

Abstract
Objective:
Protein disulfide isomerase A1 (PDI) was reported to support Nox1 NADPH oxidase (Nox1) activation mediated by growth factors in vascular smooth muscle cells. Our aim was to investigate the molecular mechanism by which PDI activates Nox1 and the functional implications of PDI in Nox1-activation in vascular disease.
Approach and Results:
Using recombinant proteins, we identified a redox interaction between PDI and the cytosolic subunit p47phox in vitro. Mass spectrometry of crosslinked peptides confirmed redox-dependent disulfide bonds between cysteines of p47phox and PDI, and an intramolecular bond between Cys 196 and 378 in p47phox. PDI catalytic Cys 400 and p47phox Cys 196 were essential for the activation of Nox1 by PDI in vascular smooth muscle cells. Transfection of PDI resulted in the rapid oxidation of a redox-sensitive protein linked to p47phox whereas PDI mutant did not promote this effect. Mutation of p47phox Cys 196, or the redox active cysteines of PDI, prevented Nox1 NADPH oxidase complex assembly and vascular smooth muscle cell migration. Proximity ligation assay confirmed the interaction of PDI and p47phox in murine carotid arteries after wire injury. Moreover, in human atheroma plaques, a positive correlation between the expression of PDI and p47phox occurred only in PDI family members with the a’ redox active site.
Conclusions:
PDI redox cysteines facilitate Nox1 complex assembly, thus identifying a new mechanism through which PDI regulates NADPH oxidase activity in vascular disease
Keywords: NADPH oxidase, vascular smooth muscle cells, protein disulfide isomerase, redox-signaling, Vascular Biology, Oxidant stress, Cell biology/structural biology, Mechanisms
TOC category:
Basic
TOC subcategory:
Vascular Biology
Introduction
Reactive oxygen species (ROS) are essential mediators of cell physiologic signaling and have been implicated in the pathogenesis of cardiovascular disease. Although several cellular sources of ROS have been identified, the primary source of ROS in vascular cells are the NADPH oxidases (Nox). The isoform Nox1 constitutes a catalytic core at the cell membrane with p22phox and is constitutively activated in vascular cells by the canonical system consisting of the Nox organizing subunit 1 (NoxO1) and Nox activating subunit 1 (NoxA1) 1. In contrast, p47phox and p67phox, homologous to NoxO1 and NoxA1 respectively, allow for inducible activation of Nox1. Protein kinase C dependent phosphorylation of p47phox produces a conformational change allowing its binding to p22phox and to p67phox, thus activating the enzyme complex 2, 3.
Protein disulfide isomerase (PDI), belongs to the thioredoxin superfamily of dithiol-disulfide oxidoreductases and catalyzes thiol isomerase, oxidase and reductase activities 4, 5. Our group has described an interplay between PDI and Nox1, placing the isomerase as a regulator of Nox1 activity and expression. PDI regulates Nox1 activation and promotes redox-dependent processes such as PDGF-induced vascular smooth muscle cell (VSMC) migration and TNF α-dependent angiogenesis 6–8. The overexpression of PDI in VSMC increased Nox1 activation and expression in response to angiotensin II 9. Increased PDI levels were observed in resistance arteries during the development of hypertension and correlated with Nox1 expression and ROS generation10. However, the molecular mechanism through which PDI increases Nox1 signaling remains unknown.
In this study, we identify the molecular mechanism by which PDI activates Nox1. Our data indicate that PDI interacts with p47phox, forming crosslinked disulfides between redox cysteines, facilitating p47phox phosphorylation and Nox1 activation. These findings identify a novel regulatory mechanism by which PDI regulates NADPH oxidase activity in the vascular system.
Material and Methods
The data that support the findings of this study are available from the corresponding author upon request. Please see the Major Resources Table in the Supplemental Material for additional information.
Mouse models:
All procedures were approved by the Institutional Animal Care and Use Committee and complied with the standards stated in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male and female mice were used for isolation of VSMC from aortae and male mice were used for carotid injury experiments. Animals were bred at the local facility under standard conditions with 12/12 hour dark/light cycle with free access to standard chow diet and water.
Wire-mediated vascular injury of the left common carotid artery was performed in C57Bl/6 male mice as described 11. Briefly, the mice were anesthetized with buprenorphine / Isoflurane and a curved spring wire was inserted into the left common carotid artery. The wire was rotated and passed along the common carotid artery. At postoperative day 14, both carotid arteries were excised and used for Duolink analyses.
Non-human primate model:
Aortae from Cynomolgus monkeys collected from a prior study 12 and stored at −80C were analyzed. The original collection of these tissues was approved by the University of Iowa Institutional Animal Care and Use Committee and animal characteristics previously described 12.
Cell culture:
Cultured cells included VSMC isolated from aortae C57BL/6 of Nox1 null mice (Nox1 -/y) 13, COS cells with stable expression of p22phox 14, rabbit aortic VSMC from a previously established selection-immortalized line 6, and HEK-293 cells. When indicated, cells were treated with DPI (10 µM), TNF-α (10 ng/mL), thrombin (2 U/mL), angiotensin II (100 nM), PEG SOD (50 U/mL), or rutin (100 μM).
Plasmids:
Transfection of cells with plasmids described in the Major Resources Table include PDI mutated in all four redox cysteine residues9, p47phox-roGFP 15, and p47phox mutated at cysteines C196 and C378 to alanine residues using the QuikChange Site-Directed Mutagenesis Kit per the manufacturer’s instructions. . For plasmid transfection, COS and HEK-293 cells were transfected with 1 μg DNA in Opti-MEM medium with Lipofectamine 2000. After 4 h, the medium was changed to 10% FBS medium overnight. VSMC were transfected with 5 μg DNA and Lipofectamine 2000 for 48 h, then serum deprivation for 24 h prior to the experiments.
siRNA transfection:
VSMC from male and female mice were incubated with 50nM of siRNA (Invitrogen) and Hiperfect transfection reagent (Invitrogen) in serum-free DMEM for 4 h. After transfection, medium was replaced by DMEM containing 10% serum and experiments were conducted 48h later.
Recombinant fusion protein studies:
Recombinant fusion proteins composed of GST linked to p47phox and PDI were isolated from Escherichia coli, transformed with pGEX-6P2 (GE Healthcare) plasmids containing cDNA inserts encoding the human proteins of interest. GST tags were cleaved by PreScission protease (GE Healthcare). To characterize the formation of disulfide bonds between PDI and p47phox, 10 μg of recombinant proteins were submitted to an in vitro reaction protocol in incubation buffer with or without arachidonic acid (200μM) at 37ºC for 2h. Different conditions were used during incubation: alkylation with N-ethylmaleimide (NEM, 5 mM), and oxidation and reduction with diamide (0.5 M) and DTT (1 mM) respectively. Bands were cut from gels and submitted to trypsin digestion, followed by protein identification in a MALDI-TOF-TOF mass spectrometer (Axima Performance – Kratos-Shimadzu, Manchester, UK) using Mascot Server.
Proteomic analysis:
Mapping of crosslinked cysteines was performed by incubating p47phox (10 μM) with PDI wt (10 μM) in 100μL of incubation buffer for 2 h in the presence of arachidonic acid, then 5 mM NEM was added to block free thiols. The complex was resolved by non-reducing 8% electrophoresis gel and stained by Coomassie blue. PDI, p47phox and the heterodimer containing the interaction band (~100 kDa) were cut and destained. Disulfide bridges were mapped according to identified dipeptide precursor ions and fragment spectra analyzed by Stavrox 3.4.12 using a crosslink database created between PDI and p47phox 16. Three missing cleavage peptides were tolerated after lysine and 2 after arginine, with a fragment ion precision of 0.05 Da. Mass error tolerance was set to 5 ppm and fragment ion to 0.05 Da. Cross linker disulfide (−2.01565) variable modifications: oxidation of methionine of cysteines with N-ethylmaleimide, N-ethylmaleimide hydrolyzed, di- and trioxidation. Only crosslink candidates with a false discovery rate of less than 5% were considered for further evaluation. These analyses were performed at the Functional Proteomics Core Unit Faculty of Medicine Goethe-University, Frankfurt, Germany.
Proximity ligation assay:
Duolink analysis was performed as described in the manufacturer’s protocol (Duolink II Fluorescence, OLink, Upsalla, Sweden). Briefly, mice were perfused with PBS. Dissected carotid arteries were stored overnight in 4% PFA at 4° C and embedded in paraffin and sectioned to 4 μm thickness. Paraffin slides and also VSMC slides fixed in 4% PFA were rehydrated, permeabilized with Triton X-100 (0.05% in PBS) blocked and incubated overnight with antibodies against mouse anti PDI (RL90, ThermoFisher Scientific, Dreieinch, Germany) and goat anti p47 phox (#1588, provided by Thomas Leto, NIH, Washington, USA). After washing, samples were incubated with the respective PLA-probes for one hour (37°C), washed and ligated for 30min (37°C). After an additional washing, amplification with polymerase was allowed for 100min (37°C). The nuclei were stained using DAPI. Images were acquired by confocal microscope (LSM 510, Zeiss).
Gene expression in human atherosclerosis:
Data from human atherosclerotic plaque 17 which included 32 paired samples of atheroma plaque and macroscopically intact tissue was retrieved. The mRNA expression levels were downloaded from Gene Expression Omnibus (GEO) with the corresponding GEO accession ID GSE43292. Each platform’s probe ID was mapped to the corresponding gene symbol and the expression levels were averaged over multiple probes mapped to the same gene symbol. The correlation between p47 phox was calculated for the following PDI members: AGR2, AGR3, ERp27, ERp29, ERp44, P4HB, PDIA2, PDIA3, PDIA4, PDIA5, PDIA6, PDIALT, TMX1, TMX2, TMX3, TMX4, and ERp46. Conversely, the correlation of PDI was calculated for the NADPH oxidase members (Nox1, Nox2, Nox4, p47phox, p22phox, p67phox, NoxO1, NoxA1).
Statistical analysis:
All data are expressed as mean ± SEM. All data passed both normality and equal variance test and parametric analysis was applied. For two group comparisons, non-dependent samples were analyzed by a two-tailed unpaired t-test. For multiple-group comparisons, one-way or two-way ANOVA followed by Tukey’s post-hoc test was used. Differences are considered statistically significant at probability values of p<0.05. Pearce correlation analysis was used to test linear relationship between samples using a p value of 0.01 as cut off. The analysis was performed using GraphPad Prism Software (La Jolla, CA).
Results
PDI interacts with p47phox through redox cysteines.
We have shown that the redox state of PDI is important for its interaction with p47phox in neutrophils 18. PDI is structured in a “U” shape, with b - b’ domains towards the bottom of the “U” and b´ responsible for hydrophobic interaction with proteins. The a – a’ domains contain the redox cysteines located in the thioredoxin domains (CGHC) (Fig 1A). p47phox has an N-terminal domain PX, followed by two SH3 domains in tandem (which associate with p22phox), an autoinhibitory domain that prevents complex assembly (AID), and a C-terminal proline-rich region (PRR) responsible for binding to the activator subunit. To understand PDI/p47phox interactions, recombinant proteins were subjected to an in vitro reaction protocol under different redox conditions. First, PDI and p47phox interacted (~100 kDa, Fig 1B) in the presence of arachidonic acid, which unravels the p47phox auto-inhibitory domain thereby activating p47phox. The thiol alkylator N-ethylmaleimide (NEM) prevented the interaction of PDI with p47phox, due to the alkylation of the cysteines and inhibition of the formation of the disulfide bridges between the two proteins (Fig 1B). The band at ~80 kDa represents a p47phox dimer (Supp Figure I). We also identified an intermolecular disulfide bridge only in conditions of reduced PDI and oxidized p47phox, consistent with the PDI redox state found in the normal cellular environment. We speculate that the band immediately below 100 KDa may represent an intermolecular bond between two denatured PDI proteins (Fig 1C). Mutation of PDI in all four redox cysteines prevented interaction with p47phox, indicating that PDI/p47phox dimer formation is dependent on these cysteines (Fig 1D). We also identified higher molecular weight bands, suggestive of the formation of higher molecular weight complexes between PDI and p47phox, particularly after the addition of AA (Fig 1D). The reduction of both PDI and p47phox prevented interaction between these proteins (Fig 1E). All described protein interactions were confirmed by mass spectrometry (data not shown). These data identify the redox cysteines in PDI as essential for the interaction between PDI and p47phox.
Figure 1. PDI interacts with p47phox through its redox active sites.
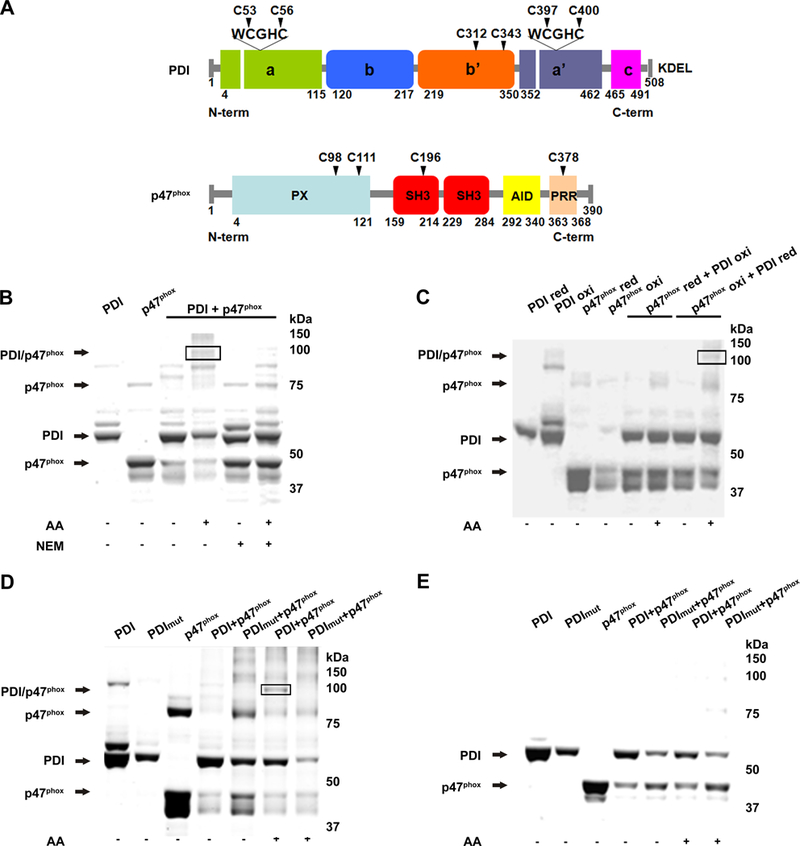
(A) Cysteine positions within domains of PDI and p47phox. (B) Non-reducing polyacrylamide gel stained with Coomassie blue shows monomers and dimers following incubation of recombinant wt PDI and p47phox with and without NEM. (C) Combinations of reduced PDI (PDI-red) and oxidized p47phox (p47phox-oxi), and oxidized PDI and reduced p47phox were analyzed as in B. (D) PDI mutated at the four redox cysteines (PDI mut) was reacted with p47phox and resolved in non-reducing (D) and reducing (E) polyacrylamide gels. AA: arachidonic acid. n=2.
PDI levels are increased in vascular disease and activation of Nox1 is dependent on PDI redox cysteines.
We next assessed whether PDI may contribute to Nox1 activation in vascular disease. We have previously reported that NADPH oxidase activity and Nox1 protein expression are increased in vessels isolated from monkeys on an atherogenic diet. 12,19 We found that PDI protein was also increased in the aorta from monkeys on an atherogenic diet as compared to a control diet (Fig 2A). We have also previously shown that Nox1-derived superoxide 20 regulates the activation of the extracellular signal-regulated kinase (ERK)1/2 and that phosphorylation of ERK1/2 is increased in atherosclerotic aortae 19. We next analyzed if PDI regulated the activation of ERK1/2. Angiotensin II induced ERK1/2 phosphorylation in VSMC which was abolished after PDI silencing (Fig 2B, Supp Fig II).
Figure 2. PDI is increased in atherosclerosis and regulates Nox1 NADPH oxidase-mediated signaling.
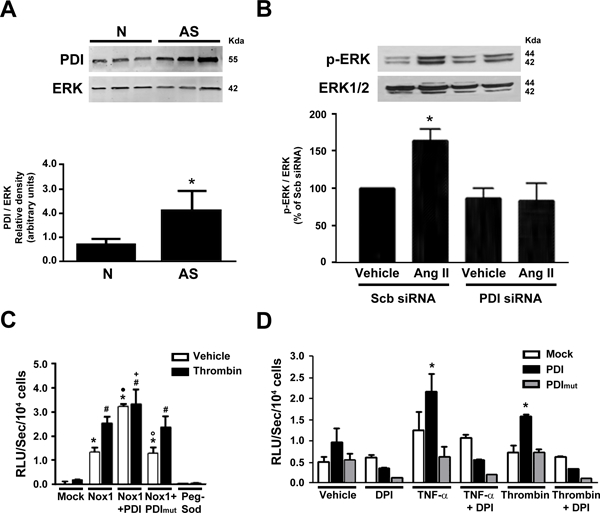
(A) Expression of PDI in non-human primate aorta (N: Normal, AS: atherosclerotic) n=3, *p<0.05 vs N. Data normalized to total ERK2 levels. (B) Ang II-induced ERK 1/2 phosphorylation after PDI silencing in VSMC. Quantification normalized to total ERK 1/2 levels. n = 3, *p<0.05 vs scr. (C) Superoxide levels in HEK-293 cells after transfection with PDI or PDI mut treated with DPI, n=3, *p<0.05 vs mock. (D) Superoxide levels measured by L-012 chemiluminescence (RLU) in Nox1-/y VSMC after transfection with Nox1, PDI or PDI mut and stimulated with thrombin. *p<0.05 vs Mock vehicle, # p<0.05 vs Mock thrombin, • p<0.05 vs Nox1 vehicle, ◦ p<0.05 vs Nox1/PDI vehicle, + p<0.05 vs PDI thrombin, n=3.
We next tested the importance of the redox-active cysteines of PDI for Nox1 activation. Superoxide generation was evaluated using L-012-enhanced chemiluminescence. In previous studies, L-012 was found to be reliable for detecting Nox-derived superoxide because, unlike other chemiluminescent probes, L-012 was not subject to redox cycling 21. In contrast Zielonka and co-workers showed that both peroxidase activity and superoxide are responsible for the overall L-012 luminescent signal intensity 22. Using a heterologous transfection system, we have measured basal chemiluminescence under various conditions. Although it is possible that a component of the measured chemiluminescence signal may not be derived from superoxide, the signal was nearly abolished in VSMC that were deficient in Nox1 (Nox1-/y) and rescued after transfection with Nox1 (Fig 2C). The co-expression of Nox1 and PDI markedly increased the levels of ROS even in the absence of an agonist and this effect was prevented by transfection of PDI mutated in all four redox cysteine residues (PDI mut). Furthermore, expression of PDI WT, but not PDI mut, in HEK293 cells increased Nox1-dependent superoxide generation in response to TNF-α and thrombin, and this effect was abolished by the flavoenzyme inhibitor DPI (Fig 2D). These data indicate that increased levels of PDI, as observed in non-human primate atherosclerotic aortae, is sufficient to augment Nox1 activation, and is dependent on PDI reactive cysteines.
PDI interacts with p47phox to activate Nox1
Nox1 requires the recruitment of NADPH oxidase cytosolic subunits to its membrane location for activation. We next determined whether PDI interacts with the cytosolic subunit p47phox and if this interaction contributes to Nox1 activation. We first tested the interaction of endogenous PDI and p47 phox in cultured VSMC. Proximity ligation assay (PLA) demonstrated that an interaction between p47phox and PDI increased in response to thrombin (Fig 3A).
Figure 3. PDI increases Nox1 activation through a redox dependent interaction with p47phox.
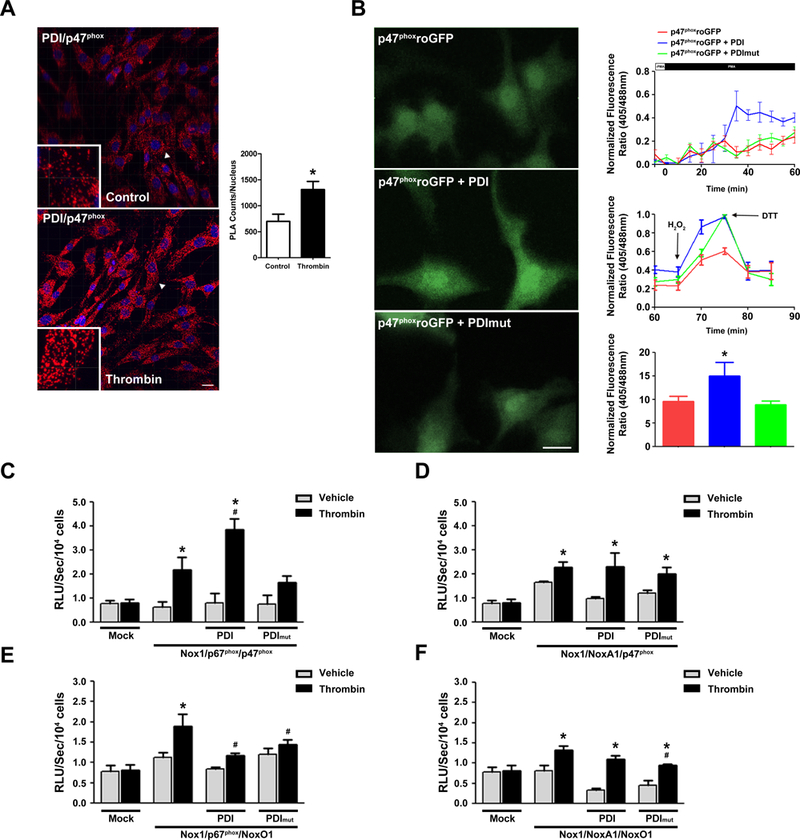
(A) Representative Z-stack images of Duolink analyses of PDI interaction with p47phox in VSMC stimulated or not with Thrombin (Imaris, Bitplane, Version 7.6.5). Positive signals demonstrating an interaction of the indicated proteins are shown as red dots, DAPI (blue), n=3. 41 slices were quantified using ImageJ. *p< 0.05 vs Control (B) Representative time course of p47-roGFP in rabbit VSMC. Cells were dually excited with 405 and 488 laser lines and the emitted fluorescence at 505–550 nm was captured at 20s sampling intervals. Representative data of at least 15 cells for each condition. * p< 0.05 vs p47-roGFP. (C-F) COS p22 phox cells were transfected with indicated subunits and superoxide levels measured by L-012 chemiluminescence (RLU). n=6 for each condition, * p< 0.05 vs mock, # p< 0.05 vs thrombin with no PDI.
Next, the ability of PDI to increase Nox1 activation by PMA was investigated using a redox-sensitive GFP (roGFP) linked to p47phox (p47-roGFP). PDI transfection resulted in the rapid oxidation of p47ro-GFP, whereas PDI mut did not promote this effect (Fig 3B). The stimulation of the cells with PMA increased p47ro-GFP oxidation only in VSMC transfected with PDI wt but not with PDI mut. After 60 min, cells were treated with hydrogen peroxide to maximally oxidize, followed by addition of DTT to maximally reduce, the roGFP. Together, these findings indicate that PDI directly interacts with the p47phox subunit via redox active cysteines and activates Nox1.
PDI redox regulation of Nox1 requires p47 phox
We next evaluated the specificity of PDI’s interaction with the NADPH oxidase cytosolic subunits for activation of Nox1. Thrombin caused Nox1-dependent superoxide production in COS p22phox cells with each combination of the cytosolic homologs (p67/p47; NoxA1/p47; p67/NoxO1; NoxA1/NoxO1) (Fig 3C-F). PDI increased superoxide generation by Nox1 only with p67/p47, and PDI mut attenuated the generation of superoxide. Taken together, these data suggest that PDI interacts with p47phox in a resting state and recruitment to the membrane and activation of Nox1 is dependent on PDI redox cysteines. Furthermore, PDI activation of Nox1 requires p47phox and is potentiated by its combination with p67phox.
Disulfide bridging of PDI and p47phox.
Because our findings suggested the importance of PDI cysteines for its interaction with p47phox, we next assessed potential inter- and intramolecular cross-linking of these two proteins. Disulfide bonds in isolated heterodimers of PDI and p47phox were evaluated using the cross-linking mass spectroscopy analysis tool StavroX (Fig 4A). P47phox cysteines 98 and 111 cross-linked with PDI cysteines 312 and 343. p47phox C196 was identified as a candidate for PDI interaction and formed an intermolecular disulfide with C400 located within the a’ domain of PDI and in the C-terminal domain (Fig 4B). p47phox Cys196 also formed an intramolecular disulfide bridge with C378 (Fig 4C). Spectra obtained were consistent with covalent adducts between peptides SESGWWFCQMKAK-KNVFVEFYAPWCGHCKQLAPIWDK (Supp Fig III), CSESTK- SESGWWFCQMK (Supp Fig IV), and KEECPAVR-SESGWWFCQMK (Supp Fig V), corresponding to the intermolecular disulfide between Cys 196 of p47phox and Cys 400 of PDI; intramolecular disulfide between Cys 196 and Cys 378 of p47phox; and intermolecular disulfide between Cys 312 of PDI and Cys 196 of p47phox. Of note, the C378-C196 p47phox intramolecular disulfide bond was not seen in the p47phox monomer but only in the PDI/p47phox dimer.
Figure 4. Identification of PDI - p47phox disulfide bonds.
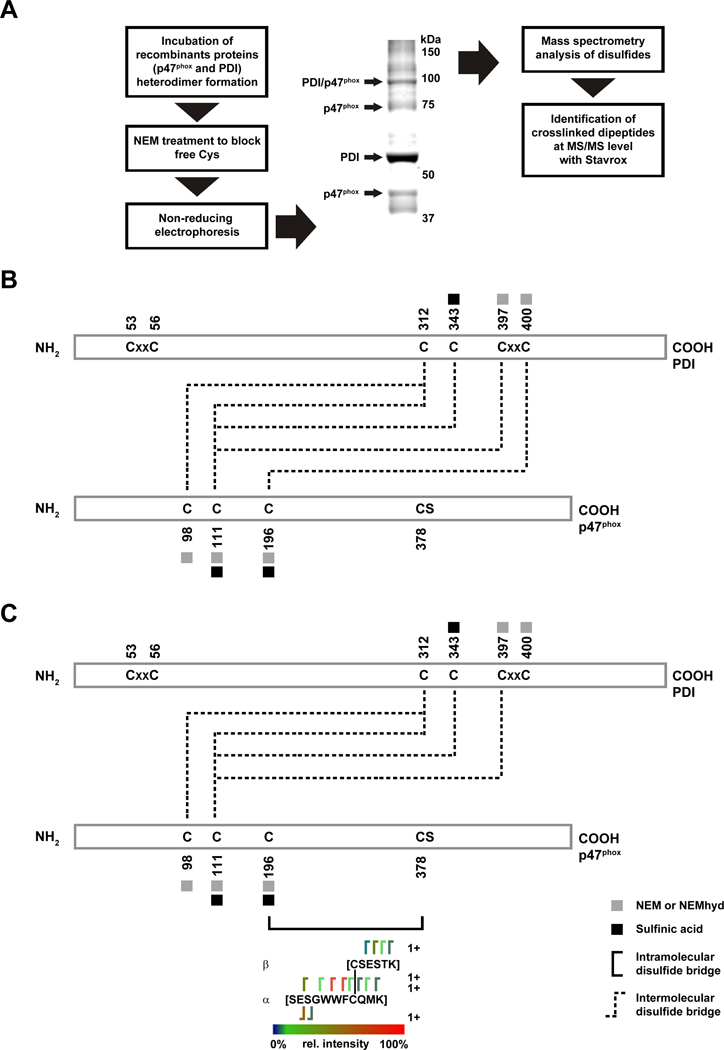
(A) Outline of analysis of samples to evaluate the PDI/p47phox complex. (B) Stravox identified the presence of disulfide bonds and indicated linkages between PDI C312 and C343 with p47phox C98 and C111; PDI C400 and p47phox C196; (C) PDI C397 with p47phox C111, and an intramolecular bond between p47phox C196 and C378.
Cys 196 p47phox is necessary for superoxide production and its recruitment to the membrane.
Based on these findings, we next examined whether C196 of p47phox is necessary for its interaction with PDI. Similar to the results in Fig 3C, expression of p47phox , p67phox and Nox1 in COS p22phox cells supported generation of superoxide to thrombin, and this was augmented by PDI (Fig 5A). However, expression of C196A p47phox did not support Nox1-dependent superoxide production in response to thrombin (Fig 5A). Interestingly, PDI overexpression in this system resulted in lower superoxide levels, similar to that observed with NoxO1 (Fig 3E). The expression of C196A p47phox in HEK-293 cells also attenuated superoxide production as compared to wt p47phox (Fig 3B). Although wt PDI did not augment Nox1 activation in this system, both wt and mut PDI interfered with superoxide production with C196A p47phox. These results indicate that Cys196 is necessary for normal function of p47phox and is involved in the interaction of p47phox and PDI. Superoxide levels were measured with lucigenin as an additional technique. The thrombin-stimulated superoxide production was attenuated upon mutation of the cysteine 196 in p47phox (Supp Figure VI). Nevertheless, with reconstitution of the Nox1 complex, we observed a significant increase in the signal (Figure 3C), which was almost totally abolished in VSMC from Nox1 KO mice and could be inhibited by SOD (Figure 2D), indicating that the majority of the signal detected was, in fact derived from Nox1 activity. Altogether, these results confirm our previous observations and indicate that p47C196 mutation decreases Nox1 activity.
Figure 5. PDI interaction with p47phox requires Cys196 for Nox1 activation.
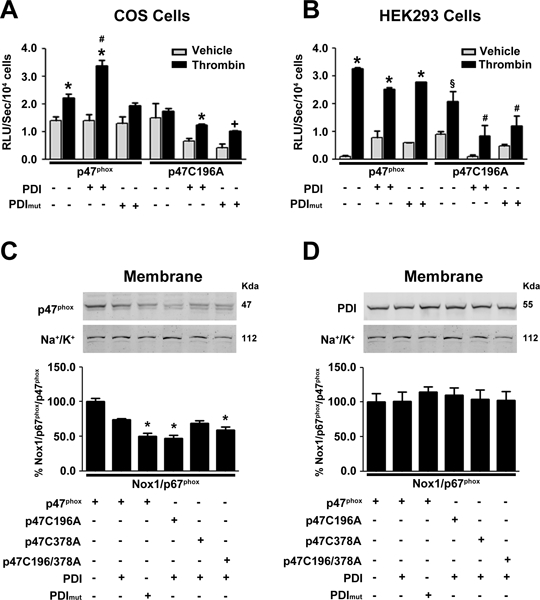
Superoxide levels in (A) COS p22 phox or (B) HEK-293 cells transfected as indicated and stimulated with thrombin, n=4. * p< 0.05 vs vehicle, # p< 0.05 vs Nox1, PDI mut stimulated with thrombin, Nox1/p47C196A, p47C196A/PDI stimulated with thrombin and p47C196A/PDI mut. § p<0.05 vs vehicle and Nox1 stimulated with thrombin, # p< 0.05 vs Nox1p47C196A. Western blot of p47phox (C) and PDI (D) in membrane fractions isolated from COS p22phox lysate transfected with PDI and p47phox or p47phox mutants after thrombin stimulation n=5. * p <0.05 vs p47phox.
Next, we evaluated the role of p47phox and PDI redox-sensitive cysteines on the recruitment of p47phox to the membrane. In reconstituted COS p22phox cells, thrombin causes the translocation of p47phox from the cytosol to the membrane (Fig 5C). Serine phosphorylation of p47phox is essential for p47phox recruitment to the membrane 23. The p47phox C196A interferes with its translocation to the membrane, whereas p47phox C378A is recruited to the membrane. Expression of PDI mut is also sufficient to attenuate recruitment of p47phox to the membrane (Fig 5C).
PDI is transported from the endoplasmic reticulum (ER) to the cell surface and its retention there involves interaction with other proteins, such as integrins 24. Recent studies indicate that PDI levels in the membrane are tightly regulated 25. We next evaluated the effect of redox-sensitive cysteines of p47phox and PDI on the membrane localization of PDI. COS p22phox cells were transfected with Nox1, p67phox , p47phox and PDI. The amount of PDI at the membrane was not altered by the expression of mutant p47phox or mutant PDI (Fig 5D). These data suggest that p47phox Cys196 is critical for its interaction with PDI and translocation to the membrane in the activation of Nox. However, additional PDI does not appear to be recruited to the membrane during this process.
Cys 196 p47phox is necessary for PDI and Nox1-mediated VSMC migration
We have previously shown that VSMC migration under several stimuli is mediated by Nox1 13, 20, 26 and that PDI regulates cellular migration via Nox1 activation 7. Therefore, we tested the requirement of p47phox C196 on VSMC migration. Migration of VSMC to thrombin was dependent on Nox1 (Fig 6A), while the overexpression of PDI had no effect. However, PDI mut attenuated migration. In rabbit VSMC, PDI increased migration in response to angiotensin II whereas PDI mut inhibited migration (Fig 6B). Additionally, p47phox expression increased migration and this effect was abolished in the presence of rutin, a PDI inhibitor. The mutation of p47phox C196, but not of C378, attenuated VSMC migration. These results implicate p47phox Cys 196 in PDI and Nox1-dependent migration of VSMC.
Figure 6. p47phox interacts with PDI in vivo.
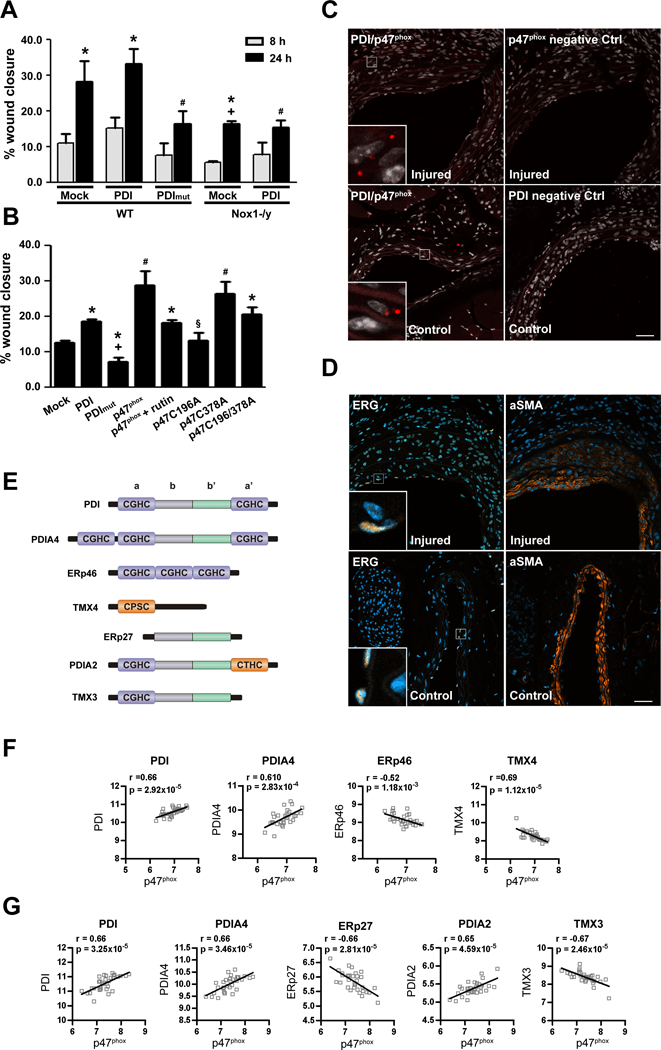
(A) VSMC migration to thrombin is dependent on Nox1 (n=3 * p<0.05 vs 8h, # p<0.05 vs PDI 24h in WT and + vs mock 24h). (B) Rabbit aortic VSMC were transfected as shown and 48 hours later stimulated with angiotensin II for 8 hours. In some experiments, rutin (100 μM), a PDI-inhibitor, was added prior to angiotensin II. (n=5, * p<0.05 vs p47phox wt, #p<0.05 vs mock, + p<0.05 vs PDI, § p<0.05 vs p47phox, p47phox C378A). (C) Interaction of PDI and p47phox after carotid artery wire-injury, postoperative day 14. Representative images of Duolink analyses of the interaction of PDI and p47phox and the respective controls as indicated. Positive signals demonstrating an interaction of the indicated proteins are shown as red dots. Co-staining with DAPI (white) to show the nucleus. Auto fluorescence shows the elastic lamina (red). The scale bars represent 40μm. n= 3. (D) Representative immunofluorescence images of carotid artery wire-injury, postoperative day 14. Staining against endothelial cell marker ERG (brown) and alpha-smooth muscle actin (α-SMA, brown) are shown as indicated. Co-staining with DAPI (blue) to show the nucleus. (E) PDI family of thiol isomerases structures. Pearson correlation analysis of p47phox and PDI family members using mRNA expression levels from 32 paired samples of macroscopically intact tissue (F) and atheroma plaque (G) was retrieved from Gene Expression Omnibus (GEO, GSE43292).
Evidence of PDI and p47phox interaction in vivo
We demonstrated the interaction of endogenous p47phox and PDI in cultured cells (Fig 3A), so we next extended these studies to assess the interaction of these proteins in intact arterial segments. Proximity ligation assay confirmed the interaction of p47phox with PDI in mice carotid arteries after wire-injury (Figure 6C). Furthermore, this interaction occurred predominantly in smooth muscle cells (Figure 6D).
We further evidence to support a relationship between PDI and p47phox by assessing mRNA levels in human vascular tissue17. There was a positive correlation between the expression levels of p47phox and of PDI and PDIA4 in human intact (Fig 6F) and atheroma (Fig 6G) tissue, but a negative correlation was obtained for p47phox and all other PDI family members. The analysis of the structure of these different PDI family members revealed that the positive correlation with p47phox only occurred between p47phox and PDI family members with the a’ redox active site (Fig 6E).
PDI levels were positively correlated to the NADPH oxidase members Nox2, p47phox, p22phox and p67phox and a negative association with Nox4 (Supp Fig VII). NoxO1 and NoxA1 had a much weaker association with PDI. Collectively, these results highly indicate a positive correlation of PDI and p47phox in human vascular tissue.
Discussion
We describe a redox dependent interaction between PDI and p47phox and identify p47phox cysteine 196 as necessary for the activation of Nox1 NADPH oxidase by PDI. The major novel findings of this study are: 1) PDI protein levels are increased in atherosclerotic aortas of nonhuman primates; 2) PDI silencing decreases redox signaling in VSMC; 3) PDI forms a disulfide bond with p47phox that is dependent on the redox status of each protein; 4) PDI catalytic cysteine 400 and p47phox cysteine 196 are essential for the activation of Nox1 by PDI; 5) PDI association with p47phox increases serine phosphorylation of p47phox and contributes to Nox1 activation and VSMC migration and 7) PDI and p47phox interaction is increased in vascular disease.
An increase in PDI levels in atherosclerosis is consistent with our previous findings of increased Nox1 expression observed in the same nonhuman primate model of atherosclerosis19. Nox1 has an important role in the migration and proliferation of VSMC13, 20, and Nox1 deficiency attenuates intimal size in murine models of atherosclerosis 27 and wire injury 28. In recent studies from our group, PDI has been implicated in Nox1-mediated VSMC migration and vascular remodeling, but to our knowledge, this is the first study to implicate the regulatory subunit p47phox in these PDI effects.
PDI interacts with p47phox in neutrophils to support Nox2 activity by acting as a redox-dependent enzyme complex organizer 18. Using recombinant proteins, we confirmed a redox-dependent interaction between PDI and p47phox. NADPH oxidase activation is sensitive to several thiol modifiers, including N-ethylmaleimide, phenylarsine oxide (PAO) and the fungal metabolite gliotoxin 21, 29. For example, photoaffinity labeling of PAO suggested its interaction with Nox2 30 and gliotoxin appears to inhibit p47phox phosphorylation by preventing its association with PKC on the membrane 29. We extend the observations that thiols affect NADPH oxidase activity 31 and identify PDI as an additional target by which these thiol modifiers may inhibit complex assembly and Nox activation.
The PDI-p47phox dimer was formed only between reduced PDI and oxidized p47phox. Additionally, the incubation of arachidonic acid was necessary for the formation of the dimer, suggesting that p47phox needs to be in an unfolded, open conformation for the p47phox cysteines to be exposed and interact with the redox cysteines of PDI. Since our previous data indicated that an oxidizing environment (3:1 GSH/GSSG ratio) increases the in vitro association between p47phox and PDI 18, it is likely that under oxidizing conditions the formation of a disulfide bond between the two proteins is favored. Therefore, in this situation PDI needs to be in a reduced state in order to attack the intramolecular disulfide bond within p47phox and form a disulfide bond.
Thiol/disulfide oxidoreductases interact with Nox2 regulatory subunits in leukocytes. For example, p40phox was identified as a thioredoxin-1 (TRX-1) binding protein 32 and we recently showed that this interaction modifies the redox state of TRX-1 to regulate NFκB activation in sepsis33. Similarly, we show here the regulation of Nox1 signaling by PDI through its redox association with p47phox. This interaction was abolished with mutation of PDI´s catalytic a and a´ domains, which are known to destabilize the a´ domain and indirectly affect occupancy of the b´ domain by its substrates 34. Both catalytic domains contain the CGHC motifs, which enable the protein to exert its disulfide reduction and oxidation activities. Nevertheless, PDI undergoes oxidation of the active site vicinal thiols within the a domain but the a´ domain remains in the reduced state 35 and this is further confirmed in other studies that showed differences between the behavior of the a and a´ domains 36. Therefore, we performed an LC-MS to determine the formation of a covalent and reversible adduct between PDI and p47phox. Our data revealed the participation of C400 at PDI´s active site a´, as critical for the binding to cysteine 196 of p47phox. In addition, C312 in the b´domain interacts with C196 in p47phox. C312 is not part of the redox active domain, but is also susceptible to redox regulation and thus its oxidation can affect the reducing potential of PDI active cysteines 35 which are essential for interaction with p47phox 18. Therefore, in addition to changes within the a domain active site, C312 within the b´ domain is also sensitive to redox regulation and potentially capable of modulating PDI activities. This could explain why the PDI/p47phox dimer was not found when we oxidized PDI. In fact, our data indicates that C312 could be one of the redox cysteines involved in PDI interaction with p47phox.
Cysteine residues are important for the function of many proteins, including catalytic groups in redox enzymes. Four cysteines residues are found in p47phox which are located at positions 98, 111, 196 and 378. PDI contains 6 cysteines located at positions 53, 56, 312, 343, 397 and 400. Our results indicated Cys 98, 111 and 196 as the site for PDI interaction with p47phox. Cys196 is the only cysteine located within the N–terminal SH3 domain and points to an important functional role in the oxidase activation. One of the roles of p47phox is to facilitate translocation of the cytoplasmic subunit p67phox to the membrane and correctly position it with respect to p22phox. Translocation and anchoring to the membrane is achieved through an interaction between the two SH3 domains of p47phox and a conventional proline rich motif (PxxP) in the cytoplasmic portion of p22phox. p47phox also associates with p67phox, which is essential for NADPH oxidase activation through its C-terminal consensus PxxP and the second SH3 domain of p67phox (p67SH3B). PDI binding to Cys 98,111 and 196 in p47phox could increase the affinity between the carboxi terminal of p47phox and the SH3 domain of p67phox thus favoring the non-canonical activation of Nox1. This is further confirmed by the fact that the mutation of C196 in p47phox decreased Nox1 activation by PDI in a Nox1 reconstituted system.
We identified a disulfide bridge between C196 and C378 in p47phox. Our data indicates that PDI and p47phox interact through PDI’s Cys 312 and 343 which are located in the substrate binding site of PDI with Cys 111 and 98 in p47phox which are located in the PX domain. The interaction of the PX domains of p47phox with phospholipids constitutes an additional mechanism to orchestrate the association of the regulatory subunits with the membrane 37. Our data also revealed an intramolecular bond between Cys 196 and 378 located in the second SH3 domain and in the auto inhibitory region of p47phox, respectively. Since this intramolecular disulfide bond was not present in p47phox alone, it is reasonable to suggest that PDI could first interact with p47phox via its PX domain, exposed during the phosphorylation of p47phox and then attack Cys196-Cys378 disulfide bond in p47phox allowing further phosphorylation of the protein. A similar mechanism was demonstrated by Araki et al 38. These authors suggested a collaborative redox coupling between PDI and Ero1, where PDI induces a conformational change in Ero1α whereby the redox cysteines at the a’ domain of PDI interact with Ero1α Cys 94 and 99, facilitating intramolecular electron transfer to the active site within Ero1α. The reduced shuttle dithiols move back to transfer electrons to the catalytic disulfide, which in turn transfers it to oxygen. In the present study we show the formation of a disulfide bond between oxidized p47phox and reduced PDI that increases p47phox translocation and Nox1 dependent superoxide production. We show that PDI interacts with Cys in p47phox PX and SH3 domains and that during this interaction a disulfide bridge between Cys 196 and 378 which is located in the C-terminal of p47phox is formed. Therefore, altogether our disulfide analysis data is indicative that PDI and p47phox redox interaction could create a regulatory loop in the control of Ser 379 phosphorylation, an essential event in p47phox activation 23. In fact, mutation of PDI redox cysteines from the a and a´ domains of p47phox of C196 or Cys 196 and 378, decreased p47phox translocation to the membrane and Nox1 dependent migration, an effect which was not observed when p47phox C378 alone was mutated. A similar mechanism could be applied to the intramolecular p47phox disulfide 196–378 which, when reduced by PDI, could enable the phosphorylation of p47phox and favor activation of Nox1.This is further confirmed by the fact that PMA increased the oxidation of p47phox ro-GFP in vascular smooth muscle cells overexpressing PDI.
Complex assembly is the primary mechanism for NADPH oxidase activation. The critical step in the activation of p47phox-dependent ROS production is the phosphorylation of p47phox. Activation of p47phox is mediated by PKC phosphorylation on multiple serine residues located in the autoinhibitory domain 39. Phosphorylation events are required to relieve auto-inhibition and allow translocation of p47phox to the membrane, where it binds to p22phox 2, 40. Little is known about the role of p47phox cysteines in the NADPH oxidase assembly. Cys196 appeared to negatively regulate oxidase activation in a cell-free system using cytosol from deficient B lymphocytes 41. The mutation of C196 to alanine increased superoxide production to more than twice that observed with wild type p47phox. On the other hand, the mutation of C378A did not produce this effect. The authors suggested that the –SH groups of these cysteines may be interacting in a mechanism dependent of oxidation to regulate enzyme assembly 41. In fact, Park showed that the phosphorylation of p47phox produced a substantial decrease in the labeling of Cys 378 by alkylating agents indicating a conformational change in p47phox C-terminal upon phosphorylation 42. Cys378 is located in the C-terminal region of p47phox vicinal to serine 379 which is phosphorylated and this promotes an H-bond in the C-terminal tail of p47phox disrupting the interaction between the AID and the SH3 domains, allowing p47phox interaction with p22phox, and oxidase activation in both leukocytes and in vascular cells 23, 43. PDI does have the potential to behave as a sensor of specific intramolecular redox switches or through intermolecular complex formation via redox and non-redox dependent mechanisms.
In this context, the existence of a disulfide bond between Cys 196 and 378 in the PDI/p47phox dimer raises the possibility that these cysteines may be interacting with each other and participating in a redox switch to control Ser379 phosphorylation, a process that is facilitated by interaction with PDI. In fact, the effects of PDI on p47phox translocation were more pronounced after thrombin stimulation, when p47phox is in its most phosphorylated form. Additionally, the transfection of a redox active PDI increased the PMA inducible oxidation of roGFP p47phox in vascular cells, suggesting that PDI interaction with p47phox facilitates p47phox phosphorylation and Nox1 activation in these cells. Consistent with this interpretation, we found that interaction of PDI and p47phox requires p47phox phosphorylation in neutrophils and precedes Nox2 activation 18.
Nox1 is constitutively activated by NoxO1, which lacks the autoinhibitory domain found in p47phox. Despite sharing similar domain arrangements, p47phox and NoxO1 are only 25% homologous. We are unaware of any data suggesting involvement of NoxO1 cysteines in the protein’s regulation. NoxO1 contains seven cysteines, among them three are located at the C-terminal region, and the position of Cys at the N-SH3 domain is different from that of Cys196 in the p47phox molecule.
Our finding of an interaction between PDI with p47phox suggests a cytosolic pool of PDI. In support of this conclusion, PDI has been shown to interact with the cytosolic proteins CuZnSOD 44, p47phox in neutrophils 18 and soluble guanylyl cyclase 45. Furthermore, there is considerable evidence that PDI regulates cytoskeletal organization, including direct interaction with actin and Rho-GTPases 7. The existence of a cytosolic PDI pool and the slow pattern of reaction of PDI with oxidants as compared to peroxiredoxins 46, 47 has led our group to propose that in the cytosol, PDI qualifies as a redox adaptor or organizer rather than a typical mass-effect sensor as previously reviewed 48, 49. Similarly, to what we described with PDI and p47phox, PDI promotes a redox switch that modulates the function of a polyadenylate-binding protein by changing its redox state or controlling its phosphorylation 50.
Signaling effects of PDI can be specific and independent of its overall importance in cell homeostasis. For example, cytosolic PDI is associated with specific signaling effects in vascular cells. We have observed that silencing of PDI in endothelial cells impairs ERK signaling by TNFα but not by other signaling pathways such as JNK, p38, NFκB or ICAM 8 . We have previously shown that PDI is overexpressed in the development of hypertension, and that PDI contributes to an increase in ERK signaling in response to angiotensin II in rat mesenteric resistance arteries 51. Tanaka et al., have recently shown that PDI is overexpressed during vascular repair 52. Our findings of a redox interaction between PDI and p47phox in injured arteries and a positive correlation between the expression of PDI and p47phox in human atheroma plaques extend these findings and propose PDI as an important redox activator of Nox1 complex in vascular disease.
In summary, we identify specific cysteine residues of PDI and p47phox necessary for the direct interaction of these proteins, the assembly of the Nox1 complex and subsequent activation of Nox1 NADPH oxidase in vascular disease. These observations suggest the novel role of PDI as a Nox1 complex organizer and potential therapeutic target in vascular disease.
Supplementary Material
{kind=link}
Highlights.
An intermolecular disulfide bond between PDI and p47phox increases the activation of Nox1 in VSMC.
PDI catalytic cysteine 400 and p47phox cysteine 196 are essential for the activation of Nox1 by PDI.
PDI facilitates p47phox mediated recruitment of Nox1 regulatory subunits to the plasma membrane.
PDI is an important redox regulator of Nox1 in vascular disease.
Acknowledgments
Sources of Funding
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo grant numbers 2012/12033–8, 2013/18300–0, 2013/07937–8, 2009/54764–6, 2013/03520–5. Conselho Nacional de Desenvolvimento Científico Tecnológico (CNPq) Grant 475609/2013–1. MG was the recipient of a FAPESP fellowship 2012/12033–8. LRL is a member of the CEPID Redoxoma FAPESP. FJM is supported by the Office of Research and Development, Department of Veterans Affairs [2I01BX001729] and the National Institutes of Health [HL130039]. BS is supported by the American Heart Association [PRE29980013]. RPB is supported by DFG SFB815 TPA01 and Z01.
Abbreviations:
- Cys or C
Cysteine
- DPI
diphenylene iodonium
- DTT
dithiothreitol
- ERK
extracellular signal-regulated kinase
- HEK
Human Embryonic Kidney
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NEM
N-Ethylmaleimide
- Nox1
NADPH Oxidase 1
- NoxA1
Nox activating subunit 1
- NoxO1
Nox organizing subunit 1
- PDGF
platelet derived growth factor
- PDI
Protein disulfide isomerase
- PDI mut
PDI mutated in redox cysteines C53, C56, C397, C400
- PMA
phorbol 12-myristate 13-acetate
- SOD
superoxide dismutase
- TNF
tumor necrosis factor
- VSMC
Vascular smooth muscle cells
- WT
wildtype
Footnotes
Disclosures
None.
References
- 1.Banfi B, Clark RA, Steger K and Krause KH. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. The Journal of biological chemistry. 2003;278:3510–3. [DOI] [PubMed] [Google Scholar]
- 2.el Benna J, Faust LP and Babior BM. The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. The Journal of biological chemistry. 1994;269:23431–6. [PubMed] [Google Scholar]
- 3.Bedard K and Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological reviews. 2007;87:245–313. [DOI] [PubMed] [Google Scholar]
- 4.Ellgaard L and Ruddock LW. The human protein disulphide isomerase family: substrate interactions and functional properties. EMBO Rep. 2005;6:28–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilkinson B and Gilbert HF. Protein disulfide isomerase. Biochim Biophys Acta. 2004;1699:35–44. [DOI] [PubMed] [Google Scholar]
- 6.Janiszewski M, Lopes LR, Carmo AO, Pedro MA, Brandes RP, Santos CX and Laurindo FR. Regulation of NAD(P)H oxidase by associated protein disulfide isomerase in vascular smooth muscle cells. The Journal of biological chemistry. 2005;280:40813–9. [DOI] [PubMed] [Google Scholar]
- 7.Pescatore LA, Bonatto D, Forti FL, Sadok A, Kovacic H and Laurindo FR. Protein disulfide isomerase is required for platelet-derived growth factor-induced vascular smooth muscle cell migration, Nox1 NADPH oxidase expression, and RhoGTPase activation. The Journal of biological chemistry. 2012;287:29290–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Camargo LdL, Babelova A, Mieth A, Weigert A, Mooz J, Rajalingam K, Heide H, Wittig I, Lopes LR and Brandes RP. Endo-PDI is required for TNF alpha-induced angiogenesis. Free Radical Biology and Medicine. 2013;65:1398–1407. [DOI] [PubMed] [Google Scholar]
- 9.Fernandes DC, Manoel AH, Wosniak J Jr, and Laurindo FR. Protein disulfide isomerase overexpression in vascular smooth muscle cells induces spontaneous preemptive NADPH oxidase activation and Nox1 mRNA expression: effects of nitrosothiol exposure. Archives of biochemistry and biophysics. 2009;484:197–204. [DOI] [PubMed] [Google Scholar]
- 10.Androwiki AC, Camargo Lde L, Sartoretto S, Couto GK, Ribeiro IM, Verissimo-Filho S, Rossoni LV and Lopes LR. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Front Chem. 2015;3:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner N, Junk S, Laufs U, Link A, Walenta K, Bohm M and Nickenig G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circulation research. 2003;93:e17–24. [DOI] [PubMed] [Google Scholar]
- 12.Hathaway CA, Heistad DD, Piegors DJ and Miller FJ Jr., Regression of atherosclerosis in monkeys reduces vascular superoxide levels. Circulation research. 2002;90:277–83. [DOI] [PubMed] [Google Scholar]
- 13.Zimmerman MC, Takapoo M, Jagadeesha DK, Stanic B, Banfi B, Bhalla RC and Miller FJ Jr., Activation of NADPH oxidase 1 increases intracellular calcium and migration of smooth muscle cells. Hypertension. 2011;58:446–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price MO, McPhail LC, Lambeth JD, Han CH, Knaus UG and Dinauer MC. Creation of a genetic system for analysis of the phagocyte respiratory burst: high-level reconstitution of the NADPH oxidase in a nonhematopoietic system. Blood. 2002;99:2653–61. [DOI] [PubMed] [Google Scholar]
- 15.Pal R, Basu Thakur P, Li S, Minard C and Rodney GG. Real-time imaging of NADPH oxidase activity in living cells using a novel fluorescent protein reporter. PLoS One. 2013;8:e63989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gotze M, Pettelkau J, Schaks S, Bosse K, Ihling CH, Krauth F, Fritzsche R, Kuhn U and Sinz A. StavroX--a software for analyzing crosslinked products in protein interaction studies. J Am Soc Mass Spectrom. 2012;23:76–87. [DOI] [PubMed] [Google Scholar]
- 17.Ayari H and Bricca G. Identification of two genes potentially associated in iron-heme homeostasis in human carotid plaque using microarray analysis. J Biosci. 2013;38:311–5. [DOI] [PubMed] [Google Scholar]
- 18.de APAM, Verissimo-Filho S, Guimaraes LL, Silva AC, Takiuti JT, Santos CX, Janiszewski M, Laurindo FR and Lopes LR. Protein disulfide isomerase redox-dependent association with p47(phox): evidence for an organizer role in leukocyte NADPH oxidase activation. Journal of leukocyte biology. 2011;90:799–810. [DOI] [PubMed] [Google Scholar]
- 19.Stanic B, Pandey D, Fulton DJ and Miller FJ Jr., Increased epidermal growth factor-like ligands are associated with elevated vascular nicotinamide adenine dinucleotide phosphate oxidase in a primate model of atherosclerosis. Arteriosclerosis, thrombosis, and vascular biology. 2012;32:2452–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jagadeesha DK, Takapoo M, Banfi B, Bhalla RC and Miller FJ Jr, Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc Res. 2012;93:406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Daiber A, August M, Baldus S, Wendt M, Oelze M, Sydow K, Kleschyov AL and Munzel T. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free radical biology & medicine. 2004;36:101–11. [DOI] [PubMed] [Google Scholar]
- 22.Zielonka J, Lambeth JD and Kalyanaraman B. On the use of L-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of NADPH oxidase: a reevaluation. Free radical biology & medicine. 2013;65:1310–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Faust LR, el Benna J, Babior BM and Chanock SJ. The phosphorylation targets of p47phox, a subunit of the respiratory burst oxidase. Functions of the individual target serines as evaluated by site-directed mutagenesis. J Clin Invest. 1995;96:1499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Swiatkowska M, Szymanski J, Padula G and Cierniewski CS. Interaction and functional association of protein disulfide isomerase with alphaVbeta3 integrin on endothelial cells. FEBS J. 2008;275:1813–23. [DOI] [PubMed] [Google Scholar]
- 25.Araujo TLS, Fernandes CG and Laurindo FRM. Golgi-independent routes support protein disulfide isomerase externalization in vascular smooth muscle cells. Redox biology. 2017;12:1004–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Streeter J, Schickling BM, Jiang S, Stanic B, Thiel WH, Gakhar L, Houtman JC and Miller FJ Jr., Phosphorylation of Nox1 regulates association with NoxA1 activation domain. Circulation research. 2014;115:911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B and Miller FJ Jr., Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis. 2011;216:321–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B and Griendling KK. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arteriosclerosis, thrombosis, and vascular biology. 2009;29:480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsunawaki S, Yoshida LS, Nishida S, Kobayashi T and Shimoyama T. Fungal metabolite gliotoxin inhibits assembly of the human respiratory burst NADPH oxidase. Infection and immunity. 2004;72:3373–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Doussiere J, Poinas A, Blais C and Vignais PV. Phenylarsine oxide as an inhibitor of the activation of the neutrophil NADPH oxidase--identification of the beta subunit of the flavocytochrome b component of the NADPH oxidase as a target site for phenylarsine oxide by photoaffinity labeling and photoinactivation. Eur J Biochem. 1998;251:649–58. [DOI] [PubMed] [Google Scholar]
- 31.Souza HP, Souza LC, Anastacio VM, Pereira AC, Junqueira ML, Krieger JE, da Luz PL, Augusto O and Laurindo FR. Vascular oxidant stress early after balloon injury: evidence for increased NAD(P)H oxidoreductase activity. Free radical biology & medicine. 2000;28:1232–42. [DOI] [PubMed] [Google Scholar]
- 32.Nishiyama A, Ohno T, Iwata S, Matsui M, Hirota K, Masutani H, Nakamura H and Yodoi J. Demonstration of the interaction of thioredoxin with p40phox, a phagocyte oxidase component, using a yeast two-hybrid system. Immunol Lett. 1999;68:155–9. [DOI] [PubMed] [Google Scholar]
- 33.Trevelin SC, Dos Santos CX, Ferreira RG, de Sa Lima L. et al. Apocynin and Nox2 regulate NF-kappaB by modifying thioredoxin-1 redox-state. Sci Rep. 2016;6:34581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hatahet F and Ruddock LW. Protein disulfide isomerase: a critical evaluation of its function in disulfide bond formation. Antioxid Redox Signal. 2009;11:2807–50. [DOI] [PubMed] [Google Scholar]
- 35.Kozarova A, Sliskovic I, Mutus B, Simon ES, Andrews PC and Vacratsis PO. Identification of redox sensitive thiols of protein disulfide isomerase using isotope coded affinity technology and mass spectrometry. J Am Soc Mass Spectrom. 2007;18:260–9. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez-Perilli L, Mastrogiovanni M, de Castro Fernandes D, Rubbo H, Laurindo F and Trostchansky A. Nitroarachidonic acid (NO2AA) inhibits protein disulfide isomerase (PDI) through reversible covalent adduct formation with critical cysteines. Biochim Biophys Acta. 2017;1861:1131–1139. [DOI] [PubMed] [Google Scholar]
- 37.Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC and Yaffe MB. The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol. 2001;3:675–8. [DOI] [PubMed] [Google Scholar]
- 38.Araki K, Iemura S, Kamiya Y, Ron D, Kato K, Natsume T and Nagata K. Ero1-alpha and PDIs constitute a hierarchical electron transfer network of endoplasmic reticulum oxidoreductases. J Cell Biol. 2013;202:861–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gimenez M, Schickling BM, Lopes LR and Miller FJ Jr., Nox1 in cardiovascular diseases: regulation and pathophysiology. Clin Sci (Lond). 2016;130:151–65. [DOI] [PubMed] [Google Scholar]
- 40.Ago T, Nunoi H, Ito T and Sumimoto H. Mechanism for phosphorylation-induced activation of the phagocyte NADPH oxidase protein p47(phox). Triple replacement of serines 303, 304, and 328 with aspartates disrupts the SH3 domain-mediated intramolecular interaction in p47(phox), thereby activating the oxidase. The Journal of biological chemistry. 1999;274:33644–53. [DOI] [PubMed] [Google Scholar]
- 41.Inanami O, Johnson JL and Babior BM. The leukocyte NADPH oxidase subunit p47PHOX: the role of the cysteine residues. Arch Biochem Biophys. 1998;350:36–40. [DOI] [PubMed] [Google Scholar]
- 42.Park JW and Babior BM. Activation of the leukocyte NADPH oxidase subunit p47phox by protein kinase C. A phosphorylation-dependent change in the conformation of the C-terminal end of p47phox. Biochemistry. 1997;36:7474–80. [DOI] [PubMed] [Google Scholar]
- 43.Meijles DN, Fan LM, Howlin BJ and Li JM. Molecular insights of p47phox phosphorylation dynamics in the regulation of NADPH oxidase activation and superoxide production. The Journal of biological chemistry. 2014;289:22759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS and Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. The Journal of biological chemistry. 2006;281:30152–65. [DOI] [PubMed] [Google Scholar]
- 45.Heckler EJ, Kholodovych V, Jain M, Liu T, Li H and Beuve A. Mapping Soluble Guanylyl Cyclase and Protein Disulfide Isomerase Regions of Interaction. PLoS One. 2015;10:e0143523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karala AR, Lappi AK, Saaranen MJ and Ruddock LW. Efficient peroxide-mediated oxidative refolding of a protein at physiological pH and implications for oxidative folding in the endoplasmic reticulum. Antioxidants & redox signaling. 2009;11:963–70. [DOI] [PubMed] [Google Scholar]
- 47.Cox AG, Peskin AV, Paton LN, Winterbourn CC and Hampton MB. Redox potential and peroxide reactivity of human peroxiredoxin 3. Biochemistry. 2009;48:6495–501. [DOI] [PubMed] [Google Scholar]
- 48.Laurindo FR, Pescatore LA and Fernandes Dde C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radic Biol Med. 2012;52:1954–69. [DOI] [PubMed] [Google Scholar]
- 49.Trevelin SC and Lopes LR. Protein disulfide isomerase and Nox: new partners in redox signaling. Curr Pharm Des. 2015;21:5951–63. [DOI] [PubMed] [Google Scholar]
- 50.Kim J and Mayfield SP. Protein disulfide isomerase as a regulator of chloroplast translational activation. Science. 1997;278:1954–7. [DOI] [PubMed] [Google Scholar]
- 51.Androwiki ACD, Camargo LdL, Sartoretto S, Couto GK, Ribeiro IMR, Verissimo-Filho S, Rossoni LV and Lopes LR. Protein disulfide isomerase expression increases in resistance arteries during hypertension development. Effects on Nox1 NADPH oxidase signaling. Frontiers in Chemistry. 2015;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanaka LY, Araujo HA, Hironaka GK, Araujo TL, Takimura CK, Rodriguez AI, Casagrande AS, Gutierrez PS, Lemos-Neto PA and Laurindo FR. Peri/Epicellular Protein Disulfide Isomerase Sustains Vascular Lumen Caliber Through an Anticonstrictive Remodeling Effect. Hypertension. 2016;67:613–22. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.