

Most sequenced bacterial genomes contain genes encoding proteins of unknown or hypothetical function. To identify a phenotype for mutations in such genes, deletion is the preferred method for mutagenesis because it reduces the likelihood of polar effects, although it does not eliminate the possibility. Allelic exchange to produce deletions is dependent on the length of homologous regions used to generate merodiploids. Shorter regions of homology resolve at lower frequencies. The work presented here demonstrates the utility of inducing DNA double-strand breaks to increase the frequency of merodiploid resolution in Clostridium difficile. Using this approach, we reveal the roles of two genes, encoding homologues of AddAB, in survival following DNA damage. The method is readily applicable to the production of deletions in C. difficile and expands the toolbox available for genetic analysis of this important anaerobic pathogen.
KEYWORDS: AddAB, Clostridium difficile, deletions, nosocomial, SOS system, allelic exchange, double-strand-break repair, fliC, mutagenesis
ABSTRACT
Clostridium difficile is an important nosocomial pathogen associated with potentially fatal disease induced by the use of antibiotics. Genetic characterization of such clinically important bacteria is often hampered by lack of availability of suitable tools. Here, we describe the use of I-SceI to induce DNA double-strand breaks, which increase the frequency of allelic exchange and enable the generation of markerless deletions in C. difficile. The usefulness of the system is illustrated by the deletion of genes encoding putative AddAB homologues. The ΔaddAB mutants are sensitive to ultraviolet light and the antibiotic metronidazole, indicating a role in homologous recombination and the repair of DNA breaks. Despite the impairment in recombination, the mutants are still proficient for induction of the SOS response. In addition, deletion of the fliC gene, and subsequent complementation, reveals the importance of potential regulatory elements required for expression of a downstream gene encoding the flagellin glycosyltransferase.
IMPORTANCE Most sequenced bacterial genomes contain genes encoding proteins of unknown or hypothetical function. To identify a phenotype for mutations in such genes, deletion is the preferred method for mutagenesis because it reduces the likelihood of polar effects, although it does not eliminate the possibility. Allelic exchange to produce deletions is dependent on the length of homologous regions used to generate merodiploids. Shorter regions of homology resolve at lower frequencies. The work presented here demonstrates the utility of inducing DNA double-strand breaks to increase the frequency of merodiploid resolution in Clostridium difficile. Using this approach, we reveal the roles of two genes, encoding homologues of AddAB, in survival following DNA damage. The method is readily applicable to the production of deletions in C. difficile and expands the toolbox available for genetic analysis of this important anaerobic pathogen.
INTRODUCTION
Clostridium difficile, also known as Clostridioides difficile (1), is a Gram-positive obligately anaerobic spore-forming bacillus originally isolated in 1935 from the fecal microbiota of healthy neonates (2). C. difficile was first associated with human infections in 1962 (3), but its role as the pathogen responsible for antibiotic-associated pseudomembranous colitis was not confirmed until the late 1970s (4–6). C. difficile is currently one of the most commonly reported pathogens in nosocomial infections in the United States and the European Union (7–11). The bacterium is acquired through ingestion of vegetative cells or spores, which are ubiquitous in the environment, and although not part of the normal gut microbiota of humans, 1% to 3% of adults are carriers (12). Exposure to antibiotics is a major risk factor for C. difficile infection (CDI) (13, 14). Disruption of the normal gut microbiota leads to a loss of colonization resistance which, together with spore germination due to exposure to bile salts, results in proliferation of C. difficile (15–17). The organism adheres to the mucus layer covering the epithelial surface of the gastrointestinal (GI) tract via multiple adhesins, and then it penetrates the mucus and adheres to enterocytes, marking the beginning of the first phase of the pathogenic process (18, 19). The second important phase of pathogenesis is toxin production (18, 19). Toxigenic C. difficile strains produce two major toxins, toxin A (TcdA) and toxin B (TcdB), which are encoded on a 19.6-kb chromosomal region termed the pathogenicity locus (PaLoc) and are recognized as primary virulence factors (20, 21).
Although treatment of CDI depends on the clinical presentation of disease (13, 22), the first step is usually the discontinuation of the inciting antibiotic. Metronidazole is the first choice for mild to moderate CDI (23), while vancomycin is preferred as the first-line drug for moderate to severe CDI (23, 24). Due to an increased rate of failure of metronidazole and vancomycin treatments and recurrence of CDI (25), alternatives, such as fidaxomicin, a narrow-spectrum antibiotic (26, 27), rifaximin, a broad-spectrum nonabsorbable antibiotic (28), and nitazoxanide, a broad-spectrum antiparasitic (29), have been tested against C. difficile.
Metronidazole targets the DNA of bacterial cells. The antibiotic enters cells by passive diffusion, and the prodrug is activated in the cytoplasm. The molecule is converted to a nitroso free radical, which interacts with DNA to cause single- or double-stranded chromosomal breaks, resulting in DNA degradation and cell death (30, 31). In Bacteroides fragilis, resistance to metronidazole can result from the presence of nim genes, which inactivate the nitroso radicals causing DNA damage (32). Alternatively, resistance can be mediated by deficiency of the ferrous iron transporter FeoAB, which potentially interferes with metronidazole activation (33) or increased production of proteins involved in homologous recombination, such as RecA (34) and RecQ (35). Increased expression of other genes involved in DNA repair, such as recF, recN, and uvrA, has been observed during the growth of B. fragilis under subinhibitory concentrations of metronidazole, indicating the importance of recombination as a response to the drug (36). Similar mechanisms of resistance have also been described for C. difficile, including an increase in RecA production by metronidazole-resistant strains (37, 38).
The C. difficile genome has a large number of integrated and extrachromosomal mobile genetic elements, including conjugative and nonconjugative transposons and bacteriophages (39, 40), which illustrates the importance of horizontal gene transfer during the evolution of this bacterium (41, 42). Conjugative transposon-mediated transfer of the PaLoc from toxigenic to nontoxigenic C. difficile strains has also been described (43). It has been hypothesized that the chromosomal transfer and recombination events require the action of the relaxosome on the oriT of the conjugative transposon without prior excision of the element, a mechanism similar to the well-characterized Hfr conjugation mediated by an integrated F plasmid in Escherichia coli, albeit occurring at a much lower frequency (43). Integration of transferred chromosomal fragments requires homologous recombination with processing of double-stranded DNA ends to enable loading of RecA. However, little is currently known about homologous recombination in C. difficile.
Reverse genetics requires methodologies for generating defined mutations. The lack of selectable markers in multidrug-resistant strains of bacteria poses a particular problem for tool development, while insertional inactivation of genes using antibiotic resistance makes the production of multiple mutations problematic. Markerless deletions are therefore preferred because they are less likely to produce polar effects, and the resistance determinant, used during construction, can be recycled for multiple rounds of mutagenesis. Deletions can be generated by allelic replacement, where a construct lacking the gene of interest is first integrated into the genome of the target strain (Fig. 1). Integrants can be selected based on antibiotic resistance; however, resolution of the merodiploid can be a rare event which makes the process potentially laborious. To increase the frequency of resolution events, a method originally described for E. coli, involving the yeast homing endonuclease I-SceI, can be used (44, 45). This approach utilizes site-specific cleavage by the endonuclease of an 18-bp cognate recognition site present in the vector carrying the construct of interest, resulting in loss of the vector at high frequencies and selecting for bacteria in which homologous recombination has successfully repaired the double-stranded break (Fig. 1). Induction of double-strand breaks has been used successfully in pathogens which were once considered genetically intractable, such as Bacteroides fragilis (46). In this study, the application of I-SceI-mediated DNA cleavage to create markerless mutants in C. difficile is described. Further, markerless mutants of addAB homologues and fliC in C. difficile are described to gain insight into homologous recombination in this bacterium and to demonstrate the utility of this method, respectively.
FIG 1.

Schematic illustrating the generation of markerless deletions in C. difficile by induction of double-strand breaks. (A) DNA flanking the addAB genes (LF, left flank; RF, right flank) was amplified and ligated into a plasmid containing an I-SceI recognition sequence (pES271). Homologous recombination between the LF sequences on the plasmid and chromosome led to integration of the vector. The introduction of an I-SceI-expressing plasmid generates a double-strand break in the chromosome which must be repaired for the cell to survive. Recombination between LF sequences regenerates the original chromosome configuration of addAB, while recombination between RF sequences generates a deletion of the intervening DNA. Small arrows above LF and below RF represent the positions of primers used to differentiate between wild-type and deletion genotypes. (B) Example of an agarose gel showing PCR products for genotyping-resolved integrants. Primers used in the reaction correspond to the small arrows at LF and RF shown in panel A. The wild-type amplicon is 8.2 kb, while the deletion amplicon is ∼1 kb. Lane 1, 1-kb size ladder; lane 2, negative PCR control; lane 3, deletion amplicon from pES271; lane 4, wild-type addAB amplicon from 630Δerm; lanes 5 and 6, products from two independent ΔaddAB mutant strains; lane 7, product from resolved wild-type strain.
RESULTS
Enhancing allelic exchange using double-strand breaks.
To facilitate the generation of markerless mutants, an erythromycin-sensitive strain, C. difficile 630Δerm, was used for mutagenesis. Sequence analysis of the parental strain (GenBank accession number AM180355) identified two adjacent genes annotated as encoding ATP-dependent helicase/DNase subunits, suggesting that they were involved in double-strand-break repair. The proteins encoded by genes CD630DERM_RS05940 and CD630DERM_RS05945 share 34% identity (399/1,182 residues) and 38% identity (484/1,279 residues) with AddB and AddA of Bacillus subtilis, respectively (here referred to as addAB). In most Gram-positive and many Gram-negative bacteria, the AddAB heterodimer is involved in degradation of double-stranded DNA as part of the presynaptic processing step of homologous recombination (47, 48).
Approximately 500-bp sequences flanking both sides of addAB were amplified and fused by PCR. The fusion product was inserted into an unstable conjugative vector, pES185, which is based on pJIR1456 (49) but modified to contain an I-SceI recognition sequence. The resulting plasmid containing the deletion construct, pES271, was conjugated into C. difficile 630Δerm using the RP4 functions provided by E. coli S17-1λpir. Transconjugants were selected on brain heart infusion (BHI) plates containing thiamphenicol, followed by analysis using PCR to confirm presence of the deletion construct present in the plasmid. Potential integrants resulting from a single crossover between the plasmid and chromosome were selected on thiamphenicol after multiple passages in broth without selection. Thiamphenicol-resistant integrants were screened by PCR using primers that annealed to plasmid and chromosomal DNA to generate a unique amplicon. Integration can occur either between sequences on the left flank (LF) or the right flank (RF) of the deletion construct. Note that Fig. 1A illustrates integration via a single crossover at the left flank.
To generate double-strand breaks, a plasmid containing the gene encoding I-SceI under the control of the constitutive fdx promoter from Clostridium sporogenes (50) was constructed by ligation into pMTL82254. This I-SceI-expressing plasmid, pES288, was introduced into merodiploids by conjugation, and erythromycin-resistant (Ermr) colonies were screened for sensitivity to thiamphenicol. The resolution frequency from three independent matings varied from 7% to 33%. In contrast, screening of 80 transconjugants with a vector that did not contain the gene encoding I-SceI failed to yield any thiamphenicol-sensitive strains following the same procedure. Thus, expression of I-SceI enhanced the second recombination event, which was required for resolution of the merodiploids.
Phenotypic analysis of C. difficile ΔaddAB mutants.
Recombination to resolve integrants should theoretically produce an equal ratio of wild-type to deletion genotypes, provided there are no confounding factors, such as the presence of chi sites or growth defects of the mutants which might affect the outcome. Screening by PCR of thiamphenicol-sensitive strains allowed the identification of mutants derived from the addAB merodiploid (Fig. 1B). However, the frequency of ΔaddAB mutants (4% to 7%) present within all the resolved strains indicated some form of bias against the deletion genotype during or after the resolution event. Such a bias could reflect the frequency of chi sites in the sequences flanking the double-strand break; however, the sequence of potential chi sites in C. difficile has not been determined. Sequencing of PCR products spanning the addAB region in the deletion strains showed that 7.25 kb of DNA had been deleted precisely at the point defined by the sequence of the primers used to generate the construct in pES271 (data not shown).
Previous studies in other species have demonstrated growth defects in mutants defective in presynaptic processing of double-strand breaks. In E. coli, recBC mutants have reduced viability and increased doubling times (51). This is also true for addAB mutants of Streptococcus pneumoniae and Bacteroides fragilis (52, 53). The growth of two independently derived ΔaddAB mutants was studied in anaerobic investigation medium (AIM) broth, and both strains had an average doubling time of ∼45 min, similar to that of the parental strain 630Δerm (∼40 min). This suggests that in C. difficile, the deletion of addAB has a minor effect on viability.
The role of AddAB in DNA repair was tested by exposing cells to the DNA-damaging agents ultraviolet (UV) light and metronidazole. Exposure of the ΔaddAB mutant strains to 10 J/m2 of UV resulted in a 1,000-fold decrease in viability, while exposure of the parental strain to the same dose of UV had little effect (Fig. 2A). A merodiploid that had resolved to produce the wild-type genotype (R-WT) behaved in a manner similar to the parental strain. Growth of the parental strain 630Δerm on subinhibitory concentrations (0.06 μg/ml) of metronidazole was unaffected compared to that with absence of the antibiotic, but the growth of the two ΔaddAB mutant strains was reduced by 1,000-fold in the presence of metronidazole (Fig. 2B).
FIG 2.

Phenotypic analysis of ΔaddAB mutant strains. (A) Exponentially growing cultures of parental (630Δerm), ΔaddAB mutant (ΔaddAB-1 and ΔaddAB-2) and resolved wild-type (R-WT) strains were diluted, spotted onto AIM agar, and irradiated at 10J/m2 with UV light (bottom images). The no-UV control is shown in the top images. Dilution values are shown at the top of the panel. Note the similar viability of the parental and ΔaddAB mutant strains in the absence of DNA damage. Undil, undiluted. (B) Exponentially growing cultures of all strains were diluted (shown at the top of the panel) and spotted onto AIM agar with and without 0.06 μg/ml metronidazole (Mz). (C) Micrographs showing random fields of cells (×400 magnification), illustrating the effect of 0.125 μg/ml metronidazole or 1 μg/ml levofloxacin on cell morphology for parental and ΔaddAB mutant strains. The micrographs on the left show cells grown in the absence of antibiotics, while the images on the right show cell filamentation for both strains when metronidazole or levofloxacin was present.
The presence of single-stranded DNA resulting from inhibition of replication or following DNA damage leads to induction of the SOS response in many bacteria, including C. difficile (54). By microscopy, cultures of 630Δerm grown in the presence of 0.125 μg/ml metronidazole for 4 h showed the presence of filamentous cells, consistent with division inhibition associated with the SOS response (Fig. 2C). Growth of the ΔaddAB mutant strains in the same concentration of metronidazole also produced similar filamentous cells, suggesting that AddAB is not required for induction of the SOS response. To confirm that inhibition of division was due to the SOS response, we treated cells with levofloxacin, which has previously been shown to induce filamentation associated with a LexA-regulated pathway (54). Treatment of the 630Δerm strain with a subinhibitory concentration of levofloxacin (1 μg/ml) for 6 h led to an increase in length of a subpopulation of cells (Fig. 2C). The average length of 630Δerm cells in an untreated culture was 5.1 ± 0.99 μm, while the addition of levofloxacin increased the average cell length to 11.3 ± 8.9 μm. The addition of levofloxacin to the ΔaddAB mutant strain had a similar effect (Fig. 2C). The average cell length of the ΔaddAB mutant strain was 5.3 ± 1.6 μm, while treated cells had an average length of 18.8 ± 9.4 μm. Together, these data suggest that the AddAB homologues present in C. difficile are dispensable for induction of the SOS response.
Deletion and complementation of fliC.
Despite the success of the deletion strategy, screening for integrants was a time-consuming process. To improve the efficiency of the system, another plasmid, pMTL83151, was used, which was reported to show segregational instability and therefore had the identifiable phenotype of two different colony sizes. In the presence of thiamphenicol, smaller colonies appear to result from a loss of plasmid in the population and so have fewer resistant cells, while larger colonies have a chromosomally integrated plasmid and are thus resistant to the antibiotic (55, 56). This plasmid was modified by introducing an I-SceI recognition sequence to produce vector pES242. To validate this approach, a deletion of the fliC gene (CD630DERM_RS01750), which encodes the major structural protein of the flagellum, was generated. This target was chosen because of the previously described loss of motility in a C. difficile strain in which the fliC gene had been insertionally inactivated (57).
Approximately 500-bp sequences flanking both sides of fliC were amplified and fused by PCR, followed by cloning into pES242 to produce the allelic replacement vector pES2921. This plasmid was transferred into 630Δerm by conjugation. When streaked onto thiamphenicol BHI plates, two colony sizes were evident, suggesting that integrants were present. Putative integrants were cultured in the absence of thiamphenicol and restreaked onto agar with thiamphenicol until no small colonies were evident. Integration of pES2921 was confirmed by PCR (data not shown). The I-SceI-expressing plasmid pES288 was introduced by conjugation, and Ermr colonies were screened for loss of thiamphenicol resistance. The resolution frequencies of integrants derived from two independent matings were 2% (3/144) and 3.6% (4/111). The introduction of pMTL82254 into one of the integrants did not produce any thiamphenicol-sensitive colonies, again indicating the action of I-SceI on the merodiploids to enable resolution. Screening of resolved strains by PCR showed that approximately half the colonies were wild type, and half contained deletions of fliC (Fig. 3A). Motility assays in soft agar demonstrated that two independently derived ΔfliC mutant strains (strains 88 and 383) were incapable of penetrating the medium beyond the site of inoculation (Fig. 3B), a phenotype indicative of the deletion genotype. Transmission electron microscopy (TEM) of cells grown on AIM showed the production of peritrichous flagella (observed as numerous thread-like extensions from the cell surface) in the parental strain and an absence of flagella in the ΔfliC mutant strains (Fig. 3C), again confirming successful deletion of the target gene. In addition, we note that resolution of the integrants also produced cells that were motile and contained a wild-type copy of the fliC gene (Fig. 3B).
FIG 3.

Identification and phenotypic analysis of ΔfliC mutants. (A) Example of an agarose gel showing PCR screening of resolved merodiploids. PCR primers used will amplify a 1.9-kb sequence which includes the fliC gene. Lane 1, 1-kb size ladder; lanes 2, 3, and 6, resolution to the wild type; lanes 4, 5, 7, and 8, deletions of fliC; lanes 9 and 10, controls amplifying the fliC region in 630Δerm and the deletion construct; lane 11, PCR negative control. (B) Motility stab assay using 0.175% soft agar. The parental strain 630Δerm and resolved wild-type (R-WT) strains showed penetration of the medium from the initial inoculum. Two independent fliC deletion mutants (88 and 383) grew at the site of inoculation but failed to spread into the agar. (C) Transmission electron micrographs of the parental strain (1) and a resolved wild-type strain (2) show the presence of thread-like flagella (examples indicated by arrows) associated with the cells. There was no evidence of flagella on the cell surfaces of either of the fliC mutants (3 and 4).
One potential advantage of markerless deletions, compared to insertion mutations, is the reduced possibility of polar effects on the expression of downstream genes. Since the fliC gene is within a gene cluster required for flagellar assembly and function, we tested complementation of the deletion using a plasmid-borne copy of the gene. The native fliC promoter and gene, as previously identified (57), were amplified and ligated into pMTL84151 to produce pES196, followed by conjugation of the plasmid into the ΔfliC mutant strains and the parental 630Δerm strain. Electron microscopy showed visible production of flagella by the ΔfliC mutant strains and the parental strain when they contained pES196 (Fig. 4A). When the control plasmid pMTL84151 was present, flagella were only produced by the parental strain and not in the ΔfliC mutant strains. These data demonstrate successful complementation of the ΔfliC mutation by the presence of assembled flagella on the cell surface. Despite the observations made by TEM, motility assays in soft agar showed that the complemented ΔfliC mutant strains were not capable of penetrating the medium (Fig. 4B), and the inoculum had the same appearance as the stab tubes from the deletion strains (Fig. 3B). Microscopy of cells in a wet mount showed that ΔfliC cells containing pES196 had a tumbling behavior rather than the linear swimming motion of the parental strain (data not shown). Together, these data indicate that the flagella observed on the complemented ΔfliC mutant strains were functionally defective, possibly due to either misassembly of the structure or polarity affecting the expression of downstream genes.
FIG 4.
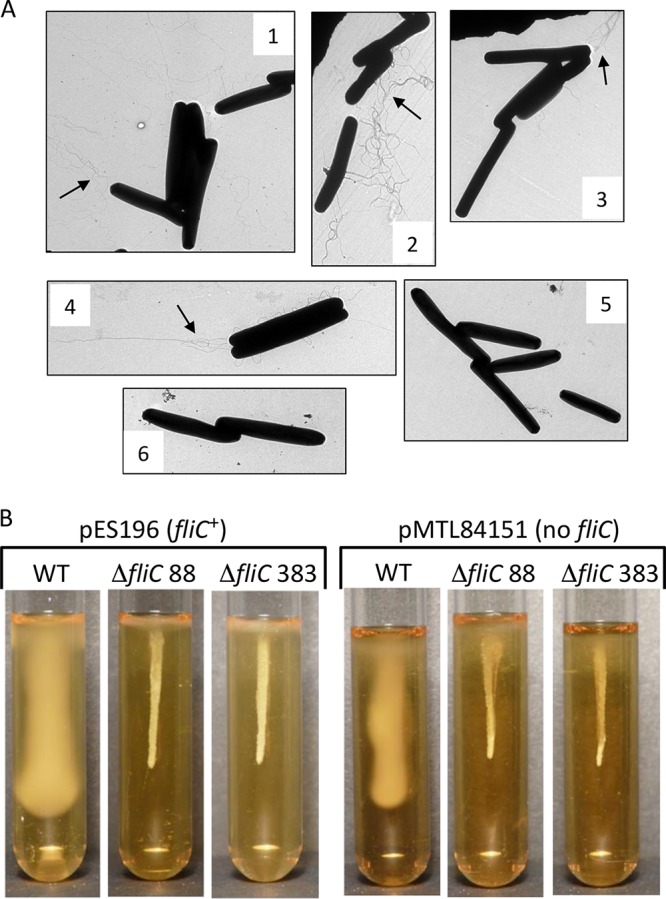
Complementation of ΔfliC mutants. (A) Transmission electron micrographs of strains containing either pES196 (fliC+) or the vector alone (pMTL84151). 1, 630Δerm/pES196; 2, ΔfliC 383/pES196; 3, ΔfliC 88/pES196; 4, 630Δerm/pMTL84151; 5, ΔfliC 383/pMTL84151; 6, ΔfliC 88/pMTL84151. Flagella are indicated by arrows. (B) Motility stab assay for strains containing either the fliC complementing plasmid (pES196) or the vector alone (pMTL84151). The strain genotype is indicated above each tube, with the presence of each plasmid shown at the top of each set of tubes. Only wild-type strains (630Δerm) containing a chromosomally carried fliC gene showed penetration of the medium.
DISCUSSION
The abundance of genome sequence data has facilitated the application of reverse genetics to study the function of predicted genes in bacteria. Genetic manipulation of model organisms, such as E. coli, is achieved with relative ease primarily due to the “domestication” of strains by removal of restriction-modification (R/M) systems which degrade foreign incoming DNA (58). Manipulation of wild-type bacteria with medical or industrial importance is hampered both by a lack of tools and the presence of multiple R/M systems, which makes transformation and conjugation inefficient when using DNA derived from E. coli. In C. difficile 630, there are at least five type II R/M systems and one type IV restriction system (41), which partially explains the difficulties in introducing unmodified DNA into this strain.
A predominant method for generating mutations in C. difficile is the ClosTron system, which was originally designed to make insertional mutations, but this is limited by the number of selectable markers subsequently available to produce multiple mutants (59). Alternative methods to produce markerless deletions either require prior mutagenesis of strains, e.g., pyrE mutants to allow selection of integrants using 5-fluoroorotic acid (5-FOA) (60), or screening of multiple colonies to identify spontaneous resolution of integrants (61). A novel allelic exchange procedure using CRISPR-Cas9 has been described (62), but each deletion will require cloning of regions of homology and an appropriate single guide RNA (sgRNA). The frequency of spontaneous resolution for regions of homology between 300 and 600 bp have been reported to be low and inconsistent (61), presumably reflecting the stochastic nature of DNA damage or replication errors that occur within the repeat regions, which subsequently require recombination for repair. In our experiments, we were unable to detect resolution of merodiploids that contained 500-bp regions of homology unless I-SceI was expressed in the cells. Using this method with induction of double-strand breaks, we were able to generate a large deletion covering the putative addAB genes and a deletion of the fliC gene. The heterodimer of AddAB is involved in processing of double-strand breaks in a number of Gram-positive and Gram-negative bacteria. Our previous single-molecule observations for AddAB from B. fragilis demonstrated a translocation rate of 250 bp per s with up to 40 kb being unwound and degraded from a double-strand end (53). The sensitivity of our ΔaddAB mutant strains to UV light and metronidazole is consistent with the encoded proteins performing the same functions in C. difficile. The model organism E. coli is the paradigm for homologous recombination and the SOS response in prokaryotes. Regulation of the SOS response in other bacteria has been relatively understudied in comparison. In C. difficile, the SOS regulon is controlled by a LexA homologue which modulates not only responses to DNA damage, but also regulates other processes, including motility and biofilm formation (54). Induction of the SOS response in E. coli requires the generation of single-stranded DNA which activates RecA to facilitate self-cleavage by LexA (63, 64). This single-stranded DNA is produced by the action of the RecBCD complex preferentially degrading one DNA strand; therefore, recBCD mutants do not show an SOS response (65). In B. subtilis, the SOS response is reported to be greatly reduced in the absence of AddAB (66). In contrast, our C. difficile ΔaddAB mutant strains demonstrated activation of SOS, as indicated by cell filamentation when grown in subinhibitory concentrations of metronidazole. These data suggest there is potential redundancy in the pathway and that another exonuclease is responsible for generating single-stranded DNA from a double-strand break in the absence of AddAB. One potential candidate could be the single-stranded exonuclease RecJ, since recJ mutations increase the sensitivity of addAB mutants to DNA-damaging agents in B. subtilis (67). The action of other proteins on double-strand breaks in the absence of AddAB would also be consistent with the minor effect of the addAB deletion on the growth rate of our strains. This is also in contrast to addAB mutants of B. subtilis, which show a 50% reduction in viability (68).
The second target for demonstrating the utility of I-SceI-induced double-strand breaks was fliC. The deletion of fliC led to the expected phenotype of loss of motility, as reported previously for insertional mutations generated using the ClosTron system (57). Complementation of the ΔfliC mutation enabled the production of flagella, as visualized by TEM; however, the strains were not able to penetrate soft agar, which indicated a defect in flagellar function. One possible explanation is that overexpression of FliC had a detrimental effect on flagellar assembly or function in the ΔfliC mutant strain, although overexpression in the parental strain had no apparent effect. The fliC deletion in our strains extended 21 bp beyond the stop codon of the reading frame. The next downstream gene (CD630DERM_RS01755) is a glycosyltransferase which is separated from fliC by 92 bp. Flagella in C. difficile are posttranslationally modified with N-acetylhexosamine by the action of the glycosyltransferase encoded downstream of fliC. Insertional disruption of the gene RS01755 using ClosTron produced cells with reduced numbers of flagella and a nonmotile phenotype (69). The canonical −10 (TATAGT) promoter sequence of gene RS01755 is identifiable, while the putative −35 sequence (TTATTC) is more divergent from the consensus. The deletion spanning fliC, however, does not disrupt the putative RNA polymerase binding site for RS01755. We therefore suggest that our ΔfliC mutant strain is also defective in modification of flagellin as a consequence of reduced or altered expression of the glycosyltransferase. While one advantage of markerless deletions is the reduced possibility of polarity, in this case, deletion of 21 bp after the fliC gene exposed a potential regulatory sequence which is involved in controlling or modulating expression of the downstream gene.
Together, these results demonstrate that induction of double-strand breaks by expression of I-SceI is a useful and generally applicable tool for generating markerless deletions in C. difficile. The absence of the 18 bp I-SceI recognition sequence in all Clostridium species, and the ability of plasmids with pBP1 and pCB102 origins of replication to function in diverse clostridia, makes this approach suitable for a wide range of important organisms. Additional advantages are that amplification and cloning of large regions of homology, and counterselection, are not required, which will make the generation of deletions in wild-type strains more practical.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
C. difficile 630Δerm (70) was used for mutagenesis throughout this study. Cultures were routinely grown at 37°C in an atmosphere containing 10% CO2, 10% H2, and 80% N2 within an anaerobic cabinet (Don Whitley). The medium used for cultivation of C. difficile was either brain heart infusion (BHI) or anaerobic investigation medium (AIM), while LB was used for E. coli. Antibiotics were used at the following concentrations: 10 μg/ml erythromycin, 15 μg/ml thiamphenicol, 250 μg/ml d-cycloserine, and 8 μg/ml cefoxitin for C. difficile, and 500 μg/ml erythromycin and 30 μg/ml chloramphenicol for E. coli. The plasmids used in this study are shown in Table 1.
TABLE 1.
Plasmids used in this work
| Plasmid | Descriptiona | Reference or source |
|---|---|---|
| pJIR1456 | E. coli-C. perfringens shuttle vector (pIP404 replicon, catP marker), Tmr Cmr | 49 |
| pMTL83151 | E. coli-Clostridium shuttle vector (pCB102 replicon, catP marker), Tmr Cmr | 55 |
| pMTL82254 | E. coli-Clostridium shuttle vector (pBP1 replicon, ermB marker), Ermr | 55 |
| pMTL84151 | E. coli-Clostridium shuttle vector (pCD6 replicon, catP marker), Tmr Cmr | 55 |
| pES185 | pJIR1456 + I-SceI recognition site (SacI site), Tmr Cmr | This study |
| pES271 | pES185 + addBA deletion cassette (SphI site), Tmr Cmr | This study |
| pES242 | pMTL83151 + I-SceI recognition site (SacI site), Tmr Cmr | This study |
| pES2921 | pES242 + fliC deletion cassette (FspI site), Tmr Cmr | This study |
| pES288 | pMTL82254 + Pfdx::I-sceI (SbfI site), Ermr | This study |
| pES196 | pMTL84151 + fliC gene and native promoter (NotI/XhoI site), Tmr Cmr | This study |
Tmr, thiamphenicol resistant; Cmr, chloramphenicol resistant.
Oligonucleotides.
A list of oligonucleotides used to generate deletions is given in Table 2. PCR amplicons representing deletion products were ligated into the appropriate vectors following digestion with the relevant restriction endonuclease using standard procedures.
TABLE 2.
Sequences of oligonucleotides used to generate amplicons for allelic replacement vectors pES271 and pES2921
| Primer | Sequence |
|---|---|
| SphI_addBA1 | TTCCGCATGCTAAATGGGGATATAATACAGGC |
| addBA2 | CCTAAGTCCCATAAATTTCCG |
| addBA2_addBA3 | CGGAAATTTATGGGACTTAGGTGGAGTTGATGAAGCTGTTTG |
| SphI_addBA4 | TTCCGCATGCTAGCAACCACAATATTTTCTCC |
| SphI-fliC1 | TTCCGCATGCTTCAGCTTTAGAGTCTTTGTTG |
| fliC2 | CTCCTTAGTATAGTTGACATCC |
| fliC3 | GGATGTCAACTATACTAAGGAGAAAAGAAAGGATAAGGCTTTGC |
| SphI-fliC4 | TTCCGCATGCTGGTTGTTCATGAACTTTCCC |
| cmFliCFora | CCCTGGCGGCCGCAACTTTATGATAGTATGGAGC |
| cmFliCReva | CCCTGCTCGAGCTATCCTAATAATTGTAAAACTC |
Primers cmFliCFor and cmFliCRev were used to produce the fliC complementing plasmid pES196.
Conjugation of plasmids into C. difficile.
Deletion constructs and the plasmid expressing I-SceI were introduced into C. difficile by conjugation from E. coli S17-1λpir using a modification of a previously described method (59). Briefly, exponential cultures of donor and recipient cells were centrifuged, washed, and resuspended in a 1/10 volume of fresh medium. Different donor-to-recipient ratios were mixed, spread onto BHI agar plates, and incubated anaerobically for 16 to 24 h. The conjugation mixture was harvested in prereduced phosphate-buffered saline (PBS) and spread onto plates containing thiamphenicol, d-cycloserine, and cefoxitin.
Metronidazole and UV sensitivity tests.
Cultures of control and deletion strains were grown to an optical density at 600 nm (OD600) of 0.3 in AIM and serially diluted in prereduced PBS. Five-microliter aliquots were then spotted onto AIM plates. For metronidazole sensitivity, plates contained 0.25, 0.125, or 0.06 μg/ml metronidazole. For UV sensitivity, plates were irradiated at 10, 15, or 20 J/m2. All plates were incubated anaerobically for 48 h, and all experiments were performed in triplicate.
Motility assay.
An inoculum from a single colony of each strain was stabbed into a glass tube containing AIM with 0.175% agar. The swim agar tube was incubated anaerobically at 37°C for 24 h.
Microscopy and transmission electron microscopy.
For phase-contrast microscopy, cells were fixed with 20% formaldehyde before being placed on slides coated with poly-l-lysine. Images were captured on a Metalux II microscope using a Hamamatsu digital camera with the Improvision Openlab software. For TEM, a colony of each strain was resuspended in prereduced PBS before the addition of 3% glutaraldehyde. A 20-μl sample was placed on a carbon-coated grid followed by staining with 1% phosphotungstic acid. Grids were examined using a Philips CM120 BioTWIN transmission electron microscope.
ACKNOWLEDGMENTS
We thank Neil Fairweather for providing 630Δerm, Julian Rood for providing plasmid pJIR1456, and Nigel Minton for providing pMTL82254, pMTL84151, and pMTL83151.
This work was partly supported by a Wellcome Trust grant (090288/Z/09/ZA) to G.W.B.
E.-S.T., P.V., and G.W.B. performed experimental work. G.W.B., M.P.G., and I.R.P. planned the experiments and interpreted the data. G.W.B., P.V., and I.R.P. wrote and prepared the manuscript.
REFERENCES
- 1.Oren A, Rupnik M. 2018. Clostridium difficile and Clostridioides difficile: two validly published and correct names. Anaerobe 52:125–126. doi: 10.1016/j.anaerobe.2018.07.005. [DOI] [PubMed] [Google Scholar]
- 2.Hall I, O'Toole E. 1935. Intestinal flora in newborn infants with a description of a new pathogenic anaerobe, Bacillus difficilis. Am J Dis Child 49:390–402. doi: 10.1001/archpedi.1935.01970020105010. [DOI] [Google Scholar]
- 3.Smith LD, King EO. 1962. Occurrence of Clostridium difficile in infections of man. J Bacteriol 84:65–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bartlett JG, Chang TW, Gurwith M, Gorbach SL, Onderdonk AB. 1978. Antibiotic-associated pseudomembranous colitis due to toxin-producing clostridia. N Engl J Med 298:531–534. doi: 10.1056/NEJM197803092981003. [DOI] [PubMed] [Google Scholar]
- 5.George RH, Symonds JM, Dimock F, Brown JD, Arabi Y, Shinagawa N, Keighley MR, Alexander-Williams J, Burdon DW. 1978. Identification of Clostridium difficile as a cause of pseudomembranous colitis. BMJ 1:695. doi: 10.1136/bmj.1.6114.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson HE, Price AB, Honour P, Borriello SP. 1978. Clostridium difficile and the aetiology of pseudomembranous colitis. Lancet 1:1063–1066. doi: 10.1016/S0140-6736(78)90912-1. [DOI] [PubMed] [Google Scholar]
- 7.Desai K, Gupta SB, Dubberke ER, Prabhu VS, Browne C, Mast TC. 2016. Epidemiological and economic burden of Clostridium difficile in the United States: estimates from a modeling approach. BMC Infect Dis 16:303. doi: 10.1186/s12879-016-1610-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans CT, Safdar N. 2015. Current trends in the epidemiology and outcomes of Clostridium difficile infection. Clin Infect Dis 60:S66–S71. doi: 10.1093/cid/civ140. [DOI] [PubMed] [Google Scholar]
- 9.Jones AM, Kuijper EJ, Wilcox MH. 2013. Clostridium difficile: a European perspective. J Infect 66:115–128. doi: 10.1016/j.jinf.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez C, Fernandez J, Van Broeck J, Taminiau B, Avesani V, Boga JA, Vazquez F, Delmee M, Daube G. 2016. Clostridium difficile presence in Spanish and Belgian hospitals. Microb Pathog 100:141–148. doi: 10.1016/j.micpath.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Davies K, Freeman J, Mayor S. 2018. Update on Clostridium difficile from the European Congress of Clinical Microbiology and Infectious Diseases (ECCMID), Vienna, Austria, April 22–25, 2017. J Hosp Infect 98:1–3. doi: 10.1016/j.jhin.2017.11.016. [DOI] [PubMed] [Google Scholar]
- 12.Barbut F, Braun M, Burghoffer B, Lalande V, Eckert C. 2009. Rapid detection of toxigenic strains of Clostridium difficile in diarrheal stools by real-time PCR. J Clin Microbiol 47:1276–1277. doi: 10.1128/JCM.00309-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ananthakrishnan AN. 2011. Clostridium difficile infection: epidemiology, risk factors and management. Nat Rev Gastroenterol Hepatol 8:17–26. doi: 10.1038/nrgastro.2010.190. [DOI] [PubMed] [Google Scholar]
- 14.Freeman J, Bauer MP, Baines SD, Corver J, Fawley WN, Goorhuis B, Kuijper EJ, Wilcox MH. 2010. The changing epidemiology of Clostridium difficile infections. Clin Microbiol Rev 23:529–549. doi: 10.1128/CMR.00082-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denève C, Bouttier S, Dupuy B, Barbut F, Collignon A, Janoir C. 2009. Effects of subinhibitory concentrations of antibiotics on colonization factor expression by moxifloxacin-susceptible and moxifloxacin-resistant Clostridium difficile strains. Antimicrob Agents Chemother 53:5155–5162. doi: 10.1128/AAC.00532-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vedantam G, Clark A, Chu M, McQuade R, Mallozzi M, Viswanathan VK. 2012. Clostridium difficile infection: toxins and non-toxin virulence factors, and their contributions to disease establishment and host response. Gut Microbes 3:121–134. doi: 10.4161/gmic.19399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sorg JA, Sonenshein AL. 2009. Chenodeoxycholate is an inhibitor of Clostridium difficile spore germination. J Bacteriol 191:1115–1117. doi: 10.1128/JB.01260-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll KC, Bartlett JG. 2011. Biology of Clostridium difficile: implications for epidemiology and diagnosis. Annu Rev Microbiol 65:501–521. doi: 10.1146/annurev-micro-090110-102824. [DOI] [PubMed] [Google Scholar]
- 19.Denève C, Janoir C, Poilane I, Fantinato C, Collignon A. 2009. New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrob Agents 33:S24–S28. doi: 10.1016/S0924-8579(09)70012-3. [DOI] [PubMed] [Google Scholar]
- 20.Davies AH, Roberts AK, Shone CC, Acharya KR. 2011. Super toxins from a super bug: structure and function of Clostridium difficile toxins. Biochem J 436:517–526. doi: 10.1042/BJ20110106. [DOI] [PubMed] [Google Scholar]
- 21.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 22.Kachrimanidou M, Malisiovas N. 2011. Clostridium difficile infection: a comprehensive review. Crit Rev Microbiol 37:178–187. doi: 10.3109/1040841X.2011.556598. [DOI] [PubMed] [Google Scholar]
- 23.Jarrad AM, Karoli T, Debnath A, Tay CY, Huang JX, Kaeslin G, Elliott AG, Miyamoto Y, Ramu S, Kavanagh AM, Zuegg J, Eckmann L, Blaskovich MA, Cooper MA. 2015. Metronidazole-triazole conjugates: activity against Clostridium difficile and parasites. Eur J Med Chem 101:96–102. doi: 10.1016/j.ejmech.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Debast SB, Bauer MP, Kuijper EJ. 2014. European Society of Clinical Microbiology and Infectious Diseases: update of the treatment guidance document for Clostridium difficile infection. Clin Microbiol Infect 20:1–26. doi: 10.1111/1469-0691.12418. [DOI] [PubMed] [Google Scholar]
- 25.Vardakas KZ, Polyzos KA, Patouni K, Rafailidis PI, Samonis G, Falagas ME. 2012. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents 40:1–8. doi: 10.1016/j.ijantimicag.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 26.Poxton IR. 2010. Fidaxomicin: a new macrocyclic, RNA polymerase-inhibiting antibiotic for the treatment of Clostridium difficile infections. Future Microbiol 5:539–548. doi: 10.2217/fmb.10.20. [DOI] [PubMed] [Google Scholar]
- 27.Chaparro-Rojas F, Mullane KM. 2013. Emerging therapies for Clostridium difficile infection–focus on fidaxomicin. Infect Drug Resist 6:41–53. doi: 10.2147/IDR.S24434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oldfield EC IV, Oldfield EC III, Johnson DA. 2014. Clinical update for the diagnosis and treatment of Clostridium difficile infection. World J Gastrointest Pharmacol Ther 5:1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Musher DM, Logan N, Bressler AM, Johnson DP, Rossignol JF. 2009. Nitazoxanide versus vancomycin in Clostridium difficile infection: a randomized, double-blind study. Clin Infect Dis 48:e41–e46. doi: 10.1086/596552. [DOI] [PubMed] [Google Scholar]
- 30.Land KM, Johnson PJ. 1999. Molecular basis of metronidazole resistance in pathogenic bacteria and protozoa. Drug Resist Updat 2:289–294. doi: 10.1054/drup.1999.0104. [DOI] [PubMed] [Google Scholar]
- 31.Lofmark S, Edlund C, Nord CE. 2010. Metronidazole is still the drug of choice for treatment of anaerobic infections. Clin Infect Dis 50:S16–S23. doi: 10.1086/647939. [DOI] [PubMed] [Google Scholar]
- 32.Reysset G. 1996. Genetics of 5-nitroimidazole resistance in Bacteroides species. Anaerobe 2:59–69. doi: 10.1006/anae.1996.0008. [DOI] [PubMed] [Google Scholar]
- 33.Veeranagouda Y, Husain F, Boente R, Moore J, Smith CJ, Rocha ER, Patrick S, Wexler HM. 2014. Deficiency of the ferrous iron transporter FeoAB is linked with metronidazole resistance in Bacteroides fragilis. J Antimicrob Chemother 69:2634–2643. doi: 10.1093/jac/dku219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steffens LS, Nicholson S, Paul LV, Nord CE, Patrick S, Abratt VR. 2010. Bacteroides fragilis RecA protein overexpression causes resistance to metronidazole. Res Microbiol 161:346–354. doi: 10.1016/j.resmic.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul L, Patrick S, Nord CE, Abratt V. 2011. The role of Bacteroides fragilis RecQ DNA helicases in cell survival after metronidazole exposure. FEMS Microbiol Lett 319:125–132. doi: 10.1111/j.1574-6968.2011.02271.x. [DOI] [PubMed] [Google Scholar]
- 36.de Freitas MCR, Resende JA, Ferreira-Machado AB, Saji GDRQ, de Vasconcelos ATR, da Silva VL, Nicolás MF, Diniz CG. 2016. Exploratory investigation of Bacteroides fragilis transcriptional response during in vitro exposure to subinhibitory concentration of metronidazole. Front Microbiol 7:1465. doi: 10.3389/fmicb.2016.01465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chong PM, Lynch T, McCorrister S, Kibsey P, Miller M, Gravel D, Westmacott GR, Mulvey MR, Canadian Nosocomial Infection Surveillance Program (CNISP). 2014. Proteomic analysis of a NAP1 Clostridium difficile clinical isolate resistant to metronidazole. PLoS One 9:e82622. doi: 10.1371/journal.pone.0082622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moura I, Monot M, Tani C, Spigaglia P, Barbanti F, Norais N, Dupuy B, Bouza E, Mastrantonio P. 2014. Multidisciplinary analysis of a nontoxigenic Clostridium difficile strain with stable resistance to metronidazole. Antimicrob Agents Chemother 58:4957–4960. doi: 10.1128/AAC.02350-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mullany P, Allan E, Roberts AP. 2015. Mobile genetic elements in Clostridium difficile and their role in genome function. Res Microbiol 166:361–367. doi: 10.1016/j.resmic.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Amy J, Johanesen P, Lyras D. 2015. Extrachromosomal and integrated genetic elements in Clostridium difficile. Plasmid 80:97–110. doi: 10.1016/j.plasmid.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 41.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 42.He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, Holt KE, Seth-Smith HM, Quail MA, Rance R, Brooks K, Churcher C, Harris D, Bentley SD, Burrows C, Clark L, Corton C, Murray V, Rose G, Thurston S, van Tonder A, Walker D, Wren BW, Dougan G, Parkhill J. 2010. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci U S A 107:7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brouwer MS, Roberts AP, Hussain H, Williams RJ, Allan E, Mullany P. 2013. Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat Commun 4:2601. doi: 10.1038/ncomms3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pósfai G, Kolisnychenko V, Bereczki Z, Blattner FR. 1999. Markerless gene replacement in Escherichia coli stimulated by a double-strand break in the chromosome. Nucleic Acids Res 27:4409–4415. doi: 10.1093/nar/27.22.4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plessis A, Perrin A, Haber JE, Dujon B. 1992. Site-specific recombination determined by I-Scei, a mitochondrial group-I intron-encoded endonuclease expressed in the yeast nucleus. Genetics 130:451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patrick S, Houston S, Thacker Z, Blakely GW. 2009. Mutational analysis of genes implicated in LPS and capsular polysaccharide biosynthesis in the opportunistic pathogen Bacteroides fragilis. Microbiology 155:1039–1049. doi: 10.1099/mic.0.025361-0. [DOI] [PubMed] [Google Scholar]
- 47.Chédin F, Kowalczykowski SC. 2002. A novel family of regulated helicases/nucleases from Gram-positive bacteria: insights into the initiation of DNA recombination. Mol Microbiol 43:823–834. doi: 10.1046/j.1365-2958.2002.02785.x. [DOI] [PubMed] [Google Scholar]
- 48.Zuniga-Castillo J, Romero D, Martinez-Salazar JM. 2004. The recombination genes addAB are not restricted to Gram-positive bacteria: genetic analysis of the recombination initiation enzymes RecF and AddAB in Rhizobium etli. J Bacteriol 186:7905–7913. doi: 10.1128/JB.186.23.7905-7913.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyras D, Rood JI. 1998. Conjugative transfer of RP4-oriT shuttle vectors from Escherichia coli to Clostridium perfringens. Plasmid 39:160–164. doi: 10.1006/plas.1997.1325. [DOI] [PubMed] [Google Scholar]
- 50.Heap JT, Cartman ST, Kuehne SA, Cooksley C, Minton NP. 2010. ClosTron-targeted mutagenesis. Methods Mol Biol 646:165–182. doi: 10.1007/978-1-60327-365-7_11. [DOI] [PubMed] [Google Scholar]
- 51.Capaldo FN, Ramsey G, Barbour SD. 1974. Analysis of the growth of recombination-deficient strains of Escherichia coli K-12. J Bacteriol 118:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Halpern D, Gruss A, Claverys JP, El-Karoui M. 2004. rexAB mutants in Streptococcus pneumoniae. Microbiology 150:2409–2414. doi: 10.1099/mic.0.27106-0. [DOI] [PubMed] [Google Scholar]
- 53.Reuter M, Parry F, Dryden DT, Blakely GW. 2010. Single-molecule imaging of Bacteroides fragilis AddAB reveals the highly processive translocation of a single motor helicase. Nucleic Acids Res 38:3721–3731. doi: 10.1093/nar/gkq100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walter BM, Cartman ST, Minton NP, Butala M, Rupnik M. 2015. The SOS response master regulator LexA is associated with sporulation, motility and biofilm formation in Clostridium difficile. PLoS One 10:e0144763. doi: 10.1371/journal.pone.0144763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Heap JT, Pennington OJ, Cartman ST, Minton NP. 2009. A modular system for Clostridium shuttle plasmids. J Microbiol Methods 78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 56.Heap JT, Ehsaan M, Cooksley CM, Ng YK, Cartman ST, Winzer K, Minton NP. 2012. Integration of DNA into bacterial chromosomes from plasmids without a counter-selection marker. Nucleic Acids Res 40:e59. doi: 10.1093/nar/gkr1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dingle TC, Mulvey GL, Armstrong GD. 2011. Mutagenic analysis of the Clostridium difficile flagellar proteins, FliC and FliD, and their contribution to virulence in hamsters. Infect Immun 79:4061–4067. doi: 10.1128/IAI.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blakely GW, Murray NE. 2008. DNA restriction and modification, p 538–549. In Schaechter M. (ed), The encyclopedia of microbiology, 3rd ed. Elsevier, Oxford, United Kingdom. [Google Scholar]
- 59.Heap JT, Pennington OJ, Cartman ST, Carter GP, Minton NP. 2007. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods 70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 60.Ehsaan M, Kuehne SA, Minton NP. 2016. Clostridium difficile genome editing using pyrE alleles. Methods Mol Biol 1476:35–52. doi: 10.1007/978-1-4939-6361-4_4. [DOI] [PubMed] [Google Scholar]
- 61.Faulds-Pain A, Wren BW. 2013. Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in Clostridium difficile and Streptococcus suis. Appl Environ Microbiol 79:4768–4771. doi: 10.1128/AEM.01195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McAllister KN, Bouillaut L, Kahn JN, Self WT, Sorg JA. 2017. Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci Rep 7:14672. doi: 10.1038/s41598-017-15236-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Little JW. 1991. Mechanism of specific LexA cleavage: autodigestion and the role of RecA coprotease. Biochimie 73:411–421. doi: 10.1016/0300-9084(91)90108-D. [DOI] [PubMed] [Google Scholar]
- 64.Cox MM. 2007. Regulation of bacterial RecA protein function. Crit Rev Biochem Mol Biol 42:41–63. doi: 10.1080/10409230701260258. [DOI] [PubMed] [Google Scholar]
- 65.Chaudhury AM, Smith GR. 1984. Escherichia coli recBC deletion mutants. J Bacteriol 160:788–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kidane D, Sanchez H, Alonso JC, Graumann PL. 2004. Visualization of DNA double-strand break repair in live bacteria reveals dynamic recruitment of Bacillus subtilis RecF, RecO and RecN proteins to distinct sites on the nucleoids. Mol Microbiol 52:1627–1639. doi: 10.1111/j.1365-2958.2004.04102.x. [DOI] [PubMed] [Google Scholar]
- 67.Sanchez H, Kidane D, Castillo Cozar M, Graumann PL, Alonso JC. 2006. Recruitment of Bacillus subtilis RecN to DNA double-strand breaks in the absence of DNA end processing. J Bacteriol 188:353–360. doi: 10.1128/JB.188.2.353-360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sciochetti SA, Blakely GW, Piggot PJ. 2001. Growth phase variation in cell and nucleoid morphology in a Bacillus subtilis recA mutant. J Bacteriol 183:2963–2968. doi: 10.1128/JB.183.9.2963-2968.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Twine SM, Reid CW, Aubry A, McMullin DR, Fulton KM, Austin J, Logan SM. 2009. Motility and flagellar glycosylation in Clostridium difficile. J Bacteriol 191:7050–7062. doi: 10.1128/JB.00861-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Δerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J Med Microbiol 54:137–141. doi: 10.1099/jmm.0.45790-0. [DOI] [PubMed] [Google Scholar]