

SUMMARY
Nutritional supplementation with probiotics can prevent pathologic bone loss. Here we examined the impact of supplementation with Lactobacillus rhamnosus GG (LGG) on bone homeostasis in eugonadic young mice. Micro-computed tomography revealed that LGG increased trabecular bone volume in mice, which was due to increased bone formation. Butyrate produced in the gut following LGG ingestion, or butyrate fed directly to germ-free mice, induced the expansion of intestinal and bone marrow (BM) regulatory T (Treg) cells. Interaction of BM CD8+ T cells with Treg cells resulted in increased secretion of Wnt10b, a bone anabolic Wnt ligand. Mechanistically, Treg cells promoted the assembly of a NFAT1-SMAD3 transcription complex in CD8+ cells, which drove expression of Wnt10b−/−. Reducing Treg cell numbers, or reconstitution of TCRβ−/− mice with CD8+ T cells from Wnt10b−/− mice, prevented butyrate-induced bone formation and bone mass acquisition. Thus, butyrate concentrations regulate bone anabolism via Treg cell-mediated regulation of CD8+ T cell Wnt10b production.
In Brief
Tyagi et al. show that oral supplementation with the widely used probiotic, Lactobacillus rhamnosus GG (LGG), increases bone mass in mice by increasing the serum levels of the short chain fatty acid butyrate. LGG or butyrate increase the frequency of regulatory T (Treg) cells in the intestine and in the bone marrow. Treg cells stimulate CD8+ T cells to secrete the Wnt ligand Wnt10b, which stimulates bone formation by activating Wnt signaling in osteoblasts. Therefore, LGG and butyrate may represent new interventions for the prevention and treatment of osteoporosis.
Graphical Abstract
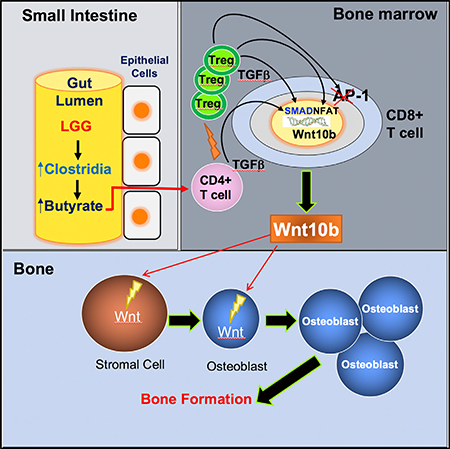
INTRODUCTION
Fractures due to osteoporosis have devastating consequences, with complications of hip fractures leading to mortality rates of 24%–30% during the first year following injury, and almost 50% rates of permanent disability (Burge et al., 2007). Many FDA-approved drugs are used for the treatment of osteoporosis. However, most cases of osteoporosis remain untreated or ineffectively treated due to the cost and side effects of presently available drugs (Khosla and Shane, 2016). Therefore, there is an exigent need to identify and substantiate new, inexpensive, safe, and effective interventions for both the prevention and treatment of osteoporosis.
Nutritional supplementation with probiotics, defined as viable microorganisms that confer a health benefit when administered in adequate quantities, may represent one such strategy. Indeed, small-scale human studies report positive results from supplementing the diet of osteoporotic patients with probiotics (Jafarnejad et al., 2017; Nilsson et al., 2018). In animals, probiotics prevent the bone loss induced by periodontal disease (Messora et al., 2013), diabetes (Zhang et al., 2015), and estrogen deficiency (Li et al., 2016; Ohlsson et al., 2014). In spite of these reports, knowledge is still scant on whether probiotics can improve postnatal skeletal development or delay skeletal involution in healthy subjects. Earlier investigations by our group report that the widely used probiotic Lactobacillus rhamnosus GG (LGG) protects mice from ovariectomy-induced stimulated bone resorption and bone loss (Li et al., 2016). In the same study, we observe that LGG drives bone formation and increases bone volume in eugonadic mice (Li et al., 2016), although the functional elements responsible for this response remained enigmatic.
The enzymatic activity harbored within some taxa of gut microbiota can digest carbohydrates to generate millimolar concentrations of the short-chain fatty acids (SCFAs) butyrate, propionate, and acetate (Bach Knudsen, 2015). Although LGG alone, does not produce SCFAs, LGG expands intestinal butyrate-producing bacteria (Berni Canani et al., 2016), indicating that LGG may indirectly increase production of SCFAs by the gut microbiota. SCFAs selectively support the development of peripheral regulatory T (Treg) cells (Arpaia et al., 2013; Furusawa et al., 2013; Smith et al., 2013). Reports highlight the bone-regulating capacities of Treg cells, describing mechanisms where Treg cells blunt bone resorption (Kelchtermans et al., 2009; Kim et al., 2007), stimulate bone formation by promoting the differentiation of osteoblasts (Lei et al., 2015), and are pivotal for parathyroid hormone (PTH)-stimulated bone formation (Yu et al., 2018).
Here we examined the role of LGG-induced butyrate in the regulation of bone homeostasis in young eugonadic mice. We found that LGG supplementation increased intestinal and systemic butyrate concentrations and stimulated bone formation. Increased butyrate also resulted in higher numbers of Treg cells. Examination of the role of Treg cells in bone formation revealed a pathway whereby interaction of Treg cells with BM CD8+ T cells regulated the production of Wnt10b, which acts on stromal cells and osteoblasts to promote bone formation.
RESULTS
LGG Supplementation Increases Butyrate Levels and the Number of Treg Cells
Conventionally raised 10-week-old female mice were supplemented by oral gavage with 1 × 109 CFU LGG or vehicle control for 4 weeks. LGG supplementation resulted in a change in microbial diversity in the intestinal lumen and an expansion in the proportion of SCFA producing clostridia (Figures 1A and 1B). Furthermore, LGG supplementation of mice induced the enrichment of transcripts of a bacterial gene coding for butyryl-CoA: acetate CoA-transferase (Figure 1C), an enzyme involved in butyrate production by lactate-utilizing bacteria in the gut (Duncan et al., 2004; Hippe et al., 2011). Consistent with these findings, elevated levels of butyrate but not propionate were detected in small intestine (SI) tissue and in the serum of LGG-treated mice (Figures 1D–1G). Butyrate induces Treg cell differentiation (Arpaia et al., 2013; Furusawa et al., 2013; Smith et al., 2013). Accordingly, we found that feeding of LGG or butyrate increased the absolute and relative number of BM and splenic Treg cells (Figures 1H–1K and S1A). Because CD25 antibody (Ab) depletes Treg cells in vivo (Setiady et al., 2010; Yu et al., 2018), Treg cells induction was blunted in mice injected with an anti-CD25 Ab but not in control mice injected with irrelevant Ab (Irr. Ab) (Figures 1H–1K). Moreover, treatment with LGG or butyrate increased the CD4/CD8+ BM T cell ratio, both in mice treated with Irr. Ab and those treated with anti-CD25 Ab (Figures S1B and S1C), indicating that LGG and butyrate independently regulated Treg cell differentiation and CD4+/CD8+ T cell ratio.
Figure 1. LGG Increases the Relative Frequency of Clostridia in the Gut, the Levels of Butyrate (But) in the Small Intestine and Serum, and the Number of BM and Splenic Treg Cells.

(A and B) Detailed relative abundance of bacterial taxa at the class level within fecal pellets collected from mice treated with LGG or vehicle control for 4 weeks. (legend continued on next page)
(C) Measurement of transcript levels of butyryl-CoA:acetate CoA-transferase in the luminal contents of the ileum in mice administered LGG or vehicle control for 4 weeks.
(D–G) Butyrate and propionate concentrations in small intestine tissue and serum of mice administered LGG or vehicle control for 4 weeks.
(H and I) Relative and absolute frequency of BM Treg cells.
(J and K) Relative and absolute frequency of splenic Treg cells.
Data were expressed as mean ± SEM. All data were normally distributed according to the Shapiro-Wilk normality test. n = 6–7 mice per group in (A) and (B); n = 9 mice per group in (C)–(G); n = 12–13 mice per group in (H)–(K). Data (B–G) were analyzed by unpaired t tests. All other data were analyzed by two-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to the indicated group. ns = not significant.
Partial Treg Cell Depletion by Anti-CD25 Ab Abrogates the Bone Anabolic Activity of Butyrate
In vivo prospective μCT measurements of the spine revealed that in mice treated with Irr. Ab, administration of LGG or butyrate caused a marked increase in bone volume fraction (BV/TV) compared to baseline, at both 2 and 4 weeks of treatment. By contrast, neither LGG nor butyrate increased BV/TV in mice depleted of Treg cells by anti-CD25 Ab (Figure 2A). Moreover, LGG or butyrate supplementation induced significant changes in the vertebral indices of bone structure: trabecular number (Tb.N), trabecular separation (Tb.Sp), and trabecular thickness (Tb.Th), in Irr. Ab-treated mice but not in anti-CD25 Ab-treated mice (Figures S2A–S2C). Tb.N is a measure of the number of trabecular rods within trabecular bone, Tb.Th is a measure of the average thickness of the trabecular rods, while Tb.Sp is a measure of the average distance between rods. In vitro μCT analysis of femurs harvested at sacrifice confirmed that LGG and butyrate equally increased femoral BV/TV in mice treated with Irr. Ab but not in mice treated with anti-CD25 Ab (Figure 2B). Femoral Tb.N and Tb.Sp were also altered by LGG and butyrate in mice treated with control Ab but not in those treated with antiCD25 Ab (Figures S2D and S2E). However, LGG and butyrate failed to increase femoral Tb.Th (Figure S2F) and indices of cortical structure (Figures S2G–S2J), indicating that LGG and butyrate had no effects on cortical bone, but rather improved trabecular bone volume by increasing the number of trabeculae.
Figure 2. Treatment with Anti-CD25 Ab Abrogates the Bone Anabolic Activity of LGG and Butyrate (But).

(A) Prospective measurements of vertebral trabecular bone volume fraction (BV/TV) by in vivo μCT scanning.
(B) Cross-sectional measurements of femoral BV/TV by in vitro mCT scanning.
(C) Mineral apposition rate (MAR).
(D) Bone formation rate per mm bone surface (BFR/BS).
(E) Number of osteoclasts per mm bone surface (N.Oc/BS).
(F) Percentage of bone surface covered by osteoclasts (Oc.S/BS).
(G) Serum levels of P1NP, a marker of bone formation.
(H) Serum levels of type 1 cross-linked C-telopeptide (CTX), a marker of bone resorption.
(I–K) Wnt10b mRNA levels in whole BM, BM CD8+ T cells, and BM CD4+ T cells.
n = 10–17 mice per group. Data were expressed as mean ± SEM. All data were normally distributed according to the Shapiro-Wilk normality test. Data in (A) were analyzed by ANOVA for repeated-measures. ****p < 0.0001 compared to baseline, ####p < 0.0001 compared to Irr. Ab vehicle. All other data were analyzed by two-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to the indicated group. ns = not significant. Irr. Ab = Irrelevant antibody.
Analysis of femoral cancellous bone by histomorphometry revealed that LGG and butyrate increased the indices of bone formation: mineral apposition rate (MAR), bone formation rate (BFR/ BS), osteoblast number (N.Ob/BS), and osteoblast surfaces (Ob.S/BS) in control mice but not in those treated with antiCD25 Ab (Figures 2C, 2D, S2K, and S2L). Indices of bone resorption (N.Oc/BS and Oc.S/BS) were not affected by LGG and butyrate in all groups (Figures 2E and 2F). However, Treg cell-depleted groups did have higher indices of resorption as compared to Treg cell-replete mice, supporting previous reports that Treg cells also function to suppress bone resorption. Serum P1NP, a marker of bone formation, was increased by LGG and butyrate in Treg cell-replete mice (Figure 2G). Serum CTX, a marker of bone resorption, did not increase in response to LGG or butyrate, although Treg cell-depleted mice did have higher levels of CTX, confirming the capacity of Treg cells to suppress bone resorption (Figure 2H). Moreover, LGG or butyrate did not alter the expression of RANKL mRNA in BM or sorted Treg cells (Figures S3A and S3B), nor did they alter the expression of OPG in the BM (Figure S3C), all of which are critical regulators of osteoclastogenesis and bone resorption. Another factor produced by Treg cells that regulates bone resorption is IL-10. We found that LGG or butyrate did not alter the transcript levels of IL-10 in the BM or in sorted Treg cells (Figures S3D and S3E).
Osteoblasts derive from BM stromal cells (SCs). Administration of either LGG or butyrate increased the proliferation of SCs in mice treated with Irr. Ab but not in mice treated with anti-CD25 Ab (Figure S3F), while no effects on the rate of SC apoptosis were detected in these groups (Figure S3G). Measurement of transcript enrichment of SC genes involved in osteoblast differentiation, including type 1 collagen (Col-I), runt related transcription factor 2 (Runx2), osterix (Osx), bone sialoprotein (BSP), and osteocalcin (OCN), revealed that LGG and butyrate treatment amplified osteoblast differentiation in Treg cell-replete mice, while inducing no response on SCs in Treg cell-depleted mice (Figure S3H). Transcript levels of seven measured Wntresponsive genes were amplified by LGG or butyrate in SCs of Treg cell-replete mice, but not in SCs from mice where Treg cells were diminished by anti-CD25 Ab treatment (Figure S3I). These data indicated that LGG and butyrate activated Wnt signaling in SCs, through a Treg cell-dependent mechanism. A potent activator of Wnt signaling is the Wnt ligand Wnt10b, and CD8+ T cells are a major source of Wnt10b in the BM (Bedi et al., 2012; Terauchi et al., 2009). We found that both butyrate and LGG increased Wnt10b transcript levels in whole BM (Figure 2I). This response was ascribed to CD8+ T cells (Figure 2J) but not to conventional CD4+ T cells (Figure 2K). The LGG or butyrate-induced responses on Wnt10b production were inhibited by anti-CD25 Ab treatment (Figures 2I–2K). Increased Wnt10b transcript levels were also detected in CD8+ T cells (eGFP-CD8+ T cells) but not in conventional CD4+ cells (eGFP-CD4+ T cells) or in Treg cells (eGFP+CD4+ T cells) sorted from Foxp3.eGFP reporter mice treated with LGG or butyrate (Figures S3J–S3L).
Since some inflammatory cytokines blunt bone formation, the capacity of anti-CD25 Ab treatment to block LGG- and buty-rate-induced bone anabolism could be ascribed to increased production of inflammatory cytokines, rather than the detected increase in Treg cell differentiation and the resulting Wnt10b secretion by CD8+ T cells. This possibility is unlikely because BM cells from groups of mice treated with anti-CD25 Ab expressed similar levels of IL-1β, TNFα, IL-6, IL-17A, IL-4, and IFNγ mRNAs, as BM cells from mice treated with Irr Ab (Figure S3M). In addition, evidence against a nonspecific inhibitory effect of partial Treg cell depletion on bone formation was corroborated by the finding that treatment with anti-CD25 Ab did not decrease bone formation in vehicle-treated mice (Figures 2C, 2D, and 2G).
Partial Treg Cell Depletion in DEREG Mice Prevents the Bone Activity of Butyrate
Additional experiments were conducted using DEREG mice, a strain that expresses a fusion protein of the human diphtheria toxin (DT) receptor (hDTR) and eGFP under the control of the Foxp3 promoter (Lahl et al., 2007). Foxp3+ Treg cells can be selectively depleted upon DT administration to DEREG mice, whereas Treg cells in WT mice are insensitive to DT. Furthermore, DT is known to not cause toxic effects in mice (Klingenberg et al., 2013). Whereas Treg cell ablation in DEREG mice causes scurfy-like symptoms in newborn animals, older mice do not develop autoimmune diseases (Klingenberg et al., 2013). We treated DEREG mice with DT (0.5 μg/mouse, i.p. 2 times per week for 4 weeks), a dose titrated to block the increase in Treg cells induced by LGG and butyrate. Mice were also treated with vehicle, LGG, or butyrate for 4 weeks. Controls included DEREG mice not treated with DT and WT littermate (LM) mice treated with DT. LGG and butyrate expanded the number of BM Treg cells, splenic Treg cells, and Peyer’s patches (PP) Treg cells in control mice, but not in DEREG + DT mice (Figures 3A–3C). Since the calculation of the absolute number of PP Treg cells is inaccurate because of the variability of the size of the collected PP tissue, PP Treg cells were shown only as percentage.
Figure 3. Treatment of DEREG Mice but Not of WT Littermates (WTLM) with Diphtheria Toxin (DT) Prevents the Increase in the Number of Treg Cells and the Bone Anabolic Activity Induced by LGG and Butyrate (But).

(A and B) Absolute number of Treg cells in the BM and the spleen.
(C) Relative number of Treg cells in Peyer’s patches (PP). Since the enumeration of the absolute number of PP Treg cells is inaccurate, PP Treg cells are shown as percentage.
(D) Prospective measurements of vertebral trabecular bone volume fraction (BV/TV) by in vivo μCT scanning.
(E) Cross-sectional measurements of femoral BV/TV by in vitro μCT scanning.
(F) Mineral apposition rate (MAR).
(G) Bone formation rate (BFR).
(H) The number of osteoclasts per mm bone surface (N.Oc/BS).
(I) The percentage of bone surface covered by osteoclasts (Oc.S/BS).
(J) Serum levels of osteocalcin (OCN), a marker of bone formation.
(K) Serum levels of type 1 cross-linked C-telopeptide of collagen (CTX), a marker of bone resorption.
(L) Wnt10b mRNA levels in whole BM.
(M) Wnt10b mRNA levels in BM CD8+ T cells.
For this assay, samples from 2 mice per group were randomly pooled together to generate a sufficient amount of mRNA. n = 6–13 mice per group. Data were expressed as mean ± SEM. All data were normally distributed according to the Shapiro-Wilk normality test. Data in (A) were analyzed by ANOVA for repeatedmeasures. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to baseline, #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001 compared to Irr. Ab vehicle. Data in (B)–(L) were analyzed by two-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to the indicated group. ns = not significant.
In vivo prospective μCT measurements revealed that LGG and butyrate increased vertebral BV/TV, Tb.N, Tb.Sp, and Tb.Th compared to baseline in control mice, but not in DEREG mice treated with DT (Figures 3D and S4A). Analysis by μCT of femurs harvested at sacrifice showed that LGG and butyrate increased BV/TV in the two control groups but not in DT-treated DEREG mice (Figure 3E). Tb.N and Tb.Th were increased by LGG and butyrate in Treg cells replete but not in Treg cell-depleted mice. By contrast, LGG and butyrate failed to increase Tb.Sp and indices of cortical structure in all groups of mice (Figure S4B). Analysis of femoral cancellous bone by histomorphometry revealed that LGG and butyrate increased bone formation in the two control groups but not in DT-treated DEREG Treg cell-depleted mice (Figures 3F, 3G, and S4C). Indices of bone resorption were higher in DEREG + DT mice compared to controls due to the partial Treg cell depletion, whereas LGG and butyrate did not further increase bone resorption in any group of mice (Figures 3H and 3I). Measurement of biochemical markers of bone formation and resorption generated results similar to the histomorphometric analysis. In fact, DEREG + DT mice had lower osteocalcin levels and higher CTX levels than control mice (Figures 3J and 3K), due to the partial Treg cell depletion. Moreover, LGG and butyrate increased osteocalcin levels in control but not DEREG + DT mice, although not affecting CTX levels in any group of mice. Finally, LGG and butyrate could not increase the expression of Wnt10b in whole BM or CD8+ T cell in DEREG + DT mice, whereas they induce significant increases in Wnt10b transcript levels in BM and CD8+ T cells in Treg cell-replete control groups (Figures 3L and 3M). Altogether, these data supported the conclusion that an increase in the number of BM Treg cell is required for LGG and butyrate to enhance bone formation.
The Bone Anabolic Activity of LGG Requires the Presence of Gut Flora
Since butyrate exerted a bone anabolic activity equal to that of LGG, the data suggested that LGG supplementation may result in increased production of butyrate, which in turn stimulates bone formation. However, an indirect mechanism is likely because LGG does not produce butyrate, nor does it harbor genes in its chromosome coding for enzymes that generate butyrate (Douillard et al., 2013; Kankainen et al., 2009). To test the hypothesis that the presence of SCFA-generating bacteria in the intestine was required for LGG to induce bone anabolism, germ-free (GF) mice were treated with LGG or butyrate for 4 weeks. Analysis of femurs harvested at sacrifice by μCT revealed that butyrate increased BV/TV, while LGG did not (Figure 4A). In addition, serum levels of osteocalcin (Figure 4B) and the number of Treg cells in the BM and spleen were also increased by butyrate but not by LGG (Figures 4C and 4D). The pool of PP Treg cells was expanded by both LGG and butyrate, but the activity of butyrate was significantly greater than that of LGG (Figure 4E). Analysis of whole BM and BM CD8+ T cells harvested from GF mice at sacrifice revealed that butyrate increased Wnt10b transcript levels, whereas LGG did not (Figures 4F and 4G). These findings demonstrate that LGG requires the presence of intestinal flora to induce bone anabolism.
Figure 4. Butyrate (But), but Not LGG Induces Bone Anabolic Effects in Germ-Free Mice.

(A) Femoral trabecular bone volume fraction (BV/TV) as measured by in vitro mCT scanning.
(B) Serum levels of osteocalcin, a marker of bone formation.
(C and D) Absolute number of Treg cells in the BM and the spleen.
(E) Relative number of Treg cells in Peyer’s patches (PP). Since the enumeration of the absolute number of PP Treg cells is inaccurate, PP Treg cells are shown as percentage.
(F) Wnt10b mRNA levels in whole BM.
(G) Wnt10b mRNA levels in BM CD8+ T cells.
For (G), samples from 2 mice per group were randomly pooled together to generate a sufficient amount of mRNA. n = 5–10 mice per group. Data are expressed as mean ± SEM. All data were normally distributed according to the Shapiro-Wilk normality test. Data were analyzed by one-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. *p < 0.05, **p < 0.01, and ****p < 0.0001 compared to the indicated group. ns = not significant.
Butyrate Increases Bone Volume through T Cell-Produced Wnt10b
To conclusively demonstrate that Wnt10b produced by CD8+ T cells mediates the anabolic activity of LGG and butyrate, we assessed the effects of LGG and butyrate in mice specifically lacking Wnt10b production by CD8+ T cells. In these experiments, TCRβ−/− mice, a strain completely devoid of ab T cells, were reconstituted with sorted CD8+ T cells from global Wnt10b−/− mice and CD4+ T cells from WT mice. These mice are herein referred to as Wnt10bCD4ΔCD8 mice. Controls included non-reconstituted TCRβ−/− mice and TCRβ−/− mice reconstituted with WT CD4+ T cells and WT CD8+ T cells (Wnt10bCD4CD8 mice). Two weeks after adoptive transfer, which is a period sufficient for engraftment and expansion of both WT and Wnt10b−/− T cells (Li et al., 2014), all mice were treated with either vehicle, LGG, or butyrate for 4 weeks. LGG or butyrate increased whole BM Wnt10b transcript levels in Wnt10bCD4CD8 mice, but not in Wnt10bCD4ΔCD8, or in non-reconstituted TCRβ−/− mice (Figure 5A). These findings indicated that LGG and butyrate specifically regulate CD8+ T cell production of Wnt10b. Confirming this hypothesis, measurements of Wnt10b transcript levels in BM CD8+ T cells sorted at the end of the treatment period revealed that LGG and butyrate increased Wnt10b transcript levels in CD8+ T cells purified from Wnt10bCD4CD8 mice (Figure 5B). As expected, Wnt10b transcripts were undetectable in CD8+ T cells sorted from Wnt10bCD4ΔCD8 mice. LGG and butyrate treatment equally increased the numbers of Treg cells in the BM, spleen, and PPs of Wnt10bCD4CD8 mice and Wnt10bCD4ΔCD8 mice (Figure S5A), demonstrating that LGG and butyrate expand Treg cells via a Wnt10b-independent mechanism. However, despite increasing Treg cells numbers, in vivo prospective μCT measurements revealed that LGG and butyrate only caused a significant increase in BV/TV in Wnt10bCD4CD8 mice and not in Wnt10bCD4ΔCD8 mice nor in TCRβ−/− mice (Figure 5C). These findings indicated that CD8+ T cell-produced Wnt10b is required for the bone anabolic activity of both LGG and butyrate. Furthermore, LGG and butyrate supplementation increased vertebral Tb.N and decreased Tb.Sp in Wnt10bCD4CD8 mice, but not in Wnt10bCD4ΔCD8 mice and TCRb−/− mice. By contrast, LGG and butyrate did not alter Tb.Th in all groups of mice (Figure S5B). Analysis of distal femurs by in vitro μCT confirmed that LGG and butyrate increase femoral BV/TV in Wnt10bCD4CD8 mice but not in Wnt10bCD4DCD8 mice and TCRβ−/− mice (Figure 5D). In the femur, Tb.N was increased by LGG and butyrate in Wnt10bCD4CD8 mice but not in Wnt10bCD4ΔCD8 mice and TCRβ−/− mice (Figure S5C). LGG and butyrate did not increase femoral Tb.Th, Tb.Sp, or indices of cortical structure in all groups of mice (Figure S5C). Finally, attesting to a link between Wnt10b and bone formation, LGG and butyrate increased serum osteocalcin in Wnt10bCD4CD8 mice but not in Wnt10bCD4ΔCD8 mice and TCRβ−/− mice (Figure 5E). Together, these data showed that LGG and butyrate-induced bone anabolism was dependent on a functional Wnt10b gene within CD8+ cells.
Figure 5. LGG and Butyrate (But) Do Not Induce Bone Anabolic Effects in Mice that Are Deficient in T Cell Production of Wnt10b.

TCRβ−/− mice were adoptively transferred with WT CD4+ T cells and WT CD8+ T cells, or WT CD4+ T cells and Wnt10b−/− CD8+ T cells.
(A) Whole BM Wnt10b transcript levels.
(B) Wnt10b transcript levels in CD8+ T cells purified from reconstituted TCRβ−/− mice at the end of the LGG or butyrate (but) treatment period.
(C) Prospective measurements of vertebral trabecular bone volume fraction (BV/TV) by in vivo μCT scanning.
(D) Femoral BV/TV as measured by in vitro μCT scanning at the end of the treatment period.
(E) Serum levels of osteocalcin, a marker of bone formation.
n = 5–12 mice per group. In (B), samples from 2 mice were pooled together. Data are expressed as mean ± SEM. All data were normally distributed according to the Shapiro-Wilk normality test. Data were analyzed by two-way ANOVA for repeated-measures and post hoc tests applying the Bonferroni correction for multiple comparisons. *p < 0.05, **p < 0.01, and ****p < 0.0001 compared to the indicated group, or baseline. ##p < 0.01, ###p < 0.001 compared to vehicle. ns = not significant.
Wnt10b Production Is Induced by NFAT and SMAD Signaling in CD8+ T Cells
The Wnt10b gene promoter region harbors three DNA binding motifs for NFAT transcription factors located adjacent to binding sites for SMADs, the TGFβ signaling proteins. This organization suggests that Wnt10b transcription may be regulated by the binding of NFAT/SMAD dimers to the Wnt10b promoter. To assess the function of SMADs and NFATs in the control of Wnt10b gene expression, we first measured Wnt10b transcript levels in purified splenic CD8+ T cells after in vitro stimulation with the SMAD inducer TGFβ and the NFAT activator ionomycin. We found that concurrent stimulation of both TGFβ and ionomycin synergistically increased Wnt10b transcript levels, whereas stimulation with either TGFβ or ionomycin alone had a significant but less potent inducing effect (Figure 6A), indicating a potential cooperative function for SMAD and NFAT in the induction of Wnt10b gene expression. Indeed, ChIP assays carried out using BM CD8+ T cells isolated from mice treated in vivo with LGG or butyrate, and/or anti-CD25 Ab, revealed that CD8+ T cells from LGG- and butyrate-treated mice had higher binding of NFAT1 and SMAD3 to the Wnt10b promoter, compared to CD8+ T cells from vehicle-treated mice (Figures 6B–6E). By contrast, and attesting to specificity, LGG and butyrate did not increase the binding of NFAT2 and SMAD2 to the Wnt10b promoter. Blockade of the increase in the number of Treg cells by treatment with anti-CD25 Ab prevented LGG and butyrate from increasing the binding of NFAT1 and SMAD3 to the Wnt10b promoter, indicating that elevated numbers of Treg cells are required for LGG and butyrate to stimulate the binding of these transcription factors to the Wnt10b promoter.
Figure 6. LGG and Butyrate (But) Increase Wnt10b Transcription by Promoting the Binding of NFAT/SMAD Complexes to the Wnt10b Promoter.

(A) Measurement of Wnt10b transcript levels in splenic CD8+ T cells treated with the NFAT activator ionomycin and TGFβ.
(B–E) ChIP assays measuring LGG and butyrate induced binding of NFAT1, SMAD3, NFAT2, and SMAD2, to the Wnt10b promoter in BM CD8+ T cells. Cells from 4–5 mice were pooled to generate 1 sample.
(F) Diagrammatic representation of the Wnt10b promoter and effects of Wnt10b promoter deletion on the activity of Wnt10b-luciferase reporter constructs in primary splenic CD8+ T cells. Cells were stimulated with ionomycin (500 ng/mL) and TGFβ1 (5 ng/mL) for 24 hr to induce reporter activity. n = 3 samples per group.
(G) Effects of mutation of the SMAD and NFAT binding sites on the Wnt10b promoter on the activity of a luciferase-Wnt10b reporter construct in primary splenic CD8+ T cells.
Data were expressed as mean ± SEM. Data were analyzed by Kruskal-Wallis and Dunn’s multiple comparisons non-parametric tests, as they were not normally distributed as assessed by Shapiro-Wilk normality test. In (A), n = 5 per group, **p < 0.01 and ****p < 0.0001 compared to the indicated groups. In (B)–(F), n = 3 samples per group, *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to Veh Irr.Ab or empty vector. In (G), n = 3 samples per group, ****p < 0.0001 compared to all other groups.
To investigate the functional relevance of NFAT/SMAD binding to the Wnt10b promoter, we cloned a 2,000-bp upstream region of Wnt10b, including the region harboring the three putative NFAT/SMAD binding sites, into a luciferase reporter plasmid. The Wnt10b-luc reporter plasmid and the TK-pRL renilla transfection efficiency control vector were transfected into primary murine CD8+ T cells. Transfected cells were stimulated with ionomycin and TGFb for 24 hr to activate Wnt10b transcription. Cell lysates were then assessed by luciferase assay, as an index of promoter activity. We found that stimulation with ionomycin and TGFβ increased Wnt10b promoter activity in the full length cloned 2,000-bp WT promoter (Figure 6F). Deletion of the two most distal NFAT/SMAD binding sites did not alter the activity of the reporter. However, deletion of all three NFAT/SMAD binding sites abolished the activity of the reporter, suggesting that the NFAT/SMAD binding site critical for Wnt10b transcription is located between bases 705 and 272 of the Wnt10b promoter region. Site-directed mutagenesis experiments confirmed that the critical binding sites are located between 705 bp and 272 bp in the Wnt10b promoter (Figure 6G). Mutation of the NFAT binding site alone, the SMAD site alone, or of both the SMAD and the NFAT sites lowered the activity of the Wnt10-bluc reporter in primary murine CD8+ T cells to baseline levels.
We next investigated how LGG and butyrate regulate NFAT and SMAD signaling. Sorting of BM cells from Foxp3.eGFP reporter mice revealed that LGG and butyrate treatment increased TGFβ1 levels in Treg cells (eGFP+CD4+ cells) and conventional CD4+ cells (eGFPCD4+ T cells) but not in CD8+ T cells (eGFPCD8+ T cells) (Figures 7A–7C). These data indicated that LGG and butyrate increased the amount of TGFβ produced by Treg cells and conventional CD4+ T cells within their microenvironments and thus the amount of TGFβ available to increase SMAD levels in the nuclei of adjacent CD8+ T cells. Moreover, LGG and butyrate increased the nuclear levels of pSMAD2, pSMAD3, and NFAT1, but not NFAT2, in BM CD8+ T cells (Figure 7D), while treatment with anti-CD25 Ab blocked these responses, confirming that Treg cells play an essential role in activating NFAT and SMAD signaling in CD8+ T cells.
Figure 7. Effects of LGG and Butyrate (But) on TGFb1 Production by BM T Cells, on NFAT1/2 and SMAD2/3 Activation, and on PI3K and Akt Signaling in BM CD8+ T Cells.

(A–C) TGFb1 mRNA expression by FACS-sorted BM conventional eGFPCD4+ T cells, eGFPCD8+ T cells, and eGFP+ Treg cells. DEREG (eGFP.Foxp3) reporter mice were treated with vehicle, LGG, or butyrate for 4 weeks. BM cells were sorted at the end of the treatment period.
(D) Immunoblot analysis for the detection of NFAT1, NFAT2, pSMAD2, and pSMAD3 in purified BM CD8+ T cells.
(E) Immunoblot analysis for the detection of c-Jun and c-Fos in purified BM CD8+ T cells.
(D and E) Fresh BM CD8+ T cells were pooled together and then used for obtaining nuclear and cytoplasmic fractions. Laminin B1 was used as nuclear loading control. Tubulin was used as cytoplasmic loading control.
(F) Immunoblot analysis for the detection of Phospho-PI3K p85 in the whole lysate from BM CD8+ T cells.
(G and H) pAKT levels in BM CD8+ T cells and percent of pAKT+ BM CD8+ T cells, as determined by flow cytometry.
(D–F) Conventionally raised WT mice were treated with vehicle, LGG, or butyrate for 4 weeks. BM CD8+ T cells were purified at the end of the treatment period using the EasySep Mouse CD8+ T Cell Isolation Kit. One representative experiment of 3 experiments.
Data were expressed as mean ± SEM, n = 5–10 mice per group in (A)–(C), (G), and (H). Data were analyzed by two-way ANOVA and post hoc tests applying the Bonferroni correction for multiple comparisons. **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared to vehicle or the indicated group. ns = not significant.
In activated T cells, which express AP-1 at high levels, AP-1 acts as the preferred partner of NFAT. Treg cells lowers the production of AP-1, forcing NFAT to bind to other partners such as SMADs (Macian, 2005). Accordingly, we found that LGG and butyrate did not increase the nuclear levels of c-Fos and c-Jun proteins in CD8+ T cells (Figure 7E). By contrast, treatment with anti-CD25 Ab resulted in higher nuclear levels of c-Fos and c-Jun in all groups, suggesting that an increase in Treg cells favored the association of NFAT with SMADs, and antagonized the association of NFAT with AP-1.
Treg cells suppress conventional T cell activation via numerous mechanisms (Shevach, 2009). Among them is the capacity of Treg cells to blunt CD28 signaling (Wing et al., 2008). LGG and butyrate reduced the level of phosphorylated PI3K (Figure 7F) and pAKT in BM CD8+ T cells (Figures 7G and 7H), which are kinases downstream of CD28 (Mosenden and Taske´ n, 2011). These effects were prevented by treatment with anti-CD25 Ab, suggesting that the increase in Treg cells induced by LGG and butyrate may blunt CD28 signaling in CD8+ T cells. Furthermore, LGG and butyrate decreased the number of CD80+ mature dendritic cells (DCs) (CD11c+MHC-II+CD40+CD80+ cells) and the number of CD86+ mature DCs (CD11c+MHC-II+CD40+ CD86+ cells) (Figures S6A–S6D). This was relevant because repression of the CD28 ligands, CD80/86 in DCs is a mechanism whereby Treg cells blunt CD28 signaling (Shevach, 2009).
The CD8+ T cells of OT-I mice all carry a transgenic TCR responsive to ovalbumin (OVA) peptide. To further investigate the role of Treg cells, OT-I CD8+ T cells were co-cultured for 24 hr with OVA peptide-pulsed CD11c+ DCs, both in the presence and absence of Treg cells. ChIP assays revealed that in these in vitro conditions, the addition of Treg cells to the co-cultures resulted in a significant increase in the binding of NFAT1, NFAT2, and SMAD3, but not of SMAD2, to the Wnt10b promoter region (Figures S7A–S7D). Moreover, the inclusion of Treg cells to the co-cultures caused an increase in the production of Wnt10b by CD8+ T cells (Figure S7E). In this coculture system, the effects of Treg cell number on Wnt10b transcript level was dose dependent (Figure S7F), indicating that CD8+ T cells are regulated primarily by the number of Treg cells. Together, the data further supported the hypothesis that Treg cells increase Wnt10b gene expression by recruiting NFATs and SMAD3 to the Wnt10b promoter.
In addition, OT-I CD8+ T cells were co-cultured for 24 hr with OVA peptide-pulsed CD11c+ DCs with or without CTLA-4-Ig, an inhibitor of CD28 co-stimulation (Najafian and Sayegh, 2000). ChIP assays revealed that CTLA-4Ig treatment mimicked the effects of Treg cells, as it increased the binding of NFAT1, NFAT2, and SMAD3, but not of SMAD2, to the Wnt10b promoter (Figures S7G–S7J). Moreover, we found that CTLA-4Ig treatment also increased Wnt10b transcript levels (Figure S7K). These findings suggest that blunting of CD28 signaling stimulates NFAT-and SMAD-driven Wnt10b gene expression.
To investigate whether butyrate may have additional direct regulatory effects on the production of Wnt10b, resting and in vitro activated CD8+ T cells were cultured with butyrate for 24 hr. At dose up to 250 μM, butyrate did not increase Wnt10b transcript levels (Figure S7L), suggesting that butyrate does not directly regulate Wnt10b expression in CD8+ T cells.
DISCUSSION
We reported that oral delivery of LGG or butyrate to eugonadic young mice increased trabecular bone volume due to stimulation of bone formation. We also showed that LGG supplementation increased the levels of butyrate in the gut and in serum. This critical SCFA elevated the number of Treg cells in the BM, leading to increase production of the Wnt ligand Wnt10b, via enhanced binding of NFAT1 and SMAD3 to the Wnt10b promoter. Wnt10b is an established enhancer of osteoblast differentiation and is critical for post-natal bone development. These findings provided proof of principle that LGG and butyrate may represent a therapeutic strategy to enhance bone anabolism. Moreover, LGG is a lactobacillus, which is the most common genus of bacteria with reported probiotic activities. Therefore, the mechanism of action of LGG is likely to be generalizable to lactic acid-generating bacteria.
Butyrate was equally active in GF and conventional mice, while LGG did not increase bone mass in GF mice. These findings indicated that a key mechanism whereby LGG-induced bone anabolism is by indirectly increasing production of butyrate in the small intestine. LGG is known to expand bacteria of the clostridia taxonomic class, which are recognized as the central generators of butyrate in the intestine (Flint et al., 2015; Louis et al., 2010). In agreement with these reports, we found that LGG altered the diversity of the microbiome resulting in higher abundance of SCFAs generating Clostridia. We further found that LGG increased the levels of butyrate, but not propionate, in the small intestine and serum. We thus concluded that LGG exerted its anabolic effects by acting in concert with the extant microbiome to generate butyrate. The effects of LGG on acetate and other SCFAs and the contribution of these metabolites to the bone anabolic activity of LGG remain to be determined.
Emerging reports describe skeletal effect of SCFAs, including stimulation of the osteogenic differentiation of SCs (Chen et al., 2007) and enhancement of bone density (Weaver et al., 2011). Propionate or butyrate increase bone volume in normal mice and prevent ovariectomy and inflammation-induced bone loss by suppressing osteoclast formation, via enhancement of glycolysis at the expense of oxidative phosphorylation (Lucas et al., 2018). This metabolic reprogramming of osteoclast precursors causes cell stress, thereby preventing osteoclast differentiation (Lucas et al., 2018). We found that in intact female mice, which have a lower rate of bone turnover than ovariectomized mice, LGG and butyrate increased bone volume by stimulating bone formation. Whereas the anti-resorptive activity of propionate or butyrate described by Lucas et al. (2018) is T cell independent, we found the bone anabolic activity of butyrate and LGG to be dependent on Treg cells and CD8+ T cells. Although the reasons for these differences are yet unknown, in the study of Lucas et al. (2018) mice are younger and are supplemented with a higher dose of propionate or butyrate for a longer period. Furthermore, the C57BL/6 mice used in this study almost certainly had different microbiota diversity as compared to the C57BL/6 mice used by Lucas et al. (2018), as they were purchased from different vendors, housed in different facilities, and fed a different diet. In fact, Lucas et al. (2018) purchased their mice from Charles River, which harbor segmented filamentous bacteria (SFB) in their gut, which are potent activators of Th17 cells (Ivanov et al., 2009), a highly osteoclastogenic T cell population (Komatsu and Takayanagi, 2012). By contrast, we used SFB-negative mice from Jackson Laboratories.
The link between Treg cells and the bone anabolic activity of LGG and butyrate was established using two experimental models: treating WT mice with anti-CD25 Ab and treating DEREG mice with DT. In both experiments, partial Treg cells blockade prevented the increase in bone volume and bone formation induced by LGG or butyrate, demonstrating that an enlargement of the pool of inducible BM Treg cells was required for the bone anabolic activity of LGG and butyrate. Partial Treg cell blockade also prevented the increase in Wnt10b production by CD8+ T cells induced by LGG and butyrate. These findings were significant because Wnt10b activates Wnt signaling in osteoblastic cells, leading to osteoblast proliferation (Kato et al., 2002), differentiation (Bodine and Komm, 2006), and survival (Almeida et al., 2005; Bodine et al., 2005).
We also found that LGG and butyrate increased the frequency of CD4+ T cells, thus altering the CD4+:CD8+ T cell ratio in the BM. However, interventions that prevented the increase in the number of Treg cells blocked the bone anabolic activity of LGG or butyrate but did not prevent the increase in the CD4+:CD8+ T cell ratio, indicating that the bone activity of LGG or buty-rate was not driven by an increase in the CD4+:CD8+ T cell ratio.
We did not find LGG and butyrate to increase the production of IL-10 by Treg cells, nor to potentiate the activity of Treg cells. However, at least in an in vitro coculture system, Treg cells increased Wnt10b expression in CD8+ T cells in a dose-dependent manner. Accordingly, we hypothesize that LGG or butyrate increased the production of Wnt10b by CD8+ T cells solely by a mechanism involving an expansion of the numbers of Treg cells.
We sought direct evidence of the pivotal role of Wnt10b by investigating the effects of LGG and butyrate in T cell-deficient TCRβ−/− reconstituted with Wnt10b−/− T cells. This approach was preferred to directly treating Wnt10b/ mice with LGG and butyrate, because of the confounding effect of the low baseline bone mass of Wnt10b−/− mice (Bennett et al., 2005). By contrast, Wnt10b−/− T cells engraft and expand normally after transfer (Li et al., 2014). These experiments revealed that LGG and butyrate did not induce bone anabolism in mice lacking ab T cells or CD8+ T cell production of Wnt10b, demonstrating that the effects of LGG and butyrate were mediated by CD8+ T cells and specifically by their production of Wnt10b. We did not find butyrate to directly stimulate the expression of Wnt10b in BM CD8+ T cells in vitro, suggesting that butyrate stimulated Wnt10b expression and thus bone formation, indirectly, by enlarging the pool of BM Treg cells. However, we could not exclude the possibility that butyrate may have had additional effects on T cells or other BM cells, unrelated to Wnt10b generation, that may have contributed to the bone anabolic activity of LGG or butyrate.
Based on the current and previous studies (Roser-Page et al., 2014), it is likely that TCR activation is required for CD8+ T cells to express Wnt10b. In vivo, CD8 T cell activation was likely to result from mildly autoreactive CD8+ T cells that are activated in response to homeostatic T cell renewal functions (Surh and Sprent, 2000), as well as endogenous antigens of microbial origin that are physiologically absorbed from the gut. In the context of T cell activation, the preferred partner of NFAT is AP-1 (Fehr et al., 2010; Macia´ n et al., 2002). Treg cells lower the production of AP-1 and favor the binding of NFAT to SMADs (Macian, 2005). Our ChIP data indeed demonstrated that LGG and butyrate increased the binding of NFAT1 and SMAD3 to the Wnt10b promoter, but only when the number of Treg cells was increased. We further found NFAT2 to bind to the Wnt10b promoter in activated OT-1 CD8+ T cells but not in WT CD8+ T cells from mice treated with LGG or butyrate. This reflected the higher levels of activation of OT-1 cells as compared to WT CD8+ T cells.
Recently, attempts have been made to manipulate Treg cells for clinical purposes (Di Ianni et al., 2011; Trzonkowski et al., 2009). An increase in the number of Treg cells is achievable by nutritional supplementation with the probiotic LGG, butyrate or other SCFAs. Thus, an increase in the number of Treg cells via nutritional supplementation may represent a therapeutic modality for increasing bone mass and preventing osteoporosis. Moreover, the use of probiotics or butyrate to increase the number of Treg cells may find wider applications, such as in transplant medicine or as a treatment for inflammatory and autoimmune conditions.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Roberto Pacifici, MD (roberto.pacifici@emory.edu). The mouse lines obtained from other laboratories are described below and may require a Material Transfer Agreement (MTA) with the providing scientists.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Experimental Animals
All the treatment and surgical procedures were approved by the Institutional Animal Care and Use Committee of Emory University. All in vivo experiments were carried out in female mice. In vitro experiments were conducted using primary cells from female mice. Conventionally raised C57BL/6 WT, TCRβ−/−, OT-1, and DEREG mice were purchased from Jackson Laboratories east coast facility (Bar Harbor, ME). To control for facility variation, each batch of C57BL/6 mice were sourced from the same Jackson Laboratory vivarium. Female C57BL6 Germ Free (GF) mice were purchased from Taconic biosciences (Rensselaer, NY). All conventionally raised mice entering Emory University were shipped to the same room in the same vivarium within the Whitehead Biomedical Research Building. All conventionally raised mice were maintained under general housing environment and fed sterilized food (5V5R chow) and autoclaved water ad libitum. GF mice were housed in a Tecniplast ISOcage P - Bioexclusion System within the Emory Gnotobiotic Animal Core. All mice were acclimatized within our facility for 3 days before experimentation. Littermates of the same sex were randomly assigned to experimental groups. Mice were enrolled in the studies at 10 weeks of age and treated with vehicle, LGG or butyrate for 4 weeks and then sacrificed.
METHODS DETAILS
SCFAs extraction and derivatization
Measurement of Propionate and Butyrate levels were done by the Emory Integrated Lipidomics Core (EILC), a member core of the Emory Integrated Core Facilities (EICF), Briefly, to extract the SCFAs from the SI tissues, 40 mg of sample was homogenized with 1 mL of acetonitrile-water (1:1, v/v) (Fisher Chemical # A955, # A4522 HPLC grade) using the Bead Ruptor 24 Elite (Omni International, USA) with 1.4 mm ceramic beads (Fisher Scientific # 15340153). To extract the SCFAs from serum, 150 μL of serum samples was vortexed for 5 min with 1 μL of acetonitrile-water (1:1, v/v). The homogenates were centrifuged at 4000 rpm for 10 min, and the supernatants containing the SCFAs were collected. The extracted SCFAs as well as the butyrate standard (Sigma # 08089) were derivatized to their 3-nitrophenylhydrazones using 3-nitrophenylhydrazine hydrochloride (Sigma #N21804). Specifically, 2 volumes of the SCFA extracts were mixed with 1 volume of 200 mM 3-nitrophenylhydrazine hydrochloride in acetonitrile-water (1:1, v/v) and 1 volume of 120 mM N-(3-dimethylaminopropyl)-N”-ethylcarbodiimide hydrochloride (Sigma # 03449 HPLC) in acetonitrile-water (1:1, v/v) containing 6% pyridine (Sigma # 270407). The reactions were incubated at 40C for 30 min and then diluted to 1 mL with acetonitrile.
Butyrate measurements by LC MS/MS
The ExionLC AC system coupled to a triple quadrupole mass spectrometer QTRAP5500 (ABSciex, USA) was used. Chromatographic separation was established using a C18-reverse phase HPLC Accucore column (4.6 × 100mm, 2.6μm, ThermoScientific, USA) at a flow rate of 0.5 mL/min at 40°C during a 15 min gradient (0 min 15% B, 2 min 15% B, 6min 55% B, 8min 100% B, 10 min 100% B, 13 min 0% B 15 min 0% B). The mobile phase consisted of solvent A (water, 0.1% formic acid (Thermo scientific # 85178 LC-MS grade) and Solvent B (acetonitrile, 0.1% formic acid). The MRM transition of m/z 222.1 → 137.1 was used for butyrate identification. Nitrogen was used as the nebulizing and desolvation gas. Electrospray ionization was performed in the negative ion mode with optimized parameters: curtain gas 20, ionization spray voltage 4500, T probe heat temperature 650°C, ion source gas 1 60, ion source gas 2 50, declustering potential 60, collision energy 25, collision cell exit potential 11, entrance potential 10. A calibration curve was generated with the derivatized butyrate standard (6 different concentrations) to determine the concentration of the corresponding butyrate species within the samples. Peak determination, peak area integration and calibration curves for standards were performed with the MultiQuant 3.0.2 software (AB Sciex, USA).
Microbiota analysis
C57BL/6 were purchased from Jackson Laboratories, hence derived from a common origin, and acclimatized at Emory University for three days. Thereafter, the microbiome of mice was equalized for two weeks by exchange of bedding. When mice were 10-weeks old, they were randomly assigned to treatment groups. Mice were fed 1×109 CFU Lactobacillus rhamnosus GG or PBS five times a week for 4 weeks. After 4 weeks feeding, fecal pellets were collected from mice and DNA extracted from fecal samples using the MoBio DNA isolation kit. The V4 region of the 16S genes will be amplified using the methods of Caporaso et al. (Caporaso et al., 2011). Amplicons were sequenced on an Illumina MiSeq instrument at the Emory Integrated Genomics Core (EIGC). Analysis of the sequencing reads were done by the Emory Integrated Computational Core (EICC) using standard methodology for microbiome analysis. Briefly, the raw sequence was processed via QIIME, using closed-reference OTU picking and the Greengenes reference database. The resulting files were then moved into R, and analyzed using the phyloseq package. Data processing involved demultiplexing, QC filtering (Edgar et al., 2011), OTU representation (Edgar, 2010), taxonomy assignment via a reference database (Caporaso et al., 2010a; McDonald et al., 2012; Wang et al., 2007), and phylogeny and diversity analysis (Lozupone et al., 2007) using the QIIME (Caporaso et al., 2010b) and MOTHUR(Schloss et al., 2009) pipelines.
LGG and butyrate treatment
Mice were treated with LGG, 1 X 109 total bacteria, 5 days a week by oral gavage for 4 weeks starting at 10 weeks of age. Conventionally raised mice were treated with 100mM butyrate (Cayman Chemicals, Ann Arbor, MI) dissolved in drinking water for 4 weeks, which resulted in an average intake of 36 mg of butyrate/per mouse/per day. Since GF mice disliked the taste of butyrate containing water, GF mice were treated with butyrate dissolved in drinking water in increasing doses (1mM during the first week, 2.5mM in the second week, and 10mM in the last two weeks of treatment) to accustom the animals to the taste.
μCT measurements
μCT scanning and analysis was performed as reported previously (Li et al., 2015; Li et al., 2014; Robinson et al., 2015), using Scanco μCT-40 and Scanco vivaCT 40 scanners. Voxel size was 12 mm3 for in vitro measurements of femurs and 21 mm3 for in vivo measurements of the spine. For the femoral trabecular region, we analyzed 70 slices, beginning 50 slices below the distal growth plate. Femoral cortical bone was assessed using 80 continuous CT slides located at the femoral midshaft. In vivo measurements of spinal trabecular bone contours along the periosteal surfaces were drawn encompassing 50 slices of the L4 vertebra, starting at the beginning of trabecular bone within the spinal body, as described (Li et al., 2016). X-ray tube potential was 70 kVp, 114 mA, and integration time was 350 ms for the in vivo measurements and 200 ms for the in vitro measurements. We used the thresholding approach described by Bouxsein et al. (Bouxsein et al., 2010), which is recommended by Scanco, the μCT-40 manufacturer, and involves a visual inspection and comparison of preview and slice-wise gray scale 2D images. The same threshold value was used for all measurements.
Anti-CD25 Ab treatment
WT mice were injected with anti-CD25 Ab (clone PC61, 500μg/mouse/injection IP) or isotype matched irrelevant Ab (BioXCell, West Lebanon, NH) on days 2, 0, 5, 7, 12, 14, 19 and 21. Mice were further treated with Vehicle, LGG (1X 109 CFU/mice/day 5 days/week and Butyrate (100mM).
DT treatment
Diphtheria Toxin (DT) was purchased from Merck (catalog number 322326), and each lot was tested for toxicity in WT mice and titrated for potency in DEREG mice prior to use. DEREG and littermate control mice were administered 0.5 mg DT intraperitoneally on two consecutive days each week for total of four weeks, as previously described (Yu et al., 2018).
T cell purification and adoptive transfer
Splenic CD4+ and CD8+ T cells were purified by negative selection with EasySep Mouse CD4+ and CD8+ T Cell Isolation Kit (StemCell Technologies, Auburn, CA) and transferred into 4 weeks old TCRβ−/− recipient mice by IV injection (3 3 106 cells per mouse). Recipient mice were treated with vehicle, LGG or butyrate for 4 weeks starting 2 weeks after the T cell transfer.
Excision of Peyer’s patches (PP) from small intestine and preparation of single cell suspension by mechanical dislocation
Peyer’s patches (PP) cell isolation was performed as described (Lefrancois and Lycke, 2001). Briefly, the small intestine was removed and flushed of fecal content. PPs were excised and collected in 1 mL cooled RPMI1640. PPs were dissociated using the plunger of a 2.5 mL syringe and gently forced through a 70 μm cell strainer placed over a 50 mL tube. A single cell suspension was used for measuring the number of Treg cells in PP by flow cytometry.
Quantitative bone histomorphometry
The measurements, terminology and units used for histomorphometric analysis, were those recommended by the Nomenclature Committee of the American Society of Bone and Mineral Research75. Longitudinal sections of the femur were prepared and analyzed as described previously74 using the Bioquant Image Analysis System (R&M Biometrics). Briefly, mice were injected subcutaneously with calcein at day 7 and day 2 before sacrifice. Non-consecutive longitudinal sections (5 mm thick) were cut from methyl methacrylate plastic-embedded blocks along the frontal plane using a Leica RM2155 microtome and stained with Goldner’s trichrome stain for the static measurements. Additional 10 mm thick sections were left unstained for dynamic (fluorescent) measurements. Measurements were obtained in an area of cancellous bone that measured ≈2.5 mm2 and contained only secondary spongiosa, which was located 0.5–2.5 mm proximal to the epiphyseal growth cartilage of the femurs.
ELISAs
P1NP, osteocalcin and CTX were measured by ELISA (Immunodiagnostic Systems Ltd. (Boldon, UK).
Stromal cell purification
BM SCs were purified as previously described (Bedi et al., 2012; Gao et al., 2008). Briefly, unfractionated BM cells were cultured for 7 days in α-MEM medium containing 10% FBS to allow the proliferation of SCs. After discarding non-adherent cells, adherent macrophages were eliminated from the rest of the adherent cells by positive immunoselection by MACS Microbeads (Miltenyi Biotec, Auburn, CA, USA) coupled to anti-CD11c antibody. This marker is expressed on non-adherent dendritic cells and adherent monocytes and macrophages. The remaining adherent cells were defined as SCs because they express ALP, type-I collagen (COLI), and RUNX2, and have the capacity to form mineralization nodules when further cultured under mineralizing conditions.
BrdU incorporation studies
SCs proliferation was assessed by BrdU incorporation assay as previously described(Bedi et al., 2012; Gao et al., 2008), using a BrdU ELISA kit from Roche Diagnostics. SCs were pulsed with bromodeoxyuridine (BrdU) for 4 hours and assayed using a BrdU ELISA kit (Roche Diagnostics) according to the manufacturer’s instructions. SCs proliferation was measured at 450 nm with reference wavelength at 690 nm.
SC apoptosis assay
The activity of caspase-3, the key protease in the induction of apoptosis, was measured in SCs using a CaspACE Assay System (Promega Corporation) according to the manufacturer’s protocol. In Brief, SC extract was prepared by lysing and centrifuging SCs in lysis buffer provided by the kit. Prepared extracts were assayed by measuring absorbance at 405 nm.
Real-time RT-PCR
Total RNA was extracted from whole BM cells and purified BM T cells using Trizol (Invitrogen). cDNA was synthesized from 1 mg total RNA with the Revert AidTM H Minus first strand cDNA synthesis kit (Invitrogen). The mRNA expression levels of genes were analyzed by RT-PCR using an ABI Prism 7000 or One Step Plus Sequence Detection System and SYBR GREEN PCR Master Mix (Applied Biosystems, Foster City, CA, USA). Changes in relative gene expression between groups were calculated using the 2 –ΔΔCt method with normalization to 18S rRNA as previously described. All the primers used were designed by Primer Express Software v2.0 (Applied Biosystems) and most were validated in previous investigations (Li et al., 2016; Li et al., 2015; Yu et al., 2018). The primer sequences are provided in Table S1.
Flow cytometry
The following anti-mouse antibodies were used for cell surface staining: purified CD16/32, BV 421-TCRb (clone H57–597), PerCP/Cy5.5-CD4 (clone RM4–5), PE-CD25 (clone PC61), BV 711-CD8 (clone 53–6.7), PerCP/Cy5.5-F4/80 (clone BM8), BV 421-CD11c (clone N418), Alexa Fluor 700-I-A/I-E (clone M5/114.15.2), PE-CD40 (clone 3/23), APC-CD80 (clone 16–10A1) (Biolegend, San Diego, CA) and BV650-CD86 (clone GL1, BD Biosciences). For intracellular Foxp3 staining, cells were washed, fixed and permeabilized using BD Transcription Factor Buffer Set (BD Biosciences). Cells were then incubated with APC-anti-Foxp3 (clone FJK-16 s, eBioscience) antibody. For phospho-epitope analysis, fresh total BM cells were fixed in BD Cytofix Fixation Buffer for 10 min at 37°C, permeabilized with BD Phosflow Perm Buffer III for 30 min on ice, and stained with PE anti-AKT (pS473) (BD Biosciences) and cell surface antibodies. Flow cytometry was performed on a LSR II system (BD Biosciences, Franklin Lakes, NJ), the live cells were discriminated by LIVE/DEAD Fixable Yellow Dead Cell Stain Kit (ThermoFisher) and data were analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Cell sorting by Flow cytometry
DEREG (eGFP.Foxp3) reporter mice were used for cell sorting. Single cell suspensions were obtained from BM and stained with CD16/32,BV 421-TCRβ (clone H57–597), PerCP/Cy5.5-CD4 (clone RM4–5), PE-CD25 (clone PC61) and BV 711-CD8-(clone 53–6.7) (Biolegend, San Diego, CA). 1 X106 cells were sorted on a BD Arya cell sorter. The sorted cells were collected in complete RPMI-1640 media. Purified cells were then collected by centrifugation and dissolved in Trizol reagent for RNA isolation.
Western Blotting
The nuclear and cytoplasmic fractions of fresh BM CD8+ T cells were obtained using NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher Scientific). The proteins in lysates were protected by Halt Protease and Phosphatase Inhibitor Cocktail (ThermoFisher Scientific). Lysates were cleared by centrifugation and the supernatants were boiled in SDS loading buffer. The same amounts of proteins were separated on 10% Mini-PROTEAN TGX Precast Gels (Bio-rad) and electroblotted to nitrocellulose membrane (ThermoFisher Scientific). Proteins were detected by anti-NFAT1 (catalog no.MA1–025), anti-c-Jun (catalog no. 397500) (ThermoFisher Scientific), anti-NFAT2 (catalog no. sc-7294), anti-c-Fos (catalog no. sc-52-G) (Santa Cruz Biotechnology), anti-phospho-Smad2 (Ser465/467) (catalog no. 3108) or anti-phospho-Smad3 (Ser423/425) (catalog no. 9520) (Cell Signaling Technology, Danvers, MA), anti-Smad3 (phosphor S423+ S425) (catalog no. ab52903, Abcam) antibodies. Anti-beta Tubulin (catalog no. 6046, Abcam) and anti-Lamin B1 (catalog no. 12586, Cell Signaling Technology, Danvers, MA) antibodies were used to confirm the purity of subcellular fractionations, respectively. Anti-beta actin (catalog no. sc-1616) antibody was used as the loading control for the testing of phospho-PI3K p85 (catalog no. 4257) (Cell Signaling Technology, Danvers, MA) in the whole lysate from BM CD8+ T cells. Western blot analysis was conducted using Luminata Crescendo Western HRP substrate (EMD Millipore). Band intensities were quantified with Quantity One 1D Analysis Software (Bio-Rad Laboratories) and expressed relative to Lamin B1 or beta actin.
In vitro generation of induced Treg cells
Treg cells were induced in vitro as described (Fantini et al., 2007). Briefly, purified splenic CD4+CD25- T cells from 6–8 weeks old WT mice were cultured in RPMI-1640 medium were in plates coated with anti-CD3 Ab (10 μg/mL) in the presence of recombinant human IL-2 (100 U/mL), TGFβ1 (20 ng/mL) and trans-retinoic acid (1 pmol/mL) for 4 days. The percentage of total CD4+cells found to be Foxp3+ by flow cytometry at the end of the culture period was 95%.
Plasmids and site-directed mutagenesis
A plasmid containing 2000 to +216 bp DNA sequence of mouse Wnt10b promoter was constructed by inserting a PCR product of mouse genomic DNA into the pGL3.0 vector (Promega, Madison, WI). mwnt10b-luc (a plasmid containing 705 to +216 bp DNA sequence of mouse Wnt10b promoter constructed by inserting a PCR product of mouse genomic DNA into the pGL3.0 vector) was a gift from Dr. D. J. Klemm, University of Colorado, Denver. Other shortened constructs were subcloned based on the construct of mwnt10b-luc (2000 bp to +216 bp). The amplification template for all mutations is the mwnt10b-luc (705 to +216 bp). The NFAT binding site (5′-AGGAAAA-3′) at 282 to 276 bp was changed to 5′-AGcttAA-3′ using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs). Similarly, the SMAD binding site (5′-GTCTAGA-3′) at 341 to 335 bp was mutated to 5′-catagcg-3′. The primers used are provided in Table S1.
Wnt10b promoter reporter gene assays
Freshly purified spleen CD8+ T cells were transfected with either 3.6 μg pGL3 basic vector or 3.6 mg Wnt10b-luc (including WT or mutant DNA segments) reporter constructs together with 0.4 μg TK-pRL transfection control vector using the Amaxa Nucleofector system and Amaxa Mouse T Cell Nucleofector Kit (Lonza). Cells were left unstimulating for 24 h and cultured with ionomycin (0.5 mg/mL, Sigma) plus TGFβ1 (5 ng/mL, Biolegend) for another 24 h. Luciferase activity was determined by the Dual-Luciferase reporter assay kit (Promega BioSciences, San Luis Obispo, CA).
APC assay and in vitro CTLA-4Ig treatment
The APC assay was performed as previously described (Roser-Page et al., 2014). Splenic CD11c+ dendritic cells (DCs) sorted by immunomagnetic beads (Miltenyi Biotech) were used as antigen presenting cells. CD8+ T cells expressing a monoclonal ovalbumin (OVA)-specific transgenic TCR were purified from the spleens of OT-I mice by negative selection. CD11c+ DCs were pulsed for 4 hours with 1 μM antigen (OVA peptide) (SIINFEKL, InvivoGen). After two washes with medium, cells were used for APC assay. For induced Treg cells cocultures, OVA presenting dendritic cells at 150,000/well were incubated with splenic OT-I CD8+ T cells (1 million/well) with or without induced Treg cells (1 million/well or increasing dose of Treg cells from 0.5 to 4 million/well) for 24 h. CD8+ T cells were separated by EasySep Mouse CD8a positive Selection Kit II (StemCell Technologies, Auburn, CA) and dissolved in TRIzol reagent for RNA isolation and real time RT-PCR of Wnt10b mRNA. For ChIP assay, the time of coculture of OVA presenting dendritic cells, CD8+ T cells and induced Treg cells was 4 h. During CTLA-4Ig treatment, OVA presenting dendritic cells at 150,000/ well were incubated with splenic OT-I CD8+ T cells (1 million/well) with or without CTLA-4Ig (100 μg/mL) for 4h or 24 h.
ChIP Assays
ChIP assays were performed using a kit and following the manufacturer’s instructions (Pierce Agarose ChIP Kit, ThermoFisher Scientific). Fresh purified BM CD8+ T cells were fixed in 1% formaldehyde for 10 min. Chromatin was digested by micrococcal nuclease to an average length of 200–1000 bp. The similar amounts of chromatin were immunoprecipitated with 2 μg of anti-NFAT1 (clone 25A10.D6D2, ThermoFisher Scientific), anti-NFAT2 (clone 7A6, Santa Cruz Biotechnology), anti-Smad3 (catalog no. 28379, ChIP grade, Abcam), anti-Smad2 (clone D43B4, Cell Signaling Technology, Danvers, MA) or control rabbit IgG. Isolated DNAs were quantitated using ABI SYBR Green PCR master mix (Applied Biosystems) on an ABI StepOnePlus Real-Time PCR System and calculated as fold enrichment compared with background signal.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as Mean ± SEM. When data were normally distributed according to the Shapiro-Wilk normality test, they were analyzed as follows. Prospective data were analyzed by analysis-of-variance (ANOVA) for repeated-measures. Cross-sectional data were analyzed by unpaired two tailed t tests, one-way ANOVA or two-way ANOVA as appropriate. This analysis included the main effects for animal strain and treatment plus the statistical interaction between animal strain and treatment. When the statistical interaction was statistically significant (p < 0.05) or suggestive of an important interaction, then t tests were used to compare the differences between the treatment means for each animal strain, applying the Bonferroni correction for multiple comparisons. Data that were not normally distributed (as tested by Shapiro-Wilk normality test) were analyzed by Kruskal-Wallis non-parametric tests.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivomAb anti-CD25 Ab (clone PC61) | BioXCell | Cat# BE0012; RRID: AB_1107619 |
| InVivomAb anti-CD25 isotype matched irrelevant Ab | BioXCell | Cat# BE0088; RRID: AB_1107775 |
| Anti-Mouse BV 711-CD8-(clone 53–6.7) | Biolegend | Cat# 100747; RRID: AB_11219594 |
| Anti-Mouse CD16/32 (Clone 93) | Biolegend | Cat# 101302; RRID: AB_312801 |
| Anti-Mouse BV 421-TCRβ (clone H57–597), | Biolegend | Cat# 109230; RRID: AB_2562562 |
| Anti-Mouse PerCP/Cy5.5-CD4 (clone RM4–5) | Biolegend | Cat# 100540; RRID: AB_893326 |
| Anti-Mouse PE-CD25 (clone PC61) | Biolegend | Cat# 102008; RRID: AB_312857 |
| Anti-Mouse PerCP/Cy5.5-F4/80 (clone BM8) | Biolegend | Cat# 123128; RRID: AB_893484 |
| Anti-Mouse BV 421-CD11c (clone N418) | Biolegend | Cat# 117330; RRID: AB_11219593 |
| Anti-Mouse Alexa Fluor 700-I-A/I-E (clone M5/114.15.2) | Biolegend | Cat# 107622; RRID: AB_493727 |
| Anti-Mouse PE-CD40 (clone 3/23) | Biolegend | Cat# 124610; RRID: AB_1134075 |
| Anti-Mouse APC-CD80 (clone 16–10A1) | Biolegend | Cat# 104714; RRID: AB_313135 |
| Anti-Mouse BV650-CD86 (clone GL1) | BD Biosciences | Cat# 564200; RRID: AB_2738665 |
| Anti-Mouse APC-anti-Foxp3 (clone FJK-16 s) | eBioscience | Cat# 17–5773; RRID: AB_469457 |
| Anti-Mouse PE anti-AKT (pS473) | BD Biosciences | Cat# 560378; RRID: AB_1645328 |
| anti-NFAT1 | ThermoFisher Scientific | Cat# MA1–025; RRID: AB_2152763 |
| anti-c-Jun | ThermoFisher Scientific | Cat# 397500; RRID: AB_2533433 |
| anti-NFAT2 | Santa Cruz Biotechnology | Cat# sc-7294; RRID: AB_2152503 |
| anti-c-Fos | Santa Cruz Biotechnology | Cat# sc-52-G; RRID: AB_2629503 |
| anti-phospho-Smad2 (Ser465/467) | Cell Signaling Technology | Cat# 3108; RRID: AB_490941 |
| anti-phospho-Smad3 (Ser423/425) | Cell Signaling Technology | Cat# 9520; RRID: AB_2193207 |
| anti-Smad3 (phospho S423+ S425) | Abcam | Cat# ab52903; RRID: AB_882596 |
| Anti-beta Tubulin | Abcam | Cat#. 6046; RRID: AB_2210370 |
| anti-Lamin B1 | Cell Signaling | Cat# 12586; RRID: AB_2650517 |
| Anti-beta actin | Santa Cruz Biotechnology | Cat# sc-1616; RRID: AB_630836 |
| phospho-PI3K p85 | Cell Signaling Technology | Cat# 4257; RRID: AB_659889 |
| Control rabbit IgG | ThermoFisher Scientific | Prod# 1862244 |
| Anti CD3 Ab | biolegend | Cat# 100331; RRID: AB_1877073 |
| Mouse CD4+ cell isolation kit | STEMCELL Technologies | Cat# 19852A |
| Mouse CD8a Selection kit II | STEMCELL Technologies | Cat# 18953 |
| Mouse T cell isolation kit | STEMCELL Technologies | Cat# 19851A |
| Mouse CD11c isolation kit | Miltenyi Biotech | Cat# 130–108–338 |
| CTLA4Ig (Abatacept) | Bristol-Myers Squibb Company | NA |
| Bacterial and Virus Strains | ||
| Lactobacillus rhamnosus GG | American Type Tissue Collection | ATCC 53103 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Diphtheria Toxin | Merck | Cat# 322326 |
| Butyrate | Cayman Chemicals | Cat# 156–54–7 |
| acetonitrile-water (1:1, v/v), | Fisher Chemical | Cat# A4522 |
| Butyrate standard | Sigma | Cat# 08089 |
| 3-nitrophenylhydrazine hydrochloride | Sigma | Cat# N21804 |
| N-(3-dimethylaminopropyl)-N”-ethylcarbodiimide hydrochloride | Sigma | Cat# 03449 |
| 6% pyridine | Sigma | Cat # 270407 |
| 0.1% formic acid | ThermoFisher scientific | Cat # 85178 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | ThermoFisher Scientific | Cat# 78833 |
| Halt Protease and Phosphatase Inhibitor Cocktail | ThermoFisher Scientific | Cat# 78441 |
| rpmi-1640 medium | Mediatech, Inc. | Cat# 10–040-CV |
| Recombinant human IL-2 | Biolegend | Cat# 575408 |
| TGFβ1 | Peprotech | Cat# 100–21 |
| Trans-retinoic acid | ThermoFisherScientific | Cat# AC20734–0010 |
| Ionomycin | Sigma | Cat# 13909 |
| OVA peptide antigen | Andspec | Cat# AS-60193 |
| Q5 Site-Directed Mutagenesis | BioLabs | Cat# E0554S |
| Critical Commercial Assays | ||
| EasySep Mouse CD4+ and CD8+ T Cell Isolation Kit | StemCell Technologies | Cat# 19852 |
| P1NP ELISA | Immunodiagnostic Systems | Cat# AC-33F1 |
| Osteocalcin ELISA | Immunodiagnostic Systems | Cat# AC-12F1 |
| CTX ELISA | Immunodiagnostic Systems | Cat# AC-06F1 |
| BrdU ELISA kit | Roche Diagnostics | Cat# 11 647 229 001 |
| CaspACE Assay System | Promega | Cat# G7351 |
| LIVE/DEAD Fixable Yellow Dead Cell Stain Kit | ThermoFisher | Cat# L34959 |
| NE-PER Nuclear and Cytoplasmic Extraction Reagents | ThermoFisher Scientific | Cat# 78833 |
| Luminata Crescendo Western HRP substrate | EMD Millipore | Cat# WBLUR0100 |
| Amaxa Mouse T Cell Nucleofector Kit | Lonza | Cat# V4XP-3012 |
| Dual-Luciferase reporter assay kit | Promega | Cat# E1910 |
| EasySep Mouse CD8a positive Selection Kit II | StemCell Technologies | Cat# 18953 |
| Pierce Agarose ChIP Kit | ThermoFisher Scientific | Cat# 26156 |
| ABI SYBR Green PCR master mix | Applied Biosystems | Cat# 4309155 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | Jackson Laboratory | Cat# 000664;RRID:IMSR_JAX:000664 |
| Mouse: (TCRβ−/−) B6.129P2-Tcrbtm1Mom/J | Jackson Laboratory | Cat# 002118;RRID:IMSR JAX:002118 |
| Mouse: (OT-1) C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Laboratory | Cat# 003831;RRID:IMSR JAX:003831 |
| Mouse: (DEREG) C57BL/6-Tg(Foxp3-DTR/EGFP) 23.2Spar/Mmjax | Jackson Laboratory | Cat# 32050-JAX;RRID:MMRRC 032050-JAX |
| Oligonucleotides | ||
| Primers for qRT-PCR and ChIP-qPCR, see Table S1 | Invitrogen | N/A |
| Recombinant DNA | ||
| pGL3.0 vector | Promega | N/A |
| pGL3.0 –2000 to +216 bp DNA sequence of mouse Wnt10b | Roberto Pacifici, Emory University SOM (This Paper) | N/A |
| pGL3.0 | Promega | N/A |
| mWnt10b-luc (a plasmid containing 705 to +216 bp DNA sequence of mouse Wnt10b promoter constructed by inserting a PCR product of mouse genomic DNA into the pGL3.0 vector) | Gift from Dr. D. J. Klemm,University of Colorado,Denver. (PMID:18957421) | N/A |
| Software and Algorithms | ||
| MultiQuant 3.0.2 software | AB Sciex, USA | https://sciex.com/products/software/multiquant-software |
| Graphpad Prism 6 | GraphPad Software | N/A |
| Bioquant Image Analysis System | (R&M Biometrics) | https://osteo.bioquant.com/ |
| FlowJo | Tree Star, Inc. | https://www.flowjo.com/ |
| Quantity One 1D Analysis Software | Bio-Rad Laboratories | http://www.bio-rad.com/en-us/product/quantity-one-1-d-analysis-software?ID=1de9eb3a-1eb5-4edb-82d2-68b91bf360fb |
| Other | ||
| 1.4 mm ceramic beads | Fisher Scientific | Cat# 15340153 |
| C18-reverse phase HPLC Accucore column (4.6 × 100mm, 2.6mm) | ThermoScientific, USA | Cat# 17126–032130 |
| MACS Microbeads | Miltenyi Biotec | Cat# 130–106–639 |
Highlights.
Treatment with the probiotic Lactobacillus rhamnosus GG (LGG) increases bone mass in mice by stimulating the production of butyrate
LGG or butyrate expands the pool of Treg cells in the gut and the bone marrow
Treg cells upregulate the expression of osteogenic Wnt ligand Wnt10b by CD8+ T cells
Wnt10b stimulates bone formation by activating Wnt signaling in osteoblasts
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health (DK112946, DK108842, and RR028009 to R.P.; DK108735 to E.H.; DK098391 to R.M.J.; AR070091, AR068157, and AG062334 to M.N.W.). T.M.D. is a recipient of the Research Fellowship award from the Crohn’s and Colitis Foundation of America (CCFA). M.N.W. was also supported by a grant from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (5I01BX000105). The Emory Gnotobiotic Animal Core is supported by the Georgia Clinical & Translational Science Alliance and the Emory University School of Medicine. The Emory Integrated Core Facilities are supported by the Emory Neuroscience NINDS Core Facilities, the Georgia Clinical & Translational Science Alliance, and the Emory University School of Medicine.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and one table and can be found with this article online at https://doi.org/10.1016/j.immuni.2018.10.013.
REFERENCES
- Almeida M, Han L, Bellido T, Manolagas SC, and Kousteni S (2005). Wnt proteins prevent apoptosis of both uncommitted osteoblast progenitors and differentiated osteoblasts by beta-catenin-dependent and -independent signaling cascades involving Src/ERK and phosphatidylinositol 3-kinase/AKT. J. Biol. Chem 280, 41342–41351. [DOI] [PubMed] [Google Scholar]
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, and Rudensky AY (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bach Knudsen KE (2015). Microbial degradation of whole-grain complex carbohydrates and impact on short-chain fatty acids and health. Adv. Nutr 6, 206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedi B, Li JY, Tawfeek H, Baek KH, Adams J, Vangara SS, Chang MK, Kneissel M, Weitzmann MN, and Pacifici R (2012). Silencing of parathyroid hormone (PTH) receptor 1 in T cells blunts the bone anabolic activity of PTH. Proc. Natl. Acad. Sci. USA 109, E725–E733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, and MacDougald OA (2005). Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 102, 3324–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berni Canani R, Sangwan N, Stefka AT, Nocerino R, Paparo L, Aitoro R, Calignano A, Khan AA, Gilbert JA, and Nagler CR (2016). Lactobacillus rhamnosus GG-supplemented formula expands butyrate-producing bacterial strains in food allergic infants. ISME J 10, 742–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine PV, and Komm BS (2006). Wnt signaling and osteoblastogenesis. Rev. Endocr. Metab. Disord 7, 33–39. [DOI] [PubMed] [Google Scholar]
- Bodine PV, Billiard J, Moran RA, Ponce-de-Leon H, McLarney S, Mangine A, Scrimo MJ, Bhat RA, Stauffer B, Green J, et al. (2005). The Wnt antagonist secreted frizzled-related protein-1 controls osteoblast and osteocyte apoptosis. J. Cell. Biochem 96, 1212–1230. [DOI] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, and Mu€ller R (2010). Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res 25, 1468–1486. [DOI] [PubMed] [Google Scholar]
- Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, and Tosteson A (2007). Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J. Bone Miner. Res 22, 465–475. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, and Knight R (2010a). PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26, 266–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pen˜ a AG, Goodrich JK, Gordon JI, et al. (2010b). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, and Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TH, Chen WM, Hsu KH, Kuo CD, and Hung SC (2007). Sodium butyrate activates ERK to regulate differentiation of mesenchymal stem cells. Biochem. Biophys. Res. Commun 355, 913–918. [DOI] [PubMed] [Google Scholar]
- Di Ianni M, Falzetti F, Carotti A, Terenzi A, Castellino F, Bonifacio E, Del Papa B, Zei T, Ostini RI, Cecchini D, et al. (2011). Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 117, 3921–3928. [DOI] [PubMed] [Google Scholar]
- Douillard FP, Ribbera A, Kant R, Pietilä TE, Jarvinen HM, Messing M Randazzo CL, Paulin L, Laine P, Ritari J, et al. (2013). Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet 9, e1003683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan SH, Louis P, and Flint HJ (2004). Lactate-utilizing bacteria, isolated from human feces, that produce butyrate as a major fermentation product. Appl. Environ. Microbiol 70, 5810–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, and Knight R (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantini MC, Dominitzki S, Rizzo A, Neurath MF, and Becker C (2007). In vitro generation of CD4+ CD25+ regulatory cells from murine naive T cells. Nat. Protoc 2, 1789–1794. [DOI] [PubMed] [Google Scholar]
- Fehr T, Lucas CL, Kurtz J, Onoe T, Zhao G, Hogan T, Vallot C, Rao A, and Sykes M (2010). A CD8 T cell-intrinsic role for the calcineurin-NFAT pathway for tolerance induction in vivo. Blood 115, 1280–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flint HJ, Duncan SH, Scott KP, and Louis P (2015). Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc 74, 13–22. [DOI] [PubMed] [Google Scholar]
- Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, Nakanishi Y, Uetake C, Kato K, Kato T, et al. (2013). Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 504, 446–450. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wu X, Terauchi M, Li JY, Grassi F, Galley S, Yang X, Weitzmann MN, and Pacifici R (2008). T cells potentiate PTH-induced cortical bone loss through CD40L signaling. Cell Metab 8, 132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hippe B, Zwielehner J, Liszt K, Lassl C, Unger F, and Haslberger AG (2011). Quantification of butyryl CoA:acetate CoA-transferase genes reveals different butyrate production capacity in individuals according to diet and age. FEMS Microbiol. Lett 316, 130–135. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafarnejad S, Djafarian K, Fazeli MR, Yekaninejad MS, Rostamian A, and Keshavarz SA (2017). Effects of a multispecies probiotic supplement on bone health in osteopenic postmenopausal women: A randomized, double-blind, controlled trial. J. Am. Coll. Nutr 36, 497–506. [DOI] [PubMed] [Google Scholar]
- Kankainen M, Paulin L, Tynkkynen S, von Ossowski I, Reunanen J, Partanen P, Satokari R, Vesterlund S, Hendrickx AP, Lebeer S, et al. (2009). Comparative genomic analysis of Lactobacillus rhamnosus GG reveals pili containing a human- mucus binding protein. Proc. Natl. Acad. Sci. USA 106, 17193–17198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Patel MS, Levasseur R, Lobov I, Chang BH, Glass DA 2nd, Hartmann C, Li L, Hwang TH, Brayton CF, et al. (2002). Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J. Cell Biol 157, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelchtermans H, Geboes L, Mitera T, Huskens D, Leclercq G, and Matthys P (2009). Activated CD4+CD25+ regulatory T cells inhibit osteoclastogenesis and collagen-induced arthritis. Ann. Rheum. Dis 68, 744–750. [DOI] [PubMed] [Google Scholar]
- Khosla S, and Shane E (2016). A crisis in the treatment of osteoporosis. J. Bone Miner. Res 31, 1485–1487. [DOI] [PubMed] [Google Scholar]
- Kim YG, Lee CK, Nah SS, Mun SH, Yoo B, and Moon HB (2007). Human CD4+CD25+ regulatory T cells inhibit the differentiation of osteoclasts from peripheral blood mononuclear cells. Biochem. Biophys. Res. Commun 357, 1046–1052. [DOI] [PubMed] [Google Scholar]
- Klingenberg R, Gerdes N, Badeau RM, Gistera A, Strodthoff D, Ketelhuth DF, Lundberg AM, Rudling M, Nilsson SK, Olivecrona G, et al. (2013). Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Invest 123, 1323–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu N, and Takayanagi H (2012). Autoimmune arthritis: the interface between the immune system and joints. Adv. Immunol 115, 45–71. [DOI] [PubMed] [Google Scholar]
- Lahl K, Loddenkemper C, Drouin C, Freyer J, Arnason J, Eberl G, Hamann A, Wagner H, Huehn J, and Sparwasser T (2007). Selective depletion of Foxp3+ regulatory T cells induces a scurfy-like disease. J. Exp.Med 204, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefrancois L, and Lycke N (2001). Isolation of mouse small intestinal intraepithelial lymphocytes, Peyer’s patch, and lamina propria cells. Curr Protoc Immunol Chapter 3 Unit 3.19.11–13.19.16. [DOI] [PubMed] [Google Scholar]
- Lei H, Schmidt-Bleek K, Dienelt A, Reinke P, and Volk HD (2015). Regulatory T cell-mediated anti-inflammatory effects promote successful tissue repair in both indirect and direct manners. Front. Pharmacol 6, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Walker LD, Tyagi AM, Adams J, Weitzmann MN, and Pacifici R (2014). The sclerostin-independent bone anabolic activity of intermittent PTH treatment is mediated by T-cell-produced Wnt10b. J. Bone Miner. Res 29, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, D’Amelio P, Robinson J, Walker LD, Vaccaro C, Luo T, Tyagi AM, Yu M, Reott M, Sassi F, et al. (2015). IL-17A is increased in humans with primary hyperparathyroidism and mediates PTH-induced bone loss in mice. Cell Metab 22, 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JY, Chassaing B, Tyagi AM, Vaccaro C, Luo T, Adams J, Darby TM, Weitzmann MN, Mulle JG, Gewirtz AT, et al. (2016). Sex steroid deficiency-associated bone loss is microbiota dependent and prevented by probiotics. J. Clin. Invest 126, 2049–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, Young P, Holtrop G, and Flint HJ (2010). Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyrylCoA:acetate CoA-transferase gene. Environ. Microbiol 12, 304–314. [DOI] [PubMed] [Google Scholar]
- Lozupone CA, Hamady M, Kelley ST, and Knight R (2007). Quantitative and qualitative beta diversity measures lead to different insights into factors that structure microbial communities. Appl. Environ. Microbiol 73, 1576–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas S, Omata Y, Hofmann J, Böttcher M, Iljazovic A, Sarter K, Albrecht O, Schulz O, Krishnacoumar B, Krönke G, et al. (2018). Shortchain fatty acids regulate systemic bone mass and protect from pathological bone loss. Nat. Commun 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F (2005). NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol 5, 472–484. [DOI] [PubMed] [Google Scholar]
- Macián F, García-Cózar F, Im SH, Horton HF, Byrne MC, and Rao A (2002). Transcriptional mechanisms underlying lymphocyte tolerance. Cell 109, 719–731. [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, and Hugenholtz P (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messora MR, Oliveira LF, Foureaux RC, Taba M Jr., Zangerônimo MG, Furlaneto FA, and Pereira LJ (2013). Probiotic therapy reduces periodontal tissue destruction and improves the intestinal morphology in rats with ligature-induced periodontitis. J. Periodontol 84, 1818–1826. [DOI] [PubMed] [Google Scholar]
- Mosenden R, and Taskén K (2011). Cyclic AMP-mediated immune regulation–overview of mechanisms of action in T cells. Cell. Signal 23, 1009–1016. [DOI] [PubMed] [Google Scholar]
- Najafian N, and Sayegh MH (2000). CTLA4-Ig: a novel immunosuppressive agent. Expert Opin. Investig. Drugs 9, 2147–2157. [DOI] [PubMed] [Google Scholar]
- Nilsson AG, Sundh D, Bäckhed F, and Lorentzon M (2018). Lactobacillus reuteri reduces bone loss in older women with low bone mineral density: a randomized, placebo-controlled, double-blind, clinical trial. J. Intern. Med Published online June 21, 2018. 10.1111/joim.12805. [DOI] [PubMed] [Google Scholar]
- Ohlsson C, Engdahl C, Fåk F, Andersson A, Windahl SH, Farman HH, Move´ rare-Skrtic S, Islander U, and Sjögren K (2014). Probiotics protect mice from ovariectomy-induced cortical bone loss. PLoS ONE 9, e92368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JW, Li JY, Walker LD, Tyagi AM, Reott MA, Yu M, Adams J, Weitzmann MN, and Pacifici R (2015). T cell-expressed CD40L potentiates the bone anabolic activity of intermittent PTH treatment. J. Bone Miner. Res 30, 695–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roser-Page S, Vikulina T, Zayzafoon M, and Weitzmann MN (2014). CTLA-4Ig-induced T cell anergy promotes Wnt-10b production and bone formation in a mouse model. Arthritis Rheumatol 66, 990–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setiady YY, Coccia JA, and Park PU (2010). In vivo depletion of CD4+FOXP3+ Treg cells by the PC61 anti-CD25 monoclonal antibody is mediated by FcgammaRIII+ phagocytes. Eur. J. Immunol 40, 780–786. [DOI] [PubMed] [Google Scholar]
- Shevach EM (2009). Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity 30, 636–645. [DOI] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, shortchain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh CD, and Sprent J (2000). Homeostatic T cell proliferation: how far can T cells be activated to self-ligands? J. Exp. Med 192, F9–F14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi M, Li JY, Bedi B, Baek KH, Tawfeek H, Galley S, Gilbert L, Nanes MS, Zayzafoon M, Guldberg R, et al. (2009). T lymphocytes amplify the anabolic activity of parathyroid hormone through Wnt10b signaling. Cell Metab 10, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, Mysliwska J, and Hellmann A (2009). First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin. Immunol 133, 22–26. [DOI] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver CM, Martin BR, Nakatsu CH, Armstrong AP, Clavijo A, McCabe LD, McCabe GP, Duignan S, Schoterman MH, and van den Heuvel EG (2011). Galactooligosaccharides improve mineral absorption and bone properties in growing rats through gut fermentation. J. Agric. Food Chem 59, 6501–6510. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, and Sakaguchi S (2008). CTLA-4 control over Foxp3+ regulatory T cell function. Science 322, 271–275. [DOI] [PubMed] [Google Scholar]
- Yu M, D’Amelio P, Tyagi AM, Vaccaro C, Li JY, Hsu E, Buondonno I, Sassi F, Adams J, Weitzmann MN, et al. (2018). Regulatory T cells are expanded by Teriparatide treatment in humans and mediate intermittent PTH-induced bone anabolism in mice. EMBO Rep 19, 156–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Motyl KJ, Irwin R, MacDougald OA, Britton RA, and McCabe LR (2015). Loss of bone and Wnt10b expression in male type 1 diabetic mice is blocked by the probiotic Lactobacillus reuteri. Endocrinology 156, 3169–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.