

Abstract
In order to understand the genetic diversity and structure within and between the genera of Saccharum and Erianthus, 79 accessions from five species (S. officinarum, S. spontaneum, S. robustum, S. barberi, S. sinense), six accessions of E. arundinaceus, and 30 Saccharum spp. hybrids were analyzed using 21 pairs of fluorescence-labeled highly poloymorphic SSR primers and a capillary electrophoresis (CE) detection system. A total of 167 polymorphic SSR alleles were identified by CE with a mean value of polymorphic information content (PIC) of 0.92. Genetic diversity parameters among these 115 accessions revealed that Saccharum spp. hybrids were more diverse than those of Saccharum and Erianthus species. Based on the SSR data, the 115 accessions were classified into seven main phylogenetic groups, which corresponded to the Saccharum and Erianthus genera through phylogenetic analysis and principle component analysis (PCA). We propose that seven core SSR primer pairs, namely, SMC31CUQ, SMC336BS, SMC597CS, SMC703BS, SMC24DUQ, mSSCIR3, and mSSCIR43, may have a wide appicability in genotype identification of Saccharum species and Saccharum spp. hybrids. Thus, the information from this study contibites to manage sugarcane genetic resources.
Introduction
Sugarcane (Saccharum spp.) plays a vital role as a primary sugar-producing crop (sugar 80%) and has major potential as a renewable bioenergy crop (ethanol 50%) in world agriculture1. The Saccharum complex contains six main species: the two wild species are S. spontaneum and S. robustum, and the four cultivated species are S. officinarum, S. sinense, S. barberi and S. edule2. In addition, Erianthus arundinaceus is a species of Erianthus genus with strong abiotic stress tolerance and could be widely used for modern sugarcane breeding and a potential bioenergy plant3. Currently, sugarcane commercial breeding populations in the world share a narrow genetic base due to their common origins from a number of popular cultivars, such as POJ2878, Co419 and NCo310 which were achieved in the early 1900s2. Furthermore, these exotic varieties were developed from complex interspecific hybridization through Noblization Breeding process among wild clones of S. spontaneum and S. officinarum4. There is still a great attention among sugarcane breeders in broadening the genetic base of the crop and also in taping into the gene pool of the wild relatives to enhance stress-resistance and sucrose content5.
Since the late 1980s, sugarcane breeders and geneticists have discovered and use several DNA molecular markers including amplified fragment length polymorphisms (AFLP), restriction fragment length polymorphisms (RFLP), random amplification of polymorphic DNAs (RAPD), single nucleotide polymorphism (SNP), simple sequence repeats (SSRs), inter simple sequence repeat (ISSRs), and expressed sequence tag- simple sequence repeat (EST-SSRs) to improve Saccharum breeding6. Among these molecular markers, SSR (microsatellite) markers have been widely used to study sugarcane genetic diversity7, genetic mapping8, cross-transferability9, paternity analysis10, segregation analysis11, and marker-assisted selection12. SSR primer pairs are considered the most capable marker for plant genetics and breeding programs, because of co-dominant, multi-allelic nature, and relatively abundant with an excellent genome coverage13.
Early molecular marker research focused on the origin of wild Saccharum species. Lu et al.14 proposed a hybrid origin for S. barberi and S. sinense from natural hybridization between S. spontaneum and S. officinarum, based on a factorial correspondence analysis of RFLP markers. Subsequently, these results were supported by Irvine15 and Selvi et al.16 using SSR markers and by D’Hont et al.17 utilizing genomic in situ hybridization (GISH). Based on analysis of agronomic traits and mitochondrial profiles, S. barberi and S. sinense were placed in adjacent clusters, but apart from S. robustum18–20. Later on a number of reports focused on the analysis of genetic diversity and population structure among commercial Saccharum spp. hybrids varieties7,21–25 and among S. spontaneum populations with different ploidy levels in China26. Therefore, there has been an increasing interest among sugarcane breeders to investigate the genetic diversity of parental resources and to broaden the genetic base by tapping into the gene pools of the wild relatives27–29.
To better understand the genetic background of these euploid sugarcane clones, this study aimed to characterize the genetic diversity and population structure of 115 accessions belonging to S. officinarum, S. spontaneum, S. robustum, S. barberi, S. sinense, E. arundinaceus, and Saccharum spp. hybrids. The results may provide invaluable information for the better utilization of Saccharum and Erianthus wild germplasms at different ploidy levels in sugarcane breeding.
Results
Total alleles amplification of 21 SSR markers
A total of 167 SSR alleles were amplified from the DNA of 115 accessions including five Saccharum species, E. arundinaceus, and 30 clones of Saccharum spp. hybrids with the 21 fluorescence-labeled SSR primer pairs and capillary electrophoresis (CE) detection system. We could not find in our CE data the 16 SSR alleles reported earlier by Pan30, but instead, we have found 38 new SSR alleles that were never reported before (Table 1). Furthermore, the numbers of new and absent SSR alleles detected in this study were greater than the 20 new and 13 absent SSR alleles reported previously by Ali et al.7.
Table 1.
The general utility and amplification profile of 21 SSR primer pairs based on a capillary electrophoresis (CE) detection platform.
| No. | Markersa | Size range (bp) | Number of original bands | Number of detected bands | Absent alleles (bp)b | New alleles (bp)b | PIC |
|---|---|---|---|---|---|---|---|
| 1 | SMC119CG | 104–135 | 5 | 6 | ND | 104 | 0.92 |
| 2 | SMC1604SA | 105–130 | 6 | 7 | 109,124 | 105,107,110 | 0.93 |
| 3 | SMC1751CL | 132–160 | 5 | 7 | ND | 132,138 | 0.94 |
| 4 | SMC18SA | 135–150 | 5 | 6 | ND | 135 | 0.93 |
| 5 | SMC22DUQ | 125–165 | 7 | 8 | 125 | 142146 | 0.94 |
| 6 | SMC24DUQ* | 124–150 | 6 | 10 | ND | 124,133,139,150 | 0.95 |
| 7 | SMC278CS | 138–182 | 9 | 9 | 140,153,176 | 138,164,172 | 0.94 |
| 8 | SMC31CUQ* | 135–180 | 11 | 12 | 138 | 135,169 | 0.95 |
| 9 | SMC334BS | 143–165 | 6 | 7 | ND | 143 | 0.94 |
| 10 | SMC336BS* | 140–185 | 11 | 10 | 154 | 0 | 0.95 |
| 11 | SMC36BUQ | 100–125 | 3 | 4 | ND | 102 | 0.80 |
| 12 | SMC486CG | 220–245 | 5 | 5 | 227 | 235 | 0.91 |
| 13 | SMC569CS | 165–225 | 5 | 5 | 167,170,222 | 165,202 | 0.82 |
| 14 | SMC597CS* | 140–180 | 11 | 13 | ND | 150,152 | 0.95 |
| 15 | SMC703BS* | 200–225 | 9 | 11 | ND | 204,218 | 0.95 |
| 16 | SMC7CUQ | 140–170 | 6 | 7 | 158 | 143,160 | 0.90 |
| 17 | SMC851MS | 125–145 | 6 | 7 | ND | 138 | 0.94 |
| 18 | mSSCIR3* | 140–190 | 10 | 10 | 141,145 | 149,169 | 0.95 |
| 19 | mSSCIR43* | 200–255 | 9 | 10 | 209 | 203,229 | 0.95 |
| 20 | mSSCIR66 | 120–145 | 4 | 7 | ND | 125,136,142 | 0.94 |
| 21 | mSSCIR74 | 210–232 | 5 | 6 | ND | 214 | 0.94 |
| Total | 144 | 167 | |||||
| Average | 7.95 | 0.92 |
aCore primer was marked with asterisk (*).
bAbsent and new alleles detected in this study comparing with the 144 alleles Pan30; ND, no data.
The number of alleles detected by the CE system varied from as few as four (SMC36BUQ) to as many as 13 (SMC597CS), with an average of 7.95 per SSR primer pair. Seven SSR primer pairs, namely SMC24DUQ, SMC31CUQ, SMC336BS, SMC597CS, SMC703BS, mSSCIR3 and mSSCIR43, were highly polymorphic, each producing 10 to 13 alleles. Other eleven SSR primer pairs, namely, SMC119CG, SMC1604SA, SMC1751CL, SMC18SA, SMC22DU, SMC278CS, SMC334BS, SMC7CUQ, SMC851MS, mSSCIR66 and mSSCIR74, were moderately polymorphic, each producing six to nine alleles. The remaining three SSR primer pairs, namely, SMC36BUQ, SMC486CG and SMC569CS, were less polymorphic by producing less than six alleles each (Table 1). The PIC values of these primer pairs ranged from 0.80 (SMC36BUQ) to 0.95 (SMC24DUQ, SMC31CUQ, SMC336BUQ, SMC597CS, SMC703BS, mSSCIR3, mSSCIR43) with an average of 0.92 (Table 1).
The PIC values of each Saccharum and E. arundinaceus species were also calculated in our study. The maximum PIC value was 0.95 for mSSCIR3 on S. spontaneum and the minmum PIC value was 0.28 for SMC119CG on E. arundinaceus. Generally, higher PIC values were found in Saccharum spp. hybrids with an average value of 0.87, followed by an average PIC value of 0.86 in S. spontaneum (Table 2).
Table 2.
Polymorphism information content (PIC) of 21 SSR primer pairs analysed using 115 accessions from Saccharum, Erianthus, and Saccharum spp. hybrids.
| No. | SSR markers | S. spontaneum | S. officinarum | S. barberi | S. robustum | S. sinense | Saccharum spp. hybrids | E. arundinaceus |
|---|---|---|---|---|---|---|---|---|
| 1 | SMC119CG | 0.82 | 0.91 | 0.79 | 0.73 | 0.76 | 0.86 | 0.28 |
| 2 | SMC7CUQ | 0.88 | 0.84 | 0.67 | 0.85 | 0.62 | 0.84 | 0.38 |
| 3 | SMC18SA | 0.86 | 0.82 | 0.80 | 0.81 | 0.70 | 0.91 | 0.50 |
| 4 | SMC22DUQ | 0.88 | 0.89 | 0.83 | 0.73 | 0.61 | 0.88 | 0.59 |
| 5 | SMC24DUQ | 0.85 | 0.88 | 0.83 | 0.73 | 0.67 | 0.88 | 0.55 |
| 6 | SMC31CUQ | 0.90 | 0.88 | 0.64 | 0.83 | 0.81 | 0.91 | 0.76 |
| 7 | SMC36BUQ | 0.79 | 0.68 | 0.59 | 0.61 | 0.63 | 0.82 | 0.72 |
| 8 | SMC278CS | 0.93 | 0.77 | 0.65 | 0.81 | 0.81 | 0.86 | 0.78 |
| 9 | SMC334BS | 0.91 | 0.72 | 0.84 | 0.71 | 0.67 | 0.84 | 0.38 |
| 10 | SMC336BS | 0.92 | 0.88 | 0.84 | 0.71 | 0.71 | 0.88 | 0.73 |
| 11 | SMC486CG | 0.77 | 0.72 | 0.80 | 0.68 | 0.72 | 0.86 | 0.45 |
| 12 | SMC569CS | 0.61 | 0.77 | 0.49 | 0.73 | 0.61 | 0.83 | 0.55 |
| 13 | SMC597CS | 0.83 | 0.86 | 0.82 | 0.72 | 0.35 | 0.93 | 0.78 |
| 14 | SMC703BS | 0.82 | 0.60 | 0.56 | 0.77 | 0.64 | 0.92 | 0.88 |
| 15 | SMC851MS | 0.89 | 0.91 | 0.83 | 0.72 | 0.83 | 0.88 | 0.79 |
| 16 | SMC1604SA | 0.89 | 0.83 | 0.66 | 0.82 | 0.62 | 0.86 | 0.74 |
| 17 | SMC1751CL | 0.88 | 0.84 | 0.79 | 0.80 | 0.72 | 0.82 | 0.78 |
| 18 | mSSCIR3 | 0.95 | 0.91 | 0.78 | 0.86 | 0.85 | 0.89 | 0.89 |
| 19 | mSSCIR43 | 0.92 | 0.90 | 0.82 | 0.85 | 0.58 | 0.91 | 0.88 |
| 20 | mSSCIR66 | 0.86 | 0.86 | 0.82 | 0.82 | 0.80 | 0.82 | 0.76 |
| 21 | mSSCIR74 | 0.92 | 0.92 | 0.81 | 0.81 | 0.78 | 0.83 | 0.79 |
| Average | 0.86 | 0.83 | 0.75 | 0.77 | 0.69 | 0.87 | 0.66 |
Genetic variability
Using the CE detection system, an average of 138 polymorphic SSR bands was observed in each Saccharum or E. arundinaceus species. Among the five species of Saccharum, one species of Erianthus, and Saccharum spp. hybrids, both the highest number of polymorphic loci (NPL) and the highest percentage of polymorphic loci (PPL) were observed in Saccharum spp. hybrids population (NPL = 165, PPL = 98.8%), followed by S. spontaneum (NPL = 159, PPL = 95.21%), while the lowest number and percentage of polymorphic loci were found in E. arundinaceus (NPL = 93, PPL = 55.69%) (Fig. 1a). The highest number of observed alleles (Na = 1.98) was found in Saccharum spp. hybrids, while the lowest number of observed alleles (Na = 1.55) was found in E. arundinaceus (Fig. 1b). Morever, the highest number of effective alleles (Ne = 1.70) was found in the Saccharum spp. hybrids, followed by S. spontaneum (Ne = 1.64). The lowest number of effective alleles (Ne = 1.30) was observed in E. arundinaceus (Fig. 1c). Shannon’s index information of different populations ranged from 0. 28 (E. arundinaceus) to 0.57 (Saccharum. spp. hybrids). Analysis of Shannon’s index (I) showed that Saccharum spp. hybrids and S. spontaneum were different from the rest of other Sccharum species by sharing the highest shannon’s index value of 0.57. The lowest shannon’s diversity index value of 0.28 was observed in S. sinense (Fig. 1d). The Nei’s gene diversity (h) of the seven populations ranged from 0.21 to 0.39. The higher genetic diversity values of 0.39, 0.36 and 0.34 were observed in Saccharum spp. hybrids, S. spontaneum and S. officinarum populations, respectivaly; while the E. arundinaceus and S. barberi populations had the lower genetic diversity values of 0.21 and 0.26 (Fig. 1e).
Figure 1.

Statistical analysis of genetic variability among Saccharum, Erianthus and Saccharum spp. hybrids populations based on SSR data. Polymorphism index (PI) (a), Number of observed alleles (Na) (b), Number of effective alleles (Ne) (c), Shannon’s index (I) (d), and Nei’s genetic diversity (h) (e).
Principal Component Analysis (PCA)
Principal component analysis (PCA) data for all 115 accessions are shown in Fig. 2. The analysis classified these accessions into evelen groups involving different Saccharum and Erianthus species to some extent, i.e., Group I-A and I-B (Saccharum spp. hybrids), Group II-A and II-B (S. spontaneum), Group III (S. barberi), Group IV-A and IV-B (S. robustum), Group V-A and V-B (S. sinense), Group VI (S. officinarum), and Group VII (E. arundinaues) (Fig. 2). The amount of variance accounted for by the globle three-dimensional plot is 13.4% of Dim1, 7.12% of Dim2, and 6.51% of Dim3, with a total of 27.03% for three dimensions. This is an acceptale fit, given the small amount of variability from the large number of accessions and SSR alleles used in the analysis.
Figure 2.
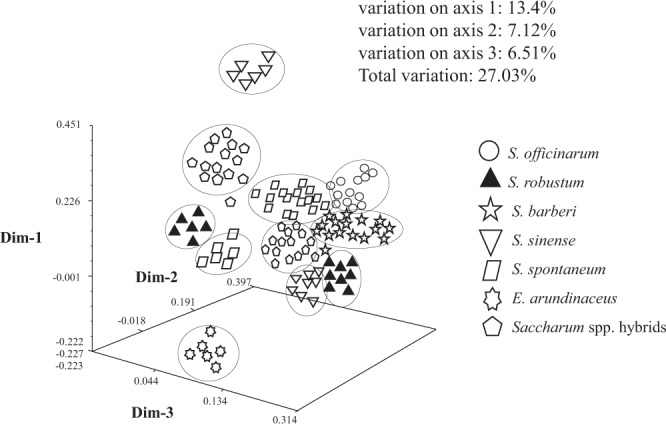
Three-dimensional principal component analysis (PCA) plot of Saccharum, Erianthus, and Saccharum spp. hybrids based on SSR data.
Phyolgenetic analysis
A phylogenetic tree is shown in Fig. 3. Based on phylogenetic analysis, the 115 accessions were clearly clustered at Saccharum and Erianthus genera level into seven major clades, also involving different Saccharum and Erianthus species to some extent. Clade-I contained 27 accessions from S. officinarum, S. robustum, S. barberi and S. sinense. Clade-II included 16 accessions from S. spontaneum. Clade-III comprised of three accessions of S. officinarum, three accessions of S. robustum, and three accessions of S. barberi. Clade-IV and Clade-V held 22 accessions of Saccharum spp. Hybrids. Clade-VI clustered 13 accessions of S. robustum and S. spontaneum and five accessions of E. arundinaceus. However, one E. arundinaceus accession, Guizhou 78-I-24 (Earu05), was clustered with six S. spontaneum accessions. Finally, Clade-VII contained eight accessions of Saccharum spp. hybrids, four accessions of S. officinarum, five accessions of S. barberi, and six accessions of S. sinense.
Figure 3.

Phylogenetic trees of Saccharum, Erianthus, and Saccharum spp. hybrids based on SSR data. A distance tree was constructed in MEGA 6 using the UPGMA method. Robustness of the node of the phylogenetic tree was assessed from 1000 bootstrap replicates and bootstrap values of >60% are shown.
To verify some core SSR primer pairs out of the 21 primer pairs, we compared two phylogenetic trees constructed based on CE-data of 21 SSR primer pairs vs 7 SSR primer pairs and of 21 SSR primer pairs vs 6 SSR primer pairs with the Robinson-Foulds distance. Further analysis with Dendextend showed a higher cophenetic correlation coefficient value (0.93) between 21 SSR primer pairs and 7 SSR primer pairs than the 0.91 cophenetic correlation coefficient value between 21 SSR primer pairs and 6 SSR primer pairs. The plots of two phylogenetic trees based on the CE-data of 21 SSR primer pairs vs 7 SSR primer pairs are shown in Fig. 4 with tanglegrams.
Figure 4.

Two phylogenetic trees constrcuted using SSR data derived from 21 SSR primer pairs vs 7 SSR primer pairs with tanglegrams.
Genetic identity analysis
Percent of genetic identity was estimated between and within the seven phylogenetic groups. Percent genetic identity between phylogenetic groups ranged from 26.9% (Saccharum spp. hybrids and S. spontaneum) to 96.4% (E. arundinaceus and S. spontaneum). Percent genetic identity within phylogenetic groups ranged from 38.9% (within S. barberi or Saccharum spp. hybrids) to 100% (within S. robustum) (Table 3).
Table 3.
Genetic identity (%) among five Saccharum species, one Erianthus species, and Saccharum spp. hybrids based on SSR data.
| Populations | S. spontaneum | S. officinarum | S. barberi | S. robustum | S. sinense | Saccharum spp. hybrids | E. arundinaceus |
|---|---|---|---|---|---|---|---|
| S. spontaneum | 98.8–39.5 | ||||||
| S. officinarum | 74.8–34.7 | 97.6-39.5 | |||||
| S. barberi | 73.0–37.7 | 83.2–36.5 | 99.4–38.9 | ||||
| S. robustum | 79.0–36.5 | 83.2–37.1 | 79.0–37.1 | 100–431 | |||
| S. sinense | 73.6–42.5 | 74.2–46.7 | 78.4–41.3 | 74.2–44.3 | 99.4–43.7 | ||
| Saccharum spp. hybrids | 73.6–26.9 | 79.6–29.9 | 72.4–33.5 | 70.0–28.1 | 69.4–35.9 | 98.8–38.9 | |
| E. arundinaceus | 96.4–40.1 | 56.2–38.3 | 53.8–39.5 | 59.2–40.7 | 57.4–37.1 | 57.4–39.5 | 96.4–50.8 |
Discussion
Since 1950s, wild accessions of Saccharum and Erianthus have been continuously collected on mainland China and maintained in the Sugarcane Germplasm Nurseries in Yacheng, Hainan province or Kaiyuan, Yunnan Province, China. However, the genetic relationship and molecular identification between these two germplasm collections have never been entirely examined. Molecular markers are considered to be most effective in analyzing the genetic diversity, population structure, and phylogenetic relationship within sugarcane germplasm31. In recent years, SSR markers are proven to be very useful for a variety of applications in plants, including linkage maps analysis, segregation analysis, population structure analysis, marker-assisted selection, assessment of genetic relationships between individuals, mapping genes of interest, and marker-assisted backcrosses, population genetics and phylogenetic studies32,33.
In this study, we investigated the genetic diversity and population structure for 115 accessions of Saccharum and Erianthus genera that originated from two collections on mainland China and a local collection in the USA by 21 SSR primer pairs. The 21 primer pairs primed the amplification of 167 polymorphic SSR alleles detectable by the CE platform, of which 38 alleles have never been reported before. Every primer pair was able to amplify varying numbers of SSR alleles from all accessions tested, regardless of their geographical origins. Seven core SSR primer pairs, namely, SMC24DUQ, SMC31CUQ, SMC336BS, SMC597CS, SMC703BS, mSSCIR3, and mSSCIR43, produced more than ten alleles among the 115 accessions, while four of the seven core primer pairs, namely, SMC31CUQ, SMC336BS, SMC597CS, and mSSCIR3, also primed the amplification of more than 10 alleles among 92 Chinese commercial sugarcane varieties7. Therefore, these seven core SSR primer pairs would have a priority of choice in identifying clones either from Saccharum species or Saccharum spp. hybrids.
The number of polymorphic SSR alleles detected in this study was higher than the 144 alleles reported by Pan30 or the 151 alleles reported by Ahmad et al.7, but lower than the 205 polymorphic alleles reported by You et al.25. We considered that the differences were due to different Saccharum clones being used in previous studies or to different scoring criteria. The differences may also be due to the complex genomes of Saccharum and Erianthus on one hand and relatively narrow genetic base of commercial sugarcane varieties on the other hand.
In this study, we observed different levels of genetic variations among accessions of Saccharum and Erianthus tested. In general, Saccharum spp. hybrids and S. spontaneum accessions had a higher genetic diversity than S. sinense and E. arundinaceus accessions. However, the highest number of observed alleles, number of effective alleles and polymorphism index were observed in accessions of Saccharum spp. hybrids, which are polyploidy with genome contributions from several Saccharum species. Historically, the modern Saccharum spp. hybrids were developed from crosses between the “Noble” cane S. officinarum and its relatives, namely, S. spontaneum, S. sinense, or S. barberi in the early 20th century34,35. The overall genetic variation values from this study were higher than those reported by You et al.24,25. We hypothesize that this phenomenon was due to the utilization of a larger number of SSR primer pairs and the large number of accessions from diverse Saccharum and Erianthus species in our study.
It is worthnoting that the 21 SSR primer pairs worked well in clustering Saccharum, Erianthus, and Saccharum spp. hybrids clones during phylogenetic analysis process. Two Saccharum spp. hybrids clones [(R570 (Sspp17)] from France and [(Q124 (Sspp18)] from Australia were clustered into a sub-clade in Clade-VII with four accessions of S. officinarum. The reason could be that R570 and Q124 varieties may have a closer affiliation with S. officinarum. In addition, the six accessions of E. arundinaceus were clustered with accessions of S. robustum and S. spontaneum in Clade-VI rather than forming a separate clade. This was because all the 21 SSR primer pairs were designed from the genomic DNA sequences of two cultivars, either Q124 or R-57030. Unlike some consensus primers that are able to prime the PCR amplification of plant genomic sequences36, these SSR primer pairs may not be able to amplify Erianthus genomic DNA at equivalent efficiency as they do to the Saccharum genomes. Another reason is that it is now generally accepted that Noble cultivars might directly emerge from S. robustum. It also has hypothesized that S. robustum be evolved from complex introgressions between S. spontaneum and other genera, particularly Erianthus and Miscanthus sharing close genetic affiliation37,38. The genetic diversity results from our study were in general conformity with the evolutionary course of the sugarcane cultivars in that the order of contributing species in today’s accessions is S. officinarum, S. spontaneum, S. robustum, S. barberi, S. sinense, E. arundinaceus and Saccharum spp. hybrids. PCA analysis also revealed a similar pattern of phylogeny to some extent.
Today, China holds more than 2,000 accessions of Saccharum and Erianthus, among which some are wild types. These accessions are either China-born or through foreign introductions. As the size of sugarcane germplasm grows, the genetic information among accessions becomes more critical for maintaining and utilization strategies designed to establish cross parentages in China’s breeding programs. We conclude that the estimation of genetic diversity and population structure of 115 accessions of Saccharum and Erianthus genera using SSR primer pairs may provide more accurate information to sugarcane breeders than the classical pedigree method. The 21 SSR primer pairs used in our study may also be of potential value for further research on genetic mapping, segregation analysis, marker-assisted selections, QTL mapping and gene tagging in sugarcane. In addition, further study with consensus PCR primers may be needed to assess the phylogenetic status of the Erianthus genus within the “Saccharum Complex”38.
Materials and Methods
Plant materials
One hundred and fifteen asseccions were used in this study, including 12 accessions from S. officinarum, 22 from S. spontaneum, 14 from S. robustum, 17 from S. barberi, 14 from S. sinense, 30 from Saccharum spp. hybrids, and six from E. arundinaceus. The leaf samples of all the clones were collected either from the Sugarcane Germplasm Nursery in Yacheng, Hainan, China or a local collection at the USDA-ARS, Sugarcane Research Unit, Houma, Louisiana, USA (Table 4). The leaf samples were collected, wiped off with 75% ethanol, and kept at −80 °C until DNA extraction.
Table 4.
A list of 115 accessions from Saccharum, Erianthus, and Saccharum spp. hybrids.
| No. | Accessions name | Sample no. | Species | No. | Accessions name | Sample no. | Species |
|---|---|---|---|---|---|---|---|
| 1 | 48Mouna | Soff01 | S. officinarum | 59 | Djatiroto | Sspo02 | S. spontaneum |
| 2 | Badila | Soff02 | S. officinarum | 60 | Fujian 79-I-1 | Sspo03 | S. spontaneum |
| 3 | Bandjermasin Hitam | Soff03 | S. officinarum | 61 | Guangdong 29 | Sspo04 | S. spontaneum |
| 4 | Barwhspt | Soff04 | S. officinarum | 62 | Guangdong 51 | Sspo05 | S. spontaneum |
| 5 | EK02 | Soff05 | S. officinarum | 63 | IMP9068 | Sspo06 | S. spontaneum |
| 6 | Fiji1 | Soff06 | S. officinarum | 64 | IMP9089 | Sspo07 | S. spontaneum |
| 7 | IN84-068B | Soff07 | S. officinarum | 65 | IND81-080 | Sspo08 | S. spontaneum |
| 8 | LA Purple | Soff08 | S. officinarum | 66 | Mol1032A | Sspo09 | S. spontaneum |
| 9 | Muntok Java | Soff09 | S. officinarum | 67 | Mpth97-107 | Sspo10 | S. spontaneum |
| 10 | NG21-003 | Soff10 | S. officinarum | 68 | Mpth97-233 | Sspo11 | S. spontaneum |
| 11 | NG57-223 | Soff11 | S. officinarum | 69 | PCANOR84-2A | Sspo12 | S. spontaneum |
| 12 | Striped Cheribon | Soff12 | S. officinarum | 70 | PCAV84-12A | Sspo13 | S. spontaneum |
| 13 | 51NG208 | Srob01 | S. robustum | 71 | PQ84-3 | Sspo14 | S. spontaneum |
| 14 | 51NG63 | Srob02 | S. robustum | 72 | S66-084A | Sspo15 | S. spontaneum |
| 15 | IJ76-339 | Srob03 | S. robustum | 73 | S66-121A | Sspo16 | S. spontaneum |
| 16 | IN84-045 | Srob04 | S. robustum | 74 | SES323A | Sspo17 | S. spontaneum |
| 17 | IN84-076 | Srob05 | S. robustum | 75 | SPONT24 | Sspo18 | S. spontaneum |
| 18 | M3035-66 | Srob06 | S. robustum | 76 | SPONT37 | Sspo19 | S. spontaneum |
| 19 | NG28-289 | Srob07 | S. robustum | 77 | Yacheng 11 | Sspo20 | S. spontaneum |
| 20 | NG57-055 | Srob08 | S. robustum | 78 | Yacheng 12 | Sspo21 | S. spontaneum |
| 21 | NG77-004 | Srob09 | S. robustum | 79 | Yunnan 82-114 | Sspo22 | S. spontaneum |
| 22 | NG77-1 | Srob10 | S. robustum | 80 | Fijian 87-II-5 | Earu01 | E. arundinaceus |
| 23 | NG77-159 | Srob11 | S. robustum | 81 | Guangxi 83-27 | Earu02 | E. arundinaceus |
| 24 | NG77-235 | Srob12 | S. robustum | 82 | Hainan 92-79 | Earu03 | E. arundinaceus |
| 25 | NG77-75 | Srob13 | S. robustum | 83 | Hainan 92-105 | Earu04 | E. arundinaceus |
| 26 | Teboe Salak Toewa | Srob14 | S. robustum | 84 | Guizhou 78-I-24 | Earu05 | E. arundinaceus |
| 27 | Agoule | Sbar01 | S. barberi | 85 | Sichuan 79-I-13 | Earu06 | E. arundinaceus |
| 28 | Chunnee | Sbar02 | S. barberi | 86 | HoCP01-517 | Sspp01 | S. spp. hybrids |
| 29 | Dhaula | Sbar03 | S. barberi | 87 | HoCP85-845 | Sspp02 | S. spp. hybrids |
| 30 | Hatuni | Sbar04 | S. barberi | 88 | HoCP91-555 | Sspp03 | S. spp. hybrids |
| 31 | HulluKabbu | Sbar05 | S. barberi | 89 | HoCP96-540 | Sspp04 | S. spp. hybrids |
| 32 | Kacai | Sbar06 | S. barberi | 90 | L01-283 | Sspp05 | S. spp. hybrids |
| 33 | Keari | Sbar07 | S. barberi | 91 | L01-299 | Sspp06 | S. spp. hybrids |
| 34 | Khagzi | Sbar08 | S. barberi | 92 | L97-128 | Sspp07 | S. spp. hybrids |
| 35 | Mungo | Sbar09 | S. barberi | 93 | L99-233 | Sspp08 | S. spp. hybrids |
| 36 | Nagans | Sbar10 | S. barberi | 94 | LCP85-384 | Sspp09 | S. spp. hybrids |
| 37 | NEWRA | Sbar11 | S. barberi | 95 | MEX85-6196 | Sspp10 | S. spp. hybrids |
| 38 | Pansahi | Sbar12 | S. barberi | 96 | TCP93-4245 | Sspp11 | S. spp. hybrids |
| 39 | Pathri | Sbar13 | S. barberi | 97 | TCP97-4442 | Sspp12 | S. spp. hybrids |
| 40 | Rounda | Sbar14 | S. barberi | 98 | TCP98-4445 | Sspp13 | S. spp. hybrids |
| 41 | Ruckri | Sbar15 | S. barberi | 99 | TCP98-4447 | Sspp14 | S. spp. hybrids |
| 42 | Sewari | Sbar16 | S. barberi | 100 | US01-40 | Sspp15 | S. spp. hybrids |
| 43 | Sunnabile | Sbar17 | S. barberi | 101 | US02-99 | Sspp16 | S. spp. hybrids |
| 44 | Binchuanxiaozhe | Ssin01 | S. sinense | 102 | Q124 | Sspp17 | S. spp. hybrids |
| 45 | ChukChe | Ssin02 | S. sinense | 103 | R570 | Sspp18 | S. spp. hybrids |
| 46 | Guangdongzhuzhe | Ssin03 | S. sinense | 104 | ROC10 | Sspp19 | S. spp. hybrids |
| 47 | Guangxizhuzhe | Ssin04 | S. sinense | 105 | ROC16 | Sspp20 | S. spp. hybrids |
| 48 | Merehi | Ssin05 | S. sinense | 106 | ROC20 | Sspp21 | S. spp. hybrids |
| 49 | MiaLan | Ssin06 | S. sinense | 107 | ROC22 | Sspp22 | S. spp. hybrids |
| 50 | Nepal3 | Ssin07 | S. sinense | 108 | ROC25 | Sspp23 | S. spp. hybrids |
| 51 | TanzhonBamboo | Ssin08 | S. sinense | 109 | ROC27 | Sspp24 | S. spp. hybrids |
| 52 | Tanzhouzhuzhe | Ssin09 | S. sinense | 110 | FN41 | Sspp25 | S. spp. hybrids |
| 53 | TekchaOkinawa | Ssin10 | S. sinense | 111 | GT40 | Sspp26 | S. spp. hybrids |
| 54 | UbaDelNatal | Ssin11 | S. sinense | 112 | MT01-77 | Sspp27 | S. spp. hybrids |
| 55 | UbaIndia | Ssin12 | S. sinense | 113 | LC05-136 | Sspp28 | S. spp. hybrids |
| 56 | UbaNaquin | Ssin13 | S. sinense | 114 | YT00-236 | Sspp29 | S. spp. hybrids |
| 57 | Wenshanzhuzhe | Ssin14 | S. sinense | 115 | YZ05-51 | Sspp30 | S. spp. hybrids |
| 58 | Coimbatore | Sspo01 | S. spontaneum |
DNA extraction
Genomic DNA was extracted from leaf tissues using a modified cetyl tri-methyl ammonium bromide (CTAB) method39 as previously described by Ahmad et al.7. The quality and concentration of DNA were measured using UV absorbance assay with a Synergy™ H1 Multi-Mode Reader (BioTek, Winooski, VT, USA) and 0.8% agarose gel electrophoresis with ethidium bromide staining.
SSR markers and SSR reactions
The 21 polymorphic SSR primer pairs from Pan30 were used in this study based on their high PIC values of greater than 0.787,40,41. All forward primers were labeled with the fluorescence dye, 6-carboxy-fluorescein (FAM). Serials of PCR-cycling conditions were performed to detect the SSR DNA fingerprints7,30. The PCR products for the capillary electrophoresis (CE) were conducted on ABI 3730XL DNA Analyzer (Applied Biosystems Inc., Foster City, CA, USA) following the manufacturer’s instructions to generate GeneScan files.
Marker scoring
The GeneScan files were analyzed with the GeneMarker™ software (version 1.80) (SoftGenetics LLC®, State College, PA, USA, www.softgenetics.com) to reveal capillary electrophoregrams of PCR amplified SSR-DNA fragments. Fragment sizes were computed automatically against the GS500 DNA size standards (Applied Biosystems, Inc., Foster City, CA, USA). SSR alleles were manually assigned to unique, true “Plus-adenine” DNA fingerprints that gave quantifiable fluorescence values. Irregular peaks and stutters peaks were not scored according to Pan et al.41. Data were scored manually in a binary format into a data matrix file, with the presence of a band scored as “1” or “A” and its absence scored as “0” or “C”. The polymorphism information content (PIC) values were calculated using the formula of Liu et al.23.
Where Pij is the frequency of jth allele for ith locus and summation extends over n alleles.
Data analysis
The allelic data matrix of “1” or “0” was used to calculate the population genetic analysis using POPGENE version 1.3242, including number of observed alleles (Na), and number of effective alleles (Ne). Nei’s genetic diversity (h), polymorphism index (PI) and Shannon’s index (I) were computed for each Saccharum and Erianthus populations based on the obtained allele frequencies. The allelic data matrix of “A” or “C” was used to perform phylogenetic analysis. Phylogenetic tree was constructed with MEGA 6 using UPGMA statistical method with substitution model of Maximum Composite Likelihood43. Robustness of the node of the phylogenetic tree was assessed from 1000 bootstrap replicates. To find out the core-primer pairs of 21 SSR primer pairs, two other phylogenetic trees were constructed using SSR data from six or seven highly polymorphic SSR primer pairs. Then, the three phylogenetic tree files were calculated and Robinson-Foulds distances of 21 SSR vs 7 SSR and 21 SSR vs 6 SSR determined with Phangorn Package44 and cophenetic correlation coefficients of the topological distance were analyzed with Dendextend45. To better view the comparison between trees, Dendextend were used to plot two trees with tanglegrams. Genetic identity matrix was calculated using BioEdit Sequence Alignment Editor Version 7.1.946. Genetic similarity coefficients among Saccharum and Erianthus populations were estimated with the SIMQUAL subprogram using the Jaccard’s coefficient, followed by principal component analysis (PCA) with the DICE subprogram as implemented in NTSYS-pc version 2.10e47.
Acknowledgements
This work was supported by Sugar Crop Research System, CARS (No. CARS-170302) and the Major Science and Technology Project of Fujian Province, China (No. 2015NZ0002-2).
Author Contributions
San-Ji Gao conceived the project and designed the experiments. Ali Ahmad performed the experiments, analyzed the data, and drafted the manuscript. Yong-Bao Pan collected the leaf samples, extracted the DNA in Louisiana, USA, interpreted SSR data, and critically revised the manuscript. Qin-Nan Wang collected the leaf samples from the Sugarcane Germplasm Nursery in Hainan, China and drafted the manuscript. Jin-Da Wang and Jun-Lü Chen participated in processing of SSR data and revision of the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Ahmad Ali, Yong-Bao Pan and Qin-Nan Wang contributed equally.
References
- 1.Waclawovsky AJ, Sato PM, Lembke CG, Moore PH, Souza GM. Sugarcane for bioenergy production: an assessment of yield and regulation of sucrose content. Plant Biotechnol J. 2010;8:263–276. doi: 10.1111/j.1467-7652.2009.00491.x. [DOI] [PubMed] [Google Scholar]
- 2.Hunsigi, G. Production of Sugarcane Theory & Practice. Berlin Heidelberg: Springer-Verlag. pp. 12–28 (1993).
- 3.Yan J, et al. Ploidy level and DNA content of Erianthus arundinaceus as determined by flow cytometry and the association with biological characteristics. PLoS ONE. 2016;11:e0151948. doi: 10.1371/journal.pone.0151948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fukuhara S, et al. Identification and characterization of intergeneric hybrid of commercial sugarcane (Saccharum spp. hybrid) and Erianthus arundinaceus (Retz.) Jeswiet. Euphytica. 2013;189:321–7. doi: 10.1007/s10681-012-0748-3. [DOI] [Google Scholar]
- 5.Cai Q, et al. Assessment of the genetic diversity in a collection of Erianthus arundinaceus. Genet Resour Crop Evol. 2012;59:1483–91. doi: 10.1007/s10722-011-9776-4. [DOI] [Google Scholar]
- 6.Parida SK, et al. Informative genomic microsatellite markers for efficient genotyping applications in sugarcane. Theor Appl Genet. 2009;118:327–38. doi: 10.1007/s00122-008-0902-4. [DOI] [PubMed] [Google Scholar]
- 7.Ahmad A, et al. Molecular identification and genetic diversity analysis of Chinese sugarcane (Saccharum spp. hybrids) varieties using SSR markers. Trop Plant Biol. 2017;10:194–203. doi: 10.1007/s12042-017-9195-6. [DOI] [Google Scholar]
- 8.Singh RK, et al. Identification of putative candidate genes for red rot resistance in sugarcane (Saccharum species hybrid) using LD-based association mapping. Mol Genet Genomics. 2016;291:1363–1377. doi: 10.1007/s00438-016-1190-3. [DOI] [PubMed] [Google Scholar]
- 9.Singh RK, Singh RB, Singh SP, Sharma ML. Identification of sugarcane microsatellites associated to sugar content in sugarcane and transferability to other cereal genomes. Euphytica. 2011;182:335–54. doi: 10.1007/s10681-011-0484-0. [DOI] [Google Scholar]
- 10.Tew TL, Pan Y. Microsatellite (simple sequence repeat) marker–based paternity analysis of a seven-parent sugarcane polycross. Crop Sci. 2010;50:1401–1408. doi: 10.2135/cropsci2009.10.0579. [DOI] [Google Scholar]
- 11.Lu X, et al. Segregation analysis of microsatellite (SSR) markers in sugarcane polyploids. Genet Mol Res. 2015;14:18384–18395. doi: 10.4238/2015.December.23.26. [DOI] [PubMed] [Google Scholar]
- 12.Liu X, et al. Identification of progenies from sugarcane × Narenga porphyrocoma (Hance) Bor. by SSR marker. Southwest China J Agri Sci. 2012;25:38–43. [Google Scholar]
- 13.Aitken KS, Jackson PA, McIntyre CL. A combination of AFLP and SSR markers provides extensive map coverage and identification of homo(eo) logous linkage groups in a sugarcane cultivar. Theor Appl Genet. 2005;110:789–801. doi: 10.1007/s00122-004-1813-7. [DOI] [PubMed] [Google Scholar]
- 14.Lu YH, D’Hont A, Walker DIT, Rao PS. Relationships among ancestral species of sugarcane revealed with RFLP using single copy maize nuclear probes. Euphytica. 1994;78:7–18. doi: 10.1007/BF00027520. [DOI] [Google Scholar]
- 15.Irvine JE. Saccharum species as horticultural classes. Theor Appl Genet. 1999;98:186–194. doi: 10.1007/s001220051057. [DOI] [Google Scholar]
- 16.Selvi A, Nair NV, Balasundaram N, Mohapatra T. Evaluation of maize microsatellite markers for genetic diversity analysis and fingerprinting in sugarcane. Genome. 2003;46:394–403. doi: 10.1139/g03-018. [DOI] [PubMed] [Google Scholar]
- 17.D’Hont A, Paulet F, Glaszmann JC. Oligoclonal interspecific origin of ‘North Indian’ and ‘Chinese’ sugarcanes. Chromosome Res. 2002;10:253–262. doi: 10.1023/A:1015204424287. [DOI] [PubMed] [Google Scholar]
- 18.Brown, J. S., Schnell, R. J., Tai, P. Y. P. & Miller, J. D. Phenotypic evaluation of Saccharum barberi, S. robustum, and S. sinense germplasm from the Miami, FL, USA world collection. Sugarcane International. 3–16, Sept-Oct (2002).
- 19.D’Hont A, Paulet F, Glaszmann JC. Cytoplasmic diversity in sugarcane revealed by heterologous probes. Sugar Cane. 1993;1:12–15. [Google Scholar]
- 20.Glaszmann, J. C., Jannoo, N., Grivet, L. & D’Hont, A. Sugarcane. In: Hamon, P., Sejuin, M., Perrier, X. & Glaszmann, J. C. (eds) Genetic diversity of tropical plants. CIRAD, Montpellier, France, and Science Publishers, Inc., Enfield, NH, USA. pp. 337 (2003).
- 21.Chen PH, Pan YB, Chen RK, Xu LP, Chen YQ. SSR marker-based analysis of genetic relatedness among sugarcane cultivars (Saccharum spp. hybrids) from breeding programs in China and other countries. Sugar Tech. 2009;11:347–354. doi: 10.1007/s12355-009-0060-2. [DOI] [Google Scholar]
- 22.Liu XL, et al. Establishment of DNA fingerprint ID in sugarcane cultivars in Yunnan, China. Acta Agronomica Sinica. 2010;36:202–210. [Google Scholar]
- 23.Liu P, et al. Highly polymorphic microsatellite DNA markers for sugarcane germplasm evaluation and variety identity testing. Sugar Tech. 2011;13:129–136. doi: 10.1007/s12355-011-0077-1. [DOI] [Google Scholar]
- 24.You Q, Xu LP, Zheng YF, Que YX. Genetic diversity analysis of sugarcane parents in Chinese breeding programmes using gSSR markers. Sci World. 2013;J2013:613062. doi: 10.1155/2013/613062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.You Q, et al. Genetic diversity analysis of sugarcane germplasm based on fluorescence-labeled simple sequence repeat markers and a capillary electrophoresis-based genotyping platform. Sugar Tech. 2016;18:380–390. doi: 10.1007/s12355-015-0395-9. [DOI] [Google Scholar]
- 26.Liu XL, Li XJ, Xu CH, Lin XQ, Deng ZH. Genetic diversity of populations of Saccharum spontaneum with different ploidy levels using SSR molecular markers. Sugar Tech. 2016;18:365–372. doi: 10.1007/s12355-015-0399-5. [DOI] [Google Scholar]
- 27.Schenck S, et al. Genetic diversity and relationships in native Hawaiian Saccharum officinarum sugarcane. Heredity. 2004;95:327–331. doi: 10.1093/jhered/esh052. [DOI] [PubMed] [Google Scholar]
- 28.Singh RK, et al. Development, cross-species/genera transferability of novel EST-SSR markers and their utility in revealing population structure and genetic diversity in sugarcane. Gene. 2013;524:309–329. doi: 10.1016/j.gene.2013.03.125. [DOI] [PubMed] [Google Scholar]
- 29.Nayak SN, et al. Promoting utilization of Saccharum spp. genetic resources through genetic diversity analysis and core collection construction. PLoS One. 2014;9:e110856. doi: 10.1371/journal.pone.0110856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pan Y-B. Databasing molecular identities of sugarcane (Saccharum spp.) clones constructed with microsatellite (SSR) DNA markers. Am J Plant Sci. 2010;1:87–94. doi: 10.4236/ajps.2010.12011. [DOI] [Google Scholar]
- 31.Tao LA, et al. Identification of genuine hybrids from the cross of chewing cane and Saccharum spontaneum in Yunnan by SSR Markers. J Plant Genet Resour. 2009;10:132–135. [Google Scholar]
- 32.Dillon SL, et al. Domestication to crop improvement: genetic resources for Sorghum and Saccharum (Andropogoneae) Ann Bot. 2007;100:975–989. doi: 10.1093/aob/mcm192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kalia RK, Rai MK, Kalia S, Singh R, Dhawan AK. Microsatellite markers: an overview of the recent progress in plants. Euphytica. 2011;177:309–334. doi: 10.1007/s10681-010-0286-9. [DOI] [Google Scholar]
- 34.Brandes, E. W. & Sartoris G. B. Sugarcane: its origin and improvement. In: Year book of Agriculture. US Government Print Office, Washington D.C. pp. 561–611 (1936).
- 35.Simmonds, N. W. S. In: Simmonds, N. W. (eds) Evolution of Crop Plants. Longmans, London. pp. 104–108 (1976).
- 36.Pan Y-B, Burner DM, Legendre BL. An assessment of the phylogenetic relationship among sugarcane and related taxa based on the nucleotide sequence of 5S rRNA intergenic spacers. Genetica. 2000;108:285–295. doi: 10.1023/A:1004191625603. [DOI] [PubMed] [Google Scholar]
- 37.Daniels JP, Smith, Paton N, Roach BT. The taxonomic status of Saccharum robustum Brandes & Jeswiet ex Grassl. Proceedings of the International Society of Sugar Cane Technologists. 1978;16:71–84. [Google Scholar]
- 38.Daniels, J. & Roach B. T. Taxonomy and evolution. In: Heinz, D. J. (eds) Sugarcane Improvement through Breeding. Elsevier Press, Amsterdam. pp. 7–84 (1987).
- 39.Hoisington, D., Khairallah, M. & Gonzalez-de-Leon, D. Laboratory Protocols: CIMMYT Applied Molecular Genetics Laboratory. Mexico, D.F. (1994).
- 40.Cordeiro GM, Taylor GO, Henry RJ. Charaterisation of microsatellite markers for sugarcane (Saccharum sp.), a highly polyploid species. Plant Sci. 2000;155:161–168. doi: 10.1016/S0168-9452(00)00208-9. [DOI] [PubMed] [Google Scholar]
- 41.Pan YB, Tew TL, Schnell RJ, Viator RP. Microsatellite DNA marker-assisted selection of Saccharum spontaneum cytoplasm-derived germplasm. Sugar Tech. 2006;8:23–29. doi: 10.1007/BF02943737. [DOI] [Google Scholar]
- 42.Yeh, F. C., Yang, R. C., Boyle, T., Ye, Z. H. & Mao, J. X. POPGENE, version 1.32: the user friendly software for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Edmonton, AB, Canada (1999).
- 43.Tamura K, Glen S, Daniel P, Alan F, Sudhir K. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schliep KP. Phangorn: phylogenetic analysis in R. Bioinformatics. 2011;27:592–593. doi: 10.1093/bioinformatics/btq706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Galili T. Dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hall TA. Bioedit: a user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucl Acids Symp Ser. 1999;41:95–98. [Google Scholar]
- 47.Rohlf, F. J. NTSYS-pc Numerical Taxonomy and Multivariate Analysis System, version 2.10e. Exeter Publishing Ltd., Setauket, New York (1988).