

Abstract
A catalytic, enantioselective method for the preparation of chiral, non-racemic, alkylboronic esters bearing two vicinal stereogenic centers is described. The reaction proceeds via a 1,2-migration of a zwitterionic thiiranium–boronate complex to give exclusively anti carbosulfenylation products. A broad scope of aryl groups migrate with good yield and excellent enantioselectivity (up to 99:1 e.r.). Similarly, a range of di- and trisubstituted alkenylboronic esters are competent reaction partners. This method provides access to both secondary and tertiary chiral alkylboronic esters.
Chiral, non-racemic, alkylboronic esters are valuable synthetic intermediates in modern organic chemistry.1,2 These compounds undergo stereospecific functional group interconversions to afford alcohols, amines, and halides, and serve as useful partners for a myriad of transition metal-catalyzed cross-coupling reactions.3 Consequently, numerous methods have been reported for the synthesis of enantiomerically enriched secondary and tertiary alkylboronic esters.4 Many of these are reductive, oxidative, or isohypsic transformations of alkenylboronic esters, which are themselves readily prepared by hydroboration of abundant, inexpensive alkynes.5 Of particular utility are the “conjunctive” coupling methods recently reported by Morken et al.6 (Scheme 1A), in which a tetracoordinate alkenylboronate complex undergoes a 1,2-metalate rearrangement7,8 in the presence of an aryl-palladium species. The net result is a secondary alkylboronic ester, and the entire process is rendered highly enantioselective by a chiral ligand. This process is attractive owing to its modular nature and broad scope. However, previous methods have not been widely used to access products with two vicinal stereogenic centers.9
Scheme 1.

Stereoselective Construction of Chiral Alkylboranes by 1,2-Migration of Alkenylboronate Complexes
In fact, diastereospecific 1,2-migrations of alkenylboronate complexes have been known for many decades. Zweifel et al. first reported the synthesis of olefins from trans-alkenylboranes in the presence of iodine and aqueous base (Scheme 1B).10 This reaction proceeds through a zwitterionic iodonium–boronate complex. Because nucleophilic opening of haliranium ions is stereospecific, 1,2-migration of an alkyl group from the boronate complex results in an α-iodinated secondary borane as a single anti diastereomer. Aggarwal et al. have recently disclosed the synthesis of α-selenylated secondary boronic esters which proceeds through a very similar mechanism (Scheme 1C).11 Treating an alkenylboronate complex with phenylselenyl chloride forms a zwitterionic seleniranium ion, which is opened by 1,2-migration to afford exclusively anti products. This transformation accommodates a broad scope of substrates which lead to bench-stable and synthetically useful products, but all in racemic form. An enantioselective variant has not yet been reported.
The activation of Lewis acids by chiral Lewis bases for enantioselective, electrophilic functionalization of olefins has been extensively developed in this laboratory12 and is ideally suited to address this challenge (Scheme 1D). Combining chiral selenophosphoramide catalyst (S)-5 with an appropriate Group 16 Lewis acid of type 4 (“sulfenylating agent”) leads to a cationic donor–acceptor complex which is highly electrophilic at sulfur. This species effects the generation of enantiomerically enriched thiiranium ions with unactivated alkenes as well as more nucleophilic alkenes such as enoxysilanes.13 The thiiranium ion is opened diastereospecifically by oxygen, nitrogen, and carbon nucleophiles, resulting in 1,2-anti-sulfeno-functionalized products. The specific challenge was whether alkenylboronate complexes could also serve as viable reactants in this process. Such highly reactive, anionic boronate complexes would likely be incompatible with the conditions used to generate the electrophilic sulfenium ions needed. Even if conditions could be found, the resulting thiiranium ions would need to trigger the subsequent 1,2-migration without configurational mutation to afford α-thiolated boranes in highly enantio- and diastereo-selective fashion.
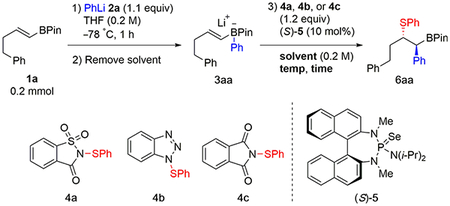
Orienting experiments employed boronate complex 3aa, generated from boronic ester 1a and phenyllithium 2a, in THF, and its reactivity was examined under a variety of conditions (Table 1).14 Different sulfenylating agents (4a, 4b, or 4c) and solvents were evaluated in combination with catalyst (S)-5. The anticipated incompatibility of anionic complex 3aa with the typical acidic promoters required to activate reagents 4 led to the initial investigation of saccharin-derived reagent 4a, which can transfer its sulfenyl group to catalyst 5 without the aid of acid.13 In dichloromethane, the desired product 6aa was formed in good yield after 3 h at cryogenic temperature (entries 1 and 2). However, this observed reactivity simply resulted from background reaction between 3aa and 4a, leading to nearly racemic 6aa. To mitigate this background reactivity, less reactive sulfenylating agents 4b and 4c were tested (entries 3–6). Although the background reaction was suppressed, no catalysis was observed.15
Table 1.
Reaction Optimization
| Entry | 3 | Cat | Solvent | Temp (°C) | Time (h) | Yield (%)a,b | e.r. |
|---|---|---|---|---|---|---|---|
| 1 | 4a | none | CH2C12 | −78 | 3 | 56a | -- |
| 2 | 4a | (S)-5 | CH2C12 | −78 | 3 | 68a | 55:45 |
| 3 | 4b | none | CH2C12 | −78 | 18 | llb | -- |
| 4 | 4b | (S)-5 | CH2C12 | −78 | 18 | 16b | 57:43 |
| 5 | 4c | none | CH2C12 | −20 | 36 | 31b | -- |
| 6 | 4c | (S)-5 | CH2C12 | −20 | 36 | 37b | 53:47 |
| 7 | 4a | none | MeOH | −60 | 24 | 10b | -- |
| 8 | 4a | (S)-5 | MeOH | −60 | 24 | 47a | 94:6 |
| 9 | 4a | none | EtOH | −60 | 24 | 14b | -- |
| 10 | 4a | (S)-5 | EtOH | −60 | 24 | 80a | 98:2 |
| 11 | 4b | none | EtOH | −20 | 24 | 7b | -- |
| 12 | 4b | (S)-5 | EtOH | −20 | 24 | 10b | -- |
| 13 | 4b | none | TFE | −20 | 2 | 10b | -- |
| 14 | 4b | (S)-5 | TFE | −20 | 2 | 32b | 85:15 |
| 15 | 4c | none | HFIP | 0 | 2 | 0c | -- |
| 16 | 4c | (S)-5 | HFIP | 0 | 2 | 27a,c | 83:17 |
Yield of isolated alcohol product 7aa from oxidation. Conditions: NaBO3 (4 equiv), THF/H2O, 25 °C.
Yield of pinacolborane 6aa by 1H NMR integration with an internal standard.
Some decomposition of boronate complex observed.
A recent report from this laboratory on Lewis base-catalyzed polyene sulfenocyclization revealed the salutary effect of hexafluoroisopropyl alcohol (HFIP) as a solvent for these reactions by obviating the need for acidic activators.16 Inspired by these results, we explored the effects of polar protic solvents on the present system. Thus, employing 4a in either methanol or ethanol led to the formation of 6aa in synthetically useful yield and excellent enantioselectivity, with very little background reaction (entries 7–10). Although no reaction was observed with 4b in ethanol (pKa = 16),17 a modest yield of 6aa was obtained in tetrafluoroethanol (TFE) (pKa = 12)17 with moderate enantioselectivity (entries 11–14). Likewise, a modest yield was observed with 4c in HFIP (pKa = 9)17 with similar enantioselectivity (entries 15 and 16), although significant decomposition of the boronate complex was observed in this more acidic medium. These results suggest that, whereas protic solvents can attenuate the background reactivity of boronate complex 3aa, only the most active sulfenylating agent 4a is capable of sulfenyl group transfer to catalyst 5 in higher-pKa alcohols, which are necessary to avoid decomposition of the boronate. Therefore, the conditions in entry 10 were selected to evaluate the scope of this transformation. The attenuation of boronate reactivity in protic solvents likely arises from hydrogen-bonding interactions that stabilize the anionic character of the pinacolate complex 3aa.
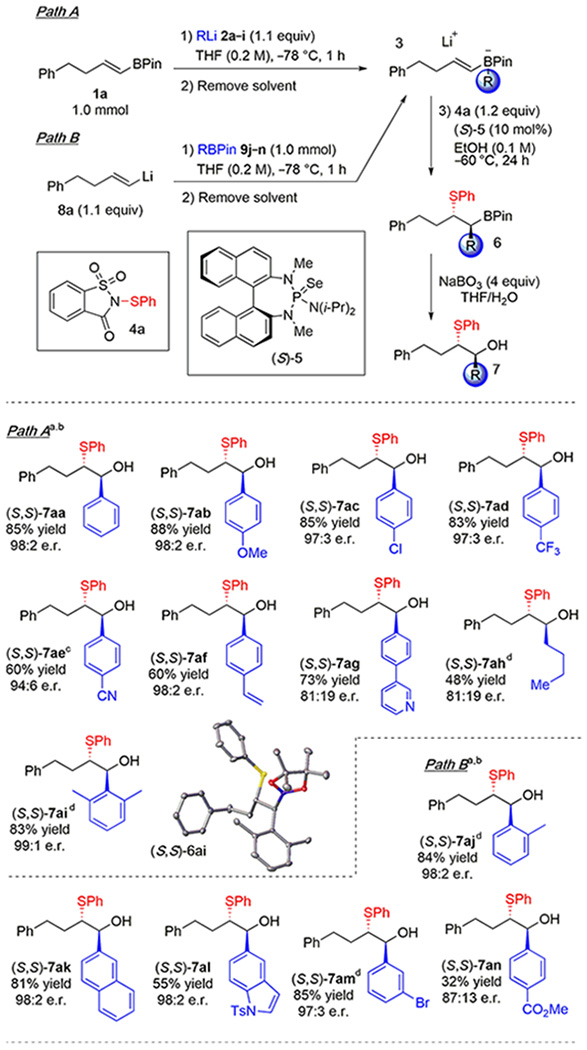
The exploration of reaction scope began with an examination of the migrating groups (Table 2). Throughout this paper, compounds are identified by the nomenclature Nxy where N is the compound class (3 = boronate complex, 6 = functionalized borane product, and 7 = alcohol resulting from borane oxidation), x is the alkenyl fragment being functionalized, and y is the migrating group. Tetracoordinate boronate complexes 3aa–3ai were accessed by addition of organolithium reagents 2a–2i to alkenylpinacolborane 1a (Path A). Aryllithium reagents 2a–2d, bearing electronically diverse 4-substituents, added and migrated efficiently to afford products 7aa–7ad in high yields and excellent enantioselectivities after oxidation of the alkylboranes. To prevent self-condensation of 4-cyanophenyllithium 2e, this reagent was generated in situ in the presence of 1a to ensure immediate formation of “ate” complex 3ae. Subsequent migration and oxidation afforded product 7ae in good yield and high enantioselectivity. Complex 3af led to the desired product 7af in acceptable yield (60%), demonstrating that styrenyl olefins do not react at an appreciable rate under the reaction conditions. Using pyridinyl-substituted phenyllithium 2g afforded product 7ag in acceptable yield but diminished enantiomeric purity. Employing n-butyllithium as the nucleophile resulted in modest yield and enantioselectivity of 7ah, demonstrating that alkyl groups migrate less efficiently.
Table 2.
Organolithium Scope
|
Isolated yields of analytically pure material.
Enantiomeric ratio determined by chiral stationary phase NP-HPLC or SFC.
2e generated in the presence of 1a.
Conditions for oxidation of 6 to 7: NaOH/H2O2/THF, 0 °C.
Because the alkenylboronate complexes are sterically and electronically distinct from any previously investigated class of alkenes, it was deemed prudent to establish the absolute stereochemical course of this reaction. This circumspection was prescient, as an X-ray crystallographic structure determination18 of intermediate 6ai (derived from 2,6-dimethyl-phenyllithium 2i, and en route to 7ai) revealed that it possessed the (S,S)-configuration, opposite to that expected on the basis of our previous results and predictive models for facial selectivity.12b Evidently, the modes of interaction between the catalytic donor–acceptor complex and the boronate are much different than those which exist between this complex and simple alkenes.
Alternatively, boronate 3 can be generated from alkenyllithium reagent 8a and arylboronic esters 9j–9n (Path B). Products 7aj and 7ak were accessed in this manner from 2-tolylpinacolborane 9j and 2-napththylpinacolborane 9k, respectively. Path A could not be used to access boronate 3al because halogen–lithium exchange of 5-bromo-N-tosylindole resulted in lithiation at multiple positions. Path B circumvented this problem, allowing the isolation of 7al in 55% yield and 98:2 enantiomeric ratio (e.r.). Likewise, Path B was required to form boronate complex 3am containing a 3-bromophenyl group, which afforded product 7am in 85% yield after migration. Finally, even using Path B, product 7an bearing a methyl ester was isolated in only 32% yield, owing to the incompatibility of this functional group with organolithium reagents.19
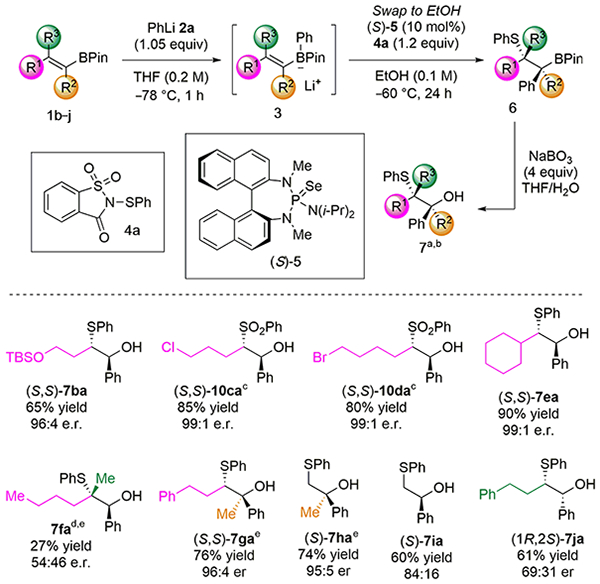
The second stage of exploration of reaction scope focused on those alkenylboranes which could engage in carbosulfenylation (Table 3). It has been previously demonstrated that trans-1,2-disubstituted alkenes are optimal substrates for catalyst 5, affording sulfeno-functionalized products in high yields and enantioselectivities. Accordingly, all trans-1,2-alkenylboronates 3ba–3ea are excellent substrates for the present transformation. Pendant silyl ethers, primary alkyl chlorides, and primary alkyl bromides are compatible with the reaction conditions (products 7ba–7da). A more congested trans-alkenylboronate 3ea still affords product 7ea in high yield and enantioselectivity. 1,2,2-Trisubstituted alkenylboronate 3fa was not an effective substrate for this transformation, as product 7fa was isolated in low yield and poor enantio-selectivity.20a In contrast, a 1,1,2-trisubstituted alkenylboronate 3ga reacted quite efficiently to form product 7ga in 76% yield and 96:4 e.r. Boronate 3ha also reacted efficiently to form product 7ha in 74% yield and 95:5 e.r. This outcome was unexpected, as geminal 1,1-disubstituted olefins are traditionally very poor substrates for catalyst 5. Nevertheless, products 7ga and 7ha highlight the utility of this method for generating chiral, non-racemic, tertiary alcohols. Unsubstituted vinyl pinacolboronate 3ia reacted to form product 7ia in good yield but more modest enantioselectivity (84:16).20b As expected from previous work, a cis-alkenylboronate 3ja was not well-recognized by catalyst 5, and product 7ja was isolated in acceptable yield but poor enantioselectivity (69:31).21
Table 3.
Alkenylboronate Scope
|
Isolated yields of analytically pure material.
Enantiomeric ratio determined by chiral stationary phase NP-HPLC or SFC.
7 oxidized to sulfone 10 with m-CPBA prior to isolation.
Tentative absolute configuration shown.
Conditions for oxidation of 6 to 7: NaOH/H2O2/THF, 0 °C.
The stability of the sulfenyl pinacolborane products provides an ideal opportunity to examine their synthetic utility (Scheme 2). Reduction of enantiomerically enriched (99:1 e.r.) α-sulfenylated borane 6aa led to different products depending on the exact conditions used. Addition of lithium metal to a solution of 6aa and tert-butanol in ammonia afforded C–S cleavage product 11aa in good yield, provided the reaction is quenched in a timely fashion. Alternatively, if 6aa was treated with LDMAN (lithium N,N-dimethyl-1-aminonaphthalenide)22 and the reaction aged for 1 h in the absence of any electrophile, an unusual rearrangement product 12aa was observed, which displayed a modest erosion in e.r. (82:18).23 This rearrangement likely proceeds through a boratirane ion intermediate.24,25 Finally, treating 6aa with LDMAN and an electrophilic reagent (isopropoxy pinacolborane) in the same pot afforded diborylated compound 13aa in 68% yield, albeit with poor diastereoselectivity.26 All attempts to oxidize α-sulfenylated borane 6aa to a sulfoxide resulted only in elimination to form a trans-olefin.27 Of course, 6aa could first be oxidized to alcohol 7aa using sodium perborate, and subsequent treatment with hydrogen peroxide afforded α-hydroxy sulfoxide 14aa in 93% yield as a mixture of diastereomers. Treating this mixture with sodium carbonate in refluxing xylenes formed elimination product 15aa in 89% yield with predominantly (E)-geometry. Finally, mesylation of 7aa followed by treatment with 4-methoxyaniline afforded secondary amine 16aa in 87% yield. The displacement is net retentive, indicating that the reaction proceeds through a thiiranium ion intermediate.28
Scheme 2.

Product Manipulations
In conclusion, an enantio- and diastereoselective, Lewis base-catalyzed carbosulfenylation of alkenylboronates has been described. The reaction proceeds by 1,2-boronate migration to open a thiiranium ion, affording chiral, non-racemic alkyl boronic esters with two vicinal stereogenic centers.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the National Institutes of Health (GM R35 127010) for generous financial support. We also thank the UIUC SCS support facilities (microanalysis, mass spectrometry, and NMR spectroscopy) for their assistance.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b10288.
Experimental procedures and characterization data for all new compounds (PDF)
X-ray crystallographic data for (S,S)-6ai (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Brown HC; Singaram B The Development of a Simple General Procedure for Synthesis of Pure Enantiomers via Chiral Organoboranes. Acc. Chem. Res 1988, 21, 287–293. [Google Scholar]
- (2).Scott HK; Aggarwal VK Highly Enantioselective Synthesis of Tertiary Boronic Esters and Their Stereospecific Conversion to Other Functional Groups and Quaternary Stereocentres. Chem. - Eur. J 2011, 17, 13124–13132. [DOI] [PubMed] [Google Scholar]
- (3).Sandford C; Aggarwal VK Stereospecific Functionalizations and Transformations of Secondary and Tertiary Boronic Esters. Chem. Commun 2017, 53, 5481–5494. [DOI] [PubMed] [Google Scholar]
- (4).Collins BSL; Wilson CM; Myers EL; Aggarwal VK Asymmetric Synthesis of Secondary and Tertiary Boronic Esters. Angew. Chem., Int. Ed 2017, 56, 11700–11733. [DOI] [PubMed] [Google Scholar]
- (5).Miyaura N Hydroboration, Diboration, Silylboration, and Stannylboration In Catalytic Heterofunctionalization; Togni A, Grützmacher H, Eds.; Wiley-VCH: 2001; pp 1–45. [Google Scholar]
- (6).(a) Zhang L; Lovinger GJ; Edelstein EK; Szymaniak AA; Chierchia MP; Morken JP Catalytic Conjunctive Cross-Coupling Enabled by Metal-Induced Metallate Rearrangement. Science 2016, 351, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lovinger GJ; Aparece MD; Morken JP Pd-Catalyzed Conjunctive Cross-Coupling between Grignard-Derived Boron “Ate” Complexes and C(sp2) Halides or Triflates: NaOTf as a Grignard Activator and Halide Scavenger. J. Am. Chem. Soc 2017, 139, 3153–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Edelstein EK; Namirembe S; Morken JP Enantioselective Conjunctive Cross-Coupling of Bis(alkenyl)borates: a General Synthesis of Chiral Allylboron Reagents. J. Am. Chem. Soc 2017, 139, 5027–5030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chierchia M; Law C; Morken JP Nickel-Catalyzed Enantioselective Conjunctive Cross-Coupling of 9-BBN Borates. Angew. Chem., Int. Ed 2017, 56, 11870–11874. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Lovinger GJ; Morken JP Ni-Catalyzed Enantioselective Conjunctive Coupling with C(sp3) Electrophiles: a Radical-Ionic Mechanistic Dichotomy. J. Am. Chem. Soc 2017, 139, 17293–17296. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Myhill JA; Zhang L; Lovinger GJ; Morken JP Enantioselective Construction of Tertiary Boronic Esters by Conjunctive Cross-Coupling. Angew. Chem., Int. Ed 2018, 57, 12799–12803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Leonori D; Aggarwal VK Lithiation-Borylation Methodology and its Application in Synthesis. Acc. Chem. Res 2014, 47, 3174–3183. [DOI] [PubMed] [Google Scholar]
- (8).Thomas SP; French RM; Jheengut V; Aggarwal VK Homologation and Alkylation of Boronic Esters and Boranes by 1,2-Metallate Rearrangement of Boron Ate Complexes. Chem. Rec 2009, 9, 24–39. [DOI] [PubMed] [Google Scholar]
- (9).(a) During galley proof preparation, Morken et al. reported the synthesis of alkylboronic ester bearing two vicinal stereogenic centers from trans-alkenylboronic esters by employing a bulky ester to disfavor direct Suzuki–Miyaura coupling. See: Myhill JA; Wilhelmsen CA; Zhang L; Morken JP Diastereoselective and Enantioselective Conjunctive Cross-Coupling Enabled by Boron Ligand Design. J. Am. Chem. Soc 2018, DOI: 10.1021/jacs.8b09909. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) For 1,2-difunctionalization of indoles, see: Panda S; Ready JM Tandem Allylation/1,2-Boronate Rearrangement for the Asymmetric Synthesis of Indolines with Adjacent Quaternary Stereocenters. J. Am. Chem. Soc 2018, 140, 13242–13452. [DOI] [PubMed] [Google Scholar]
- (10).Zweifel G; Arzoumanian H; Whitney CC A Convenient Stereoselective Synthesis of Substituted Alkenes via Hydroboration-Iodination of Alkynes. J. Am. Chem. Soc 1967, 89, 3652–3653. [Google Scholar]
- (11).(a) Armstrong RJ; Aggarwal VK 50 Years of Zweifel Olefination: a Transition-Metal-Free Coupling. Synthesis 2017, 49, 3323–3336. [Google Scholar]; (b) Armstrong RJ; Sandford C; García-Ruiz C; Aggarwal VK Conjunctive Functionalization of Vinyl Boronate Complexes with Electrophiles: a Diastereoselective Three-Component Coupling. Chem. Commun 2017, 53, 4922–4925. [DOI] [PubMed] [Google Scholar]
- (12).(a) Kalyani D; Kornfilt DJP; Burk MT; Denmark SE Lewis Base Catalysis: a Platform for Enantioselective Addition to Alkenes using Group 16 and 17 Lewis Acids (n → σ*) In Lewis Base Catalysis in Organic Synthesis; Vedejs E, Denmark SE, Eds.; Wiley-VCH: 2016; pp 1153–1212. [Google Scholar]; (b) Denmark SE; Hartmann E; Kornfilt DJP; Wang H Mechanistic, Crystallographic, and Computational Studies on the Catalytic, Enantioselective Sulfenofunctionalization of Alkenes. Nat. Chem 2014, 6, 1056–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Denmark SE; Rossi S; Webster MP; Wang H Catalytic, Enantioselective Sulfenylation of Ketone-Derived Enoxysilanes. J. Am. Chem. Soc 2014, 136, 13016–13028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).The solid “ate” complex 3 obtained after solvent removal, while isolable, is not stable to air or moisture.
- (15).Using THF as the reaction solvent resulted in diminished yield (4a) or no conversion (4b, 4c). Various additives (MgBr2, LiClO4, 12-crown-4, n-Bu4NBr, n-Bu4NOMs) had no significant influence on yield or enantioselectivity.
- (16).Tao Z; Robb KA; Zhao K; Denmark SE Enantio-selective, Lewis Base-Catalyzed Sulfenocyclization of Polyenes. J. Am. Chem. Soc 2018, 140, 3569–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).pKa values (in water):HFIP and TFE: Filler R; Schure RM Highly Acidic Perhalogenated Alcohols. A New Synthesis of Perfluoro-tert-butyl Alcohol. J. Org. Chem 1967, 32, 1217–1219.MeOH and EtOH: Ballinger P; Long FA Acid Ionization Constants of Alcohols. II. Acidities of some Substituted Methanols and Related Compounds. J. Am. Chem. Soc 1960, 82, 795–798.
- (18).CCDC 1866767 contains the crystallographic data for compound 6ai. These data can be obtained free of charge from the Cambridge Crystallographic Data Center (www.ccdc.cam.ac.uk).
- (19).The slight drop in enantioselectivity (87:13) observed for product 7an was perplexing and could not be explained solely on the basic of electronic effects, because similarly electron-deficient aryl groups migrated with high e.r. (see products 7ac–7ae). The only difference between the reaction mixtures in Path A and Path B is the presence of LiBr. Unlike lithium–halogen exchange of a bromoarene to form 2 (accomplished with n-BuLi), lithium–halogen exchange of an alkenyl bromide to form 8a requires tert-BuLi, which generates LiBr. Indeed, addition of exogenous LiBr to the reaction mixtures in Path A causes a drop in enantioselectivity, but only for electron-deficient migrating groups, whereas electron-rich migrating groups are unaffected by this additive.
- (20).(a) The thiiranium ion opens to form the stabilized tertiary carbocation, which can eliminate boron to form an alkenyl sulfide, the major observed byproduct. See ref 11b.; (b) We have also examined the reactivity of (E)-2-styryl(pinacolborane) and did not observe the desired product. Only the corresponding vinyl sulfide product was obtained. This substrate suffers from the same effect observed for the transformation of 3fa to 6fa described above.
- (21).Denmark SE; Jaunet A Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. J. Am. Chem. Soc 2013, 135, 6419–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cohen T; Matz JR Reductive Lithiation of some Thioketals using Lithium 1-(dimethylamino)naphthalenide. Synth. Commun 1980, 10, 311–317. [Google Scholar]
- (23).The absolute configuration and e.r. of borane 12aa were determined by oxidation to 23 (44% yield over two steps from 6aa). Refer to the Supporting Information for details.
- (24).For the intermediacy of similar species, see:Kropp MA; Baillargeon M; Park KM; Bhamidapaty K; Schuster GB Photochemistry of Alkynyl-, Alkenyl-, and Cyclopropyl-Substituted Borate Salts: The Di-π- and Cyclopropyl-π-Borate Rearrangements. J. Am. Chem. Soc 1991, 113, 2155–2163.Denmark SE; Nishide K; Faucher A-M On the Generation and Configurational Stability of (2S, 3S)-1,2,3-Triphenylborirane. J. Am. Chem. Soc 1991, 113, 6675–6676.
-
(25).A kinetic preference exists for formation of the anti-boratirane ion, which leads to (S)-12aa upon quenching.
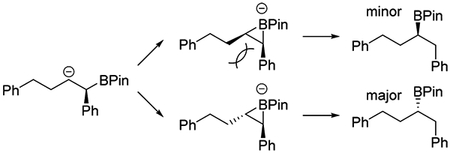
- (26).Both syn and anti diastereomers of 13aa display a modest erosion in enantioselectivity, similar in magnitude to that observed for 12aa. It can be inferred that the conversion of 6aa to 13aa also proceeds through a boratirane ion intermediate and that the benzylic boryl group originates from i-PrOBPin.
- (27).Armstrong RJ; García-Ruiz C; Myers EL; Aggarwal VK Stereodivergent Olefination of Enantioenriched Boronic Esters. Angew. Chem., Int. Ed 2017, 56, 786–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).We have attempted cross coupling reactions of 6aa to forge a C–C bond using two different conditions: (1) traditional Suzuki conditions (with iodobenzene) and (2) coupling with a lithiated furan: Bonet A; Odachowski M; Leonori D; Essafi S; Aggarwal VK Enantiospecific sp2–sp3 coupling of secondary and tertiary boronic esters. Nat. Chem 2014, 6, 584–589.In both cases, only the elimination product (1,4-diphenyl-1-butene) was observed. The thioether must be removed before the pinacolboronic ester can engage in productive cross coupling.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.