

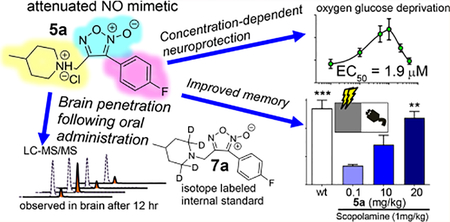
Abstract
Nitric oxide (NO) mimetics and other agents capable of enhancing NO/cGMP signaling have demonstrated efficacy as potential therapies for Alzheimer’s disease. A group of thiol-dependent NO mimetics known as furoxans may be designed to exhibit attenuated reactivity to provide slow onset NO effects. The present study describes the design, synthesis, and evaluation of a furoxan library resulting in the identification of a prototype furoxan, 5a, which was profiled for use in the central nervous system. Furoxan 5a demonstrated negligible reactivity toward generic cellular thiols under physiological conditions. Nonetheless, cGMP-dependent neuroprotection was observed, and 5a (20 mg/kg) reversed cholinergic memory deficits in a mouse model of passive avoidance fear memory. Importantly, 5a can be prepared as a pharmaceutically acceptable salt and is observed in the brain 12 h after oral administration, suggesting potential for daily dosing and excellent metabolic stability. Continued investigation into furoxans as attenuated NO mimetics for the CNS is warranted.
Graphical Abstract
INTRODUCTION
Nitric oxide (NO) is a fundamental regulator of synaptic function. The ability of NO to enhance synaptic plasticity is chiefly attributed to activation of NO-sensitive soluble guanylyl cyclase (NO-sGC), resulting in increased production of cyclic guanosine monophosphate (cGMP) and kinase signal transduction, primarily via protein kinase G (PKG), leading to phosphorylation of cAMP response element binding protein (CREB) at Ser-133.1–3 CREB phosphorylation is required for synaptic plasticity and memory consolidation. Prolonged transcriptional activation of CREB results in the production of neurogenic gene products, such as brain-derived neurotrophic factor (BDNF), and the formation of new dendritic spines, ensuring the capacity for morphological flexibility in synaptic networks.4 Regulation of CREB was first identified as being controlled by cAMP/PKA kinase signaling but actually involves numerous coregulators. Importantly, inhibition of any component of the NO/cGMP/PKG system suppresses synaptic signal. This pathway is directly inhibited by amyloid beta (Aβ) in Alzheimer’s disease (AD).5–8 Reversal of Aβ-induced deficits via agents that activate NO/cGMP signaling results in improved cognitive function, as demonstrated for nitrates, sGC activators, and phosphodiesterase inhibitors (PDEi).9–13
NO-related biological chemistry is complex, and the term “NO donor” is often used to describe multiple chemical classes, some of which do not directly yield molecular NO•. Multiple “NO donor” classes exhibit NO mimetic activity through one of several NO species (such as NO•, HNO, NO−, NO+, NO2−, or HONOO−). Here, we describe a class of synthetic, thiol-dependent NO mimetics known as furoxans. Furoxans have been investigated as potential therapeutics for a plethora of disease states.14–19 The majority of furoxan development has focused on relatively reactive furoxans that harness the potent vasodilatory or cytotoxic effects associated with large transient fluxes of NO. Indeed, of approximately 158 “furoxan”-themed original research articles in medicinal chemistry journals in the past 20 years, only two instances focused on the use of furoxans for the central nervous system (CNS) (see Supplemental Table 1). The pioneering effort by Schiefer et al. used the furoxan moiety with a putative peptidomimetic motif (Figure 1A) and demonstrated that (1) reactivity and propensity for NO mimetic activity can be modulated via a predictable structure−activity relationship (SAR), (2) furoxans protect against oxygen glucose deprivation (OGD) in primary cortical cell culture via a NO/cGMP-dependent mechanism, and (3) furoxans rescue oligomeric Aβ-induced deficits in long-term potentiation (LTP) (i.e., sustained strengthening of synaptic signal).20 Additionally, Chegaeve et al. reported restored LTP and disruption of Tau and Aβ aggregation using thiacarbocyanine-furoxan hybrids (Figure 1B).21 Each of these studies incorporated the furoxan moiety within hybrid scaffolds and confirmed efficacy with in vitro cell cultures and ex vivo hippocampal slices. Furoxans have not hitherto been investigated in vivo for CNS applications.
Figure 1.
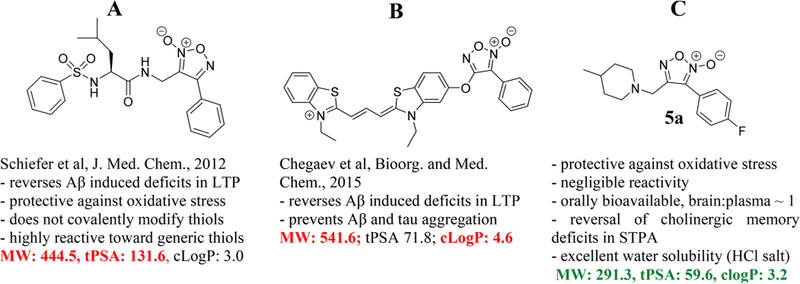
Overview of furoxan development for the CNS. The furoxans described here have more favorable physiochemical properties as potential CNS drugs than previously described furoxans. Furoxans have not been studied in the brain previously. *Abbreviations: MW = molecular weight; tPSA = total polar surface area; cLogP = calculated partition coefficient; Aβ = oligomeric amyloid beta; LTP = long-term potentiation; STPA = step-through passive avoidance.
In this paper, a focused furoxan library was designed with rigorous consideration of physiochemical properties for CNS penetration. Incorporating varying electron-donating or -withdrawing groups probed electronic sensitivity of the thiophilic furoxan ring with the goal of fine-tuning reactivity toward endogenous thiols and thus the rate of NO release. The furoxan 5a demonstrated negligible reactivity toward generic cellular thiols under physiological conditions. Nonetheless, NO/cGMP-dependent neuroprotection was observed with a hyperbolic concentration response curve, and 5a was shown to penetrate the blood brain barrier via multiple administration routes and was capable of reversing cholinergic memory blockade in a mouse model of passive avoidance memory.
RESULTS AND DISCUSSION
Library Design.
The design motif for novel furoxans is summarized in Figure 2A. Groups at GlaxoSmithKline, Eli Lilly, and others have described favorable CNS penetration as resulting from several inter-related properties including (1) membrane permeability and solubility, (2) low propensity for efflux by P-glycoproteins (Pgp), and (3) adequate brain partitioning to provide a sufficient level of unbound drug at the site of action.22–24 Behavior with regard to these properties is highly dependent upon the physiochemical properties of the molecule being studied (i.e., molecular weight [MW], partition coefficient [logP], and total polar surface area [tPSA]). On the basis of these considerations, all of our compounds obey the following design criteria: (1) MW < 400 g/mol, (2) clogP =2.5−4.5, and (3) tPSA = 45−85. A para-substituted aryl system was used to modulate furoxan reactivity while blocking potential metabolic para-hydroxylation, the main route of metabolism of phenyl-containing drugs.25 4-Methylpiperdine was incorporated as a cap group because it is small, relatively hydrophobic, and contains a tertiary amine (related 4-alkylpiperdine reported pKa ~ 9.0),26 which allows preparation of pharmaceutically acceptable salts that possess excellent water solubility. Importantly, despite predominantly existing as a charged species at physiological pH, similar 4-alkylpiperidines are present in several orally bioavailable CNS drugs, including haloperidol, risperidone, and the AD standard of care donepezil.
Figure 2.
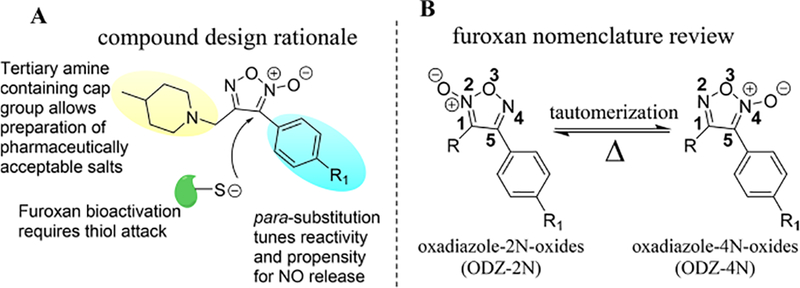
(A) Design justification for furoxan library. (B) Review of furoxan designation nomenclature. Furoxans tautomerize when heated sufficiently (at least 100 °C for a sustained period of time depending upon substitution pattern) but do not interconvert at physiologically relevant temperatures.
Synthesis.
Novel furoxans were synthesized from readily available cinnamic acids (Scheme 1) beginning with anhydride formation and reduction with sodium borohydride to produce the corresponding cinnamyl alcohols (1a−h). The cinnamyl alcohols were cyclized using an optimized cyclization reaction with excess sodium nitrite performed in a 1:1 mixture of acetic acid:dimethylformamide with gradual heating to yield 2a−h. A traditional Appel reaction afforded the alkyl bromides 3a−h, and subsequent substitution with 4-methylpiperidine yielded the oxadiazole-2N-oxides (ODZ-2N) 4a−h. The final step of the synthesis involved tautomerization of the furoxan in refluxing toluene to yield oxadiazole-4N-oxides (ODZ-4N). Separation of the ODZ-2N/ODZ-4N tautomers was realized via reverse-phase flash chromatography to yield the ODZ-4N compounds 5a−h. Initial in vitro testing nominated 5a for in vivo evaluation (vide infra). Accordingly, substitution of 3a with isotope-labeled 4-methylpiperidine-2,2,6,6-d4 followed by tautomerization provided 7a as an appropriate internal standard for LC-MS/MS studies (Scheme 2). Additionally, treatment of 5a with HCl in diethyl ether provided the water-soluble HCl salt for in vivo investigations.
Scheme 1. Library Synthesisa.
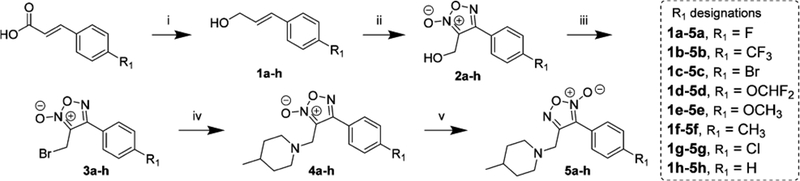
a(i) Ethyl chloroformate, triethylamine, sodium borohydride, 0 °C to r.t.; (ii) acetic acid:dimethylformamide (1:1), sodium nitrite, 0 to 60 °C; (iii) triphenylphosphine, carbon tetrabromide, 0 °C to r.t.; (iv) 4-methylpiperidine, triethylamine, r.t. to 60 °C; (v) toluene, reflux, 7 d.
Scheme 2. Synthesis of the LC-MS/MS Internal Standarda.
a(i) 4-Methylpiperidine-2,2,6,6-d4, triethylamine, r.t. to 60 °C; (ii) toluene, reflux, 7 d.
Crystallographic examination of 5a (Figure 3A) confirmed the presence of the ODZ-4N structure (see Supplemental Crystallographic Report). HPLC purity analysis using a photodiode array detector (PDA) showed a noticeable shift in the λmax of absorption for ODZ-2N analogues (~246−261 nm) versus that of ODZ-4N analogues (279−293 nm) with a representative example shown for 4a and 5a in Figure 3B (see Supplemental Characterization for PDA data for all compounds). This phenomenon may be used to easily distinguish furoxan tautomeric forms. In all instances, retention time (Rt) comparisons indicated that, despite possessing greater aromaticity based on λmax, the ODZ-4N analogues are more polar than the corresponding ODZ-2N analogues. Furoxan 5a was also profiled by 2D-NMR (see Supporting Information for homonuclear correlation spectroscopy [COSY] and hetero-nuclear multiple quantum coherence [HMQC]).
Figure 3.
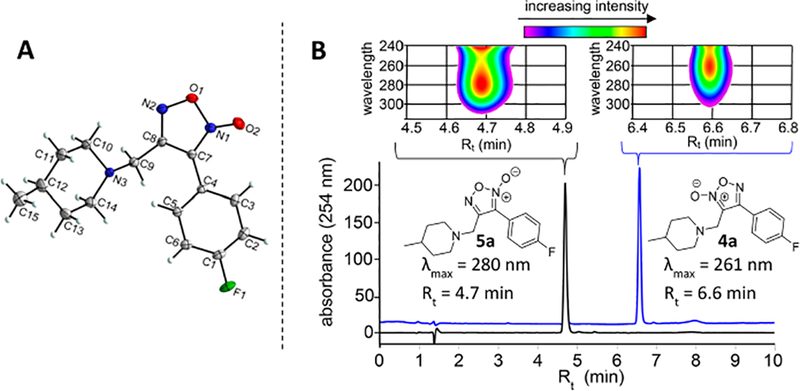
(A) Crystal structure of 5a confirming the oxadiazole-4N-oxide ring configuration. (B) HPLC-PDA analysis demonstrating differences in relative polarity and UV absorption properties for ODZ-2N and ODZ-4N furoxans 4a and 5a, respectively. Chromatograms represent overlay of UV signal at 254 nm with PDA analysis of the representative portion of the chromatogram shown above each peak.
Neuroprotection.
Oxygen glucose deprivation (OGD) provides a cellular model of oxidative stress resulting in apoptotic cell death, which provides a model of neuronal death across neurodegenerative diseases, including AD.27 Expert perspectives suggest that experimental paradigms in drug discovery should model well-understood target pathways rather than specific disease state.28,29 NO mimetics have been benchmarked as being protective against oxidative stress induced by OGD via multiple proposed mechanisms including(1) antioxidant effects, (2) mediating phosphorylation of prosurvival kinase signaling, and (3) S-nitrosylation of proapoptotic caspases.30,31 Neuroprotection was evaluated by 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MTT) assays of cell viability in nondifferentiated PC-12 cell cultures subjected to OGD for 90 min followed by reoxygenation and replacement with nutrient-rich media containing furoxans screened at three concentrations (1, 10, and 25 μM) for 24 h. Neuroprotection is reported as percent change relative to vehicle (DMSO)-treated controls.
The ODZ-2N compounds (except 4d) showed negligible neuroprotection (Table 1). A few ODZ-4N analogues provided noticeable neuroprotective activity, including compounds 5a (4-F), 5c (4-Br), and 5d (4-OCHF2). The 4-OCHF2 tautomers (4d and 5d) showed robust potency with significant efficacy at 1 μM. Furoxans 4d and 5d are considered to exhibit “outlier-like” behavior compared to the rest of the library based upon(1) 4d being the only active ODZ-2N from our series and (2) the potency of these compounds being an order of magnitude lower than any other compound. Antioxidant effects, such as those traditionally seen for NO mimetics, result in potent in vitro neuroprotection against oxidative stress associated with OGD. In NO-related drug discovery, the most potent compounds in primary assays may not survive hit-to-lead optimization due to deleterious effects in animals. Furoxans 4d and 5d warrant additional investigation; however, they were not considered for proof-of-concept in vivo investigation of attenuated furoxans because the impetus for this work was to produce NO mimetics that activate NO/cGMP signaling without producing significant fluxes of NO, thereby reducing the potential for side effects. Furoxans 5a and 5c demonstrate statistically equivalent efficacy and potency. Furoxan 5a, rather than 5c, was chosen for in vivo evaluation for multiple reasons, including the smaller molecular weight (a critical factor in brain penetration and ligand efficiency)32 and future potential for incorporation of 18F for radioimaging. Furoxan 7a was synthesized for use as an LC-MS/MS internal standard and was also tested in the primary assay. Similar activity was expected compared to nondeuterated 5a; however, 7a possessed no neuroprotective effects. UPLC separation suggests 7a is slightly more polar than 5a based on Rt (Supplemental Figure S3). LC-MS/MS was used to measure membrane permeability in the primary assay (i.e., extracellular concentration versus intracellular concentration) of 5a and 7a in the media (supernatant) and cytosol of control (no OGD) and OGD-treated PC12 cells following 24 h incubation via subcellular fractionation. Each compound was found in greatest amounts in the cytosol, and submitting to OGD insult did not significantly effect distribution (Supplemental Figure S4). These data demonstrate (1) furoxans readily cross cell membranes in the primary assay and (2) significant amounts of furoxan after 24 h incubation with PC12 cells, providing further evidence for the relatively unreactive nature of this furoxan library.
Table 1.
Protection against Oxygen Glucose Deprivation (OGD)a
| structure | tautomer | % change in viability relative to vehicle mean | ||
|---|---|---|---|---|
| 1 μM | 10 μM | 25 μM | ||
| R1 = F | 4a (2N) | 11% ± 2 | 11% ± 3 | 5% ± 3 |
| 5a (4N) | −3% ± 4 | 46%*** ± 7 | 41%*** ± 5 | |
| R1 = CF3 | 4b (2N) | −9% ± 7 | 10% ± 7 | 0% ± 14 |
| 5b (4N) | 2% ± 23% | −16% ± 7 | −22% ± 10 | |
| R1 = Br | 4c (2N) | −7% ± 2 | −2% ± 1 | 19% ± 5 |
| 5c (4N) | 25% ± 6 | 77%*** ± 5 | 96%*** ± 19 | |
| R1 = OCHF2 | 4d (2N) | 54%** ± 11 | 86%*** ± 15 | 108%*** ± 24 |
| 5d (4N) | 67%*** ± 7 | 61%*** ± 7 | 53%** ± 2 | |
| R1 = OCH3 | 4e (2N) | 14% ± 6 | 10% ± 6 | −1% ± 9 |
| 5e (4N) | 11% ± 6 | 12% ± 14 | 5% ± 17 | |
| R1 = CH3 | 4f (2N) | 7% ± 12 | 12% ± 15 | 10% ± 11 |
| 5f (4N) | 22% ± 20 | 13% ± 18 | 8% ± 18 | |
| R1 = Cl | 4g (2N) | 35% ± 16 | 7% ± 8 | 25% ± 16 |
| 5g (4N) | −10% ± 12 | 19% ± 19 | 4% ± 18 | |
| R1 = H | 4h (2N) | 9% ± 11 | 19% ± 6 | 16% ± 15 |
| 5h (4N) | −21% ± 7 | 6% ± 23 | −11% ± 6 | |
| R1 = F d4 analogue | 6a (2N) | 11% ± 8 | 6% ± 7 | −1% ± 12 |
| 7a (4N) | 8% ± 11 | 20% ± 13 | 7% ± 9 | |
Assay workflow: expose PC12 cells to OGD for 90 min → reoxygenate and add furoxan (1, 10, or 25 μM) → incubate for 24 h → MTT readout. Data represented as mean ± SEM (n = 3) of triplicate experiments.
p < 0.05,
p < 0.01,
p < 0.001 compared to vehicle control using one-way ANOVA analysis with Dunnett’s posthoc test. The viability of the OGD vehicle group is 51.4 ± 4.1% compared to vehicle group. This number is based on N = 20 OGD/vehicle trials, reported as mean ± SEM.
An expanded dose−response assay of OGD-exposed PC12 cells treated with 5a revealed a bell-shaped (i.e., hyperbolic) efficacy curve from which an EC50 = 1.9 μM was calculated (Figure 4A). For concentration response studies a resorufin based dye was used as the viability read-out due to a significantly shorter incubation time before colorimetric measurement, which allows more direct observation and increased throughput compared to MTT. The hyperbolic response curve is in agreement with previous studies that identified a correlation between production of NO2− by furoxans in the presence of cysteine (i.e., reactivity) with protection against OGD. Notably, although protective effects of 5a are lost at higher concentrations, no toxicity was observed at the highest concentration tested (50 μM). Co-incubation with ODQ (1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one), a selective sGC inhibitor, completely blocked the neuroprotective effects of 5a (Figure 4B), providing evidence for enhanced NO/cGMP signaling as being responsible for neuroprotective efficacy of 5a. This result is in agreement with previous reports of ODQ blocking the ability of furoxans or nitrates to protect against oxidative stress in cell culture.33 The presence of a basic nitrogen in the structure of the furoxan may lead to the interaction with a number of different targets in CNS. Hence, 5a (active) and 7a (inactive) were sent to the Psychoactive Drug Screening Program for binding studies to assess off-target effects as described previously.34,35 As shown in Supplementary Table 2, 5a does not interfere with binding of any radioligands for all receptors screened, including GABAA, serotonin receptors, dopamine receptors, and muscarinic and nicotinic acetylcholine receptors. No hERG binding was observed. For the most part 7a demonstrated a similar profile but significantly inhibited binding at the delta opioid receptor and benzodiazepine (BZP) rat brain site.
Figure 4.
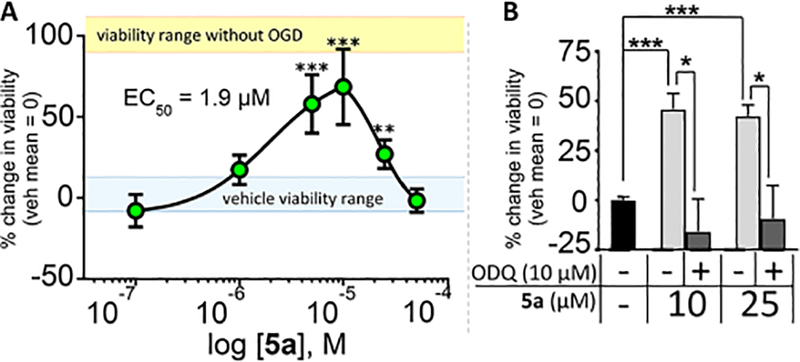
(A) Neuroprotection concentration response curve for 5a. PC12 cells were submitted to OGD for 90 min followed by addition of 5a (0.1, 1, 5, 10, 25, or 50 μM) and incubated under normoxic conditions for 24 h. Data represent percent change in viability relative to vehicle-treated control mean = 1, Prestoblue readout. Vehicle control viability range (assay baseline [blue]) and viability range control plates not submitted to OGD are shown in yellow. (B) Neuroprotection of 5a is dependent on sGC signaling. ODQ (10 μM) abolished neuroprotective activity of 5a in a PC12-OGD assay using MTT as a viability readout. Data represented as mean ± SEM (n = 3) of duplicate experiments. *p < 0.05, **p < 0.01, ***p < 0.001 compared to vehicle control using one-way ANOVA analysis with Dunnett’s posthoc test. Average viability of the OGD vehicle cells was51.4% (±4.1) that of vehicle cells based on 20 OGD/vehicle trials, represented as mean ± SEM.
Reactivity.
Furoxan 5a was incubated with N-acetyl cysteine (5 mM) in PBS (50 mM, pH 7.4) to examine the stability of the furoxan ring in the presence of excess free thiols (see Supplemental Figures S10 and S11). We hypothesized reactivity with excess thiol would correlate with neuroprotection in the primary assay. However, no reaction was observed by HPLC-PDA analysis of all compounds, whereas control incubations using the electrophile N-ethylmaleimide (NEM) showed complete reaction of NEM after 2 and 12 h. In agreement with the observed attenuated reactivity, negligible NO production was observed from 5a in the form of NO2− in the primary assay (Supplemental Figure S11).
Improved Memory in Step-Through Passive Avoidance (STPA).
Pilot behavioral studies evaluated the ability of 5a to reverse scopolamine-induced deficits in STPA, which assesses the ability of drugs to reverse hippocampal-dependent aversive memory deficits.36–38 Scopolamine is a well-characterized pan-muscarinic antagonist that produces significant amnesic effects when administered 30 min prior to training in STPA.36,39,40 Reversal of scopolamine-induced amnesia is not a sophisticated model, but it is well understood and remains useful as a resource-efficient screen for procognitive agents.41 Importantly, reversal of scopolamine-induced cholinergic deficits in STPA via NO/cGMP enhancement is well characterized and has been benchmarked for nitrates, sGC activators, PKG activators, and several phosphodiesterase inhibitors (PDEi).42–46 The STPA apparatus consists of an acrylic box with an illuminated compartment connected to a darkened compartment by a small guillotine door. Mice are placed in the illuminated side and trained against their natural tendency to translocate to the dark side using an aversive stimulus during double trial acquisition. Memory consolidation is measured as the latency to enter the dark compartment 24 h after training (Figure 5). Testing latency 24 h after treatment avoids potential convolution from lethargy caused by drug exposure, and lethargy is not reported for agents acting via cGMP signaling as shown for sildenafil in the open field test.47
Figure 5.
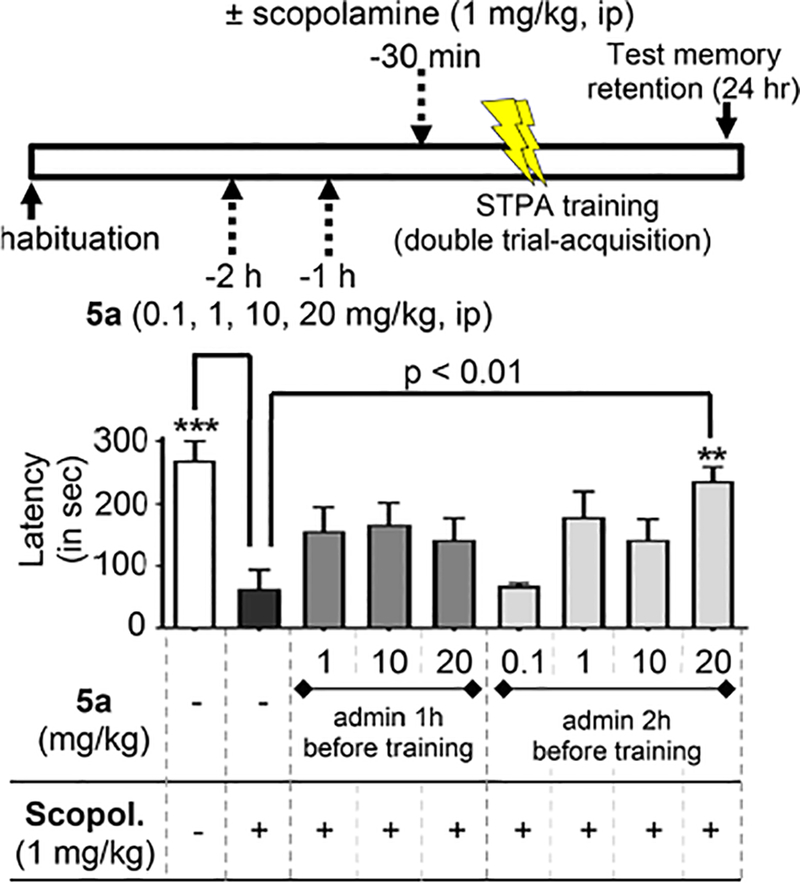
Reversal of cholinergic memory deficits in STPA by 5a. Male C57BL/6 mice administered either vehicle or 5a (0.1, 1, 10, or 20 mg/kg) via ip injection at 1 or 2 h prior to training. Scopolamine (1 mg/kg) was administered 30 min prior to training. Memory was measured as latency to enter a dark chamber 24 h after training. Data represented as mean and SEM (n = 8−9) for n = 8, see exclusion criteria in Experimental Section. **p < 0.01 and ***p < 0.001 compared to negative scopolamine-treated control using one-way ANOVA analysis with Dunnett’s posthoc test.
Furoxans have never been studied in the CNS. Hence, choices for dosing and administration timing were not evident at the onset. A NO-chimeric nitrate reversed scopolamine-induced deficits in STPA when administered 20 min prior to training but had no effect when given 60 min prior to training, demonstrating the short biological half-life of nitrates resulting in rapid onset of neuromodulatory efficacy at a low dose (1 mg/kg).9 On the basis of the knowledge that attenuated furoxans likely have greater metabolic stability, we anticipated that a longer pretraining treatment duration would be required to allow sufficient time for slow-onset NO mimetic effects. With these considerations in mind, we examined varying doses at two pretraining time points (1 or 2 h). Scopolamine-treated mice showed significantly less latency to enter the dark compartment compared to that of wild-type mice (scopolamine-treated: 63.5 ± 30 s [SEM] versus vehicle-treated: 270 ± 29 s [SEM], p < 0.001). Furoxan 5a was capable of reversing scopolamine-induced amnesia with a statistically significant effect observed when administered 2 h prior to training at 20 mg/kg (latency = 237 ± 22 s [SEM], p < 0.01). These data demonstrate, for the first time, the ability of a furoxan to improve memory in mice by reversing cholinergic memory blockade.
In Vivo Drug Stability, Brain Penetration, and Effect on Protein Phosphorylation.
To understand the STPA behavioral results, we attempted to correlate the amount of 5a in the forebrain with modulation of a hypothesized marker of efficacy, CREB phosphorylation. The forebrain contains the primary neurocircuitry responsible for learning and memory, particularly the hippocampus. Pilot studies analyzed the entire forebrain to ensure enough tissue was available for both ELISA and LC-MS/MS analysis from the same animal (Figure 6A). Furoxan 5a was measured by LC-MS/MS analysis of plasma and forebrain samples after intraperitoneal (ip) and oral (po) administration at varying time points (1, 2, or 12 h [n = 4]). CREB phosphorylation was also analyzed in the forebrain samples as the pCREB:tCREB ratio. Selected sacrifice times were intended to provide a preliminary assessment of the in vivo t1/2 while providing a pharmacodynamic measurement in a system mimicking behavioral studies in which 5a (20 mg/kg) reversed scopolamine-induced memory deficits when administered 2 h prior to behavioral training.
Figure 6.
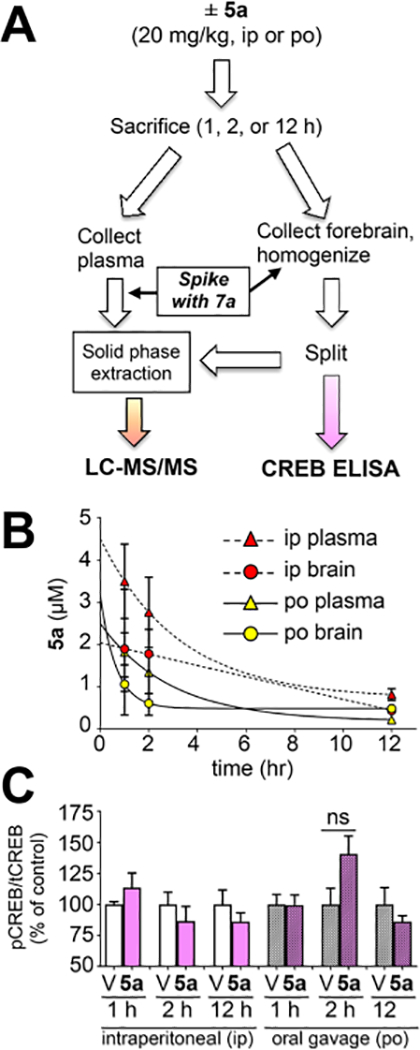
(A) Male C57BL/6 mice (N = 4 per cohort) were treated with vehicle or 5a (20 mg/kg), and at the specified time, plasma and forebrain were collected and processed as indicated. (B) At the specified time (1, 2, or 12 h), plasma (triangles) and forebrain (circles) were collected, spiked with 7a, and extracted. Oral gavage (po) is indicated by solid lines and yellow symbols. Intraperitoneal injection (ip) is indicated by dotted lines and red symbols. Quantitation based on the ratio of the AUC of the MRM transitions for 5a versus 7a relative to a calibration curve. Concentrations (μM) were calculated after normalizing for tissue weight (assuming tissue/plasma density =1). Data shown as mean ± SEM (n = 4) for all cohorts except 1 h ip (n = 3), see exclusion criteria in Experimental Section. (C) Forebrains were homogenized, normalized by bradford, and analyzed for pCREB and tCREB by ELISA (V = veh, D = drug [5a]). The ratio of absorbance values was normalized to Veh mean = 1 for each treatment group. Data represent mean ± SEM (n = 4), analysis by one-way ANOVA with Tukey’s test.
Assessment of brain:plasma ratio was carried out via MRM (multiple reaction monitoring) using LC-MS/MS. The fore-brain and plasma samples were processed using solid-phase extraction (SPE). Furoxan 5a was observed in all drug-treated animals, including detectable levels after 12 h (Figure 6). Deuterated internal standards are desirable for absolute quantitation via LC-MS/MS because they have similar physiochemical properties to their nondeuterated analogue but may be distinguished based on molecular weight. Inclusion of 7a allowed for absolute quantitation of 5a in the forebrain of treated mice. Extraction methodology and tandem LC-MS/MS parameters (triple quadrupole utilizing optimized MRM transitions) were developed and validated via extraction of untreated brain tissue spiked with varying concentrations of 5a and a fixed concentration of 7a to develop a calibration curve. Systemic t1/2 for brain and plasma measurements was between 50 and 110 min depending on the administration route. There were no significant differences in levels of 5a in the brain (B) versus plasma (P). Furthermore, there were no significant differences in levels in the brain for po versus ip administration, suggesting a consistent and favorable biodistribution profile with either administration route. Differences in B:P based on administration route trended toward but did not reach significance (p = 0.16 using unpaired t test) with an average oral administration B:P ratio of 1.49 (±0.52, SEM) and intraperitoneal administration average B:P ratio of 0.68 (±0.03, SEM). We anticipate 5a accesses the brain via passive diffusion based on a low molecular weight (291.1 g/mol) and a clogP of ~3.1, both of which are predicted to allow passive membrane permeability.48 These findings are novel for several reasons, including being the first report (1) demonstrating a furoxan crossing the blood brain barrier, (2) to our knowledge, demonstrating the first NO mimetic observed in the brain 12 h after administration (ip or po), and (3) establishing a B:P ratio for a furoxan.
Given the knowledge that 5a reverses scopolamine-induced deficits in STPA, we expected 5a to elicit a change in CREB phosphorylation (pCREB). However, no effect on pCREB was observed for any cohorts treated with 5a alone based on analysis by ELISA (Figure 6C). A more rigorous evaluation was performed in follow-up investigations studying hippocampal protein phosphorylation by Western blotting. Mice were treated in an identical manner to STPA studies except the animals were sacrificed at the time-point previously used for training in STPA (i.e., at the time of memory acquisition). These studies included four cohorts (N = 8 per cohort [with experimenters blinded to treatment]) with each receiving one ip injection 2 h prior to sacrifice (either 5a [20 mg/kg] or vehicle) and one ip injection 30 min prior to sacrifice (either scopolamine [1 mg/kg] or vehicle). Blots for either phosphorylated protein [pProtein] or total protein [tProtein] were normalized to GAPDH loading controls prior to calculating the phosphorylation ratio as pProtein:tProtein analyzed by one-way ANOVA. Despite these efforts, statistically significant changes in hippocampal protein phosphorylation were not observed in any cohorts for pCREB:tCREB or pErk:tErk (Supplementary Figure S16). Importantly, we did not observe significant changes in protein phosphorylation for mice administered only scopolamine, the negative control group. Most articles utilizing acute scopolamine-induced memory deficits do not report measurements of protein phosphorylation, and those that do, agree with our findings. Indeed, acute scopolamine induces impaired learning and memory but has no effect on protein phosphorylation in normal mice (C57BL/6).49,50 Furthermore, neither acute nor long-term administration of PDEi’s affects hippocampal pCREB in normal mice.39,51 Hence, our findings for furoxans are in line with literature precedent for agents acting via NO/cGMP signaling. Long-term administration (>3 weeks) of nitrates or PDEi’s to AD transgenic (AD-Tg) mice results in increases in hippocampal pCREB relative to vehicle-treated controls.10,12,13 Hippocampal pCREB is most appropriate as a marker of efficacy when studying long-term administration of these agents in AD transgenic mice (such as APP/PS1, 5xFAD, 3xTg).
We provide evidence for furoxan effects associated with activation of NO/cGMP signaling including (1) protection against oxidative stress, which is abolished upon blocking cGMP production with a selective sGC inhibitor, and 2) reversal of scopolamine-induced memory impairment reliably shown for NO mimetics, PDEI’s, and sGC activators. Furoxans have been extensively described as thiol-dependent NO mimetics. Our pursuit of furoxans with attenuated reactivity has resulted in the identification of efficacious furoxans that lack reactivity with generic thiols. Characterizing the reaction of an attenuated furoxan with thiol is difficult due to the lack of reactivity. The specific thiol(s) responsible for and mechanism of bioactivation is not known at present. On the basis of our data, we present two proposed scenarios: (A) a subset of yet to be identified sufficiently reactive cellular thiols attack the furoxan ring to activate NO release, or (B) efficacious furoxans described here are metabolized in a CYP-independent manner (based on activity in PC12 cells) to a yet to be identified active metabolite. In scenario B, the active metabolite would contain the intact furoxan but would have greater thiophilic reactivity. In medicinal chemistry, it is well-known that instances in which a deuterated derivative lacks efficacy of an equivalent nondeuterated molecule may be explained by a requirement for metabolic activation for efficacy (i.e., an active metabolite is responsible for efficacy). Hence, the lack of activity of the deuterated (d4) analogue, 7a, provides support for the notion of an active metabolite. Additional supportive data are provided by the STPA behavioral model in which activity was observed when 5a was administered 2 h prior to training but not 1 h. The foundational efforts described here provide a reasonable starting point for exploration of these scenarios going forward.
CONCLUSIONS
The neuroprotective properties of novel attenuated furoxans have been explored. The furoxan 5a elicits a hyperbolic concentration response curve for protection against oxidative stress that is completely blocked by coincubation with ODQ, a selective sGC inhibitor, thus implicating neuroprotection resulting from a cGMP-dependent mechanism. The incorporation of a tertiary amine into the cap group provides access to pharmaceutically acceptable salts. Fortunately, the presence of the tertiary amine [calculated pKa ~9], which is likely charged at physiologic pH, does not hinder the ability to cross biological membranes based on levels of 5a in the cytosolic fractions of PC12 cells and brain penetration following administration by oral gavage. Memory improvement was observed for 5a when administered 2 h prior to training in STPA, showing statistically significant recovery of cognitive function after an induced deficit by scopolamine. Cumulatively, our data indicates that 5a is a relatively stable attenuated (i.e, weakly reactive) NO mimetic based on (1) negligible reactivity with generic thiols at physiological pH and temperature, (2) the presence of 5a in PC12 cells 24 h after treatment, and (3) the presence of 5a in the brain and plasma 12 h after po and ip administration. Furthermore, we hypothesize 5a works via a NO/cGMP/CREB-dependent mechanism based upon (1) eliminated neuroprotection upon sGC blockage, (2) a hyperbolic concentration response curve, and (3) reversal of cholinergic memory deficits in STPA. Our data suggest that free thiols alone are not capable of activating release of NO from attenuated furoxans. Identification of the protein target(s) or microenvironment(s) that facilitate bioactivation would allow for the development of a quantitative structure−activity relationship (QSAR) for analogue refinement leading to candidate nomination for IND enabling studies. Further investigation is warranted to elucidate the exact mechanism of furoxan bioactivation leading to NO release.
EXPERIMENTAL SECTION
All chemicals and reagents were purchased from Sigma-Aldrich (St. Louis, MO) or Fisher Scientific unless stated otherwise. All protocols and procedures involving the use of animals were approved by the Institutional Animal Care and Use Committee at the University of Toledo.
General Methods.
Reagents and solvents were purchased from common commercial suppliers (Fisher Scientific or Sigma-Aldrich) and used as received. Isotope labeled starting materials were purchased from Cambridge Isotope Laboratories. All reactions were carried out under atmospheric conditions at room temperature unless otherwise indicated. Reactions were monitored by thin-layer chromatography (TLC, LuxPlate silica gel 60 F254 plates) and revealed by UV light (254 nm). Column chromatography was performed using Teledyne Combiflash Rf with RediSepRf Gold columns and RediSep Rf C18 columns for tautomerization purification. HPLC analysis was performed using a Shimadzu Prominence HPLC (LCD-20AD) with temperature-controlled autosampler (SIL-20AC), refractive index (RID-20A), and PDA (SPD-M20A) detectors. LC-MS/MS analysis was carried out with a Nexera XR UPLC coupled with a Shimadzu 8050 triple quadrupole mass spectrometer (ESI, positive mode). Separations utilized a Phenomenex Kinetix core column (2.6 μm, C18, 100 Å, 100 × 4.6 mm column). HPLC conditions: mobile phase A = water (0.1% formic acid [FA]) and mobile phase B = acetonitrile(0.1% FA); 1.0 mL/min at 30% B for 1 min followed by gradient increase to 95% B over 5 min followed by 1 min at 95% B and re-equilibration at 30% B for 4 min resulting in a total run time = 10 min. UPLC-MS/MS utilized the same conditions substituting aqueous ammonium formate (25 mM, 0.1% FA) as mobile phase A and a flow rate of 0.4 mL/min. Purity of tested compounds was found to be >95% pure at two wavelengths, 254 and 282 nm, unless otherwise indicated. NMR (1H, 13C) was taken using a Bruker Avance 600 MHz spectrometer (cryoprobe). High-resolution mass spectra (HRMS) were recorded using a Waters Synapt high-definition mass spectrometer (HDMS) equipped with nano-ESI source in positive mode.
Crystallography.
Furoxan 5a was chosen for crystallographic examination. A clear, colorless, irregular prism-like specimen of C15H18FN3O2, with approximate dimensions 0.058 mm × 0.156 mm × 0.270 mm, was used for the X-ray crystallographic analysis. The X-ray intensity data were measured. The integration of the data using a monoclinic unit cell yielded a total of 26380 reflections to a maximum θ angle of 33.11° (0.65 Å resolution) of which 5531 were independent (average redundancy 4.769, completeness = 95.1%, Rint = 4.72%, Rsig = 4.02%) and 4041 (73.06%) were greater than 2σ(F2). The final cell constants of a = 11.3374(14) Å, b = 7.2234(9) Å, c = 18.825(2) Å, β =97.601(2)°, volume = 1528.1(3) Å3 are based upon the refinement of the XYZ-centroids of reflections above 20 σ(I). The final anisotropic full-matrix least-squares refinement on F2 with 255 variables converged at R1 = 5.17% for the observed data and wR2 = 14.18% for all data. The methyl group (C15) is disordered. The occupancy of the C15 atoms was refined to be 50% and then fixed. Data were refined with isotropic atomic displacement parameters; the bonded hydrogen atoms were calculated on idealized hydrogens and refined as riding atoms. The goodness-of-fit was 1.012. The largest peak in the final difference electron density synthesis was 0.487 e−/Å3, and the largest hole was −0.540 e−/Å3 with an RMS deviation of 0.058 e−/Å3. On the basis of the final model, the calculated density was 1.266 g/cm3 and F(000) of 616 e−.
Oxygen Glucose Deprivation (OGD) Neuroprotection Assay.
Viability is reported as % change relative to vehicle-treated control over at least three separate trials. Undifferentiated PC12 cells (passage 12−14) were exposed to OGD as described previously.52 Briefly, 1 × 106 PC12 cells were plated overnight in growth medium (DMEM/F12 1:1 containing 5% FBS, 5% HS, penicillin/streptomycin). The medium was replaced with glucose-free buffered salt solution (HBSS phenol red medium), and the plate was placed in an anaerobic chamber under inert atmosphere (argon) for 1.5 h in a 37 °C incubator. After OGD, the glucose-free medium was removed, and fresh growth medium was added along with drug or vehicle (DMSO) treatment for 24 h at 37 °C. Cell viability was assessed by MTT dye or PrestoBlue cell viability reagent (ThermoFisher) according to the manufacturer instructions. MTT (0.5 mg/mL) was added to each well for 4 h; the medium was aspirated, and then the dye was resuspended in DMSO and read by microplate reader (absorbance, 540 nm). Alternatively, PrestoBlue (10%) was added to each well for 30 min and then read by a microplate reader (fluorescence, excitation/emission 560/590 nm).
Reactivity Analysis.
The appropriate furoxan (250 μM) or N-ethylmaleimide (250 μM) was incubated ±N-acetylcysteine (5 mM) in PBS (50 mM, pH 7.4) at 37 °C and aliquots taken at 2 and 12 h. The mixtures were analyzed by HPLC-PDA analysis according to general methods. Peak integration determined the area under the curve (AUC) from UV absorbance spectra at the λ-max of each analyte provided the relative abundance of each starting material. The results can be found in Supplemental Figures S10 and S11.
LC-MS/MS Study of Cell Permeability.
Because of the different in vitro activities between 5a and its d4-counterpart 7a, the cellular permeability of each compound was investigated by LC-MS/MS. Briefly, 1 × 106 PC12 cells were exposed to OGD as described in the Neuroprotection Assay section and treated with each analyte (25 μM) or vehicle control for 24 h. A control plate, which was not submitted to OGD, was also studied. The supernatant (1 mL) fractions were collected and stored at −80 °C until further analysis. Cells were lysed using fractionation buffers for the cytosolic fraction. To each sample was added 150 μL of buffer A (HEPES [10 mM], MgCl2 [1.5 mM], KCl [10 mM], 1% NP-40, dithiothreitol [0.5 mM], phenyl-methylsulfonyl fluoride [0.2 mM], sodium orthovanadate [1 mM], NaF [50 mM], sodium pyrophosphate [10 mM], protease inhibitor cocktail [Thermo]), which was then vortexed and maintained at 0 °C for 15 min. The resulting solution was centrifuged, and the supernatant was collected as cytosolic fraction. Fractions were stored at −80 °C until further analysis. Supernatant (extracellular) and cytosolic (intracellular) samples (100 μL) were diluted with 200 μL of 2% formic acid (FA) in dH2O and spiked with a corresponding internal standard (7a for vehicle and 5a-treated samples; 5a for 7a-treated samples). Analysis was carried out as described in the UPLCMS/MS method described in the General Methods section. The AUCs for the MRM transitions of the analytes were integrated and normalized to their respective internal standards. The result can be found in Supplemental Figure S4.
Step-Through Passive Avoidance.
In the early morning (within 1 h of the light “on” cycle beginning), male C57BL/6 mice were habituated in the STPA apparatus to confirm the photophobic phenotype. Translocation in <30 s was considered normal. All mice exhibited normal photophobic behavior. Mice were weighed and administered either vehicle (0.9% NaCl with 1% DMSO) or the HCl salt of 5a (0.1, 1, 10, or 20 mg/kg) via ip injection at 60 or 120 min prior to training in STPA. Scopolamine (1 mg/kg) or vehicle (0.9% NaCl) was administered 30 min prior to training via ip injection. Mice were placed in the illuminated side of the passive avoidance apparatus (Ugo Basile), and entry into the dark chamber (monitored by sensors in the STPA apparatus) resulted in automatic closing of a vertical door and administration of a mild aversive electrical shock (0.5 mA, 3 s in duration) via electrical floor grid. The mouse was kept in the dark chamber for 30 s and then returned to the holding cage for 120 s before the second training session. In the second training, in instances when the mice did not translocate in 300 s, they were gently pushed into the dark compartment. Latency to enter the dark compartment was measured 24 h (±30 min) after initial training to determine memory retention. No shock was administered during the retention trial. Animals that did not translocate during the retention trial were assigned a retention latency of 300 s. Mice were euthenized by carbon dioxide poisoning and cervical dislocation within 24 h of retention testing completion. Notes: (1) Double trial acquisition increases the statistical power of data from STPA and decreases the requisite sample sizes from n = 12−14 to 7−9.41 (2) In four treatment cohorts (60 min [10 mg/kg], 60 min [20 mg/kg], 120 min [1 mg/kg], and 120 min [10 mg/kg]), upon opening the dark compartment following the second training, a mouse jumped from the apparatus and landed on the floor; these mice were excluded from further analysis, hence the n = 8 for these groups. An oversight in experimental setup resulted in the use of vehicle containing 1% DMSO; however, it should be noted that the HCl salt of 5a readily dissolves in saline at the concentration of these studies used.
LC-MS/MS Method Development.
Synthesized standards of 5a and 7a were used to study LC-MS/MS fragmentation to provide parent to daughter ion transitions for MRM analysis as follows: 5a (292.2 → 112.1, 292.2 → 99.1, 292.2 → 154.1) and 7a (296.2→ 158.1, 296.2 → 116.2, 296.2 → 102.2). MRM transitions are shown in Supplemental Figures S10 and S11. During method development, blank brain homogenate (100 μL each) was added to 200 μL of 2% aqueous FA spiked with a fixed concentration of 7a (0.5 μM, final concentration based on reconstitution in 100 μL of LC-MS load solvent) and varying amounts of 5a (0.0001, 0.001, 0.01, 0.1, 1, or 10 μM, final concentration based on reconstitution in 100 μL of LC-MS load solvent). Each mixture was vortexed briefly and applied to a solid-phase extraction resin (Evolute Express ABN cartridge; size = 30 mg/mL [Biotage]) using a Vacmaster manifold (Biotage). Cartridges were washed with dH2O (1 mL), and analytes were eluted with MeOH (1 mL) and evaporated to dryness via high flow air purge. Samples were reconstituted in 100 μL of ACN:dH2O (1:1) for LC-MS/MS analysis (injection volume of 20 μL). All samples were run in one batch, and then the entire batch was repeated. The internal standard AUC deviation was <10% for all samples. For each sample, the AUC for 5a was divided by the intrarun TIC 7a to provide 5a:7a ratios that were averaged between the two runs and plotted against the corresponding concentrations, demonstrating appropriate linearity of R2 = 0.99. The lower limit of quantitation (LLOQ) for 5a was <0.1 nM (signal-to-noise [S/N] to be >10:1). Determination of the exact limit of detection (LOD) and LLOQ was not required based on the presence of 5a at high concentrations.
Bioavailability Study.
C57BL/6 mice were weighed and administered the HCl salt of 5a (20 mg/kg) via oral gavage (vehicle = sesame oil) or intraperitoneal injection (vehicle = 0.9% NaCl with 1% DMSO). Mice were returned to holding cages and monitored prior to euthenisia by carbon dioxide poisoning and cervical dislocation after 1, 2, or 12 h. No signs of lethargy or distress were noted during monitoring. Blood was collected into K2EDTA coated tubes (BD Microtainer) by cardiac puncture, flash frozen in liquid N2, and stored at −80 °C for further analysis. Each brain was removed and immediately placed on a metal dissection surface that lay atop a bed of dry ice to minimize protein degradation and two portions dissected out: (1) the forebrain (containing cerebral cortex and hippocampus) and (2) the cerebellum. Dissected out tissue was placed in a preweighed 5 mL eppendorf tube and flash frozen with liquid N2 and stored at −80 °C for further analysis.
Each forebrain was placed in a 1.5 mL RINO tube (Next Advance Inc.) followed by addition of tissue extraction reagent (1 mL [Invitrogen], containing a protease/phosphatase inhibitor cocktail). The mixture was homogenized with The Bullet Blender Storm (Next Advance Inc.) for 1 min at 4 °C (speed 8) followed by centrifugation at 13000 rpm for 2 min at 4 °C. A homogenate aliquot of 100 μL was extracted, and the rest of the homogenate was stored at −80 °C for further analysis. Homogenate (100 μL) or plasma (50 μL) was added to 2% FA (200 μL, containing 7a [0.5 μM, final concentration upon reconstitution in 100 μL] and applied to a solid-phase extraction resin (Evolute Express ABN cartridge; size = 30 mg/mL [Biotage]) using a Vacmaster manifold (Biotage). Cartridges were washed with dH2O (1 mL); analytes were eluted with MeOH (1 mL) and evaporated to dryness via high flow air purge. Samples were reconstituted in 100 μL of load solvent for LC-MS/MS analysis (injection volume of 10 μL). All samples were run in one batch, and then the entire LC batch was repeated. For each sample, the AUC for 5a was divided by the intrarun AUC of 7a to provide 5a:7a ratios, which were averaged between the two runs. Forebrain homogenate extraction was repeated a second time, and comparison of analyte concentration from both extractions provided a strong correlation (R2 = 0.99), suggesting remarkable consistency for the solid-phase extraction method. Plasma extraction was only performed once due to the limited amount of material and the reliability shown for forebrain extractions; n = 4 for all PK data except the 1 h ip group. Note, for the 1 h ip group, we measured 5a at levels 50−100× greater than any other mouse (in plasma and brain). Experimenters hypothesized potentially mistaken bladder injection. We have not investigated this outlier further. It is possible that the alkaline pH of the bladder deprotonated the piperidine ring allowing increased membrane permeability and greater distribution.
Measuring pCREB:tCREB Ratio by ELISA.
pCREB and tCREB were measured using ELISA kits purchased from Invitrogen according to the provided protocol. Briefly, protein concentrations for forebrain homogenates from the bioavailability study were analyzed by Bradford assay. Each homogenate (4 μL) was added to standard diluent buffer (96 μL) and incubated for 2 h alongside calibration curve standard solutions. Wells were washed with wash buffer (4 × 300 μL) followed by 1 h incubation with appropriate primary antibodies (CREB [pS133] detection antibody or CREB (total) detection antibody). Wells were washed with wash buffer (4 × 300 μL) followed by 30 min incubation with secondary antibody (anti-rabbit IgG HRP). Wells were washed with wash buffer (4 × 300 μL) followed by 30 min incubation with the chromogen (100 μL) and subsequent addition of the stop solution (100 μL). Absorbance was measured at 450 nm using a Biotek Synergy H2 plate reader. Levels of pCREB and tCREB were normalized to total protein concentration to provide reported pCREB:tCREB ratios. Data represent SEM with analysis by one-way ANOVA with Tukey’s test; n = 4.
In Vivo Study of Protein Phosphorylation by Western Blotting.
Protein phosphorylation was measured according to previously described procedures.53 C57BL/6 mice were weighed and administered the HCl salt of 5a (20 mg/kg) via ip injection (vehicle =0.9% NaCl [1% DMSO]) 2 h prior to sacrifice. Scopolamine (1 mg/kg) or vehicle (0.9% NaCl) was administered 30 min prior to sacrifice via ip injection. Mice were returned to holding cages and monitored prior to euthanasia by carbon dioxide poisoning and cervical dislocation. No signs of lethargy or distress were noted during monitoring. Hippocampal tissue was dissected out and flash frozen in liquid nitrogen until transfer to −80 °C for storage. Hippocampi were homogenized with cell extraction reagent (Thermofisher) containing protease/phosphatase inhibitor cocktail and centrifuged at 13000 rpm for 5 min, and the supernatant was collected and frozen at −80 °C. Protein concentration was determined by Bradford assay, and the samples were analyzed by loading equivalent amounts of protein onto 10−20% Tris-Glycine Mini gels (Novex WedgeWell, ThermoFisher). Proteins were transferred to PVDF membranes and blocked with 5% BSA for 1 h at room temperature followed by overnight incubation at 4 °C with the following antibodies: rabbit anti-pCREB (Ser133) (1:1000, Cell Signaling), rabbit anti-CREB (1:1000, Cell Signaling), rabbit anti-p-p44/42 MAPK (ERK1/2) (Thr202/Tyr204) (1:2000, Cell Signaling), rabbit anti-p44/42 MAPK (ERK1/2) (1:1000, Cell Signaling), and rabbit anti-GAPDH (1:2000, Millipore). After washing three times with TBST (5 min/wash), membranes were incubated with secondary antibody, goat antirabbit (1:10000 in 5% milk, Jackson ImmunoResearch). Blots were imaged using Bio-Rad ChemiDoc XRS. The images were analyzed using NIH ImageJ, and the values were normalized with respect to GAPDH and expressed as the ratio of phosphorylated protein to total protein. See Supporting Information for the detailed experimental procedure.
Synthetic Chemistry.
General Reduction Procedure.
The appropriate cinnamic acid (1 equiv) was dissolved in anhydrous tetrahydrofuran (THF, 20 mL) and brought to 0 °C under argon. Triethylamine (TEA, 1 equiv) was added dropwise over 2 min. After an additional 5 min, ethyl chloroformate (1 equiv) was added dropwise over 5 min to yield a white precipitate. The solution was filtered through a glass frit, and NaBH4 (3.8 equiv) was added portionwise to the filtrate at 0 °C with stirring (in some instances, additional equivalents of NaBH4 were added as indicated for each analogue below). Thirty min after the final NaBH4 addition, the reaction was quenched with MeOH (20 mL) added over 30 min using a syringe pump, and the reaction was stirred overnight. HCl (1 N) was added dropwise over 30 min until the solution became acidic (~30 mL). The reaction was extracted with CH2Cl2 (3 × 50 mL), and the combined organic extracts were washed with brine (1 × 100 mL). The combined organic layers were concentrated in vacuo and purified by flash chromatography (SiO2, hexanes/ethyl acetate) to give the corresponding cinnamyl alcohols as described below.
(E)-3-(4-Fluorophenyl)prop-2-en-1-ol (1a).
This compound was synthesized using the general procedure with (E)-3-(4-fluorophenyl)-acrylic acid (2.06 g, 12.4 mmol), TEA (1.26 g, 12.4 mmol), ethyl chloroformate (1.35 g, 12.4 mmol), and NaBH4 (4.70 g, 124.2 mmol) to afford 1a as a white solid (762 mg, 40.3%). 1H NMR (CDCl3, 400 MHz): δ 7.36−7.33 (2H, qd, J = 12.0 Hz), 7.03−6.99 (2H, t), 6.60−6.56 (1H, d, J = 16.0 Hz), 6.32−6.25 (1H, m), 4.33−4.31 (2H, d, J =8.0 Hz).
(E)-3-(4-(Trifluoromethyl)phenyl)prop-2-en-1-ol (1b).
This compound was synthesized using the general procedure with (E)-3-(4-(trifluoromethyl)phenyl)acrylic acid (2.15 g, 9.25 mmol), TEA (936 mg, 9.25 mmol), ethyl chloroformate (1.00 g, 9.25 mmol), and NaBH4(1.29 g, 35.2 mmol) to afford 1b as a white solid (1.84 g, 91.2%). 1HNMR (CDCl3, 400 MHz): δ 7.57−7.55 (2H, d, J = 8.0 Hz), 7.47−7.45 (2H, d, J = 8.0 Hz), 6.68−6.64 (1H, d, J = 16.0 Hz), 6.48−6.42 (1H, m), 4.37−4.35 (2H, d, J = 8.0 Hz).
(E)-3-(4-Bromophenyl)prop-2-en-1-ol (1c).
This compound was synthesized using the general procedure with (E)-3-(4-bromophenyl)-acrylic acid (3.00 g, 13.2 mmol), TEA (1.34 g, 13.2 mmol), ethyl chloroformate (1.43 g, 13.2 mmol), and NaBH4 (1.85 g, 50.2 mmol) to afford 1c as a white solid (2.27 g, 81.6%). 1H NMR (CDCl3, 400 MHz): δ 7.44−7.42 (2H, d, J = 8.0 Hz), 7.25−7.23 (2H, d, J = 8.0 Hz), 6.57−6.53 (1H, d, J = 16.0 Hz), 6.38−6.31 (1H, m), 4.32−4.30 (2H, d, J = 8 Hz).
(E)-3-(4-(Difluoromethoxy)phenyl)prop-2-en-1-ol (1d).
This compound was synthesized using the general procedure with (E)-3-(4-(difluoromethoxy)phenyl)acrylic acid (1.14 g, 5.32 mmol), TEA (540 mg, 5.32 mmol), ethyl chloroformate (579 mg, 5.32 mmol), and NaBH4 (2.24 g, 60.7 mmol) to afford 1d as a white solid (923 mg, 86.3%). 1H NMR (CDCl3, 400 MHz): δ 7.34−7.32 (2H, d, J = 8.0 Hz), 7.05−7.03 (2H, d, J = 8.0 Hz), 6.68−6.49 (2H, m), 6.32−6.25 (1H, m), 4.31−4.30 (2H, d, J = 4.0 Hz).
(E)-3-(4-Methoxyphenyl)prop-2-en-1-ol (1e).
This compound was synthesized using the general procedure with (E)-3-(4-methoxyphenyl)acrylic acid (3.00 g, 16.8 mmol), TEA (1.70 g, 16.8 mmol), ethyl chloroformate (1.83 g, 16.8 mmol), and NaBH4 (4.46 g, 118 mmol) to afford 1e as a white solid (1.95 g, 70.7%). 1H NMR (DMSO-d6, 400 MHz): δ 7.37−7.35 (2H, d, J = 8.0 Hz), 6.90−6.88 (2H, d, J = 8.0 Hz), 6.52−6.48 (1H, d, J = 16.0 Hz), 6.26−6.19 (1H, m), 4.11−4.10 (2H, d, J = 4.0 Hz), 3.75 (3H, s).
(E)-3-(p-Tolyl)prop-2-en-1-ol (1f).
This compound was synthesized using the general procedure with (E)-3-(p-tolyl)acrylic acid (3.04 g, 18.7 mmol), TEA (1.90 g, 18.7 mmol), ethyl chloroformate (2.03 g, 18.7 mmol), and NaBH4 (2.69 g, 71.2 mmol) to afford 1f as a white solid (1.57 g, 56.5%). 1H NMR (CDCl3, 400 MHz): δ 7.28−7.26 (2H, d, J = 8.0 Hz), 7.13−7.11 (2H, d, J = 8.0 Hz), 6.59−6.55 (1H, d, J =16.0 Hz), 6.34−6.27 (1H, m), 4.30−4.28 (2H, d, J = 8.0 Hz), 2.33 (1H, s).
(E)-3-(4-Chlorophenyl)prop-2-en-1-ol (1g).
This compound was synthesized using the general procedure with (E)-3-(4-chlorophenyl)-acrylic acid (2.00 g, 11.0 mmol), TEA (1.11 g, 11.0 mmol), ethyl chloroformate (1.19 g, 11.0 mmol), and NaBH4 (1.57 g, 41.6 mmol) to afford 1g as a white solid (1.30 g, 70.6%). 1H NMR (CDCl3, 400 MHz): δ 7.31−7.26 (4H, d, J = 8.0 Hz), 6.58−6.54 (1H, d, J = 16.0 Hz), 6.36−6.30 (1H, m), 4.32−4.31 (2H, d, J = 4.0 Hz).
(E)-3-Phenylprop-2-en-1-ol (1h).
This compound was synthesized using the general procedure with (E)-3-phenylprop-2-enoic acid (2.00 g, 13.5 mmol), TEA (1.50 g, 14.9 mmol), and ethyl chloroformate(1.61 g, 14.9 mmol) to afford 1h as a white solid (1.46 g, 80.5%). 1H NMR (CDCl3, 400 MHz): δ 7.42−7.41 (2H, d, J = 4.0 Hz), 7.36−7.34 (2H, t), 7.29−7.28 (1H, t), 6.66−6.63 (1H, d, J = 12.0 Hz), 6.42−6.38 (1H, m), 4.36−4.35 (2H, d, J = 4.0 Hz).
General Cyclization Procedure.
The appropriate cinnamyl alcohol (1 equiv) was dissolved in AcOH:DMF (1:1) and brought to 0 °C under argon. Sodium nitrite (10 equiv) was then added. The reaction was stirred for 1 h at 0 °C. The reaction mixture was heated at 40 °C; sodium nitrite was added (5 equiv), and the mixture was heated to 80 °C over 1.5 h. The reaction was diluted with ice water (100 mL) and extracted with EA (3 × 50 mL). Combined organic layers were washed with brine (1 × 100 mL), and the organic layer was concentrated by rotary evaporation. The combined organic layers were concentrated in vacuo and purified by flash chromatography (SiO2, hexanes/ethyl acetate) to give the corresponding furoxan as indicated below.
4-(4-Fluorophenyl)-3-(hydroxymethyl)-1,2,5-oxadiazole 2-Oxide (2a).
This compound was synthesized using the general procedure with 1a (762 mg, 5.01 mmol) and NaNO2 (5.19 g, 75.2 mmol) to afford 2a as a yellow solid (278 mg, 26.4%). 1H NMR (CDCl3, 400 MHz): δ 7.92−7.88 (2H, qd, J = 16.0 Hz), 7.31−7.29 (2H, t), 4.78 (2H, s).
3-(Hydroxymethyl)-4-(4-(trifluoromethyl)phenyl)-1,2,5-oxadiazole 2-Oxide (2b).
This compound was synthesized using the general procedure with 1b (1.51 g, 7.47 mmol) and NaNO2 (5.15 g, 74.7 mmol) to afford 2b as a yellow solid (699 mg, 36.0%). 1H NMR (CDCl3, 400 MHz): δ 8.24−8.22 (2H, d, J = 8.0 Hz), 8.02−8.00 (2H, d, J = 8.0 Hz), 4.76−4.72 (2H, d, J = 16.0 Hz), 4.01 (1H, s).
4-(4-Bromophenyl)-3-(hydroxymethyl)-1,2,5-oxadiazole 2-Oxide (2c).
This compound was synthesized using the general procedure with 1c (1.11 g, 5.21 mmol) and NaNO2 (5.39 g, 78.1 mmol) to afford 2c as a reddish-brown solid (993 mg, 70.4%). 1H NMR (CDCl3, 400 MHz): δ 7.96−7.95 (2H, d, J = 4.0 Hz), 7.85−7.80 (2H, d, J = 20.0 Hz), 4.65 (2H, s).
4-(4-(Difluoromethoxy)phenyl)-3-(hydroxymethyl)-1,2,5-oxadiazole 2-Oxide (2d).
This compound was synthesized using the general procedure with 1d (923 mg, 4.61 mmol) and NaNO2 (2.47 g, 35.8 mmol) to afford 2d as a yellow solid (306 mg, 25.7%). 1H NMR (CDCl3, 400 MHz): δ 7.88−7.86 (2H, d, J = 8.0 Hz), 7.30−7.28 (2H, d, J = 8.0 Hz), 6.80−6.43 (1H, m), 4.73 (2H, s).
3-(Hydroxymethyl)-4-(4-methoxyphenyl)-1,2,5-oxadiazole 2-Oxide (2e).
This compound was synthesized using the general procedure with 1e (1.88 g, 11.5 mmol) and NaNO2 (7.89 g, 114 mmol) to afford 2e as a yellow solid (523 mg, 20.6%). 1H NMR (CDCl3, 400 MHz): δ 7.92−7.90 (2H, d, J = 8.0 Hz), 7.21−7.19 (2H, d, J = 8.0 Hz), 6.03 (1H, s), 4.57 (2H, s), 3.89 (1H, s).
3-(Hydroxymethyl)-4-(p-tolyl)-1,2,5-oxadiazole 2-Oxide (2f).
This compound was synthesized using the general procedure with 1f (2.28 g, 15.2 mmol) and NaNO2 (10.6 g, 154 mmol) to afford 2f as a yellow solid (687 mg, 21.7%). 1H NMR (CDCl3, 400 MHz): δ 7.69−7.68 (2H, d, J = 4.0 Hz), 7.33−7.31 (2H, d, J = 8.0 Hz), 4.7 (2H, s), 2.47−2.36 (4H, qd).
4-(4-Chlorophenyl)-3-(hydroxymethyl)-1,2,5-oxadiazole 2-Oxide (2g).
This compound was synthesized using the general procedure with 1g (1.67 g, 9.90 mmol) and NaNO2 (13.7 g, 198 mmol) to afford 2g as a yellow solid (495 mg, 22.1%). 1H NMR (CDCl3, 400 MHz): δ 7.81−7.79 (2H, d, J = 8.0 Hz), 7.53−7.51 (2H, d, J = 8.0 Hz), 4.72 (2H, s).
3-(Hydroxymethyl)-4-phenyl-1,2,5-oxadiazole 2-Oxide (2h).
This compound was synthesized using the general procedure with 1h (1.00 g, 7.45 mmol) and NaNO2 (5.14 g, 74.5 mmol) to afford 2h as a yellow solid (727 mg, 50.9%). 1H NMR (CDCl3, 400 MHz): δ 7.81−7.80 (2H, m), 7.59−7.56 (3H, m), 4.75 (2H, s).
General Bromination Procedure.
The appropriate furoxan (1equiv) was dissolved in anhydrous CH2Cl2 at 0 °C under argon. PPh3(1.2 equiv) was added in one portion. The reaction was stirred for 15 min followed by addition of CBr4 (1.2 equiv). The ice bath was removed, and reaction was maintained at room temp for 2 h. The combined organic layers were concentrated in vacuo and purified by flash chromatography (SiO2, hexanes/ethyl acetate) to give the corresponding furoxans.
3-(Bromomethyl)-4-(4-fluorophenyl)-1,2,5-oxadiazole 2-Oxide (3a).
This compound was synthesized using the general procedure with 2a (408 mg, 2.12 mmol), PPh3 (668 mg, 2.55 mmol), and CBr4 (844 mg, 2.55 mmol) to afford 3a as a yellow solid (306 mg, 57.8%). 1H NMR (CDCl3, 400 MHz): δ 7.83−7.79 (2H, qd), 7.31−7.29 (2H, t), 4.40 (2H, s).
3-(Bromomethyl)-4-(4-(trifluoromethyl)phenyl)-1,2,5-oxadiazole 2-Oxide (3b).
This compound was synthesized using the general procedure with 2b (877 mg, 3.37 mmol), PPh3 (1.06 g, 4.05 mmol), and CBr4 (1.34 g, 4.05 mmol) to afford 3b as a yellow solid (366 mg, 33.6%). 1H NMR (CDCl3, 400 MHz): δ 7.98−7.96 (2H, d, J = 8.0 Hz), 7.88−7.86 (2H, d, J = 8.0 Hz), 4.45 (2H, s).
3-(Bromomethyl)-4-(4-bromophenyl)-1,2,5-oxadiazole 2-Oxide (3c).
This compound was synthesized using the general procedure with 2c (772 mg, 2.85 mmol), PPh3 (897 mg, 3.42 mmol), and CBr4(1.13 g, 3.42 mmol) to afford 3c as a yellow solid (554 mg, 58.3%). 1H NMR (CDCl3, 400 MHz): δ 7.79−7.76 (2H, m), 7.60−7.57 (2H, m),4.39 (2H, s).
3-(Bromomethyl)-4-(4-(difluoromethoxy)phenyl)-1,2,5-oxadia zole 2-Oxide (3d).
This compound was synthesized using the general procedure with 2d (306 mg, 1.19 mmol), PPh3 (373 mg, 1.42 mmol), and CBr4 (579 mg, 1.42 mmol) to afford 3d as a white solid oil (334 mg, 81.0%). 1H NMR (CDCl3, 400 MHz): δ 7.81−7.79 (2H, d, J = 8.0Hz), 7.33−7.31 (2H, d, J = 8.0 Hz), 6.80−6.44 (1H, m), 4.38 (2H, s).
3-(Bromomethyl)-4-(4-methoxyphenyl)-1,2,5-oxadiazole 2-Oxide (3e).
This compound was synthesized using the general procedure with 2e (523 mg, 2.35 mmol), PPh3 (1.00 g, 3.81 mmol), and CBr4(1.26 g, 3.81 mmol) to afford 3e as a yellow oil (578 mg, 86.1%). 1H NMR (CDCl3, 400 MHz): δ 7.76−7.74 (2H, d, J = 8.0 Hz), 7.11−7.09 (2H, d, J = 8.0 Hz), 4.42 (2H, s), 3.91 (3H, s).
3-(Bromomethyl)-4-(p-tolyl)-1,2,5-oxadiazole 2-Oxide (3f).
This compound was synthesized using the general procedure with 2f (650 mg, 3.15 mmol), PPh3 (992 mg, 3.78 mmol), and CBr4 (1.25 g, 3.78 mmol) to afford 3f as a white solid (605 mg, 71.3%). 1H NMR (CDCl3, 400 MHz): δ 7.69−7.67 (2H, d, J = 8.0 Hz), 7.40−7.38 (2H, d, J = 8.0 Hz), 4.40 (2H, s), 2.46 (3H, s).
3-(Bromomethyl)-4-(4-chlorophenyl)-1,2,5-oxadiazole 2-Oxide (3g).
This compound was synthesized using the general procedure with 2g (711 mg, 3.14 mmol), PPh3 (988 mg, 3.76 mmol), and CBr4(1.25 g, 3.76 mmol) to afford 3g as a yellow oil (477 mg, 52.5%). 1H NMR (CDCl3, 400 MHz): δ 7.77−7.75 (2H, d, J = 8.0 Hz), 7.59−7.57 (2H, d, J = 8.0 Hz), 4.40 (2H, s).
3-(Bromomethyl)-4-phenyl-1,2,5-oxadiazole 2-Oxide (3h).
This compound was synthesized using the general procedure with 2h (303 mg, 1.58 mmol), PPh3 (496 mg, 1.89 mmol), and CBr4 (627 mg, 1.89 mmol) to afford 3h as a yellow solid (230 mg, 57.3%). 1H NMR (CDCl3, 400 MHz): δ 7.80−7.78 (2H, m), 7.60−7.57 (3H, m), 4.41 (2H, s).
General Substitution Procedure.
The appropriate furoxan (1 equiv) was dissolved in either dichloromethane (CH2Cl2) or dimethylformaldyhide (DMF) (solvent used is specified for each compound below) under argon and placed at either 50 °C (for reactions in CH2Cl2) or 100 °C (for reactions in DMF). 4-Methylpiperidine (1.1 equiv) was added neat in one portion immediately followed by addition of base (1.1 equiv) in one portion (triethylamine [TEA] or K2CO3, as specified for each compound below). The reaction mixture was concentrated in vacuo and purified by flash chromatography (SiO2, hexanes/ethyl acetate) to give the corresponding furoxans as indicated below. Note: using TEA as the base gave the best reaction yields.
4-(4-Fluorophenyl)-3-((4-methylpiperidin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (4a).
This compound was synthesized using the general procedure with 3a (1.60 g, 5.86 mmol) was dissolved in DMF (20 mL) at 50 °C, K2CO3 (972 mg, 7.03 mmol), and 4-methylpiperidine (697 mg, 7.03 mmol) to afford 4a as yellow oil(1.57 g, 91.5%). 1H NMR (CDCl3, 400 MHz): δ 8.11−8.07 (2H, m),7.23−7.18 (2H, m), 3.47 (2H, s), 2.83−2.80 (2H, d, J = 12.0 Hz),2.22−2.15 (2H, t), 1.67−1.64 (2H, d, J = 12.0 Hz), 1.40 (1H, m),1.26−1.19 (2H, m), 0.94−0.93 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 165.7, 163.2, 156.8, 130.5, 123.4−123.3, 116.4−116.1,53.7, 50.5, 34.2, 30.4, 21.7. Measured purity at 254 nm: 96.6%; 282 nm: 96.0%. HRMS: calcd for (C15H18FN3O2): 291.1383; found 292.1458 (M + H+).
3-((4-Methylpiperidin-1-yl)methyl)-4-(4-trifluoromethyl)phenyl)-1,2,5-oxadiazole 2-Oxide (4b).
This compound was synthesized using the general procedure with 3b (300 mg, 0.93 mmol) dissolved in DMF (2 mL) and heated to 100 °C and then K2CO3 (141 mg, 1.02 mmol) and 4-methylpiperidine (101 mg, 1.02 mmol) to afford 4b as white, crystalline solid (240 mg, 75.7%). 1H NMR (CDCl3, 400 MHz): δ 8.26−8.24 (2H, d, J = 8.0 Hz), 7.80−7.78 (2H, d, J = 8.0 Hz), 3.49 (2H, s), 2.83−2.80 (2H, d, J = 12.0 Hz), 2.23−2.17 (2H, m), 1.68−1.65 (2H, d, J = 12.0 Hz), 1.42−1.40 (1H, m), 1.25−1.18 (2H, m),0.96−0.93 (3H, m). 13C NMR (CDCl3, 100 MHz): 156.6, 132.9−132.7, 130.5, 128.7, 126.1−126.0, 124.6, 122.8, 53.7, 50.5, 34.2, 30.3,21.7. Measured purity at 254 nm: 97.2%; 282 nm: 92.2%. Mp 88−89 °C. HRMS: calcd for (C16H18F3N3O2): 341.1351; found 342.1429 (M + H+).
4-(4-Bromophenyl)-3-((4-methylpiperidin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (4c).
This compound was synthesized using the general procedure with 3c (300 mg, 0.90 mmol) dissolved in DMF (2 mL) and heated to 100 °C and then K2CO3 (137 mg, 0.99 mmol) and 4-methylpiperidine (98.0 mg, 0.99 mmol) to afford 4c as a white solid (178 mg, 56.1%). 1H NMR (CDCl3, 400 MHz): δ 7.98−7.95 (2H, m),7.67−7.64 (2H, m), 3.46 (2H, s), 2.81−2.78 (2H, d, J = 12.0 Hz),2.21−2.15 (2H, m), 1.66−1.63 (2H, d, J = 12.0 Hz), 1.42−1.38 (1H, m), 1.22−1.15 (2H, m), 0.94−0.92 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 156.9, 132.4, 129.8, 126.0−125.8, 112.9, 53.7,50.5, 34.2, 30.3, 21.8. Measured purity at 254 nm: 98.7%; 282 nm:98.4%. Mp 84−85 °C. HRMS: calcd for (C15H18BrN3O2): 351.0582; found 352.0662 (M + H+).
4-(4-(Difluoromethoxy)phenyl)-3-((4-methylpiperidin-1-yl)-methyl)-1,2,5-oxadiazole 2-Oxide (4d).
This compound was synthesized using the general procedure with 3d (270 mg, 0.84 mmol) in DMF (2 mL) heated to 100 °C and then K2CO3 (139 mg, 1.01 mmol) and 4-methylpiperidine (100 mg, 1.01 mmol) to afford 4d as a yellow solid (235 mg, 82%). 1H NMR (CDCl3, 400 MHz): δ 8.12−8.10 (2H, d, J = 8.0 Hz), 7.27−7.25 (2H, d, J = 8.0 Hz), 6.79−6.43 (1H, m), 3.47 (2H, s), 2.82−2.79 (2H, d, J = 12.0 Hz), 2.21−2.16 (2H, t), 1.67−1.64 (2H, d, J = 12.0 Hz), 1.42−1.39 (2H, m), 1.26−1.16 (2H, m), 0.94−0.93 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 156.8, 153.1, 130.1, 124.1, 119.8, 117.2, 115.5, 113.7, 113.0,53.8, 50.5, 34.2, 30.3, 21.7. Measured purity at 254 nm: 95.9%; 282 nm: 97.9%. Mp 48−49 °C. HRMS: calcd for (C16H19F2N3O3): 339.1394; found 340.1464 (M + H+).
4-(4-Methoxyphenyl)-3-((4-methylpiperdin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (4e).
This compound was synthesized using the general procedure with 3e (550 mg, 1.93 mmol) in DMF (5 mL) heated to 70 °C and then K2CO3 (497 mg, 3.60 mmol) and 4-methylpiperidine (357 mg, 3.60 mmol) to afford 4e as a white solid (563 mg, 96.2%). 1H NMR (CDCl3, 400 MHz): δ 7.91−7.89 (2H, d, J = 8.0 Hz), 6.94−6.92 (2H, d, J = 8.0 Hz), 3.79 (3H, s), 3.39 (2H, s),2.74−2.71 (2H, d, J = 12.0 Hz), 2.11−2.06 (2H, t), 1.57−1.54 (2H, d, J = 12.0 Hz), 1.32−1.29 (2H, m), 1.17−1.12 (2H, m), 0.85−0.83 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 161.8, 157.5, 129.8, 199.4, 114.5, 133.2, 55.4, 53.7, 50.5, 34.2, 30.4, 21.8. Measured purity at 254 nm: 99.1%;, 282 nm: 97.4%. Mp 67−68 °C. HRMS: calcd for (C16H21N3O3): 303.1583; found 304.1653 (M + H+).
3-((4-Methylpiperdin-1-yl)methyl)-4-(p-tolyl)-1,2,5-oxadiazole 2-Oxide (4f).
This compound was synthesized using the general procedure with 3f (178 mg, 0.66 mmol) in CH2Cl2 (1 mL) and carried out at room temperature with TEA (201 mg, 1.98 mmol) and 4-methylpiperidine (66 mg, 0.66 mmol) to afford 4f as a white solid (141 mg, 74.4%). 1H NMR (CDCl3, 400 MHz): δ 7.95−7.94 (2H, d, J = 4.0 Hz), 7.35−7.34 (2H, d, J = 4.0 Hz), 3.5 (2H, s), 2.84−2.82 (2H, d, J = 8.0 Hz), 2.46−2.44 (3H, s), 2.21−2.17 (2H, m), 1.67−1.65 (2H, d, J = 8.0 Hz), 1.41−1.26 (1H, s), 1.24−1.20 (2H, m), 0.95−0.94 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 157.8, 141.4, 129.8, 128.1, 124.2, 113.3, 53.7, 50.5, 34.2, 30.4, 21.8−21.5. Measured purity at 254 nm: 98.5%; 282 nm: 97.5%. Mp 72−73 °C. HRMS: calcd for (C16H21N3O2): 287.1634; found 288.1712 (M + H+).
4-(4-Chlorophenyl)-3-((4-methylpiperidin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (4g).
This compound was synthesized using the general procedure with 3g (476 mg, 1.64 mmol) in CH2Cl2 (1.5 mL) heated to 35 °C and with TEA (499 mg, 4.93 mmol) and 4-methylpiperidine (196 mg, 1.97 mmol) to afford 4g as pale yellow solid (400 mg, 79.1%). 1H NMR (CDCl3, 400 MHz): δ 8.15−8.13 (2H, d, J = 8.0 Hz), 7.61−7.59 (2H, d, J = 8.0 Hz), 3.52 (2H, s), 2.91−2.88 (2H, d, J = 12.0 Hz), 2.31−2.26 (2H, m), 1.76−1.73 (2H, d, J =12.0 Hz), 1.35−1.28 (1H, m), 1.04−1.02 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 156.8, 137.4, 129.6, 129.4, 125.5, 113.0,53.7, 50.5, 34.2, 30.3, 21.7. Measured purity at 254 nm: 99.7%; 282 nm: 99.9%. Mp 67−68 °C. HRMS: calcd for (C15H18ClN3O2): 307.1088; found 308.1161 (M + H+).
3-((4-Methylpiperidin-1-yl)methyl)-4-phenyl-1,2,5-oxadiaozle 2-Oxide (4h).
This compound was synthesized using the general procedure with 3h (245 mg, 0.96 mmol) in DMF [20 mL] heated to 100 °C and with K2CO3 (146 mg, 1.06 mmol) and 4-methylpiperidine (105 mg, 1.06 mmol) to afford 4h as a white solid (198 mg, 75.4%). 1H NMR (CDCl3, 400 MHz): δ 8.04−8.02 (2H, d, J = 8.0 Hz), 7.54−7.52 (2H, d, J = 8.0 Hz), 3.48 (2H, s), 2.82−2.79 (2H, d, J = 12.0 Hz),2.20−2.14 (2H, m), 1.65−1.62 (2H, d, J = 12.0 Hz), 1.39−1.37 (2H, m), 1.25−1.12 (2H, m), 0.93−0.91 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 157.7, 131.0, 129.0, 128.4, 128.2, 127.1, 113.1,53.6, 50.5, 34.2, 30.3, 21.7. Measured purity at 254 nm: 98.6%; 282 nm: 98.6%. Mp 62−63 °C. HRMS: calcd for (C15H19N3O2): 273.1477; found 274.1565 (M + H+).
General Tautomerization Procedure.
The appropriate furoxan was dissolved in toluene (15 mL) and refluxed for 7 days under argon. The resulting tautomeric mixture was purified using reverse-phase column chromatography (mobile phase: 25 mM ammonia formate, 0.1% formic acid: methanol, 0.1% formic acid). Yields are reported as “isolated yield”, which possessed the desired purity for biological testing.
3-(4-Fluorophenyl)-4-((4-methylpiperidin-1-yl)methyl)-1,2,5-oxadiaozle 2-Oxide (5a).
This compound was synthesized using the general procedure with 4a (1.56 g, 5.35 mmol) to afford 5a as a white crystalline solid (isolated yield = 556.0 mg, 35.6%). 1H NMR (CDCl3, 400 MHz): δ 8.23−8.19 (2H, m), 7.26−7.18 (2H, m), 3.55 (2H, s),2.94−2.92 (2H, d, J = 8.0 Hz), 2.20−2.14 (2H, t), 1.69−1.66 (2H, d, J = 12.0 Hz), 1.57 (1H, s), 1.44−1.42 (2H, m), 1.25−1.20 (2H, m),0.95−0.93 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 164.4, 162.7, 154.5, 130.6, 130.5, 119.3−119.2, 116.2, 116.0, 115.0, 53.6, 53.5,34.2, 30.5, 21.8. Measured purity at 254 nm: 95.6%; 282 nm: 98.5%. Free base, mp 55 °C. HCl salt, mp 167−168 °C. HRMS: calcd for (C15H18FN3O2): 291.1383; found 292.1468 (M + H+).
4-((4-Methylpiperdin-1-yl)methyl)-3-(4-(trifluoromethyl)phenyl)-1,2,5-oxadiazole 2-Oxide (5b).
This compound was synthesized using the general procedure with 4b (100 mg, 0.29 mmol) to afford 5b. 1H NMR (CDCl3, 400 MHz): δ 8.39−8.37 (2H, d, J = 8.0 Hz), 7.80−7.79 (2H, d, J = 4.0 Hz), 3.60 (2H, s), 2.97−2.95 (2H, d, J = 8.0 Hz), 1.72−1.70 (2H, d, J = 8.0 Hz), 1.46−1.45 (1H, m), 1.28−1.20 (3H, m),0.97−0.96 (2H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 154.4, 128.5, 125.8, 125.7 (2C), 114.8, 53.6, 53.5, 34.2, 30.5, 21.7. Measured purity at 254 nm: 91.7%; 282 nm: 97.7%. Mp 78−79 °C. HRMS: calcd for (C16H18F3N3O2): 341.1351; found 342.1431 (M + H+).
3-(4-Bromophenyl)-4-((4-methylpiperdin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (5c).
This compound was synthesized using the general procedure with 4c (100 mg, 0.28 mmol) to afford 5c as a white crystalline solid (isolated yield = 25.0 mg, 25.0%). 1H NMR (CDCl3, 400 MHz): δ 8.13−8.11 (2H, d, J = 8.0 Hz), 7.68−7.67 (2H, d, J = 8.0 Hz), 3.57 (2H, s), 2.95−2.94 (2H, d, J = 4.0 Hz), 2.21−2.17 (2H, t),1.71−1.69 (2H, d, J = 8.0 Hz), 1.46−1.44 (1H, m), 1.28−1.19 (2H, m), 0.97−0.96 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 154.4, 132.2, 129.7, 124.9, 122.2, 115.2, 53.6−53.5, 34.2, 30.5, 21.8. Measured purity at 254 nm: 95.0%; 282 nm: 96.8%. Mp 68−69 °C. HRMS: calcd for (C15H18BrN3O2): 351.0582; found 352.0664 (M + H+).
3-(4-(Difluoromethoxy)phenyl)-4-((4-methylpiperidin-1-yl)-methyl)-1,2,5-oxadiazole 2-Oxide (5d).
This compound was synthesized using the general procedure with 4d (235.0 mg, 0.69 mmol) to afford 5d as a white crystalline solid (isolated yield = 78.0 mg, 33.2%). 1H NMR (CDCl3, 400 MHz): δ 8.24−8.22 (2H, d, J = 8.0 Hz), 7.26−7.24 (2H, d, J = 8.0 Hz), 6.78−6.41 (1H, t), 3.55 (2H, s),2.95−2.92 (2H, d, J = 12.0 Hz), 2.20−2.15 (2H, t), 1.69−1.66 (2H, d, J = 12.0), 1.57 (2H, s), 1.25−1.20 (2H, m), 0.95−0.94 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 154.5, 152.3, 130.1, 120.3, 119.6, 117.2, 115.5, 115.0, 113.7, 53.6−53.5, 34.2, 30.5, 21.8. Measured purity at 254 nm: 93.6%; 282 nm: 96.9%. Mp 56−57 °C. HRMS: calcd for (C16H19F2N3O3): 339.1394; found 340.1764 (M + H+).
3-(4-Methoxyphenyl)-4-((methylpiperidin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (5e).
This compound was synthesized using the general procedure with 4e (250.0 mg, 0.82 mmol) to afford 5e as a white crystalline solid (isolated yield = 130.0 mg, 52.0%). 1H NMR (CDCl3, 400 MHz): δ 8.12−8.10 (2H, d, J = 8.0 Hz), 7.03−7.01 (2H, d, J = 8.0 Hz), 3.87 (3H, s), 3.54 (2H, s), 2.96−2.93 (2H, d, J = 12.0 Hz), 2.19−2.13 (2H, t), 1.69−1.66 (2H, d, J = 12.0 Hz), 1.43−1.41 (2H, m), 1.28−1.18 (2H, m), 0.95−0.93 (3H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 161.0, 154.6, 129.8, 115.6, 115.2, 114.3,55.4, 53.6−53.5, 34.2, 30.6, 21.8. Measured purity at 254 nm: 99.8%; 282 nm: 99.2%. Mp 54−55 °C. HRMS: calcd for (C16H21N3O3): 303.1583; found 304.1662 (M + H+).
4-((4-Methylpiperdin-1-yl)methyl)-3-(p-tolyl)-1,2,5-oxadiazole 2-Oxide (5f).
This compound was synthesized using the general procedure with 4f (119.0 mg, 0.41 mmol) to afford 5f as a white crystalline solid (isolated yield = 12.4 mg, 10.4%). 1H NMR (CDCl3, 400 MHz): δ 8.03−8.02 (2H, d, J = 4.0 Hz), 7.33−7.32 (2H, d, J = 4.0 Hz), 3.55 (2H, s), 2.96−2.94 (2H, d, J = 8.0 Hz), 2.42 (3H, s), 2.18−2.14 (2H, t), 1.68−1.66 (2H, d, J = 8.0 Hz), 1.42 (1H, s), 1.26−1.20 (2H, m), 0.95−0.94 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 154.7, 140.7, 129.6, 128.1, 120.2, 115.7, 53.6, 53.5, 34.2, 30.5,21.8, 21.5. Measured purity at 254 nm: 98.9%; 282 nm: 99.7%. Mp 77−78 °C. HRMS: calcd for (C16H21N3O2): 287.1634; found 288.1712 (M + H+).
3-(4-Chlorophenyl)-4-((4-methylpiperidin-1-yl)methyl)-1,2,5-oxadiazole 2-Oxide (5g).
This compound was synthesized using the general procedure with 4g (338.4 mg, 1.10 mmol) to afford 4g as a pale, yellow solid (isolated yield = 28.9 mg, 8.9%). 1H NMR (CDCl3, 400 MHz): δ 8.18−8.17 (2H, d, J = 4.0 Hz), 7.52−7.50 (2H, d, J = 8.0 Hz), 3.57 (2H, s), 2.95−2.93 (2H, d, J = 8.0 Hz), 2.21−2.17 (3H, m),1.70−1.68 (2H, d, J = 8.0 Hz), 1.46−1.42 (1H, m), 1.27−1.19 (3H, m), 0.97−0.95 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 154.4, 136.5, 129.5, 129.2, 121.7, 115.0, 53.6, 53.5, 34.2, 31.0, 30.5,21.8. Measured purity at 254 nm: 95.8%; 282 nm: 98.5%. Mp 68−69 °C. HRMS: calcd for (C15H18ClN3O2): 307.1088; found 308.1158 (M + H+).
4-((4-Methylpiperdin-1-yl)methyl)-3-phenyl-1,2,5-oxadiazole 2-Oxide (5h).
This compound was synthesized using the general procedure with 4h (250 mg, 0.91 mmol) to afford 5h as a pale, yellow solid (isolated yield = 53.0 mg, 21.2%). 1H NMR (CDCl3, 400 MHz): δ 8.15−8.14 (2H, d, J = 4.0 Hz), 7.55−7.49 (3H, m), 3.58 (2H, s),2.98−2.96 (2H, d, J = 8.0 Hz), 2.20−2.16 (2H, m), 1.70−1.68 (2H, d, J = 8.0 Hz), 1.45−1.41 (1H, m), 1.28−1.21 (2H, m), 0.97−0.95 (2H, d, J = 8.0 Hz). 13C NMR (CDCl3, 100 MHz): 154.7, 130.4, 128.9, 128.2, 123.2, 115.6, 53.6, 53.4, 34.2, 30.5, 21.8. Measured purity at 254 nm: 92.3%; 282 nm: 95.8%. Mp 36−37 °C. HRMS: calcd for (C15H19N3O2): 273.1477; found 274.1557 (M + H+).
Radiolabeled Synthesis.
Radiolabeled Substitution Procedure.
All radiolabeled reagents were purchased from commercially available sources and used without further purification. Compound 4a (255 mg, 0.93 mmol) was dissolved in CH2Cl2 (5 mL) while stirring at r.t. under argon. A stirred solution of TEA (283 mg, 2.80 mmol) and 4-methylpiperidine-2,2,6,6-d4 (100 mg, 0.93 mmol) was added to the 4a solution. The reaction was stirred for 10 min and then purified using column chromatography (mobile phase: hexanes/ethyl acetate) to give 6a as a white crystalline solid (255 mg, 91.1%).
4-(4-Fluorophenyl)-3-((4-methylpiperidin-1-yl-2,2,6,6-d4)methyl)-1,2,5-oxadiazole 2-Oxide (6a).
1H NMR (CDCl3, 400 MHz): δ 8.10−8.09 (2H, t), 7.24−7.21 (2H, t), 3.49 (2H, s), 1.67−1.64 (2H, dd), 1.43−1.39 (1H, m), 1.22−1.18 (2H, t), 0.95−0.94 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 165.2, 163.6, 156.9, 130.5, 130.5, 123.2, 123.2, 116.4, 116.2, 113.0, 53.1−52.5, 50.4, 34.1, 30.2, 21.8. Measured purity at 254 nm: 97.6%; 282 nm: 96.0%. Mp 54−55 °C. HRMS: calcd for (C15H14D4FN3O2): 295.1634; found 296.1710 (M + H+).
Radiolabeled Tautomerization Procedure.
Compound 6a (215 mg, 0.73 mmol) was dissolved in toluene (5 mL) and refluxed for 7 days to afford 7a. Separation was carried out according to the general procedure. Compound 7a was isolated as a white crystalline solid (isolated yield = 52 mg, 24.2%).
3-(4-Fluorophenyl)-4-((4-methylpiperidin-1-yl-2,2,6,6-d4)methyl)-1,2,5-oxadiazole 2-Oxide (7a).
1H NMR (CDCl3, 400 MHz): δ 8.24−8.21 (2H, m), 7.24−7.21 (2H, t), 3.57 (2H, s), 1.69−1.67 (2H, dd), 1.46−1.44 (1H, m), 1.24−1.19 (2H, t), 0.97−0.96 (3H, d, J = 4.0 Hz). 13C NMR (CDCl3, 100 MHz): 164.4, 162.7, 130.6−130.5, 119.3, 119.3, 116.2, 116.1, 115.1, 53.4, 53.0, 52.5, 34.1, 30.4, 21.8. Measured purity at 254 nm: 95.9%; 282 nm: 95.9%. Mp 52−53 °C. HRMS: calcd for (C15H14D4FN3O2): 295.1634; found 296.1712 (M + H+).
Supplementary Material
ACKNOWLEDGMENTS
LC-MS/MS was carried out at the University of Toledo Shimadzu Laboratory for Pharmaceutical Research Excellence. Ki determinations, receptor binding profiles, and HERG data were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2013-00017-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. Small-molecule X-ray crystallography and high-resolution mass spectrometry were performed by the Instrumentation Center in the Department of Chemistry and Biochemistry at the University of Toledo with special thanks to Dr. Kristin Kirschbaum and Dr. Dragan Isailovic. This work was funded in part by a New Investigator Award from the American Association of Colleges of Pharmacy (AACP), the Alzheimer’s Association (NIRG-15–363739) and National Institutes of Health R01AG057598. Student stipend support was provided by the Office of Undergraduate Research at the University of Toledo and National Science Foundation Award #1432921.
ABBREVIATIONS USED
- ACN
acetonitrile
- AD
Alzheimer’s disease
- Aβ
amyloid beta
- B:P
brain to plasma ratio
- clogP
calculated partition coefficient
- cGMP
cyclic guanosine monophosphate
- CNS
central nervous system
- COSY
homonuclear correlation spectroscopy
- CREB
cAMP response element binding protein
- EA
ethyl acetate
- FA
formic acid
- IND
investigational new drug application
- LC-MS/MS
tandem mass spectrometry
- LOD
limit of detection
- LLOQ
lower limit of quantitation
- LTP
long-term potentiation
- HMQC
heteronuclear multiple quantum coherence spectroscopy
- MRM
multiple reaction monitoring
- MTT
3-(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide
- MW
molecular weight
- NEM
N-ethylmaleimide
- NO
nitric oxide
- OGD
oxygen glucose deprivation
- ODQ
(1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one)
- ODZ-2N
oxadiazole-2N-oxide
- ODZ-4N
oxadiazole-4N-oxide
- PDA
photodiode array detector
- PDEi
phosphodiesterase inhibitors
- Pgp
P-glycoproteins
- PKA
protein kinase A
- PKG
protein kinase G
- SAR
structure−activity relationship
- S/N
signal-to-noise
- SPE
solid phase extraction
- sGC
soluble guanylyl cyclase
- STPA
step-through passive avoidance
- TEA
triethylamine
- tPSA
total polar surface area
- UPLC
ultra-high-pressure liquid chromatography
Footnotes
Supporting Information
.doc .pdf .pdf .pdf .csv The Supporting Information is available free of charge on the ACS Publications website at DOI:10.1021/acs.jmedchem.8b00389.
Molecular formula strings (CSV)
Supporting information (PDF)
Supplemental characterization (PDF)
Supplemental crystal structure report (PDF)
Supplemental pharmacokinetic data (PDF)
Accession Codes
The X-ray crystal structure of 5a has been deposited into the Cambridge Crystallographic Data Centre, ID code: CCDC 1838464.
The authors declare no competing financial interest.
REFERENCES
- (1).Lu YF; Kandel ER; Hawkins RD Nitric oxide signaling contributes to late-phase LTP and CREB phosphorylation in the hippocampus. J. Neurosci 1999, 19 (23), 10250–10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ciani E; Guidi S; Bartesaghi R; Contestabile A Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: implication for a survival role of nitric oxide. J. Neurochem 2002, 82(5), 1282–1289. [DOI] [PubMed] [Google Scholar]
- (3).Johannessen M; Delghandi MP; Moens U What turns CREB on? Cell. Signalling 2004, 16 (11), 1211–1227. [DOI] [PubMed] [Google Scholar]
- (4).Teich AF; Nicholls RE; Puzzo D; Fiorito J; Purgatorio R; Fa M; Arancio O Synaptic therapy in Alzheimer’s disease: a CREB-centric approach. Neurotherapeutics 2015, 12 (1), 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Puzzo D; Palmeri A; Arancio O Involvement of the nitric oxide pathway in synaptic dysfunction following amyloid elevation in Alzheimer’s disease. Rev. Neurosci 2006, 17 (5), 497–523. [DOI] [PubMed] [Google Scholar]
- (6).Saura CA; Valero J The role of CREB signaling in Alzheimer’s disease and other cognitive disorders. Rev. Neurosci 2011, 22 (2), 153–169. [DOI] [PubMed] [Google Scholar]
- (7).Pugazhenthi S; Wang M; Pham S; Sze CI; Eckman CB Downregulation of CREB expression in Alzheimer’s brain and in Abeta-treated rat hippocampal neurons. Mol. Neurodegener 2011, 6,60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Puzzo D; Vitolo O; Trinchese F; Jacob JP; Palmeri A; Arancio O Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J. Neurosci 2005, 25 (29), 6887–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bennett BM; Reynolds JN; Prusky GT; Douglas RM; Sutherland RJ; Thatcher GR Cognitive deficits in rats after forebrain cholinergic depletion are reversed by a novel NO mimetic nitrate ester. Neuropsychopharmacology 2007, 32 (3), 505–513. [DOI] [PubMed] [Google Scholar]
- (10).Luo J; Lee SH; VandeVrede L; Qin Z; Ben Aissa M; Larson J; Teich AF; Arancio O; D’Souza Y; Elharram A; Koster K; Tai LM; LaDu MJ; Bennett BM; Thatcher GRJ A multifunctional therapeutic approach to disease modification in multiple familial mouse models and a novel sporadic model of Alzheimer’s disease. Mol. Neurodegener 2016, 11 (1), 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chien W-L; Liang K-C; Teng C-M; Kuo S-C; Lee F-Y; Fu W-M Enhancement of long-term potentiation by a potent nitric oxide-guanylyl cyclase activator, 3-(5-hydroxymethyl-2-furyl)-1-benzylindazole. Mol. Pharmacol 2003, 63 (6), 1322–1328. [DOI] [PubMed] [Google Scholar]
- (12).Palmeri A; Privitera L; Giunta S; Loreto C; Puzzo D Inhibition of phosphodiesterase-5 rescues age-related impairment of synaptic plasticity and memory. Behav. Brain Res 2013, 240, 11–20. [DOI] [PubMed] [Google Scholar]
- (13).Puzzo D; Loreto C; Giunta S; Musumeci G; Frasca G; Podda MV; Arancio O; Palmeri A Effect of phosphodiesterase-5 inhibition on apoptosis and beta amyloid load in aged mice. Neurobiol. Aging 2014, 35 (3), 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ren Z; Gu X; Lu B; Chen Y; Chen G; Feng J; Lin J; Zhang Y; Peng H Anticancer efficacy of a nitric oxide-modified derivative of bifendate against multidrug-resistant cancer cells. J. Cell Mol. Med 2016, 20 (6), 1095–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Dutra LA; de Almeida L; Passalacqua TG; Reis JS; Torres FAE; Martinez I; Peccinini RG; Chin CM; Chegaev K; Guglielmo S; Fruttero R; Graminha MAS; dos Santos JL Leishmanicidal activities of novel synthetic furoxan and benzofuroxan derivatives. Antimicrob. Agents Chemother 2014, 58 (8), 4837–4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Tosco P; Bertinaria M; Di Stilo A; Cena C; Sorba G; Fruttero R; Gasco A Furoxan analogues of the histamine H3-receptor antagonist imoproxifan and related furazan derivatives. Bioorg. Med. Chem 2005, 13 (15), 4750–4759. [DOI] [PubMed] [Google Scholar]
- (17).Chegaev K; Cena C; Giorgis M; Rolando B; Tosco P; Bertinaria M; Fruttero R; Carrupt P-A; Gasco A Edaravone derivatives containing NO-donor functions. J. Med. Chem 2009, 52(2), 574–578. [DOI] [PubMed] [Google Scholar]
- (18).Amir M; Waseem AM; Sana T; Somakala K Furoxan derivatives as nitric oxide donors and their therapeutic potential. Int. Res. J. Pharm 2015, 6 (9), 585–599. [Google Scholar]
- (19).Velazquez C; Rao PNP; McDonald R; Knaus EE Synthesis and biological evaluation of 3,4-diphenyl-1,2,5-oxadiazole-2-oxides and 3,4-diphenyl-1,2,5-oxadiazoles as potential hybrid COX-2 inhibitor/nitric oxide donor agents. Bioorg. Med. Chem 2005, 13 (8), 2749–2757. [DOI] [PubMed] [Google Scholar]
- (20).Schiefer IT; VandeVrede L; Fa M; Arancio O; Thatcher GRJ Furoxans (1,2,5-oxadiazole-N-oxides) as novel NO mimetic neuroprotective and procognitive agents. J. Med. Chem 2012, 55 (7), 3076–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Chegaev K; Federico A; Marini E; Rolando B; Fruttero R; Morbin M; Rossi G; Fugnanesi V; Bastone A; Salmona M; Badiola NB; Gasparini L; Cocco S; Ripoli C; Grassi C; Gasco A NO-donor thiacarbocyanines as multifunctional agents for Alzheimer’s disease. Bioorg. Med. Chem 2015, 23 (15), 4688–4698. [DOI] [PubMed] [Google Scholar]
- (22).Gleeson MP Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem 2008, 51 (4), 817–834. [DOI] [PubMed] [Google Scholar]
- (23).Rankovic Z CNS drug design: balancing physicochemical properties for optimal brain exposure. J. Med. Chem 2015, 58 (6), 2584–2608. [DOI] [PubMed] [Google Scholar]
- (24).Cecchelli R; Berezowski V; Lundquist S; Culot M; Renftel M; Dehouck M-P; Fenart L Modelling of the blood-brain barrier in drug discovery and development. Nat. Rev. Drug Discovery 2007, 6 (8), 650–661. [DOI] [PubMed] [Google Scholar]
- (25).Park BK; Kitteringham NR; O’Neill PM Metabolism of fluorine-containing drugs. Annu. Rev. Pharmacol. Toxicol 2001, 41 (1), 443–470. [DOI] [PubMed] [Google Scholar]
- (26).Ruela ALM; Santos MG; Figueiredo EC; Pereira GR LC-PDA and LC-MS studies of donepezil hydrochloride degradation behaviour in forced stress conditions. J. Braz. Chem. Soc 2014, 25, 2094–2101. [Google Scholar]
- (27).Goldberg MP; Strasser U; Dugan LL Techniques for assessing neuroprotective drugs in vitro. Int. Rev. Neurobiol 1996, 40, 69–93. [DOI] [PubMed] [Google Scholar]
- (28).Shineman DW; Basi GS; Bizon JL; Colton CA; Greenberg BD; Hollister BA; Lincecum J; Leblanc GG; Lee LB; Luo F; Morgan D; Morse I; Refolo LM; Riddell DR; Scearce-Levie K; Sweeney P; Yrjanheikki J; Fillit HM Accelerating drug discovery for Alzheimer’s disease: best practices for preclinical animal studies. Alzheimer’s Res. Ther. 2011, 3 (28), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Frye SV; Arkin MR; Arrowsmith CH; Conn PJ; Glicksman MA; Hull-Ryde EA; Slusher BS Tackling reproducibility in academic preclinical drug discovery. Nat. Rev. Drug Discovery 2015, 14 (11), 733–734. [DOI] [PubMed] [Google Scholar]
- (30).Zhang L; Qu Y; Yang C; Tang J; Zhang X; Mao M; Mu D; Ferriero D Signaling pathway involved in hypoxia-inducible factor-1alpha regulation in hypoxic-ischemic cortical neurons in vitro. Neurosci. Lett 2009, 461 (1), 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Chung H; Seo S; Moon M; Park S Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J. Endocrinol 2008, 198 (3), 511–521. [DOI] [PubMed] [Google Scholar]
- (32).Di L; Rong H; Feng B Demystifying brain penetration in central nervous system drug discovery. J. Med. Chem 2013, 56 (1), 2–12. [DOI] [PubMed] [Google Scholar]
- (33).Vandevrede L; Tavassoli E; Luo J; Qin Z; Yue L; Pepperberg DR; Thatcher GR Novel analogues of chlormethiazole are neuroprotective in four cellular models of neurodegeneration by a mechanism with variable dependence on GABAA receptor potentiation. Br. J. Pharmacol 2014, 171 (2), 389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang XP; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FR; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492 (7428), 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Huang XP; Mangano T; Hufeisen S; Setola V; Roth BL Identification of human Ether-a-go-go related gene modulators by three screening platforms in an academic drug-discovery setting. Assay Drug Dev. Technol 2010, 8 (6), 727–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Angelucci L; Calvisi P; Catini R; Cosentino U; Cozzolino R; De Witt P; Ghirardi O; Giannessi F; Giuliani A; Guaraldi D; Misiti D; Ramacci MT; Scolastico C; Tinti MO Synthesis and amnesia-reversal activity of a series of 7- and 5-membered 3-acylamino lactams. J. Med. Chem 1993, 36 (11), 1511–1519. [DOI] [PubMed] [Google Scholar]
- (37).Pinza M; Farina C; Cerri A; Pfeiffer U; Riccaboni MT; Banfi S; Biagetti R; Pozzi O; Magnani M; Dorigotti L Synthesis and pharmacological activity of a series of dihydro-1H-pyrrolo[1,2-a]imidazole-2,5(3H,6H)-diones, a novel class of potent cognition enhancers. J. Med. Chem 1993, 36 (26), 4214–4220. [DOI] [PubMed] [Google Scholar]
- (38).Murphy KJ; O’Connell AW; Regan CM Repetitive and transient increases in hippocampal neural cell adhesion molecule polysialylation state following multitrial spatial training. J. Neurochem 1996, 67 (3), 1268–1274. [DOI] [PubMed] [Google Scholar]
- (39).Park SJ; Kim DH; Jung JM; Kim JM; Cai M; Liu X; Hong JG; Lee CH; Lee KR; Ryu JH The ameliorating effects of stigmasterol on scopolamine-induced memory impairments in mice. Eur. J. Pharmacol 2012, 676 (1), 64–70. [DOI] [PubMed] [Google Scholar]
- (40).Han R-W; Zhang R-S; Xu H-J; Chang M; Peng Y-L; Wang R Neuropeptide S enhances memory and mitigates memory impairment induced by MK801, scopolamine or Aβ1–42 in mice novel object and object location recognition tasks. Neuropharmacology 2013, 70, 261–267. [DOI] [PubMed] [Google Scholar]
- (41).Detrait ER; Hanon E; Dardenne B; Lamberty Y The inhibitory avoidance test optimized for discovery of cognitive enhancers. Behav Res. Meth 2009, 41 (3), 805–811. [DOI] [PubMed] [Google Scholar]
- (42).Ota KT; Pierre VJ; Ploski JE; Queen K; Schafe GE The NO-cGMP-PKG signaling pathway regulates synaptic plasticity and fear memory consolidation in the lateral amygdala via activation of ERK/MAP kinase. Learn. Mem 2008, 15 (10), 792–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Vanmierlo T; Creemers P; Akkerman S; van Duinen M; Sambeth A; De Vry J; Uz T; Blokland A; Prickaerts J The PDE4 inhibitor roflumilast improves memory in rodents at non-emetic doses. Behav. Brain Res 2016, 303, 26–33. [DOI] [PubMed] [Google Scholar]
- (44).Verhoest PR; Fonseca KR; Hou X; Proulx-Lafrance C; Corman M; Helal CJ; Claffey MM; Tuttle JB; Coffman KJ; Liu S; Nelson F; Kleiman RJ; Menniti FS; Schmidt CJ; Vanase-Frawley M; Liras S Design and discovery of 6-[(3S,4S)-4-methyl-1-(pyrimidin-2-ylmethyl)pyrrolidin-3-yl]-1-(tetrahydro-2H-pyran-4-yl)-1,5-dihydro-4H-pyrazolo[3,4-d]pyrimidin-4-one(PF-04447943), a selective brain penetrant PDE9A inhibitor for the treatment of cognitive disorders. J. Med. Chem 2012, 55 (21), 9045–9054. [DOI] [PubMed] [Google Scholar]
- (45).Van der Staay FJ; Rutten K; Barfacker L; Devry J; Erb C; Heckroth H; Karthaus D; Tersteegen A; van Kampen M; Blokland A; Prickaerts J; Reymann KG; Schroder UH; Hendrix M The novel selective PDE9 inhibitor BAY 73–6691 improves learning and memory in rodents. Neuropharmacology 2008, 55 (5), 908–918. [DOI] [PubMed] [Google Scholar]
- (46).Vardigan JD; Converso A; Hutson PH; Uslaner JM The selective phosphodiesterase 9 (PDE9) inhibitor PF-04447943 attenuates a scopolamine-induced deficit in a novel rodent attention task. J. Neurogenet 2011, 25 (4), 120–126. [DOI] [PubMed] [Google Scholar]
- (47).Zhang J; Guo J; Zhao X; Chen Z; Wang G; Liu A; Wang Q; Zhou W; Xu Y; Wang C Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav. Brain Res 2013, 250, 230–237. [DOI] [PubMed] [Google Scholar]
- (48).Summerfield SG; Stevens AJ; Cutler L; del Carmen Osuna M; Hammond B; Tang S-P; Hersey A; Spalding DJ; Jeffrey P Improving the in vitro prediction of in vivo central nervous system penetration: integrating permeability, p-glycoprotein efflux, and free fractions in blood and brain. J. Pharmacol. Exp. Ther 2005, 316(3), 1282–1290. [DOI] [PubMed] [Google Scholar]
- (49).Mao F; Wang H; Ni W; Zheng X; Wang M; Bao K; Ling D; Li X; Xu Y; Zhang H; Li J Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (ache) and phosphodiesterase 5 (pde5) for the treatment of Alzheimer’s disease. ACS Chem. Neurosci 2018, 9 (2), 328–345. [DOI] [PubMed] [Google Scholar]
- (50).Fiorito J; Vendome J; Saeed F; Staniszewski A; Zhang H; Yan S; Deng SX; Arancio O; Landry DW Identification of a novel 1,2,3,4-tetrahydrobenzo[b][1,6]naphthyridine analogue as a potent phosphodiesterase 5 inhibitor with improved aqueous solubility for the treatment of Alzheimer’s disease. J. Med. Chem 2017, 60 (21), 8858–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Puzzo D; Staniszewski A; Deng SX; Privitera L; Leznik E; Liu S; Zhang H; Feng Y; Palmeri A; Landry DW; Arancio O Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J. Neurosci 2009, 29 (25), 8075–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Madineni A; Alhadidi Q; Shah ZA Cofilin inhibition restores neuronal cell death in oxygen-glucose deprivation model of ischemia. Mol. Neurobiol 2016, 53 (2), 867–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Suneja SK; Mo Z; Potashner SJ Phospho-CREB and other phospho-proteins: improved recovery from brain tissue. J. Neurosci. Methods 2006, 150 (2), 238–241. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.