

Abstract
The role of human endogenous retroviruses (HERVs) in disease pathogenesis is unclear. We show that HERV-K is activated in a subpopulation of patients with sporadic amyotrophic lateral sclerosis (ALS) and that its envelope (env) protein may contribute to neurodegeneration. The virus was expressed in cortical and spinal neurons of ALS patients, but not in neurons from control healthy individuals. Expression of HERV-K or its env protein in human neurons caused retraction and beading of neurites. Transgenic animals expressing the env gene developed progressive motor dysfunction accompanied by selective loss of volume of the motor cortex, decreased synaptic activity in pyramidal neurons, dendritic spine abnormalities, nucleolar dysfunction, and DNA damage. Injury to anterior horn cells in the spinal cord was manifested by muscle atrophy and pathological changes consistent with nerve fiber denervation and reinnervation. Expression of HERV-K was regulated by TAR (trans-activation responsive) DNA binding protein 43, which binds to the long terminal repeat region of the virus. Thus, HERV-K expression within neurons of patients with ALS may contribute to neurodegeneration and disease pathogenesis.
INTRODUCTION
Human endogenous retroviruses (HERVs) constitute nearly 8% of the human genome and have been termed junk DNA (1). These retroviral sequences are remnants of infections that occurred over several million years, resulting in the integration of provirus genomes into the DNA of germline cells. Most HERV proviruses have accumulated numerous nonsense mutations that have rendered them defective (1). However, it is becoming increasingly apparent that endogenous retroviral sequences may get expressed under select pathological circumstances. Multiple complete sequences of the most recently acquired HERV-K are present in the human genome (2). HERV-K maybe expressed in the brain of patients with amyotrophic lateral sclerosis (ALS) (3) and reverse transcriptase activity can be found in the blood and brain tissue of these patients (4–8), but the role of HERV-K in the pathophysiology of this disease remains unknown. ALS is a progressive neurodegenerative disease and is universally fatal, except in some patients with HIV infection where an ALS-like syndrome can be reversed by antiretroviral drugs (9). However, an extensive search for exogenous retroviruses in ALS has not been successful (5). Here, we explored whether endogenous retroviral elements could be expressed in these patients and whether HERV-K could contribute to the pathophysiology of this disease.
RESULTS
HERV-K is expressed in the brain tissue of ALS patients
The HERV-K genome similar to that of other retroviruses has three major structural genes, the gag, pol, and env genes that encode the capsid, reverse transcriptase, and envelope proteins, respectively. Primer sets were used to amplify transcripts from each of these genes by reverse transcription polymerase chain reaction (RT-PCR) (Fig. 1A). We found that transcripts for all three genes were elevated in postmortem brain tissue samples from ALS patients (Fig. 1B). There was good correlation between the expression of each of these genes (Fig. 1C), confirming that the entire viral genome was expressed in these patient samples. The expression of HERV-K was also compared to the expression of several other HERVs. No significant elevation of these HERVs was noted (fig. S1A). Given that there are multiple loci that encode the HERV-K genome, we conducted RNA sequencing and analyzed the transcripts of each of the loci. The loci at chromosomes 7C and 10A were expressed in all three postmortem brain samples from patients with sporadic ALS at higher levels compared to controls (fig. S1B and table S1). No specific clinical phenotype was associated with the expression of HERV-K in ALS patients. To determine the cell types in which HERV-K was expressed, we immunostained postmortem brain tissue from patients with ALS and found expression of the env protein in the cortex of ten individuals (Fig. 1D), with a strong expression in the cytoplasm of large pyramidal neurons (Fig. 1E). Anterior horn neurons in the spinal cord also showed a similar pattern of immunostaining for HERV-K env protein (Fig. 1F and fig. S1, C and D). No immunostaining was seen in the lateral or posterior horns of the spinal cord (fig. S1D). No immunostaining was noted in the glial cells or in the white matter (Fig. 1G). Furthermore, no immunostaining was noted in the cortex or white matter of brain tissue from healthy individuals (Fig. 1H) or in postmortem brain tissue from patients with Alzheimer’s disease (Fig. 1I and fig. S1F). However, robust immunostaining for amyloid was present in postmortem brain tissue from Alzheimer’s disease patients (fig. S1E).
Fig. 1. HERV-K expression in brain tissue from ALS patients.
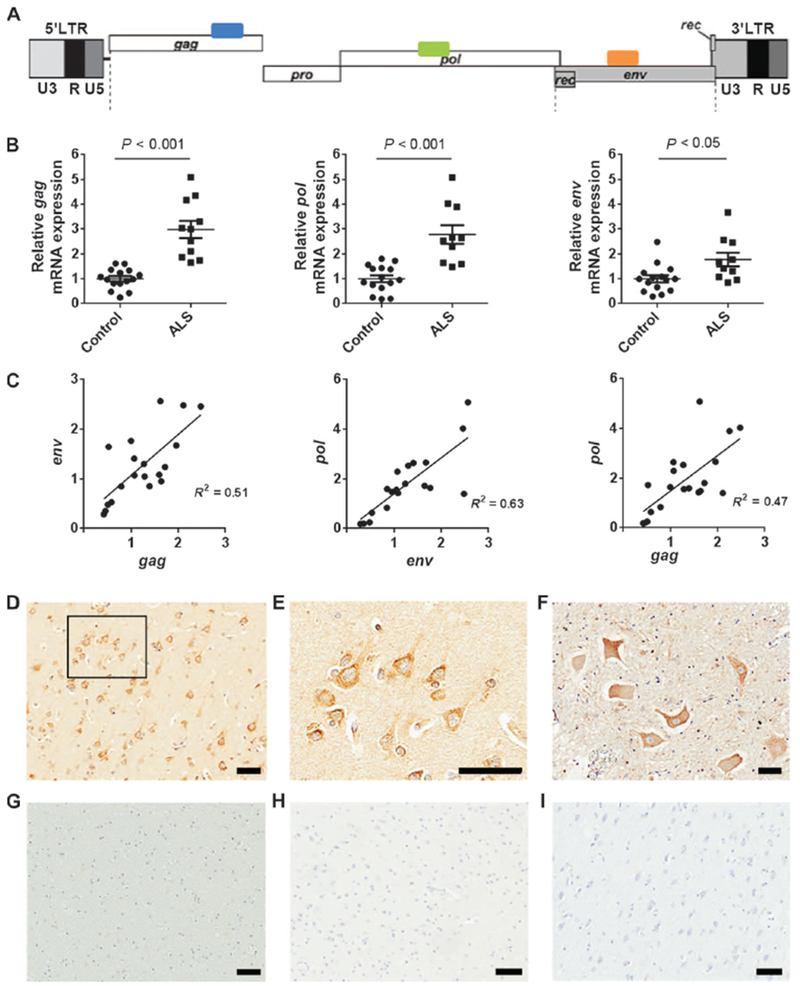
(A) HERV-K genome showing regions amplified by PCR. (B) All HERV-K genes showed increased expression in ALS patients compared to healthy controls (ALS, n = 11; control, n = 16). Brain tissue sample information is listed in table S1. Values represent means ± SEM. Significance was determined by unpaired Student’s t test. Variances were significantly different between groups. (C) Pearson correlation analyses revealed positive correlations between mRNA expression of HERV-K env, pol, and gag from autopsy brain cortical tissues. Pearson’s correlation coefficients were used to quantify the linear relationship between two variables. (D) Representative images show HERV-K env-immunoreactive neurons in the frontal cortex of a patient with ALS. (E) A higher-magnification image of the boxed area in the image shows focal accumulation at the cell membrane of cortical neurons. (F) Anterior horn motor neurons in the lumbar spinal cord of an ALS patient were also immunoreactive for HERV-K env. (G) The white matter of a patient with ALS. (H and I) Cortical neurons of an individual with a normal brain who died in a motor vehicle accident (H) and cortical neurons from a patient with Alzheimer’s disease showing no immunoreactivity for HERV-K env (I). Scale bars, 50 μm (D to I).
Expression of HERV-K in human neurons in vitro causes toxicity
To determine the relevance of HERV-K expression in neurons, we transfected the HERV-K genome and the HERV-K env gene into human neuronal cultures. Both the entire genome and the env gene caused a similar decrease in cell numbers and retraction of neurites (Fig. 2, A to C) in a dose-dependent manner (fig. S2A). The expression of the genes was confirmed by RT-PCR (fig. S2C). This suggested that the env protein could contribute to neurotoxicity and neuronal death. To determine the effect of activation of endogenous HERV-K in neurons, we used the gene-editing tool CRISPR/Cas9 (10) in which the nuclease activity of Cas9 [CRISPR (clustered regularly interspaced short palindromic repeats)-associated protein 9] was altered to contain four copies of the transcription factor VP16 (11). This was delivered to the human neurons in culture by a lentiviral vector. We designed a single guide RNA (sgRNA 8) to direct the transcription factor to the long terminal repeat (LTR) region of HERV-K. Activation of the endogenous HERV-K through the LTR resulted in neurotoxicity as evidenced by the loss of neurons (Fig. 2D) and the retraction of neurites (Fig. 2E). Activation of the viral genes was confirmed by RT-PCR, and a twofold increase in expression above controls was observed (fig. S2D). To determine whether the process of neuronal injury led to HERV-K activation, we treated the neurons with 3-nitropropionic acid, N-methyl-d-aspartate, or hydrogen peroxide. No activation of HERV-K was noted as determined by measuring viral transcripts (fig. S2B).
Fig. 2. HERV-K env-induced neuronal toxicity in vitro.
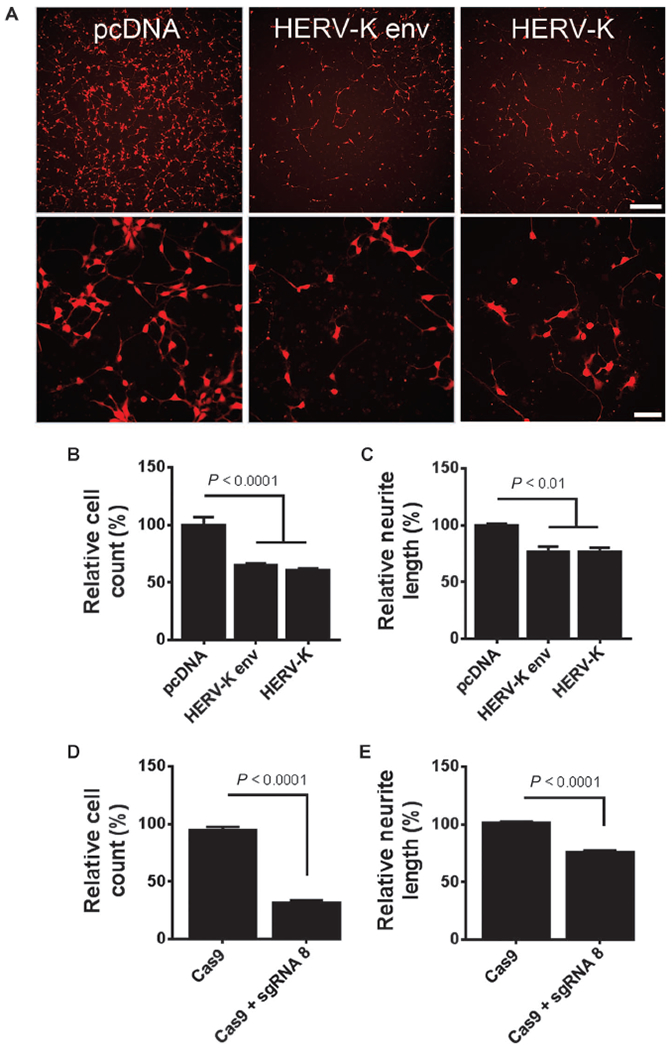
(A) The HERV-K env gene or the entire HERV-K genome was transfected into pluripotent stem cell-derived human neurons expressing tdTomato (a fluorescent marker to label neurons) and monitored for morphological changes. pcDNA was used as a control. Scale bars, 200 μm; 50 μm. (B and C) At 24 hours after transfection, (B) total cell counts and (C) neurite length were significantly decreased. Values represent means ± SEM from three independent experiments. Significance was determined by one-way analysis of variance (ANOVA) followed by Newman-Keuls post hoc comparison. Variances were significantly different between groups. (D and E) Endogenous HERV-K expression was induced using the CRISPR/Cas9 system. Stem cell–derived human neurons were transduced with a lentiviral construct encoding Cas9 fused to the transcription activation domain VP64 for 24 hours. Cells were either mock-treated (Cas9 alone) or transduced with guide RNA targeting the HERV-K promoter (sgRNA 8). Forty-eight hours after sgRNA delivery, (D) total cell counts and (E) mean neurite fiber length were significantly decreased. Values represent means ± SEM from three independent experiments. Significance was determined by unpaired Student’s t test.
Expression of HERV-K env in vivo causes degeneration of motor neurons
The findings were initially confirmed in vivo by in utero electroporation of the env gene into embryonic mouse brain, which resulted in dysmorphic changes in neurons and punctate dilatation of neuronal processes (fig. S3). We next generated transgenic animals in which the env gene was expressed in neurons (fig. S4A). The gene produced the full length and transmembrane domain of the env protein (fig. S4B). Expression of transcripts in the animals was confirmed by RT-PCR (fig. S4C). The level of expression of HERV-K env transcripts was nearly twofold higher in the transgenic animals compared to that in postmortem brain tissue of patients with ALS (fig. S4D). HERV-K env protein was detected by immunostaining and showed widespread expression in cortical neurons of transgenic mouse brain (fig. S4E). Similar to ALS patient tissues, the mouse neurons showed expression of env protein in the cytoplasm of the neuronal cell bodies and the apical dendrites (Fig. 3, A and B, and fig. S4, E to H). There was accompanying astrocytosis in regions surrounding the neurons where HERV-K env was expressed, but there was no difference in immune reactivity of microglial cells (Fig. 3, A and B, and fig. S5). Golgi staining showed decreased length, branching, and complexity of dendrites (Fig. 3, C to E). The number of dendritic spines was also decreased (Fig. 3F) and was associated with morphological changes showing loss of stalks resulting in an increase in stubby spines and a decrease in mushroom spines (Fig. 3G and fig. S6, A to D). There was also beading of the axons and dendrites (Fig. 3G and fig. S6, E and F).
Fig. 3. HERV-K-induced neuronal toxicity in vivo.
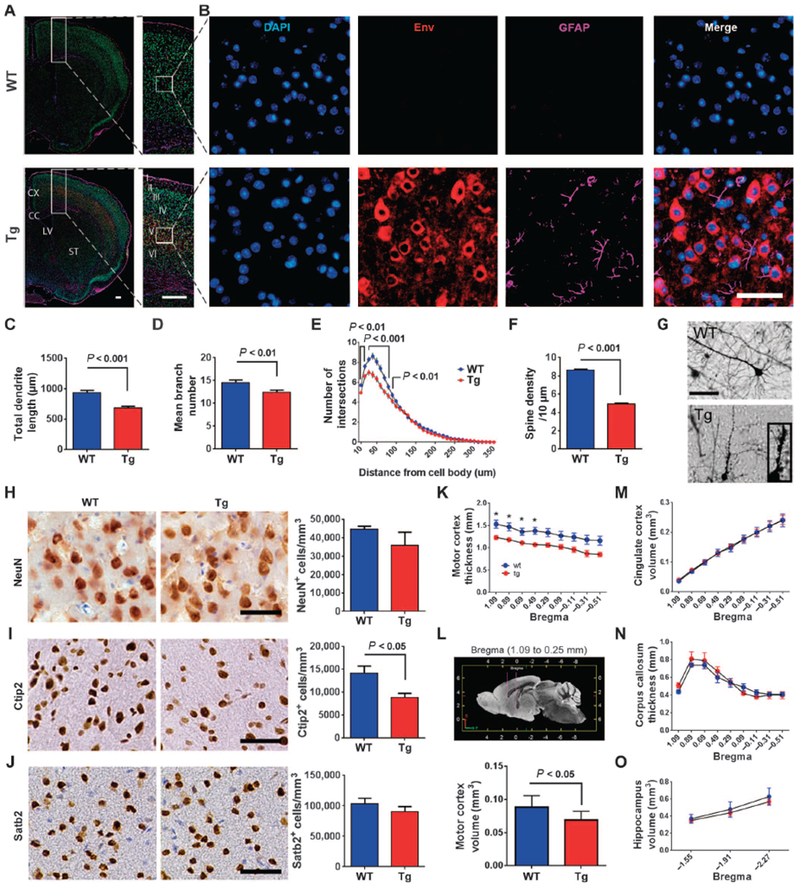
(A) Coronal sections of wild-type (WT) and HERV-K env transgenic (Tg) mice were immunostained for HERV-K env (red) and glial fibrillary acidic protein (GFAP) (purple). DAPI, 4′,6-diamidino-2-phenylindole (blue). (B) Enlarged images of cerebral cortex. (C to G) Golgi impregnated mouse pyramidal neurons show that (C) total dendrite length, (D) mean branch number, (E) dendritic complexity by Sholl analysis, and (F) spine density were significantly reduced in transgenic mice with (G) extensive dendritic beading. Values represent means ± SEM. The number of animals used for quantification was three animals per group. Significance was determined by unpaired Student’s t test. Scale bars, 500 μm (A); 50 μm (B and G). (H to J) Immunostaining for NeuN as a marker for neurons (H), Ctip2 immunostaining as a marker for corticospinal motor neurons (I), and Satb2 immunostaining as a marker for callosal projection neurons (J) in layer V of the motor cortex of wild type (n = 4) and transgenic (n = 3) mice. Values represent means ± SEM. Significance was determined by unpaired Student’s t test. Scale bars, 50 μm. Magnetic resonance images for wild type (n = 5) and transgenic (n = 5) mice were acquired on a 14-Tesla magnetic resonance imaging scanner. (K and L) Regional analysis revealed a reduction in cortical thickness (K) and volume (L) of the rostral part of the motor cortex. (M to O) There was no significant difference between wild-type and transgenic mice for the volume of the cingulate cortex (M), corpus callosum (N),and hippocampus (O). Values represent means ± SEM. Significance was determined by unpaired Student’s t test. The number of animals used for quantification was five animals in each group.
HERV-K env transgenic animals display specific loss of upper and lower motor neurons
Immunostaining of neurons expressing NeuN showed no significant change in the numbers of neurons in the frontal cortex (Fig. 3H). However, corticospinal motor neurons immunostained for Ctip2 showed a decrease in cell numbers (Fig. 3I; P < 0.05). In contrast, there was no significant change in the number of callosal projection neurons staining positive for Satb2, suggesting that the effect was specific for motor neurons (Fig. 3J). Magnetic resonance images of the brain of transgenic animals also showed a specific decrease in thickness and volume of the motor cortex (~22%) with no significant changes in the volume of the cingulate cortex and hippocampus or the thickness of the corpus callosum (Fig. 3, K to O) or the architecture of the brain (fig. S7, A to G). Immunostaining of the transgenic mouse spinal cord showed widespread expression of HERV-K env in neurons (Fig. 4, A to D).
Fig. 4. HERV-K env expression and injury to lower motor neurons.
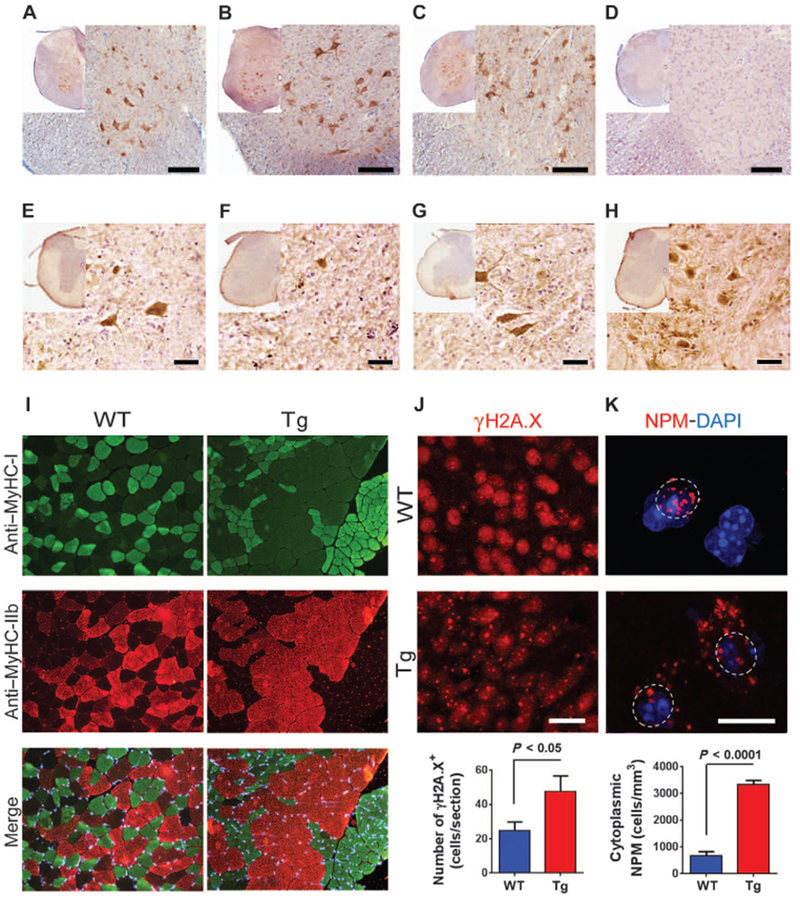
(A to C) Representative photomicrographs show HERV-K env immunoreactivity in the cervical (A), thoracic (B), and lumbar (C) spinal cord of transgenic mice. (D) This panel shows lack of HERV-K env immunoreactivity in the thoracic spinal cord of WT mice. (E to H) ChAT immunoreactivity is shown in the cervical (E), thoracic (F), and lumbar (G) spinal cord of transgenic mice and in the thoracic spinal cord of WT mice at 6 months of age (H). Intense ChAT signal seen in the motor neurons of the ventral horn in the thoracic spinal cord of WT mice (H) contrasts with very few ChAT-positive cell bodies in the ventral horn of the thoracic spinal cord of transgenic mice (F). Scale bars, 50 μm (A to H). (I) Sections of the tibialis anterior muscle from 6-month-old animals show a normal mosaic distribution of type I (green), type IIb (red), and type IIa (unstained) fibers in WT mice and fiber type grouping in the transgenic mice. Skeletal muscles from WT (n = 5) and transgenic (n = 6) mice were isolated and immunostained. MyHC, myosin heavy chain. (J) γH2A.X-positive foci in immunostained entorhinal cortex from 6-month-old WT (n = 4) and transgenic (n = 4) mice. Numbers of cells with γH2A.X-positive foci were increased in motor cortex of transgenic mice. Values represent means ± SEM. Significance was determined by unpaired Student’s t test. Scale bar, 20 μm. (K) Fluorescence micrographs showing the localization of nucleophosmin (NPM) in cells in the motor cortex of WT (n = 4) and transgenic (n = 3) mice. Numbers of cells with nucleophosmin localized to the cytoplasm were increased in the motor cortex of transgenic mice. Values represent means ± SEM. Significance was determined by unpaired Student’s t test. Scale bar, 10 μm.
However, only rare motor neurons were present in the anterior horns, with near absence of motor neurons at some levels of the spinal cord (Fig. 4, E to H). Immunostaining of the quadriceps and tibialis anterior muscles for type I and type II myosin isoforms showed fiber type grouping and examples of grouped atrophy suggestive of a chronic denervation and reinnervation process (Fig. 4I and fig. S8). There were no dystrophic changes in the muscle fibers, and the nuclei were in the periphery of the fibers, suggesting that there were no myopathic features.
Ongoing neuronal injury was also evident by the presence of double-stranded DNA breaks as seen by immunostaining for γH2A.X (12), which showed aggregated foci of the phosphorylated histone protein in the chromatin (Fig. 4J). The number of γH2A.X foci was increased in neurons in the frontal cortex (Fig. 4J). Nucleolar dysfunction has been observed in several neurodegenerative diseases including Alzheimer’s disease and Parkinson’s disease (13). We therefore evaluated whether neurons from transgenic mice showed signs of nucleolar stress. Immunostaining for the nucleolar marker nucleophosmin showed translocation from the nucleolus to the cytoplasm of cortical neurons (Fig. 4K). Together, these data suggested that disruption of nucleolar function may be a key mechanism by which HERV-K leads to neuronal dysfunction (14).
HERV-K env transgenic animals develop motor dysfunction
To determine the functional consequences of HERV-K env expression in neurons, we performed a panel of behavioral tests on the animals. These tests showed that the animals developed progressive motor dysfunction. In an open field, they traveled shorter distances and rested for longer periods (Fig. 5A). Transgenic mice fell faster in a rotarod performance test (Fig. 5B) and displayed evidence of spasticity with increased clasping of the hindlimbs (Fig. 5C and movies S1A and S1B). Y maze testing confirmed that these differences were not due to an impairment of working memory (Fig. 5D). Sensory and vestibular functions were also unimpaired (Fig. 5, E and F). Motor function in the transgenic mice showed a progressive decline from 3 to 6 months of age as evaluated in open-field testing (fig. S9, A to D) with 50% mortality by 10 months (Fig. 5G). In terminal stages, the animals developed profound weakness of the limbs and spinal muscles resulting in minimal movement and a hunched back causing decreased movement of the thoracic cage, affecting the muscles of respiration (Fig. 5G).
Fig. 5. HERV-K-induced alterations in behavioral and functional analysis of mouse phenotype.
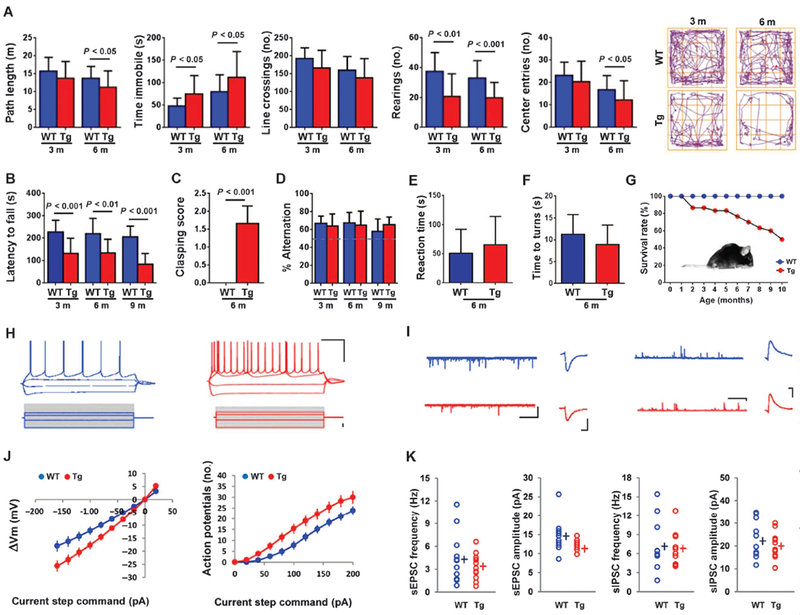
(A) Open-field testing showed that the transgenic mice were less active than WT animals as determined by decreased path length traveled, increased periods of immobility, decreased line crossings, decreased number of rearings, and decreased number of entries into the center of the field. Representative tracings are shown. There was a progressive decrease in activity overtime [n= 16(WT)and n = 15 (transgenic) at 3 months; n = 26 (WT) and n = 24 (transgenic) at 6 months]. (B) Transgenic mice spent less time on an accelerating rotarod [n = 18 (WT) and n = 17 (transgenic) at 3 and 6 months; n = 18 (WT) and n = 9 (transgenic) at 9 months]. The sample size declined at 9 months due to increased death at that age. (C) Transgenic mice showed an increased clasping reaction in the tail suspension test [n = 18 (WT) and n = 17 (transgenic)]. (D) A Y maze test shows that spontaneous alteration was not different between WT and transgenic mice [n = 18 (WT) and n = 17 (transgenic) at 3 and 6 months; n = 18 (WT) and n = 9 (transgenic) at 9 months]. (E) The time to notice adhesive tapes sticking on the palms of the hind paws in transgenic mice was not significantly different from WT mice [n = 10 (WT) and n = 10 (transgenic)]. (F) When placed on a 45° angle slope, the time to turn was not different between WT and transgenic mice [n = 8 (WT) and n = 12 (transgenic)]. (G) A cohort of transgenic animals declined from 30 to 15 animals over 10 months. Insert shows a 9-month-old terminally ill transgenic mouse with a hunched back. (H and I) Electrophysiological properties of mouse cortical pyramidal neurons [n = 3, WT (blue); n = 3, transgenic (red)]. (H) Voltage traces evoked by −160, −40,40, and 80 pA current steps (1 s). Scale bar, 200 ms/20 mV. Amplitude of the steady-state membrane potentials is plotted against each injected current step. (I) Traces of sEPSCs (scale, 5 s/100 pA) and average sEPSCs (scale, 10 pA/5 s). (J) Change in membrane potential (ΔVm) and number of action potentials evoked for a range of current injections. (K) Pooled data values of sEPSC frequency (left) and amplitude (right). Values represent means ± SEM and were analyzed by the Mann-Whitney nonparametric test.
Functional activity of the neurons was also assessed in electrophysiological recordings. Passive and active membrane properties of layer V cortical pyramidal neurons from wild-type and transgenic mouse prefrontal cortices were tested by injecting gradient steps of electrical current into the cell (Fig. 5H). Subthreshold responses (current steps, from −120 to 20 pA) reflected an increase in the global input resistance of the cell (Fig. 5I and table S2). Membrane excitability was assessed with a series of depolarizing current steps to evoke action potentials. We found that the number of action potentials was enhanced in transgenic animals, resulting in a right shift of the input-output function curve (Fig. 5I). This increase in intrinsic excitability is associated with a decrease in the first action potential latency and the rheobase, defined as the minimal current to induce an action potential (table S2). Other action potential parameters such as threshold, amplitude, rise time, width, and after-hyper-polarization amplitude remained unchanged. Finally, we examined synaptic transmission by recording spontaneous excitatory and inhibitory postsynaptic currents (sEPSCs and sIPSCs). We found that only the sEPSC amplitude was changed, with a significant decrease in the sEPSC observed in transgenic animals compared to that in wild-type mice, which could be attributed to the decrease in spine density (Fig. 5, J and K). The increase in input resistance was consistent with the decrease in neurite number and branching.
HERV-K expression is regulated by TAR DNA binding protein 43
Previously, HERV-K pol gene expression was found to correlate with TAR (trans-activation-responsive) DNA binding protein 43 (TDP-43) mRNA in postmortem brain tissue from patients with ALS (3). TDP-43 has been shown to regulate the replication of HIV (15), and it also binds to transposable elements (16). Hence, we determined whether TDP-43 could also regulate HERV-K expression. When a plasmid with TDP-43 was transfected into human neurons, HERV-K expression occurred as demonstrated by immunostaining for the env protein and measuring the viral transcripts (Fig. 6, A and B). When HERV-K and TDP-43 were cotransfected into HeLa cells, there was an increased replication of HERV-K as evidenced by the reverse transcriptase activity in the culture supernatants (Fig. 6C) and increased viral transcripts in the cell extracts (Fig. 6D). HIV-transactivator of transcription (HIV-Tat) protein is known to increase HERV-K replication, and hence it was used as a control (17). TDP-43 showed additive responses with Tat, suggesting that they may act on different sites. This was confirmed using a HERV-K LTR construct with a luciferase reporter gene (Fig. 6E). Knockdown of endogenous TDP-43 with small interfering RNA (siRNA) also decreased HERV-K expression (Fig. 6F). We next determined whether TDP-43-mediated induction of HERV-K involved direct association with the HERV-K promoter. The consensus HERV-K LTR sequence was scanned to identify pyrimidine-rich motifs associated with TDP-43 DNA binding. Putative TDP-43 binding sites, consisting of more than eight contiguous pyrimidine bases (16), were identified at five loci as indicated and were labeled according to their position relative to the first base of the HERV-K LTR (Fig. 6G and fig. S10A). Binding of TDP-43 to HERV-K LTR was confirmed by chromatin immunoprecipitation (fig. S10B). Hence, we constructed biotinylated oligomers representing each of these sites and incubated them with nuclear extracts from 293T cells followed by washing of DNA/protein complexes under low- and high-salt conditions and then analyzed the complexes by Western blots using an antibody to TDP-43. We found that TDP-43 bound to region 726–734 (5′-CCCTCTCCC-3′) with the highest affinity, suggesting that it was the critical binding site on the HERV-K LTR (Fig. 6, H and I). TDP-43 binding to HERV-K LTR was associated with increased binding of elongation-competent RNA polymerase II (fig. S10C). No effect was seen on an unrelated genomic region (fig. S10D).
Fig. 6. HERV-K activation by TDP-43 and identification of binding sites on LTR.
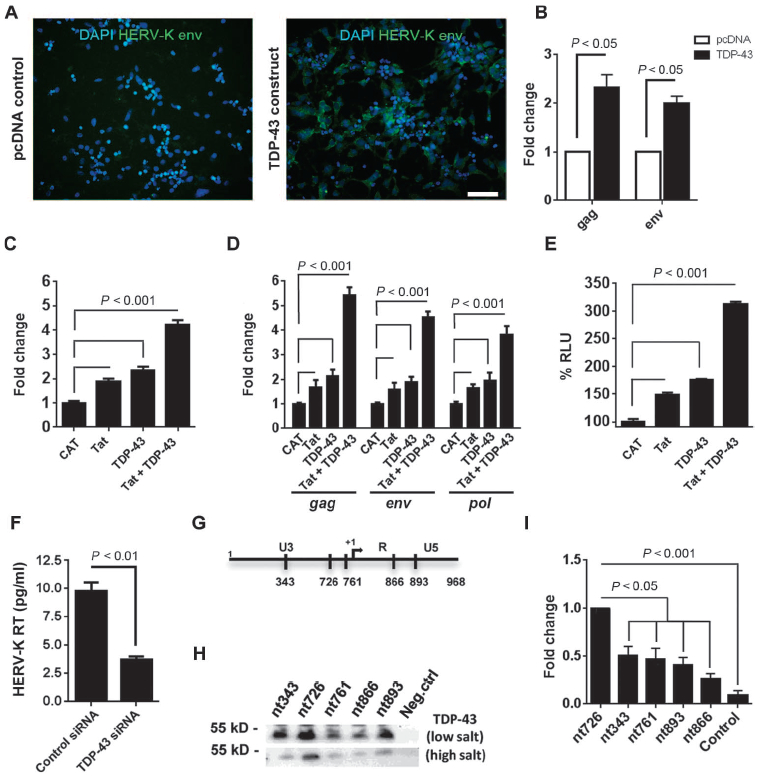
(A and B) Stem cell-derived neurons were transfected with either pcDNA control or TDP-43 expression construct. (A) Forty-eight hours after transfection, cells were fixed with PFA and stained for HERV-K env protein. Scale bar, 50 μm. (B) Twenty-four hours after transfection, cells were collected for RNA extraction, and quantitative RT-PCR was used to measure HERV-K transcripts. (C and D) HERV-K plasmid was cotransfected with chloramphenicol acetyltransferase (CAT) (control), Tat, TDP-43, or Tat and TDP-43 in HeLa cells, and 24 hours after transfection, reverse transcriptase activity (HERV-K RT) was measured in culture supernatants by the product-enhanced reverse transcriptase (PERT) assay. (D) The levels of HERV-K transcripts were measured using RT-PCR and expressed as fold change compared to CAT control. (E) HERV-K LTR-MetLuc plasmid was cotransfected with CAT, Tat, TDP-43, or Tat and TDP-43, and luciferase activity was measured. RLU, relative light units. (F) Knockdown of endogenous TDP-43 with siRNA reduced HERV-K expression. (G) Putative TDP-43 binding sites in HERV-K LTR reported relative to the first base of the LTR. (H) Binding of TDP-43 to biotinylated oligonucleotides derived from the putative binding sites under low- or high-salt conditions. (I) Quantification of (H) indicating binding affinity. Values represent means ± SEM from three independent experiments. Significance was determined by unpaired Student’s t test, nt, nucleotide.
DISCUSSION
Here, we show that an endogenous retroviral protein contributes to neurodegeneration in mammalian brain and spinal cord. Expression of HERV-K env protein in transgenic mouse neurons led to motor dysfunction with accompanying DNA damage and morphological and functional changes consistent with axonal and dendritic injury. These findings may have implications for a subgroup of ALS patients with increased HERV-K expression. Because activation of HERV-K can lead to neurodegeneration, blocking its activation and replication may affect the course of ALS.
It is becoming increasingly apparent that mutations in several different genes can lead to the same phenotypic neurodegenerative disease. Several genetic mutations have been implicated in ALS. These include superoxide dismutase (SOD-1), TDP-43, fused in sarcoma (FUS), and repeat expansions in C9ORF72 (18). However, none of these mutations explain how the disease spreads along anatomical pathways. ALS typically starts in a focal region in one of the limbs or the bulbar region and then spreads to contiguous regions. Our finding that full-length transcripts of HERV-K can be found activated in the brain of some ALS patients raises the possibility that the virus could spread from one neuron to the next and, in the process, lead to neurotoxicity through expression of the env protein. Whereas the mechanism of spread needs to be further investigated, the observation that the HERV-K env protein was found localized to the neuronal cell body suggests that the spread may occur laterally from neuron to neuron. This would be different from the transsynaptic spread seen with rabies virus (19), but would better explain the anatomical spread of ALS from one brain region to an adjacent region.
Because we studied only autopsy tissues, we considered the possibility that HERV-K was a response to neurodegeneration. However, HERV-K immunostaining could not be detected in brain tissue from patients with Alzheimer’s disease. In a previous study, we showed that HERV-K gene expression was absent in brain tissue from Parkinson’s disease patients and in brains from patients who had died in accidents (3). We also showed that treatment of human neurons in culture with toxins that induced mitochondrial or oxidative stress and excitotoxicity (pathways that have been implicated in the pathophysiology of ALS) did not induce HERV-K expression. Further, there was low basal-level HERV-K expression in neurons in culture; however, endogenous HERV-K activation resulted in neuronal injury. Our findings contrast with another study claiming that HERV-K may be neuroprotective (20). However, in this study, HERV-K was expressed in neuronal cell lines. Endogenous retroviruses including HERV-K can be expressed in tumor cells where they may have cytoprotective effects (21). In terminally differentiated cells such as neurons, however, the expression of HERV-K may be cytotoxic.
We developed a transgenic mouse model in which the HERV-K env protein was expressed under a neuronal promoter leading to widespread expression in neurons. The amount of expression of the gene was comparable to that in brain tissue from patients with ALS. Despite the widespread expression of the protein, the animals developed a progressive motor neuron disease affecting only the upper and lower motor neurons. There were no developmental abnormalities in the brain, and the cytoarchitecture of the brain appeared normal. The animals displayed progressive motor dysfunction first noticeable at 2 to 3 months of age and developed selective atrophy of the motor cortex. The neurons showed morphological changes with abnormal processes and electrophysiological abnormalities. There were changes consistent with denervation and reinnervation of the muscles due to anterior horn cell involvement. This profound weakness of the limbs eventually involved the spinal muscles and thoracic cage resulting in 50% mortality by 10 months. In contrast, the SOD-1 transgenic mouse model shows predominantly lower motor neuron involvement and a much more rapid progression (22). In our animal model, we did not control the site of insertion of the HERV-K env, however, injection of the HERV-K env gene with a cytomegalovirus promoter into the brains of animals in utero and transfection of neurons in vitro resulted in similar morphological changes in the neurons, suggesting that the phenotype in the animals was unlikely to be due to the site of insertion of the gene.
The mechanism by which the HERV-K env leads to neuronal injury is not well understood. However, we found that there was evidence of double-stranded DNA breaks in the neurons and entrapment of the nucleolar protein nucleophosmin in the cytoplasm of neurons in the transgenic animals. Nucleophosmin is a multifunctional protein that shuttles between the nucleolus and the cytoplasm (23) and is required for ribosome assembly and transport, molecular chaperoning, and DNA transcription (24). Nucleolar dysfunction has also been seen in patients with ALS who have C9ORF72 mutations, and emerging evidence suggests that nucleolar dysfunction with impairment of ribosomal synthesis may play a critical role in the pathophysiology of ALS in these patients (25).
The Cas-Br-E murine leukemia virus is a retrovirus that is also known to cause a motor neuron disease (26). In this animal model, a 372-base pair fragment in the env gene was formd to be critical for causing the motor neuron disease (27). However, the histopathology in these animals shows the presence of spongiform changes, which is unlike that of ALS or the HERV-K env transgenic mouse model (27). In the murine leukemia vims model, the virus infects glial cells, and the neurodegeneration is secondary to glial cell infection (28). The env protein of another human retrovirus, HIV, has also been shown to cause neurotoxicity. The mechanism by which it causes neurotoxicity has been extensively studied (29). However, HIV infects perivascular macrophages and microglia in the brain and not neurons. The HIV env protein gp120 is released extracellulariy from infected cells where it may interact with neurons or with astroglial cells and block glutamate uptake, setting up a cascade of events leading to neuronal injury. In contrast, we show that HERV-K env protein is expressed in neurons and leads to neuronal injury through intracellular processes that are yet to be fully characterized. The HIV-Tat protein has also been shown to be neurotoxic (30). However, HERV-K lacks a homolog to Tat.
We have shown that HERV-K rs regulated by TDP-43. TDP-43 is a multifunctional protein. It is overexpressed in ALS (3) and forms cytoplasmic aggregates (31). Its RNA binding properties, including binding to transposable elements, have been well studied. TDP-43 also has been shown to be defective in patients with frontotemporal dementia (16). We have demonstrated that TDP-43 is also capable of binding to the HERV-K LTR DNA. However, as opposed to its effects on mRNA, the DNA binding leads to transactivation of HERV-K. This is particularly important because the HERV-K LTR has five binding sites for TDP-43. Thus, TDP-43 may serve as an important regulator of HERV-K expression and contribute to subsequent neurodegeneration.
Our study has several limitations. We studied only patients with sporadic ALS, hence it remains unknown whether HERV-K activation can also occur in patients with genetic causes of ALS. Our small sample size also did not allow us to determine whether there was a clear phenotype associated with HERV-K activation in patients with ALS. Nonetheless, our study clearly demonstrates the activation of HERV-K in patients with ALS. This activation results in neuronal injury with the selective degeneration of motor neurons. The expression of HERV-K is regulated by TDP-43. Thus, our study implicates HERV-K env in ALS pathogenesis.
MATERIALS AND METHODS
Study design
We designed this study to determine whether HERV-K could play a role in the pathogenesis of ALS. We first showed the expression of each of the transcripts of HERV-K in the brains of 11 patients with ALS. Brain tissue samples from 16 individuals with no known brain disease were used as controls. The samples were matched for sex, postmortem interval, RNA integrity values, and the anatomical region of the brain studied. Immunohistochemistry showed that the expression of the virus was localized to cortical neurons in brain tissue and anterior horn cells in spinal cord obtained from 10 patients with ALS. Brain tissue samples from 10 patients with Alzheimer’s disease were used as controls. Sample sizes were based on the availability of tissues and previous experience with such assays. To determine whether the expression of the virus in neurons could cause neurotoxicity, we transfected neuronal cultures with plasmids containing the entire HERV-K genome, HERV-K env, or pcDNA and monitored them for cell counts or changes in neurite length. All experiments were done in replicates of 18 and repeated twice. Similar amounts of toxicity were seen with HERV-K and HERV-K env. Hence, in a pilot experiment, we injected the HERV-K env plasmid (n = 11) or control plasmid (n = 9) into the mouse brain in utero. Expression of HERV-K env reproduced the morphological abnormalities in the neurons. We next generated transgenic animals in which HERV-K env was expressed under a neuronal promoter. Non-transgenic littermates were used as controls. The transgenic animals developed progressive motor dysfunction over 6 months at which time nearly 50% of the animals died. Because of profound motor abnormalities in the animals, it was not possible to perform the testing in a blinded manner. However, all histopathological and radiological assessments were performed by an investigator blinded to the genotype of the animals. The transgenic animals had selective thinning of the motor cortex and morphological abnormalities in neurites of the motor neurons with DNA strand breaks and nucleolar abnormalities. There was accompanying astrocytosis. All behavioral experiments were done in sample sizes of at least 15 animals in each group, and histological studies had at least five animals in each group. The regulation of HERV-K expression was studied in vitro. These data were analyzed from at least three separate experiments. The data showed that TDP-43 binds to the LTR of HERV-K to regulate its expression.
Quantitative PCR
All human samples were obtained after the approval by the Office of Human Subjects Research Protection at the National Institutes of Health (NIH) and the Institutional Review Boards of Johns Hopkins University. All samples were analyzed by investigators blinded to the clinical condition or identity of the patients. Total RNA was extracted from frozen brain tissue (table S1) with RNeasy Plus Mini Kit (Qiagen). The RNA extracts were treated with ribonuclease-free deoxyribonuclease (Qiagen). The quality of RNA was evaluated with the Agilent 2100 Bioanalyzer. Only samples that had an RNA integrity number (RIN) >8 were used for this study to ensure that there was little or no RNA degradation in the samples. Reverse transcription was performed with 1 μg of RNA using the Superscript III first-stand kit (Invitrogen). Quantitative PCR was done using Applied Biosystems ViiA 7. The amount of RNA in brain samples was expressed as relative levels to control samples after normalization with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA. To confirm that there was no DNA contamination, control PCRs were performed with reverse transcription product in which Superscript III was omitted. Primers for quantitative PCR are listed in table S3. The HERV-K full-length primer sequences were designed to amplify the unspliced full-length HERV-K transcript without the LTRs and the spliced transcripts representing env and rec. Primers for HERV-K env and mouse GAPDH were used to quantify the relative expression level of env in the transgenic mice (table S3).
DNA constructs
The HERV-K whole genome consensus sequence (32,33) was synthesized and cloned into pcDNA3.1 vector (Invitrogen). HERV-K 5′LTR was PCR-cloned and inserted into pMetLuc reporter vector (Clontech). TDP-43 gene was obtained from Addgene and was cloned into pcDNA3.1 vector. HIV-Tat plasmids were reported previously (34). Codon-optimized HERV-K env gene was synthesized and cloned into pcDNA3.1 vector for transfection assays and in utero injections and into the Xho I site of a Thy-1 expression cassette from Addgene for the generation of transgenic mice.
Induction of endogenous HERV-K by lentiviral transduction
Lentiviral constructs encoding HERV-K LTR–targeting sgRNA or nuclease-null Cas9 linked to the transcription activator domain VP64 were generated using the ViraPower Lentiviral Packaging Mix according to the manufacturer’s protocol (Invitrogen). Lentivirus stocks obtained from transfection of human embryonic kidney 293T cells were concentrated using the Retro-X Concentrator (Clontech), and the copy number was determined using the Lenti-X qRT-PCR Titration Kit (Clontech). The dCas9-NLS-3×HA-VP64 vector (Addgene) served as the source of insert material for the Cas9 lentivirus. Briefly, neural stem cell–derived neurons were incubated with Cas9-VP64 lentiviral vector with polybrene (5 μg/ml) (Sigma). After 4 hours, the inoculum was diluted with an equal amount of complete medium, and the cells were incubated overnight. Twenty-four hours after transduction, the supernatant was removed, and the cells were incubated with the lentiviral vector expressing the sgRNA 8, with polybrene (5 μg/ml) for an additional 24 hours.
Immunohistochemistry of human autopsy brain tissue
We analyzed human autopsy tissues of 10 patients with ALS from the ALS Center at the University of Pittsburgh. Samples from the frontal cortex and the cervical spinal cord were examined. The mean age of patients at the time of death was 59.3 years (range, 45 to 73 years), and patients were 50% male and 50% female. The autopsy was done on an average of 7.1 hours after death (range, 2 to 10 hours). Sections from 10 Alzheimer’s patients served as controls. We obtained Alzheimer’s patient samples from the Department of Pathology at the University of Kentucky. The mean age of patients at the time of death was 84.5 years (range, 74 to 95 years), and patients were 50% male and 50% female. The autopsy was done on an average of 3.4 hours after death (range, 2 to 5 hours). Autopsied brains were formalin-fixed and paraffin-embedded. Five-micrometer-thick sections were obtained and stained as follows. The slides were deparaffinized and rehydrated using xylene and graded ethanol. Antigen retrieval was done by steaming in citrate buffer for 20 min. Peroxidase blocking was achieved with dual enzyme block solution (Dako), and protein blocking was done with protein block solution (Dako). Incubation with mouse anti–HERV-K env (1:500; Austral Biologicals) or anti-β amyloid (1:500; BioLegend) was done overnight at room temperature. PowerVision polymeric horseradish peroxidase (HRP) anti-mouse (Leica Biosystems) or HRP-conjugated anti-human immunoglobulin G (IgG) was applied as secondary antibodies for 2 hours at room temperature. Antibody binding was developed with 3,3′-diaminobenzidine (DAB; Vector Laboratories). Sections were counterstained with hematoxylin (Dako). Images were processed using an Aperio whole slide scanner (Leica Biosystems).
Generation of transgenic mice
The Thy1-HERV-K env transgene cassette was excised with Eco RI and Pvu I (fig. S4A). The purified fragment was injected into the pronuclei of fertilized eggs from C57BL6 mice. Surviving embryos were implanted into pseudopregnant C57BL6 mice. The transgenic founders were screened by genotyping with the primer set GT-env-F2: 5′-ACCAGCTGGCTGACCTGTAG-3′ and GT-env-R2: 5′-GGCAGCTTCATCTGTTCCTC-3′. All experiments were performed on a single heterozygous line. HERV-K env transcripts were analyzed by gel electrophoresis and real-time PCR (fig. S4, C and D). HERV-K env protein was confirmed by Western blot analysis (fig. S4B) after immunostaining for the HERV-K env, which showed staining in cortical neurons (fig. S4, E to H). For all studies in this article, littermate animals without the transgene were used as controls. All experiments involving mice were performed according to the recommendations in the Guide for the Care and Use of Laboratory Animals of the NIH. Mice were housed in a pathogen-free barrier facility with a 12-hour light/12-hour dark cycle and ad libitum access to food and water. Both male and female animals of ages 6 weeks and 3 to 9 months were used. The sample size for each experiment was determined on the basis of previous experiences with the transgenic animal models. Quantification of data from all experiments involving mice was done by an investigator blinded to the genotype of the animals.
Immunohistochemistry and confocal microscopy in transgenic animals
Mice (6 to 9 months old) were deeply anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) intraperitoneally and perfused transcardially with saline followed by 4% (w/v) paraformaldehyde (PFA). After postfixing in PFA overnight, the brains and spinal cords were immersed in a 30% (v/v) sucrose solution. On the following day, the brains and spinal cords were cryoprotected and cut in the coronal or horizontal plane into 40-μm-thick sections on a sliding microtome. The sections were washed in tris-buffered saline (TBS) [10 mM tris-HCl (pH 7.5), 150 mM NaCl]. Endogenous peroxidase activity was blocked with 3% (v/v) hydrogen peroxide before incubation in blocking solution [TBS with 0.5% (v/v) Triton X and 2.5% (v/v) donkey serum]. Mouse anti–HERV-K env (1:500; Austral Biologicals), mouse anti-NeuN (1:500; Millipore), rabbit anti-BCL11B (1:500; Novus Biologicals), or rabbit anti–choline acetyltransferase (anti-ChAT, 1:1000; Millipore) was applied overnight at 4°C. Biotinylated goat anti-mouse IgG or goat anti-rabbit IgG was used as the secondary antibody. Antibody binding was developed using the Vectastain Elite ABCKit (Vector Laboratories) and visualized with DAB (Vector Laboratories). The sections were counterstained with hematoxylin (Dako). Images were processed using an Aperio whole slide scanner (Leica Biosystems). For confocal microscopy, primary antibodies were diluted in blocking solution as follows: mouse anti–HERV-K env (1:500; Austral Biologicals), mouse anti-NeuN (1:500; Millipore), mouse anti-nucleophosmin (1:500; Millipore), mouse anti-phospho-histone H2A.X (1:500; Millipore), and rabbit anti-GFAP (1:2000; Dako). Secondary antibodies were conjugated with Alexa Fluor 488 or Alexa Fluor 594 (1:250; Invitrogen), followed by washing and counterstaining with DAPI to label all nuclei. All images were obtained using an LSM 510 META laser scanning confocal microscope (Carl Zeiss). For quantification, positively labeled cells were counted in at least three 200 × 200-μm fields from the selected brain regions for each animal evaluated to assess protein expression levels.
Multichannel fluorescence microscopy
Multichannel wide-field fluorescence microscopy was performed on 10-μm-thick mouse brain coronal sections. The sections were immunoreacted for 1 hour at room temperature using a mixture of the following primary antibodies: mouse IgG2a anti–HERV-K env (1:200; Austral Biologicals), guinea pig IgG anti-NeuN (1:500; Millipore), mouse IgGl anti-NeuN (1:500; Milipore), chicken IgG (IgY) anti-GFAP (1:500; Abcam), and rabbit IgG anti-Iba1 (1:200; Wako Chemicals). The sections were then washed in phosphate-buffered saline and 0.5% bovine serum albumin and immunoreacted using a mixture of the following fluorochrome-conjugated secondary antibodies: goat anti-mouse IgG2a–Alexa Fluor 647 (1:200; Invitrogen), goat anti-guinea pig IgG–Alexa Fluor 488 (1:200; Invitrogen), donkey anti-chicken IgG (IgY)–IRDye 800CW (1:100; Li-Cor Biosciences), and goat anti-rabbit IgG–Alexa Fluor 594 (1:200; Invitrogen). The sections were then washed in washing buffer, incubated for 5 min at room temperature in DAPI (1 mg/ml, Invitrogen) to stain the cell nuclei, rinsed in distilled water, air-dried, and cover-slipped using Immu-Mount medium (Thermo Fisher Scientific). All sections were imaged using an Axiovert 200M fluorescence microscope (Carl Zeiss) equipped with a 20× Plan-Apochromat (Phase 2) objective (Carl Zeiss), a high-resolution ORCA-ER cooled digital camera (Hamamatsu Photonics) sensitive to a wide spectrum of emission wavelengths, including those approaching infrared, a 100-W mercury arc lamp (Carl Zeiss), and excitation/dichroic/emission filter sets (Semrock) optimized to detect the following fluorophores: DAPI, Alexa Fluor 488, Alexa Fluor 594, Alexa Fluor 647, and IRDye 800CW. Each labeling reaction was captured using filtered light through an appropriate fluorescence filter set, and the images were individually digitized at 12-bit resolution using the Volocity imaging program (Improvision). An appropriate color table was applied to each image either to match its emission spectrum or to set a distinguishing color balance. The pseudocolored images were then converted into TIFF files exported to Adobe Photoshop and overlaid as individual layers to create multicolored merged composites.
Statistical analyses
No statistical methods were used for predetermining the sample size. For animal studies, the sample size was based on previous experience with transgenic animals. Because animal groups were defined by genetic status, no randomization was used. The investigators were blinded to group allocation during the experiment and when assessing the outcome. Because of profound effects in the transgenic animals, blinding for analysis was not possible for motor tasks or for histological analysis of spinal cord and muscle. For this reason, the histological data for spinal cord and muscle are provided as descriptive analyses. All other data were analyzed with GraphPad Prism version 6 (GraphPad). The Shapiro-Wilk test of normality was applied to all data sets to determine whether a data set is normally distributed. The F test or Bartlett’s test was used to determine equality of variances between groups. Differences between means were assessed by paired two-sided Student’s t test or one-way ANOVA followed by post hoc testing. In cases where the data did not demonstrate a normal distribution, the Mann-Whitney test or Kruskal-Wallis test was used. Pearson’s correlation coefficients were used to quantify the linear relationship between two variables. All available samples or animals were included for statistical analysis.
Supplementary Material
Movie S1. Hindlimb clasping in a tail suspension test. (A) Control.
Movie S1. Hindlimb clasping in a tail suspension test. (B) Transgenic animal.
Fig. S1. HERV-K expression in autopsied human brain.
Fig. S2. Induction of HERV-K expression in human neurons.
Fig. S3. Effect of HERV-K env in postnatal neurons of mice.
Fig. S4. Expression of HERV-K env in the brain of transgenic mice.
Fig. S5. Distribution of cellular proteins in wild-type and HERV-K env transgenic mouse brains.
Fig. S6. Changes in spine morphology and dendritic damage in pyramidal neurons in HERV-K env transgenic mice.
Fig. S7. Magnetic resonance imaging and regional analysis of mouse brains.
Fig. S8. Muscle pathology in HERV-K env transgenic mice.
Fig. S9. Impairment of locomotor behavior in HERV-K env transgenic mice.
Fig. S10. TDP-43 binds to the HERV-K LTR in vivo, and TDP-43 binding to HERV-K LTR correlates with the association of a processive RNA polymerase II (phosphorylated Ser2) on the LTR.
Table S1. Clinical data for the human brain samples.
Table S2. Passive and active membrane properties of medial prefrontal cortex layer V pyramidal neurons.
Table S3. Primer sequences.
Acknowledgments:
We thank the Target ALS Human Postmortem Tissue Core at Johns Hopkins University, the University of Pittsburgh ALS Research Center, the University of Kentucky Alzheimer’s Research Center, and the National Institute of Child Health and Human Development (NICHD) Brain and Tissue Bank for Developmental Disorders at University of Maryland for providing postmortem tissue samples. Funding: This work was supported by funds from the National Institute of Neurological Disorders and Stroke, National Institute of Arthritis, Musculoskeletal and Skin Diseases, and the NICHD of the NIH and the Robert Packard Center for ALS Research.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/7/307/307ra153/DC1
Materials and Methods
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Hughes JF, Coffin JM, A novel endogenous retrovirus-related element in the human genome resembles a DNA transposon: Evidence for an evolutionary link? Genomics 80, 453–455 (2002). [PubMed] [Google Scholar]
- 2.Subramanian RP, Wildschutte JH, Russo C, Coffin JM, Identification, characterization, and comparative genomic distribution of the HERV-K (HML-2) group of human endogenous retroviruses. Retrovirology 8, 90 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Douville R, Liu J, Rothstein J, Nath A, Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann. Neurol 69, 141–151 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacGowan DJL, Scelsa SN, Imperato TE, Liu K-N, Baron P, Polsky B, A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 68, 1944–1946 (2007). [DOI] [PubMed] [Google Scholar]
- 5.McCormick AL, Brown RH Jr, Cudkowicz ΜE, Al-Chalabi A, Garson JA, Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 70, 278–283 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Steele AJ, Al-Chalabi A, Ferrante K, Cudkowicz ME, Brown RH Jr., Garson JA, Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology 64, 454–458 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Andrews WD, Tuke PW, Al-Chalabi A, Gaudin P, Ijaz S, Parton MJ, Garson JA, Detection of reverse transcriptase activity in the serum of patients with motor neurone disease. J. Med. Virol. 61, 527–532 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Viola MV, Frazier M, White L, Brody J, Spiegelman S, RNA-instructed DNA polymerase activity in a cytoplasmic particulate fraction in brains from Guamanian patients. J. Exp. Med 142,483–494 (1975). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alfahad T, Nath A, Retroviruses and amyotrophic lateral sclerosis. Antiviral Res. 99, 180–187 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horvath P, Barrangou R, CRISPR/Cas, the immune system of bacteria and archaea. Science 327, 167–170 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Mali P, Esvelt KM, Church GM, Cas9 as a versatile tool for engineering biology. Nat. Methods 10, 957–963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pospelova TV, Demidenko ZN, Bukreeva EI,Pospelov VA, Gudkov AV, Blagosklonny MV, Pseudo-DNA damage response in senescent cells. Cell Cycle 8,4112–4118 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfister JA, D’Mello SR, Insights into the regulation of neuronal viability by nucleophosmin/B23. Exp. Biol. Med 240, 774–786 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kerr LE,Birse-Archbold J-L, Short DM, McGregor AL, Heron I, MacDonald DC, Thompson J, Carlson GJ, Kelly JS, McCulloch J, Sharkey J, Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene 26,2554–2562 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Ou S-HI, Wu F, Harrich D, Garcia-Martfnez LF, Gaynor RB, Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol 69, 3584–3596 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Jin Y, Prazak L, Hammell M, Dubnau J, Transposable elements in TDP-43-mediated neurodegenerative disorders. PLOS One 7, e44099 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Hernandez MJ, Swanson MD, Contreras-Galindo R, Cookinham S, King SR, Noel RJ Jr., Kaplan MH, Markovitz DM, Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J. Virol. 86, 7790–7805 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Finsterer J, Burgunder J-M, Recent progress in the genetics of motor neuron disease. Eur. J. Med. Genet. 57, 103–112 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Osakada F, Callaway EM, Design and generation of recombinant rabies virus vectors. Nat. Protoc. 8, 1583–1601 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bhat RK, Rudnick W, Antony JM, Maingat F, Ellestad KK, Wheatley BM, Tonjes RR, Power C, Human endogenous retrovirus-K(ll) envelope induction protects neurons during HIV/AIDS. PLOS One 9, e97984 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer S, Echeverria N, Moratorio G, Landoni AI, Dighiero G, Cristina J, Oppezzo P, Moreno P, Human endogenous retrovirus np9 gene is over expressed in chronic lymphocytic leukemia patients. Leuk. Res. Rep 3, 70–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. , Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775 (1994). [DOI] [PubMed] [Google Scholar]
- 23.Okuwaki M, The structure and functions of NPM1/Nucleophsmin/B23, a multifunctional nucleolar acidic protein. J. Biochem. 143, 441–448 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Murano K, Okuwaki M, Hisaoka M, Nagata K, Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol. Cell. Biol. 28, 3114–3126 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haeusler AR, Donnelly CJ, Periz G, Simko EAJ, Shaw PG, Kim M-S, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J, C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DesGroseillers L, Barrette M, Jolicoeur P, Physical mapping of the paralysis-inducing determinant of a wild mouse ecotropic neurotropic retrovirus. J. Virol. 52, 356–363 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paquette Y, Hanna Z, Savard P, Brousseau R, Robitaille Y, Jolicoeur P, Retrovirus-induced murine motor neuron disease: Mapping the determinant of spongiform degeneration within the envelope gene. Proc. Natl. Acad. Sci. USA. 86, 3896–3900 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dimcheff DE, Volkert LG, Li Y, DeLucia AL, Lynch WP, Gene expression profiling of microglia infected by a highly neurovirulent murine leukemia virus: Implications for neuropathogenesis. Retrovirology 3, 26 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaul M, Lipton SA, Mechanisms of neuronal injury and death in HIV-1 associated dementia. Curr. HIV Res. 4, 307–318 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Li W, Li G, Steiner J, Nath A, Role of Tat protein in HIV neuropathogenesis. Neurotox. Res. 16, 205–220 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Cohen TJ, Lee VMY, Trojanowski JQ, TDP-43 functions and pathogenic mechanisms implicated in TDP-43 proteinopathies. Trends Mol. Med. 17, 659–667 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee YN, Bieniasz PD, Reconstitution of an infectious human endogenous retrovirus. PLOS Pathog. 3, e10 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee YN, Malim MH, Bieniasz PD, Hypermutation of an ancient human retrovirus by APOBEC3G. J. Virol. 82, 8762–8770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agoni L, Guha C, Lenz J, Detection of human endogenous retrovirus K (HERV-K) transcripts in human prostate cancer cell lines. Front. Oncol. 3, 180 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flockerzi A, Ruggieri A, Frank O, Sauter M, Maldener E, Kopper B, Wullich B, Seifarth W, Muller-Lantzsch N, Leib-Mosch C, Meese E, Mayer J, Expression patterns of transcribed human endogenous retrovirus HERV-K(HML-2) loci in human tissues and the need for a HERV Transcriptome Project. BMC Genomics 9, 354 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Freund A, Orjalo AV, Desprez P-Y, Campisi J, Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med 16, 238–246 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haase A, Frahm J, Matthaei D, Hanicke W, Merboldt K-D, FLASH imaging: Rapid NMR imaging using low flip-angle pulses. J. Magn. Reson. 213, 533–541 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Paxinos G, Franklin KBJ, The mouse brain in stereotaxic coordinates. (Elsevier Academic Press, Amsterdam, ed. Compact 2nd, 2004). [Google Scholar]
- 39.Yamamoto A, Lucas JJ, Hen R, Reversal of neuropathology and motor dysfunction in a conditional model of Huntington’s disease. Cell 101, 57–66 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Cyr M, Beaulieu J-M, Laakso A, Sotnikova TD, Yao W-D, Bohn LM, Gainetdinov RR, Caron MG, Sustained elevation of extracellular dopamine causes motor dysfunction and selective degeneration of striatal GABAergic neurons. Proc. Natl. Acad. Sci. U.S.A 100, 11035–11040 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Metz GA, Schwab ME, Behavioral characterization in a comprehensive mouse test battery reveals motor and sensory impairments in growth-associated protein-43 null mutant mice. Neuroscience 129, 563–574 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Thiessen DD, Lindzey G, Negative geotaxis in mice: Effect of balancing practice on incline behaviour in C57BL-6J male mice. Anim. Behav. 15, 113–116 (1967). [DOI] [PubMed] [Google Scholar]
- 43.Schneider CA, Rasband WS, Eliceiri KW, NIH image to imageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meijering E, Jacob M, Sarria J-CF, Steiner P, Hirling H, Unser M, Design and validation of a tool for neurite tracing and analysis in fluorescence microscopy images. Cytometry A 58, 167–176 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Lee M-H, Amin ND, Venkatesan A, Wang T, Tyagi R, Pant HC, Nath A, Impaired neurogenesis and neurite outgrowth in an HIV-gpl20 transgenic model is reversed by exercise via BDNF production and Cdk5 regulation. J. Neurovirol 19, 418–431 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vermeire J, Naessens E, Vanderstraeten H, Landi A, lannucci V, Van Nuffel A, Taghon T, Pizzato M, Verhasselt B, Quantification of reverse transcriptase activity by real-time PCR as a fast and accurate method for titration of HIV, lenti- and retroviral vectors. PLOS One 7, e50859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kowalczy MJ, Danczak-Pazdrowska A, Szramka-Paw1ak B, Zaba R, Silny W, Osmola-Mahkowska A, Expression of selected human endogenous retroviral sequences in skin and peripheral blood mononuclear cells in morphea. Arch. Med. Sci. 8,819–825 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ahn K, Kim H-S, Structural and quantitative expression analyses of HERV gene family in human tissues. Mol. Cells 28, 99–103 (2009). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Hindlimb clasping in a tail suspension test. (A) Control.
Movie S1. Hindlimb clasping in a tail suspension test. (B) Transgenic animal.
Fig. S1. HERV-K expression in autopsied human brain.
Fig. S2. Induction of HERV-K expression in human neurons.
Fig. S3. Effect of HERV-K env in postnatal neurons of mice.
Fig. S4. Expression of HERV-K env in the brain of transgenic mice.
Fig. S5. Distribution of cellular proteins in wild-type and HERV-K env transgenic mouse brains.
Fig. S6. Changes in spine morphology and dendritic damage in pyramidal neurons in HERV-K env transgenic mice.
Fig. S7. Magnetic resonance imaging and regional analysis of mouse brains.
Fig. S8. Muscle pathology in HERV-K env transgenic mice.
Fig. S9. Impairment of locomotor behavior in HERV-K env transgenic mice.
Fig. S10. TDP-43 binds to the HERV-K LTR in vivo, and TDP-43 binding to HERV-K LTR correlates with the association of a processive RNA polymerase II (phosphorylated Ser2) on the LTR.
Table S1. Clinical data for the human brain samples.
Table S2. Passive and active membrane properties of medial prefrontal cortex layer V pyramidal neurons.
Table S3. Primer sequences.