


Abstract
The heterotrimeric eukaryotic translation initiation factor (eIF) 2 plays critical roles in delivering initiator Met-tRNAiMet to the 40S ribosomal subunit and in selecting the translation initiation site. Genetic analyses of patients with MEHMO syndrome, an X-linked intellectual disability syndrome, have identified several unique mutations in the EIF2S3 gene that encodes the γ subunit of eIF2. To gain insights into the molecular consequences of MEHMO syndrome mutations on eIF2 function, we generated a yeast model of the human eIF2γ-I259M mutant, previously identified in a patient with MEHMO syndrome. The corresponding eIF2γ-I318M mutation impaired yeast cell growth and derepressed GCN4 expression, an indicator of defective eIF2–GTP–Met-tRNAiMet complex formation, and, likewise, overexpression of human eIF2γ-I259M derepressed ATF4 messenger RNA translation in human cells. The yeast eIF2γ-I318M mutation also increased initiation from near-cognate start codons. Biochemical analyses revealed a defect in Met-tRNAiMet binding to the mutant yeast eIF2 complexes in vivo and in vitro. Overexpression of tRNAiMet restored Met-tRNAiMet binding to eIF2 in vivo and rescued the growth defect in the eIF2γ-I318M strain. Based on these findings and the structure of eIF2, we propose that the I259M mutation impairs Met-tRNAiMet binding, causing altered control of protein synthesis that underlies MEHMO syndrome.
INTRODUCTION
A key step during initiation of protein synthesis in eukaryotic cells is formation of the ternary complex (TC) composed of the translation initiation factor 2 (eIF2), GTP and initiator Met-tRNAiMet. The eIF2 itself is a heterotrimer of distinct α, β and γ subunits. The eIF2 TC binds to the 40S small ribosomal subunit, along with other translation factors, forming a 43S preinitiation complex (PIC) that binds near the 5′ end of a messenger RNA (mRNA) forming a 48S PIC. The 48S PIC then scans down the mRNA (1). The translation start site, typically an AUG codon, is selected by base-pairing interactions between the anticodon of the tRNAiMet and the mRNA. Start codon selection triggers completion of GTP hydrolysis by eIF2, release of Pi and subsequent release of eIF2–GDP (1–3). In addition to mediating delivery of Met-tRNAiMet to the ribosome, eIF2 also plays a critical role in selection of the translation start codon. Mutations have been isolated in all three subunits of yeast eIF2 that relax the stringency of start site selection and promote initiation at a near cognate start codon (4–8). Recently, multiple cases of an X-linked intellectual disability (XLID) syndrome caused by mutations in the EIF2S3 gene, encoding the eIF2γ subunit, have been identified (9–12). The acronym MEHMO denotes the characteristic symptoms of this syndrome: Mental deficiency, Epilepsy, Hypogenitalism, Microcephaly and Obesity (13). While severely affected patients exhibit all symptoms, less affected patients exhibit only a subset of the symptoms. Furthermore, it remains unknown why symptom severity is variable between patients and how each of the identified mutations impacts eIF2 function. Thus, there is a critical need to determine the underlying molecular mechanism governing the phenotype of the MEHMO patients.
Genetic analyses of MEHMO syndrome patients have identified several unique mutations in EIF2S3 that map to distinct regions of eIF2γ (9–11), suggesting that the different mutations will likely impact distinct eIF2 functions. We previously characterized the MEHMO mutation I222T, located on the back face of the eIF2γ GTP-binding domain (Figure 1B). This eIF2γ-I222T mutation lies in a cleft that forms the docking site for helix α1 of eIF2β. Accordingly, the eIF2γ-I222T mutation impaired the interaction of overexpressed eIF2γ with the endogenous eIF2β in human cells (9). Likewise, mutation of the corresponding residue (V281) in yeast eIF2γ, encoded by the GCD11 gene, impaired eIF2β binding. In addition to impairing eIF2β binding to eIF2γ, the V281T mutation in yeast eIF2γ caused modest derepression of GCN4 expression, a sensitive reporter of eIF2 function in yeast, and impaired the stringency of translation start site selection in reporter gene experiments as revealed by increased initiation at a near cognate UUG codon. A second mutation linked to MEHMO syndrome, eIF2γ-I465Sfs*4, is caused by a 4-nt deletion near the 3′ end of the eIF2γ open reading frame (10–12). The mutation introduces a frameshift and changes the C-terminal residues of eIF2γ. While the molecular defect associated with the I465Sfs*4 mutation has not been resolved, the corresponding mutation in yeast eIF2 derepresses GCN4 expression and decreases the stringency of translation start site selection (12). A third mutation in EIF2S3 linked to MEHMO syndrome changed the poorly conserved S108 residue to arginine; however, it is unclear how this mutation impacts eIF2 function and the corresponding mutation in yeast eIF2γ had no phenotype. Consistent with the lack of phenotype in yeast, the S108R mutation was associated with milder symptoms in the patient.
Figure 1.

MEHMO syndrome mutation eIF2γ-I259M lies in the transfer RNA (tRNA)-binding pocket of eIF2γ and the corresponding yeast eIF2γ-I318M mutation confers a slow-growth phenotype. (A) Ribbons representation of Sulfolobus solfataricus aIF2–GDPNP–Met-tRNAiMet TC (PDB code 3V11; (25)) using PyMOL software (Schrodinger). Components are colored as follows: tRNAiMet, green; Met, purple, aIF2α, yellow; aIF2β helix α1, slate; GTP, gold and aIF2γ, cyan. (B) Magnification of the aIF2γ tRNA-binding pocket to highlight the proximity of SsI218 (HsI259, blue) to Met-tRNAiMet. Sites of three other mutations identified in patients with MEHMO syndrome are highlighted in stick representation including SsI181 (HsI222, red), SsT69 (HsS108, brown) and a C-terminal frameshift mutation (orange). (C) Magnification of aIF2γ from panel (B). MEHMO syndrome mutation site SsI218 (ScI318, HsI259; blue) and the adjacent SsR219 (ScR319, HsR260; pink) residues are highlighted. (D) Residues forming the methionine (magenta) binding pocket in Saccharomyces cerevisiae eIF2γ, taken from the structure of the yeast 48S PIC (PDB code 3JAP; (42). (E) Serial dilutions of yeast strains expressing the indicated WT or mutant forms of eIF2γ were spotted onto SD minimal medium containing essential amino acid supplements and plates were incubated at 30°C for 2 days. (F) Whole cell extracts (WCEs) from yeast strains expressing the indicated wild-type or mutant forms of eIF2γ were subjected to (SDS-PAGE) followed by immunoblotting with rabbit polyclonal antisera against yeast eIF2γ or GCD6 (eIF2Bϵ).
A fourth mutation in eIF2γ-I259M, was identified in two male siblings with severe microcephaly, growth retardation, seizures and intellectual disability (10), and thus likely also causes MEHMO syndrome. To gain insights into the molecular impact of the I259M mutation on eIF2 function, we mutated the corresponding residue (I318) in S. cerevisiae eIF2γ. Our studies revealed that the I318M mutation in yeast eIF2γ, like other MEHMO syndrome mutations in eIF2γ, leads to derepressed GCN4 expression and relaxed stringency of translation start site selection in vivo. However, in contrast to the previously characterized MEHMO mutations in eIF2γ, the I318M impairs Met-tRNAiMet binding to eIF2 both in vivo and in vitro. Thus, these studies provide insights into the structure-function properties of eIF2 and expand our understanding into the ways in which eIF2 function can be impaired to cause MEHMO syndrome.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strain J292 (MATα leu2-3,-112 ura3-52 his3 gcn2Δ::loxP gcd11Δ::KanMX p[GCD11, URA3]) (5) was used for the analysis of eIF2γ mutants. Mutant alleles of GCD11 were generated by site-directed mutagenesis and introduced into J292 as the sole source of eIF2γ by plasmid shuffling (14). Plasmids used in this study are listed in Table 1.
Table 1.
Plasmids used in this study
| Plasmid | Description | Reference |
|---|---|---|
| p180 | GCN4-lacZ, URA3, CEN4/ARS | (20) |
| p367 | HIS4(AUG)-lacZ, URA3 | (30) |
| p391 | HIS4(UUG)-lacZ, URA3 | (30) |
| pC2872 | His8-GCD11 (eIF2γ), LEU2, CEN4/ARS | (5) |
| pC4188 | His8-gcd11-V281T in pC2872 | (9) |
| pC4187 | His8-gcd11-V281K in pC2872 | (9) |
| pC5856 | His8-gcd11-I318M in pC2872 | This study |
| pC5857 | His8-gcd11-I318L in pC2872 | This study |
| pRS423 | HIS3, 2μ | (48) |
| pC1682 | SUI2 (eIF2α) in pRS423 | (32) |
| pC4191 | SUI3 (eIF2β) in pRS423 | (9) |
| pC1683 | IMT4 (tRNAiMet) in pRS423 | (32) |
| WB759 | TK-ATF4-luc in pGL3 | (28) |
| pC4293 | pcDNA4/TO/myc-HisA | (9) |
| pC4291 | human eIF2γ in pcDNA4/TO/myc-HisA | (9) |
| pC6155 | human eIF2γ-I259M in pC4291 | This study |
Growth analysis
Derivatives of yeast strain J292 expressing WT or mutant forms of eIF2γ, either alone or with an empty vector (pRS423) or various high copy-number plasmids to overexpress eIF2α (pC1682), eIF2β (pC4191) or tRNAiMet (pC1683), were grown to saturation in minimal SD medium with required amino acid supplements. Five microliter volumes of serial dilutions (OD600 = 1.0, 0.1, 0.01, 0.001 and 0.0001) were spotted onto SD medium containing required amino acid supplements, and the plates were incubated at 30°C for 3 days.
Western blot analysis
Yeast strains expressing WT or mutant forms of eIF2γ were grown to OD600∼1 in 100 ml SD medium containing required amino acid supplements at 30°C. Cells were harvested, washed with 5 ml Breaking Buffer A (20 mM Tris–HCl (pH 7.5), 500 mM KCl, 0.1 mM MgCl2, 5 mM β-mercaptoethanol, 10% (v/v) glycerol, 1× cOmplete protease inhibitor (Sigma-Aldrich) and 1 mM [4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), Sigma]), and resuspended in 1 ml Breaking Buffer A. Cells were lysed by adding 50% (w/v) glass beads and then vigorous mixing on a vortex for 5 min at 4°C. Lysates were cleared by centrifugation at 15 700 × g for 10 min at 4°C, quantified, mixed with sodium dodecyl sulphate (SDS)-sample buffer and incubated at 70°C for 10 min. Samples were subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblotting with rabbit polyclonal antisera against yeast eIF2γ (15), eIF2β (16), eIF2α (17) and GCD6 (18).
β-galactosidase assays
For measurement of GCN4-lacZ, his4(UUG)-lacZ or HIS4(AUG)-lacZ expression, yeast cultures were grown overnight to saturation in SC medium and then used to inoculate 25 ml of SC medium at OD600 = 0.25. Cultures were grown 6 h to OD600≤1 and β-galactosidase activities were determined as previously described (19,20). Means and standard deviations of β-galactosidase activities were calculated for three independent transformants.
Cell culture and luciferase assays
HEK293T cells were obtained from American Type Culture Collection and maintained in Dulbecco Modified Eagle Medium (DMEM) containing 4.5 g/l D-glucose, L-glutamine and 110 mg/l sodium pyruvate (Gibco) that was supplemented with 10% Fetal Bovine Serum (FBS, Gibco) and 1% penicillin/streptomycin (Quality Biological). For measurements of ATF4-luc expression, ATF4-luc and a Renilla reporter plasmid were transiently co-transfected with an empty vector or overexpression plasmids encoding WT eIF2γ or eIF2γ-I259M into HEK293T cells following the Lipofectamine 3000 (Invitrogen) manufacturer’s protocol. Culture media was exchanged for fresh DMEM medium 8 h after transfection. Cell lysis and measurement of relative luciferase activities were conducted following the Promega Dual-Luciferase Reporter Assay System protocol with a Centro XS LB 960 High Sensitivity Microplate Luminometer. Means and standard deviations of relative luciferase activities were calculated for three biological replicates.
eIF2 purification
Preparation of eIF2 was performed as previously described (21) with the following modifications. Yeast strains expressing WT eIF2γ or eIF2γ-I381M were grown to OD600∼4 in 10 l yeast extract peptone dextrose (YPD) medium at 30°C. Cells were pelleted by centrifugation at 6000 × g for 10 min at 4°C, and cell pellets were frozen at −80°C overnight. The following day, cells were lysed using a Waring Blender, 50% (w/v) glass beads, in 200 ml Lysis Buffer (75 mM Tris–HCl (pH 7.5), 100 mM KCl, 0.1 mM GDP, 14 mM β-mercaptoethanol and 1× cOmplete protease inhibitor (Roche)). Lysates were clarified by centrifugation at 15 000 × g for 30 min at 4°C. The supernatant was transferred to a beaker and ammonium sulfate was added gradually to 75% saturation over 1 h at 4°C. The slurry was then pelleted by centrifugation at 15 000 × g for 30 min at 4°C, and the pellet was resuspended in Buffer A (20 mM Tris–HCl (pH 7.5), 0.5 M KCl, 0.1 mM MgCl2, 20 mM imidazole, 10% (v/v) glycerol, 10 μM GDP, 5 mM β-mercaptoethanol and 1× cOmplete protease inhibitor). Following addition of 10 ml Ni-NTA resin (Qiagen), samples were stirred for 1 h at 4°C. The Ni-NTA resin was subsequently washed with 30 ml Buffer A, and eIF2 complexes were eluted in 20 ml Buffer B (20 mM Tris–HCl (pH 7.5), 0.5 M KCl, 0.1 mM MgCl2, 500 mM imidazole, 10% (v/v) glycerol, 10 μM GDP, 5 mM β-mercaptoethanol and 1× cOmplete protease inhibitor). The elute was dialyzed overnight against 0.5 l Dialysis Buffer (20 mM Tris–HCl (pH 7.5), 100 mM KCl, 0.1 mM MgCl2, 10% (v/v) glycerol, 10 μM GDP and 2 mM dithiothreitol (DTT)) at 4°C. Samples were then clarified by centrifugation at 4000 × g for 10 min at 4°C, and the supernatant was passed through a 0.45 μm filter to clear up the solution. Samples were loaded onto an equilibrated HiTrap Heparin column (Amersham) and further processed as described previously (21). Fractions containing eIF2 were identified by SDS-PAGE, pooled, dialyzed against Storage Buffer (20 mM Tris–HCl (pH 7.5), 100 mM KAc, 0.1 mM MgAc, 10% (v/v) glycerol and 2 mM DTT), aliquoted and stored at −80°C.
Synthesis and aminoacylation of tRNAiMet
Yeast tRNAiMet was synthesized by in vitro transcription with T7 polymerase as described previously (21). His-tagged MetRS was purified from Escherichia coli XL1 Blue cells (Agilent) carrying the E. coli methionyl-tRNA synthetase (MetRS) expression plasmid pRA101 (22) as described previously (23). Aminoacylation reactions with 5 μM synthesized tRNAiMet, 2 mM adenosine triphosphate-Mg2+, 0.3 μM [35S]methionine (Perkin Elmer), 10 mM MgCl2 and 1 μM MetRS were incubated in 1× Reaction Buffer (100 mM HEPES-KOH (pH 7.5), 1 mM DTT and 10 mM KCl) for 30 min at 30°C, and aminoacylated Met-tRNAiMet was purified by phenol/chloroform extraction (21).
Met-tRNAiMet and guanine nucleotide affinity measurements
For measurements of tRNA binding, seven 2-fold serial dilutions of eIF2 with final concentrations from 3.125 to 200 nM were incubated for 15 min at 26°C in Binding Assay Buffer (25 mM HEPES-KOH (pH 7.6), 2.5 mM Mg(OAc)2, 80 mM KOAc (pH 7.6), 2 mM DTT and 0.5 mM guanosine 5′-[β,γ-imido]triphosphate (GDPNP)-Mg2+). [35S]Met-tRNAiMet was added to a final concentration of 1 nM and reactions were incubated for 15 min at 26°C, followed by vacuum filtration as previously described (21). For measurements of GDP affinity, final concentrations from 3.125 to 200 nM eIF2 were incubated for 15 min at 26°C in Binding Assay Buffer lacking GDPNP-Mg2+. [3H]GDP was added to a final concentration of 2 nM and reactions were incubated for 15 min at 26°C followed by vacuum filtration as previously described (24). Means and standard deviations for the fraction of [35S]Met-tRNAiMet or [3H]GDP bound to eIF2 were calculated for three independent experiments. Binding curves were fit using the program Kaleidagraph to the equation: fraction bound = Bmax[S] / (Kd + [S]), where Bmax is the maximum fraction bound at excess substrate [S].
eIF2γ pull-down assay
Yeast strains expressing His-tagged forms of WT or mutant eIF2γ, either alone or with an empty vector (pRS423) or high copy-number IMT4 (tRNAiMet) plasmid (pC1683), were grown to OD600 ∼ 1 in 200 ml SD medium with amino acid supplements at 30°C. Cells were harvested, washed with 10 ml Breaking Buffer B (20 mM Tris–HCl (pH 7.5), 500 mM KCl, 0.1 mM MgCl2, 10 mM imidazole, 5 mM β-mercaptoethanol, 10% (v/v) glycerol, 1× cOmplete protease inhibitor and 1 mM AEBSF), and resuspended in 2 ml Breaking Buffer B. Cells were lysed by adding 50% (w/v) glass beads and then vigorous mixing on a vortex for 5 min at 4°C. Lysates were cleared by centrifugation at 14 000 × g for 10 min at 4°C and divided into quarters. For protein input samples, one quarter of the lysate was quantified, mixed with SDS-sample buffer and incubated at 70°C for 10 min. For analysis of Met- tRNAiMet in input samples, one quarter of the lysate was quantified and subjected to RNA isolation using TRIzol LS Reagent (Invitrogen), followed by reverse transcriptase-polymerase chain reaction (RT-PCR) as described below. For eIF2γ pull-down analysis, two quarters of the lysate were independently mixed with 0.1 ml Ni-NTA resin, incubated with slow rocking on a nutator for 2 h at 4°C and then washed three times with 5 ml Breaking Buffer B. Beads were then either mixed with SDS-sample buffer or with TRIzol LS Reagent. Input and pull-down protein samples were subjected to SDS-PAGE followed by immunoblotting with rabbit polyclonal antisera against yeast eIF2γ, eIF2β and eIF2α.
For analysis of Met-tRNAiMet, RNA was extracted from the input and pull-down samples using TRIzol LS Reagent (Invitrogen) according to the manufacture’s protocol. Reverse transcription (RT) was conducted using SuperScript III First-Strand Synthesis SuperMix (ThermoFisher Scientific) with random hexamers according to the manufacturer’s protocol. For RT reactions, 2.5 μl RNA was utilized for input samples and all of the isolated RNA was utilized for pull-down samples. PCR reactions of input and pull-down samples were conducted using Phusion Hot Start II DNA Polymerase (ThermoFisher Scientific), 1× Phusion HF buffer, 200 μM deoxynucleotide triphosphates (dNTPs), 0.5 μM Forward Primer (5′-GCAGGGCTCATAACCCTGAT-3′), 0.5 μM Reverse Primer (5′-TAGCGCCGCTCGGTTTC-3′) and 1 μl RT products in 50 μl total volume. PCR was performed under the following conditions: (i) 98°C for 30 s; (ii) 15 cycles of 98°C for 10 s, 59°C for 30 s and 72°C for 2 s; and (iii) 72°C for 5 min. Reactions products were resolved by electrophoresis on a 2.5% agarose gel and stained with GelRed Nucleic Acid Stain (Biotium).
RESULTS
MEHMO syndrome mutation eIF2γ-I259M lies in the Met-tRNAiMet-binding pocket and the corresponding I318M mutation in yeast eIF2γ impairs cell growth
Recent X-chromosome exome sequencing identified a missense mutation (I259M) in the EIF2S3 gene encoding eIF2γ of two male siblings with MEHMO syndrome phenotypes, including severe microcephaly, growth retardation, seizures and intellectual disability (10). The I259 residue is located in domain II of eIF2γ and is conserved among archaeal aIF2γ and eukaryotic eIF2γ proteins, suggesting that the residue could be of functional importance. Both eIF2γ and the eIF2 complex are well conserved through evolution and structural studies on aIF2 and eIF2 complexes provide useful insights into the structure of human eIF2. High-resolution structures of the aIF2 TC, as well as of the yeast 48S PIC, reveal interactions among the eIF2α, eIF2β and eIF2γ subunits that mediate eIF2 heterotrimer formation as well as the interactions between Met-tRNAiMet and the eIF2 heterotrimer.
Based on the crystal structure of the Sulfolobus solfataricus (Ss ) aIF2 TC, composed of aIF2α, aIF2β, aIF2γ, GTP and Met-tRNAiMet (Figure 1A) (25), the Ile218 residue in aIF2γ, which is analogous to the Ile259 mutation site in Homo sapiens eIF2γ and I318 in S. cerevisiae eIF2γ, lies in the tRNA-binding pocket of aIF2γ (Figure 1B and C). Intriguingly, I218 in aIF2 is only ∼7Å from the methionine residue on Met-tRNAiMet, and the adjacent residue R219 makes direct contacts with the Met residue (Figure 1C). The proximity of I218 to a contact point with Met-tRNAiMet raises the possibility that the I259M mutation in the patients with MEHMO syndrome phenotypes might impair eIF2 TC formation by disrupting initiator Met-tRNAiMet binding to the eIF2 complex.
As it is challenging to study eIF2 mutations in mammalian systems, in part, because it has not been possible to prepare recombinant forms of mammalian eIF2 for biochemical studies, we use in vivo assays in S. cerevisiae and complementary biochemical experiments with purified wild-type and mutant yeast factors to study the impact of mutations on eIF2 function. Growth analyses were conducted for yeast strains lacking the chromosomal GCD11 gene encoding eIF2γ and expressing wild-type (WT) eIF2γ, eIF2γ-V281T, eIF2γ-V281K, eIF2γ-I318M or eIF2γ-I318L from a single copy-number plasmid. As shown previously (9) the V281T mutation, corresponding to the I222T mutation identified in a patient with MEHMO syndrome, had no impact on yeast cell growth compared to cells expressing WT eIF2γ (Figure 1E, rows 1 and 2); however, the Lys mutation at the same site (V281K) substantially impaired growth (Figure 1E, rows 1 and 3) consistent with its more severe impact on eIF2β binding to eIF2γ (9). The I318M mutation in yeast eIF2γ, corresponding to the I259M mutation in the patient, also resulted in a marked slow-growth phenotype (Figure 1E, row 4). In contrast, the I318L mutation did not impact yeast cell growth (Figure 1E, row 5). Western analyses revealed similar levels of eIF2γ in the strains, indicating that the growth defect in the strain expressing eIF2γ-I318M is due to impaired eIF2γ function and not to altered eIF2γ levels (Figure 1F).
The eIF2γ-I318M mutation derepresses GCN4 reporter expression and reduces start codon selection stringency
To further explore how the I318M mutation disrupts eIF2γ function, we examined translational control of a GCN4 reporter in the yeast strains expressing WT or mutant forms of eIF2γ. GCN4 is a transcriptional activator whose synthesis is governed by regulated translation reinitiation at upstream open reading frames (uORFs) in the leader of the mRNA (Figure 2A, top) and by changes in eIF2 TC levels. Under nonstarvation conditions, where TC levels are high, ribosomes translate the first uORF (uORF1). Following termination at the uORF1 stop codon, the 40S ribosomal subunit resumes scanning. A TC readily binds the scanning 40S subunit enabling reinitiation at one of the subsequent inhibitory uORFs (uORFs2-4), which prevents ribosomes from scanning to the GCN4 main ORF (mORF) and synthesizing GCN4 protein. Under amino acid starvation conditions, a cellular stress-response process known as general amino acid control is triggered. The protein kinase GCN2 is activated and phosphorylates eIF2α, converting eIF2 from a substrate to an inhibitor of its guanine-nucleotide exchange factor eIF2B. As GTP binding is a prerequisite for Met-tRNAiMet binding to eIF2, GCN2 phosphorylation of eIF2 impairs TC formation. The reduction in TC levels, in turn, causes ribosomes, following translation of uORF1, to scan for a longer time and thus longer distance on the GCN4 mRNA before re-binding a TC and becoming competent to initiate translation. The scanning ribosomes traverse past the inhibitory uORFs2-4 without initiating, and then acquire a TC and initiate translation at the GCN4 start codon. In the absence of eIF2 phosphorylation, such as in yeast lacking GCN2, mutations that impair TC formation or TC binding to the ribosome can also stimulate GCN4 expression, conferring a general control derepressed (Gcd−) phenotype (26,27). To quantify the impact of the eIF2γ mutations on GCN4 expression, a GCN4-lacZ reporter was introduced into the gcn2Δ strains expressing WT or mutant forms of eIF2γ. While the eIF2γ-V281T mutation slightly elevated GCN4-lacZ expression in our previous report (9), the eIF2γ-V281T mutation did not significantly impact GCN4-lacZ expression in this analysis, consistent with WT growth rate of strains expressing this mutant form of eIF2γ. In contrast, the eIF2γ-V281K mutation, which caused a significant slow-growth phenotype, resulted in an ∼8-fold increase in GCN4-lacZ expression compared to cells expressing WT eIF2γ (Figure 2A) (9). Cells expressing the eIF2γ-I318L mutant displayed a modest increase in GCN4-lacZ expression, while the most substantial GCN4 derepression was observed for the eIF2γ-I318M mutation with a 16-fold increase in GCN4-lacZ expression (Figure 2A).
Figure 2.
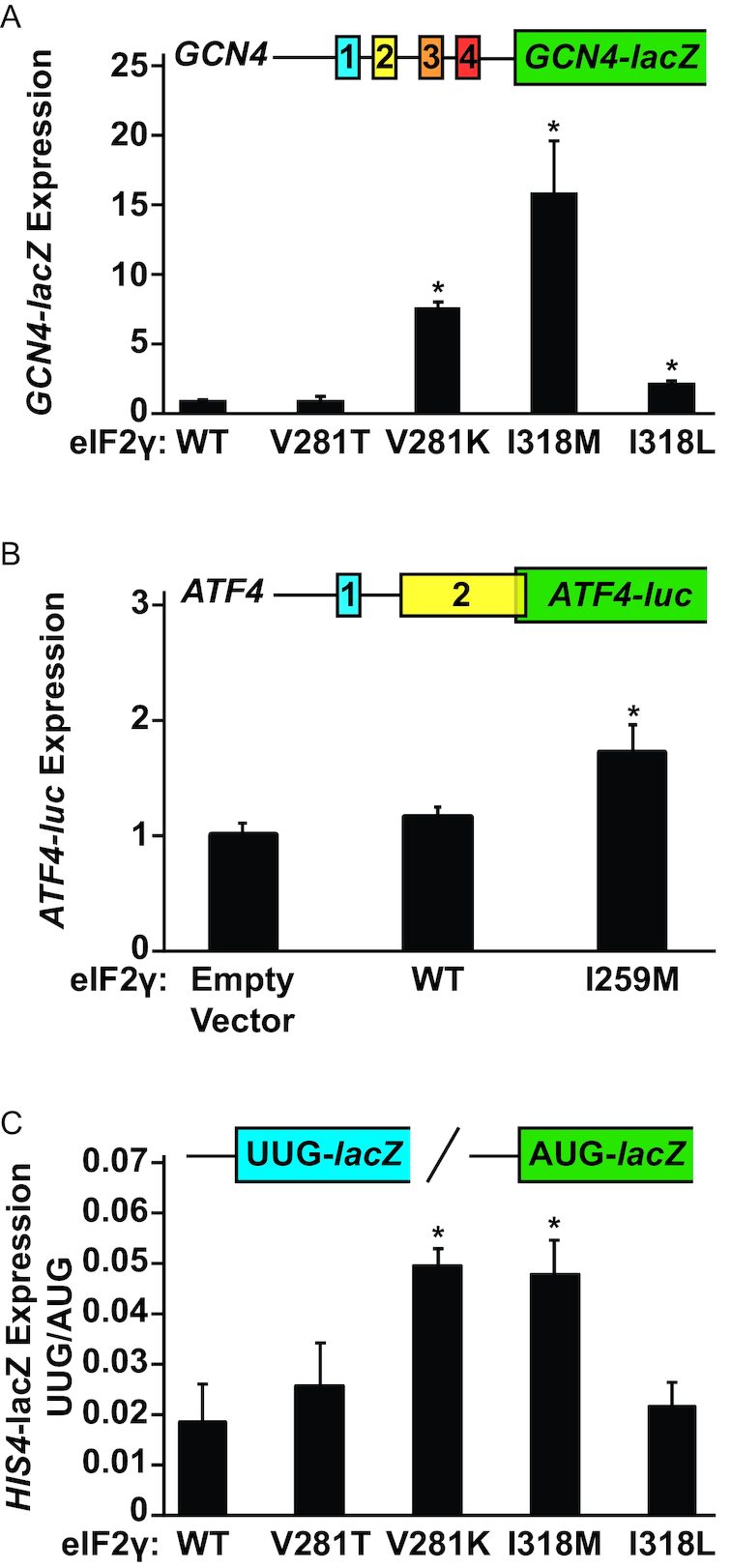
The eIF2γ-I318M mutation derepresses GCN4 reporter expression and reduces start codon selection stringency. (A) A GCN4-lacZ reporter was introduced into yeast strains expressing the indicated wild-type or mutant forms of eIF2γ. (B) An ATF4-luc and Renilla reporter plasmid were transiently co-transfected with the indicated eIF2γ overexpression plasmids into HEK293T cells. (C) his4(UUG)-lacZ and HIS4(AUG)-lacZ reporters were introduced into yeast strains expressing the indicated wild-type or mutant forms of eIF2γ. Means and standard deviations of β-galactosidase (A,C) or relative luciferase (B) activities were calculated from three independent transformants (A,C) or biological replicates (B). Statistically significant differences from WT eIF2γ β-galactosidase activities (A,C) or relative luciferase activities in cells transfected with an empty vector (B) are highlighted by an asterisk (*) and were calculated using a Student’s t-test with a P-value threshold of P = 0.05.
Expression of ATF4, the human homolog of GCN4, is also controlled by TC levels through regulated translation reinitiation at uORFs in the ATF4 mRNA leader. As in the GCN4 mRNA, the 40S ribosomal subunit readily resumes scanning following translation of uORF1 in the ATF4 mRNA. The ATF4 uORF2, which overlaps the 5′ end of the ATF4 mORF, but in a different reading frame, functionally replaces the inhibitory uORFs2-4 in the GCN4 mRNA. Under conditions of high TC levels, ribosomes initiate at uORF2 and fail to synthesize ATF4; whereas, when TC levels are lower, ribosomes bypass uORF2 and translate the ATF4 mORF (28,29). To quantify the impact of the MEHMO syndrome mutation on ATF4 expression, HEK293T cells were co-transfected with an ATF4-luc reporter and an overexpression vector encoding either WT human eIF2γ or eIF2γ-I259M. Overexpression of WT eIF2γ resulted in ATF4-luc expression similar to cells transfected with an empty vector control, while overexpression of eIF2γ-I259M derepressed ATF4-luc expression by nearly 2-fold (Figure 2B). Taken together, these results suggest that the yeast I318M and human I259M mutations in eIF2γ impair TC formation or TC binding to the scanning ribosome.
In addition to modulating GCN4 expression, mutations that impair eIF2 function have also been found to reduce the stringency of start codon selection (8). Mutations that weaken Met- tRNAiMet binding to eIF2 are thought to allow premature release of Met- tRNAiMet from eIF2 and enable ribosomes to initiate translation at non-AUG codons (a suppressor of initiation (Sui−) phenotype) (3,5,8). To determine if the fidelity of start site selection is modulated in the eIF2γ mutants, plasmid-borne HIS4-lacZ reporters with either an AUG or UUG start codon were introduced into the yeast strains expressing WT or mutant forms of eIF2γ. As observed previously (30), cells expressing WT eIF2γ displayed a high level of stringency for start site selection with HIS4-lacZ expression from the UUG-initiated reporter at ∼2%, the level obtained from the AUG-initiated reporter (Figure 2C). While the UUG/AUG initiation ratio in cells expressing eIF2γ-V281T and eIF2γ-I318L were not significantly different from cells expressing WT eIF2γ, consistent with the mild phenotypes of these mutations in the growth and GCN4 expression studies, both the V281K and I318M mutations increased the UUG/AUG initiation ratio by ∼2.5-fold (Figure 2C). This reduction in the stringency of start codon selection further supports the notion that the eIF2γ-I318M mutation impairs TC integrity, perhaps lowering the fidelity of start site selection by prematurely releasing Met-tRNAiMet at non-AUG codons during the ribosome scanning process.
Overexpression of tRNAiMet suppresses the slow growth phenotype in yeast expressing eIF2γ-I318M
As our structural analysis indicated that the I259M mutation might disrupt TC integrity by impairing Met-tRNAiMet binding to the eIF2 complex, we next tested whether overexpression of tRNAiMet could suppress the slow-growth and translation initiation phenotypes observed for the analogous eIF2γ-I318M mutant in yeast. High copy-number plasmids encoding yeast eIF2α (SUI2), eIF2β (SUI3), tRNAiMet (IMT4), or an empty vector control were introduced into yeast strains expressing WT eIF2γ, eIF2γ-V281K or eIF2γ-I318M. Cells expressing WT eIF2γ and overexpressing either eIF2α or tRNAiMet grew similarly to the vector control, while overexpression of eIF2β resulted in a slight growth defect as previously reported (Figure 3A, rows 1–4) (16). Consistent with the growth defects noted in Figure 1E, the empty vector transformants of both the eIF2γ-V281K and eIF2γ-I318M mutants exhibited severe slow-growth phenotypes (Figure 3A, rows 5 and 9) (9). As we reported previously, the eIF2γ-V281K mutation impairs eIF2β binding to eIF2γ (9). In accord with this eIF2 complex assembly defect, overexpression of eIF2β, but not eIF2α or tRNAiMet, partially suppressed the slow-growth phenotype of the eIF2γ-V281K mutant (Figure 3A, row 7) by restoring eIF2 complex levels (9). In contrast to the eIF2γ-V281K mutant, overexpression of eIF2β exacerbated the growth defect of the eIF2γ-I318M mutant (Figure 3A, row 11). The cause of this enhanced growth defect is unclear. Intriguingly, overexpression of tRNAiMet restored the growth of the eIF2γ-I318M mutant to near WT levels (Figure 3A, row 12). As western analyses indicated that overexpression of eIF2α, eIF2β or tRNAiMet did not affect the levels of the WT or mutant eIF2γ proteins (Figure 3B), we propose that overexpression of tRNAiMet suppresses the growth defect of the eIF2γ-I318M mutant by restoring eIF2 TC formation.
Figure 3.
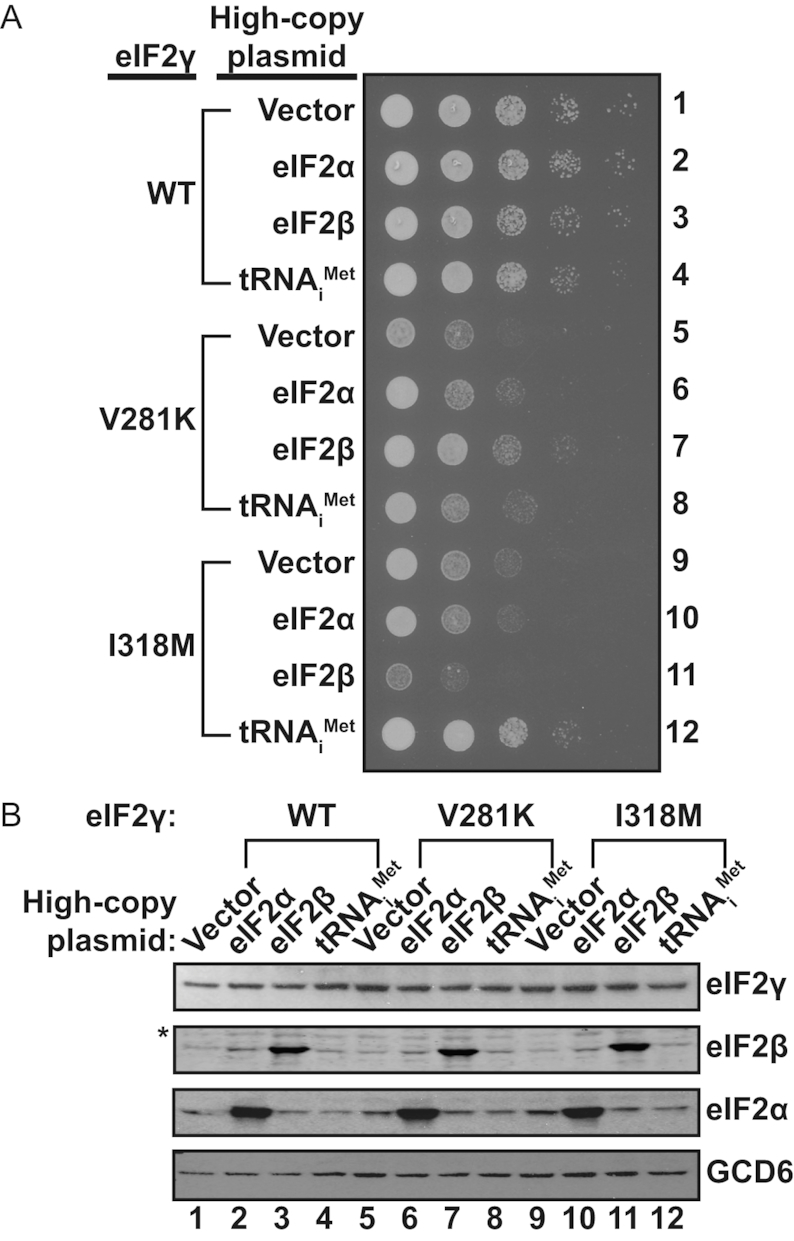
Overexpression of tRNAiMet suppresses the slow growth phenotype of yeast expressing the eIF2γ-I318M mutant. (A) 10-fold serial dilutions of yeast strains expressing the indicated wild-type or mutant forms of eIF2γ and co-transformed with an empty vector or with high copy-number plasmids containing the yeast eIF2α (SUI2), eIF2β (SUI3) or tRNAiMet (IMT4) genes were spotted onto SD minimal medium supplemented with required amino acids and incubated at 30°C for 3 days. (B) WCEs from yeast strains described in panel (A) were subjected to SDS-PAGE followed by immunoblotting with rabbit polyclonal antisera against yeast eIF2γ, eIF2β, eIF2α or GCD6 (eIF2Bϵ). The asterisk (*) indicates a non-specific immunoblot band.
To further explore the role of tRNAiMet in modulating eIF2γ-I318M function, GCN4-lacZ reporter activity was analyzed in yeast cells expressing WT eIF2γ, eIF2γ-V281K or eIF2γ-I318M with either a vector control or tRNAiMet overexpression. In accordance with our earlier observation, both the V281K and I318M mutations in eIF2γ caused robust derepression of GCN4-lacZ expression (Figure 4A, white bars). Additionally, overexpression of tRNAiMet dampened the level of GCN4 expression by roughly 50% in both the eIF2γ-V281K and eIF2γ-I318M strains (Figure 4A, black bars). These data suggest that tRNAiMet overexpression is likely sufficient to drive an increase in eIF2 TC formation by mass action in both the eIF2γ-V281K and eIF2γ-I318M mutant cells.
Figure 4.
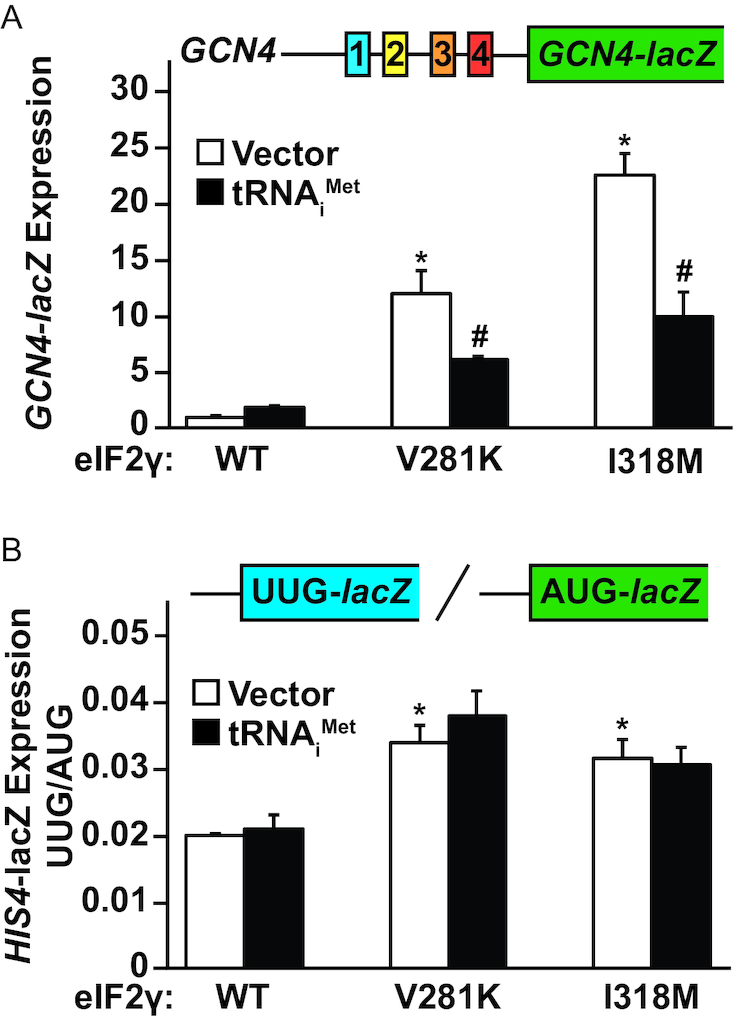
Overexpression of tRNAiMet suppresses the GCN4 induction, but not translation start codon stringency defect, caused by the eIF2γ-I318M mutation. A GCN4-lacZ reporter (A) or (B) his4(UUG)-lacZ and HIS4(AUG)-lacZ reporters were introduced into yeast strains expressing the indicated WT or mutant forms of eIF2γ with or without overexpression of tRNAiMet (IMT4). Means and standard deviations of β-galactosidase activities were calculated for three independent transformants. Statistically significant differences in β-galactosidase activities for strains expressing mutant versus WT eIF2γ with empty vector (*) or for strains overexpressing tRNAiMet versus empty vector (#) are indicated and were calculated using a Student’s t-test with a P-value threshold of P = 0.05.
We next assessed the impact of tRNAiMet overexpression on start codon selection stringency in the yeast strains expressing WT eIF2γ, eIF2γ-V281K or eIF2γ-I318M. As we observed above, both the eIF2γ-V281K and eIF2γ-I318M mutations elevated the UUG/AUG initiation ratio, indicating impaired fidelity of start codon selection (Figure 4B, white bars). Interestingly, overexpression of tRNAiMet did not attenuate the increase in UUG-lacZ expression in either of the eIF2γ mutants (Figure 4B, black bars). As will be discussed in more detail below, the translation start site fidelity defect in the eIF2γ-I318M mutant was likely due to premature release of eIF2, but not Met-tRNAiMet, from the scanning ribosome. Accordingly, overexpression of tRNAiMet and the attendant increase in TC levels was not expected to impact start site selection.
The eIF2γ-I318M mutation impairs Met-tRNAiMet binding both in vitro and in vivo
Based on the location of the I218 residue of the S. solfataricus aIF2 (corresponding to residue I318 of yeast eIF2γ) in the aIF2 TC (Figure 1A–C), the altered GCN4 mRNA translation in the eIF2γ-I318M mutant cells (Figure 2A), and the co-suppression of the cell growth and GCN4 expression defects in the mutant by overexpression of tRNAiMet (Figures 3A and 4A), we hypothesized that the I318M mutation impaired Met-tRNAiMet binding to eIF2. To assess the impact of the eIF2γ-I318M mutation on eIF2 complex integrity and TC formation, we first examined co-precipitation of the eIF2α and eIF2β subunits with His-tagged WT eIF2γ or eIF2γ-I318M. As controls, the assays were also performed using extracts from strains expressing untagged eIF2γ or His-tagged eIF2γ-V281K. Whereas no eIF2 subunits were precipitated with Ni2+ affinity resin from the lysates of cells expressing untagged eIF2γ (Figure 5A, lane 5), both eIF2α and eIF2β were readily co-precipitated with His-tagged WT eIF2γ (Figure 5A, lane 6). The eIF2γ-V281K mutation, as reported previously (9), impaired eIF2β, but not eIF2α, binding to eIF2γ (Figure 5A, lane 8); however, the eIF2γ-I318M mutant pulled-down eIF2α and eIF2β to the same extent as WT eIF2γ (Figure 5A, lane 7). Thus, the eIF2γ-I318M mutation did not affect eIF2 complex formation. We next used the same co-precipitation assays to assess TC formation by monitoring the pulldown of Met-tRNAiMet with the eIF2 complexes. Total RNA was isolated from the pellets of the pulldown assays and RT-PCR was used to specifically amplify tRNAiMet as described in the ‘Materials and Methods’ section. As shown in the bottom panel of Figure 5A, His-tagged eIF2γ-V281K pulled down lower levels of Met-tRNAiMet than WT eIF2γ, a downstream consequence of reduced eIF2 heterotrimer formation in the eIF2γ-V281K mutant (Figure 5A, lanes 6 and 8). The I318M mutation had a much more profound impact on TC levels with substantially reduced amounts of Met-tRNAiMet pulled down with the eIF2γ-I318M mutant (Figure 5A, lane 7), indicating a defect in TC formation.
Figure 5.

The eIF2γ-I318M mutation reduces eIF2–GTP–Met-tRNAiMet TC formation. (A) WCEs from yeast strains expressing the indicated WT or mutant forms of untagged or His-tagged eIF2γ were either immediately processed (Input; lanes 1–4) or incubated with Ni-NTA resin (His-eIF2γ pull-down; lanes 5–8). Input and pull-down protein samples were subjected to SDS-PAGE followed by immunoblotting (western blot, WB) with rabbit polyclonal antisera against yeast eIF2γ, eIF2β and eIF2α. Input and pull-down RNA samples were subjected to RT-PCR using oligonucleotide primers specific to yeast tRNAiMet, and reaction products were resolved by gel electrophoresis. The asterisk (*) indicates a non-specific immunoblot band migrating faster than eIF2γ. (B and C) [35S]Met-tRNAiMet plus non-hydrolyzable GDPNP (B) or [3H]GDP (C) were incubated with increasing concentrations of purified eIF2 complexes containing WT eIF2γ (black circles) or eIF2γ-I318M (blue squares), followed by vacuum filtration. Each data point represents the mean and error bars indicate the standard deviation for the fraction of [35S]Met-tRNAiMet or [3H]GDP bound to eIF2 in nitrocellulose filter bindings assays from three independent experiments. (D) WCEs from yeast strains expressing the indicated WT or mutant forms of untagged or His-tagged eIF2γ with or without overexpression of tRNAiMet were either immediately processed (Input; lanes 1–8) or incubated with Ni-NTA resin (His-eIF2γ pull-down; lanes 9–16). Input and pull-down protein samples were subjected to SDS-PAGE followed by immunoblotting (western blot, WB) with rabbit polyclonal antisera against yeast eIF2γ, eIF2β or eIF2α. Input and pull-down RNA samples were subjected to RT-PCR using oligonucleotide primers specific to yeast tRNAiMet, and reaction products were resolved by gel electrophoresis.
To further examine the impact of the eIF2γ-I318M mutation on TC formation, we purified the eIF2 complexes from strains expressing either WT eIF2γ or the eIF2γ-I318M mutant. Yeast tRNAiMet was transcribed in vitro and aminoacylated with [35S]Met using purified recombinant E. coli methionyl-tRNA synthetase. The [35S]Met-tRNAiMet was then mixed with increasing concentrations of the highly purified eIF2 complexes in the presence of the non-hydrolyzable GTP analog GDPNP. Filter binding assays were used to assess [35S]Met-tRNAiMet binding. The WT eIF2 complexes bound Met-tRNAiMet with an observed equilibrium dissociation constant (Kd) of 18 ± 2 nM (Figure 5B), which is comparable to the value obtained in previous reports (24,21). Intriguingly, the purified eIF2 complexes containing eIF2γ-I318M bound Met-tRNAiMet with ∼5-fold lower affinity (Kd = 98 ± 10 nM). As the binding of GTP and Met-tRNAiMet to eIF2 are thermodynamically coupled, the apparent Met-tRNAiMet binding defect observed for the eIF2 complexes containing eIF2γ-I318M could be due to a defect in guanine nucleotide or in Met-tRNAiMet binding. Because eIF2 has a higher affinity for GDP than for GTP (24), we used filter binding assays to monitor [3H]GDP binding to eIF2 as a means to examine the impact of the eIF2γ mutation on guanine nucleotide binding. As shown in Figure 5C, the eIF2 complexes containing WT eIF2γ (Kd = 30 ± 6) or eIF2γ-I318M (Kd = 27 ± 7) bound [3H]GDP with similar affinities that were comparable to previously reported measurements for WT eIF2 (24,21). Thus, the Met-tRNAiMet binding defect associated with the eIF2γ-I318M mutation is not due to a defect in guanine nucleotide binding. Taken together, the data from the pulldown assays and the in vitro binding assays indicate that the eIF2γ-I318M mutation in yeast eIF2, and by analogy the corresponding eIF2γ-I259M mutation identified in the patient with MEHMO syndrome, impairs eIF2 TC integrity through a defect in Met-tRNAiMet binding.
Based on the co-suppression of cell growth and GCN4 expression defects in the eIF2γ-I318M mutant by overexpression of tRNAiMet (Figures 3A and 4A), we hypothesized that tRNAiMet overexpression is likely sufficient to drive an increase in eIF2 TC formation by mass action. To assess the impact of tRNAiMet overexpression on TC formation, we introduced a high copy-number plasmid encoding yeast tRNAiMet or empty vector control into yeast strains expressing WT eIF2γ, eIF2γ-I318M or eIF2γ-V281K and then examined co-precipitation of eIF2α, eIF2β and Met-tRNAiMet with His-tagged eIF2γ. Strains expressing untagged eIF2γ were utilized as a negative control, and no eIF2 TC components were precipitated with Ni2+ affinity resin from the lysates of cells expressing untagged eIF2γ (Figure 5D, lanes 9 and 10). eIF2α, eIF2β and Met-tRNAiMet were readily co-precipitated with His-tagged WT eIF2γ independent of tRNAiMet overexpression (Figure 5D, lanes 11 and 12). As observed previously (Figure 5A), reduced levels of eIF2β and Met-tRNAiMet, but not eIF2α, were co-precipitated with the eIF2γ-V281K mutant (Figure 5D, lane 15). Overexpression of tRNAiMet slightly elevated Met-tRNAiMet binding to eIF2γ-V281K (Figure 5D, lanes 15 and 16), consistent with the partial suppression of the GCN4 expression defects in the eIF2γ-V281K mutant by overexpression of tRNAiMet (Figure 4A). The eIF2γ-I318M mutant did not affect eIF2 heterotrimer formation, as WT eIF2γ and eIF2γ-I318M pulled down similar amounts of both the eIF2α and eIF2β subunits (Figure 5D, lanes 11–14). However, the I318M mutation substantially impaired Met-tRNAiMet binding (Figure 5D, lane 13), and co-precipitation of Met-tRNAiMet with eIF2γ-I318M was restored in cells overexpressing tRNAiMet (Figure 5D, lanes 13 and 14). Thus, overexpression of tRNAiMet is sufficient to drive an increase in eIF2 TC formation by mass action in the eIF2γ-I318M mutant cells.
DISCUSSION
In this study, we address the molecular defect in eIF2 function caused by the eIF2γ-I259M mutation originally identified in a patient with MEHMO syndrome. Based on the location of the corresponding I318 residue in yeast eIF2γ (Figure 1D) and I218 residue in Ss aIF2γ (Figure 1C), we proposed that the mutation might impact Met-tRNAiMet binding to eIF2. Consistent with this hypothesis, substitution of Met for I259 in a human eIF2γ overexpression construct or for I318 in yeast eIF2γ resulted in derepression of ATF4 or GCN4 expression, respectively, which are sensitive in vivo reporters of eIF2 TC levels. The yeast eIF2γ-I318M mutation also lowered the stringency of start codon selection. Finally, biochemical studies revealed reduced binding of Met-tRNAiMet to the mutant eIF2 complexes both in crude yeast extracts and with the purified protein. Taken together these studies reveal that defective Met-tRNAiMet binding to eIF2 is a cause of MEHMO syndrome.
To date, four mutations in eIF2γ have been linked to MEHMO syndrome. While severely affected patients exhibit all MEHMO symptoms (Mental deficiency, Epilepsy, Hypogenitalism, Microcephaly and Obesity), less affected patients exhibit only a subset of symptoms. It is interesting to speculate that the impact of the mutations on distinct functions of eIF2γ may play a critical role in determining the phenotype and symptom severity of patients with MEHMO syndrome. Substitution of Thr for I222 on the back side of the GTP-binding domain of eIF2γ impaired eIF2 complex integrity by disrupting binding of the eIF2β subunit to eIF2γ (9). The corresponding yeast eIF2γ-V281T mutation caused a slight elevation in GCN4 expression (9), though not statistically significant in this report (Figure 2A), and a modest increase in translation initiation at a near cognate UUG codon (9). The milder phenotypes in the yeast eIF2γ-V281T versus the eIF2γ-I318M mutant is consistent with the more severe symptoms in the patient with the eIF2γ-I259M mutation. In further support of this correlation between yeast phenotypes and symptoms in patients, an S108R mutation near a Zn2+ binding element in eIF2γ resulted in a somewhat milder form of MEHMO syndrome and the corresponding D167R mutation in yeast had no impacts on cell growth or reporter gene expression (12). The fourth mutation in eIF2γ identified in patients with MEHMO syndrome, eIF2γ-I465Sfs*4, substitutes three new residues in place of the native eight C-terminal residues in eIF2γ. While a molecular explanation for how this mutation impacts eIF2 function has not been revealed, the corresponding mutation at the C-terminus of yeast eIF2γ mimicked the effects of the V281T mutation. A fifth eIF2γ mutation was identified upon resequencing samples from patients with undefined XLID. A V151L mutation was identified in a patient; however, because of the nature of the study it has not been possible to determine whether the mutation is linked to MEHMO syndrome. Despite this limitation, it is noteworthy that the corresponding V210L mutation in yeast eIF2γ resulted in a 12-fold increase in GCN4-lacZ expression and a 4-fold increase in the UUG/AUG initiation ratio (9). While the impacts of mutations in eIF2 on cell growth and reporter assays in single-celled yeast would not be expected to perfectly correlate with the severity of tissue-specific symptoms in patients with the corresponding mutations, our studies have demonstrated that yeast serves as a good system to model the eIF2γ mutations identified in patients with MEHMO syndrome.
The studies of the eIF2γ-I222T mutant revealed that defects in eIF2 heterotrimer formation could cause MEHMO syndrome (9). The I222T mutation lies in a cavity that forms the docking site for eIF2β to bind to eIF2γ. Consistent with this location, the mutation impaired eIF2β association with eIF2γ and the growth defect in yeast expressing the corresponding mutation could be suppressed by overexpression of eIF2β (9), as shown for the related mutation eIF2γ-V281K in Figure 3A. The eIF2γ-I318M mutation does not impair eIF2 heterotrimer formation (Figure 5A), but instead impairs the binding of Met-tRNAiMet to eIF2 (Figure 5). Examination of the structures of the isolated archaeal aIF2–GTP–Met-tRNAiMet TC (Figure 1A–C) and the analogous yeast eIF2–GTP–Met-tRNAiMet complex bound to the 40S subunit (Figure 1D) reveals that the I318 residue in yeast eIF2γ (and by analogy the I259 residue in human eIF2) lies near the methionine-binding site. A cluster of 12 residues forms a shell around the methionine docking site on eIF2γ (Figure 1D). Mutational studies in yeast have implicated four of these residues in Met-tRNAiMet binding to eIF2. The yeast eIF2γ mutations Y142H and G397A were previously isolated in screens for mutants that derepress GCN4 expression (4,31). In addition, both of these mutations reduce the fidelity of translation start site selection causing increased UUG/AUG ratios (4). Consistent with these genetic phenotypes, the slow-growth phenotype of yeast expressing eIF2γ-G397A was suppressed by overexpression of tRNAiMet (32) and the Y142H mutation in eIF2γ was shown to impair Met-tRNAiMet binding to purified eIF2 in vitro (33). As shown in this study, the slow-growth phenotype associated with eIF2γ-I318M mutation was suppressed by overexpression of tRNAiMet (Figure 3A) and the purified eIF2 complex containing eIF2γ-I318M was defective for binding Met-tRNAiMet (Figure 5). While the I318 residue is not in the shell of residues that directly form the Met binding pocket in eIF2γ (Figure 1D), the adjacent residue R319 (Figure 1D) is part of the Met binding pocket and mutation of R319 to Asp is lethal in yeast (32). We propose that the eIF2γ-R319D mutation impairs Met recognition and that the adjacent eIF2γ-I318M mutation described in this study impairs Met-tRNAiMet binding indirectly by impacting the position of R319 and its ability to contribute to Met recognition and binding. As all 12 residues forming the Met binding pocket in eIF2γ are well conserved in evolution, it is tempting to speculate that this conservation is driven by the requirement to initiate translation with Met. While mutations in the Met recognition pocket might enable eIF2 to bind alternate elongator aminoacyl-tRNAs and thereby initiate translation with residues other than Met, which could be of interest in biotechnology, the critical roles played by conserved tRNAiMet residues in translation start site selection (34) indicate that simply relaxing the stringency of Met binding is not likely to enable initiation with alternate aminoacyl-tRNAs.
The defect in Met-tRNAiMet binding and the accompanying reduced levels of eIF2 TCs in yeast expressing eIF2γ-I318M readily accounts for the elevated GCN4 expression. As previously demonstrated (27,16–17), regulated translation reinitiation at the uORFs on the GCN4 mRNA inversely couple GCN4 synthesis to TC levels. Consistent with this idea, overexpression of tRNAiMet in the eIF2γ-I318M mutant, which through mass action elevates TC levels (Figure 5D), suppresses the induction of GCN4 expression (Figure 4A). In contrast, overexpression of tRNAiMet failed to restore start codon selection stringency in the eIF2γ-I318M mutant (Figure 4B). This differential effect of overexpression of tRNAiMet on GCN4 expression and start codon selection stringency in the eIF2γ-I318M mutant (Figure 4) likely reflects the ordered pathway of translation initiation. As the TC must bind to the 40S subunit prior to mRNA binding and start codon selection, the stringency of start site selection is not expected to be sensitive to TC levels. Thus, restoring TC levels in the eIF2γ-I318M mutant failed to suppress near cognate initiation. However, this raises the question of how defects in TC formation relax the stringency of start codon selection in the eIF2γ-I318M mutant. One possibility is that the lowered affinity of eIF2γ-I318M for Met-tRNAiMet results in premature release of eIF2, but not Met-tRNAiMet, from the scanning ribosome. This might then allow the Met-tRNAiMet to move from its initial binding site in the Pout location that is conducive to ribosome scanning and transient inspection of the mRNA sequence for a start codon to a more fully engaged Pin location that prevents further scanning and marks selection of the translation start codon (AUG or near cognate) (1–3). A second possibility is that the I318M mutation might have a greater impact on Met-tRNAiMet binding to eIF2 in the Pout versus Pin state. Greater destabilization of the Pout state would shift the equilibrium to the Pin state and reduce the stringency of start codon selection. However, these simple models do not account for the critical role of eIF1 in translation start site selection. Genetic, biochemical and structural studies have established that eIF1 acts as a gatekeeper to block Pi and subsequently eIF2–GDP release until a codon-anticodon match is established between the mRNA and Met-tRNAiMet in the P site of the ribosome (1,2,35–43). Considering this critical role of eIF1, the eIF2γ mutation might relax start codon selection stringency by either enabling eIF2 release prior to eIF1 release or by prompting premature release of eIF1 in the absence of perfect anticodon–codon complementarity. The latter model was previously invoked to explain the relaxed start codon selection stringency of eIF2γ mutants that do not affect Met-tRNAiMet binding affinity (5). Accordingly, it was proposed that the eIF2γ mutations alter the positioning of Met-tRNAiMet in the P site to provoke eIF1 release at near cognate start codons (5). Perhaps the eIF2γ-I318M mutation in addition to weakening Met-tRNAiMet binding, alters the conformation of Met-tRNAiMet on the scanning ribosome to enable selection of a near cognate start codon and release of eIF1 in the absence of perfect base-pairing complementarity between the mRNA codon and the anticodon of tRNAiMet.
Finally, it is intriguing to compare the impacts of mutations in eIF2γ with mutations in the guanine nucleotide exchange factor eIF2B (44). eIF2B plays a critical role in determining eIF2 TC levels by regulating the levels of GTP-bound eIF2 (45). Following selection of the translation start codon by the scanning PIC, eIF2 completes hydrolysis of the bound GTP and Pi is released (2). Subsequently, eIF2 in complex with GDP is released from the PIC. In order for eIF2 to participate in additional rounds of translation initiation, the GDP bound to eIF2 must be exchanged by GTP in a reaction catalyzed by eIF2B (1). Mutations have been identified in each of the five subunits of eIF2B that cause leukoencephalopathy with vanishing white matter (VWM) (44,45). The symptoms associated with MEHMO syndrome and VWM have some overlap, as a subset of patients with MEHMO syndrome also have decreased white matter in their brains (9–11). Furthermore, yeast models of VWM and MEHMO syndrome mutations exhibit similar phenotypes with impaired cell growth and increased GCN4 expression, indicative of decreased eIF2 TC levels (Figures 1E and 2A) (9,44,46). However, it is also noteworthy that symptom presentation occurs earlier in the pathogenesis of MEHMO syndrome than VWM disease, and that eIF2γ mutations affect a broad spectrum of tissues that are not impacted by mutations in eIF2B (9–11,44,46). While it is unclear why mutations in two essential translation initiation factors result in the different symptoms described for MEHMO syndrome and VWM, one possibility is that impaired translation start site selection, as described here and in our previous analyses of MEHMO syndrome mutations, underlies the additional phenotypes observed in patients with MEHMO syndrome (Figures 2C and 4B) (9). Whereas VWM mutations impair eIF2B activity and are proposed to cause disease due to low eIF2 TC levels (46,47), MEHMO syndrome mutations result in dysregulation of translation initiation through both decreased eIF2 TC levels and reduced start codon selection stringency. How the molecular differences between eIF2γ and eIF2B mutations promote distinct patient phenotypes, and how mutations in eIF2γ, a ubiquitously expressed translation initiation factor, result in the tissue-specific phenotypes associated with MEHMO syndrome will be critical questions to address in future studies.
ACKNOWLEDGEMENTS
We are grateful to Alan Hinnebusch, Jon Lorsch, Nicholas Guydosh, and members of the Dever, Hinnebusch, Lorsch and Guydosh labs for helpful discussions. We thank Ronald Wek for kindly providing the ATF4-luc reporter plasmid.
FUNDING
Intramural Research Program, NICHD (NIH); Postdoctoral Research Associate Training (PRAT) Fellowship, National Institute of General Medical Sciences (NIGMS) [1Fi2GM123961 to S.K.Y.-B. ]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Hinnebusch A.G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 2014; 83:779–812. [DOI] [PubMed] [Google Scholar]
- 2. Hinnebusch A.G., Lorsch J.R.. The mechanism of eukaryotic translation initiation: new insights and challenges. Cold Spring Harb. Perspect. Biol. 2012; 4:a011544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hinnebusch A.G. Molecular mechanism of scanning and start codon selection in eukaryotes. Microbiol. Mol. Biol. Rev. 2011; 75:434–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dorris D.R., Erickson F.L., Hannig E.M.. Mutations in GCD11, the structural gene for eIF2γ in yeast, alter translational regulation of GCN4 and the selection of the start site for protein synthesis. EMBO J. 1995; 14:2239–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alone P.V., Cao C., Dever T.E.. Translation initiation factor 2γ mutant alters start codon selection independent of Met-tRNA binding. Mol. Cell. Biol. 2008; 28:6877–6888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Castilho-Valavicius B., Yoon H., Donahue T.F.. Genetic characterization of the Saccharomyces cerevisiae translational initiation suppressors sui1, sui2 and SUI3 and their effects on HIS4 expression. Genetics. 1990; 124:483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Donahue T.F., Cigan A.M., Pabich E.K., Castilho-Valavicius B.. Mutations at a Zn(II) finger motif in the yeast elF-2β gene alter ribosomal start-site selection during the scanning process. Cell. 1988; 54:621–632. [DOI] [PubMed] [Google Scholar]
- 8. Huang H., Yoon H., Hannig E.M., Donahue T.F.. GTP hydrolysis controls stringent selection of the AUG start codon during translation initiation in Saccharomyces cerevisiae. Genes Dev. 1997; 11:2396–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Borck G., Shin B.S., Stiller B., Mimouni-Bloch A., Thiele H., Kim J.R., Thakur M., Skinner C., Aschenbach L., Smirin-Yosef P. et al. eIF2γ mutation that disrupts eIF2 complex integrity links intellectual disability to impaired translation initiation. Mol. Cell. 2012; 48:641–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Moortgat S., Desir J., Benoit V., Boulanger S., Pendeville H., Nassogne M.C., Lederer D., Maystadt I.. Two novel EIF2S3 mutations associated with syndromic intellectual disability with severe microcephaly, growth retardation, and epilepsy. Am. J. Med. Genet. A. 2016; 170:2927–2933. [DOI] [PubMed] [Google Scholar]
- 11. Stanik J., Skopkova M., Stanikova D., Brennerova K., Barak L., Ticha L., Hornova J., Klimes I., Gasperikova D.. Neonatal hypoglycemia, early-onset diabetes and hypopituitarism due to the mutation in EIF2S3 gene causing MEHMO syndrome. Physiol. Res. 2018; 67:331–337. [DOI] [PubMed] [Google Scholar]
- 12. Skopkova M., Hennig F., Shin B.S., Turner C.E., Stanikova D., Brennerova K., Stanik J., Fischer U., Henden L., Muller U. et al. EIF2S3 Mutations Associated with Severe X-Linked Intellectual Disability Syndrome MEHMO. Hum. Mutat. 2017; 38:409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steinmuller R., Steinberger D., Muller U.. MEHMO (mental retardation, epileptic seizures, hypogonadism and -genitalism, microcephaly, obesity), a novel syndrome: assignment of disease locus to xp21.1-p22.13. Eur. J. Hum. Genet. 1998; 6:201–206. [DOI] [PubMed] [Google Scholar]
- 14. Boeke J.D., Trueheart J., Natsoulis G., Fink G.R.. 5-fluoroorotic acid as a selective agent in yeast molecular genetics. Methods Enzymol. 1987; 154:164–175. [DOI] [PubMed] [Google Scholar]
- 15. Alone P.V., Dever T.E.. Direct binding of translation initiation factor eIF2γ-G domain to its GTPase-activating and GDP-GTP exchange factors eIF5 and eIF2Bϵ. J. Biol. Chem. 2006; 281:12636–12644. [DOI] [PubMed] [Google Scholar]
- 16. Dever T.E., Yang W., Åström S., Byström A.S., Hinnebusch A.G.. Modulation of tRNAiMet, eIF-2 and eIF-2B expression shows that GCN4 translation is inversely coupled to the level of eIF-2.GTP.Met-tRNAiMet ternary complexes. Mol. Cell. Biol. 1995; 15:6351–6363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dever T.E., Feng L., Wek R.C., Cigan A.M., Donahue T.D., Hinnebusch A.G.. Phosphorylation of initiation factor 2α by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell. 1992; 68:585–596. [DOI] [PubMed] [Google Scholar]
- 18. Bushman J.L., Foiani M., Cigan A.M., Paddon C.J., Hinnebusch A.G.. Guanine nucleotide exchange factor for eIF-2 in yeast: genetic and biochemical analysis of interactions between essential subunits GCD2, GCD6 and GCD7 and regulatory subunit GCN3. Mol. Cell. Biol. 1993; 13:4618–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moehle C.M., Hinnebusch A.G.. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae. Mol. Cell. Biol. 1991; 11:2723–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hinnebusch A.G. A hierarchy of trans-acting factors modulate translation of an activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 1985; 5:2349–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Acker M.G., Kolitz S.E., Mitchell S.F., Nanda J.S., Lorsch J.R.. Reconstitution of yeast translation initiation. Methods Enzymol. 2007; 430:111–145. [DOI] [PubMed] [Google Scholar]
- 22. Alexander R.W., Nordin B.E., Schimmel P.. Activation of microhelix charging by localized helix destabilization. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12214–12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murray J., Savva C.G., Shin B.S., Dever T.E., Ramakrishnan V., Fernandez I.S.. Structural characterization of ribosome recruitment and translocation by type IV IRES. Elife. 2016; 5:e13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kapp L.D., Lorsch J.R.. GTP-dependent recognition of the methionine moiety on initiator tRNA by translation factor eIF2. J. Mol. Biol. 2004; 335:923–936. [DOI] [PubMed] [Google Scholar]
- 25. Schmitt E., Panvert M., Lazennec-Schurdevin C., Coureux P.D., Perez J., Thompson A., Mechulam Y.. Structure of the ternary initiation complex aIF2-GDPNP-methionylated initiator tRNA. Nat. Struct. Mol. Biol. 2012; 19:450–454. [DOI] [PubMed] [Google Scholar]
- 26. Dever T.E. Gene-specific regulation by general translation factors. Cell. 2002; 108:545–556. [DOI] [PubMed] [Google Scholar]
- 27. Hinnebusch A.G. Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 2005; 59:407–450. [DOI] [PubMed] [Google Scholar]
- 28. Vattem K.M., Wek R.C.. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Young S.K., Wek R.C.. Upstream open reading frames differentially regulate gene-specific translation in the integrated stress response. J. Biol. Chem. 2016; 291:16927–16935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Donahue T.F., Cigan A.M.. Genetic selection for mutations that reduce or abolish ribosomal recognition of the HIS4 translational initiator region. Mol. Cell. Biol. 1988; 8:2955–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harashima S., Hinnebusch A.G.. Multiple GCD genes required for repression of GCN4, a transcriptional activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell. Biol. 1986; 6:3990–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roll-Mecak A., Alone P., Cao C., Dever T.E., Burley S.K.. X-ray structure of translation initiation factor eIF2γ: implications for tRNA and eIF2α binding. J. Biol. Chem. 2004; 279:10634–10642. [DOI] [PubMed] [Google Scholar]
- 33. Erickson F.L., Hannig E.M.. Ligand interactions with eukaryotic translation initiation factor 2: role of the γ-subunit. EMBO J. 1996; 15:6311–6320. [PMC free article] [PubMed] [Google Scholar]
- 34. Dong J., Munoz A., Kolitz S.E., Saini A.K., Chiu W.L., Rahman H., Lorsch J.R., Hinnebusch A.G.. Conserved residues in yeast initiator tRNA calibrate initiation accuracy by regulating preinitiation complex stability at the start codon. Genes Dev. 2014; 28:502–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheung Y.N., Maag D., Mitchell S.F., Fekete C.A., Algire M.A., Takacs J.E., Shirokikh N., Pestova T., Lorsch J.R., Hinnebusch A.G.. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 2007; 21:1217–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martin-Marcos P., Nanda J.S., Luna R.E., Zhang F., Saini A.K., Cherkasova V.A., Wagner G., Lorsch J.R., Hinnebusch A.G.. Enhanced eIF1 binding to the 40S ribosome impedes conformational rearrangements of the preinitiation complex and elevates initiation accuracy. RNA. 2014; 20:150–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Martin-Marcos P., Nanda J., Luna R.E., Wagner G., Lorsch J.R., Hinnebusch A.G.. β-Hairpin loop of eukaryotic initiation factor 1 (eIF1) mediates 40 S ribosome binding to regulate initiator tRNAMet recruitment and accuracy of AUG selection in vivo. J. Biol. Chem. 2013; 288:27546–27562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maag D., Fekete C.A., Gryczynski Z., Lorsch J.R.. A conformational change in the eukaryotic translation preinitiation complex and release of eIF1 signal recognition of the start codon. Mol. Cell. 2005; 17:265–275. [DOI] [PubMed] [Google Scholar]
- 39. Algire M.A., Maag D., Lorsch J.R.. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell. 2005; 20:251–262. [DOI] [PubMed] [Google Scholar]
- 40. Passmore L.A., Schmeing T.M., Maag D., Applefield D.J., Acker M.G., Algire M.A., Lorsch J.R., Ramakrishnan V.. The eukaryotic translation initiation factors eIF1 and eIF1A induce an open conformation of the 40S ribosome. Mol. Cell. 2007; 26:41–50. [DOI] [PubMed] [Google Scholar]
- 41. Saini A.K., Nanda J.S., Lorsch J.R., Hinnebusch A.G.. Regulatory elements in eIF1A control the fidelity of start codon selection by modulating tRNAiMet binding to the ribosome. Genes Dev. 2010; 24:97–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Llacer J.L., Hussain T., Marler L., Aitken C.E., Thakur A., Lorsch J.R., Hinnebusch A.G., Ramakrishnan V.. Conformational differences between open and closed states of the eukaryotic translation initiation complex. Mol. Cell. 2015; 59:399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hussain T., Llacer J.L., Fernandez I.S., Munoz A., Martin-Marcos P., Savva C.G., Lorsch J.R., Hinnebusch A.G., Ramakrishnan V.. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell. 2014; 159:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pavitt G.D., Proud C.G.. Protein synthesis and its control in neuronal cells with a focus on vanishing white matter disease. Biochem. Soc. Trans. 2009; 37:1298–1310. [DOI] [PubMed] [Google Scholar]
- 45. Pavitt G.D. Regulation of translation initiation factor eIF2B at the hub of the integrated stress response. Wiley Interdiscip. Rev. RNA. 2018; 9:e1491. [DOI] [PubMed] [Google Scholar]
- 46. Richardson J.P., Mohammad S.S., Pavitt G.D.. Mutations causing childhood ataxia with central nervous system hypomyelination reduce eukaryotic initiation factor 2B complex formation and activity. Mol. Cell. Biol. 2004; 24:2352–2363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Li W., Wang X., Van Der Knaap M.S., Proud C.G.. Mutations linked to leukoencephalopathy with vanishing white matter impair the function of the eukaryotic initiation factor 2B complex in diverse ways. Mol. Cell. Biol. 2004; 24:3295–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Christianson T.W., Sikorski R.S., Dante M., Shero J.H., Hieter P.. Multifunctional yeast high-copy-number shuttle vectors. Gene. 1992; 110:119–122. [DOI] [PubMed] [Google Scholar]