

Abstract
Endothelial colony forming cells (ECFC) and mesenchymal stem cells (MSC) combined have great potential to be used for cell therapy of ischemic vascular diseases. However, to improve allogeneic stem cell engraftment the use of immunosuppression, such as cyclosporine has been suggested. Our aim was to assess the impact of cyclosporine on hind limb revascularisation upon MSC and ECFC combination therapy. Balb/c immunocompetent mice subjected to hind limb ischemia (right femoral artery ligation) were given both human ECFC and MSC (weekly intramuscular injections) with or without cyclosporine (daily injection). Surprisingly, mice receiving cyclosporine had a significant decrease in reperfusion based on laser Doppler imaging compared to vehicle controls and had poorer limb survival. In vitro, the downstream calcineurin target NFATC4 was highly expressed in the self‐renewing fraction of ECFCs. ECFCs cultured with cyclosporine had reduced colony formation capacity and tube formation in Matrigel. Lastly, ECFC displayed increased proliferation and loss of capacity for long term culture when in the presence of cyclosporine clearly showing a loss of quiescence and progenitor function. Our findings demonstrate the deleterious impact of cyclosporine on ECFC function, with significant impact on ECFC‐based allogeneic cellular therapy. stem cells translational medicine 2019;8:162&7
Keywords: Endothelial colony forming cell, Mesenchymal stem cells, Cyclosporine, Ischemia
Significance Statement.
This study presents evidence indicating the deleterious effect of the immunosupression agent cyclosporine on the function and survival of endothelial colony forming cells in vivo. These findings have significant clinical implications for allogeneic stem cell therapies treating ischemic disease.
Introduction
Cardiovascular disease (CVD) and in particular atherosclerosis continues to be a major disease burden globally resulting in significant morbidity and mortality of patients affected 1. Fundamental pharmacological and surgical interventions provide patients with numerous therapeutic options; however, in the case of diffuse disease, they fail to completely reperfuse the tissue, with symptoms soon returning. Over the past two decades, we have seen emergence of stem cell therapies, in particular using progenitors of endothelial or mesenchymal potency to treat ischemic vascular disease.
Endothelial colony forming cells (ECFC) are a rare subset of endothelial cells, distinct from the hematopoietic lineage, and entirely devoid of leucocyte or myeloid markers 2, 3. ECFC have self‐renewal capacity and form characteristic high‐proliferative‐potential (HPP) colonies in culture, with each having a significant capacity for long‐term culture (>15 passages) and expansion of cells with clear characteristics of endothelial cells 3. ECFC can be readily isolated from adult and fetal (umbilical cord blood) circulation but also in greater quantities from vascular structures, particularly those of the placenta 4, 5. These characteristics have recently resulted in ECFCs to be recognized as true endothelial progenitors 6. Significantly, ECFCs have a capacity to form entirely new vascular structures and repair damaged host vasculature when used in in vivo transplantation scenarios, demonstrating their potential importance in being used a cell therapy for CVD 7, 8. ECFCs' immunogenicity is an important theoretical limitation of this cell type in allogeneic (off‐the‐shelf) use and there is difficulty in accessing autologous ECFCs in abundance for cellular therapy.
MSC are known for their extensive ability to home to sites of injury in vivo and manipulate the microenvironment through extensive paracrine and immunomodulatory capacity, thereby assisting host tissue regeneration and promoting host angiogenesis 9, 10, 11. We and others have recently demonstrated the synergistic effects MSC exert onto ECFC that have either been co‐cultured or co‐injected together 12, 13. This combinatorial effect led to increased human ECFC survival and engraftment in vivo, increasing vessel formation in immunocompetent mice. However, levels of engraftment and vessel formation were even higher when MSC and ECFCs were combined in immunodeficient animals, suggesting that addition of immunosuppression therapy to the cell therapy may be beneficial
Cyclosporine (C/S) is the gold‐standard drug of choice for inducing immunosuppression in the context of allogeneic transplantation by affecting the activity of T‐cells via inhibiting calcineurin, leading specifically to the downregulation of NFAT family members (nuclear factor of activated T Cells) 14, 15, 16. In the present study we tested if C/S use in combination with xenogeneic MSC and ECFCs could improve tissue perfusion in immunocompetent animals subjected to hind‐limb ischemia. To our surprise, C/S use resulted in significantly poorer outcomes. Detailed evaluation of the effect of C/S on ECFCs revealed that this drug directly affected ECFC self‐renewal and tube formation potential through NFATc4
Materials and Methods
Murine Hind Limb Ischemia
We obtained Balb/c and nu/nu (on balb/c background) from the Australian Animal Resource Centre. Mice from 6 to 10 weeks and 17–22 g were anesthetized in accordance with institutional ethics and guidelines for the care of experimental animals. The proximal and distal portions of the right femoral artery were ligated, followed by isolation and resection of the artery as per previously published protocol conducted by our lab. Stable ischemia of the right limb was verified by hind limb blood perfusion measured using laser Doppler perfusion imager (LDPI) system (Moor Instruments Ltd., Devon, UK) immediately after the surgery and 2 days postsurgery.
ECFC and MSC in vitro Culture
Human placental fetal ECFC and MSC were isolated using our previously published protocol. The use of human tissue was granted by the human ethics boards of The University of Queensland and the Royal Brisbane and Women's Hospital. Placental ECFC (PL‐ECFC) and MSC (PL‐MSC) were cultured on rat tail collagen coated tissue‐culture flasks in Endothelial Growth Medium (EGM2) (Lonza) with 10% of fetal bovine serum.
In Vivo Stem Cell Transplantation
Two days after femoral artery ligation nu/nu mice were administered weekly (one dose) intramuscularly around the femoral ligation site 106 PL‐MSC with or without 106 PL‐ECFC in 1:1. Saline was used in the vehicle control group. To investigate the effects of C/S, 2 days after femoral artery ligation wild‐type Balb/c mice were administered weekly (one dose) intramuscularly around the femoral ligation site a combination of 106 PL‐MSC and 106 PL‐ECFC with or without daily intraperitoneal C/S (5 mg/kg) injections. Corn oil was used as the vehicle control group.
Reperfusion assessment was conducted at days 0, 2, 8, 15, and 22 using the LDPI system. Blood perfusion were expressed as the ratio of perfusion in the right (ischemic) limb versus the left (nonischemic) limb to eliminate confounding effect from the environment and interindividual variations.
PL‐ECFC in vitro Culture with C/S
PL‐ECFC were cultured for 24 hours prior to C/S treatment. After 24 hours, cells were cultured in EGM2/10% containing either vehicle (Ethanol) or 250 ng/ml of C/S (Sigma) with daily changes of fresh medium. Cell proliferation, ECFC colony forming capacity (HPP and low‐proliferative potential [LPP]) and cell expansion capabilities (over eight passages) were assessed.
In Vitro Tube Formation Assay
Twenty‐four‐well plate was precoated with 50 μl of Matrigel (In Vitro Technologies) and incubated for 2 hours before the assay. Then, 5 × 104 PL‐ECFC were passaged and seeded into each well culturing in EGM2/10% containing either vehicle (Ethanol) or 250 ng/ml of C/S. Bright field images were taken every 4 hours interval using Incucyte Zoom (Essen BioScience, Ann Arbor, MI). Number of tubes formed per field of view was counted for each time point.
RNA Isolation and Quantitative Polymerase Chain Reaction
Total RNA from CsA treated PL‐ECFC and vehicle control PL‐ECFC were isolated using Qiagen RNesay Mini Plus isolation kit (QIAGEN). mRNA were reverse transcript into cDNA using Tetro cDNA synthesis kit (Bioline). Genomic DNA were removed using DNase I (Invitrogen) digestion prior to cDNA synthesis. Quantitative polymerase chain reaction was performed using Quanstudio Flex 7 (Applied Biosystem) in triplicates. Primer sequences for genes of interest are listed in Table 1.
Table 1.
Primer sequences
| Gene | Forward | Reverse |
|---|---|---|
| HPRT | CCTGGCGTCGTGATTAGTGAT | AGACGTTCAGTCCTGTCCATAA |
| NFATC4 | CTTCTCCGATGCCTCTGACG | CGGGGCTTGGACCATACAG |
| ID2 | AGTCCCGTGAGGTCCGTTAG | AGTCGTTCATGTTGTATAGCAGG |
| TRIM47 | CTGAGCAGTCCAAAGTCCTGA | CTACGGCTGCACTCTTGATG |
| CDKN1A (P21) | CGGAACAAGGAGTCAGACATT | AGTGCCAGGAAAGACAACTAC |
| CDKN1C (P57) | GCGGCGATCAAGAAGCTGT | GCTTGGCGAAGAAATCGGAGA |
| CDKN2A (P16) | AGCTGTCGACTTCATGACAAG | GAGCTTTGGTTCTGCCATTTG |
| IL33 | CCACTGAGGAAAGAGCCATAG | TGAGCCTATCGTTTGGAACTG |
Flow Cytometry and FACS
We used directly conjugated antibodies for flow cytometry. Antibodies included human CD34‐PE (AbDSerotec, Raleigh), CFSE and mouse CD3‐PB, CD4‐Texas‐red, CD8‐PerCP‐Cy5.5, CD25‐PE, and FOXP3‐A647. Cells were incubated for 20 minutes at 4° with a dilution of 1:200.
Immunofluorescence
Dissected tissues were fixed for 2 hours in 4% PFA. The fixative was removed with 3× washes of PBS (Amresco, E404). Tissues were subsequently infused with sucrose before cryo‐embedding. For specific antigen staining, cryo‐sections were permeabilized in 0.5% Triton‐x‐100 (Chem Supply, Australia) before blocking with 20% normal goat serum (Gibco). Paraffin‐sections were deparaffinized and rehydrated. For this study, rat anti‐mouse CD31 (BD Biosciences) was used. Excess and unbound antibody was then removed with 3× 5 minutes washes in a solution containing 1× PBS/0.1% Tween‐20 (Amresco). Secondary antibodies conjugated with Alexa‐fluor 568 or 647 (Invitrogen) were used for fluorescence detection. Briefly, sections were incubated with secondary antibodies for 40 minutes at room temperature. Excess antibody was removed by 3 × 5 minutes washes in PBS/0.1% Tween‐20. Nuclear staining was revealed in specimens mounted with ProLong® Gold mounting media containing DAPI (Invitrogen). Confocal images were acquired with a Zeiss LSM 710 microscope equipped with Argon 561–10 nm DPSS and 633 nm HeNe lasers, and a 405–30 nm diode. We used the 405 nm diode laser for DAPI (detector 1, main beam filter MBS‐405, 414–463‐nm barrier filter), a 514‐nm Argon line for GFP (detector 3, main beam filter MBS‐458/514, 512‐570‐nm barrier filter) and the 561‐nm photodiode laser for Alexa‐568 (detector 3, main beam filter MBS‐488/561, 562‐611‐nm barrier filter). Images were acquired using 20× or 40× objectives.
Statistical Analysis
Analyses were performed using GraphPad Prism software, version 7. Descriptive statistics were evaluated using Mann‐Whitney t tests and a p value <.05 was considered significant.
Results
Murine Hind‐Limb Ischemia Reperfusion Assessment after Cell Therapy
To assess the combinatorial effect of using ECFC and MSC as a cell therapy for treating ischemic disease, we first used an immunocompromised murine model of hind‐limb ischemia. Here athymic nu/nu mice were provided either PL‐MSC, PL‐MSC+ PL‐ECFC together or saline as a vehicle control (Supporting Information Fig. S1A). At day 15 (D15) postfemoral artery ligation, a significant increase in reperfusion was observed in mice provided with PL‐MSC + PL‐ECFC (68.27% ± 4.85 vs. PL‐MSC 52.58% ± 5.5 and Saline 33.21 ± 6.9; **p < .01) (Supporting Information Fig. S1B, S1C). Importantly, the combination of PL‐MSC + PL‐ECFC resulted in no feet or toes being lost, whereas over 50% of mice with saline treatment had some form of limb deformity (**p < .01 Saline vs. PL‐MSC + PL‐ECFC) (Supporting Information Fig. S1D). This ensured that the combination cellular therapy was superior to MSC therapy alone in a model where no T cell was present.
We next tested the combination therapy of PL‐MSC + PL‐ECFC using wild‐type immunocompetent mice with or without (vehicle only) daily injections of C/S (Fig. 1A). Strikingly, already by D8 a significant improvement in reperfusion in the ischemic limb was observed in mice not treated compared to mice treated with C/S (53.61% ± 3.4 vs. 39.83% ± 4.1; *p < .05). This superiority in reperfusion without C/S treatment continued at D15 (72.13% ± 3.9 vs. 33.4% ± 2.8; **p < .01) and D22 (74.77% ± 2.9 vs. 46.1 ± 3.4; **p < .01) (Fig. 1B, 1C). Importantly, the use of C/S resulted in more mice (>50%) having feet or toes being lost, whereas all untreated mice had reperfused and normal feet/toes (**p < .01) (Fig. 1D). To assess the impact of C/S on individual stem cell populations, additional experiments were conducted. PL‐ECFC injected without C/S resulted in a significant increase in reperfusion by D15 (31.65 ± 3.5) and D22 (43.9 ± 4.4) compared to mice given C/S or vehicle only (D15 5.05 ± 3.9; D22 26.57 ± 5.8; **p < .01 ***p < .001). Importantly, no difference in the level of reperfusion was observed between mice given C/S or vehicle only (Supporting Information Fig. S1E). This clearly demonstrated the direct effect of C/S on ECFC function. However, when PL‐MSC was injected with or without C/S, no difference in reperfusion level was observed (Supporting Information Fig. S1F). Overall these findings indicate that C/S has deleterious effects on the benefit of ECFC therapy.
Figure 1.
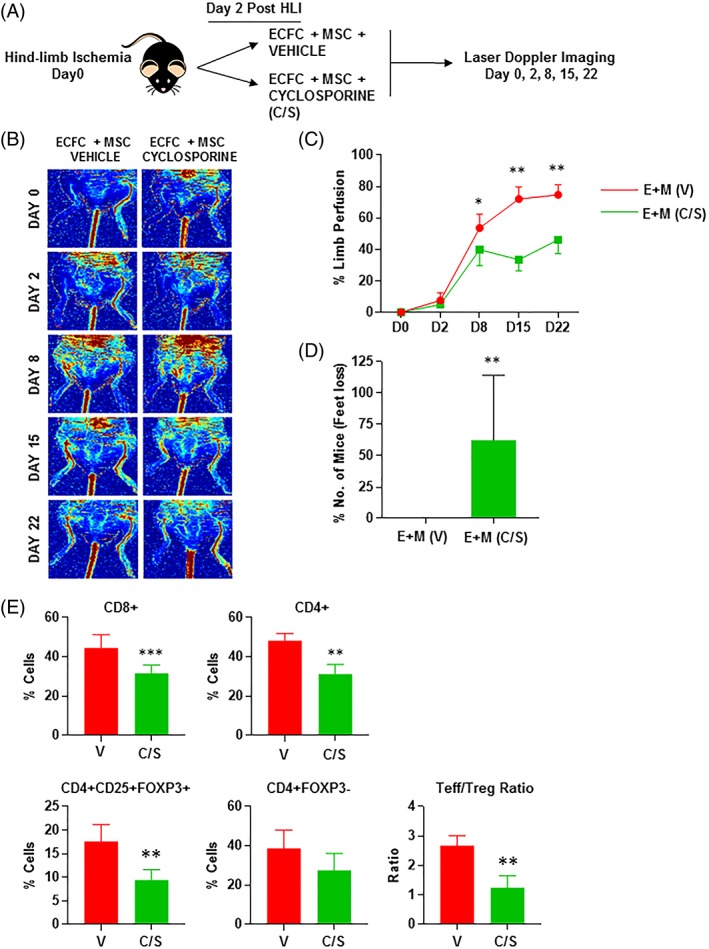
(A): schematic representation of the murine hind‐ischemia model and cell therapy strategy adopted. (B): high‐resolution laser Doppler images taken demonstration complete ischemia postsurgery (day 0) and improvement in reperfusion over time. (C): quantification of the limb reperfusion as measured by the laser Doppler (*p < .05, **p < .01). Red line, ECFC + MSC + vehicle; green line, ECFC + MSC + C/S. (D): quantification of fee lost in mice treated with or without C/S (**p < .01). (E): assessment of T‐cell infiltrate with or without C/S treatment (**p < .01; ***p < .001). All error bars represented as standard deviation. Abbreviations: C/S, cyclosporine; ECFC, endothelial colony forming cells; HLI, hind limb ischemia; MSC, mesenchymal stem cells.
Assessment of cell engraftment into ischemic site was performed by GFP through immunofluorescence given that ECFCs were GFP tagged. Staining against mouse CD31 was also used to determine if any chimeric vessels between human and mouse cells were being formed. At D28, only nucleated GFP positive cells were observed in muscle from animals treated with C/S (white arrows), although this event was rare. Additionally, GFP+ cells identified colocalized with mouse CD31 (white asterix) (Supporting Information Fig. S2).
To ensure mice being treated with C/S were being adequately immunosuppressed, draining lymph nodes adjacent to the ischemic limb were assessed for T‐Cell populations under flow cytometry conditions. In each case CD8+ (***p < .001), CD4+ (**p < .01) were significantly decreased as expected in mice after daily treatment of C/S. Although regulatory CD4 + CD25 + FOXP3+ T‐cells were less frequent (*p < .01) the ratio of T‐Effector/T‐Regulatory also significantly decreased in mice treated with C/S (**p < .01) confirming the reduction in T cell responses upon treatment (Fig. 1E). Overall, these findings suggested that mice treated with C/S had adequate immunosuppression that should have allowed better engraftment of the delivered progenitors and better leg perfusion.
In Vitro Assessment of ECFC Function Treated with C/S
In previous work, we have isolated the self‐renewing fraction of ECFCs (CD34+) in culture and characterized their gene expression signature 7. A key gene identified as differentially expressed between self‐renewing ECFCs (CD34+) and control ECFCs (CD34−) was NFATC4, a target of the calcineurin pathway targeted by C/S (Supporting Information Fig. S3A). We therefore took on to evaluate the effect of C/S on ECFC cultures in vitro. PL‐ECFC were cultured with or without 250 μM of C/S to firstly assess changes in cell proliferation over short‐term experiment of 36 hours (Fig. 2A, 2B). Strikingly, PL‐ECFC cultured with C/S demonstrated significant increase in proliferation and confluency after just 6 hours of treatment compared to control (29.3% ± 2.5 vs. 19.5% ± 1.7; *p < .05). This significant increase in proliferation was observed also after 12 (55.1% ± 2.2 vs 22.2 ± 3.4; **p < .01) and 18 hours (71.1 ± 3.4 vs. 33 ± 4.1; **p < .01) of C/S treatment (Fig. 2A). DNA content analysis reflecting cell cycling stage under flow cytometry conditions after 18 hours of treatment further confirmed these observations. Following C/S treatment PL‐ECFC were less likely to be in G0/1 phase (55% ± 6 vs. 10% ± 2; **p < .01), whereas a significant increase of PL‐ECFC in the S/G2M proliferative phase was observed (45% ± 2 vs. 90 ± 5; **p < .01) (Fig. 2B). Increase in proliferation after C/S treatment was further confirmed by CFSE flow cytometry (a measure of cell replication) compared to controls. Cells that shift to the left have lost the CFSE dye as replication occurs. It is clear that after 24 and 48 hours of C/S treatment there was an increase in proliferation of the CD34+ ECFC (4.57% and 14.2% respectively) compared to CD34+ ECFC without C/S (2.72% and 10.3%, respectively) (Supporting Information Fig. S4A, S4B).
Figure 2.
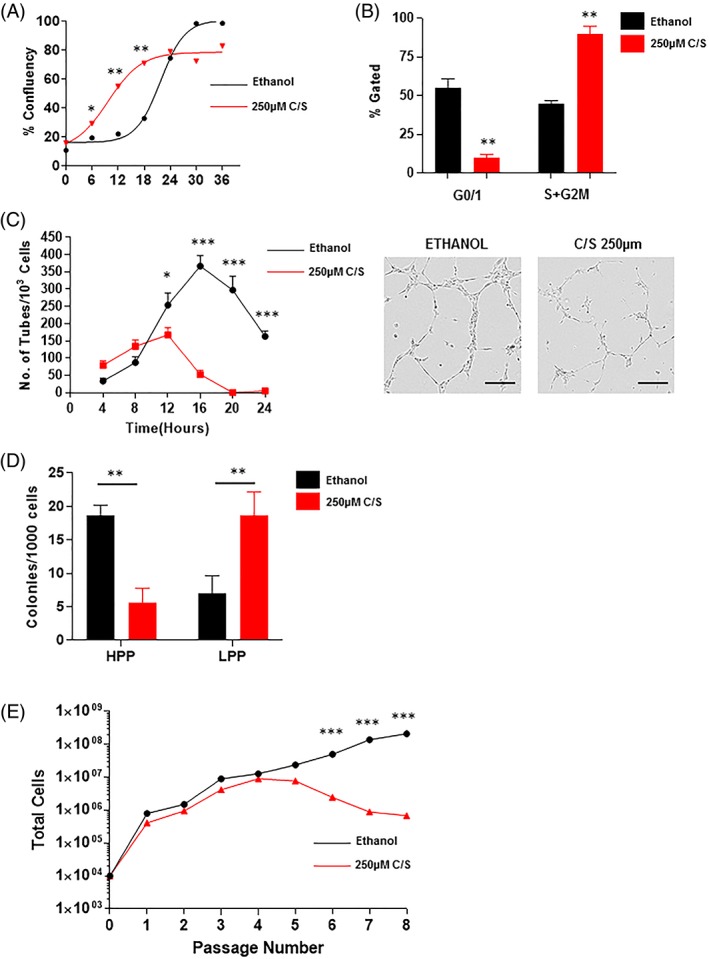
(A): quantification of in vitro ECFC culture to reach confluency. Treatment of ECFC with C/S (red) significantly increase confluency rate compared to ethanol controls (black) (*p < .05; **p < .01). (B): assessment of cell cycle under flow cytometry showed significantly fewer ECFC in G0/1 phase and significantly more ECFC in S + G2M phase when treated with C/S (**p < .01). (C): quantification of tube formation in Matrigel with or without C/S (*p < .05; ***p < .001). (D): quantification of HPP colonies and LPP ECFC colonies with or without C/S (**p < .01). (E): assessment of long‐term passaging of ECFC with or without C/S (***p < .001). All error bars represented as standard deviation. Abbreviations: C/S, cyclosporine; ECFC, endothelial colony forming cells; HPP, high‐proliferative potential; LPP, low‐proliferative potential.
In conjunction, RNA was isolated from cells treated with C/S for 18 hours to assess cell cycle gene expression changes. First, we expectedly observed a significant decrease in the expression of target genes of calcineurin pathway such as TRIM47 (10 fold decrease; *p < .001) and NFATC4 (nine fold decrease; ***p < .001) in PL‐ECFC treated with C/S (Supporting Information Fig. S3B). Importantly, significant reduction was observed in genes important for the quiescence of ECFCs: CDKN1C P57 (2‐fold decrease; *p < .05) and IL33 (12‐fold decrease; ***p < .001) previously reported by us 7 as being important in this process (Supporting Information Fig. S3C). The loss of quiescence has been shown to result in a loss of self‐renewal and progenitor activity. We therefore examined self‐renewal and differentiation of ECFCs.
We next assessed if this increase in proliferation resulted in changes in tube formation capacity using Matrigel. Clearly, after only 12 hours of treatment with C/S, a significant decrease in the number of tubes formed was observed (254 ± 29 vs. 167 ± 17; *p < .05) (Fig. 2C). Furthermore, the capacity to form tubes over time was significantly reduced, whereby after 20 hours of treatment no tubes could be observed (297 ± 33 vs. 0 ± 0; ***p < .001). These findings point to a rapid differentiation of ECFCs and a loss of progenitor activity.
The final assessment was to determine changes in ECFC colony formation as counted by the number of HPP and LPP after 14 days in culture, which is the gold‐standard for ECFC self‐renewal evaluation. Following C/S treatment, a significant reduction in HPP colony formation per 1,000 cells plated was observed (18.6 ± 3 vs. 5.6 ± 3; **p < .01), whereas a significant increase in LPP colonies per 1,000 cells plated was observed (7 ± 3.5 vs. 19 ± 5; **p < .01) (Fig. 2D). These data suggest that C/S treatment not only increases proliferation but causes the loss of ECFC colony forming capacity. The consequence of this was a loss of ability to maintain an ECFC culture over long‐term cultures and numerous passages (passage 8; >100 days in culture) as the HPP progenitor pool is being depleted. By passage 6 a clear significant divergence in cell numbers that can been produced is observed (***p < .001) (Fig. 2E).
Discussion
Endothelial progenitor therapy has been an essential goal of regenerative medicine for decades. To date, over 100 clinical trials are registered exploring some form of endothelial progenitors have been reported. However, unlike MSCs, allogeneic transplantation is challenging due to the immunogenicity of the cells, encouraging the exploration of immunosuppression therapy as a means to increase engraftment and therefore function. In the present study, we explored this path using C/S, the most commonly used calcineurin inhibitor in addition to ECFCs and MSCs in combination. We demonstrate that not only C/S did not improve revascularisation but it was deleterious despite resulting in significant down‐turn of the immune system. Exploring this further, we found that C/S had direct effects on endothelial but not mesenchymal cells. Indeed C/S's target calcineurin pathway and especially its effector NFATc4 is overexpressed in the quiescent self‐renewing fraction of ECFCs. Upon inhibition of this pathway, C/S accelerated cell proliferation and differentiation as measured by tube formation at the expense of a loss in self‐renewal, and colony formation ability resulting overall in poor revascularisation in vivo and an inability to expand over long term in vitro.
Previous studies have reported the benefits of associating MSCs and ECFCs to improve ECFC engraftment both in immunosuppressed and immunocompetent recipients 13, 17. We here confirm for the first time that MSC‐ECFC combinations outperform MSCs alone and are effective at improving hind limb ischemia and preventing amputation. The mechanisms at play in this improvement could be twofold. Indeed MSCs seem to have a direct influence on the recipient's immune system 7. However, they also have additional effects beyond their immune role on ECFCs 13. Indeed, ECFCs in culture has been shown to be primed upon co‐culture with MSCs 12. As a result of this priming, despite having reduced proliferation and colony forming potential, they had much improved engraftment in vivo, resistance to serum starvation and tube formation in vitro. This has been at least in part the result of sustained Notch signaling. Whether the use of C/S to some extent counteracts the priming effect of MSCs remains to be investigated. Overall, our findings strongly suggest that C/S is not required in MSC‐ECFC combinations for hind limb ischemia therapy.
In previous studies, we and others have identified that ECFC cultures have a self‐renewing fraction that highly expresses CD34 7. This allowed to characterize for the first time the gene expression signature of self‐renewing versus differentiating and proliferating cells. Self‐renewing CD34+ cells in ECFC cultures are quiescent, express high levels of cell cycle regulators CDKN1C or IL33 as well as high levels of Notch target genes or Notch ligands. Of importance, loss of quiescence through Notch inhibition or CDKN1C inhibition resulted in accelerated proliferation but an inability to form new high proliferative potential colonies mimicking progenitor exhaustion. NFATC4 is a key gene highly expressed in the CD34hi fraction of ECFCs suggesting its importance in quiescence. In other settings, NFAT family members play similar roles in maintaining stem cell quiescence and function 18. Inhibition of NFATC4 using C/S not only affected the pathway's target genes but also affected cell cycle regulators and IL33, accelerating cell division and therefore resulting in the loss of progenitor capacity as measured by colony formation. Of note, we have previously reported that ECFC preparation that are devoid of their self‐renewing fraction are less likely to engraft and result in smaller size vessels 7 .
Although our study has detailed the effects of C/S on transplanted cells, we have not yet examined the potential alterations of the host vasculature. Indeed, we have described a hierarchy of progenitor to differentiated cells in the endothelium of both mice and human tissues and have shown through RNA sequencing that NFATC4 is also present and overexpressed in the endothelial progenitors of murine aortas 19. Whether C/S treatment resulted in more global progenitor depletion in the mouse and hence reduced further the recovery post hind‐limb arterial ligation remains a possibility. This is supported by the pro‐inflammatory effect of C/S on murine endothelial cells 20. It is also supported by the fact that we did not observe significant engraftment of the delivered ECFCs. In accordance long‐term treatment with C/S resulted in increased atherosclerosis 21. Indeed, previous clinical reports have reported poorer vascular outcome in patients with limb ischemia treated with C/S 22, 23.
Conclusion
The better definition of endothelial progenitors and their ease of access through the human term placenta, gives the research community new hope for the delivery of these cells as an off‐the‐shelf product 5, 6. The combination of ECFCs with donor matched MSCs further improves their efficacy and in the present study we show that the use of immunosuppression therapy such as C/S is not only dispensable but is deleterious for future clinical trials of ischemic disorders using ECFCs.
Author Contributions
S.S: conception and design, collection and assembly of data, data analysis and interpretation, manuscript writing, final approval; J.A and E.R: collection and assembly of data, final approval; A.S: data analysis, final approval; K.K and J.P: conception and design, financial support, data analysis and interpretation, manuscript writing, final approval.
Disclosure of Potential Conflicts of Interest
J.P. and K.K. are coinventors on a patent relating to the isolation of endothelial progenitors from the placenta.
Supporting information
Supplementary Figure 1: (A) Schematic representation of the murine hind‐ischemia model and cell therapy strategy adopted. (B) High resolution Laser Doppler images taken demonstration complete ischemia post‐surgery (Day 0) and improvement in reperfusion over time. (C) Quantification of the limb reperfusion as measured by the Laser Doppler (**p < 0.01). Blue line: ECFC+MSC; Green line: MSC; Black line: Saline. (D) Quantification of feet lost between treatment groups (**p < 0.01). (E&F) Quantification of the limb reperfusion as measured by the Laser Doppler (**p < 0.01; ***p < 0.001). All error bars represented as standard deviation.
Supplementary Figure 2: (A) Immunofluorescence of ischemic muscle tissue from mice treated with and without cyclosporine (C/S). White arrows: GFP+ cells. White asterix: GFP + CD31+ human/mouse chimeric vessel. Scale bar represents 50 μm.
Supplementary Figure 3: (A) Assessing the expression of NFACT4 between the self‐renewing population of ECFC (CD34+) and differentiated ECFC (CD34‐) in culture. (B) Assessment of cyclosporine target genes (***p < 0.001). (C) Assessment of cell cycle and quiescent endothelial marker IL33 (*p < 0.05; ***p < 0.001). All error bars represented as standard deviation.
Supplementary Figure 4: (A) Flow cytometry plot demonstrating positive and negative CFSE staining of PL‐ECFC. (B) Increase in proliferation was observed in PL‐ECFC treated with cyclosporine (red) compared to ethanol controls (black) at 24 and 48 hours.
Acknowledgments
The authors would like to thank the Translational Research Institute flow cytometry facility for their assistance. Funding sources: K.K. salary was supported by NHMRC Career Development Fellowship (APP1125290). J.P. salary was supported by the ARC DECRA Research Fellowship (DE180100984).
References
- 1. Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: A review of global methodologies of mortality measurement. Circulation 2013;127:749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ingram DA, Mead LE, Moore DB et al. Vessel wall‐derived endothelial cells rapidly proliferate because they contain a complete hierarchy of endothelial progenitor cells. Blood 2005;105:2783–2786. [DOI] [PubMed] [Google Scholar]
- 3. Ingram DA, Mead LE, Tanaka H et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood 2004;104:2752–2760. [DOI] [PubMed] [Google Scholar]
- 4. Shafiee A, Fisk NM, Hutmacher DW et al. Fetal endothelial and mesenchymal progenitors from the human term placenta: potency and clinical potential. Stem Cells Translational Medicine 2015;4:419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Patel J, Seppanen E, Chong MS et al. Prospective surface marker‐based isolation and expansion of fetal endothelial colony‐forming cells from human term placenta. Stem Cells Translational Medicine 2013;2:839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Medina RJ, Barber CL, Sabatier F et al. Endothelial progenitors: A consensus statement on nomenclature. Stem Cells Translational Medicine 2017;6:1316–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Patel J, Wong HY, Wang W et al. Self‐renewal and high proliferative colony forming capacity of late‐outgrowth endothelial progenitors is regulated by cyclin‐dependent kinase inhibitors driven by notch signaling. Stem Cells 2016;34:902–912. [DOI] [PubMed] [Google Scholar]
- 8. Patel J, Donovan P, Khosrotehrani K. Concise review: Functional definition of endothelial progenitor cells: A molecular perspective. Stem Cells Translational Medicine 2016;5:1302–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Copland IB. Mesenchymal stromal cells for cardiovascular disease. J Cardiovasc Dis Res 2011;2:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schaun MI, Eibel B, Kristocheck M et al. Cell therapy in ischemic heart disease: Interventions that modulate cardiac regeneration. Stem Cells Int 2016;2016:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood 2007;110:3499–3506. [DOI] [PubMed] [Google Scholar]
- 12. Shafiee A, Patel J, Wong HY et al. Priming of endothelial colony‐forming cells in a mesenchymal niche improves engraftment and vasculogenic potential by initiating mesenchymal transition orchestrated by NOTCH signaling. FASEB J 2017;31:610–624. [DOI] [PubMed] [Google Scholar]
- 13. Shafiee A, Patel J, Lee JS et al. Mesenchymal stem/stromal cells enhance engraftment, vasculogenic and pro‐angiogenic activities of endothelial colony forming cells in immunocompetent hosts. Sci Rep 2017;7:13558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borel JF. History of the discovery of cyclosporin and of its early pharmacological development. Wien Klin Wochenschr 2002;114:433–437. [PubMed] [Google Scholar]
- 15. Rao A, Luo C, Hogan PG. Transcription factors of the NFAT family: Regulation and function. Annu Rev Immunol 1997;15:707–747. [DOI] [PubMed] [Google Scholar]
- 16. Tedesco D, Haragsim L. Cyclosporine: a review. J Transplant 2012;2012:230386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Souidi N, Stolk M, Rudeck J et al. Stromal cells act as guardians for endothelial progenitors by reducing their immunogenicity after co‐transplantation. Stem Cells 2017;35:1233–1245. [DOI] [PubMed] [Google Scholar]
- 18. Horsley V, Aliprantis AO, Polak L et al. NFATc1 balances quiescence and proliferation of skin stem cells. Cell 2008;132:299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patel J, Seppanen EJ, Rodero MP et al. Functional definition of progenitors versus mature endothelial cells reveals key soxf‐dependent differentiation process. Circulation 2017;135:786–805. [DOI] [PubMed] [Google Scholar]
- 20. Rodrigues‐Diez R, Gonzalez‐Guerrero C, Ocana‐Salceda C et al. Calcineurin inhibitors cyclosporine A and tacrolimus induce vascular inflammation and endothelial activation through TLR4 signaling. Sci Rep 2016;6:27915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roselaar SE, Schonfeld G, Daugherty A. Enhanced development of atherosclerosis in cholesterol‐fed rabbits by suppression of cell‐mediated immunity. J Clin Invest 1995;96:1389–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fletcher F, Ain M, Jacobs R. Healing of foot ulcers in immunosuppressed renal transplant patients. Clin Orthop Relat Res 1993;296:37–42. [PubMed] [Google Scholar]
- 23. Davenport A, Parkin A. The effect of cyclosporin on lower limb blood flow in renal transplant recipients. Transpl Int 1991;4:239–242. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: (A) Schematic representation of the murine hind‐ischemia model and cell therapy strategy adopted. (B) High resolution Laser Doppler images taken demonstration complete ischemia post‐surgery (Day 0) and improvement in reperfusion over time. (C) Quantification of the limb reperfusion as measured by the Laser Doppler (**p < 0.01). Blue line: ECFC+MSC; Green line: MSC; Black line: Saline. (D) Quantification of feet lost between treatment groups (**p < 0.01). (E&F) Quantification of the limb reperfusion as measured by the Laser Doppler (**p < 0.01; ***p < 0.001). All error bars represented as standard deviation.
Supplementary Figure 2: (A) Immunofluorescence of ischemic muscle tissue from mice treated with and without cyclosporine (C/S). White arrows: GFP+ cells. White asterix: GFP + CD31+ human/mouse chimeric vessel. Scale bar represents 50 μm.
Supplementary Figure 3: (A) Assessing the expression of NFACT4 between the self‐renewing population of ECFC (CD34+) and differentiated ECFC (CD34‐) in culture. (B) Assessment of cyclosporine target genes (***p < 0.001). (C) Assessment of cell cycle and quiescent endothelial marker IL33 (*p < 0.05; ***p < 0.001). All error bars represented as standard deviation.
Supplementary Figure 4: (A) Flow cytometry plot demonstrating positive and negative CFSE staining of PL‐ECFC. (B) Increase in proliferation was observed in PL‐ECFC treated with cyclosporine (red) compared to ethanol controls (black) at 24 and 48 hours.