

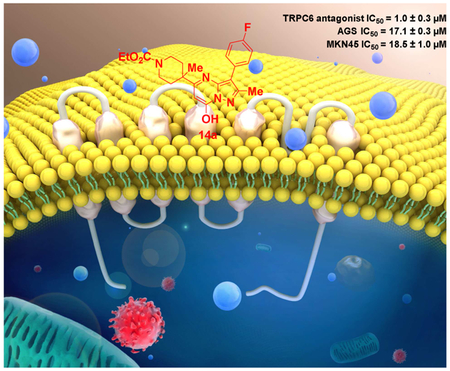
Abstract
Transient receptor potential canonical 6 (TRPC6) proteins form receptor-operated Ca2+-permeable channels, which have been thought to bring benefit to the treatment of diseases, including cancer. However, selective antagonists for TRPC channels are rare and none of them has been tested against gastric cancer. Compound 14a and analogs were synthesis by chemical elaboration of previously reported TRPC3/6/7 agonist 4o. 14a had very weak agonist activity at TRPC6 expressed in HEK293 cells but exhibited strong inhibition on both 4o-mediated and receptor-operated activation of TRPC6 with an IC50 of about 1 μM. When applied to the culture media, 14a suppressed proliferation of AGS and MKN45 cells with IC50 values of 17.1 ± 0.3 and 18.5 ± 1.0 μM, respectively, and inhibited tube formation and migration of cultured human endothelial cells. This anti-tumor effect on gastric cancer was further verified in xenograft models using nude mice. This study has found a new tool compound which showed excellent therapeutic potential against human gastric cancer most likely through targeting TRPC6 channels.
Keywords: Nonselective cation channels, TRPC channels, Ca2+ signaling, anticancer, drug discovery
Graphical Abstracts
Introduction
Gastric cancer is globally the fourth most common cancer and the third leading cause of cancer deaths [1]. The 5-year survival rate of gastric cancer is less than 25% [2]. Tremendous efforts have been made to combat the advanced gastric cancer [3–10]. However, neither the therapeutics nor the molecular understanding of mechanisms underlying tumorigenesis for gastric cancer has matched that in many other cancers. Thus, identification of new molecular targets of gastric cancer and development of effective treatment approaches based on these targets are major focus areas of clinical or translational cancer research.
Transient receptor potential canonical 6 (TRPC6) proteins form receptor-operated Ca2+-permeable channels, which have been implicated in kidney [11–13], pulmonary [14–18] and cardiacdiseases [19,20]. Therefore, drugs that target TRPC6 are thought to bring benefit to the treatment of diseases, including cancers [21–26]. Functional TRPC6 channels were found to be overexpressed in prostate, breast, neuroglia, liver, kidney, oesophagus, stomach and lung tumors, as compared to the low or undetectable levels in the corresponding normal tissues [21, 24–26]. TRPC6 was also shown to play a vital role in cancer-related cell signaling pathways, including proliferation, differentiation, and apoptosis [21]. The channel may contribute significantly to most hepatic and gastric cancers given its up/mis-regulation in the tumorigenic tissues [21]. In a recent report, TRPC6-mediated cytosolic Ca2+ elevation was shown to contributed to the development of human gastric cancer. Thus, targeting TRPC6 channel function may be a viable approach to treat gastric cancer. Over the past decades, very few small molecular TRPC6 antagonists [15,16,25,27–32] (examples shown in Chart 1) have been reported, and some of them have shown strong growth-inhibitory effects on cancer both in vitro and in vivo. Among them, vitamin D3 effectively reduced calcium-induced early-stage prostate tumorigenesis by down-regulating TRPC6 and the calcium sensing receptor both in vitro and in vivo [25]. The use of SKF96365, albeit being nonspecific, supported the results from siRNA silencing on the essential roles of TRPC6 channels in G2/M phase progression of gastric cancer cells and tumor formation in the xenograft model [27]. However, despite these progresses, the utility of the above substances has been limited, partly because most of them have low potency, poor selectivity or low oral bioavailability. More importantly, their possible effects on gastric cancer have not been explored.
Chart 1.

Chemical structures of TRPC6 antagonists
We have previously reported the identification of a novel pyrazolopyrimidine-based TRPC6 lead agonist (4n) with an EC50 of ~1 μM [33]. Here, we report further modifications of 4n to provide additional analogues and among them, we identified a potent TRPC6 antagonist, compound 14a, which not only effectively inhibited gastric cancer cell growth in culture, but also suppressed xenograft tumor formation in nude mice with excellent bioavailability, demonstrating the therapeutic potential of TRPC6 antagonists in treating gastric cancer.
Materials and methods
Materials
Pyrazolopyrimidines 4a-4h, 4o, 5a-5c, 9a, 14a-14h, 16, 17a, 17b, 19a-19j were prepared via similar synthetic routes as reported previously [33] and/or described in the Supporting Information. Human gastric cancer cell lines (AGS and MNK-45) and normal human kidney cells (HK-2) were obtained from Prof. Yizheng Wang and Prof. Xiaoguang Chen cultured in RPMI-1640 media supplemented with 10 % fetal bovine serum. Human umbilical vein endothelial cells (HUVEC) were purchased from ATCC and cultured in RPMI-1640 media supplemented with 10 % fetal bovine serum.
Cell lines and cell culture
HEK293 cells growth and stable cell lines that express human TRPC3, mouse TRPC6 plus M5 muscarinic receptor, mouse TRPC4 plus μ opioid receptor, mouse TRPV3, and mouse TRPM8 were established as described previously [33,34]. For those that co-expressed M5 muscarinic or μ opioid receptor, 100 μg/mL hygromycin B (Calbiochem) was also included in the culture medium. Stable cell lines that inducibly express human TRPC7, TRPA1, and TRPV1 were established as described previously [33,35] and maintained in the medium supplemented with 100 μg/mL hygromycin B and 5 μg/mL blasticidin [26]. The expression of TRP channels was induced by the addition of 0.1 μM doxycycline to the culture medium 24 h prior to the assay.
Fluorescence Ca2+ and membrane potential assays
The detailed protocols for the Flexstation FMP and Ca2+ assays have been described previously [33,35]. Briefly, the extracellular solution for all FlexStation assays contained (in mM): 140 NaCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, with pH adjusted to 7.4 with NaOH. Probenecid (2 mM) was included in all Ca2+ assays except for TRPV1. Assays were run at 32°C. For all fluorescence measurement, changes in the fluorescence intensity were displayed as a ratio of fluorescence change to the fluorescence before the application of stimulating or test compound (ΔF/F0).
Electrophysiology recordings
The detailed protocols for whole-cell patch clamp recordings have been described previously [33,35]. The currents were recorded at 5 kHz. Voltage ramps of 100 ms to −100 mV after a brief (20-ms) step to +100 mV from holding potential of 0 mV were applied every 1 s. Cells were continuously perfused with the bath solution through a gravity-driven multi-channel system with the desired channel opening placed about 50 μm away from the cell being recorded. Drugs were diluted in the bath solution and applied to the cell through perfusion.
Cell proliferation assays
The viability of cells was detected by MTT assay. Briefly, the cells were seeded in wells of 96-well plates overnight, and then treated with tested compounds. MTT solution was added into the wells and incubated for 2 h. After the medium was removed, DMSO was added into each well. The plates were gently agitated and OD570 was determined using the Molecular Devices SpectraMax M5 (Sunnyvale, USA). The 50% inhibitory concentration (IC50) values were calculated.
Tube formation and cell migration assays
The spontaneous formation of capillary-like structures in vitro on standard matrigel was used to investigate the anti-angiogenic activity of 14a. The 24-well plate was coated with 200 mL matrigel (BD Biosciences) for 30 min at 37˚C. HUVECs (3×104 cells/well) were seeded on the matrigel bed. Cells were treated with SKF96365 (5.0 and 10 μM) or 14a (2.5, 5 and 10 μM) in the presence of VEGF (0.3 nmol/L) for 24 h [36]. Tube formations was determined in three random fields (×100) from each well. Data were analyzed as tubule lengths quantified by Image-Pro Plus software (IPP, Media Cybernetics) and compared with that in untreated control wells. The effects of 14a on the migration of HUVECs were examined by wound-healing assay. Cells were seeded in 6-well plates and incubated overnight. Monolayers were scratched with a 200-μL pipette tip. Cells were exposed to vehicle or test agents for 24 h. Three randomly selected fields were photographed at the beginning and end of the treatment. Migration distance was calculated using IPP software.
Preparation of liposome 14a
Liposomes 14a were prepared using a film dispersion method followed our published protocol [37]. Briefly, 14a, lecithin and cholesterol were dissolved in the chloroform, and then the organic solvent was evaporated to obtain a membrane. The membrane was then dissolved by the addition of PBS to obtain the liposome solution.
Primary pharmacokinetic study
The pharmacokinetics of 14a liposomes were explored using Sprague-Dawley rats after a single intraperitoneal injection at a dose of 30 mg/kg. Briefly, around 500 μL of blood from the posterior orbit was collected into a heparinized vacutainer tube before compound administration and at 0.25, 0.5, 1, 2, 4, 8, 12 h post administration. The blood samples were centrifuged at 5000 rpm for 10 min at 4˚C to obtain plasma samples. Concentrations of 14a in the plasma samples were quantified using a Diamonsil-C18 (4.6 mm × 250 mm, 5mm) HPLC column at the room temperature. The mobile phase was 60:40 (methanol: water) at a flow rate of 1 mL/min. The effluent was detected at 254 nm and the area under the peak was used for quantification.
Xenograft studies
Nude mice (4–6-week-old, BALB/c) were used to establish the AGS xenograft tumors following the published protocol [36] Briefly, the AGS cells (5×106) were implanted in the dorsum of the recipient mouse by means of subcutaneous injection. Once a tumor had reached around 300 mm3 in size and was obvious, the animals were randomized into the vehicle control, and two treatment groups with 3 mice per group. The control mice received the vehicle. Liposome 14a was administrated by intraperitoneal injection in two treatment groups at doses of 100 mg/kg and 200 mg/kg twice a day separately for two consecutive weeks. Tumor growth and body weight were measured every 3 days during the treatment. At the end of the treatment, mice were sacrificed and tumors were removed and weighed. The use of animals was approved by the Animal Experimentation Ethics Committee of Wuhan University and Yantai University in accordance with the guidelines for ethical conduct in the care and use of animals.
Data analysis
All data were analyzed and plotted using Origin 7.5 (OriginLab) and Graphpad prism (V 5.01). Summary data are presented as the mean ± S.E.M. The statistical significance between different groups was determined with analysis of variance (ANOVA) and Student’s t test. The difference was considered statistically significant when P < 0.05.
Results
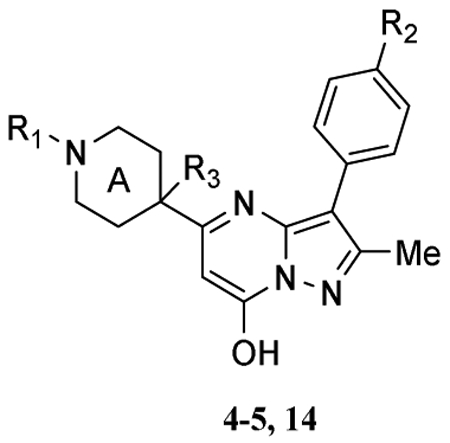
Structure-activity relationship (SAR) analysis
Previously, we reported the identification and resynthesis of compound 4o as a TRPC6 agonist [33], which exhibited an EC50 value of ~4.7 μM in a cell-based fluorescence Ca2+ assay performed in microtiter plates using an HEK293 cell line that stably co-expressed mouse TRPC6 and M5 muscarinic receptors (TRPC6-HEK293). Using the same assay, we found that although most compounds reported here (Table 1) did not significantly induce intracellular Ca2+ elevation in the TRPC6-HEK293 cells, a few of them inhibited the Ca2+ elevation evoked by compound 4o (10 μM), which was introduced at either 2.5 (see later in Fig. 2) or 33 min following the application of the test compound (Fig. 1a). The concentration response relationships were determined for 5a and 14a, which yielded IC50 values of 8.4 ± 1.1 μM and 1.0 ± 0.3 μM, respectively (Table 1 entries 10 and 13).
Table 1.
Effect of compounds 4a-4h, 4o, 5a-5c, 9a, 14a-14h on TRPC6 channels
| Compd | R1 | R2 | R3 | TRPC6 IC50(?M)a |
|---|---|---|---|---|
| 4a | MeNHCS | F | H | >130 |
| 4b | MeCO | F | H | >130 |
| 4c | Me(CH2)7CH=CH(CH2)7CO | F | H | NA |
| 4d | BocNH(CH2)2CO | F | H | NA |
| 4e |  |
F | H | NA |
| 4f |  |
F | H | ~100 |
| 4g | Ph2CHCO | F | H | ~100 |
| 4h | 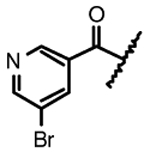 |
F | H | NA |
| 4o | EtOCO | F | H | 4.7 ± 0.03b |
| 5a | Boc | F | H | 8.4 ± 1.1 |
| 5b | Boc | Cl | H | NA |
| 5c | Boc | CF3 | H | NA |
| 14a | EtOCO | CF3 | Me | 1.0 ± 0.3 |
| 14b | Boc | NO2 | Me | agonistc |
| 14c | 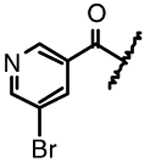 |
F | Me | NA |
| 14d | 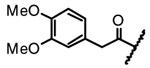 |
F | Me | NA |
| 14e | 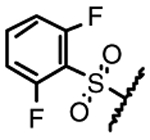 |
F | Me | NA |
| 14f | 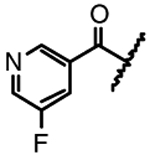 |
F | Me | NA |
| 14g | 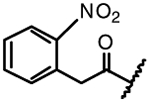 |
F | Me | NA |
Effects on TRPC6 or IC50 against activation by 10 μM 4o, estimated based on one experiment with triplicates.
EC50 determined based on Ca2+ assay.
EC50 cannot be accurately determined because of the self-fluorescence of the compound, NA: No activity.
Fig. 2.

Compound 14a inhibited agonist-evoked TRPC3/6/7 activity. (A-C) 14a inhibited [Ca2+]i increases induced by 4o in HEK293 cells that expressed mouse TRPC6 (A), mouse TRPC7 (B) and human TRPC3 (C). Similar to Fig. 1A, with cells seeded in wells of 96-well plates loaded with Fluo-8 and fluorescence read in a microplate reader, except that 4o was added 2.5 min after the application of different concentrations of 14a to a final concentration of 5 (A), 1 (B), or 3 (C) μM. (D-F) Representative traces of whole-cell currents at +100 (blue traces) and -100 mV (red traces) evoked by GSK1702934A (1 μM) in HEK293 cells that expressed TRPC6 (D), C7 (E), and C3 (F) and their inhibition by 14a at concentrations as indicated. I-V curves obtained from voltage ramps for selected conditions are shown below the traces.
Fig. 1.

Compounds 5a and 14a are partial agonists of TRPC6. (A) 14a Inhibited the 4o-evoked Ca2+ response in TRPC6-HEK293 cells. Cells seeded in wells of a 96-well plate were loaded with Fluo-8 and fluorescence changes read in a microplate reader while 14a and 4o were added as indicated by horizontal bars on top of the traces. Note there was a 30 min break before the addition of 4o. Traces represent averages of fluorescence changes (ΔF) normalized to the fluorescence at the beginning of the reading (F0) of triplicate measurements from one experiment, which was repeated three times. (B) Similar to (A) but the cells were loaded with FLIPR membrane potential (FMP) dye and treated with 14a, which was then followed by carbachol (CCh). (C-G) Representative traces of whole-cell currents at +100 (blue traces) and -100 mV (red traces) in TRPC6-HEK293 cells. I-V curves obtained from voltage ramps for selected conditions are shown at right of (C, D, F, G) or below (E) the traces. (C, F) Currents activated by 14a (20 μM, C) and 5a (20 μM, F). (D, G) Currents activated by CCh (30 μM) and their inhibition by 14a (20 μM, D) and 5a (20 μM, G). (E) Currents activated by OAG (30 μM) and their inhibition by 14a (30 μM).
As summarized in Table 1, any substitution of the ethyl carbamate at R1 position of 4o resulted in the loss of agonist activity of the original lead compound. Previously, we showed that a carboxybenzyl group at this position was well tolerated, indicating a limited flexibility at this area for the stimulatory action of the pyrazolopyrimidine compounds on TRPC6. Intriguingly, the substitution of the ethyl carbamate by tert-butyl carbamate (5a) converted the agonist into an antagonist, despite the moderate potency (Fig. 1F, 1G). The substitution of fluoride at R2 position by chloride (5b) and trifluoromethyl group (5c) eliminated the inhibitory effect. All other substitutions at R1 (4a – 4i) led to either a complete lack of effect or very weak inhibition at TRPC6, despite the presence of fluoride at R2.
We then explored the perturbation of the piperidine by introducing a methyl group at position C-19 (Table 1). The addition of the methyl group to 4o converted the agonist into a potent antagonist of TRPC6 (Table 1 entry 13, 14a, Fig. 1A–1E). Interestingly, the ethyl carbamate at R1 still appears to be crucial for the antagonistic action of the pyrazolopyrimidine compounds as all substitutions at this position resulted in the loss of function on TRPC6.
The importance of the piperidine (A ring) in 4o or 14a on TRPC6 activity was also explored (Table 2). Replacing the piperidine scaffold (A ring) with alpha-substituted ethyl acetates (16, 17a-17b) or N-substituted acetamide analogs (19a-19j) at position C-18 abolished the agonistic action of 4o on TRPC6. Some of N-acetamide substituted pyrazolopyrimidines (19a-19j) exhibited inhibitory actions on 4o-induced TRPC6 activity, but these were only detected at concentrations higher than 30 μM (Table 2). These series of compounds were, therefore, not further exploited.
Table 2.
Effect of compounds 16, 17a, 17b, 19a-19j on TRPC6 channels
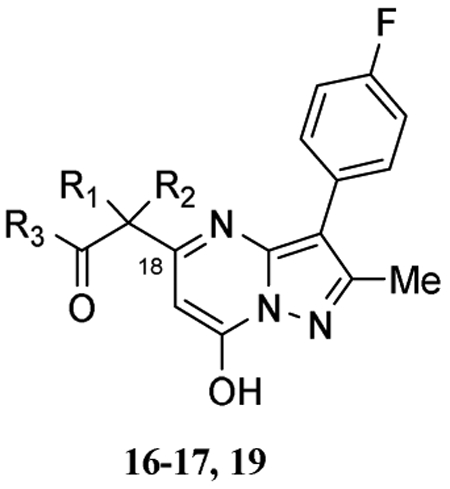
| Compd | R1 | R2 | R3 | TRPC6 IC50(μ(Ha)) |
|---|---|---|---|---|
| 16 | H | H | MeCH2O | NA |
| 17a | Me | Me | MeCH2O | NA |
| 17b | Et | Et | MeCH2O | NA |
| 19a | H | H | 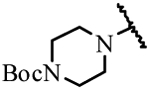 |
> 30 |
| 19b | Me | Me | 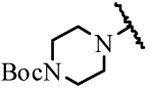 |
> 100 |
| 19c | Et | Et | 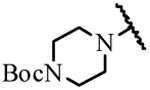 |
> 100 |
| 19d | H | H | 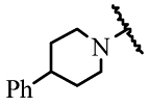 |
> 30 |
| 19e | H | H | 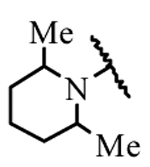 |
> 30 |
| 19f | H | H | 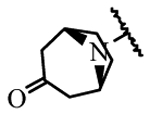 |
> 100 |
| I9g | H | H | 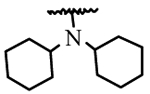 |
NA |
| 19h | H | H | 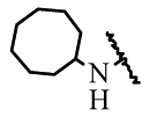 |
> 50 |
| 19i | H | H | 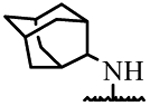 |
> 50 |
| 19j | H | H | 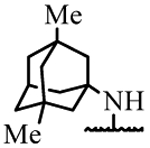 |
> 50 |
Effects on TRPC6 or IC50 against activation by 10 μM 4o, estimated based on one experiment with triplicates, NA: No activity.
Functional characterization of 14a on TRPC3, TRPC6, and TRPC7 channels
From the above SAR study using the fluorescence Ca2+ assay, compound 14a was found to be the most potent antagonist of the TRPC6 channel. In assays that used FLIPR membrane potential (FMP) dye to assess TRPC-mediated membrane potential changes [33], we found that compound 14a still evoked membrane depolarization in TRPC6-HEK293 cells however, it also inhibited the response to the subsequence application of carbachol (CCh, 10 μM) (Fig. 1B), which activated TRPC6 through stimulation of the co-expressed M5 muscarinic receptor and the consequent G protein-mediated activation of PLC. This indicates that the inhibitory effect of 14a was not restricted to activation by the structurally analogous compound 4o, but rather quite general, including receptor-operated TRPC6 activation. The fact that 14a exhibited stimulatory effect in the membrane potential assay but not the Ca2+ assay suggests that the compound, not totally surprising since it derived from a potent TRPC3/6/7 agonist, remains to be a weak TRPC6 agonist that allows Na+ influx. The Ca2+ influx might be too small and/or buffered too quickly by cellular proteins to be detected by the Ca2+ indicator dye.
Supporting the agonistic action of 5a and 14a, both compounds (at 20 μM) elicited whole-cell currents in TRPC6-HEK293 cells (Fig. 1C and 1F). The small amplitudes and very strong outward rectification of the evoked currents are typical of weakly activated TRPC channels. The stimulatory effect of 5a is also much weaker than that of 14a. However, both compounds caused immediate suppression of CCh (30 μM)-evoked TRPC6 currents, which was removable upon washout (Fig. 1D and 1G). In addition, 14a (30 μM) also caused instantaneous inhibition of TRPC6 currents activated by 1-oleoyl-2-acetyl-sn-glycerol (OAG, 30 μM) (Fig. 1E), a synthetic cell-permeable analog of diacylglycerols. These results indicate that compounds 5a and 14a are partial TRPC6 agonists that activate the channel very weakly on their own but inhibit the channel no matter it is activated by a direct agonist or through receptor stimulation.
We then examined the effect of 14a on the closely related TRPC3 and TRPC7 channels. Different from TRPC6, where 14a did not elicit detectable intracellular Ca2+ concentration ([Ca2+]i) change (Fig. 2A), in TRPC7-HEK293 cells, 14a concentration-dependently evoked increases in fluorescence signals in the Ca2+ assay, but the maximal levels reached were lower than that evoked by 1 μM of compound 4o (Fig. 2B). Importantly, the treatment with 14a inhibited the response to the subsequent application of 4o (1 μM), indicating that 14a also acts as a partial agonist that moderately activates TRPC7 but inhibits the channel’s response to a true agonist (Fig. 2B). For TRPC3, 14a did not elicit a discernible Ca2+ response on its own, but at high concentrations (>10 μM), it suppressed the small increase of [Ca2+]i evoked by 4o (3 μM) (Fig. 2C). The weak response of TRPC3 to 4o in the Ca2+ assay has been reported before [33]. These results indicate that 14a exerts inhibitory actions on all members of the TRPC3/6/7 subgroup, although affinity differences may exist and some moderate stimulatory functions are maintained, in line with the fact that 14a is derived from a TRPC3/6/7 agonist (4o).
Next, we performed whole-cell voltage clamp experiments with the currents elicited by a structurally different TRPC3/6/7 common agonist, GSK1702934A, in order to avoid inhibition that strictly resulted from competition between structural analogs for the same binding site. With [Ca2+]i clamped at 400 nM by 10 mM BAPTA in the pipette solution, GSK1702934A (1 μM) evoked marked increases in currents at both negative and positive potentials, which were then suppressed by the application of 14a (0.03 – 0.3 μM) (Fig. 2D). Compound 14a also instantaneously suppressed GSK1702934A-evoked currents in cells that expressed TRPC7 (Fig. 2E) and TRPC3 (Fig. 2F), showing that the compound can inhibit cation currents mediated by TRPC3/6/7 channels stimulated by the structurally distinct agonist. These data suggest that 14a is a common inhibitor of TRPC3/6/7 channels, with the potency order of TRPC6 > C7 > C3.
The above data clearly demonstrate the antagonistic activity of the pyrazolopyrimidine compound 14a on the TRPC3/6/7 subgroup of TRPC channels. To verify the selectivity, we tested the effects of 14a on a stable HEK293 cell line that co-expressed TRPC4 and μ opioid receptor using the fluorescence membrane potential assay, and on HEK293 cell lines that expressed TRPA1, TRPM8, TRPV1 and TRPV3 using the Ca2+ assay. For TRPC4, 14a only partially inhibited membrane depolarization evoked by DAMGO (0.1 μM), through activation of the μ receptor, at 30 and 100 μM, indicating a very weak inhibitory effect. For TRPA1-, TRPM8-, TRPV1-, and TRPV3-expressing cells seeded in wells of 96-well plates and loaded Fluo-8, compound 14a (30 μM) evoked neither Ca2+ response on its own, nor it significantly affected the Ca2+ response evoked by the agonist of the corresponding channel (Fig. 3). In the presence of 30 μM 14a, the response to 100 μM flufenamic acid for TRPA1 was 77.8 ± 8.1% (n = 5), to 100 μM menthol for TRPM8 was 81.5 ± 5.6% (n = 5), to 0.15 μM capsaicin for TRPV1 was 91.1 ± 3.2% (n = 4), and to 200 μM 2-APB for TRPV3 was 98.0 ± 1.3% (n= 5) of the corresponding control without 14a. Therefore, compound 14a specifically inhibits TRPC3/6/7, with a very weak effect on TRPC4 and no significant effect on other TRP channels.
Fig. 3.

Compound 14a has no significant effect on related TRP channels. Stable HEK293 cell lines expressing hTRPA1, hTRPV1, mTRPV3 and mTRPM8 were seeded in wells of 96-well plates and loaded with Fluo-8. Fluorescence changes were read in a microplate reader while 14a (30 μM) were added at 150 sec before the application of the corresponding agonists: FFA (100 μM) for TRPA1, capsaicin (0.15 μM) for TRPV1, 2-APB (200 μM) for TRPV3, and menthol (100 μM) for TRPM8. Data are means ± SEM for n = 5 measurements normalized to control (in the absence of 14a).
Anti-cancer effects of TRPC3/6/7 antagonists
Anti-proliferation activity of 5a and 14a
Having identified 5a and 14a as novel pyrazolo[1,5-a]pyrimidine compounds that inhibit TRPC6 channels, we further explored their potential utility as chemotherapeutic agents against gastric cancer by testing their in vitro cytotoxicity against two gastric cancer cell lines, AGS and MKN45, using the MTT assay [38]. SKF96365 was used as the positive control. Compounds 14a suppressed proliferation of AGS and MKN45 cells in a concentration-dependent manner (Fig. 4A, 4B) with IC50 values of 17.1 ± 0.3 and 18.5 ± 1.0 μM, respectively (Table 3). These values are comparable to or better (in the case of AGS) than that of SKF96365. However, compound 14a did not exhibit cytotoxicity on the normal human kidney cell line, HK-2 cells, when used at up to 100 μM (Fig. 4C), indicating a specific cytotoxic effect against cancer cells. Compound 5a also displayed anti-gastric cancer cell activities but required higher concentrations (Table 3).
Fig. 4.

In vitro Cytotoxicity of 14a against AGS and MKN45 gastric cancer cells and HK-2 normal cells. AGS (A), MKN45 (B) or HK-2 (C) cells were treated with indicated concentrations of compound 14a or SKF96365 for 72 h before viability was assessed by MTT assay. Data were normalized to that in the absence of drug treatment from the same experiment and are means ± SEM of three experiments.
Table 3.
IC50 Values (μM) of TRPC6 antagonists on AGS and MKN45 cells
| Compd | AGS (μM)a | MKN45 (μM)a |
|---|---|---|
| 5a | 24.0 ± 3.8 | 34.6 ± 3.9 |
| 14a | 17.1 ± 0.3 | 18.5 ± 1.0 |
| SKF96365 | 27.8 ± 1.1 | 13.7 ± 0.8 |
Results obtained using MTT assay performed in triplicates.
Inhibition of endothelial tube formation and cell migration by 14a
TRPC6 channels have been implicated in vascular endothelial growth factor (VEGF)-induced proliferation and tube formation of cultured human umbilical vein endothelial cells (HUVECs)36. Angiogenesis, the growth of the new blood vessels, is essential for human cancer progression, and metastasis. The HUVEC tube formation assay is a dynamic in vitro assay that informs the key steps of angiogenesis, including vascular endothelial cell proliferation and the formation of tube-like vascular structures. To further explore the anti-cancer potential of TRPC6 antagonists, we tested the effect of compound 14a on HUVEC tube formation using concentrations that exhibited minimal effects on HUVEC proliferation (2.5, 5 and 10 μM). The treatment with 14a for 24 h inhibited HUVEC cord formation in a concentration-dependent manner, as assessed by the tubule length (Fig. 5A and 5B). Clear vascular disruption was evident at all three 14a concentrations used (Fig. 5A). To evaluate the effect of 14a on cell migration, the wound-healing assay was used and the results showed that 14a inhibited the HUVEC cell migration at nontoxic concentrations lower than 10 μM (Fig. 5C and 5D). In both the tube formation and wound-healing assays, 14a displayed similar or slightly stronger potency than SKF96365
Fig. 5.

Compound 14a inhibited capillary tube formation and migration of HUVECs. (A, B) Effect of 14a on capillary tube formation of HUVECs. Cells were treated with SKF96365 and 14a at indicated concentrations for 24 h before images were taken (representatives shown in A) and tubule lengths of the capillary tubes measured (summary data shown in B). (C, D) Effect of 14a on HUVEC migration in wound-healing assay. Cells were treated with SKF96365 and 14a at indicated concentrations. Scratches were made and images taken at 0 and 24 h. Shown are representative images (C) and summary data of migration distances (D). Summary data are means ± SD of results from n = 3 experiments. **p < 0.01 versus control group determined by one-way analysis of ANOVA followed by Dunnett’s test.
In vivo antitumor growth effect of 14a
Finally, we explored the effect of compound 14a on gastric tumor growth in a xenograft model. AGS cells (5×106) were implanted in the dorsum of the recipient BALB/c nude mouse by subcutaneous injection. Once a tumor was visible and reached the size of 300 mm3, the mouse was treated with either vehicle control or 14a. Due to its poor aqueous solubility, liposome 14a was prepared according to the previously described procedure [37]. Intraperitoneal injection of liposome 14a at two doses, 100 mg/kg and 200 mg/kg, twice a day for two consecutive weeks, resulted in the reduction of tumor growth by 38.5% and 61.5%, respectively (Fig. 6A and 6B, Table 4). Compound 14a did not cause significant body weight loss at either dose (Table 4). These data indicate that compound 14a was efficacious in inhibiting the growth of TRPC6 over-expressing gastric tumor in vivo without obvious global toxicity. The pharmacokinetics of 14a in the liposome was studied in Sprague-Dawley rats. Following a single intraperitoneal (i.p.) dose of compound 14a to rats at 30 mg/kg, the mean plasma Cmax of 14a was 5.4 ± 2.9 μg/mL, the mean AUC was 28.3 ± 14.8 μg h/mL and Tmax was 4 h (Fig. 7).
Fig. 6.

Liposome 14a inhibited AGS tumor growth in a Xenograft model. (A) Growth curves of xenografted gastric tumors after intraperitoneal injection of 14a (100 mg/kg and 200 mg/kg) or 0.9% NaCl (Control) solution into the previously tumor-implanted nude mice. Data are means ± SD from 5 mice per group. ** p < 0.01 versus Control. (B) Representative images of nude mice bearing xenografted tumors with or without 14a treatment at two different doses for 14 days. Images of the dissected tumors are shown at right.
Table 4.
Liposome14a inhibited AGS tumor growth in vivo without global toxicitya.
| Group | Dosage | Body Weight (g) | Tumor | % | |
|---|---|---|---|---|---|
| (mg/kg) | Weight (g) | Inhibition | |||
| Control | 0 | 23.3 ± 1.3 | 22.8 ± 0.8 | 1.3 ± 0.3 | |
| 14a | 100 | 22.3 ± 0.8 | 23.1 ± 0.6 | 0.8 ± 0.3 | 38 |
| 200 | 21.8 ± 0.4 | 20.9 ± 2.0 | 0.5 ± 0.2* | 62 | |
Data are means ± SD.
p < 0.05, compared with the control group.
Fig. 7.

Plasma concentration–time profile of liposome 14a in Sprague-Dawley rats following a single i.p. dose at 30 mg/kg. Data are expressed as means ± SD (n = 3).
Discussion
While TRPC6 has been validated as a potential target, identification of selective antagonists for TRPC6 channels against gastric cancer remains challenging. We have previously reported the identification of a novel pyrazolopyrimidine-based TRPC6 lead agonist (4o), its resynthesis, and structure-activity relationship (SAR) analysis of the initial set of its analogs on TRPC3, TRPC6, and TRPC7 channels. With minor modifications, we obtained a relatively selective strong agonist (4n) for TRPC3, exhibiting an apparent affinity at the low nanomolar range (EC50 < 20 nM), whereas for TRPC6 compound 4n had an EC50 of ~1 μM. We report herein a new series of structurally-related TRPC6 antagonists, sharing in pyrazolopyrimidine scaffold. Our study also demonstrates that a small perturbation of the pyrazolo[1,5-a]pyrimidine agonist compounds can lead to dramatic changes in their activity on TRPC3/6/7 channels. These include in many cases the loss of function on the channel, but in some cases a change in the functional consequence. For the latter, the binding by the compounds, likely at the same site as the original lead, appears to stabilize the closed or inactive conformation of the channel rather than the open conformation. In more sensitive assays measuring TRPC6-mediated membrane depolarization or nonselective cation currents, 5a and 14a exhibited weak stimulatory effects, suggesting that the compounds could promote opening of the channel but this becomes quickly desensitized or inactivated, giving rise to an overall inhibitory phenotype. More importantly, compounds 5a and 14a inhibited activation of TRPC6 channels by other known stimuli including receptor-operated activation through G proteins and PLC. They also displayed inhibitory activities on proliferation of gastric cancer cells, MKN45 and AGS. Compound 14a emerged as the most promising molecule and exhibited strong inhibition on both 4o-mediated and receptor-operated activation of TRPC6 with an IC50 of about 1 μM. When applied to the culture media, 14a suppressed proliferation of AGS and MKN45 cells with IC50 values of 17.1 ± 0.3 and 18.5 ± 1.0 μM, respectively, and inhibited tube formation and migration of cultured human endothelial cells. This anti-tumor effect on gastric cancer was further verified in xenograft models using nude mice in vivo with 14a applied through intraperitoneal injection.
Our findings indicate that the lead pyrazolo[1,5-a]pyrimidine based TRPC6 inhibitor may have the potential to be developed as a novel anticancer drug to target gastric cancer that over-expresses TRPC6. Compound 14a combines high potency with a good intraperitoneal pharmacokinetic profile and reasonable selectivity versus other TRP subfamilies, which makes this compound a valuable tool for further evaluation of TRPC channel pharmacology in vivo. Clearly, more work is needed to fully explore the mechanism of action and potential therapeutic utility of 14a and other TRPC channel blockers. Given the compelling data associating TRPC6 channels with diseases such as cancers and kidney disease, we believe that such efforts are worthy of pursuing.
Supplementary Material
Highlights.
A potent TRPC6 antagonist 14a with the pyrazolopyrimidine skeleton was identified with an IC50 of ~ 1 μM.
14a suppressed proliferation of AGS and MKN45 cells as well as gastric tumor growth in a xenograft model for the first time.
14a specifically inhibited TRPC3/6/7 (TRPC6 > C7 > C3) with a very weak effect on TRPC4 and no effect on other TRP channels.
Acknowledgements
This work was partially supported by NSFC (81773674, 81728020, 81573383, 21390402), IS&TCPC (2015DFA30440, 2014cFB30020), US National Institutes of Health (NS056942, NS092377), the Applied Basic Research Programs of Scientific and Technologic Council of Suzhou (SYG201521), Key Research Project of Shandong Province (2017GSF18177), Natural Science Foundation of Jiangsu Province (BK20160387), Natural Science Foundation of Hubei Province (2017CFA024, 2017CFB711, 2016ACA126), Shenzhen Science and Technology Research Grant (JCYJ20170303170809222), NSFTAG (2016ZR-15–9), FUYTTAR (QCZ2016–10), the Fundamental Research Funds for the Central Universities and the Open Research Fund Program of the Hubei Province Engineering and Technology Research Center for Fluorinated Pharmaceuticals.
Abbreviations:
- [Ca2+]i
Intracellular Ca2+ concentration
- CCh
Carbachol
- EC50
Half maximal stimulation
- FMP
FLIPR membrane potential
- PLC
phospholipase C
- SAR
Structure-activity relationship
- TRPA
Transient receptor potential ankyrin
- TRPC
Transient receptor potential canonical
- TRPM
Transient receptor potential melatatin
- TRPV
Transient receptor potential vanilloid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no competing financial interest.
References
- [1].Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A, Global cancer statistics 2012, CA Cancer J. Clin 65 (2015) 87–108. [DOI] [PubMed] [Google Scholar]
- [2].Cutsem EV, Sagaert X, Topal B, Haustermans K, Prenen H, Gastric cancer, Lancet 388 (2016) 2654–2664. [DOI] [PubMed] [Google Scholar]
- [3].Lordick F, Janjigian YY, Clinical impact of tumour biology in the management of gastroesophageal cancer, Nat. Rev. Clin. Oncol 13 (2016) 348–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Xu W, Yang Z, Lu N, Molecular targeted therapy for the treatment of gastric cancer, J. Exp. Clin. Cancer Res 35 (2016) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Zheng Y, Duan Y, Ma J, Xu R, Zi X, Lv W, Wang M, Ye X, Zhu S, Mobley D, Zhu Y, Wang J, Li J, Wang Z, Zhao W, Liu H, Triazole–dithiocarbamate based selective lysine specific demethylase 1 (LSD1) inactivators inhibit gastric cancer cell growth, invasion, and migration, J. Med. Chem 56 (2013) 8543–8560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ma Li., Zheng Y, Wang S, Wang B, Wang Z, Pang L, Zhang M, Wang J, Ding L, Li J, Wang C, Hu B, Liu Y, Zhang X, Wang J, Wang Z, Zhao W, Liu H, Design, synthesis, and structur-activity relationship of novel lsd1 inhibitors based on pyrimidine–thiourea hybrids as potent, orally active antitumor agents, J. Med. Chem 58 (2015) 1705–1716. [DOI] [PubMed] [Google Scholar]
- [7].Ugolini A, Kenigsberg M, Rak A, Vallée F, Houtmann J, Lowinski M, Capdevila C, Khider J, Albert E, Martinet N, Nemecek C, Grapinet S, Bacqué E, Roesner M, Delaisi C, Calvet L, Bonche F, Semiond D, Egile C, Goulaouic H, Schio L, Discovery and pharmacokinetic and pharmacological properties of the potent and selective met kinase inhibitor 1-{6-[6-(4-fluorophenyl)-[1,2,4]triazolo[4,3-b]pyridazin-3-ylsulfanyl]benzothiazol-2-yl}−3-(2-morpholi n-4-ylethyl)urea (SAR125844), J. Med. Chem 59 (2016) 7066–7074. [DOI] [PubMed] [Google Scholar]
- [8].Li W, Hwang D Chen C, Shen C, Huang C, Chen T, Lin C, Chang Y, Chang Y, Lo Y, Tseng H, Lin C, Song J, Chen HC, Chen SJ, Wu S, Chen C, Synthesis and biological evaluation of n-heterocyclic indolyl glyoxylamides as orally active anticancer agents. J. Med. Chem 46 (2003) 1706–1715. [DOI] [PubMed] [Google Scholar]
- [9].Brameld KA, Owens TD, Verner E, Venetsanakos E, Bradshaw JM, Phan VT, Tam D, Leung K, Shu J, LaStant J, Loughhead DG, Ton T, Karr DE, Gerritsen ME, Goldstein DM, Funk JO, Discovery of the irreversible covalent FGFR inhibitor 8-(3-(4-acryloylpiperazin-1-yl)propyl)-6-(2,6-dichloro-3,5-dimethoxyphenyl)-2-(methylamino)pyrido[2, 3-d]pyrimidin-7(8H)-one (PRN1371) for the treatment of solid tumors, J. Med. Chem 60 (2017) 6516–6527. [DOI] [PubMed] [Google Scholar]
- [10].Ishikawa T, Seto M, Banno H, Kawakita Y, Oorui M, Taniguchi T, Ohta Y, Tamura T, Nakayama A, Miki H, Kamiguchi H, Tanaka T, Habuka N, Sogabe S, Yano J, Aertgeerts K, Kamiyama K, Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo[3,2-d]pyrimidine scaffold, J. Med. Chem 54 (2011) 8030–8050. [DOI] [PubMed] [Google Scholar]
- [11].Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB, A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis, Science 308 (2005) 1801–1804. [DOI] [PubMed] [Google Scholar]
- [12].Reiser J, Polu KR, Möller CC, Kenlan P, Altintas MM, Wei C, Faul C, Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, Brown D, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR, TRPC6 is a glomerular slit diaphragm-associated channel required for normal renal function, Nat. Genet 37 (2005) 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Riehle M, Büscher AK, Gohlke BO, Kaßmann M, Kolatsi-Joannou M, Bräsen JH, Nagel M, Becker JU, Winyard P, Hoyer PF, Preissner R, Krautwurst D, Gollasch M, Weber S, Harteneck C, TRPC6 G757D Loss-of-function mutation associates with FSGS. J. Am. Soc. Nephrol 2016, 27, 2771–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tsvilovskyy VV, Zholos AV, Aberle T, Philipp SE, Dietrich A, Zhu MX, Birnbaumer L, Freichel M, Flockerzi V, Deletion of TRPC4 and TRPC6 in mice impairs smooth muscle contraction and intestinal motility in vivo, Gastroenterology 137 (2009) 1415–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Urban N, Hill K, Wang L, Kuebler WM, Schaefer M, Novel pharmacological TRPC inhibitors block hypoxia-induced vasoconstriction, Cell Calcium 51 (2012) 194–206. [DOI] [PubMed] [Google Scholar]
- [16].Li W, Chen X, Riley AM, Hiett SC, Temm CJ, Beli E, Long X, Chakraborty S, Alloosh M, White FA, Grant MB, Sturek M, Obukhov AG, Long-term spironolactone treatment reduces coronary TRPC expression, vasoconstriction, and atherosclerosis in metabolic syndrome pigs, Basic Res. Cardiol 112 (2017) 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS, Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension, Circ. Res 95 (2004) 496–505. [DOI] [PubMed] [Google Scholar]
- [18].Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX, Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension, Proc. Natl. Acad. Sci. U.S.A 101 (2004) 13861–13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Seo K, Rainer PP, Hahna VS, Lee D, Jo S, Andersen A, Liu T, Xu X, Willette RN, Lepore JJ Jr., Marino JP, Birnbaumer L, Schnackenberg CG, Kass DA, Combined TRPC3 and TRPC6 blockade by selective small-molecule or genetic deletion inhibits pathological cardiac hypertrophy, Proc. Natl. Acad. Sci. U.S.A 111 (2014) 1551–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Onohara N, Nishida M, Inoue R, Kobayashi H, Sumimoto H, Sato Y, Mori Y, Nagao T, Kurose H, TRPC3 and TRPC6 are essential for angiotensin II-induced cardiac hypertrophy, EMBO J. 25 (2006) 5305–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dietrich A, Gudermann T, TRPC6: Physiological function and pathophysiological relevance. In Mammalian Transient Receptor Potential (TRP) Cation Channels Nilius B, Flockerzi VB, Ed., Springer-Verlag Berlin Heidelberg: Handbook of Experimental Pharmacology 222 (2014) 157–187. [DOI] [PubMed] [Google Scholar]
- [22].Wang J, Lu R, Yang J, Li H, He Z, Jing N, Wang X, Wang Y, TRPC6 specifically interacts with APP to inhibit its cleavage by γ-secretase and reduce Aβ production, Nat. Commun 6 (2016) 8876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang SL, Cao Q, Zhou KC, Feng YJ, Wang YZ, Transient receptor potential channel C3 contributes to the progression of human ovarian cancer, Oncogene 28 (2009) 1320–1328. [DOI] [PubMed] [Google Scholar]
- [24].Ding X, He Z, Shi Y, Wang Q, Wang Y, Targeting TRPC6 channels in oesophageal carcinoma growth, Expert Opin. Ther. Targets 14 (2010) 513–527. [DOI] [PubMed] [Google Scholar]
- [25].Bernichtein S, Pigat N, Delongchamps NB, Boutillon F, Verkarre V, Camparo P, Reyes-Gomez E, M_ejean A, Oudard SM, Lepicard EM, Viltard M, Souberbielle J, Friedlander G, Capiod T, Goffin V, Vitamin D3 prevents calcium-induced progression of early-stage prostate tumors by counteracting TRPC6 and calcium sensing receptor upregulation, Cancer Res. 77 (2017) 355–365. [DOI] [PubMed] [Google Scholar]
- [26].Rodrigues T, Sieglitz F, Bernardes GJL, Natural product modulators of transient receptor potential (TRP) channels as potential anti-cancer agents, Chem. Soc. Rev 45 (2016) 6130–6137. [DOI] [PubMed] [Google Scholar]
- [27].Cai R, Ding X, Zhou K, Shi Y, Ge R, Ren G, Jin Y, Wang Y, Blockade of TRPC6 channels induced G2/M phase arrest and suppressed growth in human gastric cancer cells, Int. J. Cancer 125 (2009) 2281–2287. [DOI] [PubMed] [Google Scholar]
- [28].Schleifer H, Doleschal B, Lichtenegger M, Oppenrieder R, Derler I, Frischauf I, Glasnov TN, Kappe CO, Romanin C, Groschner K, Novel pyrazole compounds for pharmacological discrimination between receptor-operated and store-operated Ca2+ entry pathways. Br. J. Pharmacol 167 (2012) 1712–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Washburn DG, Holt DA, Dodson J, McAtee JJ, Terrell LR, Barton L, Manns S, Waszkiewicz A, Pritchard C, Gillie DJ, Morrow DM, Davenport EA, Lozinskaya IM, Guss J, Basilla JB, Negron LK, Klein M, Willette RN, Fries RE, Jensen TC, Xu X, Schnackenberg CG, Marino JP, The discovery of potent blockers of the canonical transient receptor channels, TRPC3 and TRPC6, based on an anilino-thiazole pharmacophore, Bioorg. Med. Chem. Lett 23 (2013) 4979–4984. [DOI] [PubMed] [Google Scholar]
- [30].Maier T, Follmann M, Hessler G, Kleemann HW, Hachtel S, Fuchs B, Weissmann N, Linz W, Schmidt T, Lohn M, Schroeter K, Wang L, Rutten H, Strubing C, Discovery and pharmacological characterization of a novel potent inhibitor of diacylglycerol-sensitive TRPC cation channels, Br. J. Pharmacol 172 (2015) 3650–3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Miehe S, Kleemann HW, Struebing C, Use of norgestimate as a selective inhibitor of TRPC3, TRPC6 and TRPC7 ion channels, U.S. Patent 8, 455, 469, June 4, 2013. [Google Scholar]
- [32].Urban N, Wang L, Kwiek S, Rademann J, Kuebler WM, Schaefer M, Identification and validation of larixyl acetate as a potent TRPC6 inhibitor, Mol. Pharmacol 89 (2016) 197–213. [DOI] [PubMed] [Google Scholar]
- [33].Qu C, Ding M, Zhu Y, Lu Y, Du J, Miller MR, Tian J, Zhu J, Xu J, Wen M, AGA Er-Bu, Wang J, Xiao Y, Wu M, McManus OB, Li M, Wu J, Luo H, Cao Z Shen B Wang H Zhu MX Hong X Pyrazolopyrimidines as potent stimulators for transient receptor potential canonical 3/6/7 channels, J. Med. Chem, 60 (2017) 4680–4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miller M, Shi J, Zhu Y, Kustov M, Tian JB, Stevens A, Wu M, Xu J, Long S, Yang P, Zholos AV, Salovich JM, Weaver CD, Hopkins CR, Lindsley CW, McManus O, Li M, Zhu MX, Identification of ML204, a novel potent antagonist that selectively modulates native TRPC4/C5 ion channels, J. Biol Chem 286 (2011) 33436–33446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhu Y, Lu Y, Qu C, Miller M, Tian J, Thakur DP, Zhu J, Deng Z, Hu X, Wu M, McManus OB, Li M, Hong X, Zhu MX, Luo HR, Identification and optimization of 2-aminobenzimidazole derivatives as novel inhibitors of TRPC4 and TRPC5 channels, Br. J. Pharmacol 172 (2015) 3495–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ge R, Tai Y, Sun Y, Zhou K, Yang S, Cheng T, Zou Q, Shen F, Wang Y, Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. 283 (2009) 43–51. [DOI] [PubMed] [Google Scholar]
- [37].Wang H, Zhang Q, Lv G, Ma J, Ma P, Du G, Wang Z, Tian J, Fang W, Fu F, Preparation, pharmacokinetics, biodistribution, antitumor efficacy and safety of Lx2–32c-containing liposome, PLoS ONE 9 (2014) e114688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.