

Abstract
Cerebral ischemia is known to activate the repressor element-1 (RE1) silencing transcription factor (REST) which silences neural genes via epigenetic remodeling and promotes neurodegeneration. We presently determined if REST inhibition de-represses target genes involved in synaptic plasticity and promotes functional outcome after experimental stroke. Following transient focal ischemia induced by middle cerebral artery occlusion (MCAO) in adult rats, REST expression was upregulated significantly from 12h to 1 day of reperfusion compared to sham control. At 1 day of reperfusion, REST protein levels were increased and observed in the nuclei of neurons in the peri-infarct cortex. REST knockdown by intracerebral REST siRNA injection significantly reduced the post-ischemic expression of REST and increased the expression of several REST target genes, compared to control siRNA group. REST inhibition also decreased post-ischemic markers of apoptosis, reduced cortical infarct volume and improved post-ischemic functional recovery on days 5 and 7 of reperfusion compared to the control siRNA group. REST knockdown resulted in a global increase in synaptic plasticity gene expression at 1 day of reperfusion compared to the control siRNA group and significantly increased several synaptic plasticity genes containing RE-1 sequences in their regulatory regions. These results demonstrate that direct inhibition of the epigenetic remodeler REST prevents secondary brain damage in the cortex and improves functional outcome potentially via de-repression of plasticity related genes after stroke.
Keywords: Cerebral ischemia, transcription factor, synaptic plasticity, neurodegeneration, BDNF, neuron-restrictive silencing factor
INTRODUCTION
Epigenetic alterations induced following cerebral ischemia play a major role in the regulation of neuronal gene expression and pathophysiological outcome after stroke [1]. Ischemic injury has been shown to activate the transcription factor REST, which represses the expression of many target genes in neural cells through epigenetic remodeling [2–5]. REST binds to the chromatin modifying proteins Sin3A and coREST and this complex binds to RE-1 sequences located in the regulatory regions of target genes involved in various neuronal-specific functions [6]. The REST-Sin3a-coREST repressor complex leads to the subsequent deacetylation of histone proteins and hence inhibition of gene expression [7]. REST also transcriptionally controls the expression of noncoding RNAs, such as microRNAs (miRNAs) and long noncoding RNAs (lncRNAs), representing additional REST-dependent regulatory mechanisms [8,9].
During embryogenesis, REST is essential for controlling neuronal fate by repressing neuronal genes involved in axonal guidance, synaptogenesis and synaptic plasticity [10,11]. In mature neurons, REST levels are low and sequestered in the cytoplasm [12–14]. However, REST levels and nuclear translocation was shown to increase with age [15]. In the adult brain, REST has been shown to regulate neuronal excitability and neurotransmitter release by controlling expression of pre- and post-synaptic proteins as well as genes encoding channels and transporters. For example, REST governs the expression of proteins critical for neurotransmitter receptors, SNARE proteins involved in synaptic vesicle trafficking and neurotrophin signaling [11,14]. While REST is important for neuronal development and maintenance of neuronal physiology, aberrant REST activation leads to neurological dysfunction. Several studies have implicated the involvement of REST in neurodegenerative disorders such as epilepsy, Alzheimer’s disease, Huntington’s disease and Parkinson’s disease [14–18].
In vitro studies from various cell lines have indicated that REST is highly responsive to hypoxic conditions [19–22]. In HEK cells, hypoxia promotes REST accumulation in the nucleus leading to repression of the hypoxic transcriptome [20,21]. In global cerebral ischemia, the epigenetic remodeling induced by the REST complex is critical for mediating neuronal death. Previous studies showed that hippocampal CA1 neurons destined to die express REST after global ischemia and REST knockdown protected CA1 neurons against post-ischemic death [2,3]. The REST complex has been shown to induce ischemic brain damage by decreasing the expression of its downstream genes such as NFk L chain enhancer of activated B cells 2 (NFkB2), glutamate receptor, ionotropic, N-methyl d-aspartate 1 (GRIN1), and AMPA receptor subunit GluR2 (GRIA2) [2–4].
Our laboratory and others have shown that focal ischemia induces the expression of REST and its corepressors [4,23,24]. We also showed that a IncRNA called FosDT induced after stroke plays an essential role in scaffolding REST, coREST and Sin3A and inhibition of FosDT, leading to a disruption of REST scaffolding, prevented secondary brain injury after focal ischem ia [4]. However, as the direct role of REST in the post-ischemic cerebral cortex has not been assessed, we presently evaluated the effect of REST knockdown on de-repression of synaptic proteins and functional outcome after focal ischemia in the adult rat brain.
MATERIALS AND METHODS
Focal ischemia.
All surgical procedures were approved by the Research Animal Resources and Care Committee of the University of Wisconsin-Madison, and the rats were cared for in accordance with the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services Publication 86–23, revised). One hour of transient MCAO was induced in adult, male spontaneously hypertensive (SHR) rats (280–300g; Charles River Laboratories) under isoflurane anesthesia by an intraluminal suture method using a 6–0 silicon-coated monofilament (Doccol Corporation) as described previously [4,25]. Rectal temperature was maintained at 37.0 ± 0.5°C during surgery. Physiological parameters (pH, PaO2, PaCO2, hemoglobin, and blood glucose) and regional cerebral blood flow were monitored and used to determine inclusion in the study as described previously [22,25,26]. Cohorts of rats were euthanized at 12h, 1 day, 3 days or 7 days of reperfusion.
REST knockdown.
REST was knocked down with a cocktail of three in vivo grade siRNAs targeting non-overlapping regions of REST (ThermoFisher Scientific). A non-targeting negative control siRNA was used as control. The siRNA cocktail (8nmol in 3.2μΙ buffer + 0.8μΙ invivofectamine) was injected intracerebrally into the cerebral cortex with a Hamilton syringe (from bregma −0.2 mm posterior, 3 mm dorsoventral and 4.5 mm lateral) (0.5μΙ/min) 12h prior to transient MCAO as described previously [4,22].
Real-time PCR.
RNA was extracted from the peri-infarct region of the cortex using an AllPrep DNA/RNA mini kit (Qiagen) and reverse transcribed into cDNA with the Reverse Transcription System (Promega). Rat REST (NM_031788.1), Scg2 (NM_022669.1), Grin1 (NM_017010.2), Nefh (NM_012607.2), GRIA2 (NM_017261.2), Nppa (NM_012612.2), and NFkB2 (NM_001008349.1) were evaluated with real-time PCR (RT-PCR) by SYBR Green method using 18S rRNA and GAPDH mRNA as internal controls as described previously [4].
Western Blotting.
Protein samples from peri-infarct cortical tissue were subjected to electrophoresis, transferred to nitrocellulose membranes, blocked with 5% BSA in a tris-buffered saline with 0.1% Trizol, and then probed with antibodies against REST (1:500; Millipore) followed by HRP-conjugated anti-rabbit IgG secondary antibody (1:5000; Cell Signaling Technology). Blots were re-probed with ß-actin (Cell Signaling Technology) followed by HRP-conjugated anti-mouse secondary antibody (1:5000; Cell Signaling Technology). Enhanced chemiluminescence (Life Technologies) was used to develop blots, which were quantified with Image Studio software (LI-COR Biotechnology).
Immunohistochemistry.
Rats were euthanized on day 1 or day 3 of reperfusion by transcardiac 4% paraformaldehyde (PFA) perfusion fixation. Brains were post-fixed in 4% PFA, cryoprotected and sectioned (coronal: 40μm thickness). Brain sections were then immunostained with primary antibodies against REST (1:300; Proteintech), NeuN (1:300; Millipore), cleaved caspase-3 (1:400; Cell Signaling), and phosphorylated dynamin-related protein 1 (Drp1; 1:300; Cell Signaling) followed by donkey Alexa Fluor 488 or Alexa Fluor 594 secondary antibodies (1:300; Invitrogen).
RT-PCR Array.
RNA was extracted from the peri-infarct area of the ipsilateral cortex at 24h of reperfusion using an AllPrep DNA/RNA mini kit (Qiagen). From each sample, 1μg of RNA was reverse transcribed into cDNA using the RT2 First Strand Kit (Qiagen). Samples were probed using RT2 Profiler PCR array for Synaptic Plasticity (Qiagen) as per manufacturer instructions. Briefly, cDNA samples and RT2 SYBR Green/ROX PCR master mix (Qiagen) were applied to the PCR array plates containing primers and cycled using the QuantStudio 3 platform (ThermoFisher Scientific). Data was analyzed using the Qiagen Data Analysis Center.
Motor function tests.
Rats were trained for 3 days prior to MCAO in the rotarod test and adhesive removal test and then tested on days 1, 3, 5, and 7 days of reperfusion as described previously [25]. Briefly, motor coordination and learning were assessed using rotarod test by measuring the latency of time to fall from a rotating cylinder at 8 RPM. In the adhesive removal test, time to remove a small adhesive tape placed on the forepaw was measured.
Infarct Volume Estimation.
Serial brain sections (coronal: 40μm thickness) from each rat euthanized on day 7 of reperfusion by transcardiac perfusion fixation were stained with cresyl violet and used to measure infarct volume using NIH Image J software as described previously [25].
Statistics.
Mann Whitney U test and two-way ANOVA with Sidak’s post-hoc test were used to compare differences between 2 groups or multiple comparisons between two groups, respectively. Values shown are mean ± SEM and p <0.05 was used for significance cut-off. An investigator blinded to the study groups performed the behavioral and histological analyses.
RESULTS
Transient MCAO induced REST expression in the peri-infarct area of the cortex.
As the peri-infarct area is the region of potential recovery after stroke, we estimated REST mRNA and protein expression in that area. REST mRNA expression was significantly induced at 12h and 1 day of reperfusion following transient MCAO (by 3.4 to 4.6 fold, p<0.05) compared to sham (Fig. 1a). REST protein levels were also significantly increased at 1 day of reperfusion (by 2 fold, p<0.05) compared to sham (Fig. 1b). Immunohistochemical analysis of DAPI (nuclear stain) and NeuN (mature neuronal nuclear marker) showed that at 1 day of reperfusion, REST was localized in the neuronal cells and translocated into the neuronal nuclei (Fig. 1c).
Fig. 1. Focal ischemia induced REST expression in the peri-infarct cortex.
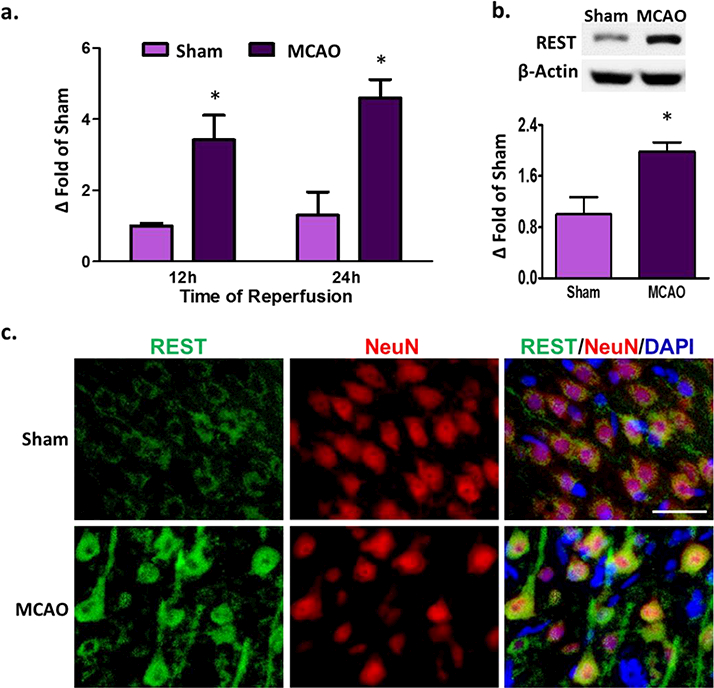
Real-time PCR analysis showed increased REST mRNA expression at 12h and 24h of reperfusion compared to sham control (n= 3 to 4/group) (a). Western blot analysis showed increased REST protein levels at 24h of reperfusion compared to sham (n= 5 to 6/group) (b). Increased REST protein levels were observed to be localized in the neurons at 24h of reperfusion (n=3/group). Scale bar = 30 μm (c). Values are mean ± SEM. *p<0.05 versus sham.
REST knockdown de-repressed the expression of REST-responsive neuroprotective genes.
To assess the role of REST on pathophysiological mechanisms after experimental stroke, we knocked-down REST in the ipsilateral cerebral cortex with an siRNA cocktail. REST siRNA treatment given 12h prior to MCAO significantly prevented the post-ischemic REST mRNA expression (by 39%; p <0.05) at 1 day of reperfusion compared to the control siRNA group (Fig. 2a). REST siRNA treated rats also showed curtailed neuronal post-ischemic REST protein expression at 3 days of reperfusion as assessed by immunohistochemical staining (Fig. 2b). To test the functional effectiveness of REST siRNA treatment, we examined the post-ischemic expression of genes that have previously been shown to display REST enrichment in their promoters [3]. REST knockdown increased the post-ischemic expression of secretogranin II (Scg2; by 140%, p<0.05), Grin1 (by 286%; p <0 .05), neurofilament heavy polypeptide (Nefh; by 250%, p<0.05), GRIA2 (by 130%, p < 0.05), natriuretic peptide A (Nppa; by 180%, p<0.05) and NFkB2 (by 90%, p<0.05), compared with control siRNA group at 1 day of reperfusion following transient MCAO (Fig. 2c). This indicates that REST knockdown leads to de-repression of its target genes. Interestingly, many of these REST target genes (Scg2, Grin1, Nefh, Gria2 and Nppa) have been associated with neuroprotection against ischemia-induced neuronal death [3,4,27].
Fig. 2. REST knockdown de-repressed REST-responsive genes after focal ischemia.
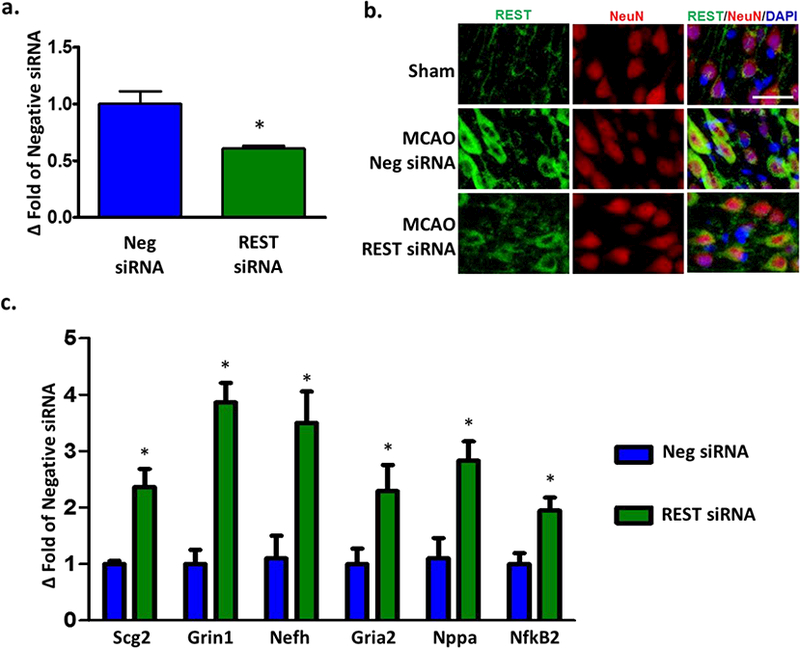
Cortical REST expression at 24h of reperfusion following transient MCAO in siRNA and negative control siRNA groups (n=3/group) (a). Immunohistochemical staining showed co-localization of REST (green) with NeuN (red) and DAPI (blue) at 3 days of reperfusion (n=3/group). Scale bar = 30 μm (b). Real-time PCR of REST target genes at 24h of reperfusion (n=3/group) (c). Values are mean ± SEM. *p<0.05 versus negative siRNA.
REST knockdown decreased apoptosis, infarct volume and improved motor function recovery.
Since REST inhibition led to de-repression of neuroprotective genes, we next examined the effect of REST knockdown on cell death in the peri-infarct cortex after transient MCAO. Immunohistochemical analyses showed decreased protein levels of cleaved caspase-3 (marker for apoptosis) and phosphorylated DRPi (marker for mitochondrial fission) in NeuN+ neurons (Fig. 3a and b) at 3 days of reperfusion following transient MCAO. Rats treated with REST siRNA also had significantly less cortical degeneration compared with control siRNA group at day 7 of reperfusion as assessed by infarct volume (by 26%; p<0 .05; Fig. 4a and 4b). To determine whether the decreased cell death correlated with improved functional recovery, we next tested whether REST knockdown regulates motor function recovery after experimental stroke. The REST siRNA group showed significantly ameliorated post-ischemic motor dysfunction compared with the control siRNA group on days 5 and 7 of reperfusion as measured by rotarod test and adhesive removal test (Fig. 4c and 4d).
Fig. 3. REST knockdown decreased apoptosis and mitochondrial damage after focal ischemia.
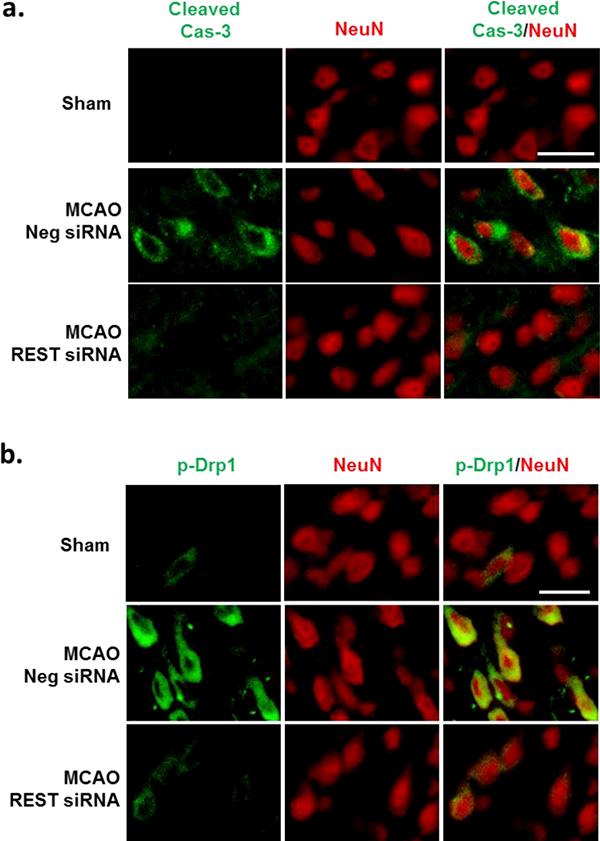
Immunohistochemical staining with NeuN (red) and the apoptosis markers cleaved caspase-3 (Cas-3) (green) and phosphorylated DRP1 (p-DRP1) (green) at 3 days of reperfusion (a and b). MCAO-induced increase in cleaved Cas-3 and p-Drp1 fluorescence was mitigated with REST siRNA treatment (n=3/group). Scale bar = 30μm.
Fig. 4. REST knockdown decreased cortical infarct volume and improved functional recovery after focal ischemia.
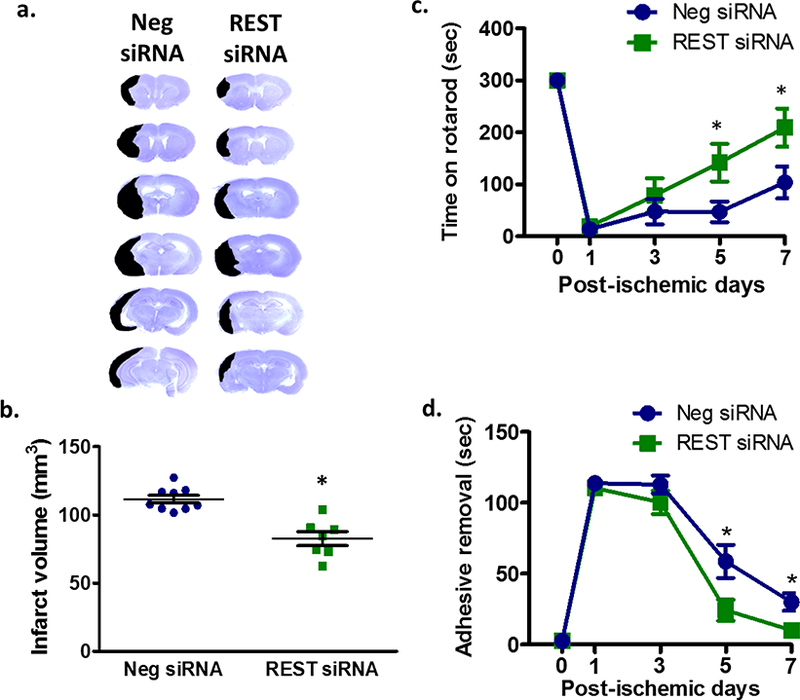
Cresyl violet-stained serial sections from representative rats from the REST siRNA and control siRNA groups at 7 days of reperfusion (a). Infarct quantification of cresyl violet-stained sections (n= 7 to 9/group) (b). Motor learning and coordination performed up to 7 days of reperfusion were assessed by measuring latency to time to stay on rotarod (c) and to remove an adhesive patch from forepaws (d). Values are mean ± SEM. *p < 0.05, compared with respective reperfusion time point.
REST knockdown de-repressed synaptic plasticity-related gene expression
The maintenance and strengthening of synaptic plasticity plays an integral role in improving functional recovery after stroke [28,29]. Previously, REST has been shown to repress a large number of neuronal genes involved in synaptic plasticity [10,14]. Therefore, we determined whether REST inhibition modulates the expression of synaptic plasticity-related genes following transient MCAO. Using real-time PCR arrays, we evaluated the expression of 84 genes involved in various aspects of synaptic plasticity. At 24h of reperfusion following transient MCAO, the REST siRNA group showed significantly enhanced expression of 21 synaptic plasticity-related genes compared to the control siRNA group (Fig. 5a-d). Several of these upregulated genes contained the RE-1 binding motif in their promoters (Fig. 5d). These genes are known to be involved in various aspects of synaptic plasticity including long-term potentiation (LTP), synaptic structure, and neuronal receptors involved in synaptic transmission (Table 1).
Fig. 5. REST knockdown increased the expression of synaptic plasticity-related genes.
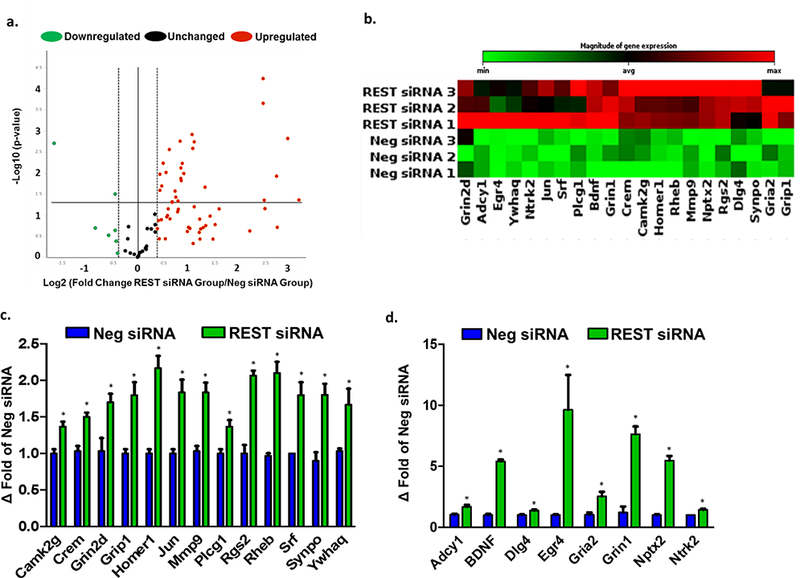
Changes in synaptic plasticity-related gene expression after focal ischemia in REST siRNA group normalized to negative control siRNA group. Volcano plot identifying significantly changed genes (a). Clustergram shows heat map of significantly increased genes (b). Histogram of significantly upregulated genes (c). Histogram showing significantly upregulated genes containing RE-1 sequences (d). Values are mean ± SEM. *p<0.05 versus control siRNA group (n=3/group).
Table 1.
Synaptic Plasticity Genes Significantly Upregulated After Focal Ischemia with REST Inhibition
| Class | Gene | RE-1 Binding Site | Fold change |
|---|---|---|---|
| Immediate-Early Response Genes (lEGs) | Early growth response 4 (Egr4) | yes | 9.6 |
| Neuronal pentraxin 2 (Nptx2) | yes | 5.5 | |
| Homer scaffolding protein 1 (Homerl) | - | 2.2 | |
| Ras homolog enriched in brain (Rheb) | - | 2.1 | |
| Regulator of G-protein signaling 2 (Rgs2) | - | 2.1 | |
| Jun proto-oncogene (Jun) | - | 1.8 | |
| Serum response factor (Srf) | - | 1.8 | |
| cAMP response element modulator (Crem) | - | 1.5 | |
| Long Term Potentiation (LTP) | Brain derived neurotrophic factor (Bdnf) | yes | 5.4 |
| Tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein theta (Ywhaq) | - | 1.7 | |
| Adenylate cyclase 1 (Adcyl) | yes | 1.7 | |
| Calcium/calmodulin-dependant protein kinase II gamma (Camk2g) | - | 1.4 | |
| Phospholipase C gamma 1 (Plcgl) | - | 1.4 | |
| Long Term Depression (LTD) | Glutamate receptor interacting protein 1 (Gripl) | - | 1.8 |
| Extracellular Matrix (ECM) Molecules | Matrix metallopeptidase 9 (Mmp9) | - | 1.8 |
| Neuronal Receptors | Glutamate ionotropic receptor NMDA type subunit 1 (Grinl) | yes | 7.6 |
| Glutamate ionotropic receptor AMPAtype subunit 2 (Gria2) | yes | 2.5 | |
| Glutamate ionotropic receptor NMDA type subunit 2d (Grin2d) | - | 1.7 | |
| Neurotrophic receptor tyrosine kinase 2 (Ntrk2) | yes | 1.4 | |
| Postsynaptic Density (PSD) | Synaptopodin (Synpo) | - | 1.8 |
| Discs large homolog 4 (Dlg4) | yes | 1.4 | |
All values shown are mean of n = 3/group. All fold changes are statistically significant between the REST siRNA and control siRNA groups.
DISCUSSION
REST has previously been shown to play a role in several neurological disorders, but the direct role of REST in the post-ischemic cortex had previously not been assessed. In the present study, we show that REST is induced in the cerebral cortex and its inhibition leads to de-repression of genes associated with neuronal survival after focal ischemia. Our results also demonstrate that REST plays a critical role in mediating cortical tissue damage after focal ischemia. Furthermore, we show for the first time that REST inhibition not only prevents neurodegeneration, but also modulates wide-scale changes in synaptic plasticity genes and improves functional outcome after ischemic injury.
Genomic studies have identified >2,000 putative REST target genes in the mammalian genome containing RE-1 sites [30–32]. In the brain, REST functions as a master regulator of neuronal physiology by governing genes involved in neurogenesis, synaptic output, and neuronal survival [11,14]. However, REST has been shown to regulate transcriptional networks in a highly selective manner in response to various stressors and changes in cellular environment. In fact, genome-wide sequencing analyses showed that differential REST interactions with individual RE-1 sites can be cell-type specific as well as context-dependent within particular cell types [21,32,33]. Indeed, this makes the role of REST in disease processes quite complex as REST has been associated with both protection and dysfunction depending on the disease model. For example, in neurotoxin-induced Parkinsonian phenotypes, REST had deleterious effects in a dopaminergic cell line, whereas REST knockout mice displayed exacerbated dopaminergic neuronal loss [34,35]. Additionally, accumulation of nuclear REST is associated with Huntington’s disease [12], while cytoplasmic sequestration of REST has been observed in an Alzheimer’s disease model [15]. Studies in the brain and various cell lines have consistently shown an induction of REST following hypoxia or cerebral ischemia, which has also been associated with worsening of injury in neuronal cells [2– 4,19–22]. Concordantly, our current study indicates that focal ischemia induces REST in neurons of the cortical penumbra, which promotes cortical injury and motor dysfunction. Importantly, this can be used to develop stroke therapies as REST knockdown protected the brain and promoted functional recovery after ischemia.
Following ischemic injury, REST has been shown to bind to promoters and inhibit the expression of a number of genes involved in excitotoxicity and neuronal stress response [3]. We found that REST knockdown de-repressed several genes (such as Scg2, GRIA2, Nppa and NFkB2) that protect brain in ischemic conditions. Scg2 is a secretory protein found in large dense core vesicle of neurons that secretes secretoneurin, a neuropeptide shown to protect the brain against stroke injury by preventing apoptosis and promoting angiogenesis [27,36]. GRIA2 encodes the GluR2 subunit of AMPA-specific glutamate receptors that prevent Ca2+ entry into neuronal cells. Downregulation of GRIA2 increases Ca2+ permeability through AMPA receptors thereby promoting excitotoxicity and neuronal death after ischemia [37]. Nppa (also known as atrial natriuretic peptide) is a peptide hormone that has been shown to reduce brain edema after ischemic brain injury [38]. NFkB2 gene encodes the NF-kB2 p100/p52 protein, which has been shown to reduce inflammation and cell death after cerebral ischemia by preventing nuclear translocation of NF-κΒ (RelA/p65) [39]. De-repression of these REST-targeted genes by REST knockdown may contribute to the neuroprotection observed in the cortex after ischemic injury.
The penumbra, which consists of the peri-infarct region, is damaged during the ischemic phase of stroke, but has the ability to recover during reperfusion. Synaptic plasticity mechanisms represent an important physiological process critical for re-wiring and attaining lost function after stroke [28]. REST has been shown to play a critical role in fine-tuning synaptic plasticity-related gene expression both during development and in the postnatal brain [10,14]. Here, we show that REST inhibition led to significant increases in several genes involved in various aspects of both structural and functional brain plasticity, majorly LTP-related and lEGs [40–42]. Of particular note is the de-repression of BDNF, which has a well-established role in promoting post-ischemic neuronal survival and functional recovery after stroke [43–45]. BDNF modulates key aspects of synaptic transmission through its interaction with TrkB and p75 receptors that induce various downstream intracellular signaling cascades [46]. BDNF activation of various pathways such as protein kinase C (PKC) and mitogen activated protein kinase/extracellular signal-regulated kinases (MAPK/ERK) signaling is likely responsible for the observed increase in several synaptic plasticity genes lacking the RE-1 motif following REST knockdown. For example, BDNF has been shown to induce expression of Camk2g [47], Grip1 [48], Homer 1 [49], Jun [50], and MMP9 [51] and is responsible for activation of Srf and Rheb [51,52].
In conclusion, our study shows that REST is a promoter of post-stroke brain damage and its knockdown prevents neuronal death after cerebral ischemic injury. Mechanistically, modulation of several key genes involved in synaptic plasticity by REST may account for the robust changes in motor function following focal ischemia. Thus, the current results show that the epigenetic regulator REST is a potential therapeutic target for stroke.
Acknowledgments
Acknowledgements: This study was funded by National Institute of Health grants R21NS095192, RO1 NS099531 and RO1 NS101960.
REFERENCES
- 1.Hu Z, Zhong B, Tan J, Chen C, Lei Q, Zeng L (2016) The Emerging Role of Epigenetics in Cerebral Ischemia. Mol Neurobiol. doi: 10.1007/s12035-016-9788-3 [DOI] [PubMed] [Google Scholar]
- 2.Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS (2003) Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci 23 (6):2112–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS (2012) Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proceedings of the National Academy of Sciences of the United States of America 109 (16):E962–971. doi: 10.1073/pnas.1121568109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta SL, Kim T, Vemuganti R (2015) Long Noncoding RNA FosDT Promotes Ischemic Brain Injury by Interacting with REST-Associated Chromatin-Modifying Proteins. J Neurosci 35 (50):16443–16449. doi: 10.1523/jneurosci.2943-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Z, Liu M, Niu G, Cheng Y, Fei J (2009) Genome-wide identification of target genes repressed by the zinc finger transcription factor REST/NRSF in the HEK 293 cell line. Acta Biochim Biophys Sin (Shanghai) 41 (12):1008–1017 [DOI] [PubMed] [Google Scholar]
- 6.Schoenherr CJ, Paquette AJ, Anderson DJ (1996) Identification of potential target genes for the neuron-restrictive silencer factor. Proc Natl Acad Sci U S A 93 (18):9881–9886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang Y, Myers SJ, Dingledine R (1999) Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci 2 (10):867–872. doi: 10.1038/13165 [DOI] [PubMed] [Google Scholar]
- 8.Qureshi IA, Mehler MF (2009) Regulation of non-coding RNA networks in the nervous system--what’s the REST of the story? Neurosci Lett 466 (2):73–80. doi: 10.1016/j.neulet.2009.07.093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J, Xie X (2006) Comparative sequence analysis reveals an intricate network among REST, CREB and miRNA in mediating neuronal gene expression. Genome Biol 7 (9):R85. doi: 10.1186/gb-2006-7-9-r85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G (2005) REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 121 (4):645–657. doi: 10.1016/j.cell.2005.03.013 [DOI] [PubMed] [Google Scholar]
- 11.Abrajano JJ, Qureshi IA, Gokhan S, Molero AE, Zheng D, Bergman A, Mehler MF (2010) Corepressor for element-1-silencing transcription factor preferentially mediates gene networks underlying neural stem cell fate decisions. Proc Natl Acad Sci U S A 107 (38):16685–16690. doi: 10.1073/pnas.0906917107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuccato C, Tartari M, Crotti A, Goffredo D, Valenza M, Conti L, Cataudella T, Leavitt BR, Hayden MR, Timmusk T, Rigamonti D, Cattaneo E (2003) Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet 35 (1):76–83. doi: 10.1038/ng1219 [DOI] [PubMed] [Google Scholar]
- 13.Paquette AJ, Perez SE, Anderson DJ (2000) Constitutive expression of the neuron-restrictive silencer factor (NRSF)/REST in differentiating neurons disrupts neuronal gene expression and causes axon pathfinding errors in vivo. Proc Natl Acad Sci U S A 97 (22):12318–12323. doi: 10.1073/pnas.97.22.12318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baldelli P, Meldolesi J (2015) The Transcription Repressor REST in Adult Neurons: Physiology, Pathology, and Diseases(1,2,3). eNeuro 2 (4). doi: 10.1523/eneuro.0010-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiacovo MP, Yankner BA (2014) REST and stress resistance in ageing and Alzheimer’s disease. Nature 507 (7493):448–454. doi: 10.1038/nature13163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zuccato C, Belyaev N, Conforti P, Ooi L, Tartari M, Papadimou E, MacDonald M, Fossale E, Zeitlin S, Buckley N, Cattaneo E (2007) Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J Neurosci 27 (26):6972–6983. doi: 10.1523/jneurosci.4278-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ooi L, Wood IC (2007) Chromatin crosstalk in development and disease: lessons from REST. Nat Rev Genet 8 (7):544–554. doi: 10.1038/nrg2100 [DOI] [PubMed] [Google Scholar]
- 18.McClelland S, Flynn C, Dube C, Richichi C, Zha Q, Ghestem A, Esclapez M, Bernard C, Baram TZ (2011) Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol 70 (3):454–464. doi: 10.1002/ana.22479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lin TP, Chang YT, Lee SY, Campbell M, Wang TC, Shen SH, Chung HJ, Chang YH, Chiu AW, Pan CC, Lin CH, Chu CY, Kung HJ, Cheng CY, Chang PC (2016) REST reduction is essential for hypoxia-induced neuroendocrine differentiation of prostate cancer cells by activating autophagy signaling. Oncotarget 7 (18):26137–26151. doi: 10.18632/oncotarget.8433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Scholz CC, Cummins EP, Cheong A, Taylor CT (2015) REST mediates resolution of HIF-dependent gene expression in prolonged hypoxia. Sci Rep 5:17851. doi: 10.1038/srep17851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Keogh CE, Fabian Z, Scholz CC, Nolan KA, Rocha LM, Tambuwala MM, Brown S, Wdowicz A, Corbett D, Murphy KJ, Godson C, Cummins EP, Taylor CT, Cheong A (2016) REST is a hypoxia-responsive transcriptional repressor. Sci Rep 6:31355. doi: 10.1038/srep31355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pandi G, Nakka VP, Dharap A, Roopra A, Vemuganti R (2013) MicroRNA miR-29c down-regulation leading to de-repression of its target DNA methyltransferase 3a promotes ischemic brain damage. PLoS One 8 (3):e58039. doi: 10.1371/journal.pone.0058039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Formisano L, Guida N, Valsecchi V, Cantile M, Cuomo O, Vinciguerra A, Laudati G, Pignataro G, Sirabella R, Di Renzo G, Annunziato L (2015) Sp3/REST/HDAC1/HDAC2 Complex Represses and Sp1/HIF-1/p300 Complex Activates ncx1 Gene Transcription, in Brain Ischemia and in Ischemic Brain Preconditioning, by Epigenetic Mechanism. J Neurosci 35 (19):7332–7348. doi: 10.1523/jneurosci.2174-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Formisano L, Guida N, Valsecchi V, Pignataro G, Vinciguerra A, Pannaccione A, Secondo A, Boscia F, Molinaro P, Sisalli MJ, Sirabella R, Casamassa A, Canzoniero LM, Di Renzo G, Annunziato L (2013) NCX1 is a new rest target gene: role in cerebral ischemia. Neurobiol Dis 50:76–85. doi: 10.1016/j.nbd.2012.10.010 [DOI] [PubMed] [Google Scholar]
- 25.Nakka VP, Lang BT, Lenschow DJ, Zhang DE, Dempsey RJ, Vemuganti R (2011) Increased cerebral protein ISGylation after focal ischemia is neuroprotective. J Cereb Blood Flow Metab 31 (12):2375–2384. doi: 10.1038/jcbfm.2011.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mehta SL, Pandi G, Vemuganti R (2017) Circular RNA Expression Profiles Alter Significantly in Mouse Brain After Transient Focal Ischemia. Stroke 48 (9):2541–2548. doi: 10.1161/strokeaha.117.017469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shyu WC, Lin SZ, Chiang MF, Chen DC, Su CY, Wang HJ, Liu RS, Tsai CH, Li H (2008) Secretoneurin promotes neuroprotection and neuronal plasticity via the Jak2/Stat3 pathway in murine models of stroke. J Clin Invest 118 (1):133–148. doi: 10.1172/jci32723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy TH, Corbett D (2009) Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci 10 (12):861–872. doi: 10.1038/nrn2735 [DOI] [PubMed] [Google Scholar]
- 29.Pekna M, Pekny M, Nilsson M (2012) Modulation of neural plasticity as a basis for stroke rehabilitation. Stroke 43 (10):2819–2828. doi: 10.1161/strokeaha.112.654228 [DOI] [PubMed] [Google Scholar]
- 30.Johnson DS, Mortazavi A, Myers RM, Wold B (2007) Genome-wide mapping of in vivo protein-DNA interactions. Science 316 (5830):1497–1502. doi: 10.1126/science.1141319 [DOI] [PubMed] [Google Scholar]
- 31.Mortazavi A, Leeper Thompson EC, Garcia ST, Myers RM, Wold B (2006) Comparative genomics modeling of the NrSf/REST repressor network: from single conserved sites to genome-wide repertoire. Genome Res 16 (10):1208–1221. doi: 10.1101/gr.4997306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ (2004) Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A 101 (28):10458–10463. doi: 10.1073/pnas.0401827101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson R, Teh CH, Kunarso G, Wong KY, Srinivasan G, Cooper ML, Volta M, Chan SS, Lipovich L, Pollard SM, Karuturi RK, Wei CL, Buckley NJ, Stanton LW (2008) REST regulates distinct transcriptional networks in embryonic and neural stem cells. PLoS Biol 6 (10):e256. doi: 10.1371/journal.pbio.0060256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu M, Cai L, Liang M, Huang Y, Gao H, Lu S, Fei J, Huang F (2009) Alteration of NRSF expression exacerbating 1-methyl-4-phenyl-pyridinium ion-induced cell death of SH-SY5Y cells. Neurosci Res 65 (3):236–244. doi: 10.1016/j.neures.2009.07.006 [DOI] [PubMed] [Google Scholar]
- 35.Yu M, Suo H, Liu M, Cai L, Liu J, Huang Y, Xu J, Wang Y, Zhu C, Fei J, Huang F (2013) NRSF/REST neuronal deficient mice are more vulnerable to the neurotoxin MPTP. Neurobiol Aging 34 (3):916–927. doi: 10.1016/j.neurobiolaging.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 36.Posod A, Wechselberger K, Stanika RI, Obermair GJ, Wegleiter K, Huber E, Urbanek M, Kiechl-Kohlendorfer U, Griesmaier E (2017) Administration of secretoneurin is protective in hypoxic-ischemic neonatal brain injury predominantly in the hypoxic-only hemisphere. Neuroscience 352:88–96. doi: 10.1016/j.neuroscience.2017.03.055 [DOI] [PubMed] [Google Scholar]
- 37.Oguro K, Oguro N, Kojima T, Grooms SY, Calderone A, Zheng X, Bennett MV, Zukin RS (1999) Knockdown of AMPA receptor GluR2 expression causes delayed neurodegeneration and increases damage by sublethal ischemia in hippocampal CA1 and CA3 neurons. J Neurosci 19 (21):9218–9227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naruse S, Aoki Y, Takei R, Horikawa Y, Ueda S (1991) Effects of atrial natriuretic peptide on ischemic brain edema in rats evaluated by proton magnetic resonance method. Stroke 22 (1):61–65 [DOI] [PubMed] [Google Scholar]
- 39.Schneider A, Martin-Villalba A, Weih F, Vogel J, Wirth T, Schwaninger M (1999) NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat Med 5 (5):554–559. doi: 10.1038/8432 [DOI] [PubMed] [Google Scholar]
- 40.Lenz M, Vlachos A, Maggio N (2015) Ischemic long-term-potentiation (iLTP): perspectives to set the threshold of neural plasticity toward therapy. Neural Regen Res 10 (10):1537–1539. doi: 10.4103/1673-5374.165215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minatohara K, Akiyoshi M, Okuno H (2015) Role of Immediate-Early Genes in Synaptic Plasticity and Neuronal Ensembles Underlying the Memory Trace. Front Mol Neurosci 8:78. doi: 10.3389/fnmol.2015.00078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lanahan A, Worley P (1998) Immediate-early genes and synaptic function. Neurobiol Learn Mem 70 (1–2):37–43. doi: 10.1006/nlme.1998.3836 [DOI] [PubMed] [Google Scholar]
- 43.Schabitz WR, Berger C, Kollmar R, Seitz M, Tanay E, Kiessling M, Schwab S, Sommer C (2004) Effect of brain-derived neurotrophic factor treatment and forced arm use on functional motor recovery after small cortical ischemia. Stroke 35 (4):992–997. doi: 10.1161/01.str.0000119754.85848.0d [DOI] [PubMed] [Google Scholar]
- 44.Schabitz WR, Sommer C, Zoder W, Kiessling M, Schwaninger M, Schwab S (2000) Intravenous brain-derived neurotrophic factor reduces infarct size and counterregulates Bax and Bcl-2 expression after temporary focal cerebral ischemia. Stroke 31 (9):2212–2217 [DOI] [PubMed] [Google Scholar]
- 45.Berretta A, Tzeng YC, Clarkson AN (2014) Post-stroke recovery: the role of activity-dependent release of brain-derived neurotrophic factor. Expert Rev Neurother 14 (11):1335–1344. doi: 10.1586/14737175.2014.969242 [DOI] [PubMed] [Google Scholar]
- 46.Lu B, Nagappan G, Lu Y (2014) BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol 220:223–250. doi: 10.1007/978-3-642-45106-5_9 [DOI] [PubMed] [Google Scholar]
- 47.Glorioso C, Sabatini M, Unger T, Hashimoto T, Monteggia LM, Lewis DA, Mirnics K (2006) Specificity and timing of neocortical transcriptome changes in response to BDNF gene ablation during embryogenesis or adulthood. Mol Psychiatry 11 (7):633–648. doi: 10.1038/sj.mp.4001835 [DOI] [PubMed] [Google Scholar]
- 48.Jourdi H, Kabbaj M (2013) Acute BDNF treatment upregulates GluR1-SAP97 and GluR2-GRIP1 interactions: implications for sustained AMPA receptor expression. PLoS One 8 (2):e57124. doi: 10.1371/journal.pone.0057124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sato M, Suzuki K, Nakanishi S (2001) NMDA receptor stimulation and brain-derived neurotrophic factor upregulate homer 1a mRNA via the mitogen-activated protein kinase cascade in cultured cerebellar granule cells. J Neurosci 21 (11):3797–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu CL, Yin JH, Hwang CS, Chen SD, Yang DY, Yang DI (2012) c-Jun-dependent sulfiredoxin induction mediates BDNF protection against mitochondrial inhibition in rat cortical neurons. Neurobiol Dis 46 (2):450–462. doi: 10.1016/j.nbd.2012.02.010 [DOI] [PubMed] [Google Scholar]
- 51.Kuzniewska B, Rejmak E, Malik AR, Jaworski J, Kaczmarek L, Kalita K (2013) Brain-derived neurotrophic factor induces matrix metalloproteinase 9 expression in neurons via the serum response factor/c-Fos pathway. Mol Cell Biol 33 (11):2149–2162. doi: 10.1128/mcb.00008-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Takei N, Inamura N, Kawamura M, Namba H, Hara K, Yonezawa K, Nawa H (2004) Brain-derived neurotrophic factor induces mammalian target of rapamycin-dependent local activation of translation machinery and protein synthesis in neuronal dendrites. J Neurosci 24 (44):9760–9769. doi: 10.1523/jneurosci.1427-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]