated with 50μl trypsin-EDTA (Corning; Cat.#25–053-Cl ) per well and fixed with 4% paraformaldehyde (PFA) (EMS; Cat.#15710) for 15 min at room temperature. For microscopy, cells were fixed without trypsin treatment. Cells were washed 3 times with 1X PBS, dipped in distilled water, and mounted on slides using FLUORO-GEL II. THP1 cells, immortalized wild-type and caspase-1/11−/− and primary human monocytes were treated with 100ng/ml LPS (O26:B6) (Sigma Aldrich Cat.#L2654) for 3–4hrs. Following LPS treatment cells were stimulated with 5mM ATP (Sigma Aldrich Cat.#A3377) and 10μM nigericin for 30 min. FLICA staining was performed as described earlier but cells were incubated in FLICA-free media for 1hr instead of washing with 1X wash buffer. For ISXII acquisition, cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature, permeabilized with 0.1% TritonX-100 (Fisher Scientific Cat.#BP151–100) for 10 mins at room temperature and blocked in PBS containing 5% fish gelatin (Sigma Aldrich Cat.#G7765 ), 1% BSA (Fisher Scientific Cat.#BP1605–100), and 0.05% Triton X-100 for 1hr at room temperature. After blocking, cells were stained with Rabbit anti-ASC (N15)-R (1:250) or Mouse anti-ASC (D-3) (1:500) (Santa Cruz) in wash buffer (PBS containing 1% fish gelatin, 1% BSA, and 0.5% Triton X-100) for 2hrs. Cells were washed 3 times with wash buffer, followed by incubation with Alexa Fluor®488 goat-anti-rabbit IgG (Life Technologies Cat.#A11034) (1:500) or Alexa Fluor®594 goat-anti-rabbit IgG2a (Life Technologies Cat.#A21135) (1:1000) in wash buffer for 1hr.
Preparation of samples for acquisition by the Amnis ImageStream®X Mark II (ISXII)
Fixed cells were harvested and stained with 1μg/ml DAPI in 1X PBS supplemented with 0.5mM EDTA (ISXII cell suspension buffer) for 10 min at room temperature (RT). Cells were washed once with 1X PBS and resuspended in 50μl ISXII cell suspension buffer by gently tapping the tubes (use of a pipette or vortex mixer should be avoided at this stage). The samples were then acquired on the ISXII. For THP1 cells, ASC localizes to nucleus (33) and thus DAPI staining was avoided to prevent interference with ASC staining.
ImageStream®X acquisition parameters
An Amnis ImageStream®X Mark II (ISXII) ((MilliporeSigma, Inc.) equipped with 405, 488, and 642 nm lasers with a single camera (6 channels) was used to acquire experimental samples using the INSPIRE® software. The flow rate was set to minimum and the objective magnification was set to 60X for all samples (0.33μm per pixel resolution). A multi-fluorophore labeled sample was used to determine accurate laser settings and avoid over-saturation. Masks are defined region of interests that are computationally calculated by INSPIRE®. A mask defines a specific region of an image which can be used for specific feature calculations. The saturation of an individual fluorophore in its corresponding channel was determined by plotting the Raw Max pixel feature versus the Area feature of the default mask of that channel. Laser settings used for acquisition are shown in Table-I. Aspect ratio feature measures circularity, thus helping to distinguish between singlets, doublets and cell clumps when used together with the Area feature. Area versus Aspect Ratio of the default mask on the bright field was used during acquisition to ensure collection of single cell events. A region (R1) was created to gate for debris. Two other regions, R2 and R3, were created to gate for all cells and singlet population respectively. 10,000 events were recorded in R3 for every sample. The R2 population was determined as the collection population and all the events in that gate were recorded until R3 reached 10,000. For the compensation control, 1000 events for single stained samples were acquired using the same setting while turning the Ch04 (Bright field) and 785nm laser (Side-scatter; SSC) off. All samples and controls were acquired as raw image files (.rif).
Table 1:
AMNIS instrument laser power setting used. Channel 1 was used for DAPI, Channel 2 was used for GFP, Channel 3 was turned off for acquisition, Channel 4 was used for bright field, Channel 5 was used for FLICA, and Channel 6 was used for side-scatter.
| Emission Channel |
Fluorophore | Excitation Laser (nm) |
Laser Power (mW) |
|---|---|---|---|
| 1 | DAPI | 405 | 10 |
| 2 | GFP | 488 | 10 |
| 3 | Blank | --- | --- |
| 4 | Bright field | --- | --- |
| 5 | FLICA | 642 | 150 |
| 6 | Side scatter (SSC) | 785 | 2 |
Image analysis by Image Data Exploration & Analysis Software (IDEAS®)
All sample and compensation analysis were done using the IDEAS® software. Individual compensation controls of single stained samples were loaded into the compensation wizard in IDEAS®. A compensation matrix file (.ctm) was generated and applied to a positive control (Nigericin treated sample) raw data file (.rif) to generate both a compensated image file (.cif) and a corresponding data analysis file (.daf). A data analysis template (.ast) was developed by analyzing the .daf file of positive control sample. We applied the template along with the compensation matrix to the rest of the experimental samples using the multiple file batch tool in IDEAS®.
Masking strategy
The ISXII instrument allows the user to combine the power of quantitative imaging with high-throughput sample collection. ISXII data can be used for quantitative imaging by developing appropriate masks in IDEAS® (MilliporeSigma, Inc.). A mask delineates a specific portion of image that can be used in calculations pertaining to the correlating cellular feature, which can then be quantified using high-throughput imaging.
Focused cell and nucleus mask
Gradient RMS measures the average contrast of the image. Low contrast (low gradient RMS) represents out of focus images and high gradient RMS reflects better-focused images. An object mask was applied to default bright field mask (M04) to accurately calculate the area of the cell and improve Gradient_RMS of the mask (Fig. 1A). Similarly, a morphology mask was applied to default channel 1 mask (M01) to accurately calculate the area of nucleus (Fig. 1B). Once these masks were created, features like area, gradient_RMS, aspect ratio etc. were recalculated using these masks.
Figure 1: Object and morphology masks that identify focused cells, nuclei, and permit gating for single cells (HEK293T).
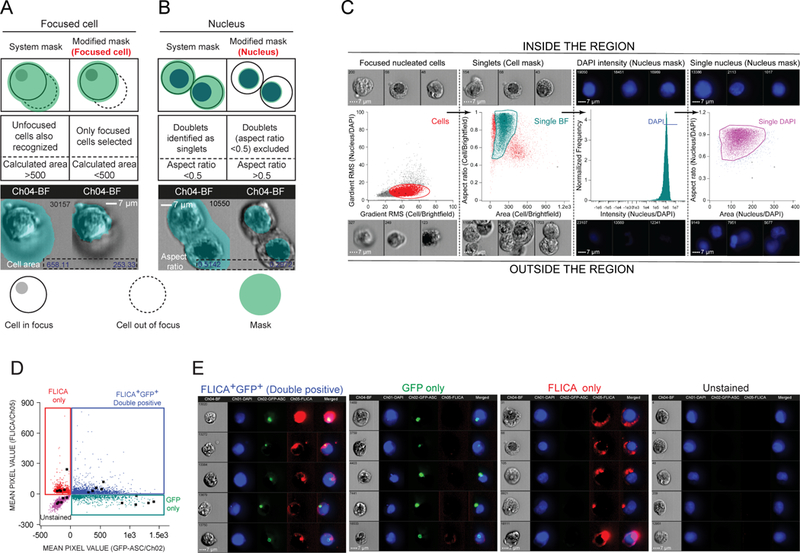
A. Identifying focused cells. Schematic representation of two cells in the field of view (top), one in focus (solid line) and other unfocused (dashed line) before and after applying the modified mask (green). The default mask (M04: brightfield) incorrectly detects the unfocused cell (left) as shown in the micrograph (bottom). Applying object mask “Focused cell” excludes the unfocused cell. B. Identifying single nuclei. Schematic representation of a doublet in the field of view before and after application of the modified mask. Applying morphology mask “Nucleus” resolves the two closely located nuclei identified as a single event by the default system mask (M02:DAPI) and reports two distinct nuclei of expected near circular morphology as shown in the micrograph (Bottom). C. Gating strategy to identify single cells. Micrographs show events inside (top) and outside (bottom) the selected region. From left to right: dotplot showing Gradient_RMS of Focused cell mask vs. that of Nucleus mask with a region set to return focused cells (cells; red), dotplot of Area vs. the Aspect Ratio of the Focused cell mask with a region set to exclude non-single cell events (single-brightfield; cyan), histogram of the Intensity of DAPI staining within the Nucleus mask with a region set to exclude dim events (DAPI; blue), and dotplot of Area vs. Aspect Ratio of the Nucleus mask with region set to exclude non-single nuclei events (Single DAPI; purple). (D-E) Identifying ASC (GFP) and active caspase-1 (FLICA) double positive cells. D. Dot plot of mean pixel values for GFP vs. FLICA for HEK293T cell events in the Single DAPI gate. Single positive, double positive, and double negative quadrants are indicated. E. Micrographs for representative events from each quadrant (solid squares) in D.
ASC speck mask
An ASC speck mask was carefully developed from Ch02 default system mask for GFP (M02). Details of the individual masks used in developing ASC speck mask are shown in Table-II. Several function masks along with combination masks were made keeping the preceding masks as input. A spot mask differentiates between punctate and diffused staining. ASC speck1 and ASC speck2 are two spot masks made using the system mask (M02) with different levels of stringency for parameters of spot to cell count and area. ASC speck1 accommodated smaller aggregates, whereas ASC speck2 has higher stringency for spot to cell count. The Boolean operator OR was used to combine both Speck1 and Speck2 masks together and named ASC speck3. A levelset mask identifies pixels in non-homogeneous regions into three different levels: dim, middle, and bright. ASC speck3 was used as the input for a levelset mask. In this instance, the levelset function with the middle and bright setting was used to mask for fairly bright and bright spots respectively. Two different levelset masks were created (ASC speck4 and ASC speck5) and combined into a single mask (ASC speck6) using the Boolean OR operation. Since the levelset mask function can generate non-contiguous masks due to low pixel intensities, the mask operation fill was used to fill in regions with very dim pixels to eliminate variations within the mask (ASC speck7). ASC speck7 mask could not differentiate between large aggregates and specks. To overcome this problem, we applied the mask operation range to the ASC speck7 mask to select for masks with an area between 15–500 pixels and an aspect ratio of 0.4–1 resulting in the ASC speck8 mask. To further minimize the background, the intensity mask function was applied to ASC speck8, which allows the user to set a minimum and maximum intensity threshold. The threshold was set to 750–4095 to eliminate the masking of all pixels with intensity less than 750. This mask is referred to as ASC speck9. Setting intensity limit at 750 made the mask smaller than the size of ASC speck. Additional dilate and fill mask functions were applied to generate ASC speck10 and 11 masks to capture appropriate sized ASC specks. The threshold mask function was applied to ASC speck 11 to further increase mask stringency by eliminating artificially small aggregate signals (ASC speck12). Another range operation was applied to ASC speck12 with the setting of area of 15–450 and aspect ratio of 0.6–1 to select for round specks (ASC speck13). Even after such an elaborate masking strategy, some non-speck like aggregation of ASC could not be eliminated. To overcome this problem, the ASC speck13 mask was then divided into individual component masks. We used the component mask function to rank the ASC speck13 individual masks by area sorted from highest to lowest (component 1 = largest area). This ensures the largest cluster of GFP signal in the cell represents the ASC speck. The component mask, ASC speck14 (Component 1 mask), with highest area was selected as ASC speck mask. For native ASC specks, order of masks was not altered, but setting was fined tuned to provide a better fit for speck characteristics (Table-II; column 3).
Table 2:
Masking strategy for ASC speck. Mask name denotes the name given to individual mask. Function defines the type of mask used. Setting defines the parameters set for individual masks to achieve ASC speck mask. Setting for reconstituted and native mask s are shown in different columns. ASC Mask 14 was used as the ASC speck mask. Spot count on ASC speck 14 was used to define ASC speck (spot count one).
| Mask name | Reconstituted ASC specks | Native ASC Specks | |
|---|---|---|---|
| HEK293T (ASC-GFP) | THP-1 | Primary human monocytes |
|
| ASC Mask 1 | Spot (M02, Ch02-GFP-ASC, Bright, 2, 10, 3) | Spot (M02, Ch02-AF488, Bright, 3, 5, 1) | Spot (M05, Ch05-AF594, Bright, 3, 5, 1) |
| ASC Mask 2 | Spot (M02, Ch02-GFP-ASC, Bright, 1, 20, 3) | Spot (M02, Ch02-AF488, Bright, 3, 3, 1) | Spot (M05, Ch05-AF594, Bright, 3, 3, 1) |
| ASC Mask 3 | ASC Mask 1 Or ASC Mask 2 | ASC Mask 1 Or ASC Mask 2 | ASC Mask 1 Or ASC Mask 2 |
| ASC Mask 4 | LevelSet (ASC Mask 3, Ch02-GFP-ASC, Middle, 5) | LevelSet (ASC Mask 3, Ch02-AF488, Middle, 5) | LevelSet (ASC Mask 3, Ch05-AF594, Middle, 5) |
| ASC Mask 5 | LevelSet (ASC Mask 3, Ch02-GFP-ASC, Bright, 5) | LevelSet (ASC Mask 3, Ch02-AF488, Bright, 5) | LevelSet (ASC Mask 3, Ch05-AF594, Bright, 5) |
| ASC Mask 6 | ASC Mask 4 Or ASC Mask 5 | ASC Mask 4 Or ASC Mask 5 | ASC Mask 4 Or ASC Mask 5 |
| ASC Mask 7 | Fill (ASC Mask 6) | Fill (ASC Mask 6) | Fill (ASC Mask 6) |
| ASC Mask 8 | Range (ASC Mask 7, 15–500, 0.4–1) | Range (ASC Mask 7, 0–200, 0.3–1) | Range (ASC Mask 7, 0–200, 0.5–1) |
| ASC Mask 9 | Intensity (ASC Mask 8, Ch02-GFP-ASC, 750–4095) | Intensity (ASC Mask 8, Ch02-AF488, 300–4095) | Intensity (ASC Mask 8 Ch05-AF594, 750–4095) |
| ASC Mask 10 | Dilate (ASC Mask 9, 1) | Dilate (ASC Mask 9, 1) | Dilate (ASC Mask 9, 1) |
| ASC Mask 11 | Fill (ASC Mask 10) | Fill (ASC Mask 10) | Fill (ASC Mask 10) |
| ASC Mask 12 | Threshold (ASC Mask 11, Ch02-GFP-ASC, 80) | Threshold (ASC Mask 11, Ch02-AF488, 52) | Threshold (ASC Mask 11, Ch05-AF594, 52) |
| ASC Mask 13 | Range (ASC Mask 12, 15–450, 0.6–1) | Range (ASC Mask 12, 2–18, 0.5–1) | Range (ASC Mask 12, 5–20, 0.6–1) |
| ASC Mask 14 | Component (1, Area, ASC Mask 13, Descending) | Component (1, Area, ASC Mask 13, Descending) | Component (1, Area, ASC Mask 13, Descending) |
Active caspase-1 (FLICA) mask
A FLICA mask was developed similarly to the ASC speck mask by modifying the default system mask for channel 05 (M05). Details of individual masks are shown in Table-III. Spot mask function was applied to default FLICA mask to differentiate between diffused and aggregate staining pattern (FLICA-I). The FLICA-I mask was further refined using the intensity mask function by setting the lower intensity threshold to 150. This mask is referred to as FLICA-II, which can distinguish between intense aggregate-like signal and low intensity aggregates. The mask was further refined for eliminating small aggregates by using erode function to generate FLICA-III mask. FLICA-III was then dilated by 3 pixels (FLICA-IV) to accurately fit the FLICA aggregates. The Range mask was applied on FLICA-IV to select for masks with an area between 20–1000 pixels and an aspect ratio of 0.6–1 to find spots that met the proper size and circularity to be defined as a proper FLICA aggregate (FLICA-V). Diffused FLICA staining was attributed to those samples that had zero spot count on FLICA-V mask.
Table 3:
Masking strategy for FLICA aggregates. Mask name denotes the name given to individual mask. Function defines the type of mask used. Setting defines the parameters set for individual masks to achieve FLICA spot mask. FLICA-V mask identified all the FLICA aggregates in a cell. Spot count on FLICA-V was used to define diffused FLICA staining (spot count zero).
| Mask name | Function | Setting |
|---|---|---|
| FLICA-I | Spot | Spot (M05, Ch05-FLICA, Bright, 5, 5, 2) |
| FLICA-II | Intensity | Intensity (FLICA-I, Ch05-FLICA, 150–4095) |
| FLICA Spot-III | Erode | Erode (FLICA-II, 1) |
| FLICA-IV | Dilate | Dilate (FLICA-III, 3) |
| FLICA-V | Range | Range (FLICA-IV, 20–1000, 0.6–1) |
Gating strategy
Gradient RMS for brightfield (Ch4) and gradient RMS for DAPI were plotted and a region (Cells) was drawn to gate both brightfield and nuclear images that were in focus. The focused “Cells” population was further gated for specific area and aspect ratio to identify single cell population (Single BF). “Single BF” was further gated based on intensity of nuclear stain to select for the population with optimum nuclear staining (DAPI). Cells where the process cytokinesis is just starting are analyzed as single cells by this strategy. To overcome such an issue, we further gated “DAPI” population for the area and aspect ratio of the nucleus mask to gate for single nucleus containing cells. Hence, events where karyokinesis was approaching completion but cytokinesis was just starting were not counted as singlets because of their low aspect ratio of nucleus mask. This mask was designated as “Single_DAPI”. Beginning with all acquired events, the goal of the analysis through a series of gates was to resolve single, focused cells positive for FLICA only, GFP only and double positive (DP) sub-populations. Plotting mean pixel for GFP and FLICA staining helped segregate populations based on their fluorophore signals. For THP1 cells, DAPI staining was not performed. We gated for cells in focus by plotting gradient RMS for the bright field channel (Cells) and identified singlets as described above. These were further gated for positive signal for Alexa Fluor®488.
Time of Flight Inflammasome Evaluation (TOFIE)
The sample left after acquisition on the ISXII was diluted to 200μl by ISXII cell suspension buffer and acquired on LSRII flow cytometer equipped with 405nm, 488nm, and 642nm lasers with long-pass filter of 505nm and band-pass filters of 450/50nm, 530/30nm, and 660/20nm. Acquisition was done using BD DIVAS software. Data was analyzed using FlowJo. Samples were gated to exclude debris and cell doublets. Singlet population was further gated for GFP staining. A stop gate of 104 cells was set on the GFP-positive gate. The percentage of cell containing ASC specks was determined by analyzing the height (H), width (W), and area (A) of the GFP pulse area (high H:A and low W:A indicates speck positive cells) as described previously by Sester and group (25, 30).
Immunofluorescence microscopy
Fixed cells were mounted on the slide using 10μl of FLUORO-GEL II (EMS; Cat.#17985–51) mounting medium with DAPI and visualized using an Axio Observer Z1 fluorescence microscope (Zeiss). Images were acquired using ZEN-Blue at an optimal setting to avoid saturation. Image acquisition was run at identical setting. Image analysis was performed using Fiji-ImageJ software. ASC-positive cells containing specks were counted manually from randomly selected fields acquired at 20X or 40X magnifications. The percentage of speck containing cells was calculated as the fraction of ASC-positive cells containing specks. The experiment was repeated three times and a minimum of 500 ASC positives cells were visualized.
Statistical analysis
For microscopy, more than 6 fields of view from individual experiments were used for imaging with a minimum of 500 cells inspected for specks. For the ISXII experiments a minimum of 104 cells were acquired in individual experiments. For TOFIE, 104 cells were acquired in three independent experiments. Data is represented as Mean ± SEM of three independent experiment, unless otherwise mentioned. Statistical analysis was performed using GraphPad Prism7 using either Student’s t-test or 2way ANOVA followed by Tukey’s multiple comparison test with p<0.05 being considered significant.
RESULTS
Strategy to identify and gate single nucleated cells
To automate the analysis of inflammasome-associated specks, it was first necessary to accurately define and gate the cells to be analyzed. IDEAS®, the software that analyzes ImageStream®X data, provides tools to evaluate image regions (masks) and perform calculations (features). However, the default settings are not optimized for specific analyses and without modification, feature calculations of these masks are inaccurate (34). We modified the default masks and used features to identify and gate single nucleated HEK293T cells. The default mask for bright field images (M04) identifies the presence of a signal but fails to discriminate between focused and unfocused cells (Fig. 1A). Applying an Object mask to the default mask (termed Focused cell) eliminates unfocused cells and captures only focused cells. By removing pixels of unfocused cells from analysis, the Focused cell mask also accurately calculates cell area. Similarly, the default system mask for nuclei (M02) recognizes two closely located nuclei as a single event, reporting the area of such events as larger than the cells analyzed (Fig. 1B). Applying a Morphology mask (termed Nucleus) resolves such events and precisely calculates the aspect ratio and area of individual nuclei. The Gradient_RMS (root mean square) function measures the sharpness quality of an image, making it useful for selection of focused objects. To identify focused nucleated cells, we gated for cell events with a medium Gradient_RMS for the Focused cell and Nucleus masks (Fig. 1C) to eliminate unfocused cells (low Gradient_RMS) and small debris (high Gradient_RMS). Focused cells in the Cells gate were further evaluated to identify single cells by plotting area vs. aspect ratio for the Focused cell mask. Events with an aspect ratio of 0.5–1.0 and an area ranging from 50–500 units (Single-BF) represent nearly circular cells allowing selection of single cells by the gate. In contrast, events with aspect ratios less than 0.5 represent cell-doublets and those with areas greater than 500 represent clumps even if when the aspect ratio approaches one, excluding non-singlet cells from analysis. The population in the Single-BF population was further evaluated for optimum DAPI staining by plotting the intensity of DAPI staining with a region set to exclude dim events (DAPI). Re-evaluation of the DAPI-gated population for aspect ratio (0.5 – 1) and area (40 – 275 units) with the Nucleus mask yields cells with a single nucleus, thus eliminating cells with two nuclei resulting from incomplete cytokinesis (Single DAPI). The presence of the expected cell populations within and outside the gate (Fig. 1C, micrographs), illustrates the accuracy of each gating strategy.
Strategy to identify cells with both ASC and active Caspase-1
Identification of cells expressing ASC and possessing Caspase-1 activity is necessary to evaluate formation of the ~1μm ASC inflammasome-associated speck and associated Caspase-1 activity. Use of GFP-ASC allows ready visualization of expressed ASC and any ASC-containing structures in the cell (25). Active Caspase-1 can be visualized with the fluorescent substrate FLICA (27). To develop a strategy to visualize and analyze cells containing both ASC and active caspase-1, the Single-DAPI population was segregated into sub-populations of GFP only, FLICA only and double positives (DP) by plotting the mean pixel values of the GFP and FLICA signals (Fig. 1D). Representative images from individual gates indicate the expected staining pattern (Fig. 1E). Similar results can be obtained by plotting histograms of intensity of individual fluorophore.
Validating masking strategy for ASC specks
IDEAS® allows development of requirement-specific masks (or regions of interests) to calculate features such as area, shape, distribution, frequency. We developed a strategy and the requirement-specific masks to automate detection of bona fide ASC specks and eliminate non-speck like aggregates. To establish a test population of cells, we selected cells (approximately 50–70 events) based on their visual GFP-ASC signal pattern to include those with speck like morphology, non-speck like aggregates, and cytoplasmic diffused GFP-ASC (Fig. 2A). Each mask was validated on the sample population before developing the subsequent mask. Such selective sampling allowed us to develop a masking strategy accurately reflecting speck morphology while eliminating false-positives and false-negatives (type I and type II errors). To achieve maximum accuracy our masking strategy is divided into three separate steps. Specifically, separation of punctate from diffuse signal (Fig. 2B), discrimination of circular from irregular structures (Fig. 2C) and selection based on area, circularity and number (Fig. 2D). To segregate bright, punctate GFP-ASC in ASC specks from diffused cytoplasmic GFP-ASC, the Spot mask was modified to create masks to select smaller and larger puncta (ASC Mask 1&2), these were combined to accommodate all punctate signals (ASC Mask 3) (Fig. 2B). To separate bright ASC-speck puncta from the dim puncta of random aggregates, Levelset mask was modified to create a combined mask (ASC Mask 6) composed of masks to select for medium intensity (ASC Mask 4) and bright (ASC Mask 5) puncta (Fig. 2B). ASC specks are highly organized circular structures, whereas aggregates are typically irregular. Another series of masks was designed to discriminate circular puncta from non-circular aggregates (Fig. 2C). A Fill mask was applied to ASC Mask 6 to remove variations within the masks (ASC Mask 7) and an Intensity mask was used with parameters set to select for high intensity signals (ASC Mask 8). Applying a Range mask with parameters set to identify less elongated (more elliptical to circular) structures further eliminated random aggregates with aspect ratio greater than 0.4 (ASC Mask 9). The mask was further refined for its size by using the Dilate mask (ASC Mask 10) and smoothed with an additional Fill mask (ASC Mask 11) (Fig. 2D). Finally, GFP has a weak tendency to oligomerize (35–37) that could result in false-positives. However, GFP aggregates are expected to be random in shape with less signal intensity, whereas ASC specks are more organized and circular in appearance. To select for circular puncta with high signal intensity and eliminate non-speck, aggregate artifacts, we generated two masks. As ASC specks are organized and expected to contain more ASC than random aggregates, a Threshold mask based on the intensity of visually inspected specks was used to exclude dimmer objects (ASC Mask 12), followed by a refined Range mask to more stringently select for circularity (aspect ratio >0.6) and further constrain the signal area to within approximately 30μm2 (ASC Mask 13). A speck in our analysis is defined as those ASC objects meeting the combined mask criteria of ASC Mask 13. Further, ASC Mask 13 does not exclude cells with more than one speck. Thus, to ensures that cells with multiple ASC specks are not counted multiple times which would inflate the frequency of speck-containing cells, we defined a Component mask to rank the area of the specks that (Fig. 2D) returns them in rank order from largest to smallest. Superimposing ASC Mask 14 on ASC speck images confirmed that this mask accurately reflects speck morphology (Fig. 2E). ASC Mask 14 can thus discriminate between Speck-positive and Speck-negative cells by bimodally distributing the population to distinguish cells with specks (1 or more) from those without (0 specks) by only counting the largest structure (ASC Mask 14). (Fig. 2F). Less than 0.5% of cells defined by ICCE as Speck-negative contained structures visually indistinguishable from ASC specks (Fig. S1), indicating that false-negative errors are exceedingly rare. Hereafter, ASC Speck mask refers to ASC Mask 14.
Figure 2: ASC speck masking strategy and calculation for frequency of cells with specks.
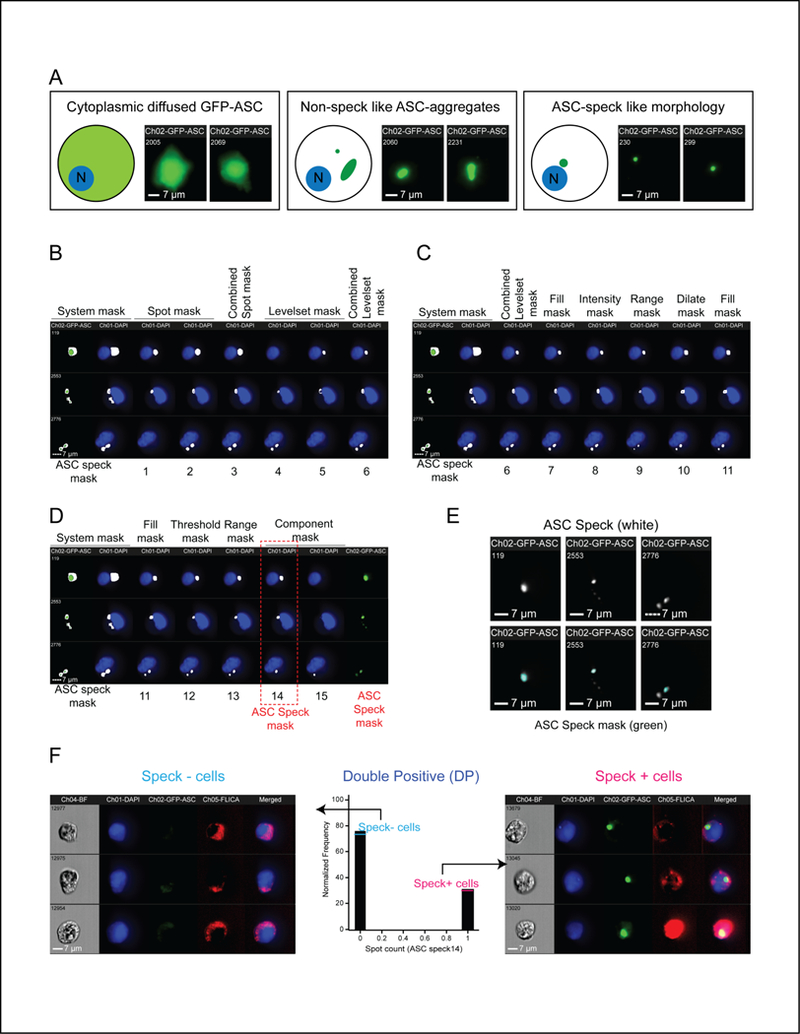
A. Schematic and representative images of sample HEK293T cell populations used to generate ASC speck mask. B-D. ASC Speck masking strategy. DAPI (blue), GFP-ASC (green) and corresponding ASC speck mask (white) are shown. “ASC speck mask” columns indicate the default system mask on the GFP channel alone (left) or with this mask superimposed on the DAPI channel (right). Numbers 1–15 represent different ASC masks (described in Table-II) sequentially applied to modify the default mask. B. Identifying bright, punctate staining. Spot masks: Recognize single punctate staining (ASC Mask 1). Identify multiple puncta (ASC Mask 2). Combined Spot mask 1 and 2 (ASC Mask 3). Levelset masks based on GFP signal threshold: Detect dimmer signals (ASC Mask 4). Detect brighter signals (ASC Mask 5). Combined Levelset masks 4 and 5 (ASC Mask 6). C. Differentiating GFP aggregates from ASC specks. A Fill mask was applied to make ASC mask 6 uniform (ASC Mask 7) with an Intensity mask to select pixels above a selected intensity (ASC Mask 8). A Range mask was applied to define minimum diameter and near circularity criteria (ASC Mask 9) and a Dilate mask was used to refine the size of ASC Mask 9 (ASC Mask 10) with another Fill mask for uniformity (ASC Mask 11). D. Final refinements to differentiate ASC specks from artifacts. A Threshold mask was used to eliminate GFP aggregates (ASC Mask 12) and was improved with a Range mask for area and circularity (ASC Mask 13). Some non-speck like aggregates were further sorted in terms of area (Component mask). The mask with the highest area (ASC Mask 14; highlighted by red box) is referred to as “ASC speck mask” as described in the study. E. ASC speck mask accurately reflects speck morphology. Micrograph of ASC specks (top). ASC specks with superimposed ASC speck mask (green; bottom). F. Determining ASC speck frequency. Histogram of cells evaluated with the “Spot count” function on the ASC speck mask (center; Spot count of 0 = speck negative (–), 1 = speck positive (+)). Representative images from speck- (left) and speck+ (right) sub-populations
Detection of ASC speck formation in response to NLRP3 agonist
The masks described above apply the same heuristic to all cells. Thus, “specks” fit the criteria defined by the mask and “aggregates” are those excluded by the mask. We previously established by microscopy that HEK293T cells transfected with 50ng of ASC-GFP and 100ng of NLRP3 minimized background speck formation and maximal nigericin speck induction. Using ICCE, we tested a range of ASC-GFP concentrations (10–500ng) to evaluate spontaneous speck formation, aggregate formation, and nigericin induction of specks. Background speck formation was minimized with 50–100ng of ASC-GFP and 100ng of NLRP3. Maximal nigericin induction of speck formation was also observed using these quantities (Fig S2A). Aggregates smaller, larger, sufficiently non-circular, or dimmer than specks are present in about 20% of cells receiving 10–50 ng of ASC-GFP and spontaneous/background specks are infrequent (~10%). With 250–500 ng, aggregates diminish to <5%, but spontaneous specks are common (~30%) (Fig S2B). These results are consistent with, and provide cell-based experimental corroboration for, biophysical studies demonstrating that speck formation is an all-or-none phenomenon and that the metastable protein ASC spontaneously aggregates at “supercritical” concentrations (>450nM) (39–41).
To validate the efficiency of our strategy, the frequency of ASC speck-containing cells detected by ICCE was evaluated and compared with flow cytometry-based TOFIE. Following nigericin treatment, 30% of NLRP3-expressing cells contained ASC specks using ICCE, a three-fold increase over untreated controls (Fig. 3A). In the absence of NLRP3, only 1% of nigericin-treated cells had ASC specks. In contrast, using the same samples for TOFIE, 60% of NLRP3-expressing cells contained ASC specks, a 1.6-fold increase above the untreated control (Fig. 3B). This difference suggested that either ICCE or TOFIE inaccurately identifies ASC specks. In the initial description of TOFIE, microscopic evaluation of speck frequency was compared to the TOFIE method. To make a similar comparison, we transfected HEK293T cells as before and counted ASC specks using fluorescent microscopy. By microscopic evaluation, 27% of NLRP3 expressing cells contained specks following nigericin treatment (Fig. 3C), a four-fold increase over untreated control. This frequency and fold increase are comparable to our results using ICCE. As ASC aggregates would have a decreased width:area profile (the key parameter for determining specks in TOFIE) we examined the speck-negative population from our ICCE experiments to evaluate the frequency of non-speck, ASC aggregates. In addition to small, dim ASC accumulations that are unlikely to be specks, other types of ASC aggregates were observed in speck-negative cells, including cells with nuclear accumulation of ASC and massive aggregations several fold larger than the canonical 1μm ASC speck (Fig. 3D and2A). These features were observed by microscopy as well (Fig. 3E) but are considered artifacts (red arrows) rather than bona fide ASC specks (white arrows). These phenomena result in decreased width:area ASC pulse profiles that fall within the parameters defined as a speck by TOFIE, leading to false-positives. In our hands, these artifacts accounted for about 2-fold more speck detection when using TOFIE versus microscopy or ICCE. This demonstrates that ICCE more precisely standardizes the definition of an ASC speck and distinguishes them from ASC artifacts, yielding accurate determinations comparable to careful microscopic evaluation.
Figure 3: Validation of ICCE with TOFIE and microscopy.
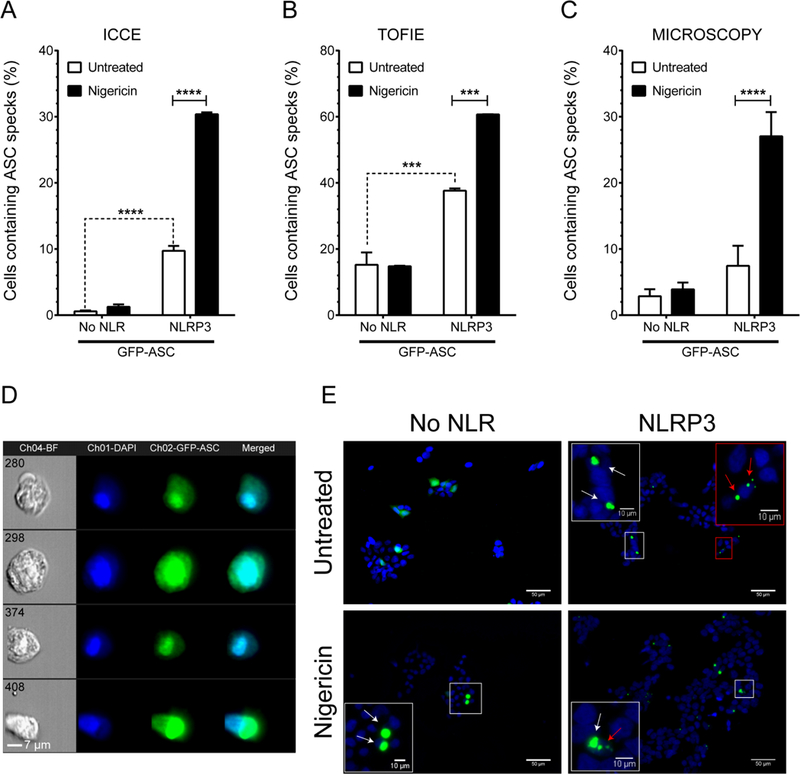
A. ICCE quantitation of ASC speck-containing cells. ICCE-calculated frequency of specks in HEK293T cells expressing GFP-ASC and pro-Caspase-1 in the absence or presence of NLRP3. Mean of pooled data from three independent transfections. B. TOFIE quantitation of ASC speck-containing cells. TOFIE analysis of cells from the corresponding samples shown in A. Mean of pooled data from 3 independent transfections. C. Microscopy quantitation of ASC speck- and GFP-ASC aggregate-containing cells. Fixed HEK293T cells transfected as in A and B were analyzed by microscopy and ASC specks were counted manually. Data represented as Mean ± SEM for three independent experiments. ***, p<0.001, ****, p<0.0001, 2-way ANOVA followed by Tukey’s multiple comparison test. D. GFP-ASC aggregates in nigericin-treated NLRP3 containing cells. ICCE “speck negative” events representative of GFP-ASC aggregates in nigericin treated cells. E. Micrographs of representative fields showing staining of nuclei (blue) and ASC (green). Selected ROI is shown in inset. ASC specks are indicated (red arrow), non-speck ASC aggregates (white arrow). Scale bar for the figure is 50 μm and for the inset is 10 μm.
Detection of endogenous ASC speck formation in response to NLRP3 agonist
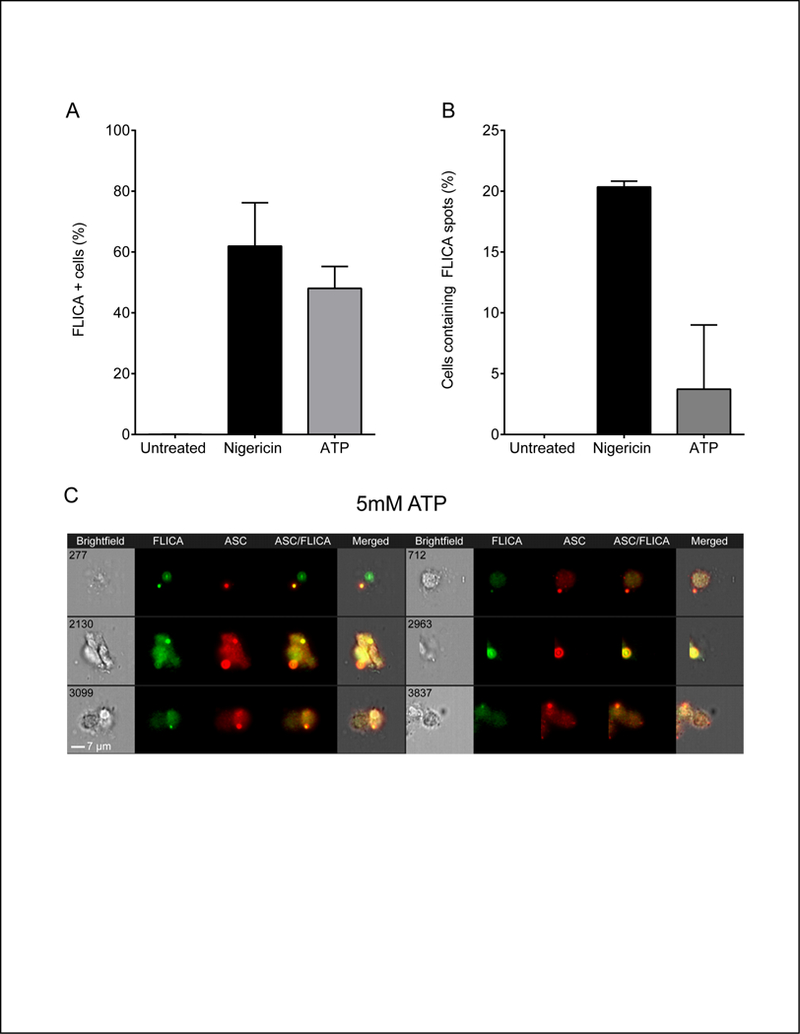
Although inflammasome reconstitution in HEK293T cells is a useful method to study the molecular basis of ASC speck formation and inflammasome function, evaluation of these events in macrophages is essential/desirable. HEK293T do not express inflammasome components which make them as most accepted model for studying the effects of various factors in inflammasome function. Whereas, the leukemic monocyte cell line THP1 provides a more physiological model of inflammasome activation and is thus widely used for screening triggers and inhibitors of inflammasome function. To establish whether our strategy can enumerate and localize specks in macrophages, we activated LPS primed THP1 with ATP and applied our masking strategy for speck identification. ASC primarily localizes to nucleus in resting THP1. Since staining the nucleus with DAPI interferes with detection of ASC staining, DAPI was not used for THP1 cells and the strategy to identify single, focused cells for analysis required modification. To identify focused cells, a threshold Gradient RMS (Cell mask; brightfield) value was selected to exclude unfocused cells (Cells). Focused Cells were further evaluated for singlets by plotting area vs. aspect ratio for “Cell” mask following the strategy described for Fig. 1 (Single-BF). Single-BF population was further evaluated for optimal ASC staining by plotting the histogram of intensity of Alexafluor488® staining (Secondary antibody staining for ASC) with a region set to exclude dim events (AF488+). (Fig. S3A). The specks formed in THP1 cells appear smaller than reconstituted specks containing GFP-ASC (17, 19, 20). Therefore, the ASC Speck mask settings were altered to detect these smaller specks, but the order of masks in the masking strategy was unchanged (see Table-II). Around 8% of the untreated cells contained ASC specks. Following ATP treatment, 32% of THP1 contained specks, a 3.5-fold increase in specks compared to untreated control (Fig. 4A). Less than 1% of cells deemed speck-negative contained structures resembling ASC specks. Also, less than 1% of cells defined as speck positive contained structures which would not be classified by microscopy as ASC specks. We also validated our assay on primary human monocytes. The image properties of the speck were different from THP-1, likely due to the use of different primary and secondary antibodies. Thus, we fine-tuned the masking strategy to account for those changes without altering the order of the masks (see Table-II). Cells containing ASC and active caspase-1(double positives) were identified by plotting intensities of FAM-FLICA and Alexafluor594® staining of the secondary antibody for ASC (Fig. S3B). Untreated primary human monocytes had no specks within the double positive population. Treatment with nigericin and ATP induced specks in 32% and 22% cells, respectively (Fig. 4B). We analyzed the same samples by TOFIE for speck positive cells. Similar to ICEE, no specks positive cells were detected in untreated cells. However, both nigericin and ATP treated cells returned specks in 49% cells (Fig. 4C). The 1.5–2-fold higher frequency of speck positive cells reported by TOFIE was similar to as observed in HEK293T cells. Visual inspection of nigericin and ATP stimulated cells lacking specks revealed the presence of ASC aggregates (Fig. 4D) which are likely to considered as events with low width to area profile by TOFIE, confirming that ASC aggregates are also considered positive by TOFIE which can be eliminated by ICCE. Thus, the same masking strategy can be applied to detect specks in primary monocytes and macrophage cell lines.
Figure 4: ICCE analysis of endogenous ASC specks in THP1 and primary human monocytes.
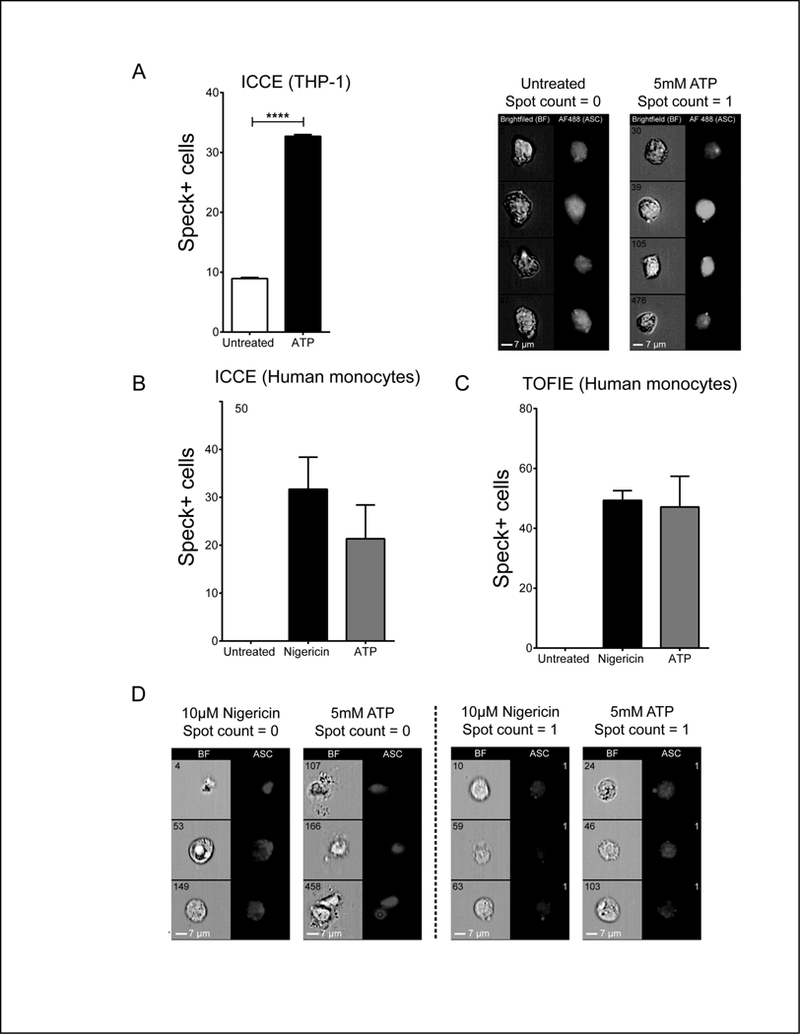
LPS primed THP1 (A) and primary human monocytes cells (B) either left untreated or stimulated with 5mM ATP or 10μM nigericin for 30mins were fixed and analyzed by ICCE (A-B) and TOFIE (C) for ASC specks A. ICCE detection of ASC specks in LPS-primed THP1 cells stimulated with ATP. Frequency of speck containing LPS-stimulated THP1 cells without and with ATP stimulation (left panel). Representative images of cells, Amnis brightfield and AF488 channel (ASC) before (Spot count=0, middle panel) and after ATP stimulation (Spot count=1, right panel). Note: ASC is detected in THP1 cell nuclei, but ASC speck mask 14 only detects appropriately sized, bright, punctate events (specks). Mean ± SEM of data from 26 groups of 1000 cells each, ****, p<0.0001, student’s t-test. B. ICCE detection of ASC specks in LPS-primed primary human monocytes stimulated with ATP and nigericin. Frequency of speck containing LPS-primed primary human monocytes cells without and with nigericin or ATP stimulation. C. TOFIE quantitation of ASC speck-containing primary human monocytes cells. Frequency of cells with specks from the corresponding samples in B. Mean ± SEM (n=2). D. Amnis brightfield and AF594 channel (ASC) of representative cells without specks (Spot count=0, left panels) and those with specks after nigericin and ATP stimulation (Spot count=1, right panels). Note: LPS-primed but unstimulated primary human monocytes cells lack active caspase-1staining and were absent from the double-positive gate.
Calculation of ASC speck area
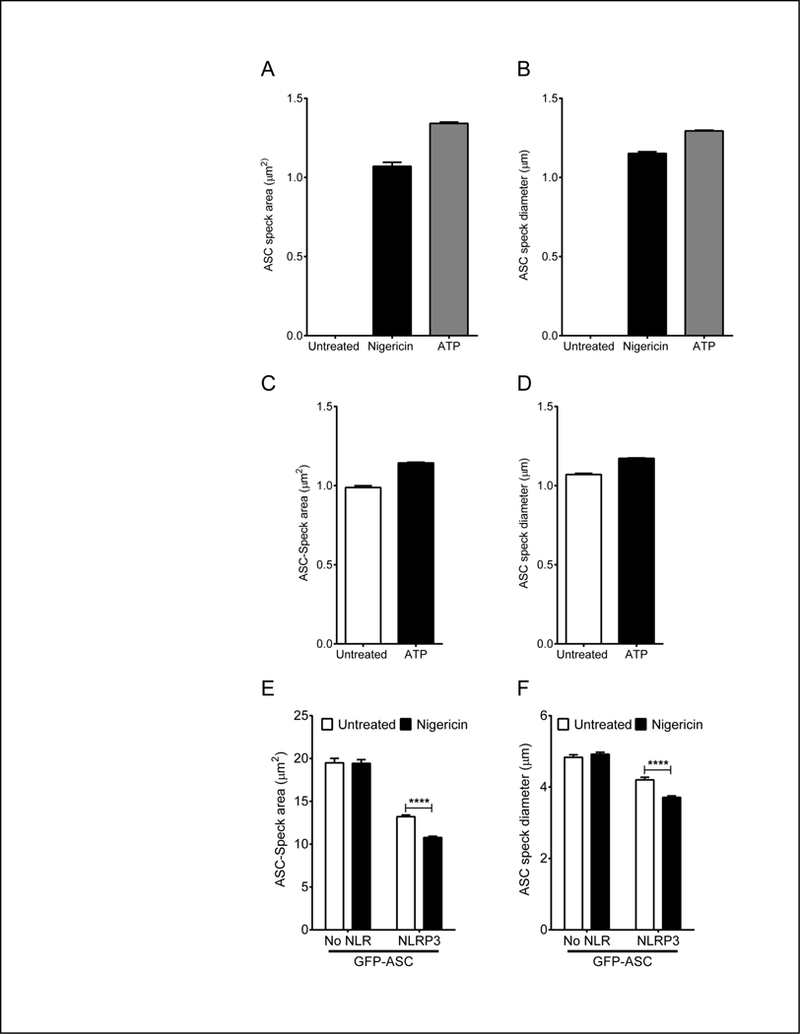
Enzymatically inactive caspase-1 activates receptor-interacting protein kinase-2 (RIP-2) to drive inflammation independently of the inflammasome (42). Moreover, the area of ASC specks is larger when caspase-1 is enzymatically inactive (43). Thus, speck area provides qualitative information regarding potential speck function and the type of the pathway involved in the inflammatory response. Morphological features such as area can be precisely calculated if the mask accurately resembles the structure in question. Applying the area feature to our defined ASC Speck mask (Fig. 2D) allows calculation of ASC speck area. Speck diameter can also be evaluated by applying the diameter feature to the ASC speck mask. In primary human monocytes, ASC speck area was approximately 1μm2 (Fig. 5A) with a diameter of about 1μm (Fig. 5B), consistent with previous studies of speck size. In THP1 cells, ASC speck area was also around 1μm2 (Fig. 5C). ASC speck area after ATP treatment of LPS-primed cells was fractionally larger than that of random ASC specks in cells without ATP addition. As expected, ASC speck diameter was also 1μm, consistent (Fig. 5D). In unstimulated HEK293T cells, speck area was significantly decreased in the presence of NLRP3 (Fig. 5E). Moreover, speck area was significantly reduced in NLRP3 expressing cells following nigericin treatment. However, ASC speck area was unaltered in nigericin treated cells lacking NLRP3. The GFP-ASC fusion is larger than ASC alone and as expected the diameter of ASC specks in NLRP3 expressing cells was larger than in THP-1 cells, about 4μm and notably smaller after nigericin treatment (Fig. 5F). When only ASC is expressed, the diameter of ASC specks is larger at approximately 5μm. Thus, ICCE can be used to evaluate speck area and diameter in a high-throughput system yielding quantitative results similar to those reported by others (43, 44). These changes in speck area suggest a possible higher order organization of the speck upon NLRP3 activation and implicate NLRP3 activation in dictating features of such organization (Fig. 5E-F).
Figure 5: ICCE analysis of ASC speck area and diameter in primary human monocytes, THP1 and HEK293T.
Cells were treated as described in Fig. 4. A-B. ICCE quantitation of ASC speck-area and diameter in primary human monocytes cells. ASC speck area (A) and speck diameter (B) in speck-positive in primary human monocytes cells without and with nigericin or ATP stimulation. Means ± SEM (n=2). C-D. ICCE quantitation of ASC speck-area and diameter in THP1 cells. ASC speck area (C) and speck diameter (D) in speck-positive THP1 cells without and with ATP stimulation. Mean ± SEM (26 groups of 1000 cells). ****, p<0.0001, student’s t-test. E-F. ICCE quantitation of ASC speck area and diameter in HEK293T cells. ASC speck area (E) and diameter (F) for speck-positive HEK293T expressing GFP-ASC and caspase-1 in the presence or absence of NLRP3 and either untreated or treated with 5μM nigericin. Mean ± SEM (10 groups of 1000 cells each). *, p<0.01, 2-way ANOVA followed by Tukey’s multiple comparison test.
Characterizing distribution and area of active caspase-1
Active inflammasomes are characterized by the presence of active capsase-1 which can be visualized with FLICA, a fluorescent inhibitor which binds irreversibly to the caspase-1 active site (38). FLICA exhibits minimal background staining above cellular autofluorescence (Fig S4A-B) and its mean fluorescence intensity (MFI) increases above background when caspase-1 is activated (Fig S4C). Further, NLRP3 inflammasome activation can be assessed by ImageStream using the change in FLICA MFI alone (no specific mask) (Fig S4D). A small number of microscopy studies have used FLICA to evaluate active caspase-1 in inflammasome specks (27, 38). However, this approach is labor intensive and no high-throughput approach has been developed. This limitation can be overcome with our ImageStream®X approach by constructing an appropriate FLICA mask to differentiate between diffuse and localized caspase-1 activity (Fig. 6A). To distinguish cells with bright punctate FLICA staining, Spot mask was applied to the default FLICA mask (FLICA-I). An Intensity mask was used to select for high intensity signals (FLICA-II). Like ASC specks, we expected FLICA spots to be highly organized. Thus, random small aggregates of FLICA were eliminated from the analysis by applying an Erode mask which uniformly reduces the size of mask by a defined number of pixels and excludes events below that pixel size (FLICA-III). A Dilate mask was applied to further refine the mask size (FLICA-IV). Finally, a Range mask was used to select for elliptical to circular shaped structures resembling a more organized accumulation of active caspase-1 (FLICA-V). This masking strategy differentiates punctate FLICA staining from diffuse staining and more precisely defines clusters of FLICA signal. Superimposing FLICA-V on FLICA spot images confirmed that this mask accurately reflects FLICA spot morphology (Fig. 6B). In HEK293T cells, even without caspase-1, some cells appear in the FLICA+ gate. To control for this background, we established a stringent threshold equal to the 130% of FLICA+ cells in the no caspase-1 control. This value was subtracted from FLICA-V mask counts for both spot negative and spot positive populations before calculating frequencies of FLICA spot containing cells. The number of FLICA spots within cells was determined by applying the spot count feature to the FLICA-V and returns the distribution frequency by number of FLICA spots per cell. A region was drawn to measure frequency of cells showing diffuse FLICA staining pattern (Spot count 0) and cells with FLICA aggregates (Spot count 1). Approximately 60–70% cells had diffuse FLICA staining indicating distributed cytoplasmic caspase-1 activity (Fig. 6C). FLICA spots were not above threshold levels in untreated cells expressing NLRP3, ASC, and caspase-1; however, following nigericin treatment, FLICA spots were present in 22–32% cells (Fig. 6D). These frequencies are similar to what we observe for ASC specks in NLRP3 and ASC expressing cells lacking Caspase-1 (Fig. 3A). Because active caspase-1 binds FLICA, the number of FLICA stained cells and FLICA intensity can be used as measures of inflammasome activation. Nigericin treatment resulted in a 4–20-fold increase in the number of cells with FLICA staining which was proportional to amount of transfected pro-caspase-1 encoding plasmid (Fig. 6E). This increase in FLICA+ cells correlates well with inflammasome activation. We also evaluated overall FLICA intensity. Background fluorescence of samples lacking caspase-1 was subtracted from all samples to provide a corrected FLICA intensity (CFI) for both untreated and nigericin treated samples. The CFI of nigericin treated samples was increased versus 4-fold over untreated samples when 20ng of Caspase-1 was transfected (Fig. 6F). However, with 50ng of caspase-1 plasmid, background FLICA intensity increased almost 2-fold over cells receiving 20ng of caspase-1 plasmid. Nigericin-treatment yielded FLICA intensities indistinguishable from cells receiving 20ng of caspase-1 plasmid. Thus, FLICA intensity correlates well with inflammasome activation at lower quantities, but the utility of this measurement decreases when more caspase-1 is introduced. As expected, cells with ASC specks typically have coincident FLICA spots (Fig. 6G). In cells lacking caspase-1, minimal FLICA intensity was detected at ASC specks while speck-associated FLICA intensity increases markedly in cells with active caspase (Fig. S4E-F). To assess ASC speck and FLICA co-occurrence in a cell, we measured the frequency of cells containing a FLICA spot in speck+ and speck- population. Cells without NLRP3 had no concurrent ASC specks and FLICA spots beyond the threshold levels in cells lacking caspase-1 (not shown). Approximately 30% of cells containing ASC specks had concomitant FLICA spots (Fig. 6H), the remainder have diffused FLICA staining. This result is consistent with caspase-1 being activated at the inflammasome followed by release of the FLICA-bound tetramer into the cytoplasm. We verified our masking strategy for detection of endogenous active caspase-1 in primary human monocytes. Active caspase-1 was not detected in untreated human monocytes (Fig 7A). Activation of NLRP3 inflammasome by nigericin and ATP reported around 60% FLICA-positive cells (Fig 7A). NLRP3 inflammasome activation also resulted in increase in cells containing FLICA spots, with 20% cells reported to show FLICA spots in nigericin treated cells and 4% with ATP treatment (Fig. 7B). Thus, our masking strategy and methodology can be used to detect the presence of ASC speck and active caspase-1 simultaneously. Moreover, this strategy can be used to detect the distribution pattern of caspase-1, evaluate inflammasome activation by FLICA intensity and assess co-incidence of ASC specks and FLICA spots.
Figure 6: FLICA spot masking strategy and evaluation of inflammasome activation by FLICA in HEK293T cells.
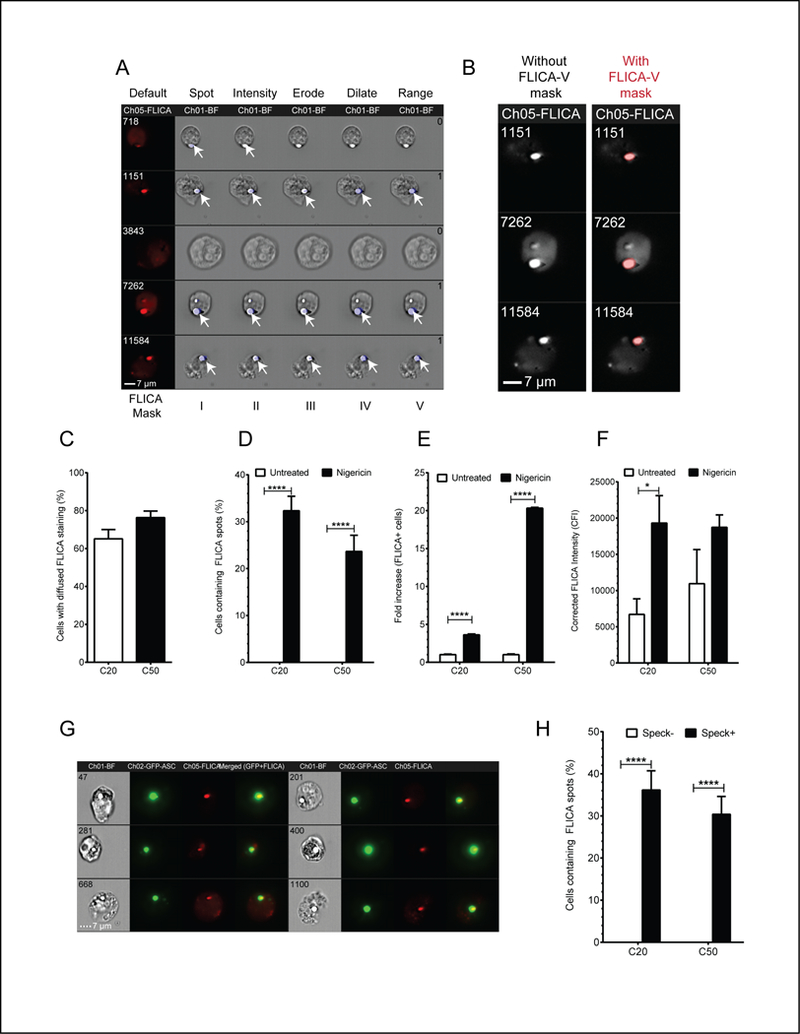
Brightfield (gray), FLICA and corresponding FLICA mask (blue, arrow) are shown. “Default” indicates the default system mask on the FLICA channel (left). FLICA I-V represent the different FLICA masks (described in Table-III) applied to modify the default mask. A. Identifying FLICA, punctate and diffused staining. Spot mask: Identify punctate staining (FLICA-I). Intensity: Select pixels above a selected intensity (FLICA-II). Erode mask: Remove mask from low intensity signals (FLICA-III). Dilate mask: Refining mask better fit the FLICA spot morphology (FLICA-IV). Range mask: Selects more organized accumulation of active caspase-1(FLICA-V). Arrows show the position of mask in a micrograph and number of FLICA spots detected are shown in number on FLICA-V micrograph. B. FLICA-V mask accurately reflects punctate FLICA morphology. Micrograph of FLICA spots (left). FLICA spots with superimposed FLICA-V mask (red; right). C. Diffused FLICA is the major staining pattern, irrespective of amount of transfected caspase-1. D. Cells containing FLICA spots were calculated for using spot count feature (ND: Not Detected). FLICA aggregation was induced above threshold level with nigericin treatment and was reduced in high caspase-1 transfected samples. E. ASC speck-containing cells positive for FLICA were calculated as described in the result section. For each condition where caspase-1 was transfected, data was normalized to respective untreated control. Nigericin-treated samples showed increased FLICA staining. F. Corrected FLICA Intensity (CFI) was calculated as described in the result section. Nigericin treatment induced greater caspase-1 activation with increased background for higher caspase-1 transfected samples. G. ASC speck positive cells showing co-visualization of ASC speck and FLICA spots. H. ASC Speck and FLICA spot association. Co-occurrence of ASC speck and FLICA spot was calculated as described in the result section (ND: Not Detected). Data represented as Mean ± S.E.M of pooled data from 13 groups of 1000 cells each. *, p<0.05, ****, p<0.0001, 2-way ANOVA followed by Tukey’s multiple comparison test.
Figure 7: ICCE quantification of endogenous active caspase-1.
(A) Frequency of FLICA-positive primary human monocytes without or with nigericin or ATP stimulation detected using the double positive gate (described in text and Fig. S3B). (B) Frequency of ASC speck-containing cells positive for FLICA spots within the double positive gate. Data represented as Mean ± SEM (n=2). Cells treatments as described in Fig 5B.
DISCUSSION
Here we describe the first imaging flow-cytometer-based high-throughput technique automating simultaneous image analysis of ASC specks and active caspase-1. In addition to enumerating ASC specks with active caspase-1, this technique can be used to perform quantitative image analyses to probe the dynamics of speck formation and caspase-1 activation, including parameters such as shape, area, and relative concentrations of ASC and caspase-1 activity. Further, the instrumentation also allows calculation of spatial coincidence between ASC specks and active caspase-1 which has not been previously studied. Combining high-throughput sample collection with fluorescent cell imaging makes this technique a powerful tool not only for the study of inflammasome activation in cell populations, but also to facilitate single-cell analysis to better understand the dynamics of ASC speck formation and caspase-1 activation.
Currently, inflammasome specks and active caspase-1 are evaluated using microscopy and/or flow cytometry. Both approaches present certain drawbacks. Flow cytometric techniques, although having the capacity to assay large numbers of cells, are constrained by the inability to detect colocalized enzymatic activity and evaluate morphological features of the speck structure together with quantitative fluorescence analysis at the single-cell level. Microscopy-based evaluation of inflammasome specks allows for quantitative and qualitative image analysis to characterize these structure in inflammatory settings. However, image acquisition and analysis by microscopy is time-consuming. Consequently, sample sizes tend to be small. Despite being useful for single-cell analysis, microscopy often fails to truly represent population data (28). Moreover, microscopy-based analysis is also subject to human bias and errors (29). Thus, automating the detection of specks and evaluation of caspase-1 activation eliminates human bias from the experiment analysis and offers a more accurate representation of the data.
In this study, we utilized computational image analysis methods to develop a gating and masking strategy to evaluate the features of ASC specks and associated caspase-1 activity. This strategy thoroughly excludes false positive events detected by TOFIE and manual microscopy approaches, increasing the accuracy of the analysis. The default masks supplied with the IDEAS® software report any signal. These masks fail to eliminate background and decrease signal to noise ratios (34) which can lead to inaccurate feature calculations. Therefore, we computationally created both brightfield and nucleus channels masks to accurately define cellular and nuclear boundaries which select cells containing single nuclei, eliminating cell clumps, doublets and karyokinetic cells. By gating for shape and area of cell as well as nucleus only single cells were exclusively selected for analysis (Fig. 1). Similarly, cells with GFP-ASC and active caspase-1 (positive for FLICA staining) were selected. (Fig. 1D-E). The ability to verify masks and gates with corresponding images substantially increases selection of true positives in a bias-free manner. Previously, two groups attempted to utilize ImageStream®X instruments to automate speck detection (20, 45). We used each strategy in a separate analysis of our data and found that both fail to eliminate non-speck aggregates (data not shown), demonstrating the significant limitations of these earlier approaches and the necessity for a more complex masking and gating strategy. Karmakar et al. used the spot-count wizards available in IDEAS® (45). This wizard fails to exclude cells with multiple ASC specks and those with speck-like structures resulting from random protein aggregation or altered cellular morphology due to pyroptotic signals. Tzeng et al. developed a gating strategy using the basic principle applied by TOFIE where the ratio of area of brightfield and GFP channel was used to determine the formation of specks (20). In their approach, the ratio of brightfield cell image area to the area of ASC-citrine will be greater than one when cells are stimulated and form specks. Selecting ASC specks by this method fails to exclude aggregates considerably larger than the expected ~1μm speck, which is also reflected by the disparity in our comparative analysis of the same samples by ICCE and TOFIE (Fig. 3). In contrast, the extensive masking strategy we describe sequentially segregates diffuse staining patterns from punctate structures and distinguishes circular to elliptical structures from those with an irregular shape. Further, the ICCE masking strategy allows differentiation between such artifacts and singular ~1 μm ASC structures consistent with the established physical features of ASC specks (Fig. 2&5). Moreover, generation of mask defining ASC specks equip the user to further calculate features such as shape, area and number of ASC specks in a cell. Thus, in addition to being faster and less biased than microscopic approaches, this complex masking strategy makes ICCE-based speck enumeration more robust, sensitive and accurate (error rate <0.5%) than TOFIE.
ICCE is not limited to detecting ASC specks in a reconstitution system but can also detect native inflammasomes in macrophages and primary human monocytes making it applicable in physiological settings (Fig. 4). However, native specks are smaller than reconstituted specks and require adjustments to the individual mask settings. Settings for both reconstituted and native specks are provided in Table-II (columns 3–5). ICCE can also be used for qualitative assessment of ASC speck features such as area, which is not possible using TOFIE. Variations in ASC speck size have recently been associated with the presence of active caspase-1, and the duration of caspase-1 activity (43, 44). Further, in the absence of NLRP3, hypotonic conditions lead to ASC speck formation without caspase-1 activation (46), reminiscent of the specks formed when ASC is overexpressed (17). Using ICCE we demonstrate that ASC speck size is reduced in the presence of NLRP3 (Fig. 5). Speck size is further reduced following nigericin treatment, which may reflect the smaller speck sizes associated with activation of caspase-1. ICCE can be used to accurately correlate speck size with inflammasome function at both the single-cell level and population levels. Although we demonstrate that our approach can be used for HEK293T, THP1 and primary human monocytes cells with results comparable to critical manual microscopic evaluation, we have not yet evaluated primary macrophages. However, our study is limited to the highest resolution of the objective magnification of the ImageStream®X which is 0.33μm. Thus, to study the detailed morphology of ASC specks and association of ASC molecules with caspase-1, high-resolution cryo-EM and super-resolution microscopy-based techniques should be employed. Imaging flow cytometers allow examination of altered cell morphology which is impossible using traditional flow cytometers. Examining altered cell morphology after stimulation with different inflammasome agonists may reveal qualitative differences between the triggers that drives caspase-1-mediated cytokine processing and/or cell death.
The available options for intracellular detection of active caspase-1 are limited. A common challenge encountered with the use of caspase-1 probes, including FLICA, is the need to reduce background noise and correct for non-specific binding (47). Our gating and masking strategy eliminates background noise, corrects for non-specific staining and determines the presence and distribution pattern (diffused or punctate) of active caspase-1. According to the manufacturer’s protocol, fluorescence measurements for FLICA stained samples must be acquired within 16hrs. A bi-fluorescent caspase-1 activation reporter that can be fixed and stained with a labelled antibody with the appropriate emission profile might further improve the potential of ICCE (48). The ImageStream®X is available in a single as well as a two-camera system. The two-camera system provides a larger selection of fluorophores which may further enhance ICCE by facilitating labelling of multiple inflammasome components. However, as antibody-coupled fluorochromes have different intensities, compensation may become an issue when staining for multiple inflammasome components. Whether this can be avoided by using the two-camera system remains to be seen. Ours is the first attempt to characterize the distribution, intensity, and morphology of active caspase-1. This is also the first high-throughput imaging-based method which co-visualizes speck formation together with active caspase-1.
In summary, the described assay allows rapid and unbiased quantification of native as well as reconstituted specks. ICCE not only enumerates speck and cells with active caspase-1, but also determines area, diameter, and distribution for ASC and active caspase-1. High-throughput imaging combined with quantitative imaging analysis makes simultaneous single-cell and population-based analyses of inflammasome activation a viable approach, with significant advantages over other techniques which only provide population level information. Additionally, simultaneous detection of active caspase-1 and specks in single cells facilitates quantitative evaluation of inflammasome activation. This may have broad application, for example, assessing novel inflammasome stimuli and regulators as well as performing comparative analyses. Further, ICCE can be used to study the dynamics and morphology of speck formation and/or caspase-1 activation under various activating conditions or to compare wild-type and mutated inflammasome component proteins. Finally, our masking/gating strategy can be further customized to accommodate other cell-types allowing further development as needed. The ability to study the inflammasome at both the single-cell and population level, while simultaneously providing quantitative image analysis of ASC specks and caspase-1 makes ICCE a highly powerful and versatile tool that is superior to other available techniques for detecting inflammasome activation.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Drs. Katherine Fitzgerald and Bibhuti B. Mishra for the kind gift of immortalized WT and Caspase-1/11−/− macrophages and guidance in using these cells.
This work was supported in part by NIH Grants R01AI072259 and P01AI056320 (subproject 2) to J.A.H. from the National Institute of Allergy and Infectious Diseases.
NON-STANDARD ABBREVIATION
- ASC
Apoptosis-associated Speck-like protein containing a Caspase recruitment domain
- NLRP3
NOD-like receptor containing pyrin domain 3
- ICCE
Inflammasome and Caspase-1 activity Characterization and Evaluation
- TOFIE
Time of Flight Inflammasome Evaluation
Footnotes
FOOTNOTE
REFERENCES:
- 1.Broz P, and Dixit VM. 2016. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16: 407–420. [DOI] [PubMed] [Google Scholar]
- 2.Latz E, Xiao TS, and Stutz A. 2013. Activation and regulation of the inflammasomes. Nat Rev Immunol 13: 397–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, and Han J. 2015. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res 25: 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, and Shao F. 2015. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665. [DOI] [PubMed] [Google Scholar]
- 5.Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, Navia MA, Murcko MA, Chambers SP, Aldape RA, Raybuck SA, and et al. 1994. Structure and mechanism of interleukin-1 beta converting enzyme. Nature 370: 270–275. [DOI] [PubMed] [Google Scholar]
- 6.Elliott EI, and Sutterwala FS. 2015. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol Rev 265: 35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lamkanfi M, and Dixit VM. 2014. Mechanisms and functions of inflammasomes. Cell 157: 1013–1022. [DOI] [PubMed] [Google Scholar]
- 8.Martinon F, Burns K, and Tschopp J. 2002. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10: 417–426. [DOI] [PubMed] [Google Scholar]
- 9.Tschopp J, and Schroder K. 2010. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 10: 210–215. [DOI] [PubMed] [Google Scholar]
- 10.Faustin B, Lartigue L, Bruey JM, Luciano F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I, and Reed JC. 2007. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol Cell 25: 713–724. [DOI] [PubMed] [Google Scholar]
- 11.Matusiak M, Van Opdenbosch N, Vande Walle L, Sirard JC, Kanneganti TD, and Lamkanfi M. 2015. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc Natl Acad Sci U S A 112: 1541–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis BK, Wen H, and Ting JP. 2011. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 29: 707–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menu P, and Vince JE. 2011. The NLRP3 inflammasome in health and disease: the good, the bad and the ugly. Clin Exp Immunol 166: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mortimer L, Moreau F, MacDonald JA, and Chadee K. 2016. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol 17: 1176–1186. [DOI] [PubMed] [Google Scholar]
- 15.Ting JP, Kastner DL, and Hoffman HM. 2006. CATERPILLERs, pyrin and hereditary immunological disorders. Nat Rev Immunol 6: 183–195. [DOI] [PubMed] [Google Scholar]
- 16.Yang CA, and Chiang BL. 2015. Inflammasomes and human autoimmunity: A comprehensive review. J Autoimmun 61: 1–8. [DOI] [PubMed] [Google Scholar]
- 17.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, Hidaka E, Katsuyama T, Higuchi T, and Sagara J. 1999. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem 274: 33835–33838. [DOI] [PubMed] [Google Scholar]
- 18.Richards N, Schaner P, Diaz A, Stuckey J, Shelden E, Wadhwa A, and Gumucio DL. 2001. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem 276: 39320–39329. [DOI] [PubMed] [Google Scholar]
- 19.Dick MS, Sborgi L, Ruhl S, Hiller S, and Broz P. 2016. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun 7: 11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tzeng TC, Schattgen S, Monks B, Wang D, Cerny A, Latz E, Fitzgerald K, and Golenbock DT. 2016. A Fluorescent Reporter Mouse for Inflammasome Assembly Demonstrates an Important Role for Cell-Bound and Free ASC Specks during In Vivo Infection. Cell Rep 16: 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuri P, Schieber NL, Thumberger T, Wittbrodt J, Schwab Y, and Leptin M. 2017. Dynamics of in vivo ASC speck formation. J Cell Biol 216: 2891–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, Schroder GF, Fitzgerald KA, Wu H, and Egelman EH. 2014. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156: 1193–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McConnell BB, and Vertino PM. 2000. Activation of a caspase-9-mediated apoptotic pathway by subcellular redistribution of the novel caspase recruitment domain protein TMS1. Cancer Res 60: 6243–6247. [PubMed] [Google Scholar]
- 24.Richards N, Schaner P, Diaz A, Stuckey J, Shelden E, Wadhwa A, and Gumucio DL. 2001. Interaction between pyrin and the apoptotic speck protein (ASC) modulates ASC-induced apoptosis. J Biol Chem 276: 39320–39329. [DOI] [PubMed] [Google Scholar]
- 25.Sester DP, Thygesen SJ, Sagulenko V, Vajjhala PR, Cridland JA, Vitak N, Chen KW, Osborne GW, Schroder K, and Stacey KJ. 2015. A novel flow cytometric method to assess inflammasome formation. J Immunol 194: 455–462. [DOI] [PubMed] [Google Scholar]
- 26.Stutz A, Horvath GL, Monks BG, and Latz E. 2013. ASC speck formation as a readout for inflammasome activation. Methods Mol Biol 1040: 91–101. [DOI] [PubMed] [Google Scholar]
- 27.Sokolovska A, Becker CE, Ip WK, Rathinam VA, Brudner M, Paquette N, Tanne A, Vanaja SK, Moore KJ, Fitzgerald KA, Lacy-Hulbert A, and Stuart LM. 2013. Activation of caspase-1 by the NLRP3 inflammasome regulates the NADPH oxidase NOX2 to control phagosome function. Nat Immunol 14: 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Arrasate M, and Finkbeiner S. 2005. Automated microscope system for determining factors that predict neuronal fate. Proc Natl Acad Sci U S A 102: 3840–3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carter M, and Shieh JC. 2015. Guide to research techniques in neuroscience. Elsevier Acad. Press, Amsterdam. [Google Scholar]
- 30.Sester DP, Zamoshnikova A, Thygesen SJ, Vajjhala PR, Cridland SO, Schroder K, and Stacey KJ. 2016. Assessment of Inflammasome Formation by Flow Cytometry. Curr Protoc Immunol 114: 14 40 11–14 40 29. [DOI] [PubMed] [Google Scholar]
- 31.Atianand MK, Duffy EB, Shah A, Kar S, Malik M, and Harton JA. 2011. Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J Biol Chem 286: 39033–39042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.O’Connor W Jr., Harton JA, Zhu X, Linhoff MW, and Ting JP. 2003. Cutting edge: CIAS1/cryopyrin/PYPAF1/NALP3/CATERPILLER 1.1 is an inducible inflammatory mediator with NF-kappa B suppressive properties. J Immunol 171: 6329–6333. [DOI] [PubMed] [Google Scholar]
- 33.Bryan NB, Dorfleutner A, Rojanasakul Y, and Stehlik C. 2009. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J Immunol 182: 3173–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pugsley HR 2017. Quantifying autophagy: Measuring LC3 puncta and autolysosome formation in cells using multispectral imaging flow cytometry. Methods 112: 147–156. [DOI] [PubMed] [Google Scholar]
- 35.Snapp EL, Hegde RS, Francolini M, Lombardo F, Colombo S, Pedrazzini E, Borgese N, and Lippincott-Schwartz J. 2003. Formation of stacked ER cisternae by low affinity protein interactions. J Cell Biol 163: 257–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zacharias DA, and Tsien RY. 2006. Molecular biology and mutation of green fluorescent protein. Methods Biochem Anal 47: 83–120. [PubMed] [Google Scholar]
- 37.Zacharias DA, Violin JD, Newton AC, and Tsien RY. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296: 913–916. [DOI] [PubMed] [Google Scholar]
- 38.Man SM, Hopkins LJ, Nugent E, Cox S, Gluck IM, Tourlomousis P, Wright JA, Cicuta P, Monie TP, and Bryant CE. 2014. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A 111: 7403–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cai X, Chen J, Xu H, Liu S, Jiang QX, Halfmann R, and Chen ZJ. 2014. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 156: 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sahillioglu AC, Sumbul F, Ozoren N, and Haliloglu T. 2014. Structural and dynamics aspects of ASC speck assembly. Structure 22: 1722–1734. [DOI] [PubMed] [Google Scholar]
- 41.Gambin Y, Giles N, O’Carroll A, Polinkovsky ME, Johnston W, Hunter DJ, Alexandrov K, Schroder K, and Sierecki E. 2018. Single-Molecule Fluorescence Reveals the Oligomerization and Folding Steps Driving the Prion-like Behavior of ASC. J Mol Biol 430: 491–508. [DOI] [PubMed] [Google Scholar]
- 42.Heymann MC, Winkler S, Luksch H, Flecks S, Franke M, Russ S, Ozen S, Yilmaz E, Klein C, Kallinich T, Lindemann D, Brenner S, Ganser G, Roesler J, Rosen-Wolff A, and Hofmann SR. 2014. Human Procaspase-1 Variants with Decreased Enzymatic Activity Are Associated with Febrile Episodes and May Contribute to Inflammation via RIP2 and NF-kB Signaling. Journal of Immunology 192: 4379–4385. [DOI] [PubMed] [Google Scholar]
- 43.Stein R, Kapplusch F, Heymann MC, Russ S, Staroske W, Hedrich CM, Rosen-Wolff A, and Hofmann SR. 2016. Enzymatically Inactive Procaspase 1 stabilizes the ASC Pyroptosome and Supports Pyroptosome Spreading during Cell Division. J Biol Chem 291: 18419–18429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, Gomez GA, Holley CL, Bierschenk D, Stacey KJ, Yap AS, Bezbradica JS, and Schroder K. 2018. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med 215: 827–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Karmakar M, Katsnelson M, Malak HA, Greene NG, Howell SJ, Hise AG, Camilli A, Kadioglu A, Dubyak GR, and Pearlman E. 2015. Neutrophil IL-1beta processing induced by pneumolysin is mediated by the NLRP3/ASC inflammasome and caspase-1 activation and is dependent on K+ efflux. J Immunol 194: 1763–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Compan V, Martin-Sanchez F, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Verkhratsky A, Brough D, and Pelegrin P. 2015. Apoptosis-associated speck-like protein containing a CARD forms specks but does not activate caspase-1 in the absence of NLRP3 during macrophage swelling. J Immunol 194: 1261–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Edgington LE, Berger AB, Blum G, Albrow VE, Paulick MG, Lineberry N, and Bogyo M. 2009. Noninvasive optical imaging of apoptosis by caspase-targeted activity-based probes. Nat Med 15: 967–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu T, Yamaguchi Y, Shirasaki Y, Shikada K, Yamagishi M, Hoshino K, Kaisho T, Takemoto K, Suzuki T, Kuranaga E, Ohara O, and Miura M. 2014. Single-cell imaging of caspase-1 dynamics reveals an all-or-none inflammasome signaling response. Cell Rep 8: 974–982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.