

Abstract
Two 6β-N-heterocyclic naltrexamine derivatives, NAP and NMP, have been identified as peripherally selective mu opioid receptor (MOR) antagonists. To further enhance the peripheral selectivity of both compounds, the 17-amino group and the nitrogen atom of the pyridine ring in both NAP and NMP were methylated to obtain dMNAP and dMNMP, respectively. Compared with NAP and NMP, the binding affinities of dMNAP and dMNMP shifted to MOR and KOR (kappa opioid receptor) dual selective and they acted as moderate efficacy partial agonists. The results from radioligand binding studies were further confirmed by molecular docking studies. In vivo studies demonstrated that dMNAP and dMNMP did not produce antinociception nor did they antagonize morphine’s antinociceptive activity, indicating that these compounds did not act on the central nervous system. Meanwhile, both dMNAP and dMNMP significantly slowed down fecal excretion, which indicated that they were peripherally acting opioid receptor agonists. All together, these results suggested that dMNAP and dMNMP acted as peripheral mu/kappa opioid receptor modulators and may be applicable in the treatment of diarrhea in patients with bowel dysfunction.
Keywords: Methylation, naltrexamine, peripheral opioid receptor modulators, diarrhea, opioid bowel dysfunction, irritable bowel syndrome
INTRODUCTION
Opioid receptors are classified into for main types, the mu opioid receptor (MOR), kappa opioid receptor (KOR), delta opioid receptor (DOR), and the nociceptin opioid peptide receptor (NOP).1−3 These receptors are extensively located in the central nervous system (CNS) and the peripheral nervous system (PNS), such as the gastrointestinal tract (GIT). Among them, MOR is the major target for opioid medications. The activation of MOR by opioid agonists results in different pharmacological effects, such as analgesia, and numerous side effects including addiction, respiratory depression, urinary retention, and constipation.4,5 Opioid-induced constipation (OIC) is one of the most common side effects that limits the use of opioids in the clinic. It has been reported that 9.3−95% of patients suffer from OIC; as a result, most patients on opioid analgesics refrain from taking their medication.6−14 Thus, peripherally selective opioid antagonist may prevent the development of OIC without inhibiting the analgesic effects of opioid agonists. Methylnaltrexone (MNTX, 1, Figure 1), naloxogel, and Alvimopan are two peripherally selective opioid antagonists that have been developed to treat OIC.15−18 However, methylnaltrexone has a low efficiency (48%−62%) to induce spontaneous bowel movement in patients. Moreover, long-term use of Alvimopan increases the risk of developing myocardial infarction which limits its usage to treat OIC.19−22
Figure 1.

Chemical structures of MNTX (1), eluxadoline (2), NAP (3), NMP (4), dMNAP (5), and dMNMP (6).
In fact, OIC is not the only commonly reported symptom of opioid bowel dysfunction (OBD) associated with opioid use.23 Other clinical symptoms of OBD include gastric reflux, bloating, abdominal distention, and straining. The mechanism of OBD, while is still not very clear, may involve complicated actions in both the CNS and PNS. Since the clinical symptoms of OBD are mainly connected with the abnormal function of the gastrointestinal tract, peripheral opioid receptors in the GIT, especially the MOR, is considered an effective target to treat OBD.24,25 The traditional treatment for OBD includes laxatives, dietary measures, physical activity, and biofeedback therapy.26 In addition to traditional treatments, opioid receptor antagonists, such as naltrexone, naloxone, and nalmefene, have also been extensively studied to treat OBD.23 These agents competitively block the opioid receptors and reverse the CNS and PNS effects produced by opioid agonists.27,28 However, the use of these antagonists to treat OBD is limited due to their lack of selectivity to the MOR, poor lipid solubility, and/or pharmacokinetic profiles.27−29
In addition to treating OBD, opioid receptors have been identified as important targets for treating irritable bowel syndrome (IBS). IBS is considered as the most common disease of the GIT.30,31 The pathology of IBS is a complicated process, and may involve numerous factors, such as diet, the use of antibiotics, body infection, dysfunction of the GIT, immune dysfunction, and even psychological or environmental changes.30 About 3.6 million people visit the hospital all over the world every year because of IBS.31,32 Typical symptoms of IBS include diarrhea, constipation, pain, bloating, and flatulence. Based on the clinical symptoms, IBS can be categorized into three subgroups, i.e., IBS with diarrhea (IBS-D), IBS with constipation (IBS-C), and IBS with a mixed stool pattern (IBS-M). Several agents have been approved to treat IBS-D. For example, Alosetron, an antagonist of 5-HT3 receptor, was approved for treating women who suffer from IBS-D.33 Rifaximin, an approved broad-spectrum antibiotic, was used to treat IBS-D in both men and women.34 Eluxadoline (2, Figure 1), a mixed opioid receptor modulator which has mu agonist, delta antagonist, and unidentified kappa agonist properties, was approved for treating IBS-D in 2015 by the United States Food and Drug Administration (FDA).30 Studies have shown that activation of the MOR by opioid agonists such as morphine result in reduction of GIT motility and secretion.35 Interestingly, Wade et al. demonstrated that DOR antagonists can increase GIT motility.36 As a result, eluxadoline has been prescribed to treat both pain and diarrhea in IBS.37,38a Recent studies showed that eluxadoline may be associated with risk of pancreatitis.38b Apparently, developing novel and peripherally selective opioid modulators will provide more diversified clinical options to benefit patients suffering from OBD and IBS, as well as tools to further explore the potential role of opioid receptors as targets to treat GIT diseases.
Our laboratory previously designed and synthesized two 6β-N-heterocyclic substituted naltrexamine derivatives, NAP39−42 (3, Figure 1) and NMP43 (4, Figure 1). These two compounds were identified as peripheral MOR antagonists and showed potency to improve GIT motility in animal model studies. BNAP, a quaternized derivative of NAP, showed a mixed MOR antagonist/KOR agonist properties in a hypernociceptive model of visceral pain.44 Herein, we have further explored the peripheral selectivity of NAP and NMP by design, syntheses and pharmacological evaluation of two methylated NAP derivatives, dMNAP (5, Figure 1) and dMNMP (6, Figure 1). Both compounds were identified as dual MOR/KOR partial agonist with potential application to treat bowel dysfunctional patients with diarrhea.
RESULTS AND DISCUSSION
dMNAP and dMNMP were synthesized by a two-step reaction. Then, both compounds were applied for binding and functional assays to assess their binding affinity and efficacy to the opioid receptors. Meanwhile, in vivo studies were used to examine their peripheral selectivity and related functions.
Chemistry.
Based on previous work conducted in our group,39−43 two 6β-N-heterocyclic naltrexamine derivatives, dMNAP and dMNMP, were designed and synthesized, as shown in Scheme 1. Both the 17-amino group and the nitrogen atom on the pyridine ring in both NAP and NMP were methylated to obtain dMNAP and dMNMP (Supporting Information). Interestingly, no monomethylation product of NAP and NMP was obtained.
Scheme 1. Synthetic Procedure of dMNAP and dMNMPa.

aReagents and conditions: (a) TBDMSCl, (C2H5)3N, THF; (b) (i) CH3I, acetone/DMF (10/1, v/v), 6 days, 80 °C; (ii) MeOH/HCl, 4 h.
Binding and Functional Assays.
The radioligand binding assay was applied to determine the binding affinities of dMNAP and dMNMP to the opioid receptors. [3H]Naloxone was used to label the MOR while [3H]diprenorphine was used to label both the KOR and DOR. The [35S]-GTPγS functional assay was used to determine the potency and efficacy of dMNAP and dMNMP at the MOR, KOR, and DOR and the results were interpreted as potency (EC50) and efficacy (%Emax relative to DAMGO, U50,488H, and SNC80). The binding affinity, selectivity, potency, and efficacy data for dMNAP and dMNMP are summarized in Table 1.
Table 1. Radioligand Binding and Functional Assay Results for dMNAP and dMNMP at the MOR, DOR, and KORa.
| cmpds | dMNAP | dMNMP | ||
|---|---|---|---|---|
| Ki (nM) | MOR | 28.26 ± 2.47 | 2.85 ± 0.25 | |
| DOR | 9860.18 ± 1140.65 | 603.80 ± 117.88 | ||
| KOR | 20.11 ± 2.41 | 3.09 ± 0.34 | ||
| selectivity | δ/μ | 348.9 | 211.9 | |
| κ/μ | 0.7 | 1.1 | ||
| cmpds | dMNAP | dMNMP | ||
| [35S]GTPγS binding | MOR | EC50 (nM) | 239.65 ± 48.92 | 13.52 ± 0.99 |
|
Emax (% DAMGO) |
40.68 ± 2.04 | 44.63 ± 4.18 | ||
| KOR | EC50 (nM) | 142.70 ± 27.03 | 27.27 ± 5.0 | |
|
Emax (% U50,488H) |
37.72 ± 1.58 | 42.66 ± 1.28 | ||
| DOR | EC50 (nM) | ND | ND | |
|
Emax (% SNC80) |
6.76 ± 2.02 | 9.89 ± 1.56 | ||
The values are the mean ± SEM of at least three independent experiments. [3H]Naloxone was used to label the MOR while [3H]diprenorphine was used to label both DOR and KOR. The Emax value of each compound was determined as a percentage the maximal stimulation produced by a full agonist: DAMGO for MOR, U50,488H for KOR, and SNC80 for DOR (normalized to 100%).
The binding affinities for dMNAP and dMNMP at the MOR, KOR, and DOR were compared to those of NAP and NMP.39−43 As shown in Table 1, the binding affinities of dMNAP at the MOR (Ki,MOR = 28.26 ± 2.47 nM) and DOR (Ki,DOR = 9860.18 ± 1140.65 nM) decreased significantly compared to NAP (Ki,MOR = 0.37 ± 0.07 nM; Ki,DOR = 277.5 ± 8.0 nM), respectively. On the other hand, the binding affinity at KOR of dMNAP (Ki, KOR = 20.11 ± 2.41 nM) increased somewhat compared to NAP (Ki,KOR = 60.7 ± 5.6 nM). The same trend was also observed for dMNMP, where the MOR (Ki,MOR = 2.85 ± 0.25 nM) and DOR (Ki,DOR = 603.80 ± 117.88 nM) binding affinities decreased to some degree, respectively, compared to NMP (Ki,MOR = 0.58 ± 0.25 nM; Ki,DOR = 273.6 ± 1.8 nM), while the KOR binding affinity of dMNMP (Ki,KOR = 3.09 ± 0.34 nM) improved over 30-fold compared to NMP (Ki,KOR = 96.7 ± 12.2 nM). As a result, dMNAP and dMNMP seemed to act as MOR/KOR dual selective agents. The binding affinities of dMNAP and dMNMP to other GPCRs were also determined (Tables S1−S5, Supporting Information),45 and the results showed that dMNAP and dMNMP were highly selective to opioid receptors compared to other GPCRs.
The [35S]GTPγS binding assay was used to assess the potency and relative efficacy of dMNAP and dMNMP at the MOR, KOR, and DOR. As observed in Table 1, though dMNAP (EC50 = 239.65 ± 48.92 nM) and dMNMP (EC50 =13.52 ± 0.99 nM) were less potent at the MOR than NAP (EC50 = 1.14 ± 0.38 nM) and NMP (EC50 = 1.52 ± 0.26 nM), both dMNAP (%Emax = 40.683 ± 2.040) and dMNMP (%Emax = 44.63 ± 4.18) were more efficacious than NAP (%Emax = 22.72 ± 0.84) and NMP (%Emax = 30.63 ± 0.55), respectively.39−43 The MOR efficacy was determined as the percent of stimulation produced by the compounds relative to DAMGO (a MOR full agonist). The potency and efficacy of dMNAP and dMNMP were also determined at both KOR and DOR (Table 1). Apparently, dMNAP and dMNMP, not only acted as MOR/KOR dual selective ligands, but also as partial agonists with approximately 40% efficacy at both receptors and potentially as DOR antagonists but with weak binding affinity (Table 1).
From a previous study conducted on BNAP,44 which also carries a quaternized pyridine nitrogen atom, the MOR and KOR binding affinities (Ki) of BNAP were determined as 0.76 ± 0.09 nM and 3.46 ± 0.05 nM, respectively. BNAP had an Emax value of 14.6% relative to the stimulation by DAMGO at the MOR and an Emax of 45.9 ± 1.7% relative to the stimulation by U50,488H at the KOR.44 The results for dMNAP and dMNMP obtained together with those of BNAP suggested that quaternization in the “address” part of NAP or NMP may play an important role in determining the opioid receptor binding affinity, selectivity, and efficacy. On the other hand, quaternization in the “message” part may not have such a significant influence. To further support our observation, methylnaltrexone (MNTX), which has a methyl group introduced at position 17 amino group, showed very similar binding affinity, selectivity, and efficacy profiles as naltrexone.15,16
Tail Immersion Test.
As mentioned earlier, dMNAP and dMNMP were designed as peripherally selective opioid ligands. To determine whether these compounds may be centrally acting, a tail immersion assay was conducted to find out whether dMNAP and dMNMP produced antinociception or blocked morphine’s antinociceptive effects by subcutaneous injection (s.c.).39,42 As observed in Figure 2A, dMNAP at a dose of 10 mg/kg produced insignificant antinociception compared to morphine (10 mg/kg). dMNMP, on the other hand, did not produce antinociception and its %MPE (the percentage maximum possible effect) was not significantly different from saline treatment. Both dMNAP and dMNMP did not block morphine’s antinociceptive effects, indicating that these compounds did not act as opioid antagonists in the CNS (Figure 2B). Similarly, MNTX, acting as a peripherally selective opioid antagonist approved for treating OIC, was also methylated at position 17 to form a quaternary ammonium cation and lost its ability to cross the blood-brain barrier (BBB).16 Dimethylation of NAP and NMP seemed to have reinforced such an effect as well.
Figure 2.

Tail flick test for dMNAP and dMNMP as agonist (A) and antagonist (B) in mice (10 mg/kg). Morphine (10 mg/kg) and saline were used as control (n = 5, *P < 0.05; ***P < 0.0005).
Gastrointestinal Transit.
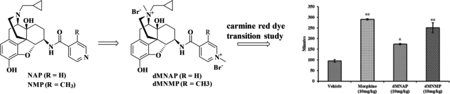
Measuring GIT transit time is a standard way of examining GIT motility.46,47 It has been reported that opioid agonists not only reduce GIT motility and secretion but also enhance reabsorption of water in the gastrointestinal tract by activating peripheral MOR.48,49 The carmine red GIT transit study was conducted to determine whether dMNAP and dMNMP had any effect on GIT motility.50−52 In this assay, the test compounds at a single dose (10 mg/kg) were first administered s.c. at time zero. Morphine (10 mg/kg) and saline were used as positive and negative controls, respectively. At 20 min after injection, carmine red dye was given by oral administration (p.o.) to the mice. The time for the appearance of red fecal pellet (maximum to 6 h) was recorded.
As shown in Figure 3, morphine (290.2 ± 2.9 min) significantly increased GIT transit time compared to vehicle (95.0 ± 6.3 min). Meanwhile, both dMNAP and dMNMP also significantly increased GIT transit time (173.4 ± 3.0 and 250.6 ± 23.3 min, respectively). As MOR/KOR partial agonists, dMNAP and dMNMP displayed the features of opioid agonist to reduce the movement of GIT. To further confirm the effect of reducing GIT transit time, a dose response study was conducted. As shown in Figure 4, both dMNAP (Figure 4A) and dMNMP (Figure 4B) significantly slowed down the extraction time of carmine red dye compared with the control group MNTX (25 mg/kg), at all doses (2 mg/kg –32 mg/kg).
Figure 3.

Transition time of carmine red dye for dMNAP and dMNMP in GIT. Morphine (10 mg/kg) and saline were used as control (n = 5, *P < 0.05; **P < 0.005).
Figure 4.

Dose response studies of dMNAP (A) and dMNMP (B) in carmine red dye assays. Morphine (10 mg/kg) and MNTX (2.5 mg/kg) were used as control (n = 5, *P < 0.05; **P < 0.005; ***P <0.0005).
Recently, it was reported that KOR agonists may exhibit analgesic effect without the peripheral side effect of MOR agonists.53,54 In addition, Fichna et al. reported that salvinorin A, a KOR agonist, showed its ability to reduce the motility of colon in an in vivo study.55 Development of peripherally selective KOR agonists is of interest because they display analgesic activity while avoiding the negative side effects associated with MOR agonists in the periphery and those associated with the central effects of KOR agonists. We have previously shown that BNAP,44 a mixed MOR/KOR modulator, induced antinociception at 3-fold lower concentration compared to morphine in a hypernociception model of visceral pain. Since both dMNAP and dMNMP acted as dual selective partial agonists at the MOR/KOR, we therefore examined whether these compounds were also effective in the acetic acid stretching assay, a model for visceral pain. As shown in Figure 5, both dMNAP and dMNMP showed no significant analgesic effect at 10 mg/kg on their own, and did not reverse morphine’s antinociception effects in periphery either at the same dose. These observations are consistent with the relatively low potency of dMNAP and dMNMP as KOR partial agonists compared to BNAP.
Figure 5.

(A) Analgesic activity of dMNAP in the acetic acid induced writhing assay in mice chronically treated with a 75 mg morphine pellet (MP) or placebo pellet (PP). (B) Analgesic activity of dMNMP in the acetic acid induced writhing assay in mice chronically treated with a 75 mg morphine pellet (MP) or placebo pellet (PP). One-way ANOVA, Tukey’s multiple comparison test, P > 0.05.
Molecular Modeling Studies.
As described above, dMNAP and dMNMP acted as MOR/KOR dual partial agonist (Figure S3, Supporting Information). Hence, both compounds may recognize and bind to the inactive as well as active states of both receptors. In order to understand the selectivity of dMNAP and dMNMP at the MOR, KOR, and DOR and their functional efficacies at the MOR and KOR, docking studies were conducted utilizing the crystal structures of the antagonist-bound MOR (PDB ID: 4DKL),56 KOR (PDB ID: 4DJH),57 and DOR (PDB ID: 4EJ4);58 agonist-bound MOR (PDB ID: 5C1M)59 and KOR (PDB ID: 6B73)60 using GOLD 5.4.61,62 The highest scored (CHEM-PLP) solutions were chosen from every docking operation as the optimal docking poses of dMNAP and dMNMP in these protein structures, respectively (Figures 6 and 7).
Figure 6.

Docking studies of dMNAP (A–C) and dMNMP (D–F) in the inactive MOR, KOR, and DOR, respectively, with highest CHEM-PLP scores. Protein shown as cartoon model in light-green (MOR), light-pink (KOR), and light-blue (DOR); dMNAP, dMNMP, and key amino acid residues shown as stick model. Carbon atoms: dMNAP (magentas); dMNMP (cyan); key amino acid residues in MOR (green), KOR (orange), and DOR (light-blue). Oxygen atoms (red); nitrogen atoms (blue). The red dashed line represents the shortest distance between the position-17 quaternary amino group and D3.32. The yellow dashed line represents the shortest distance between the side chain of D3.32 and the side chain of T2.56.
Figure 7.

Docking poses of dMNAP (A, C) and dMNMP (B, D) in the active MOR and KOR, respectively, with highest CHEM-PLP score from docking studies. Protein shown as cartoon model in light-green (MOR) and light-pink (KOR); dMNAP, dMNMP, and key amino acid residues shown as stick model. Carbon atoms: dMNAP (magentas); dMNMP (cyan); key amino acid residues in MOR (green) and KOR (orange). Oxygen atoms (red); nitrogen atoms (blue). The red dashed line represents the shortest distance between the position-17 quaternary amino group and D3.32. The yellow dashed line represents the shortest distance between the side chain of D3.32 and the side chain of T2.56.
The Selectivity of dMNAP and dMNMP at the Inactive MOR, DOR, and KOR.
For comparison purpose, three inactive receptor crystal structures were adopted to understand the binding affinity of dMNAP and dMNMP first. As displayed in Figure 6, the epoxymorphinan moiety of dMNAP and dMNMP interacted with conserved residues in the MOR, DOR and KOR by forming hydrophobic interactions with M3.36, W6.48 and H6.52, and hydrogen bonding interactions with Y3.33 in all three receptors. Meanwhile, the methylated nitrogen atom at position 17 of the epoxymorphinan moiety also formed similar ionic interactions with conserved D3.32 in the MOR, DOR and KOR. The results obtained were consistent with our previous studies in which we observed similar interactions for NAP and its derivatives.63,64 In other words, quaternization at the 17-amino group seemed to have no significant influence on opioid receptor recognition in the inactive state.
The amide linker in dMNAP and dMNMP gave the pyridine ring enough flexibility to possibly interact with either one of the two “address” domains in the three opioid receptors, which had been defined in our previous studies.63,64 Since one binding domain of the MOR, DOR and KOR is composed of highly conserved and negatively charged residues E5.35, D5.35, and D5.35, the quaternized pyridine ring in dMNAP and dMNMP seemed to prefer this domain in all three receptors due to the favorable electrostatic interactions between the compounds and the overall negatively charged binding loci (Figure 6).
In addition to the above electrostatic interactions observed for dMNAP, L219ECL2 and F221ECL2 in the MOR (Figure 6A), L212ECL2 and F214ECL2 in the KOR (Figure 6B), and L200ECL2 and F202ECL2 in the DOR (Figure 6C) seemed to be able to form hydrophobic interactions with the aromatic pyridine ring of the ligand. And the hydrophobic interactions in the MOR seemed stronger than those in the KOR and DOR. Meanwhile, Y2195.31 in the KOR could also form a hydro-phobic interaction with the methyl group on the nitrogen atom of the pyridine ring of dMNAP (Table S6, Supporting Information). That may offer an answer to why the binding affinities of dMNAP to the MOR and KOR were higher than that to the DOR.
In the case of dMNMP, several additional interactions were observed. L219ECL2 and F221ECL2 in the MOR (Figure 6D), L212ECL2 and F214ECL2 in the KOR (Figure 6E) and L200ECL2 and F202ECL2 in the DOR (Figure 6F), respectively, could form hydrophobic interaction with the methyl group on the pyridine ring of dMNMP (Table S6, Supporting Information). These molecular interactions helped explain the trend of the binding affinities observed for dMNMP as well.
Docking Studies of dMNAP and dMNMP in the Activated MOR and KOR.
Docking studies were then applied to simulate the agonist functional profiles of dMNAP and dMNMP. The binding poses of dMNAP and dMNMP in the active MOR and KOR were displayed in Figure 7. In these four binding poses, the epoxymorphinan moiety of dMNAP and dMNMP would form hydrophobic interactions with conserved M3.36, W6.48, and H6.52 and hydrogen bonding interactions with Y3.33. Obviously, these interactions were similar to dMNAP and dMNMP in the inactive MOR and KOR.
Additionally, the hydrophobic interactions between the conserved W6.48 and the cyclopropyl group of dMNAP and dMNMP also existed in the active MOR and KOR, which were consistent with the findings of Che et al.60 Moreover, the methyl group at the nitrogen atom at position-17 of both compounds may also form hydrophobic interactions with the aromatic ring of the conserved Y7.43 (Table S7). These two hydrophobic interactions with conserved W6.48 and Y7.43 may make the cyclopropylmethyl group of dMNAP and dMNMP bind to the binding pocket of the active MOR and KOR stably.
As described in the previous studies of the active MOR and KOR crystal structures, an agonist binding with the MOR or KOR could lead to TM3 moving closer to TM2 compared to the antagonist-bound receptors,56,60 as represented by the shortened distance between the side chain of D3.32 (located at TM3) and the side chain of T2.56 (located at TM2). From Figure 7, both dMNAP and dMNMP bound to the active MOR and KOR through a seemingly stronger electrostatic interaction between their position-17 quaternary amino groups and the conserved D3.32 residue compared to the inactivated receptors. Such an interaction seemed to further lead to stabilization of a closer position of TM3 to TM2, as indicated by the shortened distance between D3.32 and T2.56 in the ligand bound receptors.
In summary, dimethylated derivatives of NAP and NMP, dMNAP and dMNMP, were designed and synthesized to further strengthen their peripheral selectivity. Both compounds were identified as dual MOR/KOR partial agonists and this binding profile was further validated by docking studies. Due to their peripheral selectivity, dMNAP and dMNMP did not induce antinociception or reverse morphine’s effect on antinociception in the tail immersion test. Both compounds significantly slowed down gastrointestinal transit indicating their potential application to treat bowel dysfunctional patients including diarrhea. The present opioid crisis with the potential abuse of loperamide highlights the requirement for the development of novel antidiarrheal agents that have significant peripheral restrictions.
METHODS
Drugs and Chemicals.
Morphine (morphine sulfate pentahydrate salt), was procured from the National Institute of Drug Abuse (NIDA), Bethesda, MD and then made into a 10 μM stock solution by dissolving in distilled water which was further diluted to the desired concentrations. MNTX, NAP, and NMP were synthesized as HCl salts in our lab. Other reagents were purchased from either Sigma-Aldrich or Alfa Aesar. TLC analyses were carried out on the Analtech Uniplate F254 plates. Chromatographic purification was carried out on silica gel (230–400 mesh, Merck) columns. 1H (400 MHz) and 13C (100 MHz) nuclear magnetic resonance (NMR) spectra were recorded at ambient temperature with tetramethylsilane as the internal standard on Varian Mercury 400 MHz NMR spectrometer.
Chemical Synthesis.
The chemical synthesis procedure included three steps. First, the 3-OH of NAP or NMP was protected with tert-butyldimethylsilyl chloride. Then, the protected intermediates were methylated using iodomethane. After that, the protecting group at 3 position of NAP or NMP were removed to get dMNAP and dMNMP. The synthesis detail is discussed in the Supporting Information which includes NMR, MS, IR, melting point, and HPLC.
Animals.
Male Swiss Webster mice (25–30 g, Harlan Laboratories, Indianapolis, IN) were raised in animal care quarters and maintained at room temperature on light-dark cycle. Food and water were available ad libitum. Protocols and procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Virginia Commonwealth University Medical Center and complied with the recommendations of the IASP (International Association for the Study of Pain).
Competitive Radioligand Binding and [35S]GTPγS Functional Studies.
Monocloned mouse opioid receptors expressed in Chinese hamster ovarian (CHO) cell line were used for both experiments. [3H]naloxone was used to label the MOR, while [3H]diprenorphine was used to label both the KOR and DOR.
Membrane protein (30 μg) was incubated with the corresponding radioligand in the presence of varying concentrations of test compounds in TME buffer (50 mM Tris, 3 mM MgCl2, and 0.2 mM EGTA, pH 7.4) at 30 °C for 90 min. The bound radioligand was separated by filtration using the Brandel harvester. Specific binding at the MOR, KOR and DOR was determined as the difference in binding obtained in the absence and presence of 5 μM naltrexone, U50,488, and SNC80, respectively. Bound radioactivity was determined by liquid scintillation spectrophotometry. The IC50 values were determined and converted to Ki values using the Cheng–Prusoff equation.65
In the [35S]GTPγS functional assays, 10 μg of membrane protein (from MOR-CHO, KOR-CHO, or DOR-CHO cells) was incubated with 10 μM GDP, 0.1 nM [35S]GTPγS, assay buffer (TME + 100 mM NaCl) and varying concentrations of the compounds under investigation for 90 min at 30 °C. Nonspecific binding was determined with 20 μM unlabeled GTPγS. DAMGO, U50, 488H or SNC80 (3 μM each) was included in the assay at a maximally effective concentration as a standard full agonist for the MOR, KOR, or DOR, respectively. Percent DAMGO (U50, 488H or SNC80)-stimulated [35S]GTPγS binding was defined as [net-stimulated binding by ligand/net-stimulated binding by 3 μM DAMGO (U50, 488H, or SNC80)] × 100%.
Data Analysis of Radioligand Binding Assays.
All samples were assayed in duplicate and repeated at least 4 times for a total of ≥4 independent determinations. Results were reported as mean values ± SEM. Concentration–effect curves were fit by nonlinear regression to a one-site binding model, using GraphPad Prism software 6.0, to determine EC50 and Emax values. IC50 values were obtained from Hill plots, analyzed by nonlinear regression using GraphPad Prism 6.0 software. Binding Ki values were determined from IC50 values using the Cheng-Prusoff equation: Ki = IC50/(1 + ([L]/KD)), where [L] is the concentration of the competitor and KD is the KD of the radioligand.
Tail Immersion Test.
Swiss Webster mice were used for the experiment. The water bath temperature was maintained at 56 ± 0.1 °C. The baseline latency (control) was determined before the tested compound was injected into the mice. The average baseline latency obtained for this experiment was 3.0 ± 0.1 s and only mice with a baseline latency of 2 to 4 s were used. For the study of agonism, the time of tail immersion was 20 min (time that morphine’s antinociceptive effect starts to peak) after the test compound was injected. To prevent tissue damage, a 10 s maximum cut off time was imposed. Antinociceptive response was calculated as the percentage maximum possible effect (%MPE), where %MPE = [(test – control)/(10 – control)] × 100. For the study of antagonism, the test compound was given 5 min before morphine. The tail immersion test was then conducted 20 min after giving morphine. %MPE was calculated for each mouse using at least five mice per drug. AD50 values were calculated using the least-squares linear regression analysis followed by calculation of 95% confidence interval by Bliss method.
The Transition Study of Carmine Red Dye.
Swiss Webster mice (male) were used for carmine red GIT study. Morphine (10 mg/kg), saline, and tested compound (10 mg/kg) was given by subcutaneous (s.c.) injection to mice at the time zero in each group of five mice. Twenty minutes later, carmine red dye (0.5% Carboxal and 6% Carmine Red dye in ddH2O) was given by oral administration (p.o.) to mice. The mice were then observed until they all defecated a red fecal pellet or for 6 h according to which occurred first.
Visceral Sensitivity Assay.
Swiss Webster mice were pelleted with placebo pellets or 75 mg morphine pellets for 7 days. On day 7, mice were habituated in their home cages in testing room for at least 10 min. Following the acclimation period, vehicle (10% DMSO), dMNAP (10 mg/kg), or dMNMP (10 mg/kg) was administered to the mice via subcutaneous injection and returned to their home cages. At 15 min post subcutaneous injection, mice were given 10 mL/kg 0.6% (wt/vol) acetic acid intraperitoneally and then placed in individual testing cages. At 3 min after the acetic acid administration, the number of stretches and abdominal contractions were counted for 15 min. All values shown for abdominal stretching experiments represent separate groups of mice (n = 5) as each animal was tested only once. The mice were euthanized immediately after testing.
Statistical Analysis.
Data are presented as the mean ± SEM; values of P ≤ 0.05 were considered significant and analyzed by appropriate statistical tools using GraphPad Prism 6.0 software (Graph-Pad Software Inc.).
Molecular Modeling Studies.
The crystal structures of antagonist-bound MOR (PDB ID: 4DKL), KOR (PDB ID: 4DJH), and DOR (PDB ID: 4EJ4), and agonist-bound MOR (PDB ID: 5C1M) and KOR (PDB ID: 6B73) were downloaded from the Protein Data Bank at http://www.rcsb.org. Hydrogen atoms were added to each receptor and optimized by a 10 000 iteration minimization while holding all heavy atoms as fixed with Gasteiger-Hückel charges assigned under the Tripos force field (TAFF) in Sybyl-X 2.0 (TRIPOS Inc., St. Louis, MO). dMNAP and dMNMP were sketched in Sybyl-X 2.0, assigned Gasteiger-Hückel charges and energy minimized to a gradient of 0.05 under the TAFF.
Docking studies were conducted using GOLD 5.4 with default settings. The binding site was defined to include all atoms within 10 Å of the γ-carbon atom of D3.32 in all four crystal structures (superscript numbers follow the Ballesteros-Weinstein numbering method for GPCRs). Automated docking was conducted with a distance constraint of 4 Å between the piperidine quaternary ammonium nitrogen of the ligands’ epoxymorphinan nucleus and D3.32, and between the ligands’ dihydrofuran oxygen and the phenolic oxygen of Y3.33. Based on the fitness scores and the binding orientation of each ligand within the binding cavity, the highest scored solutions were selected and merged into the receptor. The interactions between ligand and receptor within the binding pocket were optimized: clashes and strain energy were removed by energy minimizing the receptor–ligand structure complexes (2000 iterations under TAFF) in Sybyl-X 2.0.
Supplementary Material
Funding
This work was partially supported by NIH/NIDA DA024022 and DA044855 (Y.Z.). Support for dMNAP and dMNMP binding with other GPCRs (Ki determinations) was generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is directed by Bryan L. Roth MD, Ph.D. at the University of North Carolina at Chapel Hill and Project Official Dr. Jamie Driscoll at NIMH, Bethesda MD.
ABBREVIATIONS
- MOR
mu opioid receptor
- DOR
delta opioid receptor
- KOR
kappa opioid receptor
- OIC
opioid-reduced constipation
- PNS
peripheral nervous system
- CNS
central nervous system
- MNTX
methylnaltrexone
- OBD
opioid bowel dysfunction
- GIT
gastrointestinal tract
- IBS
irritable bowel syndrome
- IBSD
IBS with diarrhea
- IBS-C
IBS with constipation
- IBS-M
IBS with mixed stool pattern
- BBB
blood-brain barrier
- % MPE
percentage maximum possible effect
- s.c.
subcutaneous injection
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschemneuro.8b00234.
Results of NMR, IR, MS, and HPLC for all the synthesized compounds (intermediates); receptor binding results of the ligands with other GPCRs (PDF)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1). Pasternak GW (2014) Opioids and their receptors: Are we there yet? Neuropharmacology 76 (Pt B), 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2). Shang Y, and Filizola M (2015) Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur. J. Pharmacol 763 (Pt B), 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3). Nutt DJ (2014) The role of the opioid system in alcohol dependence. J. Psychopharmacol 28, 8–22. [DOI] [PubMed] [Google Scholar]
- (4). McNicol E, Horowicz-Mehler N, Fisk RA, Bennett K, Gialeli-Goudas M, Chew PW, Lau J, and Carr D (2003) Management of opioid side effects in cancer-related and chronic noncancer pain: a systematic review. J. Pain 4, 231–256. [DOI] [PubMed] [Google Scholar]
- (5). Stein C (2016) Opioid Receptors. Annu. Rev. Med 67, 433–451. [DOI] [PubMed] [Google Scholar]
- (6). Thorpe DM (2001) Management of opioid-induced constipation. Curr. Pain Headache Rep 5, 237–240. [DOI] [PubMed] [Google Scholar]
- (7). Moore RA, and McQuay HJ (2005) Prevalence of opioid adverse events in chronic non-malignant pain: systematic review of randomised trials of oral opioids. Arthritis Res. Ther 7, R1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8). Sykes N (1998) The relationship between opioid use and laxative use in terminally ill cancer patients. Palliat. Med 12, 375–382. [DOI] [PubMed] [Google Scholar]
- (9). Bell TJ, Panchal SJ, Miaskowski C, Bolge SC, Milanova T, and Williamson R (2009) The prevalence, severity, and impact of opioid-induced bowel dysfunction: results of a US and European Patient Survey (PROBE 1). Pain Med 10, 35–42. [DOI] [PubMed] [Google Scholar]
- (10). Vainio A, and Auvinen A (1996) Prevalence of symptoms among patients with advanced cancer: an international collaborative study. J. Pain Symptom Manage 12, 3–10. [DOI] [PubMed] [Google Scholar]
- (11). Kalso E, Edwards JE, Moore RA, and McQuay HJ (2004) Opioids in chronic non-cancer pain: systematic review of efficacy and safety. Pain 112, 372–380. [DOI] [PubMed] [Google Scholar]
- (12). Cook S, Lanza L, Zhou X, Sweeney C, Goss D, Hollis K, Mangel A, and Fehnel S (2008) Gastrointestinal side effects in chronic opioid users: results from a population-based survey. Aliment. Pharmacol. Ther 27, 1224–1232. [DOI] [PubMed] [Google Scholar]
- (13). Candrilli SD, Davis KL, and Iyer S (2009) Impact of constipation on opioid use patterns, health care resource utilization, and costs in cancer patients on opioid therapy. J. Pain Palliat. Care Pharmacother 23, 231–241. [DOI] [PubMed] [Google Scholar]
- (14). Pappagallo M (2001) Incidence, prevalence, and management of opioid bowel dysfunction. Am. J. Surg 182, 11S–18S. [DOI] [PubMed] [Google Scholar]
- (15). Foss JF (2001) A review of the potential role of methylnaltrexone in opioid bowel dysfunction. Am. J. Surg 182, S19–S26. [DOI] [PubMed] [Google Scholar]
- (16). Kast R (2009) Use of FDA approved methamphetamine to allow adjunctive use of methylnaltrexone to mediate core anti-growth factor signaling effects in glioblastoma. J. Neuro-Oncol 94, 163–167. [DOI] [PubMed] [Google Scholar]
- (17). Relistor (methylnaltrexone bromide). [prescribing information] (2014), Salix Pharmaceuticals, Inc, Raleigh, NC. [Google Scholar]
- (18). Alvimopan (Entereg) for postoperative ileus (2008) Med. Lett. Drugs. Ther 50, 93–94. [PubMed] [Google Scholar]
- (19). Thomas J, Karver S, Cooney GA, Chamberlain BH, Watt CK, Slatkin NE, Stambler N, Kremer AB, and Israel RJ (2008) Methylnaltrexone for opioid-induced constipation in advanced illness. N. Engl. J. Med 358, 2332–2343. [DOI] [PubMed] [Google Scholar]
- (20). Slatkin N, Thomas J, Lipman AG, Wilson G, Boatwright ML, Wellman C, Zhukovsky DS, Stephenson R, Portenoy R, and Stambler N (2009) Methylnaltrexone for treatment of opioid-induced constipation in advanced illness patients. J. Support Oncol 7, 39–46. [PubMed] [Google Scholar]
- (21). Adolor GlaxoSmithKline. Entereg (alvimopan), www.entereg.com [accessed Feb 6, 2012.].
- (22). Webster L, Peppin J, Harper J, and Israel R (2016) (480) Oral methylnaltrexone does not negatively impact analgesia in patients with opioid-induced constipation and chronic noncancer pain. J. Pain 17, S94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23). Pappagallo M (2001) Incidence, prevalence, and management of opioid bowel dysfunction. Am. J. Surg 182, S11–S18. [DOI] [PubMed] [Google Scholar]
- (24). De Luca A, and Coupar IM (1996) Insights into opioid action in the intestinal tract. Pharmacol. Ther 69, 103–115. [DOI] [PubMed] [Google Scholar]
- (25). Kaufman PN, Krevsky B, Malmud LS, Maurer AH, Somers MB, Siegel JA, and Fisher RS (1988) Role of opiate receptors in the regulation of colonic transit. Gastroenterology 94, 1351–1356. [DOI] [PubMed] [Google Scholar]
- (26). Chokhavatia S, John ES, Bridgeman MB, and Dixit D (2016) Constipation in elderly patients with noncancer pain: focus on opioid-induced constipation. Drugs Aging 33, 557–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27). Ahmedzai S, and Brooks D (1997) Transdermal fentanyl versussustained-release oral morphine in cancer pain: Preference, efficacy, and quality of life. J. Pain Symptom Manage 13, 254–261. [DOI] [PubMed] [Google Scholar]
- (28). Cheskin LJ, Chamia TN, Johnson RE, and Jaffe JH (1995) Assessment of nalmefene glucuronide as a selective gut opioid antagonist. Drug Alcohol Depend 39, 151–154. [DOI] [PubMed] [Google Scholar]
- (29). Meissner W, Schmidt U, Hartmann M, Kath R, and Reinhart K (2000) Oral naloxone reverses opioid-associated constipation. Pain 84, 105–109. [DOI] [PubMed] [Google Scholar]
- (30). Levio S, and Cash BD (2017) The place of eluxadoline in the management of irritable bowel syndrome with diarrhea. Ther. Adv. Gastroenterol 10, 715–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31). Cashman MD, Martin DK, Dhillon S, and Puli SR (2016) Irritable bowel syndrome: a clinical review. Curr. Rheumatol. Rev 12, 13–26. [DOI] [PubMed] [Google Scholar]
- (32). Cash BD, Lacy BE, Rao T, and Earnest DL (2016) Rifaximin and eluxadoline—newly approved treatments for diarrhea-predominant irritable bowel syndrome: what is their role in clinical practice alongside alosetron? Expert Opin. Pharmacother 17, 311–322. [DOI] [PubMed] [Google Scholar]
- (33). Camilleri M, Northcutt AR, Kong S, Dukes GE, McSorley D, and Mangel AW (2000) Efficacy and safety of alosetron in women with irritable bowel syndrome: a randomised, placebo-controlled trial. Lancet 355, 1035–1040. [DOI] [PubMed] [Google Scholar]
- (34). Pimentel M, Lembo A, Chey WD, Zakko S, Ringel Y, Yu m J., Mareya SM, Shaw AL, Bortey E, and Forbes WP (2011) Rifaximin therapy for patients with irritable bowel syndrome without constipation. N. Engl. J. Med 364, 22–32. [DOI] [PubMed] [Google Scholar]
- (35). Lacy B (2016) Emerging treatments in neurogastroenterology: eluxadoline−a new therapeutic option for diarrhea-predominant IBS. Neurogastroenterol. Motil 28, 26–35. [DOI] [PubMed] [Google Scholar]
- (36). Wade P, Palmer J, McKenney S, Kenigs V, Chevalier K, Moore B, Mabus J, Saunders P, Wallace N, Schneider C, et al. (2012) Modulation of gastrointestinal function by MuDelta, a mixed μ opioid receptor agonist/μ opioid receptor antagonist. Br. J. Pharmacol 167, 1111–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37). Dove LS, Lembo A, Randall CW, Fogel R, Andrae D, Davenport JM, Mcintyre G, Almenoff JS, and Covington PS (2013) Eluxadoline benefits patients with irritable bowel syndrome with diarrhea in a phase 2 study. Gastroenterology 145, 329–338. e1. [DOI] [PubMed] [Google Scholar]
- (38). (a)Lembo AJ, Lacy BE, Zuckerman MJ, Schey R, Dove LS, Andrae DA, Davenport JM, McIntyre G, Lopez R, Turner L, and Covington PS (2016) Eluxadoline for irritable bowel syndrome with diarrhea. N. Engl. J. Med 374, 242–253.(b) Gawron AJ, and Bielefeldt K (2018) Risk of Pancreatitis Following Treatment of Irritable Bowel Syndrome With Eluxadoline. Clin. Gastroenterol. Hepatol 16, 378–384.e2. [DOI] [PubMed] [Google Scholar]
- (39). Li G, Aschenbach LC, Chen J, Cassidy MP, Stevens DL, Gabra BH, Selley DE, Dewey WL, Westkaemper RB, and Zhang Y (2009) Design, synthesis, and biological evaluation of 6α-and 6β-N-heterocyclic substituted naltrexamine derivatives as μ opioid receptor selective antagonists. J. Med. Chem 52, 1416–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40). Yuan Y, Li G, He H, Stevens DL, Kozak P, Scoggins KL, Mitra P, Gerk PM, Selley DE, Dewey WL, and Zhang Y (2011) Characterization of 6α-and 6β-N-heterocyclic substituted naltrexamine derivatives as novel leads to development of mu opioid receptor selective antagonists. ACS Chem. Neurosci 2, 346–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41). Mitra P, Venitz J, Yuan Y, Zhang Y, and Gerk PM (2011) Preclinical disposition (in vitro) of novel μ-opioid receptor selective antagonists. Drug Metab. Dispos 39, 1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42). Yuan Y, Stevens DL, Braithwaite A, Scoggins KL, Bilsky EJ, Akbarali HI, Dewey WL, and Zhang Y (2012) 6β-N-heterocyclic substituted naltrexamine derivative NAP as a potential lead to develop peripheral mu opioid receptor selective antagonists. Bioorg. Med. Chem. Lett 22, 4731–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43). Yuan Y, Elbegdorj O, Chen J, Akubathini SK, Zhang F, Stevens DL, Beletskaya IO, Scoggins KL, Zhang Z, Gerk PM, et al. (2012) Design, synthesis, and biological evaluation of 17-cyclopropylmethyl-3, 14β-dihydroxy-4, 5α-epoxy-6β-[(4′-pyridyl) carboxamido] morphinan derivatives as peripheral selective μ opioid receptor Agents. J. Med. Chem 55, 10118–10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44). Williams DA, Zheng Y, David BG, Yuan Y, Zaidi SA, Stevens DL, Scoggins KL, Selley DE, Dewey WL, Akbarali HI, and Zhang Y (2016) 6β-N-Heterocyclic Substituted Naltrex amine Derivative BNAP: A Peripherally Selective Mixed MOR/KOR Ligand. ACS Chem. Neurosci 7, 1120–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45). Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang X-P, Norval S, Sassano MF, Shin AI, Webster LA, et al. (2012) Automated design of ligands to polypharmacological profiles. Nature 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46). Mittelstadt S, Hemenway C, and Spruell R (2005) Effects of fasting on evaluation of gastrointestinal transit with charcoal meal. J. Pharmacol. Toxicol. Methods 52, 154–158. [DOI] [PubMed] [Google Scholar]
- (47). Mortin LI, Horvath CJ, and Wyand MS (1997) Safety pharmacology screening: Practical problems in drug development. Int. J. Toxicol 16, 41–65. [Google Scholar]
- (48). Akbarali H, Inkisar A, and Dewey W (2014) Site and mechanism of morphine tolerance in the gastrointestinal tract. Neurogastroenterol. Motil 26, 1361–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49). Wood JD, and Galligan J (2004) Function of opioids in the enteric nervous system. Neurogastroenterol. Motil 16, 17–28. [DOI] [PubMed] [Google Scholar]
- (50). Ross GR, Gabra BH, Dewey WL, and Akbarali HI (2008) Morphine tolerance in the mouse ileum and colon. J. Pharmacol. Exp. Ther 327, 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51). Kang M, Maguma HT, Smith TH, Ross GR, Dewey WL, and Akbarali HI (2012) The role of β-arrestin2 in the mechanism of morphine tolerance in the mouse and guinea pig gastrointestinal tract. J. Pharmacol. Exp. Ther 340, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52). Ross GR, Gade AR, Dewey WL, and Akbarali HI (2012) Opioid-induced hypernociception is associated with hyper-excitability and altered tetrodotoxin-resistant Na+ channel function of dorsal root ganglia. Am. J. Physiol. Cell Physiol 302, C1152–C1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53). Bourgeois C, Werfel E, Galla F, Lehmkuhl K, Torres-Gomez H. c., Schepmann D, Kögel B, Christoph T, Straßburger W, Englberger W, et al. (2014) Synthesis and pharmacological evaluation of 5-pyrrolidinylquinoxalines as a novel class of peripherally restricted κ-opioid receptor agonists. J. Med. Chem 57, 6845–6860. [DOI] [PubMed] [Google Scholar]
- (54). Riviere PJM (2004) Peripheral kappa-opioid agonists for visceral pain. Br. J. Pharmacol 141, 1331–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55). Fichna J, Schicho R, Andrews C, Bashashati M, Klompus M, McKay D, Sharkey K, Zjawiony J, Janecka A, and Storr M (2009) Salvinorin A inhibits colonic transit and neurogenic ion transport in mice by activating κ-opioid and cannabinoid receptors. Neurogastroenterol. Motil 21, 1326–e128. [DOI] [PubMed] [Google Scholar]
- (56). Sunahara R, Pardo L, Weis W, Kobilka B, Granier S, et al. (2012) Crystal structure of the mu-opioid receptor bound to a morphinan antagonist. Nature 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57). Wu H, Wacker D, Mileni M, Katritch V, Han GW, Vardy E, Liu W, Thompson AA, Huang X-P, Carroll FI, et al. (2012) Structure of the human κ-opioid receptor in complex with JDTic. Nature 485, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58). Granier S, Manglik A, Kruse AC, Kobilka TS, Thian FS, Weis WI, and Kobilka BK (2012) Structure of the δ-opioid receptor bound to naltrindole. Nature 485, 400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59). Huang W, Manglik A, Venkatakrishnan A, Laeremans T, Feinberg EN, Sanborn AL, Kato HE, Livingston KE, Thorsen TS, Kling RC, Granier S, Gmeiner P, Husbands SM, Traynor JR, Weis WI, Steyaert J, Dror RO, and Kobilka BK (2015) Structural insights into μ-opioid receptor activation. Nature 524, 315–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60). Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, et al. (2018) Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 172, 55–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61). Jones G, Willett P, Glen RC, Leach AR, and Taylor R (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol 267, 727–748. [DOI] [PubMed] [Google Scholar]
- (62). Jones G, Willett P, and Glen RC (1995) Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol 245, 43–53. [DOI] [PubMed] [Google Scholar]
- (63). Zaidi SA, Arnatt CK, He H, Selley DE, Mosier PD, Kellogg GE, and Zhang Y (2013) Binding mode characterization of 6α-and 6β-N-heterocyclic substituted naltrexamine derivatives via docking in opioid receptor crystal structures and site-directed mutagenesis studies: Application of the ‘message−address’ concept in development of mu opioid receptor selective antagonists. Bioorg. Med. Chem 21, 6405–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64). Wang H, Zaidi SA, and Zhang Y (2017) Binding mode analyses of NAP derivatives as mu opioid receptor selective ligands through docking studies and molecular dynamics simulation. Bioorg. Med. Chem 25, 2463–2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65). Cheng HC (2001) The power issue: determination of KB or Ki from IC50. A closer look at the Cheng-Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods 46, 61–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.