

Abstract
SRC3, a highly conserved member of the steroid receptor co-activator (SRC) family, is recruited by transcription factors to regulate cellular function. Previously, we demonstrated that SRC1, another highly conserved member of SRC family, interacts with RORγt to regulate of Th17 differentiation. However, the relationship between SRC1 and SRC3 in the regulation of Th17 cell function remains unknown. Here, we demonstrate that mouse SRC3 interacts with RORγt in Th17 cells but not in thymocytes. In addition, Src3−/− mice exhibited defective Th17 differentiation and induction of experimental autoimmune encephalomyelitis but normal thymocyte development. Furthermore, a lysine 313 to arginine mutation of RORγt (RORγt-K313R), which disrupts the interaction of RORγt with SRC3 but not with SRC1, impairs Th17 differentiation but not thymocyte development. These data suggest that SRC3 works with SRC1 to regulate RORγt-dependent Th17 differentiation but is not essential for RORγt-dependent thymocyte development.
Introduction
The transcription factor RORγt directs the differentiation of Th17 cells, which secrete IL-17 (1). Th17 cells participate in protective immunity but also mediate pathological immune responses involved in autoimmune conditions, such as multiple sclerosis, colitis, and autism. Thus, inhibiting Th17 cell formation and function may prevent the development and progression of these conditions (2–7). Because RORγt is required for the generation of pathogenic Th17 cells, it is an attractive drug target for controlling Th17-mediated immunological disorders (8, 9). However, mice deficient in RORγt have been found to exhibit severe defects in thymocyte development, including thymocyte apoptosis, abnormal cell cycle progression, and accumulation of immature CD8+ cells (10, 11). Thus, broadly targeting RORγt could lead to severe unintended side effects. To develop more targeted approaches to inhibit Th17 differentiation, it is important to understand the mechanisms regulating RORγt activity.
Transcription factors like RORγt, which belongs to the steroid nuclear receptor family (11), cannot regulate cellular function unless in the presence of co-factors. Co-factors do not usually have DNA-binding activity and thus depend on transcription factors to carry them to the chromatin to regulate gene expression. The highly conserved steroid receptor co-activator (SRC) family consists of three members, SRC1, SRC2, and SRC3, which are important co-factors for steroid nuclear receptor-mediated transactivation. The SRCs recruit acetyltransferases and methyltransferases that epigenetically modify histones to activate gene expression (12). Our previous study showed that RORγt recruits SRC1 to stimulate Th17 differentiation (13). However, mice deficient in SRC1 only show partially impaired Th17 differentiation (13). Furthermore, it was reported recently that SRC3 also regulates Th17 differentiation (14). The highly conserved nature of the SRC family led us to question the relationship between SRC1 and SRC3 in the function of Th17 cells.
In this study, we demonstrate that SRC3 is a co-factor for RORγt that is necessary for Th17 differentiation but not for thymic T cell development. We detected SRC3-RORγt complexes in Th17 cells but not in thymocytes. In addition, CD4+ T cells from Src3−/− mice exhibited defective Th17 differentiation and induction of passive experimental autoimmune encephalomyelitis (EAE) after adoptive transfer. In contrast, Src3−/− mice did not exhibit the defects in thymocyte development observed in RORγt-deficient mice. Furthermore, we identified a lysine to arginine mutation in RORγt (RORγt-K313R) that specifically disrupts the interaction between RORγt and SRC3 but not SRC1. Cells expressing RORγt-K313R exhibited impaired Th17 differentiation but normal thymocyte development. Therefore, whereas RORγt must interact with SRC3 to regulate Th17 differentiation, the SRC3-RORγt interaction is not essential for RORγt-regulated thymocyte development.
Materials & Methods
Mice
The Rorγt−/− (Rorc2−/−) and Src3−/− mouse strains, described previously (10, 15), were bred and housed under specific pathogen-free conditions in the Animal Resource Center at the Beckman Research Institute of City of Hope under protocols approved by the Institutional Animal Care and Use Committee. Mice were 10–12 weeks of age for EAE and 6–8 weeks for all other experiments, with littermates age-matched across experimental groups.
Antibodies, cytokines and plasmids
Antibodies against RORγt (Q31–378, BD Bioscience), SRC1 (128E7, Cell Signaling), SRC3 (ab2831, Abcam), and FLAG (M2, Sigma-Aldrich) were used for immunoblot analysis. PE-indotricarbocyanine (Cy7)-conjugated anti-CD8 (53–6.7), PE-conjugated anti-RORγt (B2D), allophycocyanin (APC)-conjugated anti-IL-17A (eBio17B7), PE-conjugated anti-Thy1.2 (53–2.1), PE-conjugated anti-CD24 (M1/69), PE-conjugated anti-TCRβ (H57–597), PE-Cy5-conjugated anti-CD19 (eBio1D3), PE-conjugated anti-CD11b (M1/70), FITC-conjugated anti-CD4 (GK1.5), APC-conjugated anti-IL-4 (11B11), and APC-conjugated anti-Foxp3 (FJK-16s) were from eBioscience. Monoclonal antibodies against mouse CD3 (145–2C11), CD28 (37.51), IL-4 (11B11), IFNγ (XMG1.2), and the p40 subunit of IL-12 and IL23 (C17.8), as well as PE-Cy7-conjugated anti-Ly6G (1A8), FITC-conjugated anti-IFNγ (XMG1.2), PE-conjugated anti-GM-CSF (MP1–22E9), FITC-Cy7-conjugated anti-CD45 (104), and PE-conjugated anti-CD25 (PC61.5) were purchased from Biolegend. Goat anti-hamster antibody was from MP Biomedicals. APC-conjugated anti-CD3 (145–2C11) and FITC-conjugated anti-CD44 (IM7) were from BD Pharmingen. Recombinant mouse IL-12, IL-4, IL-6, IL-23, and TGFβ were from Miltenyi Biotech. An empty retroviral expression plasmid murine stem cell virus (MSCV)-IRES-GFP and those encodes RORγt and SRC1 have been described previously (13). Mouse SRC3 was amplified and inserted into pMSCV-IRES-GFP. Point mutations of RORγt were generated using a site-directed mutagenesis kit from Agilent Technologies.
Retrovirus Transduction
Platinum-E retroviral packaging cells (Cell Biolabs) were plated in a 10-cm dish in 10 ml RPMI-1640 medium plus 10% FBS. 24 h later, cells were transfected with an empty pMSCV vector or the appropriate retroviral expression plasmids with BioT transfection reagent (Bioland). After overnight incubation, the medium was replaced and cultures were maintained for another 24 h. Viral supernatants were collected 48 h and 72 h later, passed through 0.4-μm filters (Millipore), and supplemented with 8 μg/ml of polybrene (Sigma-Aldrich) and 100 U/ml of recombinant IL-2 (for transducing CD4+ T cells) or 5 ng/ml of recombinant IL-7 (for transducing CD4-CD8- thymocytes). Naïve CD4+ T cells were first activated with 0.25 μg/ml hamster anti-CD3 (145–2C11; Biolegend) and 1 μg/ml hamster anti-CD28 (37.51; Biolegend) in 24-well plates pre-coated with 0.2 mg/ml goat anti-hamster antibody for 24 h, then spin-infected with viral supernatants (1200 g, 30°C for 2 h). The retroviral supernatant was also used to infect CD4-CD8- thymocytes that had been co-cultured with feeder OP9-DL4 cells (a generous gift from Dr. Ellen Rothenberg, Caltech) in the presence of recombinant IL-7 (5 ng/ml) for 24 h. After spin infection, viral supernatant was replaced with culture media containing polarizing cytokines for in vitro differentiation (for transduced CD+ T cells) or 5 ng/ml of recombinant IL-7 for in vitro T cell development (for transduced CD4-CD8- thymocytes), as described below.
In vitro differentiation
Naïve CD4+ T cells were purified from C57BL/6, Rorγt−/−, Src3+/−, or Src3−/− mice by negative selection (Miltenyi Biotec). Suspensions of 4×105 cells/ml Iscove’s modified DMEM (Cellgro) containing 2 mM L-glutamine, 50 mM 2-ME, 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% FBS were cultured in 24-well plates pre-coated with 0.2 mg/ml goat anti-hamster antibody for three days. The medium was supplemented with 0.25 μg/ml hamster anti-CD3, 1 μg/ml hamster anti-CD28, and polarizing cytokines: 2 ng/ml TGFβ, 20 ng/ml IL-6, 5 μg/ml anti-IL-4, and 5 μg/ml anti-IFNγ for Th17 differentiation; 20 μg/ml IL-12 and 5 μg/ml anti-IL-4 for Th1 differentiation; 10 ng/ml IL-4 and 10 μg/ml anti- IFNγ for Th2 differentiation; or 5 ng/ml TGFβ for Treg differentiation. For analysis, cells obtained from in vitro cultures were incubated for 4–5 h with 50 ng/ml PMA (Sigma-Aldrich), 750 ng/ml ionomycin (Sigma-Aldrich), and 10 μg/ml brefeldin A (BD Biosciences) in an incubator at 37°C, followed by intracellular cytokine staining.
In vitro T cell development
Thymocytes were stained with 7-AAD and antibodies against Thy1.2, CD4, and CD8. Specific 7-AAD-Thy1.2+CD4-CD8- populations were sorted using a FACSAria (BD Biosciences) and cultured at 5×105/ml overnight on an 80% confluent OP9-DL4 monolayer in a flat-bottom 24-well culture plates with αMEM (MEM α medium; Invitrogen Life Technologies) supplemented with 20% FBS, 100 U/ml penicillin-streptomycin, 2 mM L-glutamine (Invitrogen Life Technologies), and 5 ng/ml recombinant murine IL-7. After 72 h, co-cultures were harvested for flow cytometry analysis.
Flow cytometry
Mouse thymi were homogenized by crushing with the head of a 1-ml syringe in a petri dish, followed by straining through a 40-μm nylon filter. Red Blood Cell Lysing buffer (Sigma-Aldrich) was used for red cell lysis. Cells isolated from thymi, co-cultures harvested from in vitro development, and CD4+ T cells stimulated appropriately were stained for surface markers. Intracellular cytokines was stained with Fixation/Permeabilization solution (BD Cytofix/Cytoperm Kit; BD Biosciences). The expression of surface and intracellular markers was analyzed BD FACSCanto flow cytometry system (BD).
RNA sequencing and analysis
To assess the gene expression profile of Th17 cells, naive CD4+ T cells were polarized under Th17 conditions for three days. RNA was isolated using an miRNeasy Mini Kit (Qiagen). Quality verification, library preparation, and sequencing were performed in the City of Hope Integrative Genomics Core facility. Eluted RNAs were prepared for sequencing using Illumina protocols and sequenced on an Illumina HiSeq 2500 to generate 51-bp reads. Sequenced reads were aligned to the mouse mm10 reference genome using TopHat. Gene expression levels were quantified by HTSeq, and edgeR was utilized to identify differentially expressed genes (fold-change > 1.5 and FDR < 0.05). Gene expression abundance was quantified as fragments per kilobase of transcript per million fragments mapped (FPKM). Heat maps of differentially expressed genes were made using gplots with log2-transformed FPKM values.
Chromatin immunoprecipitation
2×107 cells were incubated with 1% formaldehyde for 5 min at room temperature to cross-link proteins with chromatin. 125 mM glycine was added to stop the cross-linking reaction. To shear genomic DNA into 200–500-bp fragments, cell lysates were sonicated using a water-bath sonicator (Covaris S200). Cell lysates were centrifuged (12000 g, 10 min) and incubated with specific antibodies or IgG controls and protein A/G beads (Millipore). After extensive washing, DNA was eluted, followed by reversion of the protein–DNA cross-linking. DNA was recovered for qRT-PCR to quantify specific DNA fragments that were precipitated.
qRT-PCR
qRT-PCR was performed using SsoFast EvaGreen Supermix (Bio-Rad) in a CFX96 Real-Time PCR Detection System (Bio-Rad) using primers as following: Il17a-F: TTTAACTCCCTTGGCGCAAAA, Il17a -R:CTTTCCCTCCGCATTGACAC; Il17f -F: TGCTACTGTTGATGTTGGGAC, Il17f -R: AATGCCCTGGTTTTGGTTGAA; Ccr6-F: CCTGGGCAACATTATGGTGGT, Ccr6-R: CAGAACGGTAGGGTGAGGACA; Ccl20-F: GCCTCTCGTACATACAGACGC, Ccl20-R: CCAGTTCTGCTTTGGATCAGC; Irf4-F: TCCGACAGTGGTTGATCGAC, Irf4-R: CCTCACGATTGTAGTCCTGCTT; Rora-F: GTGGAGACAAATCGTCAGGAAT, Rora-R: TGGTCCGATCAATCAAACAGTTC; Ahr-F: AGCCGGTGCAGAAAACAGTAA, Ahr-R: AGGCGGTCTAACTCTGTGTTC. Conditions were adjusted to optimize primer amplification efficiency for all qRT-PCR reactions. Expression was calculated using the ΔΔct method and normalized to β-actin. All measurements were performed in triplicate.
Apoptosis assays
Thymocytes were freshly isolated and cultured in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin-streptomycin, and 2 mM L-glutamine at 1×106 cells/ml. Thymocytes were incubated at 37°C with 5% CO2. Dead cells were detected using Annexin V-PE and 7-AAD staining (BD Bioscience).
Induction and assessment of experimental autoimmune encephalomyelitis (EAE)
For Th17-induced passive EAE, donor mice were subcutaneously immunized with a 200-μg myelin oligodendrocyte glycoprotein 35–55 (MOG35–55) peptide emulsion (Hooke Laboratories, Lawrence, MA). 10 days later, cells were isolated from the spleen and lymph nodes and cultured with 20 μg/ml MOG35–55 for 3 days under Th17-polarizing conditions (20 ng/ml recombinant mouse IL23). Src3+/− recipient mice were then intraperitoneally transferred 3.0×107 MOG35–55-specific Th17 cells. The severity of EAE was monitored and evaluated on a scale from 0 to 5, according to guidelines from the Hooke Laboratories: 0 = no disease; 1 = paralyzed tail; 2= hind limb weakness; 3= hind limb paralysis; 4 = hind and fore limb paralysis; 5= moribund and death. When a mouse was euthanized because of severe paralysis, a score of 5 was entered for that mouse for the rest of the experiment.
Immunoprecipitation and immunoblot analysis
Cells were lysed in lysis buffer (1% Triton X-100, 20 mM Tris-cl, pH 7.4, 150 mM NaCl, and 5 mM EDTA) supplemented with protease inhibitor cocktail (Sigma) and 1 mM PMSF. Cell extracts were incubated overnight with 1 μg of the relevant antibodies, and proteins were immunoprecipitated for an additional 1 h at 4°C with protein A/G-Sepharose beads (Millipore). After incubation, beads were washed four times with lysis buffer, resolved using SDS-PAGE, and analyzed using Western blot.
Statistical analysis
GraphPad Prism software was used for all statistical analyses. Two-tailed, unpaired Student’s t tests and one-way analysis of variance (ANOVA) were used to compare experimental groups. A P value of less than 0.05 was considered statistically significant.
Results
SRC3 interacts with RORγt in Th17 cells but not in thymocytes.
Mass spectrometric analysis of RORγt-binding proteins in Th17 cells detected both SRC1 and SRC3 (data not shown), which was confirmed by immunoprecipitation assay using HEK293T cells expressing RORγt and SRC1 or SRC3 (Fig. 1A). This was consistent with our previous finding that RORγt recruits SRC1 to regulate Th17 differentiation (13). We next monitored the interactions between RORγt and SRC3 in both Th17 cells and thymocytes, in which RORγt is known to play important roles (11). Whereas SRC1 was found to interact with RORγt in both mouse Th17 cells and thymocytes (Fig. 1B), the SRC3-RORγt interaction was only detected in Th17 cells (Fig. 1C). RORγt binds to Il17a and Il17f loci to stimulate their expression (11, 16). To determine whether RORγt recruits SRC3 to Il17a and Il17f loci, we performed a ChIP assay. Consistent with previous results (11), RORγt binding to Il17a and Il17f loci was detected in wildtype (WT) but not RORγt−/− cells differentiated under Th17 polarization conditions (Fig. 1D and1E). SRC1 is known to bind to the Il17 gene (17) and thus was used as a positive control. Indeed, both SRC1 and SRC3 were found to bind to Il17a and Il17f loci in WT Th17 cells, but binding to both loci was significantly reduced in RORγt−/− cells, suggesting that RORγt recruits both SRC1 and SRC3 to the Il17 gene. These results demonstrate that SRC3 interacts with RORγt in Th17 cells but not in thymocytes.
Figure 1.
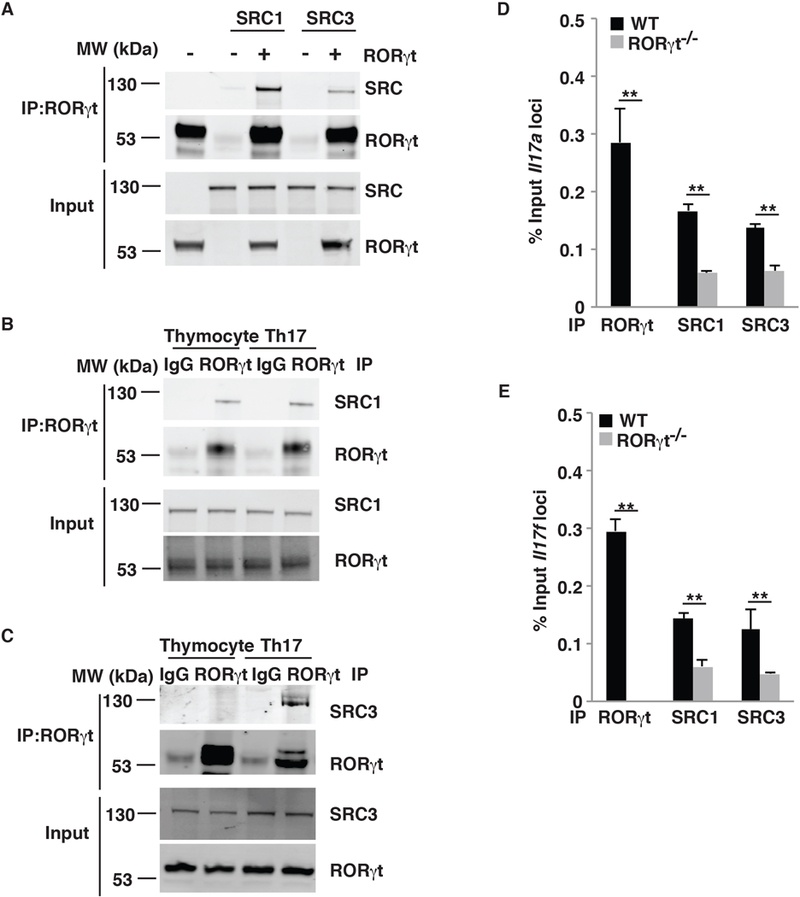
SRC3 interacts with RORγt in Th17 cells but in not thymocytes. (A) Immunoblot analysis of SRC1 and SRC3 immunoprecipitated with an anti-RORγt antibody from HEK293T cells co-transfected with plasmids to express RORγt, SRC1, and/or SRC3. The bottom images show immunoblot analysis of whole-cell lysates without immunoprecipitation (input). (B) Immunoblot analysis of SRC1 immunoprecipitated with a control IgG or anti-RORγt antibody from mouse thymocytes and differentiated Th17 cells. (C) Immunoblot analysis of SRC3 immunoprecipitated with a control IgG or anti-RORγt antibody from mouse thymocytes or differentiated Th17 cells. (D) ChIP analysis of RORγt, SRC1, and SRC3 bound to the Il17a locus in CD4+ T cells from WT or RORγt−/− mice polarized under Th17 conditions. (E) ChIP analysis of RORγt, SRC1, and SRC3 bound to the Il17f locus in CD4+ T cells WT or RORγt−/− mice polarized under Th17 conditions. Data are from one representative of three independent experiments (A–C) or are aggregated from three experiments (D–E, mean ± SEM). ** P <0.01 (t-test).
SRC3 regulates Th17 differentiation.
Because SRC3 interacts with RORγt in Th17 cells, we determined whether it plays a role in Th17 differentiation using Src3−/− mice. There were no differences in Th17 differentiation of CD4+ T cells obtained from WT and Src3+/− mice (Supplementary Fig. 1A), therefore we used Src3+/− mice as WT controls in this study. We found that Th17 but not Th1 differentiation of CD4+ T cells from Src3−/− mice was greatly reduced compared to that of CD4+ T cells from Src3+/− mice (Fig. 2A). However, the RORγt expression in Src3−/− T cells was similar to that in Src3+/− Th17 cells (Fig. 2B), suggesting that reduced Th17 differentiation is not due to changes in RORγt expression but likely due to reduced RORγt activity in the absence of SRC3. Furthermore, expression of critical Th17 genes, including Il17a, Il17f, Ccr6, and Ccl20, but not Il23r, Ahr, and Irf4, were reduced in Src3−/− T cells (Fig. 2C and Supplementary Fig. 1B), suggesting severe impairment of the Th17 differentiation program. We next performed a ChIP assay to detect the binding of SRC3 to different gene loci; SRC1 was used as a positive control, as it is known to bind Il17a and Il17f loci (17). We found that SRC3 binding to the Il23r locus is significantly lower than its binding to Il17a and Il17f loci (Fig. 2D). This difference in binding correlated with the differences in expression Il17a, Il17f, and Il23r (Fig. 2C), indicating the contribution of SRC3 to the expression of the IL-17 gene.
Figure 2.
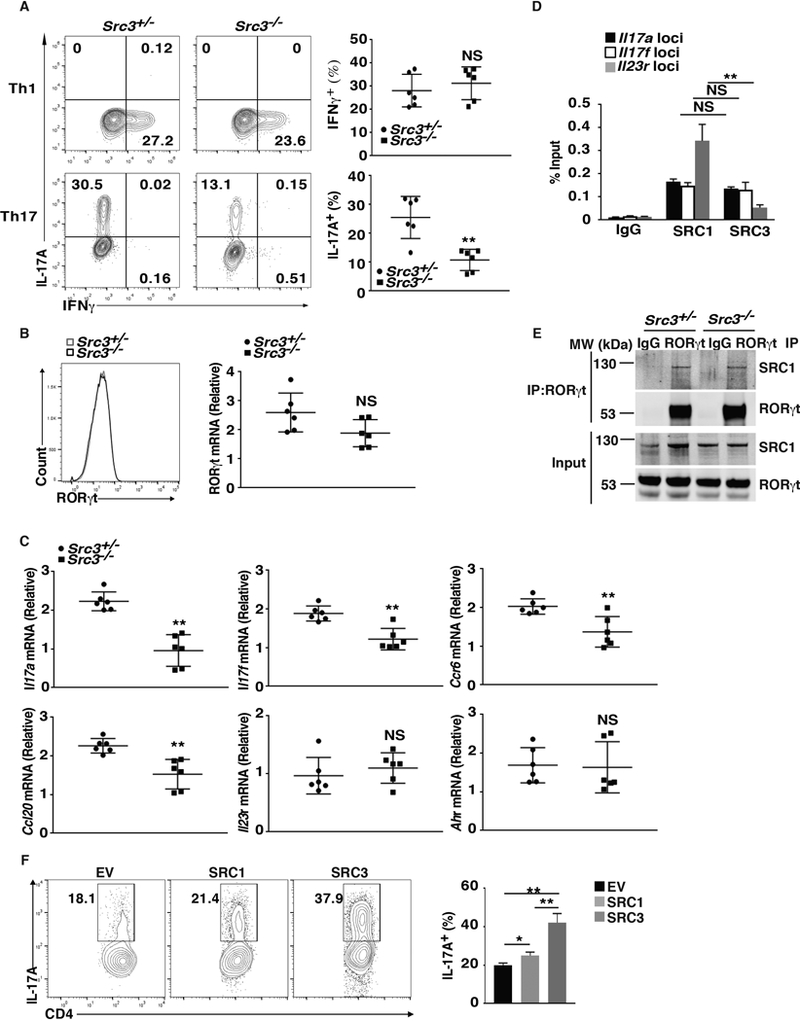
SRC3 regulates Th17 differentiation. (A) Flow cytometric analysis of intracellular IFNγ (top panels) and IL-17A (bottom panels) in naïve CD4+ T cells from Src3+/− and Src3−/− mice, cultured for three days under Th1 or Th17 priming conditions in vitro (n=6 per genotype). Numbers in each plot indicate the percentage of the cells in the gated area. Right panels present the percentages of IFNγ+ and IL-17A+ cells from individual mice. (B) Flow cytometric analysis of RORγt levels in naïve CD4+ T cells from Src3+/− and Src3−/− mice, cultured for three days under Th17 priming conditions in vitro. The plot on right shows the quantification of RORγt mRNA in Th17 cells differentiated from the CD4+ T cells of individual Src3+/− and Src3−/− mice (n=6 per genotype). (C) qPCR analysis of Il17a, Il17f, Ccr6, Ccl20, Il23r, and Ahr mRNA in Th17 cells differentiated from the CD4+ T cells of individual Src3+/− and Src3−/− mice (n=6 per genotype). (D) ChIP analysis of SRC1 and SRC3 binding to Il17a, Il17f, and Il23r loci in CD4+ T cells polarized under Th17 conditions. IgG serves as a negative control. (E) Immunoblot analysis of SRC1 immunoprecipitated with a control IgG or anti-RORγt antibody from differentiated Src3+/− and Src3−/− Th17 cells. (F) Flow cytometric analysis of IL-17A+ cells (outlined) among CD4+ T cells from Src3−/− mice transduced with a retrovirus expressing GFP alone (EV) or GFP with SRC1 or SRC3 and polarized for three days under Th17 priming conditions. The plot on the right shows the percentages of IL-17A+ cells in each group. Data are aggregated from three experiments (A and B, right panels; C; D; F; presented as mean ± SEM) or are from one representative of three independent experiments (E). NS, not significant (P>0.05); * P < 0.05 (t-test); ** P <0.01 (t-test).
Because SRC1 and SRC3 are highly conserved and both regulate Th17 differentiation, we examined whether SRC1 can compensate for deficient SRC3 function in Th17 differentiation. First, we found that the SRC1-RORγt interaction in Th17 cells was not affected by the absence of SRC3 (Fig. 2E). Furthermore, SRC3 but not SRC1 could rescue Th17 differentiation in Src3−/− T cells (Fig.2F), suggesting that the function of SRC3 does not overlap with that of SRC1 in the regulation of Th17 differentiation.
T cells from Src3−/− mice are defective in the induction of EAE.
To determine whether SRC3 plays a role in pathogenic Th17 immunity, we examined the effects of SRC3 deficiency on Th17 differentiation under pathogenic conditions. Under normal conditions (i.e., in the presence of TGFβ + IL-6), the Th17 differentiation of Src3−/− T cells was impaired compared to that of Src3+/− T cells. This difference was even more severe for Th17 differentiation under pathogenic conditions (IL-6 + IL-1 + IL-23) (Fig. 3A). Consistent with this observation, we also observed that the downregulation of critical Th17 genes, including Il7a, Il7f, Il22, Il1r1, Rora, and Il23r, but not Rorc, in Src3−/− T cells was greater under pathogenic conditions than under normal conditions (Supplementary Fig. 1C). Despite the significant effects of SRC3 deficiency, overexpression of SRC3 did not significantly affect Il1r1 expression (Supplementary Fig. 1D). We next tested the function of Src3−/− T cells in the induction of pathogenic EAE after adoptive transfer. Indeed, CD4+ T cells from Src3−/− mice polarized under Th17 conditions induced much less severe EAE compared to control CD4+ T cells from Src+/− mice (Fig. 3B). The reduced induction of EAE correlated with reduced central nervous system (CNS) infiltration by different kinds of lymphocytes, including Ly6G+ neutrophils, CD4+ and CD8+ T cells, CD19+ B cells, and CD11b+ monocytes, an indication of less inflammation (Fig. 3C and3D). In addition, we observed reduced expression of critical Th17 genes, IL17a, Il17f, Csf2, Il22, Cxcr3, and Ccl20, in CNS-infiltrating lymphocytes recovered from mice adoptively transferred with Src3−/− CD4+ T cells (Fig. 3E). These results demonstrate that SRC3 is required for Th17-mediated EAE.
Figure 3.
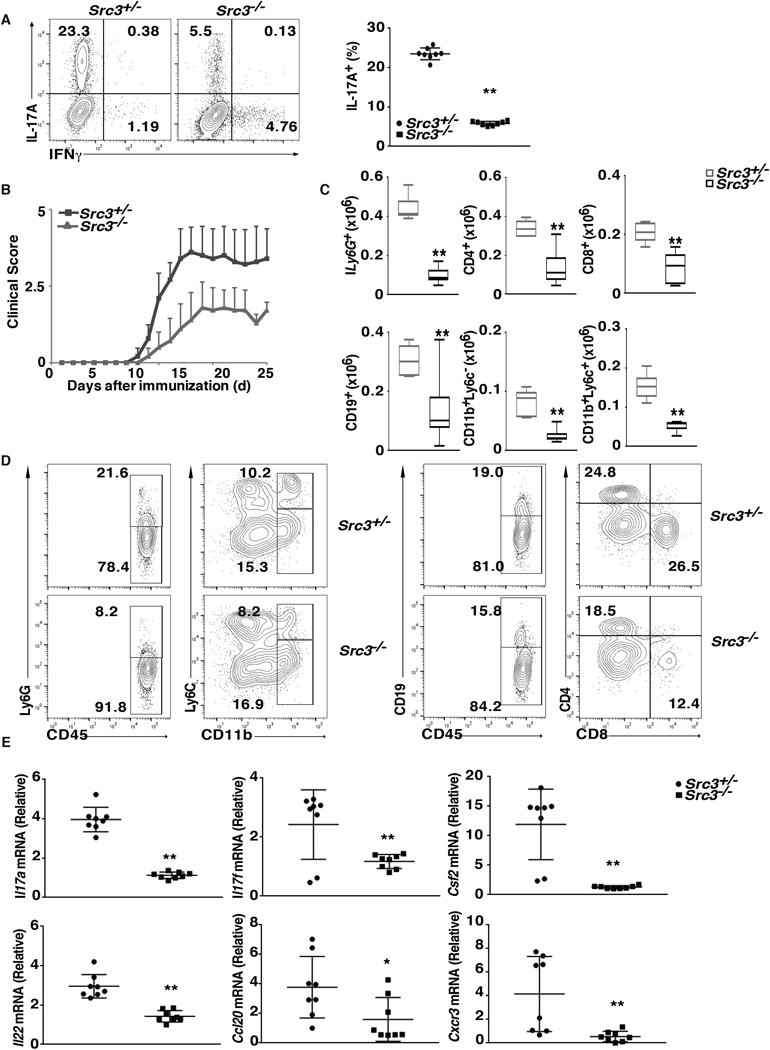
T cells from Src3−/− mice exhibit defective induction of EAE. (A) Flow cytometric analysis of intracellular IL-17A in naïve CD4+ T cells from Src3+/− and Src3−/− mice, cultured for three days under pathogenic Th17 priming conditions (TGFβ + IL-6 + IL-1 + IL-23) in vitro. Numbers in each plot indicate the percentage of the cells in the gated area. Right panels present the percentages of IL-17A+ cells from individual mice (n=7 per genotype). (B) Mean clinical EAE score of Src3+/− mice adoptively transferred with CD4+ T cells from MOG35–55-primed Src3+/− or Src3−/− mice (n = 5 per genotype) and further expanded in vitro for three days in the presence of MOG35–55 and IL-23 (20 ng/ml). (C) Quantification of cells expressing characteristic mononuclear cell surface markers among CNS-infiltrating cells in host mice adoptively transferred with Src3+/− or Src3−/− CD4+ T cells, assessed by flow cytometry at the peak of disease. (D) Gating strategy for lymphocytes shown in C. (E) qPCR analysis of Il17a, Il17f, Csf2, Il22, Cxcr3, and Ccl20 mRNA in lymphocytes that infiltrated the CNS of mice (as in B). Expression is presented relative to that of the control gene Actb. Data are from three experiments (A, right panel, and E, presented as mean ± SEM; C, presented as median [central line], the first and third quartiles [box ends], and maximum and minimum [extended lines]). * P < 0.05 (t-test); ** P <0.01 (t-test).
SRC3 is not required for thymocyte development.
In addition to Th17 differentiation, RORγt regulates thymocyte development. RORγt−/− mice had greater numbers of immature CD8+ cells and larger thymocytes that exhibited accelerated apoptosis and abnormal S phase entry (10, 18). Since SRC3 is required for RORγt-regulated Th17 differentiation, we examined whether SRC3 also plays a role in thymocyte development. There were no differences between Src3−/− mice and control Src3+/− mice in the expression of RORγt (Fig. 4A) or the size of thymocytes (Fig. 4B). Furthermore, flow cytometric analysis of surface CD4 and CD8 markers indicated that the percentages (Fig. 4C) and numbers (Fig. 4D) of CD4-CD8-, CD4+CD8+, and CD4+ and CD8+ cells, three sequential developmental stages of thymocytes, were also normal. We also did not observe increased percentages (Fig. 4E) and numbers (Fig. 4F) of immature CD8+ (i.e., CD4+CD24hiTCRβlo) cells in Src3−/− mice (Fig. 4E); differences in the number of cells with more than 2N DNA (Fig. 4F); or reduced thymocyte survival (Fig. 4G) in the absence of SRC3. RORγt is thought to control cell cycle progression and the survival of thymocytes by stimulating the expression of Bcl-xL (Bcl2l1), as overexpression of Bcl-xL prevents abnormal cell cycle progression and apoptosis in RORγt−/− thymocytes (10). Consistent with our findings that suggest that SRC3 does not play a role in RORγt-regulated cell cycle and survival during thymocyte development, we did not detect changes in the expression of Bcl2l1 in Src3−/− thymocytes (Fig. 4H). Lastly, we observed no differences in the differentiation of CD4-CD8- thymocytes from Src3−/− and Src+/− mice in vitro on stromal cells (Fig. 4I). Taken together, these data suggest that SRC3 is not required for RORγt-dependent thymocyte development.
Figure 4.
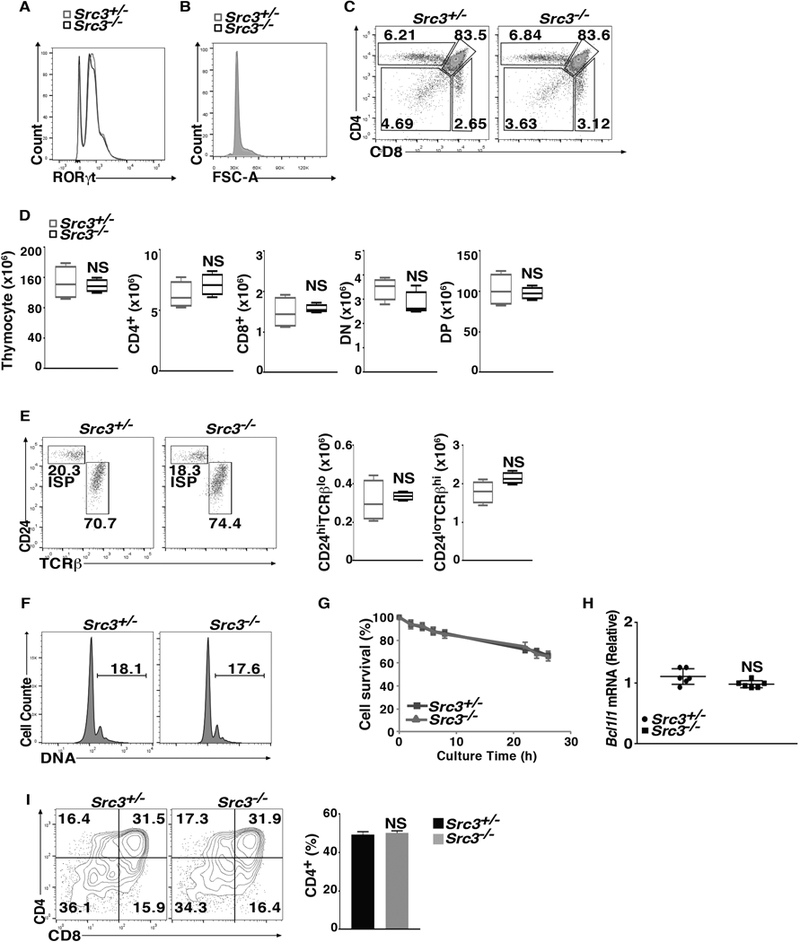
SRC3 is not required for thymocyte development. (A) Flow cytometric analysis of RORγt expression among thymocytes in Src3+/− and Src3−/− mice. (B) Flow cytometric analysis of forward scatter (FSC-A) among thymocytes in Src3+/− and Src3−/− mice. (C) Flow cytometric analysis of CD4 and CD8 on the surface of Src3+/− and Src3−/− thymocytes. (D) Quantification of total thymocytes and CD4+, CD8+, CD4-CD8- double negative (DN), and CD4+CD8+ double positive (DP) thymocytes of Src3+/− and Src3−/− mice (n = 5 per genotype). (E) Flow cytometric analysis of CD24 and TCRβ expression in CD8+ cells shown in C to assess the maturity of CD8+ thymocytes (two panels on the left) and quantification of immature TCRloCD24hi and mature TCRhiCD24lo cells (two panels on the right) (n = 5 per genotype). (F) Flow cytometric analysis of DNA content (as 7-AAD staining) of Src3+/− and Src3−/− thymocytes. The numbers in each plot indicate the percentage of cells with >2N DNA. (G) Percentage of surviving cells among thymocytes obtained from Src3+/− and Src3−/− mice (n = 5 per genotype) cultured for 0–15 h, then stained for the apoptosis marker annexin V and the membrane-impermeable DNA-interacting dye 7-AAD, analyzed by flow cytometry. (H) qPCR analysis of Bcl2l1 mRNA in thymocytes from Src3+/− and Src3−/− mice (n = 6 per genotype). (I) Flow cytometric analysis of CD4 and CD8 in cells differentiated from sorted Src3+/− and Src3−/− CD4-CD8- thymocytes co-cultured for three days with OP9-DL4 cells in the presence of IL-7 (5 ng/ml) to assess ex vivo thymocyte development (left two panels). The plot on the right shows the percentages of CD4+ plus CD4+CD8+ cells in each group. Data are from one representative of three independent experiments (A-C; F; E and I, left panel) or are aggregated from three experiments (D; G; H; E and I, right panels, presented as median [central line], maximum and minimum [box ends], and outliers [extended lines] or mean ± SEM). NS, not significant (P>0.05).
Disruption of the RORγt-SRC3 interaction impairs RORγt function in Th17 differentiation but not thymocyte development.
To separate the functions of the RORγt-SRC3 and RORγt-SRC1 interactions, we created an RORγt mutant that can bind SRC1 but not SRC3. To do this, we systematically mutated amino acids in RORγt that are predicted to contact SRCs, based on the X-ray structure of an SRC peptide-RORγt complex (Supplementary Fig. 1E) (19). Among the RORγt mutants, only the lysine 313 to arginine mutation (RORγt-K313R) significantly impaired the interaction of RORγt with SRC3 but did not affect the RORγt-SRC1 interaction (Fig. 5A and5B). We next tested the function of RORγt-K313R by introducing it retrovirally into RORγt−/− cells. Unlike WT RORγt, RORγt-K313R could not restore Th17 differentiation in RORγt−/− CD4+ cells (Fig. 5C) but could rescue thymocyte development in RORγt−/− thymocytes (Fig. 5D). RORγt−/− CD4+ cells reconstituted with RORγt-K313R also exhibited reduced expression of critical Th17 genes including Il17a, Il17f, Il22, Ccl20, and Ccr6, confirming the inability of RORγt-K313R to support Th17 differentiation (Fig. 5E and Supplementary Fig.1F). Furthermore, we conducted a ChIP assay using RORγt−/− CD4+ cells and found that RORγt-K313R had impaired affinity for both Il17a and Il17f loci compared to WT RORγt (Fig. 5F). Altogether, these data indicate that the RORγt-SRC3 interaction is essential to regulate Th17 differentiation but dispensable for thymocyte development.
Figure 5.

Disruption of the RORγt-SRC3 interaction impairs the function of RORγt in Th17 differentiation but not in thymocyte development. (A–B) Immunoblot analysis of indicated SRC immunoprecipitated with an anti-RORγt antibody from HEK293T cells co-transfected with plasmids to express WT or mutant RORγt and (A) SRC1 or (B) SRC3. (C) Flow cytometric analysis of the percentage of IL-17A+ cells (outlined) among Rorγt−/− CD4+ T cells transduced with a retrovirus expressing GFP alone (EV) or GFP with wild-type RORγ or the RORγt-K313R mutant and polarized for three days under Th17 priming conditions. The plot on the right shows the percentage of IL-17A+ cells in each group. (D) Flow cytometric analysis of CD4 and CD8 in cells differentiated from Rorγt−/− CD4-CD8- thymocytes transduced with a retrovirus expressing GFP alone (EV) or GFP with WT RORγ or RORγt-K313R and co-cultured for three days with OP9-DL4 cells in the presence of IL-7 (5 ng/ml) to assess ex vivo thymocyte development (left panels). The plot on the right shows the percentages of rescued CD4+ plus CD4+CD8+ cells in each group. (E) qPCR analysis of mRNA of Il17a, Il17f, Il22, Ccl20, Ccr6, and Ahr in Rorγt−/− CD4+ T cells transduced with WT RORγ (RORγ) or RORγt-K313R and polarized for three days under Th17 priming conditions, as shown in C. (F) ChIP analysis of RORγ or RORγt-K313R bound to Il17a (top panel) or Il17f (bottom panel) loci using Rorγt−/− CD4+ T cells transduced with WT RORγ or the RORγt-K313R mutant and polarized for three days under Th17 priming conditions, as shown in C. Data are from one representative of three independent experiments (A–B; C–D, left panel) or are aggregated from three experiments (C–D, right panels; E–F, presented as mean ± SEM). NS, not significant (P>0.05); * P < 0.05 (t-test); ** P <0.01 (t-test).
Discussion
SRC3 has long been known to be a co-factor for nuclear receptors, and it was recently found to be required for pathogenic Th17 immunity responsible for the development of EAE (14). Here, we confirmed these findings and furthermore demonstrated that SRC3 works non-redundantly with SRC1 in Th17 differentiation. Although Tanaka et al. reported that SRC3 is necessary only for pathogenic Th17 immunity, we observed defective Th17 differentiation even under normal differentiation conditions (TGFβ + IL-6). This discrepancy may be due to our use of constitutive SRC3 knockout mice instead of conditional SRC3 knockout mice (14), which likely caused developmental changes in T cells. Nevertheless, both studies support the importance of SRC3 in pathogenic Th17 immunity. We also found that Src3−/− mice have normal thymic T cell development. Specifically, we demonstrated that SRC3 interacts with RORγt in Th17 cells but not in thymocytes and disruption of the RORγt-SRC3 interaction impairs its function in Th17 differentiation but not thymocyte development.
As members of the same co-activator family, SRC1 and SRC3 are highly conserved and both interact with RORγt to stimulate Th17 differentiation, indicating that they have similar functions in Th17 cells. However, deficiency in either SRC1 or SRC3 leads to defective Th17 differentiation and Th17-dependent development of EAE (17), and thus SRC1 and SRC3 non-redundantly regulate Th17 function. Furthermore, the RORγt-K313R mutant that binds SRC1 but not SRC3 failed to fully restore Th17 differentiation in RORγt−/− T cells. These results suggest that the function of SRC1 and SRC3 do not overlap in the regulation of Th17 differentiation and T cell-mediated Th17 immunity. Our data suggest two potential mechanisms responsible for the distinct roles of SRC1 and SRC3 in Th17 cells. First, some DNA-binding sites may preferentially bind SRC1 or SRC3. Second, some genes may be regulated by both SRC1 and SRC3, whereas others are regulated by only one.
Why SRC3 is necessary as a co-factor for RORγt in Th17 cells but not in thymocytes is not understood. However, we observed that RORγt interacts with SRC3 in Th17 cells but not in thymocytes, suggesting that RORγt selectively recruits SRC3 in Th17 cells and uses different co-factors in thymocytes. Its co-factors in thymocytes remain unknown. One apparent candidate is SRC1; however, mice deficient in SRC1 have no obvious defects in thymocyte development (17), suggesting that SRC1, like SRC3, is dispensable for thymocyte development. It would thus be worthwhile to identify additional RORγt co-factors in thymocytes.
Th17 cells produce effector cytokines to mediate the pathological inflammation responsible for many types of autoimmune diseases; targeting Th17 cells is thus a potentially valuable treatment for these diseases (20). Indeed, inhibiting the Th17 pathway is effective for treating psoriasis and multiple sclerosis (21, 22). Given the essential function of RORγt in Th17 cells, pharmaceutical and academic scientists are developing RORγt inhibitors for treatment of Th17-dependent autoimmunity (6, 8, 9, 23, 24). Unfortunately, such RORγt inhibitors can induce thymic lymphoma due to inhibition of RORγt in thymocyte development (25). Although SRC3 is required for Th17 differentiation, it is not essential for regulating thymocyte development (25, 26); therefore, drugs that specifically disrupt the interaction between RORγt and SRC3 are expected to inhibit Th17-mediated pathological immunity without causing lymphoma by interference of thymocyte development. We showed that K313 of RORγt is critical for binding to SRC3, indicating that amino acids surrounding K313 can potentially be targeted to disrupt the RORγt-SRC3 interaction. Therefore, in addition to further investigating a novel function of SRC3 in RORγt-regulated Th17 differentiation, our results also facilitate the development of a new category of RORγt-based drugs that treat Th17-mediated autoimmunity without causing serious side effects.
Supplementary Material
Acknowledgements
We thank Dr. Jianming Xu (Baylor College of Medicine) for sharing the Src3 knockout mice and Dr. Ellen Rothenberg for sharing OP9-DL4 cells. We also appreciate help from the City of Hope core facilities, including the Animal Resource Center, Integrative Genomics Core, and Analytical Cytometry Core.
This work was supported by a grant from the National Institutes of Health (R01-AI109644) and institutional pilot funding. In addition, research reported in this publication was supported by the National Cancer Institute under award number P30CA33572, which includes work performed in the Animal Resource Center, Integrative Genomics Core, and Analytical Cytometry Core. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Abbreviations used in this article:
- CNS
central nervous system
- EAE
experimental autoimmune encephalomyelitis
- SRC
steroid receptor co-activator
- WT
wildtype
References
- 1.Korn T, Bettelli E, Oukka M, and Kuchroo VK. 2009. IL-17 and Th17 Cells. Annual review of immunology 27: 485–517. [DOI] [PubMed] [Google Scholar]
- 2.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, and Kuchroo VK. 2012. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol 13: 991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, and Becher B. 2011. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 12: 560–567. [DOI] [PubMed] [Google Scholar]
- 4.El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, Zhang GX, Dittel BN, and Rostami A. 2011. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 12: 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Elloso MM, Gomez-Angelats M, and Fourie AM. 2012. Targeting the Th17 pathway in psoriasis. Journal of leukocyte biology 92: 1187–1197. [DOI] [PubMed] [Google Scholar]
- 6.Skepner J, Ramesh R, Trocha M, Schmidt D, Baloglu E, Lobera M, Carlson T, Hill J, Orband-Miller LA, Barnes A, Boudjelal M, Sundrud M, Ghosh S, and Yang J. 2014. Pharmacologic inhibition of RORgammat regulates Th17 signature gene expression and suppresses cutaneous inflammation in vivo. J Immunol 192: 2564–2575. [DOI] [PubMed] [Google Scholar]
- 7.Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, and Huh JR. 2016. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 351: 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiao S, Yosef N, Yang J, Wang Y, Zhou L, Zhu C, Wu C, Baloglu E, Schmidt D, Ramesh R, Lobera M, Sundrud MS, Tsai PY, Xiang Z, Wang J, Xu Y, Lin X, Kretschmer K, Rahl PB, Young RA, Zhong Z, Hafler DA, Regev A, Ghosh S, Marson A, and Kuchroo VK. 2014. Small-molecule RORgammat antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 40: 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang Z, Xie H, Wang R, and Sun Z. 2007. Retinoid-related orphan receptor gamma t is a potential therapeutic target for controlling inflammatory autoimmunity. Expert opinion on therapeutic targets 11: 737–743. [DOI] [PubMed] [Google Scholar]
- 10.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, and Littman DR. 2000. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288: 2369–2373. [DOI] [PubMed] [Google Scholar]
- 11.He Z, Ma J, Wang R, Zhang J, Huang Z, Wang F, Sen S, Rothenberg EV, and Sun Z. 2017. A two-amino-acid substitution in the transcription factor RORgammat disrupts its function in TH17 differentiation but not in thymocyte development. Nat Immunol 18: 1128–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walsh CA, Qin L, Tien JC, Young LS, and Xu J. 2012. The function of steroid receptor coactivator-1 in normal tissues and cancer. International journal of biological sciences 8: 470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sen S, Wang F, Zhang J, He Z, Ma J, Gwack Y, Xu J, and Sun Z. 2018. SRC1 promotes Th17 differentiation by overriding Foxp3 suppression to stimulate RORgammat activity in a PKC-theta-dependent manner. Proceedings of the National Academy of Sciences of the United States of America 115: E458–E467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanaka K, Martinez GJ, Yan X, Long W, Ichiyama K, Chi X, Kim BS, Reynolds JM, Chung Y, Tanaka S, Liao L, Nakanishi Y, Yoshimura A, Zheng P, Wang X, Tian Q, Xu J, O’Malley BW, and Dong C. 2018. Regulation of Pathogenic T Helper 17 Cell Differentiation by Steroid Receptor Coactivator-3. Cell reports 23: 2318–2329. [DOI] [PubMed] [Google Scholar]
- 15.Xu J, Liao L, Ning G, Yoshida-Komiya H, Deng C, and O’Malley BW. 2000. The steroid receptor coactivator SRC-3 (p/CIP/RAC3/AIB1/ACTR/TRAM-1) is required for normal growth, puberty, female reproductive function, and mammary gland development. Proceedings of the National Academy of Sciences of the United States of America 97: 6379–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, Agarwal A, Huang W, Parkurst CN, Muratet M, Newberry KM, Meadows S, Greenfield A, Yang Y, Jain P, Kirigin FK, Birchmeier C, Wagner EF, Murphy KM, Myers RM, Bonneau R, and Littman DR. 2012. A validated regulatory network for Th17 cell specification. Cell 151: 289–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sen S, Wang F, Zhang J, He Z, Gwack Y, Xu J, and Sun Z. 2017. SRC1 promotes Th17 differentiation by overriding Foxp3 suppression to stimulate RORgammat activity in a PKC-theta dependent manner. Proceedings of the National Academy of Sciences of the United States of America In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie H, Sadim MS, and Sun Z. 2005. RORgammat recruits steroid receptor coactivators to ensure thymocyte survival. J Immunol 175: 3800–3809. [DOI] [PubMed] [Google Scholar]
- 19.Stehlin-Gaon C, Willmann D, Zeyer D, Sanglier S, Van Dorsselaer A, Renaud JP, Moras D, and Schule R. 2003. All-trans retinoic acid is a ligand for the orphan nuclear receptor ROR beta. Nat Struct Biol 10: 820–825. [DOI] [PubMed] [Google Scholar]
- 20.Yang J, Sundrud MS, Skepner J, and Yamagata T. 2014. Targeting Th17 cells in autoimmune diseases. Trends in pharmacological sciences 35: 493–500. [DOI] [PubMed] [Google Scholar]
- 21.Tonel G, Conrad C, Laggner U, Di Meglio P, Grys K, McClanahan TK, Blumenschein WM, Qin JZ, Xin H, Oldham E, Kastelein R, Nickoloff BJ, and Nestle FO. 2010. Cutting edge: A critical functional role for IL-23 in psoriasis. J Immunol 185: 5688–5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Segal BM, Constantinescu CS, Raychaudhuri A, Kim L, Fidelus-Gort R, Kasper LH, and Ustekinumab MSI. 2008. Repeated subcutaneous injections of IL12/23 p40 neutralising antibody, ustekinumab, in patients with relapsing-remitting multiple sclerosis: a phase II, double-blind, placebo-controlled, randomised, dose-ranging study. The Lancet. Neurology 7: 796–804. [DOI] [PubMed] [Google Scholar]
- 23.Huh JR, and Littman DR. 2012. Small molecule inhibitors of RORgammat: targeting Th17 cells and other applications. Eur J Immunol 42: 2232–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sheridan C 2013. Footrace to clinic heats up for T-cell nuclear receptor inhibitors. Nature biotechnology 31: 370. [DOI] [PubMed] [Google Scholar]
- 25.Guntermann C, Piaia A, Hamel ML, Theil D, Rubic-Schneider T, Del Rio-Espinola A, Dong L, Billich A, Kaupmann K, Dawson J, Hoegenauer K, Orain D, Hintermann S, Stringer R, Patel DD, Doelemeyer A, Deurinck M, and Schumann J. 2017. Retinoic-acid-orphan-receptor-C inhibition suppresses Th17 cells and induces thymic aberrations. JCI Insight 2: e91127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liljevald M, Rehnberg M, Soderberg M, Ramnegard M, Borjesson J, Luciani D, Krutrok N, Branden L, Johansson C, Xu X, Bjursell M, Sjogren AK, Hornberg J, Andersson U, Keeling D, and Jirholt J. 2016. Retinoid-related orphan receptor gamma (RORgamma) adult induced knockout mice develop lymphoblastic lymphoma. Autoimmun Rev 15: 1062–1070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.