

Abstract
Human bone marrow derived mesenchymal stromal cells (BMSCs) represent a putative cell source candidate for tissue engineering‐based strategies to repair cartilage and bone. However, traditional isolation of BMSCs by adhesion to plastic leads to very heterogeneous cell populations, accounting for high variability of chondrogenic differentiation outcome, both across donors and across clonally derived strains. Identification of putative surface markers able to select BMSC subpopulations with higher chondrogenic capacity (CC) and reduced variance in chondrogenic differentiation could aid the development of BMSC‐based cartilage and bone regeneration approaches. With the goal to identify predictive markers for chondrogenic BMSC populations, we assessed the gene expression profile of single cell‐derived clones with high and low CC. While a clustering between high and low CC clones was observed for one donor, donor‐to‐donor variability hampered the possibility to achieve conclusive results when different donors were considered. Nevertheless, increased NCAM1/CD56 expression correlated in clones derived from one donor with higher CC, the same trend was observed for three additional donors (even if no significance was achieved). Enriching multiclonal BMSCs for CD56+ expression led to an increase in CC, though still highly affected by donor‐to‐donor variability. Our study finally suggests that definition of predictive marker(s) for BMSCs chondrogenesis is challenged by the large donor heterogeneity of these cells, and by the high complexity and plasticity of the BMSCs system. Multiple pathways and external parameters may be indeed involved in determining the chondrogenic potential of BMSCs, making the identification of putative markers still an open issue. stem cells translational medicine 2019;8:194&11
Keywords: Human bone marrow derived mesenchymal stromal cells, Chondrogenesis, Selection markers, Tissue engineering, RNA sequencing, NCAM1/CD56
Significance Statement.
Based on transcriptomic data on single‐cell derived clones, this study identified CD56 as a potential predictive marker able to select human bone marrow mesenchymal stromal cells (BMSC) subpopulations with higher and more predictable chondrogenic capacity, although still highly affected by donor‐to‐donor variability. Even if selection of this single marker (i.e., CD56) for distinguishing BMSC chondrogenic subpopulations may drastically increase the clinical relevance of this approach, the intrinsic complexity and plasticity of the BMSC system challenges this conclusion. This awareness should alert the field about the difficulty of identifying univocal pre‐selection criteria following the current approaches, while possibly opening the path to innovative computational methods eventually able to process multiple variables of the BMSC system.
Introduction
Human bone marrow derived mesenchymal stromal cells (BMSCs) have been proposed as a promising cell source candidate for cartilage regeneration and bone repair through endochondral ossification 1, 2, 3, mainly due to their ease of isolation and abundance in human bone marrow 4, 5. However, their usage in the clinical practice has been so far hampered by the large donor‐to‐donor variability, resulting in an unpredictable differentiation outcome 6. Moreover, isolation of BMSCs in vitro based on their adherence to plastic results in very heterogeneous cell populations, with unpredictable ability to differentiate into cartilage, bone and adipose lineages (trilineage differentiation potential). In order to define a more homogenous starting population, several studies have recently investigated different surface markers potentially able to select within the unfractionated bone marrow BMSCs subpopulations with enhanced CFU‐F capacity, trilineage differentiation potential or self‐renewal properties. With this goal, several single surface markers for example, CD271 7, CD49a 8, and CD146 9 and surface marker combinations including CD271+CD146+ 10, CD271+MSCA‐1+CD56+ 11, CD271+CD90+CD106+ 12, and CD271+CD140a− 13, 14 were proposed to demarcate these cells. Yet, no consensus has been achieved within the scientific community on such putative marker(s). CD271+ cells isolated from fresh bone marrow were shown to have a better chondrogenic differentiation potential in vitro and at an orthotopic site compared with the cells isolated by plastic adherence, however this was demonstrated only with one donor 15. In contrast, against some previous findings suggesting a higher chondrogenic capacity (CC) of CD105+ cells compared with bulk or CD105− cells, Cleary et al. 16 demonstrated that there is no correlation between the CC and the CD105 expression level at different passages of multiclonal BMSCs. It is thus not clear yet whether any marker can delineate cell subsets with higher CC within the bulk population. Moreover, single BMSC derived clones showed a broad range of chondrogenic and osteogenic in vitro differentiation capacity, confirming the heterogeneity of the bulk population 2, 17. In a recent report, single cell RNA transcript levels of chondrogenic genes in bovine BMSCs did not correlate with their functional properties thus challenging a preselection of naïve BMSCs based on specific markers in order to improve chondrogenic outcome 18.
The goal of this study was thus to identify markers for subpopulations of BMSCs exhibiting a high and reproducible chondrogenic differentiation potential across different donors. To this end, we hypothesized the existence of a correlation between the transcriptomic profile of single cell‐derived clones and their chondrogenic differentiation capacity. Based on differentially expressed genes identified by transcriptomic profiling, we then found CD56 to be upregulated in clones with high CC. Subsequently, we sorted multiclonal BMSCs based on their CD56 protein expression in order to verify, whether the CD56+ cell subset has improved differentiation potential compared with the negative fraction and the bulk population. Even if enriching expanded BMSCs for CD56+ expression led to an increase in CC, we showed that donor‐to‐donor variability drastically challenged the possibility to achieve standardized solutions to enhance BMSC CC.
Materials and Methods
If not otherwise stated, all reagents were purchased from Sigma–Aldrich (St. Louis, MO). Cell culture media and supplements were purchased from Gibco (Grand Island, NY).
Cell Isolation and Expansion
Bone marrow aspirates and cartilage tissue biopsies were obtained from patients after informed consent during surgical procedures in accordance with the local ethical committee (University Hospital Basel; Prof. Dr. Kummer; approval date 26/03/2007 Ref Number 78/07). Human mesenchymal stromal cells were isolated from bone marrow aspirates (n = 11; mean age: 39 ± 13 years, 9:2 male : female) by plating 0.10–0.13 × 106 of nucleated cells/cm2 in alpha‐mimimum essential medium (MEM) with 10% fetal bovine serum (FBS), 100 mM HEPES buffer, 1 mM sodium pyruvate, 100 IU/ml penicillin, 100 μg/ml streptomycin and 0.29 mg/ml glutamate (complete medium, CM), supplemented with 5 ng/ml FGF2 (BioTechne, Minneapolis, MN, USA) and expanded in the same medium for one additional passages before using them for chondrogenic assays or sorting experiments.
Generation of Single‐Cell Derived Clones
The experimental set‐up for single cell‐derived clones generation is depicted in Figure 1A. In details, 10,000 nucleated cells from 4 fresh bone marrow aspirates (donor 1: male, 23 years; donor 2: male, 38 years; donor 3: female, 49 years; donor 4: female, 30 years) were seeded in 96 well plates, being the expected frequency of clonogenic cells of approximately 1/104. After 1 week, wells were checked for colony growth: wells without cells and wells with two or more colonies were discarded. Upon confluence, colonies were passaged first into three 12‐well plate wells and subsequently, according to the cell number, into culture flasks with a cell density of 500–2,000 cells/cm2. When a minimum of 0.8 × 106 cells was reached, cells were subjected to RNA isolation for transcriptomic analysis and chondrogenic assays. Senescent and nongrowing clones were discarded.
Figure 1.
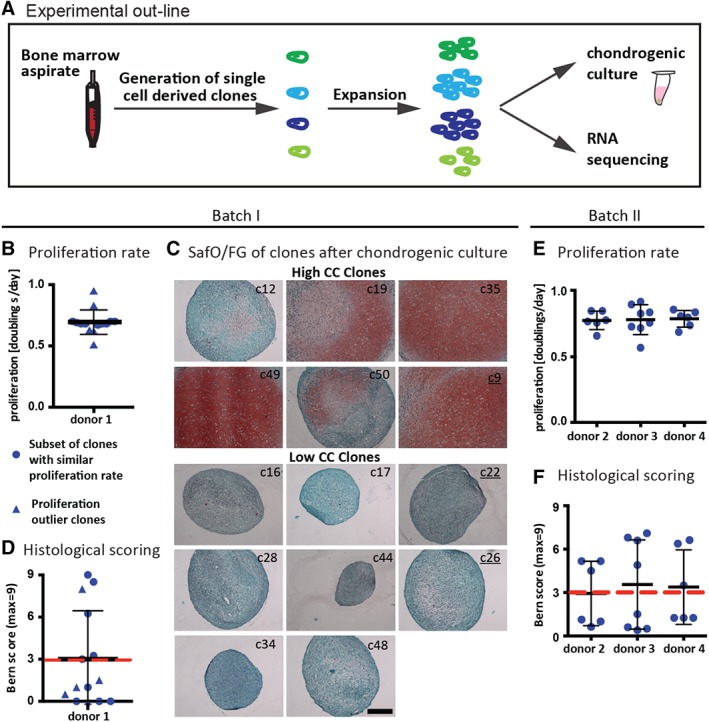
Chondrogenic capacity of single cell derived bone marrow derived mesenchymal stromal cell (BMSC) clones. (A): Fresh bone marrow aspirates were used to generate single‐cell derived clones of BMSCs. For each clone after expansion one part of the cells was subjected to chondrogenic in vitro culture and the other part to RNA sequencing. (B–D): Donor 1 was used for batch I of RNA sequencing. (B): Proliferation rate of single cell derived clones. (C): Clones were cultured as pellets (1–2 replicates) in chondrogenic medium (ChM) for 3 weeks and assessed by Safranin‐O/fast green (SafO/FG) staining. Underlined clone numbers correspond to the proliferation outliers. Scale bar: 200 μm. (D): Bern score was used to evaluate the SafO/FG stained sections. The red line indicates the threshold value to distinguish clones with high and low chondrogenic capacity (CC; c12: 3, c19: 6.25, c35: 8.5, c49: 9, c50: 3.25, c9: 8, c16: 1, c17: 0, c22: 1, c28:0, c44: 0, c26: 0.5, c34: 0, c48: 1.5). (E, F): Donors 2–4 were used for batch II of RNA sequencing. (E): Proliferation rate. (F): Bern score. The red line indicates the threshold value to distinguish clones with high and low chondrogenic capacity. The data presented excludes the remaining clones, which were not subjected to RNA sequencing.
For the clonal study of CD56+ and CD56− expressing clones, P1‐expanded cells from one donor (age: 33 years, male) were single‐cell sorted into 6 × 96 well plates based on CD56 expression. Wells with growing colonies derived from one cell were expanded subsequently as described above. From the (in total 82) generated CD56+clones, clones that did not reach confluence in 12 well plate after 20 days of expansion were discarded. After expansion, clones were tested for their capacity to differentiate toward the chondrogenic, osteogenic and adipogenic lineages, and for their expression of CD56 at mRNA level.
Chondrogenic Differentiation
For chondrogenic pellet cultures, 0.25 × 106 cells were centrifuged for 5 minutes at 1,000 rpm in serum‐free medium consisting of Dulbecco's modified Eagle's medium (DMEM), 4.5 g/l d‐glucose with 100 mM HEPES buffer, 1 mM sodium pyruvate, 100 IU/ml penicillin, 100 μg/ml streptomycin and 0.29 mg/ml glutamate, 1.25 μg/ml human serum albumin, insulin‐treansferrin‐selenium, 4.7 μg/ml linoleic acid, supplemented with 0.1 mM ascorbic acid 2‐phosphate, 10−7 M dexamethasone and 10 ng/ml TGFβ3 (Novartis; ChM). Pellets were cultured for 2–3 weeks as stated in the figure legends and the medium was changed twice a week.
Adipogenic Differentiation
Cells were seeded into 12 well plates (0.04 × 106 cells/well, 1–3 replicates depending on the gained cell number) and cultured for 2 weeks in adipogenic differentiation medium consisting in CM based on DMEM, 4.5 g/l d‐glucose supplemented with 10 μg/ml methyl‐iso buthyl xanthine, 10 μg/ml insulin Actrapid HM, 0.1 mM indomethacin and 10−6 M dexamethasone and for one additional week in CM containing insulin. After the culture, cells were fixed for 10 minutes in formalin and stained with oil red. For semiquantitative analysis, three pictures per well were taken at ×20 objective and cells clearly showing oil droplet accumulation were counted.
Osteogenic Differentiation
Cells were seeded into 12 well plates (0.04 × 106 cells/well), 1–3 replicates depending on the gained cell number) and cultured for 3 weeks in osteogenic differentiation medium consisting in CM based on alpha‐MEM supplemented with 0.1 mM ascorbic acids 2‐phosphate, 10 mM beta‐glycero‐phosphate and 10−8 M dexamethasone. Afterward, the cells were fixed for 10 minutes in formalin and stained with alizarin red. For quantitation purposes, after image acquisition the alizarin red was solubilized by incubation in 10% acetic acid, subsequently at 85°C and with 10% ammonium hydroxide neutralized supernatants were optically measured at a wavelength of 405 nm.
Cytofluorometry and Fluorescently Activated Cell Sorting
BMSCs at different passages were analyzed using the fluorophore‐conjugated antibodies against the following surface markers: CD49a (Becton, Dickinson and Company, Franklin Lakes, NJ), CD56, CD106, CD105, CD166 (BioLegendes), ROR2 (Biotechne, Minneapolis, MN, USA). Cells after trypsinization were stained for 20 minutes at 4°C in fluorescently activated cell sorting (FACS) buffer (phosphate‐buffered saline, 2% FBS, 2.5 mM EDTA) and measured with a Fortessa, BD. Data was processed in FlowJow. For sorting, the cells isolated from 11 donors were trypsinized and stained in FACS buffer for 20 minutes at 4°C with an anti‐CD56‐APC (BioLegends, clone HCD56, San Diego, CA, USA) and sorted using Aria (Becton Dickinson). Unsorted, negatively and positively sorted cells were plated at a density of 3,300 cells/cm2 and expanded until fully confluent or not more than 12 days before testing in chondrogenic assays.
Histology
Pellets after in vitro culture were fixed in 4% paraformaldehyde, dehydrated, and embedded in paraffin. Safranin‐O/fast green (SafO/FG) staining with hematoxylin (J.T. Baker) nuclear counterstaining was performed to analyze cartilage tissue formation. For semiquantitative assessment of the chondrogenesis the Bern score according to Grogan et al. 19 was determined.
Quantification of Glycosaminoglycans and DNA
Pellets were digested with 1 mg/ml protease K in 50 mM Tris with 1 mM EDTA, 1 mM iodoacetamide and 10 mg/ml pepstatin‐A for 16 hours at 56°C. For glycosaminoglycan quantification, the method of Barbosa et al. 20 was used. Briefly, diluted or undiluted digested constructs (depending on SafO intensity) were incubated with 1 ml of dimethylmethylene blue assay (DMMB) solution (16 mg/l dimethylmethylene blue, 6 mM sodium formate, 200 mM GuHCL, pH 3.0) on a shaker at room temperature for 30 minutes. Precipitated DMMB‐glycosaminoglycans (GAGs) complexes were centrifuged and supernatants were discarded. Complexes were dissolved in decomplexion solution (4 M GuHCL, 50 mM Na‐Acetate, 10% Propan‐1‐ol, pH 6.8) at 60°C, absorption was measured at 656 nm and GAG concentrations were calculated using a standard curve prepared with purified bovine chondroitin sulfate. DNA content was measured by using the CyQuant Cell Proliferation Assay Kit (Molecular Probes Inc., Eugene, OR) according to the instructions of the manufacturer.
RNA Sequencing
Based on SafO/FG staining, the clones were evaluated and divided into two groups: clones with a Bern score ≥3 were classified to have a high CC and clones with a Bern score <3 as clones of low CC. Batch I analysis included 14 clones of donor 1 (6 of high CC), while batch II comprised 6 clones of donor 2 (3 of high CC), 8 of donor 3 (4 of high CC) and 6 clones of donor 4 (3 of high CC). For mRNA isolation, the protocol of the column‐based quick‐RNA min‐prep (ZymoResearche, Orange, CA, USA) was followed. mRNA was quantified by using nanodrop and its quality was determined with the help of the 2,100 bioanalyzer (Agilent Technologies, Palo Alto, CA). Three hundred nanograms of DNase‐digested mRNA was used for library preparation with TruSeq Stranded Total RNA Kit with Ribo‐Zero Gold (Illumina, San Diego, CA, USA). Libraries were run on the fragment analyzer (Advanced Analytical, Heidelberg, Germania) for quality control and were adjusted to equal concentrations. Sequencing was performed with a HighSeq 2,500, with single end reads of 50 base lengths. Reads were aligned to the human genome (UCSC hg19) using the spliced read aligner STAR 21. Sequencing and mapping quality was assessed by the qQCReport function of the R package QuasR 22. Gene expression was quantified by the qCount function of QuasR using an exon‐union model of hg19 RefSeq genes (downloaded from UCSC 2015‐02‐09). The R package edgeR 23 was used for differential gene expression analysis. For batch I, two different approaches were tested: first only a subset of clones was compared based on similar proliferation rates, then all clones were analyzed in an unbiased way without accounting for the proliferation rate. In both cases, an additive generalized linear model including the factor “CC” (high and low) was fitted to the raw counts (function glmFit) and differential expression between samples with high and low CC was evaluated by likelihood ratio tests (function glmLRT). p‐Values were adjusted by controlling the false‐discovery rate. All genes with an adjusted p‐value <.05 were considered to be significantly differentially expressed. In case of batch II, the additive model included the additional factor “donor” (BM2, BM3, and BM4). All calculations were performed at sciCORE (http://scicore.unibas.ch/) scientific computing core facility at University of Basel. Significantly differentially expressed genes of the subset analysis of batch I were additionally compared with the panel of surface markers (QIAGEN, Hilden, Germany) to identify differentially expressed surface markers and subjected to ingenuity pathway analysis (QUIAGEN, Hilden, Germany) to identify differently activated signaling networks.
Quantitative Real‐Time Polymerase chain reaction (qPCR)
The mRNA was isolated as described in the RNAsequencing paragraph except that the DNA digestion was not performed. cDNA was generated with random primer (Promega, Madison, WI) and the kit of SuperScript reverse transcriptase III (Thermo Scientific, Waltham, MA, USA). For Quantitative Real‐Time PCR (qPCR) the TaqMan assay on demand system based on FAM and Tamra (Thermo Scientific, Waltham, MA, USA) was used. Samples were analyzed for expression of CD56 (NCAM1; Hs00941830) and as reference gene GAPDH (Hs02758991) was used.
Statistical Analysis
Statistical analysis was performed with Graph Pad Prism 6 software. For comparing one variable from different donors or single cell derived clones, unpaired t tests were applied. Pearson d'Agostino test was done to check for normality of the data sets. For multiple comparison tests of non‐normally distributed data sets, one‐way analysis of variance (ANOVA) was performed using Kruskal–Wallis test with Dunn's multiple comparison test as post hoc. Statistical significance was considered with a p‐value <.05. All the data are shown as average ± standard deviation. n indicates number of donors tested and analyzed.
Results
Isolation of Single Cell Derived Clones and Assessment of their CC
In order to find a molecular signature of BMSCs with higher CC, we isolated and expanded single cell derived clones (n = 4 donors) and assessed their gene expression and their chondrogenic in vitro differentiation potential (Fig. 1A). Overall, in 23.2% ± 11.5% of the seeded wells a single cell derived colony could be identified. Clones were expanded for 28 ± 2 days, with a mean proliferation rate of 0.7 ± 0.1 doublings/day for the different donors (Fig. 1B, 1E). As expected, chondrogenic differentiation occurred to a different extent among the clones (Fig. 1C, 1D, 1F) and the percentage of clones with high CC, defined based on a Bern score ≥3.0, ranged from 15% to 50% depending on the donor (i.e., donor 1: 14.7%, donor 2: 26.7%, donor 3: 50%, and donor 4: 21.1%). Fourteen clones generated from donor 1 (batch I) and 20 clones from the three other donors (donor 2, 3, and 4; batch II) were used for RNA sequencing.
Correlation Between Gene Expression Profile of Single Cell Derived Clones and their CC
Differential gene expression analysis on all the 14 clones selected for donor 1 (batch I) demonstrated that clones with high and low CC separated in two clusters, with 58 genes significantly differentially expressed (adjusted p‐value <.05; Fig. 2A). In order to reduce the confounding effect of the different proliferation rates, in a second analysis only clones with a similar proliferation rate (0.67–0.70) were taken into account (9 out of 14), while outliers in terms of proliferation rate were discarded. Interestingly, the clustering became more prominent and the number of significantly differentially expressed genes rose to 1,771 (Fig. 2B). This demonstrated that clones with a closer proliferation rate shared more common gene expression patterns and that the different doubling times introduced more heterogeneity, eventually masking the putative similarities among clones with equal CC. Among the significantly upregulated genes (log2‐fold‐change >0.5 in clones with high CC), there were extracellular matrix proteins which are important components of the cartilage ECM, such as hyaluronan and proteoglycan link protein (HALPN1), aggrecan (ACAN) and cartilage oligomeric matrix protein (COMP; Fig. 2C). Moreover, the gene expression of several surface markers (NCAM1/CD56, VACAM1/CD106, ALCAM/CD166, ITGA1/CD49a) was significantly higher in the clones with high CC, while the gene expression of two surface markers (ENG/CD105 and CSF1R/CD115) was significantly lower expressed in the clones with high CC (Fig. 2C). Notably, NCAM1 was the mostly upregulated surface marker by the clones with high CC (3.25 log2‐fold‐change). The significant upregulation of NCAM1 in clones with high CC was also confirmed by qPCR (Fig. 2D). Ingenuity pathway analysis (IPA) showed that downstream targets of PDGFBB signaling were downregulated and that genes implicated in chondrogenesis were rather increased in clones with high CC (Fig. 2C). According to IPA canonical pathway prediction, clones with high CC exhibited a potential inhibitory status of EIF2 pathway, while the estrogen, mTOR, eIF4, and IL8 signaling pathways were potentially activated (Fig. 2E).
Figure 2.
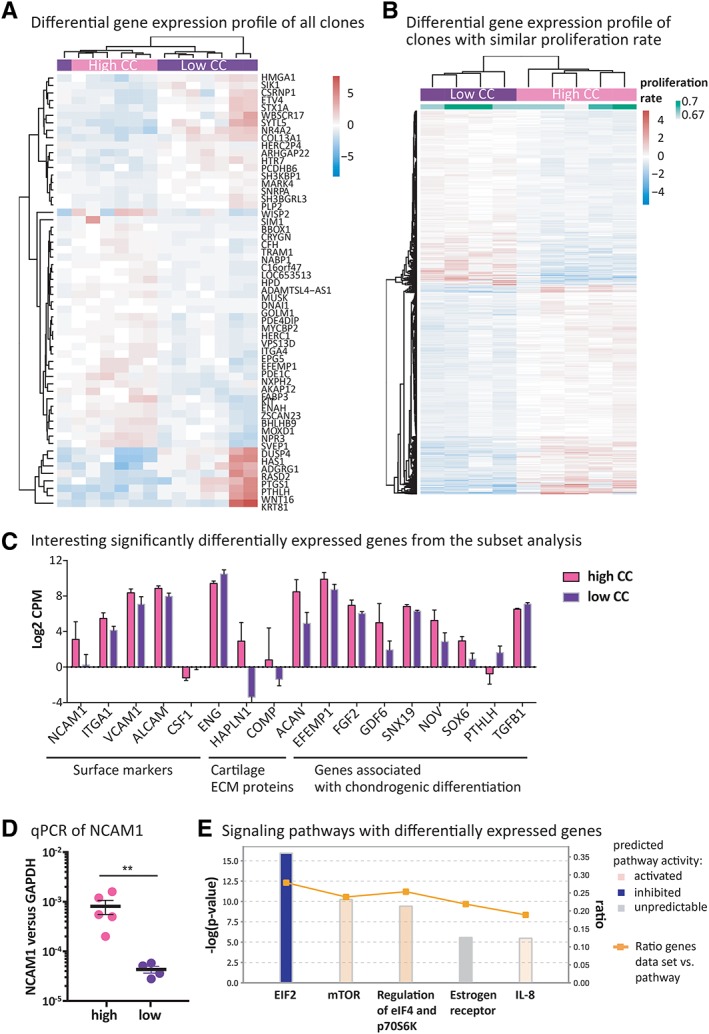
Transcriptomic analysis of single cell derived clones. (A): Heatmap of significantly differentially expressed genes (adjusted p‐value <.05) derived from differential gene expression analysis (DGA) of batch I comparing clones with high and low chondrogenic capacity (CC) when all sequenced clones were included. (B): Heatmap of significantly differentially expressed genes (adjusted p‐value <.05) derived from differential gene expression analysis comparing clones with high and low chondrogenic capacity when only the subset of clones with similar proliferation rate was included. (C): Surface markers, cartilage ECM proteins and genes associated with chondrogenic differentiation identified by ingenuity pathway analysis (IPA) that were among the significant genes of the subset analysis of batch I. (D): Gene expression of NCAM1 in clones analyzed by qPCR and normalized to GAPDH. Unpaired t test; **, p‐value <.0001. (E): Differentially expressed canonical signaling pathways between high and low chondrogenic clones determined by IPA. Bar height indicates −log(p‐value) as calculated by Fisher's exact test right‐tailed. The ratio (orange dots connected by a line) indicates the ratio of genes from the dataset that map to the pathway divided by the total number of genes that map to the same pathway.
RNA sequencing was then performed for clones combined from the three additional different donors (donors 2, 3, and 4; batch II), in order to determine if the genes identified in the first batch (donor 1) are robustly related to the BMSCs CC. Principle component analysis showed that clones preferentially grouped according to their donor origin, rather than to their CC. Moreover, clones from batch II clustered furthest apart from the clones of batch I (Fig. 3A). Since the number of samples per donor was relatively low (a lower number of clones were obtained for donor 2, 3, and 4 as compared with donor 1), the differential gene expression analysis was done for all the clones irrespective of their proliferation rate. Figure 3B shows the genes that were significantly differentially expressed between samples of high and low CC in batch II analysis. None of these six genes overlapped with the genes found in batch I (independent of whether all clones or the subset with similar proliferation rate were taken into account). Notably, a trend of increased expression of CD56 was apparent in the clones with high CC both by mRNA sequencing (Fig. 3C) and qPCR (Fig. 3D). However, the range of CD56 expression levels was too broad to lead to statistical significance.
Figure 3.
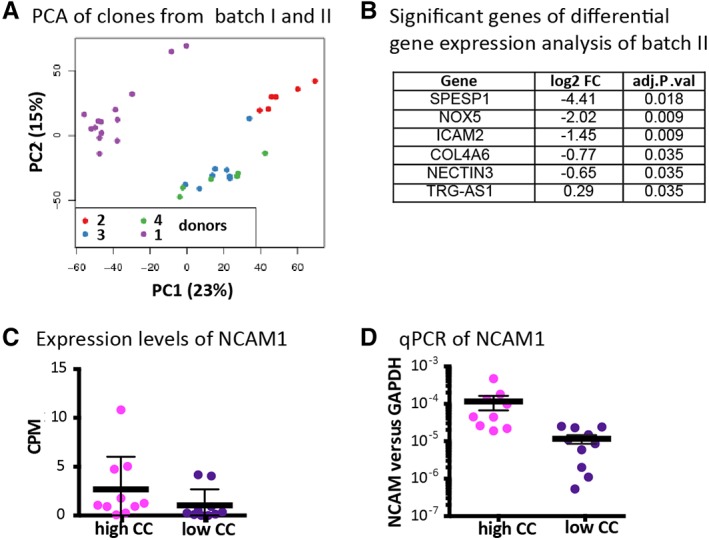
Transcriptomic analysis of more single cell derived clones from different donors. (A): Principal component analysis (PCA) comparing clones of different donors both from batch I and II. (B): Significantly differentially expressed gene of batch II (adjusted p‐value <.05). (C): NCAM1 expression by counted reads from the RNA sequencing. (D): Relative expression of NCAM1 versus GAPDH by qPCR in the clones from donors 2–4. Clones with high versus low chondrogenic capacity did not show significantly different CD56 expression as determined by unpaired t test (p‐value >.05).
Expression of Surface Markers Identified by the Transcriptomic Analysis in Expanded Multiclonal BMSCs
Aiming at finding surface marker(s) that select for BMSCs with higher CC within the bulk population, expanded multiclonal BMSCs (P1) were analyzed for the surface markers found to be upregulated in clones with high CC in the subset analysis of batch I. On average 87% ± 3% of the cells were positive for CD49a (n = 3 donors), 93.9% ± 7.1% for CD106 (n = 7 donors), while CD105 and CD166 were expressed on all of the cells (Fig. 4A). The expression of ROR2, previously correlated with chondrogenic potential of BMSC 24, was also checked. Notably, in our study BMSCs did not express ROR2 (percentage of ROR2+ cells lower than 0.1%, data not shown). The frequency of CD56+ cells instead ranged between 1% and 35%, with an average of 13% ± 11% CD56+ cells (n = 12 donors; Fig. 4B). The frequency of CD56+ cells in bulk multiclonal BMSCs slightly decreased from 19% ± 13% at P1 to 14 ± 18 at P3, while the frequency of CD56+ in the positively sorted cell subset decreased from 94% ± 2% directly after sorting to 33% ± 9% after two passages. The negatively sorted cell subset showed an increase of CD56+ cells from 0.8 ± 0.4 at P1 to 4.5 ± 2.6 at P3 (Fig. 4C). Previously, the frequency of BMSCs positive for CD56 in P2–P3 cells was determined between 24% and 89% 25. Markers exhibiting high and stable expression profiles are unlikely to be unique markers of BMSCs with high chondrogenic potential. Instead, markers with low expression, reflecting the low presence of chondrogenic clones in the total BMSCs population, are more likely to be selective markers of chondrogenic BMSCs 26. Therefore, we pursued CD56 for comparative studies of CC on bulk BMSC populations.
Figure 4.
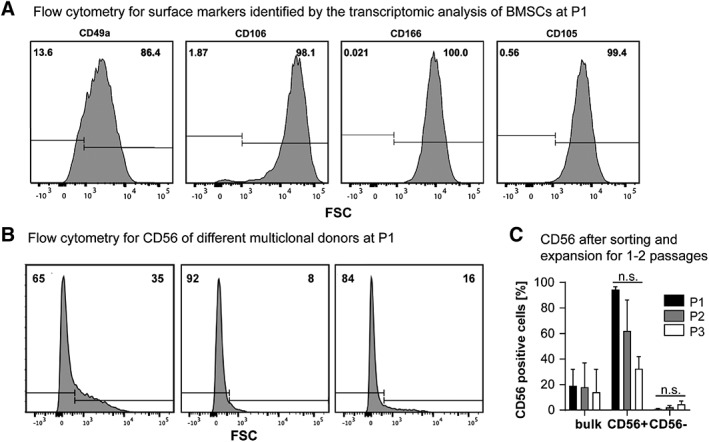
Surface marker expression identified by the transcriptomic study of batch I on P1–P3 expanded multiclonal bone marrow derived mesenchymal stromal cells (BMSCs) by FACS. (A): Representative histogram showing the frequency of CD49a, CD106, CD166, and CD105 positive and negative BMSCs by flow cytometry. (B): Expression of CD56 on P1‐expanded multiclonal BMSCs, histograms from three representative donors are shown. (C): Expression of CD56 by flow cytometry of the bulk population before sorting (P1) and after expansion (P2 and P3) and the sorted CD56 positive and negative cell subset directly after sorting (P1) and after expansion (P2 and P3). One‐way ANOVA based on Kruskal–Wallis test and Dunn's multiple comparison test, n.s.: not significant, n = 3.
Chondrogenic Potential of Sorted Multiclonal CD56+/− BMSCs
To verify the potential of CD56 to select for BMSCs with high CC, P1 expanded BMSCs were sorted and CD56 positive and negative subpopulations were tested in chondrogenic cultures, using unsorted BMSCs as a control (Fig. 5A). The gene expression levels of CD56 directly after sorting and after expansion were more than one order of magnitude higher in the CD56+ cells as compared with the negative ones and corresponded to the level found in the clones of batch I, while the unsorted cells showed an intermediate expression level (Fig. 5B). This demonstrates that the two sorted populations clearly differed in terms of CD56 expression. Importantly, trends in the expression levels among cell populations were maintained after expansion (Fig. 5B). Three out of the 11 BMSC donors were assessed in terms of chondrogenic potential by means of pellet culture (the other BMSC donors had to be discarded due to insufficient cell yield). For all BMSC donors, chondrogenically cultured CD56+ cells formed pellets with higher GAG contents as shown by SafO/FG staining (Fig. 5C). Coherently, the Bern score assessed by histological analysis was higher for the CD56+ cells as compared with either the negative or the bulk populations (Fig. 5D). On average, GAG/DNA content was significantly higher in CD56+ BMSC pellets as compared with CD56− and bulk ones (Fig. 5E). However, the GAG/DNA content of single donors (Fig. 5F) showed that the averaged results originated mainly from cells of donor 1. Thus, high donor‐to‐donor heterogeneity affected the robustness of the results.
Figure 5.
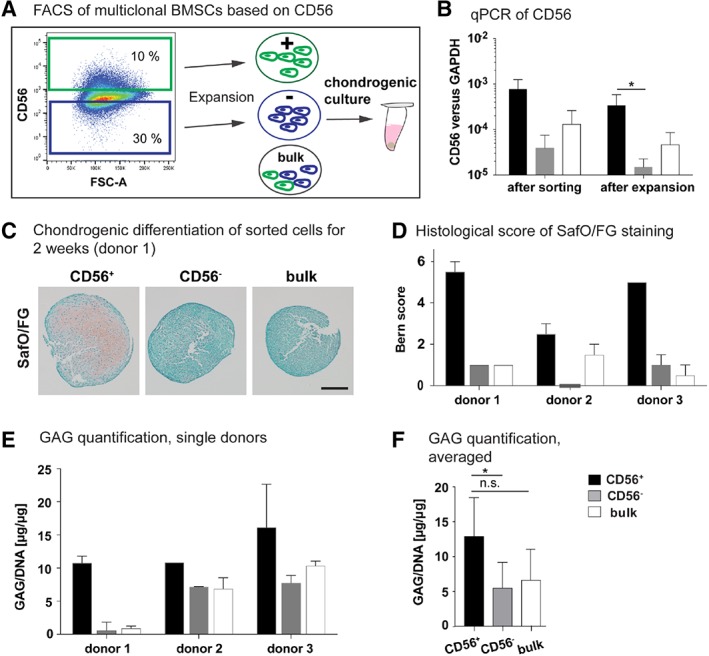
Correlation between CD56 and the chondrogenic capacity of multiclonal BMSCs. (A): P1‐expanded multiclonal bone marrow derived mesenchymal stromal cells (BMSCs) were sorted based on CD56 and after expansion subjected to 2 weeks culture in chondrogenic medium (ChM). (B): Gene expression of CD56 normalized to GAPDH of sorted cells directly after sorting (n = 3) and after 1 week expansion (n = 5). Unpaired t test; *, p‐value = .017. (C): Safranin‐O/fast green (SafO/FG) staining of paraffin sections of sorted cells after culture in ChM for 2 weeks as 3D pellets. Scale bar: 200 μm. (D): The SafO/FG stained sections were evaluated by Bern score. (E, F): The content of glycosamino glycan (GAG) of the pellets was quantified by DMMB assay and normalized to the DNA content. In (E), the separate values from each donor are shown and in (F) averaged values. One‐way ANOVA based on Kruskal–Wallis test and Dunn's multiple comparison test;*, p‐value = .024, n.s. not significant. n = 3, 1–2 replicates per cell population. Of note, donors 1–3 are different donors than used for the RNA sequencing.
Trilineage Differentiation Potential of Single Cell Derived Clones from Sorted CD56+/− BMSCs
In order to better characterize the heterogeneity within the CD56 sorted cell populations, P1‐expanded BMSCs were sorted based on CD56 protein expression and single cells were collected for clonal expansions (Fig. 6A). 10.4% of the CD56+ and 5.9% of the CD56− derived clones survived and proliferated. Clones were expanded for 24 ± 3 and 23 ± 4 days and had a mean proliferation rate of 0.81 ± 0.11 and 0.86 ± 0.14 doublings/day, respectively (Fig. 6B). The gene expression of CD56 at the end of the clonal expansion phase did not correlate with the clone origin—namely derivation from a CD56+ or a CD56− single cell—or with their chondrogenic potential (Fig. 6C). After expansion, clones were subjected to in vitro trilineage differentiation assays. Regarding chondrogenic potential, 1 clone out of 11 (i.e., 9.1%) and 4 clones out of 22 (i.e., 18.2%) showed high CC within the CD56− and CD56+ derived clones, respectively, not resulting in a significant difference between the two subpopulation derived clones (Fig. 6D). The CD56+ clones were marginally osteogenic and adipogenic, with the exception of one clone. All CD56− clones were instead osteogenic (Fig. 6E) and the majority underwent adipogenesis (Fig. 6F), as measured by alizarin red staining for calcification and oil red staining for the accumulation of fat droplets, respectively. The different potential of CD56+ and CD56− single cell‐derived clones in differentiating toward osteogenic and adipogenic linages suggests a functional distinction between these two BMSCs subpopulations.
Figure 6.
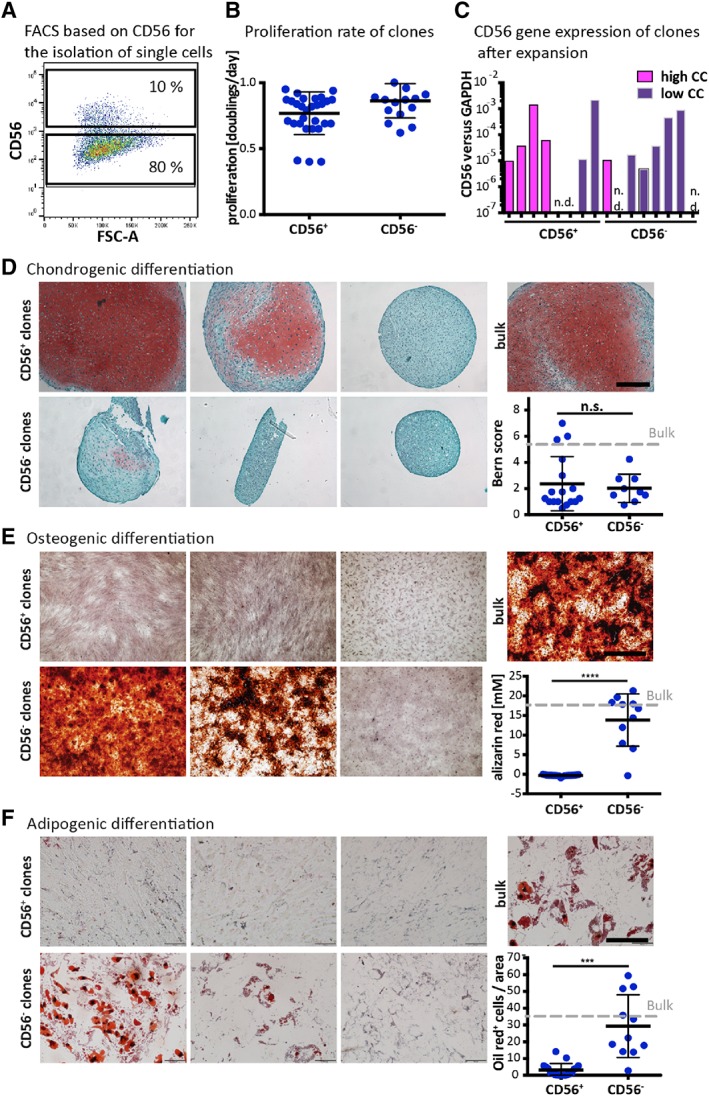
Trilineage assay of clones derived from CD56 positive or negative cells. (A): P1‐expanded bone marrow derived mesenchymal stromal cells (BMSCs) were sorted based on CD56 protein expression and single cells were collected for clonal expansions. (B): Proliferation rate of CD56 positive and negative clones. (C): Gene expression by qPCR of CD56 normalized to GAPDH after expansion. (D): Clones were cultured for 3 weeks in chondrogenic medium (ChM). Safranin‐O/fast green staining (SafO/FG) of paraffin sections of three representative CD56+ and CD56– derived clones are shown and the chondrogenic capacity was evaluated by using the Bern score. (E): Osteogenic differentiation for 3 weeks. Staining for alizarin red and colorimetric quantification thereof. (F): Adipogenic differentiation for 3 weeks. Staining for oil red and quantification by counting the cells clearly containing lipid droplets. Unpaired t test; ****, p‐value ≤.0001. The gray line corresponds to the average value measured by P3 unsorted cells. Scale bars: 200 μm.
Discussion
In this study, we explored the existence of predictive markers identifying BMSCs subsets with high CC, starting from analyzing the molecular signature of clones derived from single BMSCs. Transcriptomic data from one BMSC donor gave a clear‐cut difference between clones with high and low CC and hinted NCAM/CD56 as a marker to prospectively isolate a subpopulation of BMSCs with superior chondrogenic potential. However, results also underlined a high BMSCs donor‐to‐donor variability that challenges the possibility to identify genes regulated in a similar fashion by different donors and to simply apply a marker identified for one donor (i.e., CD56) to others.
Aiming to overcome the heterogeneity of bulk BMSCs isolated by plastic adherence, we assessed whether gene expression correlated with the CC of individual single BMSC‐derived clones. Differential gene expression analysis resulted in the segregation of clones with high and low CC, indicating a correlation between the gene expression profile and the CC of the underlying clone. Notably, the segregation of the two groups was even more prominent when clones with a different proliferation rate than the average (proliferation outliers) were excluded from the analysis. RNA sequencing was performed on three additional BMSC donors to assess whether the results found by the transcriptomic analysis of one donor could be extended to different donors. Overall, our analysis suggested that intrinsic variability among samples (either clone‐to‐clone for example, proliferation rate or donor‐to‐donor) exceeded a potential gene expression difference based on CC, masking the putative similarities among clones with similar chondrogenic potential. It could not be excluded that variable culture conditions such as the duration of expansion or different cellular confluence states at the time of harvest introduced additional variance. The difficulty in extrapolating predictive markers from differential gene expression analysis among single BMSC‐derived clones was reported previously with a bovine bone marrow donor 18. Our results highlight the requirement for a more comprehensive analysis, considering a significantly higher number of donors and clones per donor. This stands in contrast to the study of Dickinson et al. 24, in which RNA sequencing of clones derived from a single donor was performed and suggested ROR2 as a putative marker identifying an highly chondrogenic BMSCs subpopulation. Notably, chondrogenic clones derived from the donors considered in our study did not over‐express ROR2.
Interestingly, when excluding the proliferation rate as a confounding effect in the analysis of batch I, several surface markers were found to be significantly upregulated in clones with high CC and were thus considered for further analyses. Among them, CD56 was the surface marker with the highest log2‐fold‐change difference between clones with high and low CC. Moreover, CD56 was expressed at different moderate levels in different donors of BMSCs heterogeneous populations. Together with N‐cadherin, CD56 has previously been demonstrated to be an important mediator for cell–cell adhesion during prechondrogenic condensations of chondroprogenitors in limb bud development 27, 28, 29. Since the initial condensation phase is also critical for triggering in vitro chondrogenesis of adult BMSCs 27, even if no evidence support, yet the role of CD56 in this process, we hypothesized that an enrichment in CD56 could be beneficial. CD56+ sorted cells from expanded multiclonal BMSCs indeed exhibited a higher chondrogenic differentiation potential compared with the bulk or the CD56− cells. However, differences among donors affected the robustness of these results and this could be mainly attributed to the low number of tested donors. Eight out of 11 donors had to be excluded from this analysis due to low proliferation rates of corresponding cells after sorting. For this reason, future efforts should also be given in defining optimal expansion conditions for sorted BMSCs.
To further investigate the heterogeneity within the CD56 sorted cell populations, single CD56+/− cell‐derived clones were compared in trilineage differentiation assays. A clear distinction between the two subpopulations was evident in terms of osteogenic and adipogenic differentiation capacity (high in CD56− clones and limited/absent in CD56+ ones). Differences in chondrogenic potential were less conclusive, with a slightly higher fraction of Safranin‐O positive clones in the CD56+ compared with the CD56− one. Previously, Battula et al. sorted CD56+ BMSCs from fresh CD45‐depleted human bone marrow in combination with MSCA‐1 and CD271. Interestingly, the authors observed higher CFU‐Fs and better in vitro chondrogenic differentiation of CD271+MSCA‐1+CD56+ compared with the CD56− cells, along with superior adipogenic differentiation of CD56− cells at multiclonal and clonal levels 11. Our results are in line with this previous study, although different sorting times were applied. In the present work, BMSCs were expanded for one passage prior to sort CD56+ and CD56− populations, thus leading to a higher cell yield. This indicates that CD56 expression identifies a certain cell subpopulation not only in naïve BMSCs 11, but also in expanded one, extending the clinical relevance of CD56 as a prospective surface marker. Indeed, from a translational point of view, a key criterion for the applicability of a selection strategy is the possibility to couple it with the achievement of a clinically relevant cell yield.
Even if attempts of simplification through the selection of a single marker for distinguishing BMSC subpopulations may drastically increase the clinical relevance of this approach, the intrinsic complexity and plasticity of the BMSC system challenges this conclusion. First, variables related to donor‐to‐donor heterogeneity should be taken into consideration. Results obtained from one donor were not immediately transferable to other donors, highlighting the importance of considering larger donor cohorts to achieve robust and generally applicable hints.
Conclusion
Moreover, lessons from different stem cell fields (e.g., cancer stem cells, hematopoietic stem cells) suggest that a high level of complexity characterizes stem cells systems. For example, it starts to be evident that tumor progenitor cells 30, as well as hematopoietic stem cells 31, are hardly classifiable through a simple combination of markers, but are most likely characterized by spatio‐temporal plasticity. In a similar fashion, we here propose that the BMSC system may also be challenged by the same level of complexity: multiple intrinsically activated pathways and various external parameters (e.g., donor origin, cell derivation, isolation method, expansion protocol, and so forth) may be involved in determining the fate of BMSC subpopulations. This awareness should alert the field approximately the difficulty of identifying univocal preselection criteria following the current approaches, while possibly opening the path to innovative computational methods eventually able to process multiple variables of the BMSC system.
Author Contributions
C.S., P.O.: conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; F.G.: data analysis and interpretation, bioinformatics analysis of transcriptomic experiment; A.M.: provision of study material or patients; A.B., I.M.: conception and design, data analysis and interpretation, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Acknowledgments
This work was supported by the Swiss National Science Foundation (grant 31003A_156430/1 to IM). We are grateful to Philippe Demougin (University of Basel), Christian Beissel (University of Basel and ETH Zürich, Basel) for technical support with the RNA sequencing and to Lorenzo Renzi and Danny Labes (University Hospital Basel, University of Basel) with the FACS sortings.
References
- 1. Caplan AI. Mesenchymal stem cells: Cell‐based reconstructive therapy in orthopedics. Tissue Eng 2005;11:1198‐1211. [DOI] [PubMed] [Google Scholar]
- 2. Pittenger MF, Mackay AM, Beck SC et al. Multilineage potential of adult human mesenchymal stem cells. Science 1999;284:143‐147. [DOI] [PubMed] [Google Scholar]
- 3. Scotti C, Tonnarelli B, Papadimitropoulos A et al. Recapitulation of endochondral bone formation using human adult mesenchymal stem cells as a paradigm for developmental engineering. Proc Natl Acad Sci USA 2010;107:7251‐7256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nardi NB, da Silva Meirelles L. Mesenchymal stem cells: Isolation, in vitro expansion and characterization. Stem Cells 2008;174:249‐282. [PubMed] [Google Scholar]
- 5. Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007;110:3499‐3506. [DOI] [PubMed] [Google Scholar]
- 6. Somoza RA, Welter JF, Correa D et al. Chondrogenic differentiation of mesenchymal stem cells: Challenges and unfulfilled expectations. Tissue Eng Part B Rev 2014;20:596‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quirici N, Soligo D, Bossolasco P et al. Isolation of bone marrow mesenchymal stem cells by anti‐nerve growth factor receptor antibodies. Exp Hematol 2002;30:783‐791. [DOI] [PubMed] [Google Scholar]
- 8. Deschaseaux F, Gindraux F, Saadi R et al. Direct selection of human bone marrow mesenchymal stem cells using an anti‐CD49a antibody reveals their CD45med, low phenotype. Br J Haematol 2003;122:506‐517. [DOI] [PubMed] [Google Scholar]
- 9. Sacchetti B, Funari A, Michienzi S et al. Self‐renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell 2007;131:324‐336. [DOI] [PubMed] [Google Scholar]
- 10. Tormin A, Li O, Brune JC et al. CD146 expression on primary nonhematopoietic bone marrow stem cells is correlated with in situ localization. Blood 2011;117:5067‐5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Battula VL, Treml S, Bareiss PM et al. Isolation of functionally distinct mesenchymal stem cell subsets using antibodies against CD56, CD271, and mesenchymal stem cell antigen‐1. Haematologica 2009;94:173‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mabuchi Y, Morikawa S, Harada S et al. LNGFR+ THY‐1+ VCAM‐1 hi+ cells reveal functionally distinct subpopulations in mesenchymal stem cells. Stem Cell Rep 2013;1:152‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghazanfari R, Li H, Zacharaki D et al. Human non‐hematopoietic CD271pos/CD140alow/neg bone marrow stroma cells fulfill stringent stem cell criteria in serial transplantations. Stem Cells Dev 2016;25:1652‐1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li H, Ghazanfari R, Zacharaki D et al. Low/negative expression of PDGFR‐α identifies the candidate primary mesenchymal stromal cells in adult human bone marrow. Stem Cell Rep 2014;3:965‐974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mifune Y, Matsumoto T, Murasawa S et al. Therapeutic superiority for cartilage repair by CD271‐positive marrow stromal cell transplantation. Cell Transplant 2013;22:1201‐1211. [DOI] [PubMed] [Google Scholar]
- 16. Cleary MA, Narcisi R, Focke K et al. Expression of CD105 on expanded mesenchymal stem cells does not predict their chondrogenic potential. Osteoarthr Cartil 2016;24:868‐872. [DOI] [PubMed] [Google Scholar]
- 17. Muraglia A, Cancedda R, Quarto R. Clonal mesenchymal progenitors from human bone marrow differentiate in vitro according to a hierarchical model. J Cell Sci 2000;113:1161‐1166. [DOI] [PubMed] [Google Scholar]
- 18. Cote AJ, McLeod CM, Farrell MJ et al. Single‐cell differences in matrix gene expression do not predict matrix deposition. Nat Commun 2016;7:10865‐10878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grogan SP, Barbero A, Winkelmann V et al. Visual histological grading system for the evaluation of in vitro‐generated neocartilage. Tissue Eng 2006;12:2141‐2149. [DOI] [PubMed] [Google Scholar]
- 20. Barbosa I, Garcia S, Barbier‐Chassefière V et al. Improved and simple micro assay for sulfated glycosaminoglycans quantification in biological extracts and its use in skin and muscle tissue studies. Glycobiology 2003;13:647‐653. [DOI] [PubMed] [Google Scholar]
- 21. Dobin A, Davis CA, Schlesinger F et al. STAR: Ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gaidatzis D, Lerch A, Hahne F et al. Quas R: Quantification and annotation of short reads in R. Bioinformatics 2014;31:1130‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 2012;40:4288‐4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dickinson SC, Sutton CA, Brady K et al. The Wnt5a receptor, receptor tyrosine kinase‐like orphan receptor 2, is a predictive cell surface marker of human mesenchymal stem cells with an enhanced capacity for chondrogenic differentiation. Stem Cells 2017;35:2280‐2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Skog MS, Nystedt J, Korhonen M et al. Expression of neural cell adhesion molecule and polysialic acid in human bone marrow‐derived mesenchymal stromal cells. Stem Cell Res Ther 2016;7:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Narcisi R, Cleary MA, Sivasubramaniyan K et al. MSC Populations for Cartilage Regeneration. Cartilage: Springer, 2017:35‐57. [Google Scholar]
- 27. Goldring MB, Tsuchimochi K, Ijiri K. The control of chondrogenesis. J Cell Biochem 2006;97:33‐44. [DOI] [PubMed] [Google Scholar]
- 28. Tavella S, Raffo P, Tacchetti C et al. N‐CAM and N‐cadherin expression during in vitro chondrogenesis. Exp Cell Res 1994;215:354‐362. [DOI] [PubMed] [Google Scholar]
- 29. Widelitz RB, Jiang T, Murray BA et al. Adhesion molecules in skeletogenesis: II. Neural cell adhesion molecules mediate precartilaginous mesenchymal condensations and enhance chondrogenesis. J Cell Physiol 1993;156:399‐411. [DOI] [PubMed] [Google Scholar]
- 30. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med 2017;23:1124‐1134. [DOI] [PubMed] [Google Scholar]
- 31. Velten L, Haas SF, Raffel S et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol 2017;19:271‐281. [DOI] [PMC free article] [PubMed] [Google Scholar]