

Abstract
Retinal pigment epithelium (RPE) performs important functions for the maintenance of photoreceptors and vision. Malfunctions within the RPE are implicated in several retinal diseases for which transplantations of stem cell‐derived RPE are promising treatment options. Their success, however, is largely dependent on the functionality of the transplanted cells. This requires correct cellular physiology, which is highly influenced by the various ion channels of RPE, including voltage‐gated Ca2+ (CaV) channels. This study investigated the localization and functionality of CaV channels in human embryonic stem cell (hESC)‐derived RPE. Whole‐cell patch‐clamp recordings from these cells revealed slowly inactivating L‐type currents comparable to freshly isolated mouse RPE. Some hESC‐RPE cells also carried fast transient T‐type resembling currents. These findings were confirmed by immunostainings from both hESC‐ and mouse RPE that showed the presence of the L‐type Ca2+ channels CaV1.2 and CaV1.3 as well as the T‐type Ca2+ channels CaV3.1 and CaV3.2. The localization of the major subtype, CaV1.3, changed during hESC‐RPE maturation co‐localizing with pericentrin to the base of the primary cilium before reaching more homogeneous membrane localization comparable to mouse RPE. Based on functional assessment, the L‐type Ca2+ channels participated in the regulation of vascular endothelial growth factor secretion as well as in the phagocytosis of photoreceptor outer segments in hESC‐RPE. Overall, this study demonstrates that a functional machinery of voltage‐gated Ca2+ channels is present in mature hESC‐RPE, which is promising for the success of transplantation therapies. stem cells translational medicine 2019;8:179&15
Keywords: Retinal pigment epithelium, Voltage‐gated Ca2+ channels, Stem cells, Patch‐clamp, Vascular endothelial growth factor, Phagocytosis
Significance Statement.
Human stem cells provide a promising cell source for the replacement of diseased retinal pigment epithelium (RPE) in the eye, and several clinical trials with cell transplantations are ongoing. The success of these therapies is largely dependent on the correct functionality of the transplanted cells. Still, cellular ion channels, vital for the proper RPE physiology, are inadequately characterized in stem cell‐derived RPE. The results of this study demonstrate the presence and functionality of voltage‐gated Ca2+ channels in mature human embryonic stem cell‐derived RPE similar to native RPE, and provide insight into their physiological relevance. This work is a significant contribution toward a more detailed functionality confirmation of stem cell‐derived RPE.
Introduction
Retinal pigment epithelium (RPE) is a monolayer of polarized cells located in the back of the eye between the photoreceptors and the choroid, and forms a part of the blood‐retinal‐barrier 1. As a barrier, RPE regulates the transport of nutrients and ions between the bloodstream and the subretinal space. In addition, RPE performs essential functions for vision such as phagocytosis, secretion, visual cycle, and light absorption (reviewed in 2). RPE also plays a critical role in the pathogenesis of several degenerative eye diseases such as age‐related macular degeneration (AMD) 3 that is the leading cause of vision loss and blindness among the elderly worldwide 4. Stem cells provide potential for the development of transplantation therapies producing a limitless source of RPE cells for the treatment of AMD and other RPE‐originated retinal dystrophies 5. Remarkably, such therapies are already being subjected to clinical trials for AMD and Stargardt's macular dystrophy 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21 as well as to several preclinical trials 5, 22, 23, 24, 25, 26, 27, 28. Stem cell‐derived RPE has been demonstrated to resemble native tissue in many respects: it has been shown to have a proteome closely similar to the native counterpart 29, phagocytose photoreceptor outer segment (POS) fragments 27, 28, 30, 31, 32, secrete vascular endothelial growth factor (VEGF) 32, 33, 34, and participate in the functional visual cycle 35, 36. However, much is still not understood about the genetic characteristics of stem cell‐derived RPE 37 or its behavior after transplantation 38. Furthermore, there is only limited information about the functionality of ion channels 33 and Ca2+ signaling 31, 39, 40 in stem cell‐derived RPE. In particular, studies about the voltage‐gated Ca2+ (CaV) channels in these cells are lacking.
The correct operation of CaV channels is required in order for the stem cell‐derived RPE to perform its critical functions in therapeutic use, since many of the important RPE functions are related to changes in intracellular Ca2+ concentration 2. L‐type Ca2+ channels have been identified in cultured and native RPE 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, where they participate in the transport of ions and water 42 as well as the regulation of POS phagocytosis 41, VEGF secretion 43, and RPE differentiation 2. On the other hand, the malfunctioning of L‐type Ca2+ channels in RPE has been linked to the pathogenesis of certain degenerative eye diseases 45, 56. Of the L‐type Ca2+ channels, RPE has been shown to express the subtypes CaV1.1–1.3 46 with several studies suggesting that subtype CaV1.3 is the primary contributor to RPE physiology 41, 43, 46, 50, 51, 52, 53, 54, 55. Of the T‐type Ca2+ channels, RPE has been reported to express the subtypes CaV3.1 and CaV3.3, and it has been speculated that these channels participate in the regulation of VEGF secretion 46. To date, α subtypes of the third subfamily CaV2.x have not been detected in RPE 46. It is, however, unclear whether this impressive machinery of Ca2+ channels is present in stem cell‐derived RPE, and raises a question about the resemblance of human embryonic stem cell (hESC)‐derived RPE to native RPE.
To address this issue, we investigated the functionality and localization profile of CaV channels in hESC‐RPE. Here, we present our patch‐clamp recordings that reveal slowly inactivating L‐type currents in hESC‐RPE that are similar to native RPE. In some hESC‐RPE cells, fast transient currents that resemble T‐type currents were also recorded. When compared with mouse tissue, there were similarities, as well as certain differences, in the localization of CaV channels in hESC‐RPE. With regard to physiology, we show that L‐type Ca2+ channels participate in POS phagocytosis and the regulation of VEGF secretion in hESC‐RPE. Overall, our results suggest that a functional machinery of voltage‐gated Ca2+ channels is present in hESC‐RPE, and thus strengthen the potential of stem cell‐derived RPE in transplantation therapies.
Materials and Methods
Culture of hESC‐RPE
In this study, we used the previously derived hESC lines Regea08/023, Regea08/017, and Regea11/013 57. The undifferentiated hESCs were maintained, cultured, and spontaneously differentiated as described before 58. After approximately 72–124 days of differentiation in the suspension culture, the pigmented areas of the floating aggregates were manually separated. The pigmented cell clusters were dissociated with TrypLE Select (Invitrogen, UK) and seeded onto Collagen IV (5 μg/cm2, Sigma‐Aldrich, St. Louis, MO) coated 24‐well cell culture plates (Corning CellBIND; Corning, Inc., Corning, NY) with a density of 5.5 × 105 cells/cm2. The cells were cultured for approximately 22–73 days, and cells from several independent differentiation batches were used for the study.
The cells were passaged with a density of 2.5 × 105 cells/cm2 onto polyethylene terephthalate coated hanging culture inserts (pore size 1 μm, Merck Millipore) treated with Collagen IV (10 μg/cm2, Sigma‐Aldrich) or with Collagen IV and laminin (1.8 μg/cm2, LN521, Biolamina, Sweden). The cultures became confluent in 5 days on inserts, after which they were further cultured until mature monolayers were obtained (days post‐confluence presented in each figure legend). For single cell patch‐clamp experiments, the cells were detached from the inserts with TrypLE Select and let to adhere on cover slips treated with poly‐l‐lysine (Sigma‐Aldrich).
Isolation of Mouse RPE
We used C57BL/6 mice at the age of 8–12 weeks where the development and maturation of RPE had been completed 59. The mice were euthanized by CO2 inhalation and cervical dislocation. The eyes were then enucleated and bisected along the equator. The eyecups were sectioned in Ames' solution (Sigma‐Aldrich) with 10 mM HEPES and pH adjusted to 7.4, and the retina was gently removed leaving the RPE firmly attached to the eyecup. To isolate the RPE cells for patch‐clamp recordings, the eyecup was incubated at 37°C in 5% CO2 either in TrypLE Select for 15 minutes or in a solution containing (in mM) 135 TeaCl, 5 KCl, 10 HEPES, 3 EDTA‐KOH, 10 glucose, and 25 U/ml activated papain (Sigma‐Aldrich) for 30 minutes. After this, the eyecups were washed in the HEPES buffered Ames' solution supplemented with 1% bovine serum albumin (BSA; Sigma‐Aldrich). The RPE was collected by gentle trituration, stored at 37°C in 5% CO2 in the RPE culture medium and measured within 6 hours.
Ethical Issues
Approval for research with human embryos was given by the National Authority for Medicolegal Affairs, Finland (Dnro 1426/32/300/05). A supportive statement was received from the Local Ethics Committee of the Pirkanmaa Hospital District, Finland to derive and expand hESC lines from surplus embryos, and to use these cell lines for research purposes (R05116). No new cell lines were derived in this study. The procedures carried out with C57BL/6 mice were in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and the Finnish Animal Welfare Act 1986.
Patch‐Clamp Recordings
Patch‐clamp recordings were performed at room temperature (RT) on single hESC‐RPE and mouse RPE cells. Ionic currents were recorded using the standard patch‐clamp technique in whole‐cell configuration. To minimize potassium currents, patch pipettes (resistance 4–8 MΩ) were filled with a cesium based internal solution containing (in mM) 83 CsCH3SO3, 25 CsCl, 5.5 EGTA, 0.5 CaCl2, 4 ATP‐Mg, 0.1 GTP‐Na, 10 HEPES, and 5 NaCl; pH was adjusted to ~7.2 with CsOH and osmolarity was adjusted to ~290 mOsm with sucrose. The internal solution contained 2 mM lidocaine N‐ethyl chloride (Sigma‐Aldrich) to exclude the possibility of the measured fast transient currents being carried by sodium 60. The tissue was perfused with a control external solution containing (in mM): 120 NaCl, 5 TeaCl, 1.1 CaCl2, 1.2 MgCl2, 10 HEPES, 5 glucose, and 10 BaCl2. pH was adjusted to 7.4 with NaOH and the osmolarity was set to ~305 mOsm with sucrose. In some experiments, the BaCl2 concentration was decreased to 1 mM, and this was compensated by increasing the NaCl concentration to 130 mM. In the experiments that used Ca2+ channel modulators, the control bath solution contained L‐type Ca2+ channel activator 10 μM (‐)BayK8644 (Sigma‐Aldrich) or L‐type Ca2+ channel inhibitor 10 μM nifedipine (Sigma‐Aldrich). The recordings were made in voltage‐clamp mode using the Axopatch200B patch‐clamp amplifier connected to an acquisition computer via AD/DA Digidata1440 (Molecular Devices, CA). Potentials were corrected for a 10 mV liquid junction potential during data analysis. Access resistance was <25 MΩ and membrane resistance was >300 MΩ. The membrane capacitance was 33 ± 5 pF (mean ± SEM, n = 9) for hESC‐RPE cells and 23 ± 3 pF (mean ± SEM, n = 3) for mouse RPE cells. The depletion of the currents in hESC‐RPE cells in whole‐cell configuration was −11 ± 3% during 19 ± 5 minutes (mean ± SEM, n = 3) measured using a 50 ms voltage step from −100 to 10 mV. The measurements lasted for a shorter time than that of depletion. Current–voltage (IV)‐curves were obtained from the peak value of the current at given voltages. Conductance (G) was calculated as G = I/(V–V R), where V R is the reversal potential.
Indirect Immunofluorescence Staining
For immunofluorescence staining, hESC‐RPE monolayers and mouse RPE eyecups were fixed for 15 minutes with 4% paraformaldehyde. The hESC‐RPE monolayers and mouse RPE eyecup whole mount preparations were permeabilized by 15 minutes incubation in 0.1% Triton X‐100 (Sigma‐Aldrich) at RT. This was followed by incubation with 3% BSA in phosphate‐buffered saline (PBS) (Sigma‐Aldrich) at RT for 1 hour. Primary antibodies for CaV1.1, CaV1.2, CaV1.3, CaV3.1, CaV3.2, CaV3.3 (1:100; Alomone Labs, Jerusalem, Israel), cellular retinaldehyde‐binding protein (CRALBP; 1:500; Abcam, UK), zonula occludens (ZO‐1; 1:50; Life Technologies), claudin‐3 (1:80; Thermo Fisher Scientific), ezrin (1:100; Abcam, UK), acetylated α‐tubulin (1:1,000; Sigma‐Aldrich), and pericentrin (PCNT; 1:200; Abcam, UK) were diluted in 3% BSA‐PBS and incubated for 1 hour at RT. The samples were then washed four times with PBS, followed by 1 hour incubation at RT with the secondary antibodies donkey anti‐rabbit or anti‐mouse Alexa Fluor 488 and donkey anti‐rabbit or anti‐mouse Alexa Fluor 568 (1:200; Life Technologies) as well as goat anti‐rabbit or anti‐mouse Alexa Fluor 488 and goat anti‐mouse Alexa Fluor 568 (1:200; Thermo Fisher Scientific) diluted in 3% BSA‐PBS. Phalloidin was visualized using Phalloidin‐Atto 633 (1:100; Sigma‐Aldrich), an Alexa Fluor 568 conjugate (1:400; Sigma‐Aldrich) or an Alexa Fluor 647 conjugate (1:50; Life Technologies). The washes with PBS were repeated and the nuclei were stained with the 4′,6‐diamidino‐2‐phenylidole included in the mounting medium (Life Technologies).
For paraffin embedded vertical sections, the hESC‐RPE monolayers and mouse eyecups with retina attached were infused in paraffin blocks and cut into 7 μm vertical sections with a Leica SM2000 R or Leica SM2010 R sliding microtome (Leica Biosystems). The sections were then attached on glass coverslides by 1 hour incubation at 60°C. The samples were deparaffinized and hydrolyzed using xylene and ethanol series. Antigen retrieval was carried out by microwaving the samples in 10 mM sodium citrate in 0.05% Tween20 (Sigma Aldrich). The samples were blocked using 10% donkey serum and 5% BSA in tris‐buffered saline (TBS) for 1 hour at 37°C. After this, they were washed twice in 0.02% Tween20‐TBS. The CaV primary antibodies listed above, as well as Na+/K+‐ATPase (1:200; Abcam) and Bestrophin‐1 (1:500; Lagen laboratories) were diluted in 1% BSA‐TBS and incubated overnight at 4°C. The samples were then washed twice with 0.02% Tween20‐TBS. The secondary antibodies introduced above were diluted with 1% BSA‐TBS and incubated for 1 hour at RT, followed by two washes and mounting as described above.
Confocal Microscopy and Image Processing
Confocal microscopy was performed with a Zeiss LSM780 or LSM700 laser scanning confocal microscope (LSCM) on an inverted Zeiss Cell Observer microscope (Zeiss, Jena, Germany) and Plan‐Apochromat ×63/1.4 oil immersion objective. Voxel size was set to x = y = 66 nm and z = 100–200 nm and image size to 512 × 512 or 1,024 × 1,024 pixels. Reflection imaging was conducted by collecting light from the 488 nm laser line by using 20/80 dichroic beam splitter and 480–492 nm emission window at the photomultiplier tube detector. The images were saved in czi‐format and processed with ImageJ 61, adjusting only brightness and contrast, and panels were assembled using Adobe Photoshop CS6 (Adobe Systems, San Jose).
Pulse‐Chase Phagocytosis Assay
Mature hESC‐RPE monolayers on culture inserts were pre‐incubated for 24 hours at 37°C in the control medium or in the presence of the L‐type CaV modulators 10 μM (‐)BayK8644, or 10 μM nifedipine, or T‐type CaV inhibitor 5 μM ML218 (Sigma‐Aldrich). For phagocytosis assay, POS fragments were isolated and purified from fresh porcine eyes obtained from a local slaughterhouse as described before 58, 62. The POS particles were suspended to 10% fetal bovine serum (FBS) containing medium in control or in one of the drug containing conditions. In the pulse stage, equal amounts of POS containing media were added on the apical sides of the hESC‐RPE inserts and incubated for 30 minutes at 37°C. For the chase stage, the media were changed back to 10% FBS medium with or without the drugs, and the hESC‐RPE inserts were further incubated for 2 hours at 37°C. After this, the samples were fixed and stained as described above using the primary antibodies opsin (1:200; Sigma Aldrich) and ZO‐1. The samples were imaged using the Zeiss LSM780 LSCM as described above but by imaging large random fields. The number of bound and internalized POS particles that were larger than 1 μm in diameter, were counted from maximum intensity projection images after performing Gaussian blur using ImageJ. The assay was performed with three inserts in each condition and data from 5 to 6 images from each of the three inserts was pooled together resulting in n = 15–16.
Enzyme‐Linked Immunosorbent Assay for VEGF Secretion
Secretion of VEGF by mature hESC‐RPE was assessed with a commercially available human VEGF Quantikine enzyme‐linked immunosorbent assay (ELISA) kit (R&D Systems, MN) according to the manufacturer's instructions. Briefly, the polarized VEGF secretion in control conditions was studied by collecting medium samples separately from the apical and basolateral sides of the insert after 24 hours incubation with three replicates. To test the effect of CaV channel modulators on VEGF secretion, we measured the total VEGF concentration secreted through both apical and basolateral cell membranes. The inserts were incubated 24 hours in different pharmacological conditions: in 1% FBS medium in control conditions, or in this medium together with 10 μM (‐)BayK8644, 10 μM nifedipine or 5 μM ML218 with eight to nine replicates. The VEGF concentration from the collected medium samples was normalized to the number of cells based on cell counting under the Zeiss LSM780 LSCM using ×20 or ×63 objective.
Statistical Analysis
The data is stated as mean ± SEM (n, p), where n refers to the number of samples used to generate the data set and p refers to statistical significance. The data was tested for normality using the Shapiro–Wilk normality test. Some of the data sets did not meet the normality criteria. Thus, a pair‐wise comparison of the test conditions to control condition was conducted using non‐parametric Mann‐Whitney U test to confirm the possible statistical significance between the experimental conditions.
Results
Currents Through Voltage‐Gated Ca2+ Channels in hESC‐RPE
In control conditions with 10 mM extracellular Ba2+, whole‐cell voltage clamp recordings revealed voltage‐gated currents in single hESC‐RPE cells (Fig. 1A) dissociated from a mature RPE monolayer (Fig. 1B). In response to a 50 ms voltage pulse from −80 to 60 mV in 10 mV steps, nine cells showed slowly or non‐inactivating currents (Fig. 1C). Based on the normalized and averaged IV‐curve (n = 9), the current activated at low potentials reaching maximum at 10 mV (Fig. 1D). The normalized and averaged GV‐curve showed half maximum conductance at −7 ± 3 mV (n = 9) (Fig. 1E). Typical to L‐type Ca2+ channels 47, 48, diminishing the Ba2+ content from 10 to 1 mM decreased the maximum current density from 2.4 ± 0.5 pA·pF−1 (n = 9) to 1.3 ± 0.3 pA·pF−1 (n = 7) (Fig. 1F). In addition, three cells showed fast transient currents (Fig. 1G) with inactivation time constant 6 ± 1 ms (n = 3). The current pattern indicated that hESC‐RPE is likely to express both slowly inactivating L‐type currents and fast inactivating T‐type resembling currents. However, a detailed characterization of the fast inactivating currents was not possible as will be discussed later.
Figure 1.
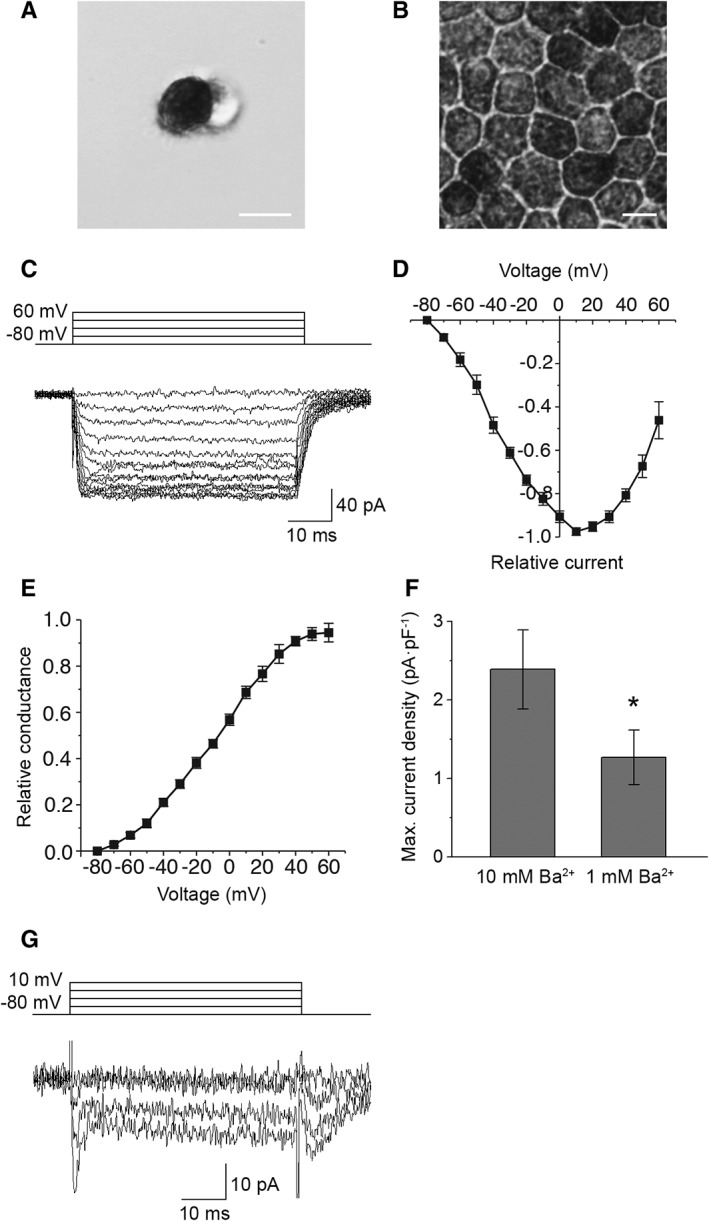
Voltage‐gated currents in hESC‐RPE. Examples of bright‐field microscopy images of (A) a single hESC‐RPE cell showing pigmented apical and non‐pigmented basal sides and (B) a mature hESC‐RPE monolayer with representative RPE morphology, scale bars 10 μm. Whole‐cell voltage clamp recordings were carried out from single hESC‐RPE cells. (C): A typical example of the slowly inactivating current elicited by 50 ms voltage steps from −80 to +60 mV in 10 mV increments. Normalized and averaged (D) IV‐curve and (E) GV‐curve of the slowly inactivating current (mean ± SEM, n = 9, cell lines 08/017 and 08/023, days post‐confluence 73–128). (F): Averaged maximum current densities (obtained at 10 mV) of the slowly inactivating current in 10 mM Ba2+ (n = 9) and in 1 mM Ba2+ (n = 7, cell lines 08/017 and 08/023, days post‐confluence 109–127). The difference in the current densities was statistically significant. (G): A typical example of the fast inactivating current elicited by 50 ms voltage steps from −80 to +10 mV in 10 mV increments (cell line 08/023, days post‐confluence 109). *Statistically significant difference with p < .05.
The Effects of L‐Type Ca2+ Channel Activator and Inhibitor
The effects of (‐)BayK8644 and nifedipine, well‐characterized activator and inhibitor of the L‐type Ca2+ channels, were tested for the slowly inactivating currents. These currents were increased by 10 μM (‐)BayK8644 (Fig. 2A, 2C) and decreased by 10 μM nifedipine (Fig. 2B, 2D). Comparison with the control current at maximum amplitude revealed that the slowly inactivating current increased after (‐)BayK8644 application by 80 ± 9% (n = 3, p < .05) (Fig. 2E) and decreased after nifedipine application by 56 ± 5% (n = 4, p < .05) (Fig. 2F). Both effects were statistically significant. These recordings confirm that the slowly inactivating currents were carried by the L‐type Ca2+ channels.
Figure 2.
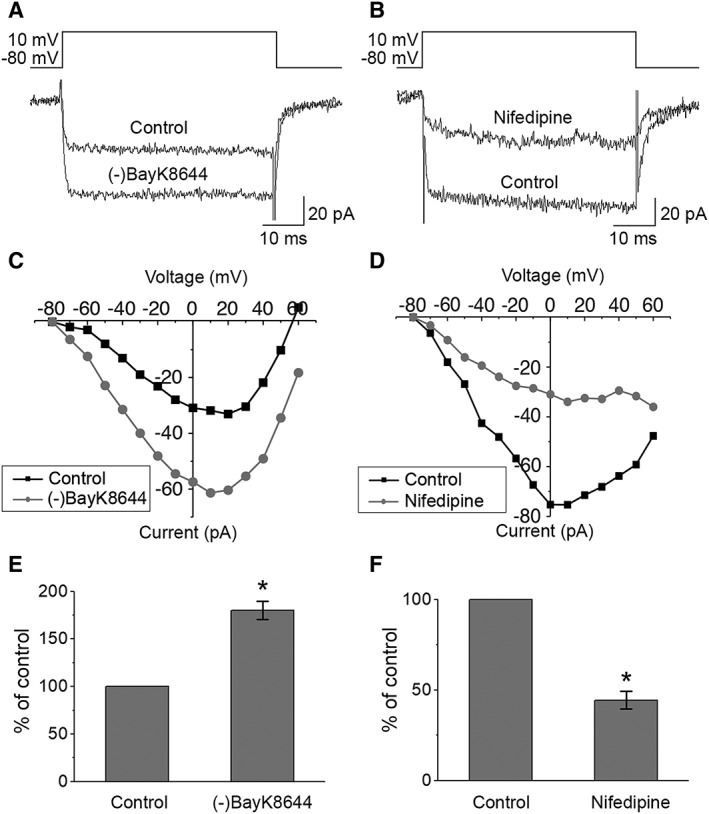
Responses of the currents to Ca2+ channel modulators. Whole‐cell measurements of currents as responses to voltage pulses from −80 to +60 mV in 10 mV increments for 50 ms duration were performed before and after the application of the specific drugs. Examples of the effects of (A) L‐type Ca2+ channel activator 10 μM (‐)BayK8644 and (B) L‐type Ca2+ channel inhibitor 10 μM nifedipine on Ba2+ currents in hESC‐RPE and (C, D) the corresponding IV‐curves, respectively. Changes in maximum current amplitudes presented as percentages from control conditions (mean ± SEM) show that both (E) activation with (‐)BayK8644 (n = 3, cell line 08/017, days post‐confluence 73–74) and (F) inhibition with nifedipine (n = 4, cell line 08/017, days post‐confluence 73–99) resulted in statistically significant changes in the recorded currents. *Statistically significant difference with p < .05.
Localization of Voltage‐Gated Ca2+ Channels in hESC‐RPE
To evaluate the localization of the CaV channels detected in the patch‐clamp measurements in hESC‐RPE, we performed antibody labeling against the L‐type Ca2+ channels CaV1.1‐CaV1.3 and the T‐type Ca2+ channels CaV3.1‐CaV3.3, together with markers for actin cytoskeleton, RPE maturity, and polarization. The hESC‐RPE showed a typical expression of CRALBP (Fig. 3A) and Na+/K+‐ATPase (Fig. 3F) on the apical side of the monolayer, as well as Bestrophin‐1 primarily on the basolateral side (Fig. 3F). Zonula occludens (ZO‐1) (Fig. 3B) and claudin‐3 (Fig. 3C) co‐localized on the cell–cell junctions with the circumferential bands of actin (phalloidin), characteristic to mature RPE 63. This data, together with the TER value of over 200 Ω cm2, strong pigmentation and cobblestone morphology (see Fig. 1B), indicate the maturity and polarization of our hESC‐RPE.
Figure 3.
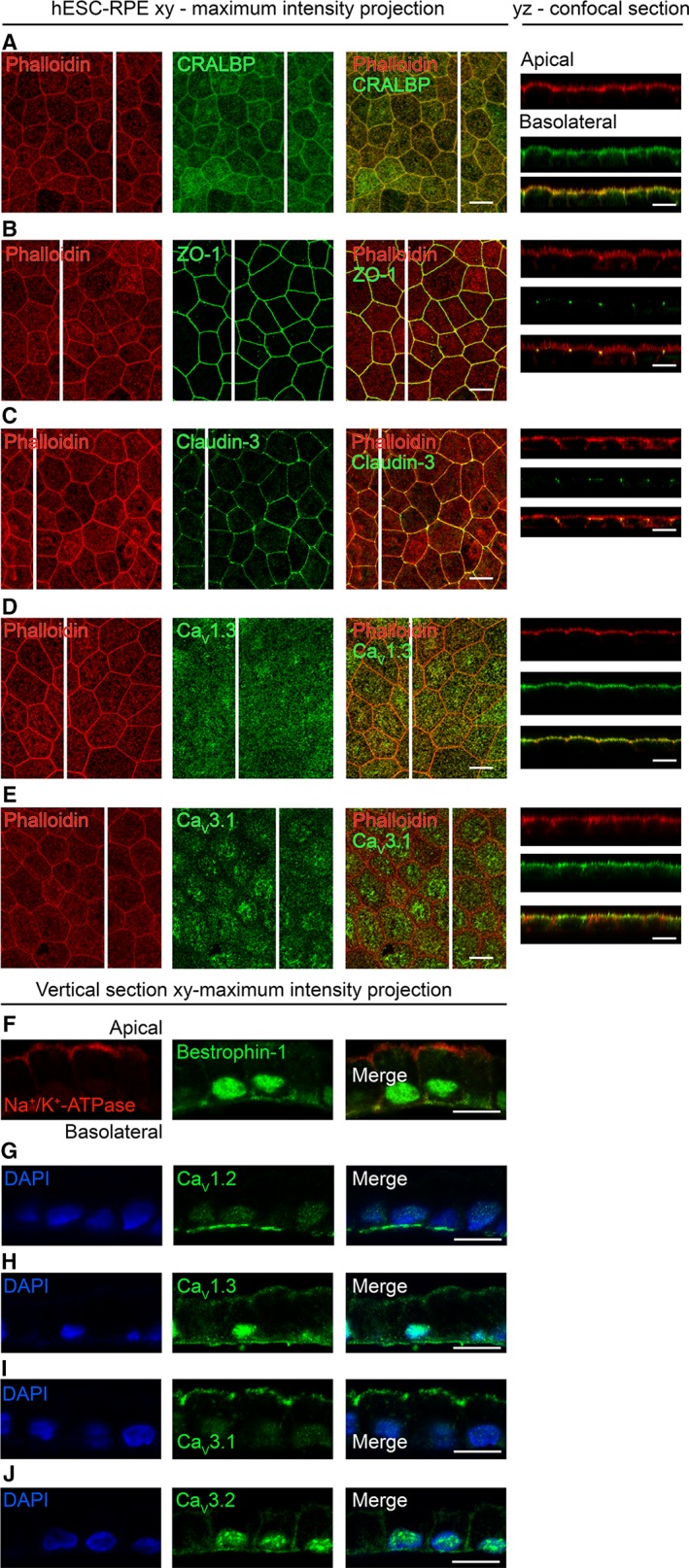
Localization of CaV channels in hESC‐RPE. Immunostainings of RPE monolayers with xy‐maximum intensity projections and yz‐confocal sections (apical side upwards, localization of the section highlighted with a white bar). Actin cytoskeleton (phalloidin, red) labeled together with (A) RPE marker CRALBP (green, cell line 08/017, days post‐confluence 91), (B) tight junction markers ZO‐1, (green, cell line 08/017, days post‐confluence 74) and (C) claudin‐3 (green, cell line 08/017, days post‐confluence 91), (D) L‐type Ca2+ channel CaV1.3 (green, cell line 08/017, days post‐confluence 109), and (E) T‐type Ca2+ channel CaV3.1 (green, cell line 08/023, days post‐confluence 66). Immunostainings of paraffin embedded hESC‐RPE vertical sections with xy‐maximum intensity projections (apical side upwards). (F): Cell polarization markers Na+/K+‐ATPase (red) and Bestrophin‐1 (green, cell line 08/023, days post‐confluence 91). Cell nuclei (DAPI, blue) together with L‐type Ca2+ channels (G) CaV1.2 (green, cell line 08/017, days post‐confluence 84) and (H) CaV1.3 (green, cell line 08/023, days post‐confluence 91), and T‐type Ca2+ channels (I) CaV3.1 (green, cell line 08/017, days post‐confluence 84) and (J) CaV3.2 (green, cell line 08/017, days post‐confluence 84). Scale bars 10 μm. Abbreviations: CaV, voltage‐gated Ca2+ channel; CRALBP, cellular retinaldehyde‐binding protein; ZO‐1, Zonula occludens; DAPI, 4′,6‐diamidino‐2‐phenylidole; hESC, human embryonic stem cell; RPE, retinal pigment epithelium.
The most prominent staining in hESC‐RPE monolayers was detected for the subtypes CaV1.3 and CaV3.1, both localizing strongly at the apical membrane (Fig. 3D, 3E). Staining of these subtypes together with RPE microvilli marker ezrin revealed the localization of CaV1.3 right below the microvilli (Supporting Information Fig. S1A) and CaV3.1 at the microvilli (Supporting Information Fig. S1B). Since pigmentation hinders the visualization of the basolateral side (see yz confocal sections in Fig. 3), we performed immunostainings on paraffin embedded vertical sections of the hESC‐RPE. This confirmed the apical localization of the subtypes CaV1.3 (Fig. 3H) and CaV3.1 (Fig. 3I) and revealed a pronounced basolateral localization of CaV1.3 (Fig. 3H). Furthermore, in hESC‐RPE, we observed basolateral localization of CaV1.2 (Fig. 3G), and basolateral and junctional localization of CaV3.2 (Fig. 3J). The CaV1.1 and CaV3.3 subtypes were not detected (data not shown).
Voltage‐Gated Ca2+ Channels in Mouse RPE
To compare the currents through voltage‐gated Ca2+ channels in hESC‐RPE with native tissue, patch‐clamp recordings were performed from the cells of freshly isolated mouse RPE (Fig. 4). An investigation of currents in whole‐cell configuration as a response to series of depolarizing voltage steps from −80 to +60 mV revealed slowly inactivating currents in the recordings (Fig. 4A). The currents activated at low potentials reaching the maximum at 20 mV in the normalized and averaged IV‐curve (n = 4) (Fig. 4B). The half maximum conductance was reached at −6 ± 3 mV (n = 4) based on the normalized and averaged GV‐curve (Fig. 4C). The maximum current density of the slowly inactivating current was 2.3 ± 0.6 pA·pF−1 (n = 4). Thus, the current characteristics of the voltage‐gated Ca2+ channels in mouse RPE were comparable to those we identified in hESC‐RPE.
Figure 4.
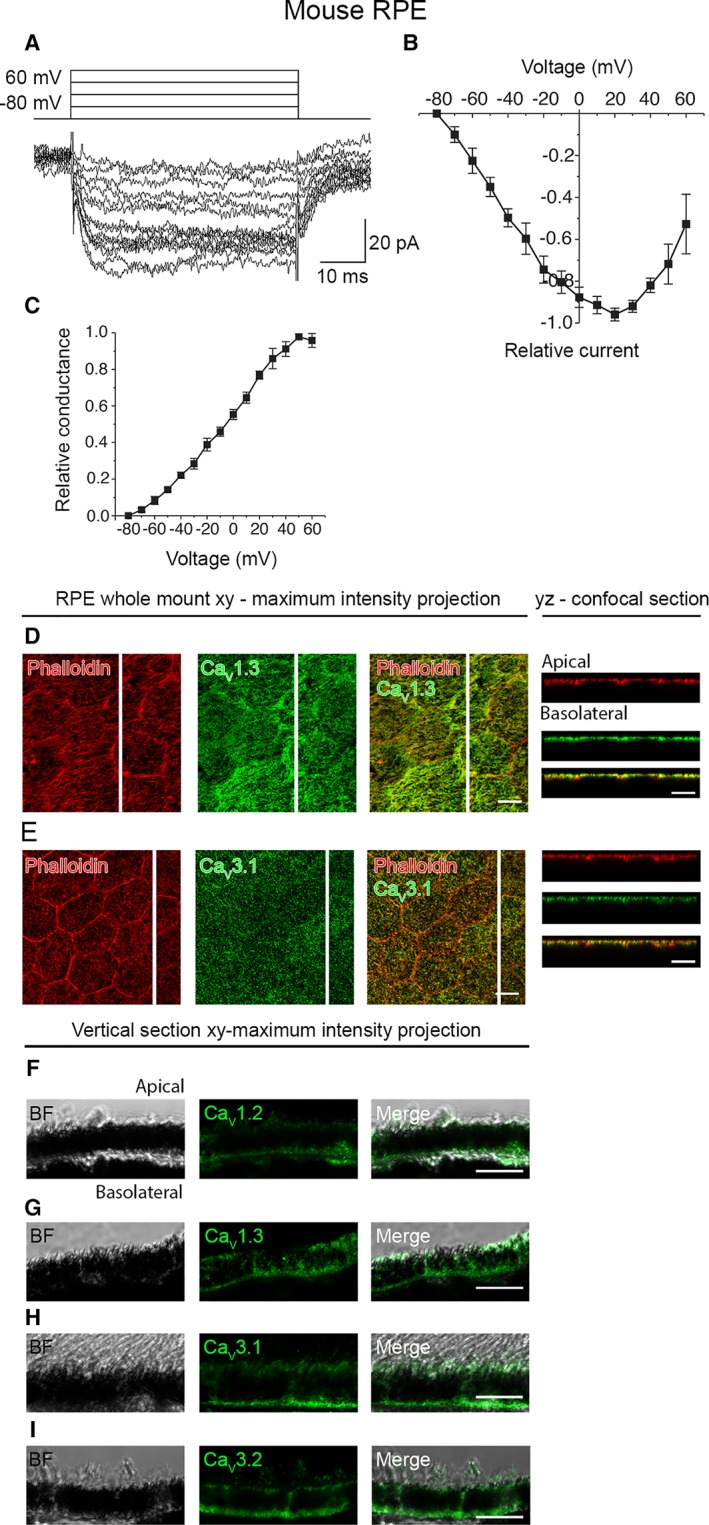
CaV channels in mouse RPE. (A): An example of the slowly inactivating L‐type current measured in whole‐cell configuration and elicited by 50 ms voltage steps from −80 to +60 mV in 10 mV increments. (B): Normalized and averaged IV‐curve of the L‐type current (mean ± SEM, n = 4). (C): Normalized and averaged GV‐curve of the L‐type current (mean ± SEM, n = 4). Localization of the CaV channels assessed by immunostainings of mouse RPE‐eyecup whole mount preparations. Confocal images show the xy‐maximum intensity projections and yz‐confocal sections of the samples (apical side upwards, localization of the section highlighted with a white bar). Actin cytoskeleton (phalloidin, red) together with (D) L‐type Ca2+ channel CaV1.3 (green), and (E) T‐type Ca2+ channel CaV3.1 (green). Immunostainings of paraffin embedded vertical sections of mouse eyecups shown as xy‐maximum intensity projections (apical side upwards). BF images together with L‐type Ca2+ channels (F) CaV1.2 (green) and (G) CaV1.3 (green), and T‐type Ca2+ channels (H) CaV3.1 (green) and (I) CaV3.2 (green). Scale bars 10 μm. Abbreviations: CaV, voltage‐gated Ca2+ channel; BF, bright‐field; RPE, retinal pigment epithelium.
Antibody labeling, similar to hESC‐RPE, was performed on mouse RPE‐eyecup whole mount preparations (Fig. 4D, 4E, Supporting Information Fig. S1C, S1D) and vertical sections of paraffin embedded eyecups (Fig. 4F–4I). The channel localization in mouse RPE followed similar characteristics as in hESC‐RPE with the exception that the apically localized CaV3.1 was also detected at the basolateral side in mouse RPE (Fig. 4H). Furthermore, it is worth pointing out, that the uniform apical staining profile of CaV1.3 observed in hESC‐RPE (Fig. 3D, 3H, Supporting Information Fig. S1A) was especially strongly detected in mouse RPE (Fig. 4D, 4G, Supporting Information Fig. S1C).
VEGF Secretion in hESC‐RPE
In hESC‐RPE, characteristic to RPE physiology, VEGF secretion was polarized. Consistent with this, we found that the amount of VEGF secreted after a 24 hour incubation in control conditions was 588 ± 37 pg/105 cells to the apical side and 1,290 ±38 pg/105 cells to the basolateral side (n = 3). Since the L‐type Ca2+ channels have been reported to play an important role in VEGF secretion 43, we investigated the effect of their pharmacological modulation on the total amount of secreted VEGF in hESC‐RPE. We followed the apical and basal secretion concurrently (Fig. 5) thus addressing the role of both apically and basally localized Ca2+ channels in the overall secretion. In control conditions, the total VEGF concentration in the medium after the 24 hour incubation was 1,950 ± 70 pg/105 cells (n = 9). Manipulation of the L‐type Ca2+ channel activity directly affected the VEGF secretion as the activator (‐)BayK8644 increased the secretion by 24 ± 9% (n = 9, p < .05) and the inhibitor nifedipine decreased the secretion by 19 ± 9% (n = 8, p < .05). Both effects were statistically significant. However, inhibition of the T‐type channels by ML218 had little effect on the VEGF secretion (8 ± 14% increase, n = 8, p > .05).
Figure 5.
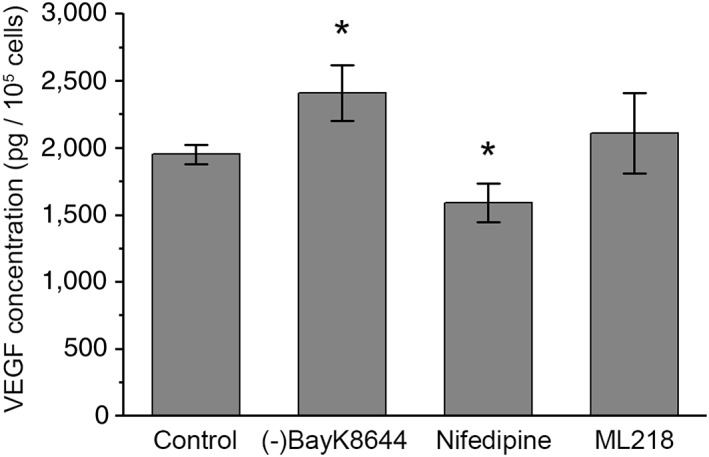
VEGF secretion from hESC‐RPE. Total concentrations of VEGF secreted by the hESC‐RPE after 24‐hour incubation in control medium alone (n = 9) as well as in control medium with L‐type Ca2+ channel activator 10 μM (‐)BayK8644 (n = 9), L‐type Ca2+ channel inhibitor 10 μM nifedipine (n = 8), or T‐type channel inhibitor 5 μM ML218 (n = 8) (mean ± SEM, cell lines 08/023 and 11/013, days post‐confluence 66–147). *Statistically significant difference with p < .05. Abbreviation: VEGF, vascular endothelial growth factor.
Voltage‐Gated Ca2+ Channels Regulate POS Phagocytosis in hESC‐RPE
Previous studies indicate that L‐type Ca2+ channels participate in the regulation of phagocytosis in RPE 41, 64. Thus, we investigated the role of CaV channels in POS phagocytosis in hESC‐RPE by pharmacologically modulating these channels during our phagocytosis assay. These experiments and subsequent labeling with opsin and ZO‐1 showed a reduction in the total number of bound and internalized POS particles in the presence of either L‐type channel activator or inhibitor, but an increase in the particle number in the presence of T‐type channel inhibitor (Fig. 6). More specifically, the median value of POS particles in a randomly taken confocal image field decreased from the control conditions (n = 16, Fig. 6A, 6E) by 30% when the L‐type channels were activated by (‐)BayK8644 (n = 16, p < .001, Fig. 6B, 6E). A higher decrease of 62% occurred when the L‐type Ca2+ channels were inhibited by nifedipine (n = 15, p < .001, Fig. 6C, 6E). Interestingly, we found that T‐type Ca2+ channel inhibitor ML218 (Fig. 6D, 6E) increased the number of POS particles by 32% (n = 16, p < .05). All the effects were statistically significant.
Figure 6.
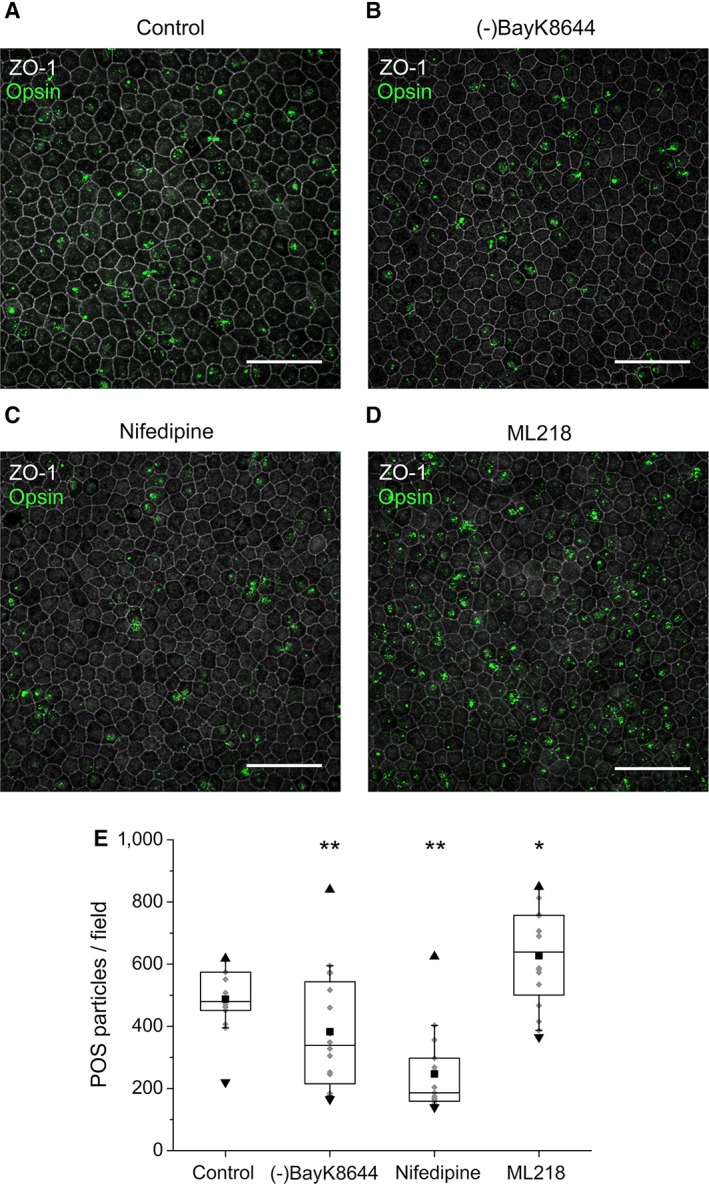
The effect of CaV channel modulators on POS phagocytosis in hESC‐RPE. Mature hESC‐RPE monolayers were incubated with purified porcine POSs in the pulse‐chase phagocytosis assay. Xy‐maximun intensity projections of the confocal images show both bound and internalized POS particles that were stained with opsin (green) together with the tight junction protein ZO‐1 (gray) in (A) control conditions, and in the presence of CaV channel modulators (B) (‐)BayK8644, (C) nifedipine, or (D) ML218. Scale bars 50 μm. (E): Quantification of POS particles in control conditions yielded the median value of 485 POS particles/field (n = 16). When modulating the CaV channels pharmacologically, the value changed in the presence of (‐)BayK8644 to 339 POS particles/field (n = 15), nifedipine to 186 POS particles/field (n = 15), and ML218 to 639 POS particles/field (n = 16). The box limits 25%–75% of the gray data points; the whiskers include 10%–90% of the data; the center line shows the median value; the black square describes the mean; the black triangles present the minimum and the maximum values. Cell line 11/013, days post‐confluence 147. Statistically significant differences with *p < .05 or **p < .001. Abbreviations: POS, photoreceptor outer segment; ZO‐1, Zonula occludens.
Localization of CaV1.3 During hESC‐RPE Maturation
It is well established that protein expression and localization change during RPE maturation 65. We addressed this considering the localization of the primary CaV channel subtype, CaV1.3, during hESC‐RPE maturation. We immunolabeled CaV1.3 together with pericentrin (PCNT), a protein localized in the centrosomes at the base of the primary cilia, that have been recently shown to be important for RPE maturation 66, 67. Figure 7 shows how the localization of these proteins changed remarkably during maturation. Fusiform hESC‐RPE cells on the first day post‐confluence (Supporting Information Fig. S2A) expressed CaV1.3 throughout the cell (Fig. 7A), and PCNT appeared as distinct puncta on the apical side. After 6 days post‐confluence, the cells gained more epithelioid morphology (Supporting Information Fig. S2B) and CaV1.3 started to localize also to the apical and basal RPE cell membranes with brighter puncta forming on the apical side (Fig. 7B). Interestingly, these puncta showed co‐localization with PCNT. The hESC‐RPE cells obtained cobblestone morphology around 31 days post‐confluence (Supporting Information Fig. S2C), and from this time point onwards, CaV1.3 was present more strongly on the apical and basolateral cell membranes (Fig. 7C). The apical side puncta were pronounced and appeared as one distinct cluster per cell co‐localizing strongly with PCNT (Fig. 7C). Immunolabeling CaV1.3 with acetylated α‐tubulin (Fig. 7E) showed the localization of CaV1.3 near the base of the primary cilia. With increasing maturation, the apical staining of CaV1.3 became more prominent and more homogeneous, while basolateral staining started to be difficult to detect due to increased pigmentation. At 84 days post‐confluence (Fig. 7D), fairly uniform apical localization of CaV1.3 was present in hESC‐RPE, although the puncta, co‐localizing with PCNT, could still be distinguished. At this time point, PCNT localized near the apical centers of the cobblestone hESC‐RPE cells (Fig. 7D, Supporting Information Fig. S2D), characteristic to mature RPE 67. This data suggest that with increasing maturation, hESC‐RPE started to gain the homogeneous apical localization of CaV1.3 detected in mouse RPE (Fig. 4D).
Figure 7.
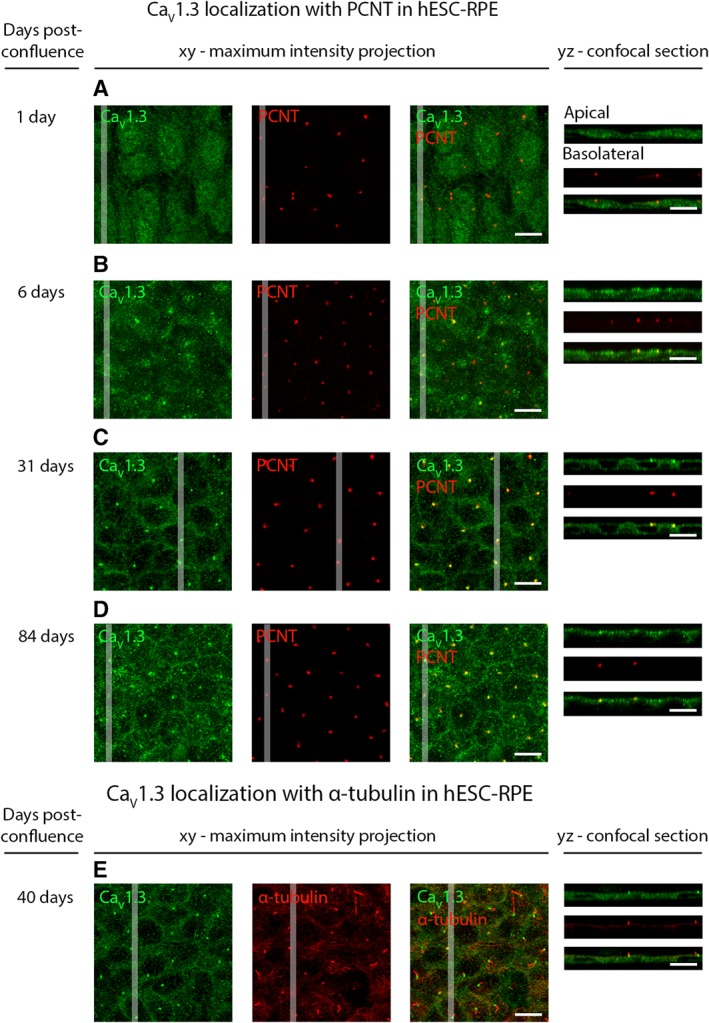
Localization of CaV1.3 during hESC‐RPE maturation. Immunolabeling of CaV1.3 (green) together with centrosome protein PCNT (red) from post‐confluence day 1 to post‐confluence day 84 at four time points: (A) day 1, (B) day 6, (C) day 31, and (D) day 84 (cell line 08/017). (E): Labeling acetylated α‐tubulin (red) together with CaV1.3 (green) shows the localization of CaV1.3 at the base of the primary cilia during maturation (cell line 08/017, days post‐confluence 40). The confocal images are shown as xy‐maximum intensity projections and yz‐confocal sections (apical side upwards, localization of the section highlighted with a white bar). Scale bars 10 μm. Abbreviations: CaV, voltage‐gated Ca2+ channel; hESC, human embryonic stem cell; RPE, retinal pigment epithelium; PCNT, pericentrin.
Discussion
Stem cell‐derived RPE provides great potential for novel cell transplantation therapies and research has already proceeded to clinical trials 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21. Essential for the success of these therapies, stem cell‐derived RPE has been shown to perform several key RPE functions 27, 28, 29, 30, 31, 32, 33, 34, 35, 36. However, the functionality of the ion channels and specifically the voltage‐gated Ca2+ channels in these cells remain poorly known, even though many of the critical RPE functions are related to Ca2+ activity. This raises the question whether the stem cell‐derived RPE destined for clinical purposes sufficiently resembles its native counterpart, and can thus replace the functions of lost cells. The present study addressed this issue by investigating the CaV channels in hESC‐RPE. Using patch‐clamp recordings and immunostainings, we showed the presence of functional L‐type Ca2+ channels in hESC‐RPE that are comparable to native mouse RPE.
In our study, two current types were detected, the slowly inactivating current and the fast inactivating current. We confirmed that the main current type, the slowly inactivating current, results from the activity of L‐type Ca2+ channels since the current responses and IV‐curves in this study resembled the previous recordings of L‐type currents from various types of native RPE 43, 46, 47, 48. Moreover, the sensitivity of the current to the L‐type Ca2+ channel activator (‐)BayK8644 45, 46, 47, 48, 49, 50, 53 and the inhibitor nifedipine 43, 44, 47, 48, 51 further indicated the presence of L‐type currents in our measurements. The recorded current is likely to be carried primarily through CaV1.3 channels. This conclusion is based on the voltage‐dependent activation of the currents at rather negative potentials 68, shape of the IV‐curve characteristic to the CaV1.3 subtype 43, 46, 55, and slow inactivation of the current. It is still likely that CaV1.2 channels contribute to the recorded current as well, since our immunostainings confirmed the presence of both of the L‐type Ca2+ channels, CaV1.2 and CaV1.3, in hESC‐RPE. To date, CaV1.3 subtype has been reported to only localize basolaterally in murine 54 and porcine 55 RPE. Our data showed that both hESC‐ and mouse RPE express the CaV1.3 subtype also on the apical cell membrane, in addition to the basolateral membrane.
It is worth noting that patch‐clamp recordings from primary RPE cultures show differences in L‐type current characteristics when compared with our recordings from hESC‐RPE 43, 45, 49, 50, 53, especially regarding the more negative activation threshold and the weaker slope of activation present in our study. The reason for this remains to be investigated, but it may be related to differences in phosphorylation, splicing variants, or the composition of the accessory subunits 69, 70. Yet, the contribution of other Ca2+ conducting channels on the currents recorded in this study for hESC‐RPE and native mouse RPE cannot be excluded. Several Ca2+‐ conducting channels, such as store‐operated Orai channels 71 and transient receptor potential (TRP) channels 41, 72, 73, 74, have important roles in the physiology of RPE. Relevant for this study, TRP channels are involved in the phagocytosis 41 and VEGF secretion 73. In addition to these other Ca2+‐conductivities, the effect of cell dissociation to patch‐clamp recordings needs to be taken into account. Cell–cell junctions break down in cell dissociation causing epithelial cells to lose their polarity, which compromises their normal ability to express and recycle proteins. This has a strong influence on the endocytotic processes that are important for the internalization of ion channels, regulating their numbers in the cell membrane 75. Therefore, after cell dissociation, ion channels can be re‐distributed to the intracellular compartments and those currents will thus be absent from the patch‐clamp recordings.
In addition to L‐type currents, our patch‐clamp recordings revealed the presence of fast transient currents in hESC‐RPE. The kinetics of these currents were comparable to those previously reported for the T‐type Ca2+ channels in cultured human RPE 46, although faster than typically reported for other cell types (reviewed in 76). Similar to the findings of the previous study 46, the fast transient currents were almost exclusively recorded in combination with the slowly inactivating current, which hindered their further analysis. In addition, TTX‐sensitive currents can also contribute to the fast transient conductance 60, 77 and make this current component extremely difficult to investigate. In immunostainings, we observed CaV3.1 and CaV3.2 in both hESC‐ and mouse RPE. CaV3.1 was localized apically at the microvilli in the both studied RPE cell types, while it was found also at the basolateral cell membrane in mouse RPE.
VEGF has a role in angiogenesis and vascular permeability, and therefore anti‐VEGF agents are commonly used in the treatment of AMD 78. In healthy RPE, VEGF secretion occurs in a polarized manner with significantly more pronounced secretion from the basal side 79, 80, as we showed here for hESC‐RPE. This secretion is regulated by several factors including hyperosmolarity 81, hyperthermia 82, oxidative stress 83, and heat‐sensitive TRPV channels 73. Particularly relevant for this study, modulating the L‐type Ca2+ channel activity has been shown to directly correlate with the VEGF secretion level 43. Our ELISA results indicated similar behavior as the activator (‐)BayK8644 increased the VEGF secretion and the inhibitor nifedipine decreased the VEGF secretion. This demonstrates that the L‐type Ca2+ channels participate in the regulation of VEGF secretion in hESC‐RPE. However, it is worth noting that the cell culture insert membrane with randomly spaced 1 μm holes may hinder both the diffusion of the drug to the basolateral cell membrane and the secretion of VEGF to the medium. Since the VEGF secretion is more pronounced in the basolateral side of the RPE, the structural constraints from the insert may lower the effect of pharmacological Ca2+ channel modulation on VEGF secretion.
Photoreceptor renewal is a critical task for RPE to maintain vision 2, and insufficient phagocytosis often leads to retinal diseases 84, 85. Several ion channels, including the L‐type Ca2+ channels, are known to have regulatory roles in phagocytosis in RPE 41, 64. We found that in hESC‐RPE, in line with the previous studies 64, inhibition of the L‐type Ca2+ channels by nifedipine decreased the phagocytosis remarkably. On the other hand, activation of these channels by (‐)BayK8644 also decreased the number of phagocytosed POS particles, although to a lesser extent. Interestingly, it was reported that in primary porcine RPE, the activation of L‐type Ca2+ channels had no effect on phagocytosis, and this was suggested to be a consequence of the regulatory effect of bestrophin‐1 setting a limit to L‐type Ca2+ channel activity 41. When comparing these results to our data, we want to point out that we used a pulse‐chase POS phagocytosis assay, while Müller et al. 41 used an assay with continuous POS supply to the RPE cells that may lead to distinct outcomes. Moreover, it is possible that bestrophin‐1 expression levels are much lower in hESC‐RPE compared to primary porcine RPE 41 thus diminishing the regulatory effect of bestrophin‐1 on L‐type Ca2+ channels in our cells. Besides, the influence of Ca2+ in phagocytosis can also be inhibitory: increase in intracellular Ca2+ and subsequent activation of protein kinase C has been shown to reduce POS ingestion 86. These observations indicate that the role for the L‐type Ca2+ channels in the regulation of POS phagocytosis is a complex process (see also 41, 64) and may include negative feedback mechanisms, especially after prolonged channel activation. Furthermore, it is known that these channels participate in the regulation of phagocytosis in concert with other ion channels including Ca2+‐dependent K+ channels, bestrophin‐1, TRPV 41, and most likely also the T‐type Ca2+ channels. Our observation about the increased number of bound or ingested POS particles following T‐type Ca2+ channel inhibition is similar to the effect of bestrophin‐1 inhibition 41. Analogous to bestrophin‐1 87, T‐type Ca2+ channels are indicated to interact with the β subunits of the L‐type Ca2+ channels 88. This implies a possible role for the T‐type channels to inhibit L‐type channels through their interaction with β subunits. Taken together, these data demonstrate a need for further studies in elucidating the concerted functioning of Ca2+ conducting channels in the regulation of phagocytosis.
Regardless of the close resemblance between stem cell‐derived and native RPE demonstrated for their proteome 29, capability of phagocytosis 27, 28, 30, 31, 32, VEGF secretion 32, 33, 34, and visual cycle 35, 36, many important differences have also been reported. These include a lower efficiency in the phagocytosis of POSs 89 as well as differences in growth factor secretion 90 and expression of adhesion junction and membrane transport genes 37. We used mouse RPE as the native counterpart for hESC‐RPE in our studies due to unavailability of live human RPE tissue. Previous work on gene and protein expression profiles of human and mouse RPE show high similarity regarding general biological functions, canonical pathways, and molecular networks 91, 92. However, there are important species‐specific differences between human and mouse RPE. These include immune regulation genes and genes related to the development of AMD and Usher syndrome 92 as well as the well‐known anatomical differences such as the absence of macula in the mouse and differences in rod and cone types and distributions.
We studied the functionality of CaV channels in mature hESC‐RPE where the localization of the primary CaV channel subtype, CaV1.3, started to resemble native RPE. During maturation, we observed significant changes in CaV1.3 localization in hESC‐RPE implying that ion channels can be highly sensitive to the level of tissue maturity. This has been previously suggested at least for Bestrophin‐1 in RPE 32, 93. We showed that in mature hESC‐RPE, CaV1.3 localized quite homogeneously on the apical and basolateral cell membranes. Intriguingly, in maturing hESC‐RPE, CaV1.3 appeared as distinct foci that co‐localized with PCNT to the base of the primary cilia. A similar punctuated appearance has been previously shown for TRP channel TRPM3 in human fetal RPE 94. PCNT is critical for the cilia formation, and relevant for our observations, PCNT is suggested to recruit protein complexes involved in cilia assembly and calcium signaling to the base of the primary cilia 66. On the other hand, primary cilia has been shown to regulate L‐type Ca2+ channel expression in mouse renal epithelial cells 95. This occurs through Wnt signaling 95, and interestingly, recent work shows the importance of the regulation of Wnt signaling not only for RPE development 96 but also for RPE maturation 67. Based on our data and taking into account these observations in the literature, it is possible, that CaV1.3 participates in ciliogenesis during RPE maturation or that its expression is coupled to the functioning of primary cilia in RPE maturation. This would not be surprising since primary cilia are important Ca2+ signaling organelles 97 with the expression of several different types of Ca2+ channels 98.
Conclusion
In this article, we demonstrate the presence of a functional machinery of voltage‐gated Ca2+ channels in hESC‐RPE, with L‐type Ca2+ channel characteristics highly resembling the native RPE. We show a regulatory role for L‐type Ca2+ channels in VEGF secretion and phagocytosis important for the hESC‐RPE functionality. We also provide novel information regarding the apical localization of CaV1.3 in RPE as well as its co‐localization near the base of the primary cilia during hESC‐RPE maturation. Our study represents an initial but significant progress toward a better understanding of CaV channels in stem cell‐derived RPE, however, further studies are needed to elucidate the specific roles for T‐type Ca2+ channels in RPE physiology. Overall, the results of the study are promising for the success of stem cell‐based RPE transplantation therapies, but highlight the need for sufficient RPE maturation as a prerequisite for its fully functional Ca2+ machinery.
Author Contributions
I.V.: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript; T.V.: collection and assembly of data, data analysis and interpretation, provision of study material, manuscript writing, final approval of manuscript; K.J.‐U.: conception and design, collection and/or assembly of data, manuscript writing, final approval of manuscript; H.U.‐J.: provision of study material, final approval of manuscript; H.S.: provision of study material, manuscript writing, final approval of manuscript; J.H.: financial support, manuscript writing, final approval of manuscript; S.N.: conception and design, financial support, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicated no potential conflicts of interest.
Supporting information
Supplementary Figure 1. Apical localization of Ca V 1.3 and Ca V 3.1 in hESC‐ and mouse RPE. Confocal microscopy xy‐maximum intensity projections and yz‐sections (apical side upwards, localization of the section highlighted with a white bar) show the immunolabeling of microvilli (ezrin, red) together with voltage‐gated Ca2+ channels A) CaV1.3 (green, cell line 08/017, days post‐confluence 50) and B) CaV3.1 (green, cell line 08/017, days post‐confluence 94) in hESC‐RPE and C) CaV1.3 and D) CaV3.1 in mouse RPE. Scale bars 10 μm.
Supplementary Figure 2. Changes in cell morphology during hESC‐RPE maturation. Reflection and bright field images of hESC‐RPE from post‐confluence day 1 to post‐confluence day 84 at four time points: A) day 1, B) day 6, C) day 31, and D) day 84 (cell line 08/017). Scale bars 5 μm.
Acknowledgments
We thank Viivi Jokinen, Julia Johansson, Elina Hurskainen, Outi Heikkilä, Marja‐Leena Koskinen, Outi Melin, Hanna Pekkanen, Outi Paloheimo, and Teemu Ihalainen for their technical assistance. The support from Tampere Imaging Facility and Tampere Facility of Electrophysiological Measurements is also greatly appreciated. This study was financially supported by the Academy of Finland (grant numbers 260375, 287287, 294054, 252225, 218050, 272808, 137801, 304909), the Emil Aaltonen Foundation, the Finnish Cultural Foundation, the Instrumentarium Science Foundation, the TEKES Human Spare Part Project and the Doctoral Programme of the President of Tampere University of Technology.
References
- 1. Strauss O. The retinal pigment epithelium in visual function. Physiol Rev 2005;85:845‐881. [DOI] [PubMed] [Google Scholar]
- 2. Wimmers S, Karl MO, Strauss O. Ion channels in the RPE. Prog Retin Eye Res 2007;26:263‐301. [DOI] [PubMed] [Google Scholar]
- 3. Ferrington DA, Sinha D, Kaarniranta K. Defects in retinal pigment epithelial cell proteolysis and the pathology associated with age‐related macular degeneration. Prog Retin Eye Res 2016;51:69‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong WL, Su X, Li X et al. Global prevalence of age‐related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta‐analysis. Lancet Glob Health 2014;2:e106‐e116. [DOI] [PubMed] [Google Scholar]
- 5. Riera M, Fontrodona L, Albert S et al. Comparative study of human embryonic stem cells (hESC) and human induced pluripotent stem cells (hiPSC) as a treatment for retinal dystrophies. Mol Ther Methods Clin Dev 2016;3:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schwartz SD, Regillo CD, Lam BL et al. Human embryonic stem cell‐derived retinal pigment epithelium in patients with age‐related macular degeneration and Stargardt's macular dystrophy: Follow‐up of two open‐label phase 1/2 studies. Lancet 2015;385:509‐516. [DOI] [PubMed] [Google Scholar]
- 7. Song WK, Park K, Kim H et al. Treatment of macular degeneration using embryonic stem cell‐derived retinal pigment epithelium: Preliminary results in Asian patients. Stem Cell Reports 2015;4:860‐872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schwartz SD, Tan G, Hosseini H et al. Subretinal transplantation of embryonic stem cell–derived retinal pigment epithelium for the treatment of macular degeneration: An assessment at 4 years. Invest Ophthalmol Vis Sci 2016;57:ORSFc1‐ORSFc9. [DOI] [PubMed] [Google Scholar]
- 9. Mandai M, Watanabe A, Kurimoto Y et al. Autologous induced stem‐cell–derived retinal cells for macular degeneration. N Engl J Med 2017;376:1038‐1046. [DOI] [PubMed] [Google Scholar]
- 10. Southwest Hospital, China . Clinical study of subretinal transplantation of human embryo stem cell derived retinal pigment epitheliums in treatment of macular degeneration diseases. Available at https://www.clinicaltrials.gov/ct2/show/NCT02749734. Accessed January 9, 2018.
- 11. Chinese Academy of Sciences . Subretinal transplantation of retinal pigment epitheliums in treatment of age‐related macular degeneration diseases. Available at https://www.clinicaltrials.gov/ct2/show/NCT02755428. Accessed January 9, 2018.
- 12. Cell Cure Neurosciences Ltd . Safety and efficacy study of OpRegen for treatment of advanced dry‐form age‐related macular degeneration. Available at https://www.clinicaltrials.gov/ct2/show/NCT02286089. Accessed January 9, 2018.
- 13. Astellas Institute for Regenerative Medicine . Long term follow up of sub‐retinal transplantation of hESC derived RPE cells in Stargardt macular dystrophy patients. Available at https://www.clinicaltrials.gov/ct2/show/NCT02445612. Accessed January 9, 2018.
- 14. Astellas Institute for Regenerative Medicine . Safety and tolerability of sub‐retinal transplantation of hESC derived RPE (MA09‐hRPE) cells in patients with advanced dry age related macular degeneration (Dry AMD). Available at https://www.clinicaltrials.gov/ct2/show/NCT01344993. Accessed January 9, 2018.
- 15. Astellas Institute for Regenerative Medicine . Sub‐retinal transplantation of hESC derived RPE(MA09‐hRPE)cells in patients with Stargardt's macular dystrophy. Available at https://www.clinicaltrials.gov/ct2/show/NCT01345006. Accessed January 9, 2018.
- 16. Astellas Institute for Regenerative Medicine . Long term follow up of sub‐retinal transplantation of hESC derived RPE cells in patients with AMD. Available at https://www.clinicaltrials.gov/ct2/show/NCT02463344. Accessed January 9, 2018.
- 17. Pfizer . Retinal pigment epithelium safety study for patients in B4711001. Available at https://www.clinicaltrials.gov/ct2/show/NCT03102138. Accessed January 9, 2018.
- 18. Federal University of São Paulo . Stem cell therapy for outer retinal degenerations. Available at https://www.clinicaltrials.gov/ct2/show/NCT02903576. Accessed January 9, 2018.
- 19. Chinese Academy of Sciences . Treatment of dry age related macular degeneration disease with retinal pigment epithelium derived from human embryonic stem cells. Available at https://www.clinicaltrials.gov/ct2/show/NCT03046407. Accessed January 9, 2018.
- 20. Pfizer . A study of implantation of retinal pigment epithelium in subjects with acute wet age related macular degeneration. Available at https://www.clinicaltrials.gov/ct2/show/NCT01691261. Accessed January 9, 2018.
- 21. da Cruz L, Fynes K, Georgiadis O et al. Phase 1 clinical study of an embryonic stem cell–derived retinal pigment epithelium patch in age‐related macular degeneration. Nat Biotechnol 2018;36:328‐337. [DOI] [PubMed] [Google Scholar]
- 22. Li Y, Tsai Y, Hsu C et al. Long‐term safety and efficacy of human‐induced pluripotent stem cell (iPS) grafts in a preclinical model of retinitis pigmentosa. Mol Med 2012;18:1312‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu B, Malcuit C, Wang S et al. Long‐term safety and function of RPE from human embryonic stem cells in preclinical models of macular degeneration. Stem Cells 2009;27:2126‐2135. [DOI] [PubMed] [Google Scholar]
- 24. Kanemura H, Go MJ, Shikamura M et al. Tumorigenicity studies of induced pluripotent stem cell (iPSC)‐derived retinal pigment epithelium (RPE) for the treatment of age‐related macular degeneration. PLOS ONE 2014;9(1):e85336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koss MJ, Falabella P, Stefanini FR et al. Subretinal implantation of a monolayer of human embryonic stem cell‐derived retinal pigment epithelium: A feasibility and safety study in Yucatan minipigs. Graefes Arch Clin Exp Ophthalmol 2016;254:1553‐1565. [DOI] [PubMed] [Google Scholar]
- 26. Galloway CA, Dalvi S, Hung SSC et al. Drusen in patient‐derived hiPSC‐RPE models of macular dystrophies. Proc Natl Acad Sci USA 2017;114:E8214‐E8223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thomas BB, Zhu D, Zhang L et al. Survival and functionality of hESC‐derived retinal pigment epithelium cells cultured as a monolayer on polymer substrates transplanted in RCS rats. Invest Ophthalmol Vis Sci 2016;57:2877‐2887. [DOI] [PubMed] [Google Scholar]
- 28. Carr A, Vugler AA, Hikita ST et al. Protective effects of human iPS‐derived retinal pigment epithelium cell transplantation in the retinal dystrophic rat. PLOS ONE 2009;4(12):e8152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hongisto H, Jylhä A, Nättinen J et al. Comparative proteomic analysis of human embryonic stem cell‐derived and primary human retinal pigment epithelium. Sci Rep 2017;7:6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subrizi A, Hiidenmaa H, Ilmarinen T et al. Generation of hESC‐derived retinal pigment epithelium on biopolymer coated polyimide membranes. Biomaterials 2012;33:8047‐8054. [DOI] [PubMed] [Google Scholar]
- 31. Singh R, Shen W, Kuai D et al. iPS cell modeling of Best disease: Insights into the pathophysiology of an inherited macular degeneration. Hum Mol Genet 2013;22:593‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brandl C, Zimmermann SJ, Milenkovic VM et al. In‐depth characterisation of retinal pigment epithelium (RPE) cells derived from human induced pluripotent stem cells (hiPSC). Neuromolecular Med 2014;16:551‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kokkinaki M, Sahibzada N, Golestaneh N. Human iPS‐derived retinal pigment epithelium (RPE) cells exhibit ion transport, membrane potential, polarized VEGF secretion and gene expression pattern similar to native RPE. Stem Cells 2011;29:825‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blenkinsop TA, Saini JS, Maminishkis A et al. Human adult retinal pigment epithelial stem cell–derived RPE monolayers exhibit key physiological characteristics of native tissue. Invest Ophthalmol Vis Sci 2015;56:7085‐7099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Maeda T, Lee MJ, Palczewska G et al. Retinal pigmented epithelial cells obtained from human induced pluripotent stem cells possess functional visual cycle enzymes in vitro and in vivo. J Biol Chem 2013;288:34484‐34493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muñiz A, Greene WA, Plamper ML et al. Retinoid uptake, processing, and secretion in human iPS‐RPE support the visual cycle. Invest Ophthalmol Vis Sci 2014;55:198‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peng S, Gan G, Qiu C et al. Engineering a blood‐retinal barrier with human embryonic stem cell‐derived retinal pigment epithelium: Transcriptome and functional analysis. Stem Cells Translational Medicine 2013;2:534‐544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Stanzel BV, Liu Z, Somboonthanakij S et al. Human RPE stem cells grown into polarized RPE monolayers on a polyester matrix are maintained after grafting into rabbit subretinal space. Stem Cell Reports 2014;2:64‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Miyagishima KJ, Wan Q, Corneo B et al. In pursuit of authenticity: Induced pluripotent stem cell‐derived retinal pigment epithelium for clinical applications. Stem Cells Translational Medicine 2016;5:1562‐1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abu Khamidakh AE, Dos Santos FC, Skottman H et al. Semi‐automatic method for Ca2+ imaging data analysis of maturing human embryonic stem cells‐derived retinal pigment epithelium. Ann Biomed Eng 2016;44:3408‐3420. [DOI] [PubMed] [Google Scholar]
- 41. Müller C, Más Gómez N, Ruth P et al. CaV1.3 L‐type channels, maxiK Ca2+‐dependent K+ channels and bestrophin‐1 regulate rhythmic photoreceptor outer segment phagocytosis by retinal pigment epithelial cells. Cell Signal 2014;26:968‐978. [DOI] [PubMed] [Google Scholar]
- 42. Wimmers S, Halsband C, Seyler S et al. Voltage‐dependent Ca2+ channels, not ryanodine receptors, activate Ca2+‐dependent BK potassium channels in human retinal pigment epithelial cells. Mol Vis 2008;14:2340‐2348. [PMC free article] [PubMed] [Google Scholar]
- 43. Rosenthal R, Heimann H, Agostini H et al. Ca2+ channels in retinal pigment epithelial cells regulate vascular endothelial growth factor secretion rates in health and disease. Mol Vis 2007;13:443‐456. [PMC free article] [PubMed] [Google Scholar]
- 44. Rosenthal R, Malek G, Salomon N et al. The fibroblast growth factor receptors, FGFR‐1 and FGFR‐2, mediate two independent signalling pathways in human retinal pigment epithelial cells. Biochem Biophys Res Commun 2005;337:241‐247. [DOI] [PubMed] [Google Scholar]
- 45. Mergler S, Steinhausen K, Wiederholt M et al. Altered regulation of L‐type channels by protein kinase C and protein tyrosine kinases as a pathophysiologic effect in retinal degeneration. FASEB J 1998;12:1125‐1134. [DOI] [PubMed] [Google Scholar]
- 46. Wimmers S, Coeppicus L, Rosenthal R et al. Expression profile of voltage‐dependent Ca2+ channel subunits in the human retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol 2008;246:685‐692. [DOI] [PubMed] [Google Scholar]
- 47. Ueda Y, Steinberg RH. Voltage‐operated calcium channels in fresh and cultured rat retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1993;34:3408‐3418. [PubMed] [Google Scholar]
- 48. Ueda Y, Steinberg RH. Dihydropyridine‐sensitive calcium currents in freshly isolated human and monkey retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 1995;36:373‐380. [PubMed] [Google Scholar]
- 49. Strauss O, Mergler S, Wiederholt M. Regulation of L‐type calcium channels by protein tyrosine kinase and protein kinase C in cultured rat and human retinal pigment epithelial cells. FASEB J 1997;11:859‐867. [DOI] [PubMed] [Google Scholar]
- 50. Strauss O, Buss F, Rosenthal R et al. Activation of neuroendocrine L‐type channels (α1D subunits) in retinal pigment epithelial cells and brain neurons by pp60c‐src. Biochem Biophys Res Commun 2000;270:806‐810. [DOI] [PubMed] [Google Scholar]
- 51. Rosenthal R, Bakall B, Kinnick T et al. Expression of bestrophin‐1, the product of the VMD2 gene, modulates voltage‐dependent Ca2+ channels in retinal pigment epithelial cells. FASEB J 2006;20:178‐180. [DOI] [PubMed] [Google Scholar]
- 52. Wollmann G, Lenzner S, Berger W et al. Voltage‐dependent ion channels in the mouse RPE: Comparison with Norrie disease mice. Vision Res 2006;46:688‐698. [DOI] [PubMed] [Google Scholar]
- 53. Rosenthal R, Thieme H, Strauss O. Fibroblast growth factor receptor 2 (FGFR2) in brain neurons and retinal pigment epithelial cells act via stimulation of neuroendocrine L‐type channels (Cav1.3). FASEB J 2001;15:970‐977. [DOI] [PubMed] [Google Scholar]
- 54. Reichhart N, Markowski M, Ishiyama S et al. Rab27a GTPase modulates L‐type Ca2+ channel function via interaction with the II–III linker of CaV1.3 subunit. Cell Signal 2015;27:2231‐2240. [DOI] [PubMed] [Google Scholar]
- 55. Reichhart N, Milenkovic VM, Halsband C et al. Effect of bestrophin‐1 on L‐type Ca2+ channel activity depends on the Ca2+ channel beta‐subunit. Exp Eye Res 2010;91:630‐639. [DOI] [PubMed] [Google Scholar]
- 56. Rohrer B, Kunchithapautham K, Genewsky A et al. Prolonged src kinase activation, a mechanism to turn transient, sublytic complement activation into a sustained pathological condition in retinal pigment epithelium cells. Adv Exp Med Biol 2014;801:221‐227. [DOI] [PubMed] [Google Scholar]
- 57. Skottman H. Derivation and characterization of three new human embryonic stem cell lines in Finland. In Vitro Cell Dev Biol Anim 2010;46:206‐209. [DOI] [PubMed] [Google Scholar]
- 58. Vaajasaari H, Ilmarinen T, Juuti‐Uusitalo K et al. Toward the defined and xeno‐free differentiation of functional human pluripotent stem cell‐derived retinal pigment epithelial cells. Mol Vis 2011;17:558‐575. [PMC free article] [PubMed] [Google Scholar]
- 59. Bodenstein L, Sidman RL. Growth and development of the mouse retinal pigment epithelium: I. Cell and tissue morphometrics and topography of mitotic activity. Dev Biol 1987;121:192‐204. [DOI] [PubMed] [Google Scholar]
- 60. Johansson JK, Ihalainen TO , Skottman H et al. Fast voltage sensitivity in retinal pigment epithelium: Sodium channels and their novel role in phagocytosis. bioRxiv 2017;223719.
- 61. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mao Y, Finnemann SC. Analysis of photoreceptor outer segment phagocytosis by RPE cells in culture. Methods Mol Biol 2013;935:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Luo Y, Zhuo Y, Fukuhara M et al. Effects of culture conditions on heterogeneity and the apical junctional complex of the ARPE‐19 cell line. Invest Ophthalmol Vis Sci 2006;47:3644‐3655. [DOI] [PubMed] [Google Scholar]
- 64. Karl MO, Kroeger W, Wimmers S et al. Endogenous Gas6 and Ca2+‐channel activation modulate phagocytosis by retinal pigment epithelium. Cell Signal 2008;20:1159‐1168. [DOI] [PubMed] [Google Scholar]
- 65. Burke JM. Epithelial phenotype and the RPE: Is the answer blowing in the Wnt? Prog Retin Eye Res 2008;27:579‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jurczyk A, Gromley A, Redick S et al. Pericentrin forms a complex with intraflagellar transport proteins and polycystin‐2 and is required for primary cilia assembly. J Cell Biol 2004;166:637‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. May‐Simera HL, Wan Q, Jha BS et al. Primary cilium‐mediated retinal pigment epithelium maturation is disrupted in ciliopathy patient cells. Cell Rep 2018;22:189‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koschak A, Reimer D, Huber I et al. a1D (Cav1.3) subunits can form L‐type Ca2+ channels activating at negative voltages. J Biol Chem 2001;276:22100‐22106. [DOI] [PubMed] [Google Scholar]
- 69. Singh A, Gebhart M, Fritsch R et al. Modulation of voltage‐ and Ca2+‐dependent gating of CaV1.3 L‐type calcium channels by alternative splicing of a C‐terminal regulatory domain. J Biol Chem 2008;283:20733‐20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Striessnig J, Pinggera A, Kaur G et al. L‐type Ca2+ channels in heart and brain. Wiley Interdiscip Rev Membr Transp Signal 2014;3:15‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cordeiro S, Strauss O. Expression of Orai genes and ICRAC activation in the human retinal pigment epithelium. Graefes Arch Clin Exp Ophthalmol 2011;249:47‐54. [DOI] [PubMed] [Google Scholar]
- 72. Martínez‐García MC, Martínez T, Pañeda C et al. Differential expression and localization of transient receptor potential vanilloid 1 in rabbit and human eyes. Histol Histopathol 2013;28:1507‐1516. [DOI] [PubMed] [Google Scholar]
- 73. Cordeiro S, Seyler S, Stindl J et al. Heat‐sensitive TRPV channels in retinal pigment epithelial cells: Regulation of VEGF‐A secretion. Invest Ophthalmol Vis Sci 2010;51:6001‐6008. [DOI] [PubMed] [Google Scholar]
- 74. Wimmers S, Strauss O. Basal calcium entry in retinal pigment epithelial cells is mediated by TRPC channels. Invest Ophthalmol Vis Sci 2007;48:5767‐5772. [DOI] [PubMed] [Google Scholar]
- 75. Balse E, Steele DF, Abriel H et al. Dynamic of ion channel expression at the plasma membrane of cardiomyocytes. Physiol Rev 2012;92:1317‐1358. [DOI] [PubMed] [Google Scholar]
- 76. Perez‐Reyes E. Molecular physiology of low‐voltage‐activated T‐type calcium channels. Physiol Rev 2003;83:117‐161. [DOI] [PubMed] [Google Scholar]
- 77. Strauss O, Wienrich M. Ca2+−conductances in cultured rat retinal pigment epithelial cells. J Cell Physiol 1994;160:89‐96. [DOI] [PubMed] [Google Scholar]
- 78. Kovach JL, Schwartz SG, Flynn HW Jr et al. Anti‐VEGF treatment strategies for wet AMD. J Ophthalmol 2012;2012:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Klettner A, Kaya L, Flach J et al. Basal and apical regulation of VEGF‐A and placenta growth factor in the RPE/choroid and primary RPE. Mol Vis 2015;21:736‐748. [PMC free article] [PubMed] [Google Scholar]
- 80. Blaauwgeers HGT, Holtkamp GM, Rutten H et al. Polarized vascular endothelial growth factor secretion by human retinal pigment epithelium and localization of vascular endothelial growth factor receptors on the inner choriocapillaris: Evidence for a trophic paracrine relation. Am J Pathol 1999;155:421‐428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hollborn M, Vogler S, Reichenbach A et al. Regulation of the hyperosmotic induction of aquaporin 5 and VEGF in retinal pigment epithelial cells: Involvement of NFAT5. Mol Vis 2015;21:360‐377. [PMC free article] [PubMed] [Google Scholar]
- 82. Faby H, Hillenkamp J, Roider J et al. Hyperthermia‐induced upregulation of vascular endothelial growth factor in retinal pigment epithelial cells is regulated by mitogen‐activated protein kinases. Graefes Arch Clin Exp Ophthalmol 2014;252:1737‐1745. [DOI] [PubMed] [Google Scholar]
- 83. Kannan R, Zhang N, Sreekumar PG et al. Stimulation of apical and basolateral vascular endothelial growth factor‐A and vascular endothelial growth factor‐C secretion by oxidative stress in polarized retinal pigment epithelial cells. Mol Vis 2006;12:1649‐1659. [PubMed] [Google Scholar]
- 84. Gal A, Li Y, Thompson DA et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet 2000;26:270‐271. [DOI] [PubMed] [Google Scholar]
- 85. Vollrath D, Yasumura D, Benchorin G et al. Tyro3 modulates mertk‐associated retinal degeneration. PLoS Genet 2015;11:e1005723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hall MO, Abrams TA, Mittag TW. ROS ingestion by RPE cells is turned off by increased protein kinase C activity and by increased calcium. Exp Eye Res 1991;52:591‐598. [DOI] [PubMed] [Google Scholar]
- 87. Milenkovic VM, Krejcova S, Reichhart N et al. Interaction of bestrophin‐1 and Ca2+ channel β‐subunits: Identification of new binding domains on the bestrophin‐1 C‐terminus. PLoS ONE 2011;6(4):e19364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Szklarczyk D, Franceschini A, Wyder S et al. STRING v10: Protein‐protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:447‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Mazzoni F, Safa H, Finnemann SC. Understanding photoreceptor outer segment phagocytosis: Use and utility of RPE cells in culture. Exp Eye Res 2014;126:51‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Sugino IK, Sun Q, Wang J et al. Comparison of FRPE and human embryonic stem cell–derived RPE behavior on aged human Bruch's membrane. Invest Ophthalmol Vis Sci 2011;52:4979‐4997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Yang X, Chung JY, Rai U et al. Cadherins in the retinal pigment epithelium (RPE) revisited: P‐cadherin is the highly dominant cadherin expressed in human and mouse RPE in vivo. PLoS ONE 2018;13(1):e0191279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bennis A, Gorgels TG, Ten Brink JB et al. Comparison of mouse and human retinal pigment epithelium gene expression profiles: Potential implications for age‐related macular degeneration. PLoS ONE 2015;10(10):e0141597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Bakall B, Marmorstein LY, Hoppe G et al. Expression and localization of bestrophin during normal mouse development. Invest Ophthalmol Vis Sci 2003;44:3622‐3628. [DOI] [PubMed] [Google Scholar]
- 94. Zhao PY, Gan G, Peng S et al. TRP channels localize to subdomains of the apical plasma membrane in human fetal retinal pigment epithelium. Invest Ophthalmol Vis Sci 2015;56:1916‐1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Muntean BS, Jin X, Williams FE et al. Primary cilium regulates CaV1.2 expression through Wnt signaling. J Cell Physiol 2014;229:1926‐1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Westenskow P, Piccolo S, Fuhrmann S. Beta‐catenin controls differentiation of the retinal pigment epithelium in the mouse optic cup by regulating Mitf and Otx2 expression. Development 2009;136:2505‐2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Delling M, DeCaen PG, Doerner JF et al. Primary cilia are specialized calcium signalling organelles. Nature 2013;504:311‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Pablo JL, DeCaen PG, Clapham DE. Progress in ciliary ion channel physiology. J Gen Physiol 2017;149:37‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Apical localization of Ca V 1.3 and Ca V 3.1 in hESC‐ and mouse RPE. Confocal microscopy xy‐maximum intensity projections and yz‐sections (apical side upwards, localization of the section highlighted with a white bar) show the immunolabeling of microvilli (ezrin, red) together with voltage‐gated Ca2+ channels A) CaV1.3 (green, cell line 08/017, days post‐confluence 50) and B) CaV3.1 (green, cell line 08/017, days post‐confluence 94) in hESC‐RPE and C) CaV1.3 and D) CaV3.1 in mouse RPE. Scale bars 10 μm.
Supplementary Figure 2. Changes in cell morphology during hESC‐RPE maturation. Reflection and bright field images of hESC‐RPE from post‐confluence day 1 to post‐confluence day 84 at four time points: A) day 1, B) day 6, C) day 31, and D) day 84 (cell line 08/017). Scale bars 5 μm.