

Abstract
In colloidal Cu2–xS nanocrystal synthesis, thiols are often used as organic ligands and the sulfur source, as they yield high-quality nanocrystals. However, thiol ligands on Cu2–xS nanocrystals are difficult to exchange, limiting the applications of these nanocrystals in photovoltaics, biomedical sensing, and photocatalysis. Here, we present an effective and facile procedure to exchange native 1-dodecanethiol on Cu2–xS nanocrystals by 3-mercaptopropionate, 11-mercaptoundecanoate, and S2– in formamide under inert atmosphere. The product hydrophilic Cu2–xS nanocrystals have excellent colloidal stability in formamide. Furthermore, the size, shape, and optical properties of the nanocrystals are not significantly affected by the ligand exchange. Water-dispersible Cu2–xS nanocrystals are easily obtained by precipitation of the nanocrystals capped by S2–, 3-mercaptopropionate, or 11-mercaptoundecanoate from formamide, followed by redispersion in water. Interestingly, the ligand exchange rates for Cu2–xS nanocrystals capped with 1-dodecanethiol are observed to depend on the preparation method, being much slower for Cu2–xS nanocrystals prepared through heating-up than through hot-injection synthesis protocols. XPS studies reveal that the differences in the ligand exchange rates are due to the surface chemistry of the Cu2–xS nanocrystals, where the nanocrystals prepared via hot-injection synthesis have a less dense ligand layer due to the presence of trioctylphosphine oxide during synthesis. A model is proposed that explains the observed differences in the ligand exchange rates. The facile ligand exchange procedures reported here enable the use of high-quality colloidal Cu2–xS nanocrystals prepared in the presence of 1-dodecanethiol in various applications.
Introduction
Copper sulfide (Cu2–xS) nanocrystals have attracted increasing attention in the past decade for their use in various applications, such as biomedical sensing,1−3 photothermal therapy,2−4 photovoltaic and plasmonic devices,2,5−8 and photo- and electrocatalysis.2,5,9,10 Cu2–xS can accommodate copper vacancies and hence can exist in a variety of compositions and crystal structures.11 The interest in Cu2–xS can be understood from its unique optoelectronic properties that depend on the size, shape, and composition of the nanocrystals. The material is a p-type semiconductor that has a stoichiometry dependent, direct band gap ranging from 1.2 eV for chalcocite (Cu2S) up to 2.0 eV for covellite (CuS).11−13 Furthermore, depending on the composition, Cu2–xS nanocrystals show a broad absorption in the near-infrared spectral region. This absorption is related to localized surface plasmon resonances (LSPR) of the holes in the top of the valence band, which are introduced in the material by copper vacancies.5−7,12 In addition, Cu2–xS is of interest for various applications due to its abundance and low toxicity.5,11,14
Due to the dependence of the optical and electronic properties on nanocrystal size, shape, and composition, the preparation of monodisperse samples with uniform properties is of great interest. Colloidal synthesis methods are known to allow for a high degree of control over the size, shape, and composition of the nanocrystals.15 For example, nanoplatelets, nanosheets, nanodisks, and spherical Cu2–xS nanocrystals have been synthesized with narrow size and shape distributions.16−22 In colloidal synthesis, nanocrystals are grown in solution by chemical precipitation reactions of copper and sulfur precursors in the presence of organic ligands. The best known colloidal synthesis technique is hot-injection, in which the precursors are rapidly injected into a preheated reaction mixture containing the other reagents. The rapid injection leads to a high availability of monomers and hence a burst of nucleation, followed by further growth of the existing nuclei. Although hot-injection synthesis protocols are well-developed and can give nanocrystals with a large range of sizes, shapes, and compositions, they usually only yield a small amount of product and cannot be easily scaled up, as factors like injection rate and mass and heat transport become less reproducible for larger reaction volumes.23 An alternative to the hot-injection method is the so-called heating-up method, where all reagents are mixed in a reaction flask and heated to induce nucleation and growth of the nanocrystals. This method allows for a more reproducible and scalable synthesis, since factors such as injection rate and nonideal mixing do not play a role.23
Organic ligands are important during the synthesis as they allow for the high control over size, shape, and composition of the nanocrystals by dynamically binding to the nanocrystal surface.15 Furthermore, after synthesis they prevent aggregation of the nanocrystals by steric stabilization. These ligands usually have a polar headgroup coordinating to the nanocrystal surface atoms through a donor atom (e.g., S, N, O) and a long apolar hydrocarbon tail that stabilizes the nanocrystals in apolar solvents.15 Commonly used ligands include phosphines, amines, and thiols. Though ligands are essential during and after synthesis, they lead to challenges when using the nanocrystals in applications. The apolar nature of the hydrocarbon chain prevents dispersion of the nanocrystals in water and other polar solvents, limiting their use in, e.g., biomedical and photocatalytic applications. Furthermore, the bulky organic ligands can restrict interparticle conductivity and can render the nanocrystal surface inaccessible to reactants, hindering the use of the nanocrystals in optoelectronic, photovoltaic, and photo- and electrocatalytic applications. As a consequence, ligand exchange is crucial to obtain colloidal nanocrystals suitable for a broad range of applications. Various procedures have been reported for ligand exchange on colloidal nanocrystals. For example, Nag et al. reported the preparation of all-inorganic nanocrystals by replacing native organic ligands with inorganic ions such as S2–, HS–, and OH–.24 Kovalenko et al. described the stabilization of colloidal nanocrystals in polar solvents by using molecular metal chalcogenide ligands, such as Sn2S64–.25,26 By a subsequent heating step, the ligands could be converted into semiconducting phases, realizing conductive arrays of nanoparticles. Aqueous phase transfers have also been extensively reported using organic ligands such as 3-mercaptopropionic acid (MPA),27−33 11-mercaptoundecanoic acid (MUA),31−35 cysteine,36 and various polymers.37−39
In Cu2–xS nanocrystal synthesis, thiols are often used as ligands as they give high quality, monodisperse products.40,41 However, no effective ligand exchange procedures have been reported for Cu2–xS and CuInS2 nanocrystals prepared via direct synthesis in the presence of thiols.40,42−47 Xie et al. reported the resistance of CuInS2 nanocrystals obtained with thiol ligands toward a ligand exchange using MPA.42 Turo et al. described the resistance of Cu2–xS nanocrystals prepared with 1-dodecanethiol (DDT) toward ligand exchange procedures and attributed this to the presence of so-called crystal-bound thiols, where the thiols occupy high coordination number sites and are thus strongly bound to the nanocrystal.40 A similar explanation has been given by other groups.43−45 More recently, Gromova et al. reported on the difficulty of surface functionalization of CuInS2 nanocrystals prepared in the presence of DDT. They attributed this to the presence of a ligand double layer on the nanocrystals surface, consisting of dodecanethiolate ligands and thioether species formed in situ during the heating-up synthesis in the presence of thiols.46
In this work, we report on ligand exchange procedures for Cu2–xS nanocrystals prepared by hot-injection and heating-up synthesis protocols in the presence of DDT. In contrast to the studies mentioned above,40,42−47 we developed procedures to replace DDT on Cu2–xS nanocrystals with MPA–, MUA–, or S2– by performing the ligand exchange procedures under inert atmosphere in a highly polar solvent (formamide). In addition, we found that the ligand exchange rates are highly dependent on both the nature of the replacing ligands (MPA–, MUA–, or S2–) and on the synthesis method used to produce the Cu2–xS nanocrystals (viz., hot-injection or heating-up). XPS studies revealed that the differences observed between the ligand exchange rates involving nanocrystals prepared by the different synthesis protocols are due to the surface chemistry of the nanocrystals, where those synthesized by the heating-up protocol have a denser native thiolate ligand layer, in comparison with those prepared by the hot-injection method in the presence of trioctylphosphine oxide (TOPO). Finally, we propose a model for ligand exchange on thiolate capped Cu2–xS nanocrystals that explains the observed differences in the ligand exchange rates. The ligand exchange protocols developed in this work present a facile way to achieve water-dispersible Cu2–xS nanocrystals with, depending on the replacing ligand, accessible nanocrystal surfaces.
Results and Discussion
Ligand Exchange Procedures on Cu2xS Nanocrystals Prepared with Thiols
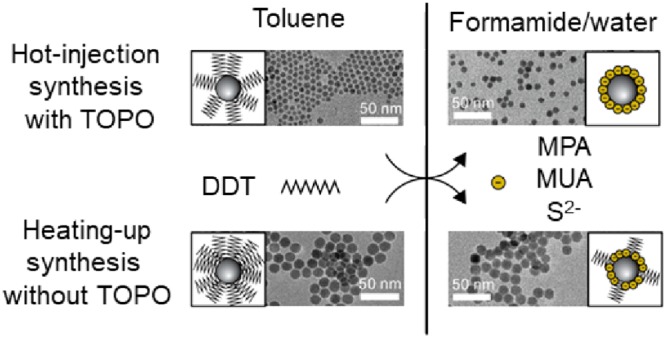
Cu2–xS nanocrystals were synthesized according to two previously described protocols, where DDT was used as both sulfur source and ligand (see Experimental Section for details). In the first method, Cu2–xS nanocrystals were synthesized using a hot-injection technique. DDT was injected into a mixture of Cu(I)acetate, trioctylphosphineoxide (TOPO), and 1-octadecene (ODE).34,48 During this synthesis, both TOPO and DDT ligands direct the growth of the nanocrystals.48 These nanocrystals will hereafter be referred to as HI_DDT/TOPO. Cu2–xS nanocrystals were also synthesized by heating CuSO4·5H2O, DDT, and oleic acid (OA) to 200 °C in a so-called heating-up synthesis.49 These nanocrystals will hereafter be referred to as HU_DDT.
To replace the native ligands on the Cu2–xS nanocrystals and obtain water-dispersible Cu2–xS nanocrystals, ligand exchange procedures using MPA, MUA, and S2– (either from (NH4)2S or Na2S) were studied. In a typical ligand exchange reaction a two-phase system was formed by combining the Cu2–xS nanocrystals in an apolar solvent (e.g., toluene) with an excess of the replacing ligand (MPA, MUA, or S2–) in polar solvent (e.g., water or formamide). The pH of the solutions containing MUA was increased to 11 using tetramethylammonium hydroxide (TMAH) in order to deprotonate the carboxylic acid group, thereby rendering the MUA water-soluble. The solutions containing S2– were also alkaline as the S2– undergoes hydrolysis, forming HS– and OH–. In contrast, the MPA solution has a low pH of 3 at the start of the ligand exchange experiments. Although not needed for the solubility of MPA in formamide or water, experiments using MPA were also performed at pH 11 to compare the different replacing ligands under identical conditions. No differences in phase transfer rate were observed between the MPA experiments carried out at low and those carried out at high pHs. Therefore, for the sake of conciseness, only experiments using MPA at low pH are further discussed.
The success of the ligand exchange was evidenced by transfer of the Cu2–xS nanocrystals from the apolar to the polar phase (Supporting Information, Figure S1). Different experimental conditions were studied for the ligand exchange experiments. First, experiments were performed in air using water as the polar solvent. Subsequently, experiments were performed in air, using formamide as the polar solvent. Lastly, experiments were performed under air-free conditions and using formamide as the polar solvent. The three different sets of experiments are discussed in more detail below.
At first, ligand exchange procedures were performed using water in air, as described elsewhere.24,34 Using this procedure, ligand exchange using MPA– and S2– (either from (NH4)2S or Na2S) as the replacing ligands was successful. However, the colloidal stability of the product nanocrystals was low, and aggregates formed during the phase transfer process. In addition, the sample obtained using (NH4)2S turned from black to blue after several hours, suggesting the formation of Cu2+–ammonia complexes. Copper has a valency of +1 in the Cu2–xS nanocrystals, but in the presence of oxygen, part of the Cu+ ions may be oxidized to Cu2+.40,50 In aqueous solution, the ammonium ion can dissociate into ammonia and a proton through NH4+ (aq) ⇌ NH3 (aq) + H+(aq). In the presence of NH3(aq), Cu2+ can then form the stable [Cu(NH3)4]2+ complex that has an intense violet-blue color.51,52 Due to the instability of the Cu2–xS nanocrystals in the presence of NH3/NH4+, (NH4)2S was not used as the S2– source in further experiments. When using S2– as the replacing ligand, both the HI_DDT/TOPO and the HU_DDT nanocrystals were transferred to the polar water phase upon stirring for about 5 h. When MPA was used as the replacing ligand, both the HI_DDT/TOPO and the HU_DDT nanocrystals were transferred to the polar aqueous MPA solution upon stirring overnight. In contrast, when MUA was used as the replacing ligand, only partial transfer of the HI_DDT/TOPO nanocrystals was observed upon stirring overnight, while for the HU_DDT nanocrystals no phase transfer was observed, even after stirring for 3 days.
As the polarity of the solvent could influence the success of the ligand exchange by stabilizing charged intermediates, the ligand exchange was also performed using formamide as the polar solvent under ambient conditions. Formamide has a higher polarity (ε = 106) than water (ε = 80).24 However, upon stirring for several hours, the color of the MUA and Na2S solution layers changed from colorless to black and then to violet-blue, while the apolar phase initially containing the Cu2–xS nanocrystals turned from black to colorless. This indicates that the Cu2–xS nanocrystals were successfully transferred to the polar phase but were not stable after the transfer. The dark blue color again suggests the formation of Cu2+ complexes, where formamide itself can act as the ligand. Complex formation between Cu2+ and formamide has been observed in oxidative and alkaline environments,53 where Cu2+ was leaking into solution from either Cu2S54 or copper foil55,56 and was quickly coordinated by formamide. The blue color was also observed when using MPA, but only after storing the MPA-capped Cu2–xS nanocrystals in formamide for several days in air. The alkaline nature of the solutions containing MUA or Na2S when compared to the more acidic MPA solution when initiating the ligand exchange experiments can possibly explain the slower complex formation between Cu2+ and formamide observed in MPA solutions.54
To prevent the oxidation of Cu+ to Cu2+ and the rapid complexation of Cu2+ by formamide, the ligand exchange experiments using formamide were performed under inert atmosphere in a nitrogen glovebox (≤6 ppm of O2). We found that under these conditions the native ligands on the Cu2–xS nanocrystals could successfully be replaced with all three ligands (MPA–, MUA–, and S2– from Na2S). Colloidal stability in the polar formamide was obtained by binding the negatively charged MPA–, MUA–, and S2– to the nanocrystal surface. The negative charge of the ligands was confirmed with ζ-potential measurements. Cu2–xS nanocrystals prepared by hot-injection synthesis gave ζ-potentials of −44.4 mV, −31.1 mV, and −34.9 mV for nanocrystals capped with S2–, MPA–, and MUA–, respectively (Supporting Information, Figure S2). For the Cu2–xS nanocrystals prepared by heating-up synthesis, the ζ-potentials were −31.3 mV, −33.9 mV, and −34.0 mV for nanocrystals capped with S2–, MPA–, and MUA–, respectively (Supporting Information, Figure S2). The resulting hydrophilic nanocrystals have a high colloidal stability and were stored in formamide inside the glovebox for several months without losing colloidal stability. To obtain water-dispersible Cu2–xS nanocrystals, the particles were precipitated using acetonitrile as antisolvent and isolated by centrifugation, followed by redispersion in water. When dispersed in water, slight turbidity developed over time (on a time scale of 10 min for S2– capped nanocrystals and days for MPA– and MUA– capped nanocrystals), indicating that the colloidal stability of the hydrophilic Cu2–xS nanocrystals is lower in water than in formamide. This difference in colloidal stability is ascribed to the polarity of the solvent, where the charged nanocrystals are better stabilized in the highly polar formamide (ε = 106) than in the less polar water (ε = 80).24
Impact of Ligand Exchange on Nanocrystal Size, Crystal Structure, and Optical Properties
Figure 1 shows transmission electron microscopy (TEM) images of the Cu2–xS nanocrystals before (Figure 1A,D) and after (Figure 1B,E) ligand exchange with S2– in formamide under an inert atmosphere. The corresponding particle size histograms (Figure 1C,F) show the size of the Cu2–xS nanocrystals without the ligand layer. Before ligand exchange, a particle size of 7.1 ± 0.8 nm was found for the nanocrystals prepared by hot-injection synthesis, whereas the nanocrystals prepared by heating-up synthesis had a size of 12.3 ± 1.2 nm. TEM analysis shows that the size and shape of the nanocrystals were not significantly affected by exchanging the native DDT ligands by any of the replacing ligands (see Figure 1 for S2–, and Supporting Information, Figure S3, for MPA and MUA).
Figure 1.

TEM images of HI_DDT/TOPO (A) before and (B) after ligand exchange with S2– and (C) corresponding particle size histograms. TEM images of HU_DDT (D) before and (E) after ligand exchange with S2– and (F) corresponding particle size histograms.
In addition, the TEM images show that the nanocrystals assembled in organized arrays when dried on the TEM grid (Figure 1). The separation between the nanocrystals in such an array can be related to the organic ligands capping the nanocrystals. The interparticle distance was 1.7 ± 0.2 nm for both the as-synthesized sample prepared by hot-injection in the presence of TOPO and by heating-up synthesis without TOPO. This distance is very similar to the DDT chain length (viz., 1.8 nm).57 Upon ligand exchange with S2– the nanocrystals come in very close proximity to each other (Figure 1B,E). This can be explained by the replacement of 1-dodecanethiol by the smaller S2–.
The crystal structure of the Cu2–xS nanocrystals before and after ligand exchange was investigated using X-ray diffraction (XRD) (Figure 2A). Prior to ligand exchange, the nanocrystals prepared by both hot-injection and heating-up synthesis can be identified as chalcocite (Cu1.997–2.0S) or djurleite (Cu1.94S). Based on the XRD patterns, no distinction can be made between these two phases. However, as the copper-deficient djurleite phase is more thermodynamically stable due to its lower crystallographic symmetry compared to the chalcocite phase, the nanocrystals are likely in the djurleite phase.8 After ligand exchange with S2– and phase transfer to formamide, the Cu2–xS nanocrystals remain in the djurleite phase (Supporting Information, Figure S4). However, after phase transfer to water and exposure to air the nanocrystals partially oxidized to a roxbyite (Cu1.81S) phase (Figure 2). This can be explained by the formation of copper vacancies under oxygen. In this phase, the copper atoms in the Cu2–xS nanocrystal remain in the +1 oxidation state, whereas the formal valency of sulfur partially evolves from −2 to −1.58 This partial oxidation was observed for both the samples prepared by hot-injection and by heating-up synthesis when replacing DDT with MUA or MPA using water as the polar solvent (Supporting Information, Figure S5).
Figure 2.
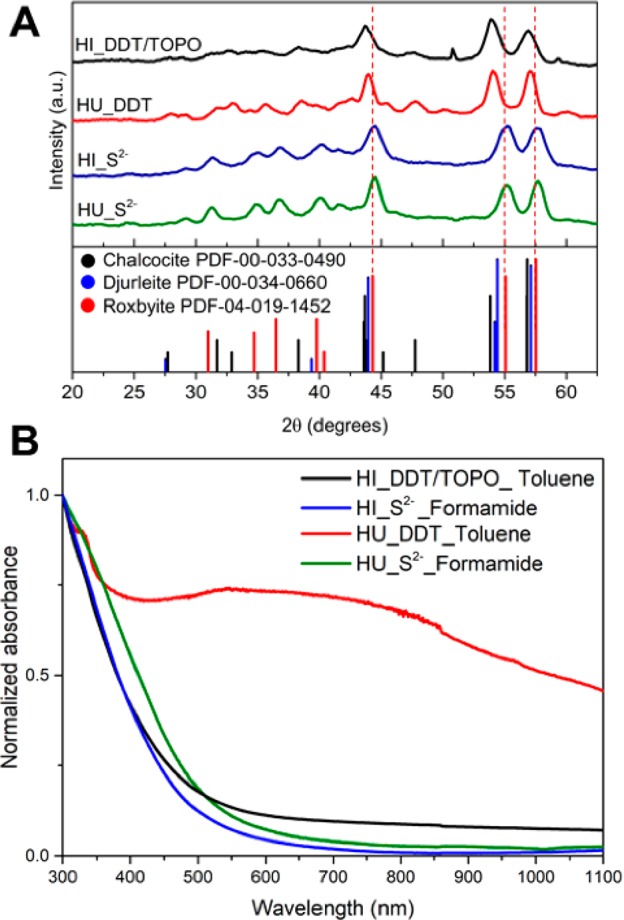
(A) X-ray diffractograms of the Cu2–xS nanocrystals before (HI_DDT/TOPO and HU_DDT) and after ligand exchange procedures with S2– and phase transfer to water (HI_S2–, HU_S2–). The dashed lines mark the position of the roxbyite peaks. (B) Absorption spectra of Cu2–xS nanocrystals before (HI_DDT/TOPO and HU_DDT) and after ligand exchange with S2–.
The absorption spectra of the Cu2–xS nanocrystals before and after ligand exchange with S2– are shown in Figure 2B. Colloidal Cu2–xS nanocrystals typically show a broad and featureless absorption with an onset in the visible region, accompanied by a lower energy tail,6,8,13 which can be ascribed to the presence of excess holes in the valence band (the so-called Urbach tail).13 Excess holes (i.e., p-doping) are commonly observed in Cu-chalcogenide nanocrystals due to Cu-vacancies and also give rise to a broad absorption band in the NIR due to localized surface plasmon resonances (LSPR) that emerge when the density of holes is sufficiently large (i.e., when the concentration of Cu-vacancies is sufficiently high).6,8,13,59 The absorption spectra of the Cu2–xS nanocrystals prepared by hot-injection indeed show a clear absorption onset due to the band-edge absorption transition and a weak tail toward longer wavelengths. The absorption in the NIR is negligible, showing that LSPR bands are not present. These observations imply that the concentration of Cu-vacancies is very low, consistent with the observed crystal structure (Figure 2A). The absorption spectrum after ligand exchange with S2– is essentially unchanged, indicating that the optical properties of the Cu2–xS nanocrystals are not affected by the ligand exchange. The absorption spectrum of the Cu2–xS nanocrystals prepared by heating-up is quite different, with a broad feature between 500 and 1000 nm that is not observed in the samples prepared by hot-injection. This feature cannot be ascribed to an LSPR band because it occurs at too short wavelengths. In addition, the spectral position of this feature shifts after the sample is placed in a sonic bath. Hence, we ascribe this spectral feature to light scattering due to the presence of nanocrystal aggregates.6 Importantly, upon ligand exchange with S2–, this feature is no longer observed in the absorption spectrum, which shows instead the characteristic absorption of nearly stoichiometric Cu2–xS nanocrystals, being very similar to that of the nanocrystals synthesized by hot-injection (Figure 2B). This is consistent with the fact that no aggregates are observed after the ligand exchange procedure in either of the samples. Similar spectra are obtained upon exchange with MUA– and MPA– (Supporting Information, Figure S6). These observations confirm that the optical properties of the Cu2–xS nanocrystals are preserved upon ligand exchange and phase transfer.
Dependence of the Ligand Exchange Rates on the Native and Replacing Ligand and Synthesis Method
The difficulty of ligand exchange on Cu2–xS nanocrystals has been ascribed to the presence of thiolate ligands at the nanocrystal surface.40,46 To verify whether other ligands are easier to replace, the ligand exchange procedures used for DDT-capped Cu2–xS nanocrystals were also performed on Cu2–xS nanocrystals capped by oleylamine. Oleylamine is expected to bind weakly to the Cu2–xS nanocrystal surface, because it is a relatively hard Lewis base and consequently will not have a strong interaction with the soft Lewis acid Cu+ in the Cu2–xS nanocrystal. First, Cu2–xS nanocrystals of ∼7 nm capped with oleylamine ligands were synthesized (Figure 3A). Similar to the Cu2–xS nanocrystals prepared with DDT, the obtained nanocrystals were in a djurleite (Cu1.94S) crystal phase (Figure 3B).
Figure 3.

(A) TEM image and (B) X-ray diffractogram of Cu2–xS nanocrystals as-synthesized capped with oleylamine. TEM images of the oleylamine capped Cu2–xS nanocrystals after ligand exchange with (C) S2– and (D) MUA.
As expected from their lower binding strength, oleylamine ligands on the Cu2–xS nanocrystals were rapidly replaced by MPA–, MUA–, and S2– using the methods described above, leading to much faster phase transfer than observed for the DDT-capped Cu2–xS nanocrystals. For example, when replacing oleylamine for S2– using Na2S in formamide under inert atmosphere, phase transfer of the Cu2–xS nanocrystals from toluene to formamide occurred within minutes, while it took about 1 h for DDT-capped Cu2–xS nanocrystals under the same conditions. However, whereas the size and shape of the nanocrystals were well preserved in ligand exchange procedures using S2– as the replacing ligand on DDT-capped Cu2–xS nanocrystals (see Figure 1 above), the product nanocrystals from the oleylamine-capped Cu2–xS nanocrystals were highly aggregated (Figure 3C). A possible explanation for this observation is the fast rate of ligand exchange for the oleylamine-capped Cu2–xS nanocrystals. The fast stripping of the native ligands during the exchange could lead to an abrupt destabilization of the nanocrystals if the incoming S2– ligands cannot provide sufficient charge stabilization of the nanocrystals fast enough, causing the nanocrystals to aggregate while at the interface between the two phases. This hypothesis is supported by the observation that when the bulkier MUA is used as the replacing ligand on the oleylamine-capped nanocrystals, the phase transfer was slower (on the order of hours) than when using S2– (on the order of minutes) and the product nanocrystals did preserve their size and shape, with negligible aggregation (Figure 3D). The negligible aggregation of the MUA–-capped Cu2–xS nanocrystals can be ascribed to the ability of MUA– ligands to provide both charge and steric stabilization, in contrast to S2– ligands. The fast phase transfer observed for oleylamine-capped Cu2–xS nanocrystals demonstrates that indeed oleylamine ligands are weakly bound to the Cu2–xS nanocrystal surface and are thus more easily replaced than DDT ligands.
The native DDT ligands on the Cu2–xS nanocrystals were replaced by all three ligands investigated when the ligand exchange was performed under inert atmosphere using formamide as polar solvent. However, a significant difference was observed in the kinetics of the ligand exchange for the three different replacing ligands. The kinetics of the ligand exchange was determined by the time it took for Cu2–xS nanocrystals to transfer from the apolar to the polar phase. This phase transfer only occurs after a sufficiently large number of apolar DDT molecules has been exchanged by polar ligands. The kinetics of the phase transfer process thus reflects the cumulative rates of a series of individual ligand exchange steps. The time it takes for phase transfer to occur can then be directly related to the ligand exchange rates, being shorter for faster exchange rates. Therefore, we will discuss the kinetics of the phase transfer in terms of ligand exchange rates. The ligand exchange was fastest when using S2–. Here, phase transfer of the Cu2–xS nanocrystals prepared by hot-injection synthesis was already observed upon stirring for 1 h. In contrast, ligand exchange was observed only after 2 hours for MPA and only after 12 hours for MUA. The same trend in ligand exchange rates with replacing ligand was observed for nanocrystals prepared by heating-up synthesis. The origin of the difference in the ligand exchange rates with the replacing ligands is discussed in more detail later. Besides the dependence of the ligand exchange rate on the replacing ligand, we found that the synthesis method of the Cu2–xS nanocrystals (hot-injection with TOPO or heating-up without TOPO) also induced differences in the ligand exchange rates. The observed trend in all experiments was that the heating-up method gives Cu2–xS nanocrystals that are more resistant toward ligand exchange than Cu2–xS nanocrystals prepared by hot-injection synthesis with TOPO. To better understand this difference, additional experiments were performed.
A possible factor influencing the success of ligand exchange could be the formation of superstructures by self-organization of monodisperse nanocrystals. These superstructures could hinder the ligand exchange as the nanocrystals that are packed in the interior of the superlattices are shielded from the surroundings and thus from the incoming ligands. Besides single nanocrystals (Figure 1A,D), TEM evidenced the presence of these superstructures in both the samples prepared by hot-injection synthesis (Figure 4A) and by heating-up synthesis. The presence of superstructures in the colloidal suspensions is verified using dynamic light scattering (DLS), where hydrodynamic sizes of 1564 ± 316 nm and 932 ± 184 nm were observed for the samples prepared by heating-up and hot-injection, respectively (Figure 4b). Assuming a DDT chain length of 1.8 nm,60 the sizes of the Cu2–xS nanocrystals including ligand layer are ∼16 nm for HU_DDT/TOPO and ∼11 nm for HI_DDT. For both samples, the hydrodynamic size found by DLS was much larger than the size of the Cu2–xS nanocrystals including the ligand shell, which indicates that large nanocrystal superstructures are present in the colloidal dispersion. The larger size of the superstructures observed for the sample prepared by heating-up synthesis is attributed to the larger particle size. As can be seen from the hydrodynamic size distribution shown in Figure 4b, the large superstructures were predominant in both samples. Considering that superstructures were present in both samples, it is unlikely that the difference in the ligand exchange rates can be ascribed to their presence.
Figure 4.
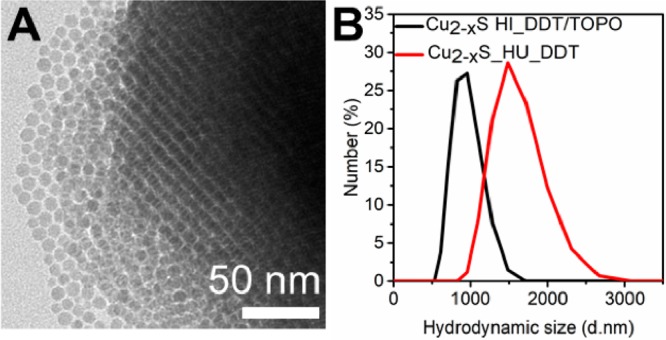
(A) TEM image of a superstructure formed from monodisperse Cu2–xS nanocrystals prepared by hot-injection synthesis. (B) Hydrodynamic size of Cu2–xS nanocrystals in toluene prepared by either hot-injection synthesis or heating-up synthesis. The large hydrodynamic size indicates the presence of nanocrystal superstructures.
Another difference between the two samples is the size of the nanocrystals. Since the bond strength between ligand and nanocrystal has been reported to be size-dependent by several groups,61,62 we investigated the influence of the nanocrystal size on the rate of ligand exchange. To make nanocrystals similar in size, Cu2–xS nanocrystals with a size of 11 nm were synthesized using the hot-injection method with TOPO by allowing for a longer reaction time (Figure 5a). Subsequently, ligand exchange procedures using MUA in water were performed. In the ligand exchange experiments discussed above, the exchange using MUA in water occurred only for the 7 nm Cu2–xS nanocrystals prepared by hot-injection synthesis in the presence of TOPO, upon stirring overnight. The 12 nm Cu2–xS nanocrystals prepared by heating-up synthesis did not transfer to the aqueous phase, even after stirring for 3 days. In contrast, the 11 nm Cu2–xS nanocrystals prepared by hot-injection in the presence of TOPO underwent phase transfer from the apolar to the polar phase upon stirring overnight (Figure 5b). The ligand exchange rate was thus similar for 7 and 11 nm Cu2–xS nanocrystals prepared by hot-injection synthesis. This demonstrates that the difference in the ligand exchange rates cannot be explained by nanocrystal size effects, nor by the presence of superstructures, and must thus be due to differences in the surface chemistry of the Cu2–xS nanocrystals prepared by the two different synthesis methods.
Figure 5.

TEM images of Cu2–xS nanocrystals of ∼11 nm prepared by hot-injection, (A) before and (B) after ligand exchange with MUA in water.
Surface Chemistry of the Cu2–xS Nanocrystals
As mentioned above, the nanocrystals prepared by hot-injection were synthesized in the presence of TOPO. TOPO is a widely used ligand in the synthesis of colloidal semiconductor nanocrystals, including Cu2–xS and CuInS2, and is generally assumed to be present as a capping ligand at the nanocrystal surface after synthesis.34,63−65 Furthermore, Wang et al. reported a shape controlling effect of TOPO in the synthesis of Cu2–xS nanocrystals, which was attributed to preferential binding of TOPO onto (001) crystal facets, thereby facilitating anisotropic growth.48 The presence of TOPO in combination with DDT at the surface of the nanocrystals synthesized by hot-injection could thus influence the ligand exchange rates. Therefore, we studied the surface species of the Cu2–xS nanocrystals in more detail by means of X-ray photoelectron spectroscopy (XPS). Due to the limited escape depth of the generated photoelectrons, XPS is a surface sensitive technique and hence will be sensitive to probe the ligand layer around the nanocrystals and (part of) the Cu2–xS nanocrystals as well.
XPS survey spectra of relevant samples are shown in the Supporting Information (Figure S7). From the survey scans, it can be readily observed that none of the phosphorus core lines are observed in the sample prepared by hot-injection, indicating the absence of TOPO at the nanocrystal surface (Supporting Information, Figure S8). In addition, liquid phase 31P NMR was used to study the presence of TOPO bound to the nanocrystal surface in a sample taken directly after synthesis and of the same sample after washing with a mixture of methanol and butanol three times (see Experimental Section for details). Figure 6a shows the 31P NMR spectra of the unwashed sample and the sample washed three times. In the unwashed sample, a sharp resonance is present at a chemical shift of 48 ppm. By using a reference solution of TOPO in ODE, the resonance at 48 ppm is ascribed to TOPO. Ligands bound to nanocrystal surfaces typically show broad resonances due to solvent exclusion from the ligand shell and shorter relaxation times originating from the restricted mobility of the ligands when bound to the nanocrystal surface.66−70 Hence, the sharp resonance observed for the unwashed sample indicates that after synthesis no significant amount of TOPO is strongly bound to the nanocrystal surface, but instead TOPO is present as free ligand in solution. The sample analyzed after three wash steps shows no resonances in the 31P NMR spectrum, indicating that washing successfully removed free TOPO ligands from the nanocrystals dispersion and also bound TOPO from the nanocrystal surface. The absence of TOPO species at the nanocrystal surface after synthesis and washing, in combination with the finding that TOPO acts as a shape-directing ligand in Cu2–xS nanocrystal synthesis,48 suggests that TOPO binds only weakly and dynamically to the nanocrystal surface during the synthesis and therefore does not end up at the nanocrystal surface in the equilibrium structure formed after synthesis and washing. The weak bond between TOPO and the nanocrystal surface can be rationalized in terms of Lewis acid–base interaction.15,24 TOPO binds through its oxygen lone pair to the Cu+ in the Cu2–xS nanocrystal. Since oxygen is a hard Lewis base and Cu+ a soft Lewis acid, the interaction between the two species will be weak. In contrast, TOPO binds to the surface of, e.g., CuInS2 nanocrystals,63,64 which can be explained by the fact that In3+ is a strong Lewis acid and will thus bind more strongly to the strong Lewis base oxygen.
Figure 6.
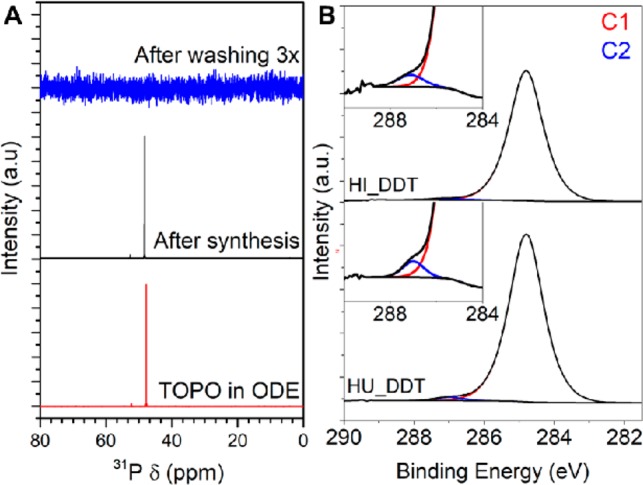
(A) 31P NMR spectra of TOPO in ODE (reference) and of as-synthesized Cu2–xS nanocrystals prepared by hot-injection in the presence of TOPO and after washing with a methanol/butanol mixture three times. For the washed sample, even upon zooming in no 31P resonances are observed. (B) XPS spectra of the C 1s region of as-synthesized Cu2–xS nanocrystals, prepared by hot-injection (top) and heating-up (bottom) synthesis protocols. The fits of the first and second carbon components (C1 and C2) are shown in red and blue, respectively.
Besides TOPO, acetate was added during the hot-injection synthesis (as Cu(I)acetate) and hence could be present as a weakly binding ligand on the Cu2–xS nanocrystals. Additionally, in the heating-up synthesis, oleic acid is used as solvent and coordinating ligand and can consequently be present in the sample. To identify the carbon species present in the as-synthesized samples high-resolution XPS studies on the carbon 1s core line were performed (Figure 6b). In the Cu2–xS nanocrystals prepared by hot-injection, carbon species can originate from the thiolate ligands, 1-octadecene, or from acetate in the copper(I)acetate precursor. For the Cu2–xS nanocrystals prepared by heating-up, carbon species can originate either from the thiolate ligands or from oleic acid used in the synthesis. Other solvents and antisolvents used during the synthesis and washing-up procedures have low boiling points and consequently will have evaporated in the ultrahigh vacuum applied during the XPS measurements.
For both samples, C 1s peaks are observed at a binding energy of 284.8 and 287.0 eV. The peak at a binding energy of 284.8 eV is ascribed to carbon atoms in an aliphatic chain (C–C) either originating from the thiolate ligands, 1-octadecene, oleic acid, or adventitious carbon.71−74 The peak at a binding energy of 287.0 eV is present for both species and is ascribed to carbon bound to sulfur in thiolates.71 Both the acetate and oleic acid species will give rise to a C 1s peak originating from carboxylate species (O=C—O–) at slightly higher binding energies in the range of 288.1–289.1 eV.71−75 As no peaks are present in this region in the C 1s spectrum of both samples, we conclude that there was no significant amount of acetate or oleic acid present. Moreover, these results show that both types of as-synthesized nanocrystals were capped solely by thiol ligands.
To further investigate the surface of the Cu2–xS nanocrystals, high-resolution XPS studies on the sulfur 2p core line were performed (Figure 7). The as-synthesized nanocrystals capped with the thiol species were compared to the S 2p regions of the nanocrystals after ligand exchange with S2–. For a single sulfur component, a set of spin–orbit coupled peaks (2p3/2 and 2p1/2) is expected with an intensity ratio of 1:2 and a spin–orbit coupling split of ∼1.2 eV. The S 2p regions of the as-synthesized samples clearly exhibited a second component and were therefore fitted using two sets of spin–orbit coupled peaks. In agreement with recent studies,40,71 we ascribe the component at an S 2p3/2 binding energy of 161.5 eV to sulfur in the Cu2–xS nanocrystal lattice and possibly crystal-bound thiolate ligands. In the crystal-bound thiolate ligands, sulfur atoms are bound into higher coordination sites within the crystal lattice and hence the binding energy is very similar to the sulfides in the Cu2–xS nanocrystal.40 The second component has a higher S 2p3/2 binding energy of 162.4 eV and hence is ascribed to a sulfur species different from the sulfur in the Cu2–xS lattice. The identical binding energy of 162.4 eV observed for the second sulfur component in the samples prepared by different synthesis methods suggests that the second sulfur component originates from the same sulfur species in both samples. This second S 2p component can be ascribed to surface-bound thiolate (i.e., thiolate bound to surface sites with lower coordination numbers), as described by Turo et al.40 Other sulfur species possibly associated with this high binding energy S 2p component are thioethers forming a ligand double layer, similar to that recently described by Gromova et al. for CuInS2 nanocrystals prepared by a heating-up synthesis in the presence of DDT.46
Figure 7.

XPS of the sulfur 2p region of Cu2–xS nanocrystals before and after ligand exchange with S2– prepared by (A) hot-injection and (B) heating-up synthesis protocols. The black lines indicate the experimental data. The fits of the first sulfur component are shown in red and orange, and the fit of the second sulfur component is shown in blue and light blue.
For the Cu2–xS nanocrystals prepared by hot-injection in the presence of TOPO, the second sulfur component holds 9% of the total sulfur species, whereas this is 28% for the nanocrystals prepared by heating-up synthesis in the absence of TOPO. This suggests a denser layer of surface-bound thiolate species or thioether species around the nanocrystals prepared by the heating-up synthesis. Figure 7 (bottom) shows the high-resolution XPS spectra in the S 2p region of the Cu2–xS nanocrystals after ligand exchange with S2–. For the nanocrystals prepared by hot-injection, the S 2p feature is best fitted with a single set of spin–orbit coupled peaks. Upon ligand exchange with S2–, the second component originating from the ligand layer is thus completely removed. In contrast, the second set of spin–orbit coupled peaks is still clearly present for the nanocrystals prepared by heating-up, indicating only partial removal of the surface-bound alkanethiols. The trend of ligand removal found with XPS studies correlates with the success and kinetics of the ligand exchange procedures discussed above, where it was found that DDT molecules on Cu2–xS nanocrystals prepared by hot-injection in the presence of TOPO were more easily exchanges by the replacing ligands.
Model for Ligand Exchange on DDT-Capped Cu2–xS Nanocrystals
Based on the observations above we propose a model for the ligand exchange that explains the observed differences in the exchange rates. In the ligand exchange protocol developed in our work a two layer system is formed in which the apolar layer contains the Cu2–xS nanocrystals capped by the native thiolate ligands and the polar formamide layer contains the replacing negatively charged ligands (MPA–, MUA–, and S2–) and, later in the process, also the negatively charged Cu2–xS nanocrystals capped with the replacing ligands. As the two solvents are immiscible and the replacing ligands and the charged nanocrystals do not disperse in the apolar phase, while the nanocrystals with the native DDT ligands do not disperse in the polar phase, the ligand exchange must necessarily take place at the interface between the polar and the apolar layers. The native DDT ligand is bound to the nanocrystal surface through its sulfur atom that donates an unshared electron pair and coordinates to the Cu+.15 In the ligand exchange reactions, the negatively charged replacing ligand (MPA–, MUA–, and S2–) will coordinate its electron-rich sulfur species to the Cu+ site on the nanocrystal,24 thereby weakening the bond between the Cu+ and the donor sulfur atom of the DDT ligands, which will eventually force the native thiolate ligand to leave the nanocrystal surface. When part of the native ligands are exchanged for charged species, the nanocrystals will be trapped at the interface since they will become charged (Figure 8). If most native ligands are exchanged for charged replacing ligands, thereby making the nanocrystal sufficiently negatively charged, the nanocrystal will transfer to the polar solvent (Figure 8). As described above, we found that the polarity of the solvent influences the success of ligand exchange. This can be understood by the good ability of the highly polar formamide to shield the charged intermediates. Similarly, the higher stability of the nanocrystals in solvents with higher polarity (e.g., formamide) results from the better shielding of the charged nanocrystals.
Figure 8.
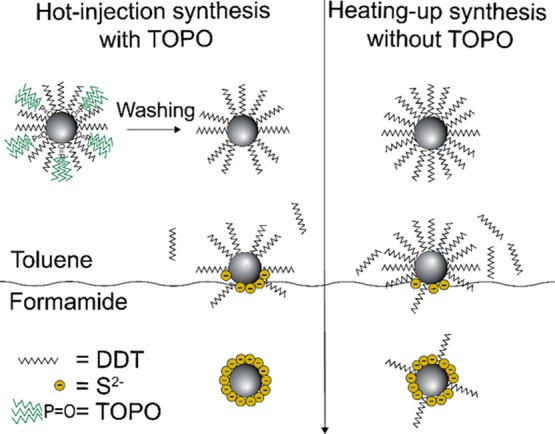
Schematic illustration of a ligand exchange of DDT for S2– and subsequent phase transfer from apolar toluene to polar formamide. The Cu2–xS nanocrystals prepared by hot-injection synthesis have a less dense ligand layer than the nanocrystals prepared by heating-up synthesis due to the removal of TOPO from the surface of the nanocrystal after synthesis and washing. Consequently, the nanocrystal surface is more accessible to the incoming ligands, resulting in a faster ligand exchange. Upon ligand exchange on nanocrystals prepared by hot-injection synthesis, all thiolate ligands are removed from the nanocrystal surface. In contrast, upon ligand exchange on nanocrystals prepared by heating-up synthesis, some thiolate ligands remain on the surface.
The observed dependence of the ligand exchange rate on the incoming ligand (viz., highest for S2–, followed by MPA– and then MUA–) can be rationalized in terms of the accessibility of the nanocrystal surface due to variable degrees of steric hindrance. The hydrophobic ligand layer at the nanocrystal surface acts as a barrier around the nanocrystal. The small S2– ions can more easily diffuse through this hydrophobic layer and reach the surface Cu+ sites, whereas the bulky MUA molecule cannot easily access the surface sites, which results in much slower ligand exchange rates. The observed difference in the ligand exchange rates for the Cu2–xS nanocrystals prepared by hot-injection or heating-up can be explained by the ligand layer at the surface of the nanocrystals. Although we cannot identify the exact nature of the ligand layer, the XPS studies discussed above indicate a denser layer of thiolate ligands around Cu2–xS nanocrystals prepared by the heating-up synthesis, i.e., in the absence of TOPO. The fact that the native ligand layer is less dense for the nanocrystals prepared through the hot-injection method can be understood from the synthesis conditions used. In the hot-injection synthesis, TOPO is used as a coordinating ligand. As has been shown by Wang et al. TOPO has a shape directing effect in the synthesis of Cu2–xS nanocrystals, implying that it dynamically binds to the nanocrystals surface during its growth.48 The bulky nature of TOPO will thus prevent the formation of a dense layer of thiolates around the growing Cu2–xS nanocrystals. As shown by the XPS and 31P NMR studies discussed above, TOPO is no longer present on the Cu2–xS nanocrystals after the synthesis, implying that it has been removed from the surface, thereby leaving vacant sites on the nanocrystal surface. Consequently, the Cu2–xS nanocrystals prepared by hot-injection synthesis in the presence of TOPO have a less dense layer of thiolate ligands at the nanocrystal surface than nanocrystals prepared by the heating-up synthesis. The denser, hydrophobic native ligand layer at the surface of the nanocrystals prepared by the heating-up method will make the surface sites less accessible for the charged incoming ligands and hence will significantly slow down the ligand exchange kinetics.
Additionally, Cu2–xS nanocrystals prepared by hot-injection show a single sulfur species upon ligand exchange with S2–, indicating that all surface-bound thiolate ligands are removed from the nanocrystal surface upon ligand exchange. In contrast, nanocrystals prepared using heating-up synthesis show residual surface-bound thiolate species at their surface upon ligand exchange (see Figure 7 and XPS discussion above). Consequently, the nanocrystals are likely capped with a combination of remaining thiolate species and sulfides, as is schematically represented in Figure 8. However, since the nanocrystals do show colloidal stability in formamide, the nanocrystal surface is covered by sufficient negatively charged sulfide anions to stabilize the nanocrystals in formamide.
Conclusions
In this paper we present effective ligand exchange procedures of DDT on Cu2–xS nanocrystals for MPA–, MUA–, and S2– from Na2S in formamide under inert atmosphere. The ligand exchange procedures did not significantly affect the size, shape, and optical properties of the nanocrystals. The resulting hydrophilic Cu2–xS nanocrystals have excellent colloidal stability in formamide. Water-dispersible Cu2–xS nanocrystals can be easily obtained by precipitation of the nanocrystals, followed by redispersion in water. In addition, the ligand exchange rates for DDT-capped Cu2–xS nanocrystals depended on the preparation method, being much slower for Cu2–xS nanocrystals prepared by heating-up than by hot-injection synthesis. XPS studies revealed that the differences in the ligand exchange rates can be attributed to the surface chemistry of the Cu2–xS nanocrystals, where nanocrystals prepared by heating-up synthesis had a denser ligand layer in comparison to nanocrystals prepared by hot-injection in the presence of TOPO. The less dense native thiolate ligand layer on the surface of the nanocrystals prepared by hot-injection originates from the presence of TOPO during the synthesis, which prevents the formation of a dense thiolate layer around the nanocrystals during their growth, thus leaving vacant surface sites after being removed upon washing after the synthesis. The dense native thiolate ligand layer makes the nanocrystal surface less accessible for the charged incoming ligands, hence significantly slowing down the ligand exchange rates. The facile ligand exchange procedures developed in our work open up opportunities for the use of high-quality colloidal Cu2–xS nanocrystals prepared with thiols in various applications, such as photo- and electrocatalysis, photothermal therapy, or solution-processable devices (flexible conductive films, sensors, solar cells).
Experimental Section
Materials
Copper(II) sulfate pentahydrate (CuSO4·5H2O, 99.999%), copper(I) acetate (CuAc, 97%), copper(I) chloride (CuCl, 99.995%), 1-dodecanethiol (DDT, ≥98%), oleic acid (OA, 90%), 1-octadecene (ODE, 90%), trioctylphosphine oxide (TOPO, 99%), oleylamine (OLAM, 70%), sodium sulfide (Na2S), 11-mercaptoundecanoic acid (MUA, 98%), 3-mercaptopropionic acid (99%), tetramethylammonium hydroxide pentahydrate (TMAH, ≥97%), formamide (FA, 98%), chloroform, anhydrous toluene, methanol, butanol, ethanol, hexane, and acetonitrile were purchased from Sigma-Aldrich. ODE and TOPO were degassed at 120 °C for 3 h prior to use. Formamide was degassed at 120 °C for 2 h prior to use. All other reagents were used as received.
Synthesis of Colloidal DDT-Capped Cu2–xS Nanocrystals by the Heating-Up Method
Cu2–xS nanocrystals of 12 nm diameter were synthesized according to the method described by van der Stam et al.49 A total of 205 mg (0.8 mmol) of CuSO4·5H2O, 7.5 mL of DDT, and 6 mL of OA were mixed in a round-bottom flask. The mixture was gradually heated to 200 °C under nitrogen protection in a Schlenk line. Subsequently, the solution was kept at 200 °C for 2 h. After that, the reaction mixture was washed three times by addition of an excess of methanol and butanol (1:1 ratio), followed by centrifugation at 2500 rpm for 5 min. The nanocrystals were redispersed in 10 mL of anhydrous toluene.
Synthesis of Colloidal Cu2–xS Nanocrystals by the Hot-Injection Method
Cu2–xS nanocrystals of various sizes were synthesized by adaptation of the method described by Xia et al.34 In a round-bottom flask, 0.13 g (1 mmol) of Cu(I)Ac and 3.6 g (9.3 mmol) of TOPO were added to 20 mL of ODE. First, the mixture was degassed at 100 °C for 1 h. Subsequently, the flask was purged with nitrogen, and subsequent steps of the synthesis were performed under nitrogen flow in a Schlenk line. The reaction mixture was heated to 210 °C. At 160 °C, 5 mL of DDT was quickly injected into the flask. The nanocrystals were allowed to grow for 1 h at 210 °C. After reaction, the nanocrystals were washed using the method described above and redispersed in 10 mL of anhydrous toluene.
Synthesis of Colloidal OLAM-Capped Cu2–xS Nanocrystals by the Heating-Up Method
OLAM-capped Cu2–xS nanocrystals of 7 nm diameter were synthesized in a Schlenk line under nitrogen according to a method described by Williamson et al.76 A S-OLAM precursor solution was prepared by dissolving 1.6 g (50 mmol) of S in 7 mL of OLAM and 3 mL of ODE at 110 °C. The copper precursor solution was prepared by dissolving 6 g (60 mmol) of CuCl in 42 mL of OLAM and 18 mL of ODE in a round-bottom flask at 110 °C. Both precursor solutions were cooled to 50 °C, after which 6 mL of the S-OLAM precursor was added to the Cu-precursor solution. The mixture was left stirring at 50 °C for 5 min. Subsequently, the temperature was increased to 185 °C in 30 min and maintained for 2 h. After that the reaction mixture was cooled using a water bath and addition of 60 mL of hexane, which also decreased the viscosity of the mixture. The product was purified by washing with ethanol, followed by centrifugation at 2500 rpm for 5 min. The Cu2–xS nanocrystals were redispersed in 75 mL of n-hexane.
Ligand Exchange Using Na2S
The phase transfer procedure using Na2S was done using an adaptation of a method described by Nag et al.24 For a typical ligand exchange, 1 mL of Na2S in formamide or demineralized water (5 mg/mL) was mixed with 1 mL of Cu2–xS nanocrystals in toluene (∼5 mg/mL). The experiments using formamide as the polar solvent were also performed under inert atmosphere in a nitrogen glovebox (≤6 ppm of O2). A biphase was formed, with the colorless Na2S solution on the bottom and the black Cu2–xS nanocrystals suspension in toluene on top (Supporting Information, Figure S1). Upon ligand exchange of the native apolar ligands to sulfide, the black Cu2–xS nanocrystals transferred to the polar formamide layer. Subsequently, the apolar layer was removed and the remaining polar phase containing the Cu2–xS nanocrystals was washed with toluene to remove the remaining apolar species. The Cu2–xS nanocrystals were precipitated by adding an excess of acetonitrile, followed by centrifugation for 2 min at 2000 rpm. The purified sulfide-capped Cu2–xS nanocrystals were redispersed in 2 mL of degassed formamide and stored under nitrogen atmosphere.
Ligand Exchange Using MUA or MPA
The phase transfer procedure using MUA or MPA was based on a method described by Xia et al.34 A total of 1.4 mmol of MUA or MPA was dissolved in 30 mL of demineralized H2O or formamide. In the case of MUA, TMAH was added to increase the pH to 11 and deprotonate the MUA. In a typical ligand exchange, 4 mL of a Cu2–xS nanocrystal suspension in toluene (∼0.75 mg/L) was mixed with 5.8 mL of the MUA or MPA solutions. A two-layer system was formed, with the Cu2–xS nanocrystals in toluene on the bottom and the polar MUA or MPA solution on top. The experiments using formamide as the polar solvent were also performed under inert atmosphere in a nitrogen glovebox (≤6 ppm of O2). The ligand exchange was successful when upon stirring the black Cu2–xS nanocrystals transferred to the polar phase. After centrifugation for 1 min at 1000 rpm, the polar layer containing the Cu2–xS nanocrystals was collected and washed by adding excess ethanol, followed by centrifugation for 5 min at 3000 rpm. The purified MUA- or MPA-capped Cu2–xS nanocrystals were redispersed in demineralized water or formamide.
Transmission Electron Microscopy (TEM)
Samples for TEM analysis were prepared by drop-casting the Cu2–xS nanocrystal suspension on a carbon-coated 200 mesh copper TEM grid. TEM analysis was performed using a FEI Tecnai-12 microscope operating at 120 kV or a FEI Technai-20 microscope operating at 200 kV.
X-ray Diffraction (XRD)
Samples for XRD analysis were prepared by precipitating the Cu2–xS nanocrystals with an antisolvent and subsequently drying the obtained powder under vacuum. The dried Cu2–xS nanocrystal powder was spread over a Si wafer. XRD measurements were performed with a Bruker D2 Phaser, equipped with a Co Kα X-ray source with a wavelength of 1.79026 Å.
ζ-Potential and Dynamic Light Scattering (DLS)
ζ-Potential and DLS measurements were performed on a Zetasizer Nano ZS from Malvern Instruments using a dip cell with palladium electrodes with 2 mm spacing in a PCS1115 cuvette and a measurement angle of 173°. Measurements were corrected by the instrument software for solvent (formamide or toluene), refractive index, temperature, and viscosity. Measurements were done in the automatic mode and repeated five times for each sample in order to obtain reliable results.
X-ray Photoelectron Spectroscopy (XPS)
Sample preparation for XPS was done under air-free conditions to prevent oxidation and chemisorption of CO2 on the samples. Samples were mounted in a glovebox and transferred into the spectrometer in a vacuum transfer module. The as-synthesized samples with the original apolar ligands were drop casted on a clean tantalum sample holder in order to avoid the influence of carbon tape. Samples after ligand exchange to S2– were dried under vacuum and stuck onto the tantalum sample holder as powder using carbon tape. XPS spectra were taken with a Thermo Scientific K-Alpha spectrometer using a 72 W monochromated Al Kα source (hν = 1486.6 eV). The X-rays are microfocused at the source to give a spot size on the sample of 400 μm in diameter. The analyzer is a double focusing 180° hemisphere with mean radius of 125 mm, run in constant analyzer energy (CAE) mode. The pass energy was set to 200 eV for survey scans and 50 eV for high resolution regions. Data analysis was done using CasaXPS software. Binding energies were calibrated by setting the lowest energy C 1s peak to 284.8 eV.
31P NMR Spectroscopy
31P NMR measurements were done on a reference solution of TOPO in ODE, the Cu2–xS nanocrystal reaction mixture directly after synthesis and after washing three times. The reaction mixture and reference solution were both diluted with CDCl3 to a concentration half of the original in order to perform a lock procedure. The washed sample was dried under vacuum overnight after the third wash step and subsequently redispersed in CDCl3. Measurements were performed on an Aligent MRF400 spectrometer at 25 °C and 161 MHz.
Acknowledgments
This work was supported by The Netherlands Center for Multiscale Catalytic Energy Conversion (MCEC), an NWO Gravitation program funded by the Ministry of Education, Culture and Science of the government of The Netherlands. P.E.d.J. acknowledges funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (ERC-2014-CoG No. 648991). The authors are grateful to Johann Jastrzebski (Utrecht University) for his assistance with 31P NMR measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemmater.8b04614.
Zeta potential measurements for Cu2–xS nanocrystals after ligand exchange. Additional TEM images and XRD patterns of Cu2–xS nanocrystals after ligand exchange. XPS survey scans for relevant samples (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Goel S.; Chen F.; Ehlerding E. B.; Cai W. Intrinsically Radiolabeled Nanoparticles: An Emerging Paradigm. Small 2014, 10, 3825–3830. 10.1002/smll.201401048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughlan C.; Ibáñez M.; Dobrozhan O.; Singh A.; Cabot A.; Ryan K. M. Compound Copper Chalcogenide Nanocrystals. Chem. Rev. 2017, 117, 5865–6109. 10.1021/acs.chemrev.6b00376. [DOI] [PubMed] [Google Scholar]
- Goel S.; Chen F.; Cai W. Synthesis and Biomedical Applications of Copper Sulfide Nanoparticles: From Sensors to Theranostics. Small 2014, 10, 631–645. 10.1002/smll.201301174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Riedinger A.; Li H.; Fu C.; Liu H.; Li L.; Liu T.; Tan L.; Barthel M. J.; Pugliese G.; et al. Plasmonic Copper Sulfide Nanocrystals Exhibiting Near-Infrared Photothermal and Photodynamic Therapeutic Effects. ACS Nano 2015, 9, 1788–1800. 10.1021/nn506687t. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Burda C. Development of Plasmonic Semiconductor Nanomaterials with Copper Chalcogenides for a Future with Sustainable Energy Materials. Energy Environ. Sci. 2012, 5, 5564–5576. 10.1039/C1EE02734D. [DOI] [Google Scholar]
- Luther J. M.; Jain P. K.; Ewers T.; Alivisatos A. P. Localized Surface Plasmon Resonances Arising from Free Carriers in Doped Quantum Dots. Nat. Mater. 2011, 10, 361–366. 10.1038/nmat3004. [DOI] [PubMed] [Google Scholar]
- Abdelhady A. L.; Ramasamy K.; Malik M. A.; O’Brien P.; Haigh S. J.; Raftery J. New Routes to Copper Sulfide Nanostructures and Thin Films. J. Mater. Chem. 2011, 21, 17888–17895. 10.1039/c1jm13277f. [DOI] [Google Scholar]
- Van Der Stam W.; Gudjonsdottir S.; Evers W. H.; Houtepen A. J. Switching between Plasmonic and Fluorescent Copper Sulfide Nanocrystals. J. Am. Chem. Soc. 2017, 139, 13208–13217. 10.1021/jacs.7b07788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao P.; Ci S.; Yi L.; Cai P.; Huang P.; Cao C.; Wen Z. Hollow CuS Microcube Electrocatalysts for CO2 Reduction Reaction. ChemElectroChem 2017, 4, 2593–2598. 10.1002/celc.201700517. [DOI] [Google Scholar]
- Zhao Z.; Peng X.; Liu X.; Sun X.; Shi J.; Han L.; Li G.; Luo J. Efficient and Stable Electroreduction of CO2 to CH4 on CuS Nanosheet Arrays. J. Mater. Chem. A 2017, 5, 20239–20243. 10.1039/C7TA05507B. [DOI] [Google Scholar]
- Van Der Stam W.; Berends A. C.; De Mello Donega C. Prospects of Colloidal Copper Chalcogenide Nanocrystals. ChemPhysChem 2016, 17, 559–581. 10.1002/cphc.201500976. [DOI] [PubMed] [Google Scholar]
- Zhuang Z.; Peng Q.; Zhang B.; Li Y. Controllable Synthesis of Cu2S Nanocrystals and Their Assembly into a Superlattice. J. Am. Chem. Soc. 2008, 130, 10482–10483. 10.1021/ja803644g. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Pan H.; Lou Y.; Qiu X.; Zhu J.; Burda C. Plasmonic Cu2–xS Nanocrystals: Optical and Structural Properties of Copper-Deficient Copper(I) Sulfides. J. Am. Chem. Soc. 2009, 131, 4253–4261. 10.1021/ja805655b. [DOI] [PubMed] [Google Scholar]
- Wadia C.; Alivisatos A. P.; Kammen D. M. Materials Availability Expands the Opportunity for Large-Scale Photovoltaics Deployment. Environ. Sci. Technol. 2009, 43, 2072–2077. 10.1021/es8019534. [DOI] [PubMed] [Google Scholar]
- Donegá C. de M Synthesis and Properties of Colloidal Heteronanocrystals. Chem. Soc. Rev. 2011, 40, 1512–1546. 10.1039/C0CS00055H. [DOI] [PubMed] [Google Scholar]
- Li X.; Shen H.; Niu J.; Li S.; Zhang Y.; Wang H.; Li L. S. Columnar Self-Assembly of Cu2S Hexagonal Nanoplates Induced by Tin(IV)-X Complex as Inorganic Surface Ligand. J. Am. Chem. Soc. 2010, 132, 12778–12779. 10.1021/ja103955s. [DOI] [PubMed] [Google Scholar]
- Sigman M. B.; Ghezelbash A.; Hanrath T.; Saunders A. E.; Lee F.; Korgel B. A. Solventless Synthesis of Monodisperse Cu2S Nanorods, Nanodisks, and Nanoplatelets. J. Am. Chem. Soc. 2003, 125, 16050–16057. 10.1021/ja037688a. [DOI] [PubMed] [Google Scholar]
- Li X.; Wang M.; Shen H.; Zhang Y.; Wang H.; Li L. S. Inorganic Sn-X-Complex-Induced 1D, 2D, and 3D Copper Sulfide Superstructures from Anisotropic Hexagonal Nanoplate Building Blocks. Chem. - Eur. J. 2011, 17, 10357–10364. 10.1002/chem.201101114. [DOI] [PubMed] [Google Scholar]
- Li W.; Shavel A.; Guzman R.; Rubio-Garcia J.; Flox C.; Fan J.; Cadavid D.; Ibáñez M.; Arbiol J.; Morante J. R.; et al. Morphology Evolution of Cu2-XS Nanoparticles: From Spheres to Dodecahedrons. Chem. Commun. (Cambridge, U. K.) 2011, 47, 10332–10334. 10.1039/c1cc13803k. [DOI] [PubMed] [Google Scholar]
- Tang A.; Qu S.; Li K.; Hou Y.; Teng F.; Cao J.; Wang Y.; Wang Z. One-Pot Synthesis and Self-Assembly of Colloidal Copper(I) Sulfide Nanocrystals. Nanotechnology 2010, 21, 285602. 10.1088/0957-4484/21/28/285602. [DOI] [PubMed] [Google Scholar]
- Van Der Stam W.; Akkerman Q. A.; Ke X.; Van Huis M. A.; Bals S.; de Mello Donega C. Solution-Processable Ultrathin Size- and Shape-Controlled Colloidal Cu2–xS Nanosheets. Chem. Mater. 2015, 27, 283–291. 10.1021/cm503929q. [DOI] [Google Scholar]
- van der Stam W.; Gradmann S.; Altantzis T.; Ke X.; Baldus M.; Bals S.; de Mello Donega C. Shape Control of Colloidal Cu2–xS Polyhedral Nanocrystals by Tuning the Nucleation Rates. Chem. Mater. 2016, 28, 6705–6715. 10.1021/acs.chemmater.6b03098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Embden J.; Chesman A. S. R.; Jasieniak J. J. The Heat-Up Synthesis of Colloidal Nanocrystals. Chem. Mater. 2015, 27, 2246–2285. 10.1021/cm5028964. [DOI] [Google Scholar]
- Nag A.; Kovalenko M. V.; Lee J. S.; Liu W.; Spokoyny B.; Talapin D. V. Metal-Free Inorganic Ligands for Colloidal Nanocrystals: S2-, HS–, Se2-, HSe–, Te2-, HTe–, TeS32-, OH–, and NH2–as Surface Ligands. J. Am. Chem. Soc. 2011, 133, 10612–10620. 10.1021/ja2029415. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Scheele M.; Talapin D. V. Colloidal Nanocrystals with Molecular Metal Chalcogenide Surface Ligands. Science 2009, 324, 1417–1420. 10.1126/science.1170524. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Bodnarchuk M. I.; Zaumseil J.; Lee J. S.; Talapin D. V. Expanding the Chemical Versatility of Colloidal Nanocrystals Capped with Molecular Metal Chalcogenide Ligands. J. Am. Chem. Soc. 2010, 132, 10085–10092. 10.1021/ja1024832. [DOI] [PubMed] [Google Scholar]
- Gugula K.; Stegemann L.; Cywiński P. J.; Strassert C. A.; Bredol M. Facile Surface Engineering of CuInS2/ZnS Quantum Dots for LED down-Converters. RSC Adv. 2016, 6, 10086–10093. 10.1039/C5RA26331J. [DOI] [Google Scholar]
- Wuister S. F.; De Mello Donegá C.; Meijerink A. Luminescence Temperature Antiquenching of Water-Soluble CdTe Quantum Dots: Role of the Solvent. J. Am. Chem. Soc. 2004, 126, 10397–10402. 10.1021/ja048222a. [DOI] [PubMed] [Google Scholar]
- Bagaria H. G.; Ada E. T.; Shamsuzzoha M.; Nikles D. E.; Johnson D. T. Understanding Mercapto Ligand Exchange on the Surface of FePt Nanoparticles. Langmuir 2006, 22, 7732–7737. 10.1021/la0601399. [DOI] [PubMed] [Google Scholar]
- Chan W. C. W.; Nie S. Quantum Dot Bioconjugates for Ultrasensitive Nonisotopic Detection. Adv. Sci. 1998, 281, 2016–2018. 10.1126/science.281.5385.2016. [DOI] [PubMed] [Google Scholar]
- Algar W. R.; Krull U. J. Luminescence and Stability of Aqueous Thioalkyl Acid Capped CdSe/ZnS Quantum Dots Correlated to Ligand Ionization. ChemPhysChem 2007, 8, 561–568. 10.1002/cphc.200600686. [DOI] [PubMed] [Google Scholar]
- Aldana J.; Wang Y. A.; Peng X. Photochemical Instability of CdSe Nanocrystals Coated by Hydrophilic Thiols. J. Am. Chem. Soc. 2001, 123, 8844–8850. 10.1021/ja016424q. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar Y.; Scotognella F.; Waiskopf N.; Kriegel I.; Dal Conte S.; Cerullo G.; Banin U. Effect of Surface Coating on the Photocatalytic Function of Hybrid CdS-Au Nanorods. Small 2015, 11, 462–471. 10.1002/smll.201402262. [DOI] [PubMed] [Google Scholar]
- Xia C.; Meeldijk J. D.; Gerritsen H. C.; de Mello Donega C. Highly Luminescent Water-Dispersible NIR-Emitting Wurtzite CuInS2/ZnS Core/Shell Colloidal Quantum Dots. Chem. Mater. 2017, 29, 4940–4951. 10.1021/acs.chemmater.7b01258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C.; Bai Z.; Liu X.; Zhang Y.; Zou B.; Zhong H. Small GSH-Capped CuInS2 Quantum Dots: MPA-Assisted Aqueous Phase Transfer and Bioimaging Applications. ACS Appl. Mater. Interfaces 2015, 7, 17623–17629. 10.1021/acsami.5b05503. [DOI] [PubMed] [Google Scholar]
- Tamang S.; Beaune G.; Texier I.; Reiss P. Aqueous Phase Transfer of InP/ZnS Nanocrystals Conserving Fluorescence and High Colloidal Stability. ACS Nano 2011, 5, 9392–9402. 10.1021/nn203598c. [DOI] [PubMed] [Google Scholar]
- Nann T. Phase-Transfer of CdSe@ZnS Quantum Dots Using Amphiphilic Hyperbranched Polyethylenimine. Chem. Commun. 2005, 1735–1736. 10.1039/b414807j. [DOI] [PubMed] [Google Scholar]
- Ben-Shahar Y.; Scotognella F.; Waiskopf N.; Kriegel I.; Dal Conte S.; Cerullo G.; Banin U. Effect of Surface Coating on the Photocatalytic Function of Hybrid CdS-Au Nanorods. Small 2015, 11, 462–471. 10.1002/smll.201402262. [DOI] [PubMed] [Google Scholar]
- Lees E. E.; Nguyen T. L.; Clayton A. H. A.; Mulvaney P. The Preparation of Colloidally Stable, Water-Soluble, Biocompatible, Semiconductor Nanocrystals with a Small Hydrodynamic Diameter. ACS Nano 2009, 3, 1121–1128. 10.1021/nn900144n. [DOI] [PubMed] [Google Scholar]
- Turo M. J.; Macdonald J. E. Crystal-Bound vs Surface-Bound Thiols on Nanocrystals. ACS Nano 2014, 8, 10205–10213. 10.1021/nn5032164. [DOI] [PubMed] [Google Scholar]
- Chen L.; Li G. Functions of 1-Dodecanethiol in the Synthesis and Post-Treatment of Copper Sulfide Nanoparticles Relevant to Their Photocatalytic Applications. ACS Appl. Nano Mater. 2018, 1, 4587–4593. 10.1021/acsanm.8b00893. [DOI] [Google Scholar]
- Xie R.; Rutherford M.; Peng X. Formation of High-Quality I-III-VI Semiconductor Nanocrystals by Tuning Relative Reactivity of Cationic Precursors. J. Am. Chem. Soc. 2009, 131, 5691–5697. 10.1021/ja9005767. [DOI] [PubMed] [Google Scholar]
- So D.; Konstantatos G. Thiol-Free Synthesized Copper Indium Sulfide Nanocrystals as Optoelectronic Quantum Dot Solids. Chem. Mater. 2015, 27, 8424–8432. 10.1021/acs.chemmater.5b03943. [DOI] [Google Scholar]
- Pan Z.; Mora-Sero I.; Shen Q.; Zhang H.; Li Y.; Zhao K.; Wang J.; Zhong X.; Bisquert J. High-Efficiency Green Quantum Dot Solar Cells. J. Am. Chem. Soc. 2014, 136, 9203–9210. 10.1021/ja504310w. [DOI] [PubMed] [Google Scholar]
- Akdas T.; Distaso M.; Kuhri S.; Winter B.; Birajdar B.; Spiecker E.; Guldi D. M.; Peukert W. The Effects of Post-Processing on the Surface and the Optical Properties of Copper Indium Sulfide Quantum Dots. J. Colloid Interface Sci. 2015, 445, 337–347. 10.1016/j.jcis.2015.01.014. [DOI] [PubMed] [Google Scholar]
- Gromova M.; Lefrancois A.; Vaure L.; Agnese F.; Aldakov D.; Maurice A.; Djurado D.; Lebrun C.; de Geyer A.; Schulli T. U.; et al. Growth Mechanism and Surface State of CuInS2 Nanocrystals Synthesized with Dodecanethiol. J. Am. Chem. Soc. 2017, 139, 15748–15759. 10.1021/jacs.7b07401. [DOI] [PubMed] [Google Scholar]
- Robinson E. H.; Turo M. J.; Macdonald J. E. Controlled Surface Chemistry for the Directed Attachment of Copper(I) Sulfide Nanocrystals. Chem. Mater. 2017, 29, 3854–3857. 10.1021/acs.chemmater.6b05080. [DOI] [Google Scholar]
- Wang Y.; Hu Y.; Zhang Q.; Ge J.; Lu Z.; Hou Y.; Yin Y. One-Pot Synthesis and Optical Property of Copper(I) Sulfide Nanodisks. Inorg. Chem. 2010, 49, 6601–6608. 10.1021/ic100473e. [DOI] [PubMed] [Google Scholar]
- Van Der Stam W.; Berends A. C.; Rabouw F. T.; Willhammar T.; Ke X.; Meeldijk J. D.; Bals S.; De Mello Donega C. Luminescent CuInS2 Quantum Dots by Partial Cation Exchange in Cu2-XS Nanocrystals. Chem. Mater. 2015, 27, 621–628. 10.1021/cm504340h. [DOI] [Google Scholar]
- Wen X.; Zhang W.; Yang S.; Dai Z. R.; Wang Z. L. Solution Phase Synthesis of Cu(OH)2 Nanoribbons by Coordination Self-Assembly Using Cu2S Nanowires as Precursors. Nano Lett. 2002, 2, 1397–1401. 10.1021/nl025848v. [DOI] [Google Scholar]
- Trevani L. N.; Roberts J. C.; Tremaine P. R. Copper(II)-Ammonia Complexation Equilibria in Aqueous Solutions at Temperatures from 30 to 250 Degrees C by Visible Spectroscopy. J. Solution Chem. 2001, 30, 585–622. 10.1023/A:1010453412802. [DOI] [Google Scholar]
- Lamble G.; Moen A.; Nicholson D. G. Structure of the Diamminecopper(1) Ion in Solution An X-Ray Absorption Spectroscopic Study. J. Chem. Soc., Faraday Trans. 1994, 90 (15), 2211. 10.1039/ft9949002211. [DOI] [Google Scholar]
- Sano M.; Maruo T.; Yamatera H. Structural Determination of Solvated Copper Species in Formamide Solution by EXAFS. J. Chem. Phys. 1988, 89, 1185–1187. 10.1063/1.455228. [DOI] [Google Scholar]
- Nisar Ansari M.; Khan S. H.; Aziz Khan A. Polarographic Studies on Copper(II)-Formamide Complex in Alkaline Medium. J. Electroanal. Chem. Interfacial Electrochem. 1970, 28, 450–452. 10.1016/S0022-0728(70)80139-5. [DOI] [Google Scholar]
- Xu J.; Xue D.; Zhu Y. Room Temperature Synthesis of Curved Ammonium Copper Molybdate Nanoflake and Its Hierarchical Architecture. J. Phys. Chem. B 2006, 110, 17400–17405. 10.1021/jp0632788. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Shao X.; Yu H.; Wang Y.; Han M. Morphosynthesis and Ornamentation of 3D Dendritic Nanoarchitectures. Chem. Mater. 2005, 17, 332–336. 10.1021/cm048436r. [DOI] [Google Scholar]
- Lou J. L.; Shiu H. W.; Chang L. Y.; Wu C. P.; Soo Y.-L.; Chen C.-H. Preparation and Characterization of an Ordered 1-Dodecanethiol Monolayer on Bare Si(111). Langmuir 2011, 27, 3436–3441. 10.1021/la103585t. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Riedinger A.; Prato M.; Casu A.; Genovese A.; Guardia P.; Sottini S.; Sangregorio C.; Miszta K.; Ghosh S.; et al. Copper Sulfide Nanocrystals with Tunable Composition by Reduction of Covellite Nanocrystals with Cu+ Ions. J. Am. Chem. Soc. 2013, 135, 17630–17637. 10.1021/ja409754v. [DOI] [PubMed] [Google Scholar]
- Kriegel I.; Jiang C.; Rodríguez-Fernández J.; Schaller R. D.; Talapin D. V.; da Como E.; Feldmann J. Tuning the Excitonic and Plasmonic Properties of Copper Chalcogenide Nanocrystals. J. Am. Chem. Soc. 2012, 134, 1583–1590. 10.1021/ja207798q. [DOI] [PubMed] [Google Scholar]
- Hinterwirth H.; Kappel S.; Waitz T.; Prohaska T.; Lindner W.; Lämmerhofer M. Quantifying Thiol Ligand Density of Self-Assembled Monolayers on Gold Nanoparticles by Inductively Coupled Plasma-Mass Spectrometry. ACS Nano 2013, 7, 1129–1136. 10.1021/nn306024a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldana J.; Lavelle N.; Wang Y.; Peng X. Size-Dependent Dissociation pH of Thiolate Ligands from Cadmium Chalcogenide Nanocrystals. J. Am. Chem. Soc. 2005, 127, 2496–2504. 10.1021/ja047000+. [DOI] [PubMed] [Google Scholar]
- Schrier J.; Wang L. W. On the Size-Dependent Behavior of Nanocrystal-Ligand Bonds. J. Phys. Chem. B 2006, 110, 11982–11985. 10.1021/jp061117y. [DOI] [PubMed] [Google Scholar]
- Asgary S.; Mirabbaszadeh K.; Nayebi P.; Emadi H. Synthesis and Investigation of Optical Properties of TOPO-Capped CuInS2 Semiconductor Nanocrystal in the Presence of Different Solvent. Mater. Res. Bull. 2014, 51, 411–417. 10.1016/j.materresbull.2013.12.059. [DOI] [Google Scholar]
- Kruszynska M.; Borchert H.; Parisi J.; Kolny-Olesiak J. Investigations of Solvents and Various Sulfur Sources Influence on the Shape-Controlled Synthesis of CuInS2 Nanocrystals. J. Nanopart. Res. 2011, 13, 5815–5824. 10.1007/s11051-011-0381-4. [DOI] [Google Scholar]
- Kruszynska M.; Borchert H.; Bachmatiuk A.; Rümmeli M. H.; Büchner B.; Parisi J.; Kolny-Olesiak J. Size and Shape Control of Colloidal Copper(I) Sulfide Nanorods. ACS Nano 2012, 6, 5889–5896. 10.1021/nn302448n. [DOI] [PubMed] [Google Scholar]
- Hens Z.; Martins J. C. A Solution NMR Toolbox for Characterizing the Surface Chemistry of Colloidal Nanocrystals. Chem. Mater. 2013, 25, 1211–1221. 10.1021/cm303361s. [DOI] [Google Scholar]
- Owen J. S.; Park J.; Trudeau P. E.; Alivisatos A. P. Reaction Chemistry and Ligand Exchange at Cadmium-Selenide Nanocrystal Surfaces. J. Am. Chem. Soc. 2008, 130, 12279–12281. 10.1021/ja804414f. [DOI] [PubMed] [Google Scholar]
- Kopping J. T.; Patten T. E. Identification of Acidic Phosphorus-Containing Ligands Involved in the Surface Chemistry of CdSe Nanoparticles Prepared in Tri-N-Octylphosphine Oxide Solvents. J. Am. Chem. Soc. 2008, 130, 5689–5698. 10.1021/ja077414d. [DOI] [PubMed] [Google Scholar]
- Becerra L. R.; Murray C. B.; Griffin R. G.; Bawendi M. G. Investigation of the Surface Morphology of Capped CdSe Nanocrystallites by 31P Nuclear Magnetic Resonance. J. Chem. Phys. 1994, 100, 3297–3300. 10.1063/1.466420. [DOI] [Google Scholar]
- De Roo J.; Yazdani N.; Drijvers E.; Lauria A.; Maes J.; Owen J. S.; Van Driessche I.; Niederberger M.; Wood V.; Martins J. C.; et al. Probing Solvent–Ligand Interactions in Colloidal Nanocrystals by the NMR Line Broadening. Chem. Mater. 2018, 30, 5485–5492. 10.1021/acs.chemmater.8b02523. [DOI] [Google Scholar]
- Berends A. C.; Van Der Stam W.; Hofmann J. P.; Bladt E.; Meeldijk J. D.; Bals S.; De Mello Donega C. Interplay between Surface Chemistry, Precursor Reactivity, and Temperature Determines Outcome of ZnS Shelling Reactions on CuInS2 Nanocrystals. Chem. Mater. 2018, 30, 2400–2413. 10.1021/acs.chemmater.8b00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts J. F.; Leadley S. R.; Castle J. E.; Blomfield C. J. Adsorption of PMMA on Oxidized Al and Si Substrates: An Investigation by High-Resolution X-Ray Photoelectron Spectroscopy. Langmuir 2000, 16, 2292–2300. 10.1021/la981558b. [DOI] [Google Scholar]
- Wilson D.; Langell M. A. XPS Analysis of Oleylamine/Oleic Acid Capped Fe3O4 Nanoparticles as a Function of Temperature. Appl. Surf. Sci. 2014, 303, 6–13. 10.1016/j.apsusc.2014.02.006. [DOI] [Google Scholar]
- Zhang L.; He R.; Gu H. C. Oleic Acid Coating on the Monodisperse Magnetite Nanoparticles. Appl. Surf. Sci. 2006, 253, 2611–2617. 10.1016/j.apsusc.2006.05.023. [DOI] [Google Scholar]
- Wagener K.; Batich C.; Kirsch B.; Wanigatunga S. Chain Propagation/Step Propagation Polymerization. III. An XPS Investigation of Poly(Oxyethylene)-b-Poly(Pivalolactone) Telechelomer. J. Polym. Sci., Part A: Polym. Chem. 1989, 27, 2625–2631. 10.1002/pola.1989.080270811. [DOI] [Google Scholar]
- Williamson C. B.; Nevers D. R.; Hanrath T.; Robinson R. D. Prodigious Effects of Concentration Intensification on Nanoparticle Synthesis: A High-Quality, Scalable Approach. J. Am. Chem. Soc. 2015, 137, 15843–15851. 10.1021/jacs.5b10006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.