

Abstract
Background
Chikungunya virus is an emerging mosquito-borne pathogen with a wide global distribution. With the severe morbidity that it causes, chikungunya virus is a major public health problem in the affected areas and poses a considerable risk for unaffected areas hosting competent vector populations. In the summer of 2017, Italy experienced a chikungunya virus outbreak that spread in the Lazio region and caused a secondary outbreak in the Calabrian village of Guardavalle, with a final case number of 436. The causative strain was recognized as an Indian Ocean lineage (IOL) virus.
Methods
To understand the underlying genetic and molecular features of the outbreak virus, viruses from mosquito pools and clinical samples were isolated in cell culture and subjected to whole-genome sequencing and genetic analyses.
Results
All 8 characterized genomes shared a high sequence identity. A distinct substitution pattern in the Italian 2017 viruses (including mutations in E1, E2, and nsP4) was partly shared with the Pakistani 2016 outbreak viruses. Evolutionary analyses indicate that these 2 recent outbreaks and several geographically widely distributed, travel-associated viruses form a cluster of rapidly emerging Indian-origin IOL viruses.
Conclusions
Our analyses show that the 2017 Italian outbreak virus belongs to a cluster of novel IOL chikungunya viruses originating in India. Their emergence calls for enhanced monitoring and strengthened preparedness measures, including vector control programs and raised awareness among general practitioners in countries potentially at risk.
Keywords: arbovirus, chikungunya virus, emerging infectious disease, Italy, molecular epidemiology, outbreak
Geographic dispersal of competent arthropod vectors and growing global transport systems have facilitated the reemergence of chikungunya virus (CHIKV), a mosquito-borne pathogen that causes substantial health and economic burdens in the affected populations. Travel to areas endemic for arboviruses is the main source of imported arbovirus infections, and imported cases are regularly reported all over Europe [1]. The dispersion and establishment of the Aedes albopictus mosquito in Southern Europe has enabled local transmission of viruses previously unencountered in the region, as demonstrated by the occurrence of autochthonous transmission of CHIKV in 2007 and 2017 in Italy, and in 2010, 2014, and 2017 in France [2–7].
CHIKV (family Togaviridae, genus Alphavirus), is an enveloped, single-stranded positive-sense RNA virus with a genome of approximately 11.8 kb, consisting of 2 open reading frames (ORFs) encoding the nonstructural “replicase” polyprotein (ORF1: nsP1-nsP2-nsP3-nsP4) and the structural polyprotein (ORF2: C-E3-E2-6K/TF-E1), with 5’ and 3’ untranslated regions (5’-3’UTR) [8]. The virus is transmitted by the broadly distributed mosquito vectors Aedes aegypti and Ae. albopictus, along with the mosquito-borne arboviruses dengue, yellow fever, and Zika virus. Humans, and in some areas nonhuman primates, are currently the only vertebrate host. Infections are characterized by a fever-rash-arthralgia syndrome and may lead to debilitating, long-lasting joint and muscle pain and rashes, especially among patients such as children, the elderly, and individuals with chronic disease. The clinical disease is associated with a high level of inflammatory responses [9].
CHIKV was first isolated in Tanzania in 1953, although the first descriptions of chikungunya-like illness in the Americas, Africa, and Asia come from the 18th century [10, 11]. Since then, CHIKV has become globally distributed and has established endemic transmission in Africa, Asia, the Americas, and the Pacific archipelago. Previous phylogenetic analyses have revealed 3 major genotypes: West African, Asian, and East/Central/South African (ECSA). The Indian Ocean lineage (IOL), a descendant of the ECSA lineage, has evolved and spread from coastal Kenya to the Indian Ocean islands in 2004 and concurrently to India in 2006, resulting in persistent transmission in Southeast Asia [12]. During dispersal in the Indian Ocean islands, the E1 A226V mutation was first observed; it was later shown to confer enhanced transmission by the Ae. albopictus vector [13–15]. This adaptive mutation has reportedly arisen in nature on at least 4 separate occasions via evolutionary convergence in response to the availability of the Ae. albopictus vector [15, 16].
In August 2017, locally acquired CHIKV infections appeared in Southeast France (Le Cannet des-Maures, Var Department) and in Anzio and Rome, on the Tyrrhenian coast, Latium region, in Central Italy. In September, a secondary outbreak was detected in Guardavalle on the Ionic coast of the Calabria region in Southern Italy. Rigorous vector control measures at the outbreak locations were applied, and concurrent with dropping daily temperatures, the Italian outbreaks were controlled, with a final laboratory-confirmed case number of 436 (297 Lazio, 132 Calabria, and cases imported from these 2 regions elsewhere: 4 Emilia-Romagna, 1 Marche, 1 Germany, 1 France) [17–20]. Partial E1 gene sequences linked the 2 Italian outbreak foci to each other, whereas the French outbreak was linked to a different source [2, 7]. The Italian 2017 outbreak virus expressing E1 226A was capable of sustained local transmission in an environment where Ae. albopictus is the sole competent CHIKV vector.
In this study, we sequenced the whole genomes of viruses isolated at different time points from the 2 outbreak foci of the Italian 2017 CHIKV outbreak to gain insight into the underlying genetic and molecular features of the outbreak virus.
METHODS
Virus Isolation
Six serum samples collected from viremic patients, 2 pools of female Ae. Albopictus mosquitoes (determined by entomologists) recovered from the residential areas of the patients (1 in Anzio and 1 in Guardavalle) and 2 serum samples from patients from the previous 2007 Italian CHIKV outbreak, were used for virus isolation. Briefly, 500 uL of clinical samples or supernatant from homogenized mosquitoes, in phosphate buffer saline supplemented with 20% heat-inactivated fetal bovine serum (Gibco, Waltham, MA) and 1% penicillin/streptomycin/amphotericin B mix (Invitrogen, Carlsbad, CA) were seeded on a 70%–80% confluent monolayer of Vero cells in 25-cm2 Corning cell culture flasks (Sigma-Aldrich Corp, Rockville, MD). After 1 hour of incubation at 37°C in 5% CO2, 6 mL of medium, consisting of Dulbecco’s MEM, 2% FBS, and 1% antibiotic-antimycotic mix (Invitrogen, Gibco), was added. The infected cell cultures were examined daily for cytopathic effect. Viral RNA was extracted from 200 mL of culture media using the QIAmp Viral RNA kit (Qiagen, Hilden, Germany) according to manufacturer instructions. The isolates were identified as CHIKV through real-time polymerase chain reaction performed using previously described CHIKV TaqMan primers and probe [5]. In total, cell culture supernatant from 8 samples from the 2017 outbreak and 2 additional samples from the 2007 Emilia Romagna outbreak (Table 1) was subjected to whole-genome sequencing by Illumina. Viral RNA was quantified using the QuantiFluor RNA system (Promega, Madison, WI). All the samples were retro-transcribed using the SuperScript III Reverse Transcriptase kit (Invitrogen). The resulting cDNA was converted into double-stranded DNA by Klenow enzyme (Roche, Basel, Switzerland) and quantified using the QuantiFluorOne dsDNA system (Promega). Each step was carried out according to the manufacturers’ instructions. For each isolate, 1 ng of DNA was used to prepare the sequencing libraries using the Nextera XT DNA library preparation kit (Illumina, San Diego, CA) following the kit protocol. The quality and concentration of the libraries were evaluated using the Agilent 2100 bioanalyzer (Agilent, Santa Clara, CA). Whole-genome sequencing was performed using the Illumina Miseq Reagent Nano Kit, V2 (2 × 150 cycles), and run on the Illumina MiSeq platform at the sequencing facility of the Scientific Department of the Army Medical Center in Rome, Italy. The reads were trimmed for quality and length (sliding window trimming with an average of 4 bases to cross and required quality of 30; the first 15 nucleotides were trimmed, and reads with minimum length of 50 were kept) in Trimmomatic [21] on the Galaxy platform run on the ISS server (https://aries.iss.it/). Reads were aligned to the reference (MG049915), and whole genomes were assembled using the CLC Genomics Workbench 11 (QIAGEN).
Table 1. .
Summary of Italian 2017 Isolates Described in This Study
| Strain | Genbank Accession No. | Sample Collection Date | Site of Origin | Source of Isolation | No. of Passages in Mouse Brain/Vero Cells | |
|---|---|---|---|---|---|---|
| 1 | Italy/Ae. albopictus/ Lazio-ISS-1/2017 | MH754507 | 09/08/2017 | Anzio, Lazio region |
Pool of 12 Aedes albopictus mosquitoes | V1 |
| 2 | Italy/Ae. albopictus/ Calabria-ISS-1/2017 | MK120194 | 09/27/2017 | Guardavalle, Calabria region | Pool of 8 Aedes albopictus mosquitoes | V1 |
| 3 | Italy/Calabria-ISS-972/2017 | MK120195 | 09/27/2017 | Guardavalle, Calabria region | Patient 972, serum sample | V1 |
| 4 | Italiy/Calabria-ISS-989/2017 | MK120196 | 09/30/2017 | Guardavalle, Calabria region | Patient 989, serum sample | V1 |
| 5 | Italy/Calabria-ISS-991/2017 | MK120197 | 10/02/2017 | Guardavalle, Calabria region | Patient 991, serum sample | V1 |
| 6 | Italy/Calabria-ISS-977/2017 | MK120198 | 10/02/2017 | Guardavalle, Calabria region | Patient 977, serum sample | V1 |
| 7 | Italy/Calabria-ISS-1011/2017 | MK120199 | 10/02/2017 | Guardavalle, Calabria region | Patient 1011, serum sample | V1 |
| 8 | Italy/Calabria-ISS-1028/2017 | MK120200 | 10/09/2017 | Guardavalle, Calabria region | Patient 1028, serum sample | V1 |
| 9 | Italia/Emilia Romagna-ISS-1/2007 | MK120201 | 08/20/2007 | Ravenna, Emilia Romagna region | Patient serum sample | M2/V4 |
| 10 | Italia/Emilia Romagna-ISS-2/2007 | MK120202 | 08/25/2007 | Ravenna, Emilia Romagna region | Patient serum sample | M2/V2 |
Features of Italian chikungunya virus isolates. Isolates 1, 2, and 3 were characterized from Ae. albopictus collected in Lazio (1 and 2) and in Calabria (3). The other isolates were derived from patient samples collected in Calabria.
Abbreviations: M, mouse brain; V, vero.
Sequence analysis was performed within MEGA 6 [22]. For phylogenetic analyses, reference strains were identified using the BLAST nucleotide search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) and drawn from GenBank (https://www.ncbi.nlm.nih.gov/genbank/). Sequence alignments of concatenated ORF1 and ORF2, excluding the noncoding regions, were generated by Clustal W and checked manually.
All full-length genomes available at the time (597 strains) were used for initial analyses (data not shown). The alignment was then reduced to 90 reference strains by removing non-IOL strains, duplicates, and sequences of low quality. Maximum likelihood phylogenies of concatenated ORF1 and ORF2 nucleotide sequence, E1 nucleotide sequence, and nsP4 amino acid sequence were constructed using the best-fit models for the alignments and testing the trees by 1000 bootstrap replicates. Trees were generated using FigTree [23].
Substitution analyses were done by comparing strains clustering closely with the Italian 2017 isolates with other full-length reference strains in the original ORF1+ORF2 alignment file. Uniqueness was verified, and additional strains were identified by performing BLASTp searches for short, 8–amino acid–long peptides spanning the location of each substitution of interests.
RESULTS
We retrieved whole-genome CHIKV sequences from the extracts of 2 mosquitos pools, from the 2 outbreak foci in Anzio, Lazio (isolate Lazio/ISS-1), and Guardavalle, Calabria (isolate Calabria/ISS-1), and from 6 serum samples from patients from Guardavalle after propagation in Vero cells (isolates ISS-S972, ISS-S989, ISS-S991, ISS-S977, ISS-S1011, and ISS-S1028) (Table 1). All the samples were collected between September 8 and October 9, 2017. In addition, 2 isolates from the 2007 outbreak in Italy were sequenced.
Maximum likelihood phylogenies of the concatenated ORF1 and ORF2 nucleotide sequences of ECSA and IOL strains showed a novel major branch of emerging IOL strains from South Asia, namely India, Pakistan, and Italy (Figure 1, blue area). This clade, representing 3 recent outbreaks and several travel-related infections, diverges from the IOL clade. In total, 7 published sequences from the recent Pakistan 2016 outbreak (MF774613, MF774614, MF774615, MF774616, MF774617, MF774618, and MF774619), 2 recent strains from India (KY057363 and KX619426), and 4 travel-associated strains were identified within the divergent Pakistani-Italian cluster (Figure 1, dark blue area). The imported strains were isolated from travelers returning to Australia from India (KY751908) and Bangladesh (MF773566), and in travelers returning to Hong Kong from New Delhi (MF499120, MF503628) [24, 25]. To further explore the degree of divergence of the Pakistani-Italian cluster, we then analyzed the amino acid sequence of nsP4, the viral RdRp (data not shown). The evolutionary divergence of this new cluster was confirmed also in this highly conserved region.
Figure 1. .
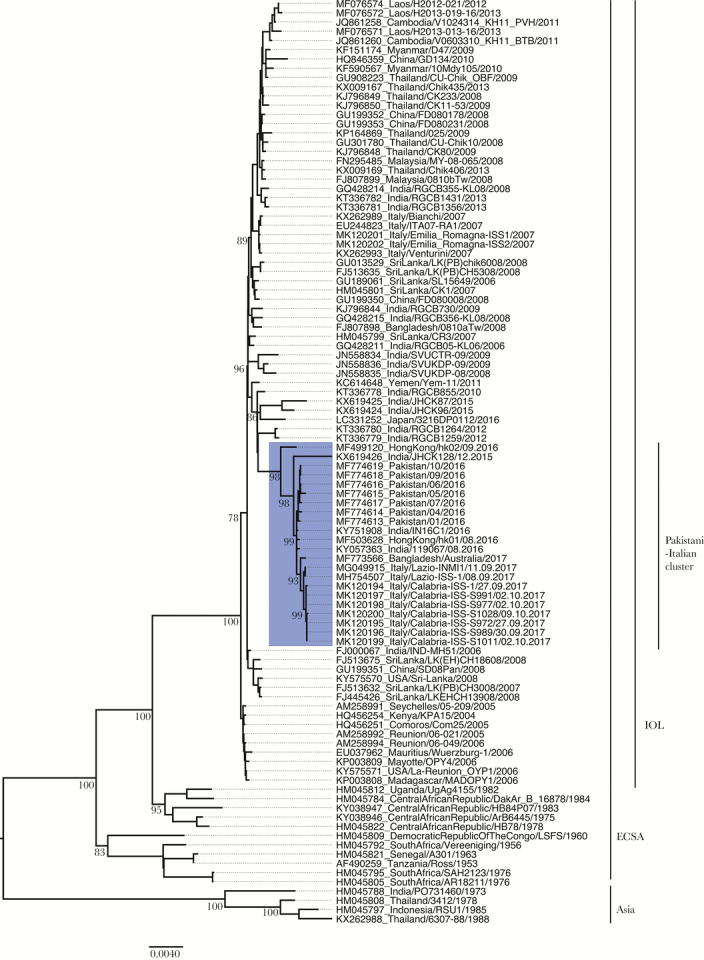
Phylogenetic analysis of ORF1+ORF2 (11.172nt) of chikungunya virus Indian Ocean lineage strains. The analysis was run on an alignment of 90 strains, in addition to the Italian isolates. The maximum likelihood phylogeny applying the GTR+G model was generated in MEGA 6, and tree support was calculated from 1000 bootstrap replicates. The tree was visualized with FigTree. Abbreviations: ECSA, East/Central/South African; IOL, Indian Ocean lineage.
The analysis of the E1 protein (Figure 2) shows distinct amino acid patterns for each of the 3 major genotypes. Unique or distinct substitutions are seen in the ECSA strains at positions 71, 142, 269, and 284; in West African unique or distinct substitutions, they are displayed at positions 145, 276, 296, 321, 347, 379, 404, and 420; and the Asian genotypes present original residues at positions 225 and 304 and heterogeneous substitutions at positions 142, 162, 305, and 397.
Figure 2. .

E1 amino acid patterns across chikungunya virus (CHIKV) evolutionary lineages. The main CHIKV clusters were analyzed to reconstruct the phylogenetic tree of the nucleotide E1 region with the maximum likelihood algorithm and the JTT+G model (500 bootstrap replication). The amino acid hot spot sites able to differentiate different lineages are shown. Abbreviations: ECSA, East/Central/South African; ITA, Italian; PAK, Pakistani; WA, West African.
Unique or distinct amino acid changes in the ORF1 and ORF2 sequences of the Italian 2017 and Pakistani 2016 isolates are summarized in Table 2. The Italian 2017 isolates display all the amino acid changes previously reported for IOL strains excluding the E1 A226V mutation, conferring improved transmission by the Ae. albopictus vector but including the E1 284E mutation at the virion surface [13, 14]. Amino acid mutations distinct to the Pakistani-Italian clade are located in nsP2 (H130Y and E145D), nsP4 (S55N and R85G), E2 (V264A), and E1 (K211E and I317V). Three mutations, nsP3 (D372E, in the hypervariable domain), C (R62C, in the nuclear localization signal), and E2 (G205S), were only observed in the Italian isolates, whereas 2 mutations, nsP1 (P476Q) and nsP4 (V281I), were observed exclusively in the Pakistani viruses. The E1 K211E and E2 V264A mutations, in conjunction with E1 226A, have been described in Indian isolates from outbreaks in New Delhi in 2010 and Tamil Nadu and Kolkata in 2011 and 2012, all of which are Ae. aegypti–dominated areas [26–28]. Interestingly, the nsP4 substitution R/K85G observed in Indian-origin viruses is unique among CHIKV’s but is present in the genomes of 2 other alphaviruses, Western equine encephalitis virus and Madariaga virus.
Table 2. .
Amino Acid Substitutions Characteristic of the Italian 2017 Cluster
| Polyprotein | ORF1 | ORF2 | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Amino acid position | 476 | 665 | 680 | 1705 | 1857 | 1918 | 1948 | 2144 | 62 | 530 | 589 | 1020 | 1035 | 1126 |
| Cleaved protein | nsP1 | nsP2 | nsP3 | nsP4 | C | E2 | E1 | |||||||
| Amino acid position | 476 | 130 | 145 | 372 | 524 | 55 | 85 | 281 | 62 | 205 | 264 | 211 | 226 | 317 |
| AF369024 S27 African prototype | P | H | E | D | R | S | R | V | R | G | V | K | A | I |
| AM258994 IOS Reunion/06.049/2006 | P | H | E | D | * | S | R | V | R | G | V | K | V | I |
| MK120201 Italy/Emilia Romagna-ISS-1/2007 | P | H | E | D | R | S | R | V | R | G | V | K | V | I |
| MK120202 Italy/Emilia Romagna-ISS-2/2007 | P | H | E | D | * | S | R | V | R | G | V | K | V | I |
| KX619426 India/JHCK128/12.2015 | P | Y | D | D | * | N | G | V | R | G | A | E | A | V |
| KY751908 India/IN16C1/2016 | P | Y | D | D | * | N | G | V | R | G | A | E | A | V |
| MF774613 Pakistan/01/2016 | P | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| MF774614 Pakistan/04/2016 | P | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| MF774615 Pakistan/05/2016 | Q | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| MF774616 Pakistan/06/2016 | Q | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| MF774617 Pakistan/07/2016 | Q | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| MF774618 Pakistan/09/2016 | Q | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| MF774619 Pakistan/10/2016 | Q | Y | D | D | * | N | G | I | R | G | A | E | A | V |
| KY057363 India/119067/08.2016 | P | Y | D | D | * | N | G | V | R | G | A | E | A | V |
| MF503628 Hong Kong/hk01/08.2016 | P | Y | D | D | * | N | G | V | R | G | A | E | A | V |
| MF499120 Hong Kong/hk02/09.2016 | P | Y | E | D | * | S | G | V | R | G | A | E | A | V |
| MF773566 Bangladesh/Australia/2017 | P | Y | D | E | * | N | G | V | R | S | A | E | A | V |
| MG049915 Italy/Lazio-INMI1/11.09.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MH754507 Italy/Lazio-ISS-1/08.09.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120194 Italy/Calabria-ISS-1/27.09.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120195 Italy/Calabria-ISS-S972/27.09.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120196 Italy/Calabria-ISS-S989/30.09.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120197 Italy/Calabria-ISS-S991/02.10.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120198 Italy/Calabria-ISS-S977/02.10.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120199 Italy/Calabria-ISS-S1011/02.10.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
| MK120200 Italy/Calabria-ISS-S1028/09.10.2017 | P | Y | D | E | * | N | G | V | C | S | A | E | A | V |
Amino acid alignment of Pakistani 2016 and Italian 2017 isolates with 600 chikungunya isolates revealed a specific substitution pattern unique to this cluster. Further BLASTp searches with short, 9–amino acid–long peptides covering the substitution site sequences identified 6 other strains deposited in GenBank that share most of these substitutions. S27 African prototype virus and the Reunion/06.049 strain representing the IOS clade are shown as the reference. The nsP3 stop codon and E1 226 substitution status for the strains are shown in addition. Amino acid substitutions are unique to the Pakistani-Italian clade. Italian 2017 and Pakistani 2016 viruses are distinguished by 3 amino acid substitutions unique to the Italian viruses (nsP3 D372E, C R62C, and E2 G205S) and 2 substitutions in the Pakistani viruses (nsP1 P476Q and nsP4 V281I), while they share 7 distinct substitutions.
*Opal stop codon.
Between the Italian 2017 CHIKV isolates, only 10, mainly silent, nucleotide changes separated the Lazio cluster from the Calabria cluster (Figure 1). The only amino acid changes within the Italian 2017 isolates are I214T in nsP1 (strain Italy/Calabria-S991/2017) and S1847 in nsP3 (strain Italy/Calabria-S1028/2017). One silent nucleotide substitution was observed between the isolate from a mosquito pool and an isolate from a viremic patient in Anzio, Lazio (described by Carletti and colleagues) [29]. Up to 8 nucleotide changes were observed by comparing isolates from the 2 Italian outbreak foci, indicating that the outbreak virus underwent molecular changes during the transmission period of a few months.
DISCUSSION
The sudden emergence of autochtonous CHIKV infections in several locations in Southern Europe highlights the vulnerability of Europe to the transmission of tropical arboviruses. The first autochthonous spread of CHIKV in Italy occurred in the Emilia Romagna region in 2007 and clearly demonstrated the ability of CHIKV to spread among the local population with the locally established Ae. albopictus mosquito vector. It was caused by a newly emerged IOL variant carrying the E1 A226V mutation [5]. Other autochthonous, Ae. albopictus–sustained, CHIKV transmission chains (involving different CHIKV strains) have been recorded in France in the last decade. In 2010, an ECSA lineage virus from India with E1 211E, E1 226A, and E2264A was detected in 1 locally acquired case [4]. In 2017, a CHIKV from Central Africa harboring the E1 A226V mutation was detected in 7 autochthonous cases [2]. Finally, in the summer of 2017, Italy experienced its second CHIKV Ae. albopictus sustained outbreak, which outnumbered the 2007 outbreak and spread to a secondary focus [7, 17–19].
Herein we described the molecular features of the recent ECSA/IOL isolates from Italy from Ae. albopictus populations and from infected patients in the 2 different locations, Anzio (Latium region), and Guardavalle (Calabria region). The virus is likely to have been initially introduced by a traveler. To assess the likely origin of the isolates and the putative molecular determinants related to Ae. albopictus adaptation and the viral evolution during the spread, we sequenced the whole genomes of the isolates collected during the Italian 2017 outbreak.
Previous analyses by us and others [7, 30] identified the origin of the outbreak to lie in viruses of the ECSA/IOL sublineage, currently circulating in India and Pakistan. Our analyses of the coding regions of CHIKV expand on this assumption and demonstrate that the Pakistani 2016 and Italian 2017 viruses, together with Bangladeshi and Indian viruses, actually form a divergent clade of emerging viruses extending from the IOL clade. Our results further show that the Italian 2017 isolates shared the closest sequence identity with the only Bangladeshi-origin virus available at the time of analysis. This clade is likely to be much wider as regrettably few sequence data were available from South Asia to represent the sequence diversity in the area. In November 2016, an outbreak of CHIKV was declared in Pakistan, and the infection is currently spreading throughout the country [31]. In Bangladesh, the first CHIKV outbreak was reported in 2008, and cases continued to occur sporadically until a major outbreak started in September 2017 [32]. As noted previously, the accuracy of CHIKV phylogenies is improved by using both the ORF 1 and ORF 2 sequences compared with the conventionally used E1 gene sequence [33]. Further confirmation of the observed divergence of viruses in the Pakistani-Italian clade was found in the analysis of the amino acid sequence of the conserved nsP4 protein.
Genomic studies of IOL have revealed a wide genetic diversity and an enhanced ability of IOL viruses to acquire adaptive mutations [33–35]. These factors are likely to play an important role in the epidemic emergence of IOL. After decades of absence, CHIKV returned to India in 2005 in the form of the newly emerged Indian Ocean lineage [36]. The E1 226V variant was the cause of a massive outbreak that occurred in the state of Kerala, where the Ae. albopictus species was the predominant vector, and the likely origin of the virus responsible for the first CHIKV outbreak in Italy in 2007 [5, 37]. Currently, both the wild-type E1 226A and the Ae. Albopictus–adapted E1 226V variants are circulating in parallel in India [36, 37]. The Italian 2017 outbreak virus, displaying E1 226A, demonstrates that the A226V mutation is not a prerequisite for sustained transmission in Southern Europe, where Ae. albopictus is the only known competent CHIKV vector. The convergence from E1 226A to 226V has been observed repeatedly during conditions where Ae. albopictus has been the dominant vector species [16]. The absence of the E1 226V substitution in the Italian 2017 outbreak virus leads to the intriguing question of whether 1 or several of the substitutions observed in the recent Pakistani-Italian cluster may confer a compensating effect on Ae. albopictus competence. As shown by vector competence studies carried out by our group [37], the 2017 isolates replicated as effectively in the Ae. albopictus vector, disregarding its lack of the E1 226V mutation, as the Italian 2007 isolate. Phylogenetic analyses and level of nucleotide identity further show an evolutionary distance between the Italian 2007 and 2017 strains indicative of separate introductions. Long-lasting persistence of CHIKV in Southern Europe is still considered unlikely because of suboptimal winter temperatures for the overwintering of the virus.
Evolution of vector-borne RNA viruses is shaped by both functional requirement of complex interacting tertiary structures in the viral particle and by bottlenecks created during infection, transmission, and spread [34, 39]. Although assessing the functional roles of individual mutations is challenging, they have the potential to significantly elucidate our understanding of transmission dynamics and pathogenesis. The role of specific mutations, especially in E1 and E2, for vector competency, transmission efficiency, and pathogenicity has been approached by several studies in attempts to identify factors that affect CHIKV fitness and dispersion. With respect of specific E1 and E2 mutations we have observed in the 2017 Italian strain, E1 211E is displayed on the surface of the E1-E2 heterodimer and has been shown to be under positive selection pressure and to contribute to weaker neutralization by human immune sera compared with the E1 211K variant commonly found among ECSA strains [28, 40]. Mosquito studies from India have shown that E1 K211E and E2 V264A confer increased replication efficiency in the Ae. aegypti mosquito; however, a similar effect in Ae. albopictus was not seen in experimental infection studies in mosquitoes [26]. The E1 211E substitution in conjunction with E1 226A has been described in IOL strains isolated in India since 2009 [27]. The E1 211E substitution is highly conserved also in the Asian genotype, including the virus that rapidly spread in the Carribean. Effects on tertiary structure and function and on vector competencies of the mutations in the emerging Pakistani-Italian IOL clade described in this study call for further studies.
CONCLUSIONS
Since the establishment of a competent arbovirus virus vector in Southern Europe, local transmission of CHIKV has occurred on several occasions, and the risk of widespread transmission has subsequently increased. South Asia, with constant mosquito activity, can act as a year-round source of arboviruses. We show that the Italian 2017 outbreak virus belonged to a cluster of viruses originating in the IOL clade, with dispersion and transmission potential demonstrated by recent outbreaks and travel-associated infections. Their emergence calls for enhanced monitoring and strengthened preparedness measures, including vector control programs and raised awareness among general practitioners in countries potentially at risk. The distinct mutations observed in the Pakistani-Italian cluster and the absence of the E1 A226V mutation in the outbreak strain warrant further studies to elucidate their functional roles in different environments and vector species.
Acknowledgments
Financial support. This work was partially supported by the Italian Ministry of the Defense.
Potential conflicts of interest. The authors declare that they have no affiliations with or involvement in any organization or entity with any financial interest or nonfinancial interest (such as personal or professional relationships, affiliations, knowledge, or beliefs) in the subject matter or materials discussed in this manuscript. All authors: no reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. European Centre for Disease Preventiona and Control (ECDC). Clusters of autochthonous Chikungunya cases in Italy. 2017. https://ecdc.europa.eu/sites/portal/files/documents/14-Sep-2017-RRA-Chikungunya-Italy-revised.pdf [Google Scholar]
- 2. Calba C, Guerbois-Galla M, Franke F, et al. Preliminary report of an autochthonous chikungunya outbreak in France, July to September 2017. Euro Surveill. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Delisle E, Rousseau C, Broche B, et al. Chikungunya outbreak in Montpellier, France, September to October 2014. Euro Surveill 2015; 20. [DOI] [PubMed] [Google Scholar]
- 4. Grandadam M, Caro V, Plumet S, et al. Chikungunya virus, southeastern France. Emerg Infect Dis 2011; 17:910–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rezza G, Nicoletti L, Angelini R, et al. ; CHIKV study group Infection with chikungunya virus in Italy: an outbreak in a temperate region. Lancet 2007; 370:1840–6. [DOI] [PubMed] [Google Scholar]
- 6. Romi R, Toma L, Severini F, Di Luca M. Twenty years of the presence of Aedes albopictus in Italy. From the annoying pest mosquito to the real disease vector. Euro Infect Dis 2008; 2:98–101. [Google Scholar]
- 7. Venturi G, Di Luca M, Fortuna C, et al. Detection of a chikungunya outbreak in central Italy, August to September 2017. Euro Surveill. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen R, Mukhopadhyay S, Merits A, et al. ICTV virus taxonomy profile: togaviridae. J Gen Virol 2018; 99:761–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gasque P, Bandjee MC, Reyes MM, Viasus D. Chikungunya pathogenesis: from the clinics to the bench. J Infect Dis 2016; 214:446–8. [DOI] [PubMed] [Google Scholar]
- 10. Halstead SB. Reappearance of chikungunya, formerly called dengue, in the Americas. Emerg Infect Dis 2015; 21:557–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ross RW. The Newala epidemic. III. The virus: isolation, pathogenic properties and relationship to the epidemic. J Hyg (Lond) 1956; 54:177–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen R, Puri V, Fedorova N, et al. Comprehensive genome scale phylogenetic study provides new insights on the global expansion of chikungunya virus. J Virol 2016; 90:10600–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schuffenecker I, Iteman I, Michault A, et al. Genome microevolution of chikungunya viruses causing the Indian Ocean outbreak. PLoS Med 2006; 3:e263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsetsarkin KA, Vanlandingham DL, McGee CE, Higgs S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog 2007; 3:e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vazeille M, Zouache K, Vega-Rúa A, et al. Importance of mosquito “quasispecies” in selecting an epidemic arthropod-borne virus. Sci Rep 2016; 6:29564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. de Lamballerie X, Leroy E, Charrel RN, et al. Chikungunya virus adapts to tiger mosquito via evolutionary convergence: a sign of things to come? Virol J 2008; 5:33, 422X-5–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. European Centre for Disease Prevention and Control (ECDC). Clusters of autochthonous chikungunya cases in Italy. 2017. https://ecdc.europa.eu/sites/portal/files/documents/RRAchikungunya-Italy-update-9-Oct-2017.pdf. Accessed January 3, 2018. [Google Scholar]
- 18. European Centre for Disease Prevention and Control (ECDC). ECDC mosquito maps. 2017. https://ecdc.europa.eu/en/diseasevectors/surveillance-and-disease-data/mosquito-maps. Accessed January 3, 2018. [Google Scholar]
- 19. European Centre for Disease Prevention and Control (ECDC), Istituto Superiore di Sanita (ISS) Surveillance atlas of infectious disease. https://www.ecdc.europa.eu/en/surveillance-atlas-infectious-diseases Accessed January 3, 2018. [Google Scholar]
- 20. Istituto Superiore di Sanita (ISS) Bulletin of the national plan of surveillance and response to arbovirus transmitted by mosquitoes (Aedes sp.), with particular reference to chikungunya, dengue and Zika viruses. 2017. http://www.salastampa.salute.gov.it/portale/temi/documenti/chikungunya/bollettino_chikungunya_20171013.pdf Accessed February 22, 2018. [Google Scholar]
- 21. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014; 30:2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 2016; 33:1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. FigTree Tree Figure Drawing Tool, version 1.4.2 (200 333 6–2014). http://tree.bio.ed.ac.uk/software/figtree/. Accessed November 15, 2018. [Google Scholar]
- 24. Huang B, Pyke AT, McMahon J, Warrilow D. Complete coding sequence of a case of chikungunya virus imported into Australia. Genome Announc. 2017; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho DTW, Chan DPC, Lam CY, et al. At the advancing front of chikungunya fever in Asia: two imported cases in Hong Kong with novel amino acid changes. J Microbiol Immunol Infect 2018; 51:419–21. [DOI] [PubMed] [Google Scholar]
- 26. Agarwal A, Sharma AK, Sukumaran D, et al. Two novel epistatic mutations (E1:K211E and E2:V264A) in structural proteins of chikungunya virus enhance fitness in Aedes aegypti. Virology 2016; 497:59–68. [DOI] [PubMed] [Google Scholar]
- 27. Shrinet J, Jain S, Sharma A, et al. Genetic characterization of chikungunya virus from New Delhi reveal emergence of a new molecular signature in Indian isolates. Virol J 2012; 9:100, 422X-9–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sumathy K, Ella KM. Genetic diversity of chikungunya virus, India 2006–2010: evolutionary dynamics and serotype analyses. J Med Virol 2012; 84:462–70. [DOI] [PubMed] [Google Scholar]
- 29. Carletti F, Marsella P, Colavita F, et al. Full-length genome sequence of a chikungunya virus isolate from the 2017 autochthonous outbreak, Lazio region, Italy. Genome Announc. 2017; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cella E, Riva E, Salemi M, et al. The new chikungunya virus outbreak in Italy possibly originated from a single introduction from Asia. Pathog Glob Health 2018; 112:93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu SQ, Li X, Zhang YN, et al. Detection, isolation, and characterization of chikungunya viruses associated with the Pakistan outbreak of 2016-2017. Virol Sin 2017; 32:511–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Melan A, Aung MS, Khanam F, et al. Molecular characterization of chikungunya virus causing the 2017 outbreak in Dhaka, Bangladesh. New Microbes New Infect 2018; 24:14–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Volk SM, Chen R, Tsetsarkin KA, et al. Genome-scale phylogenetic analyses of chikungunya virus reveal independent emergences of recent epidemics and various evolutionary rates. J Virol 2010; 84:6497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tsetsarkin KA, Weaver SC. Sequential adaptive mutations enhance efficient vector switching by chikungunya virus and its epidemic emergence. PLoS Pathog 2011; 7:e1002412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen R, Wang E, Tsetsarkin KA, Weaver SC. Chikungunya virus 3’ untranslated region: adaptation to mosquitoes and a population bottleneck as major evolutionary forces. PLoS Pathog 2013; 9:e1003591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arankalle VA, Shrivastava S, Cherian S, et al. Genetic divergence of chikungunya viruses in India (1963–2006) with special reference to the 2005–2006 explosive epidemic. J Gen Virol 2007; 88:1967–76. [DOI] [PubMed] [Google Scholar]
- 37. Kumar NP, Joseph R, Kamaraj T, Jambulingam P. A226V mutation in virus during the 2007 chikungunya outbreak in Kerala, India. J Gen Virol 2008; 89:1945–8. [DOI] [PubMed] [Google Scholar]
- 38. Fortuna C, Toma L,Remoli ME, et al. Vector competence of Aedes albopictus for the Indian Ocean lineage (IOL) chikungunya viruses of the 2007 and 2017 outbreaks in Italy: a comparison between strains with and without the E1:A226V mutation. Euro Surveill. 2018; 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Forrester NL, Guerbois M, Seymour RL, et al. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog 2012; 8:e1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chua CL, Sam IC, Merits A, Chan YF. Antigenic variation of East/Central/South African and Asian chikungunya virus genotypes in neutralization by immune sera. PLoS Negl Trop Dis 2016; 10:e0004960. [DOI] [PMC free article] [PubMed] [Google Scholar]