

Abstract
Platelets are essential for physiological hemostasis and are central in pathological thrombosis. These are their traditional and best known activities in health and disease. In addition, however, platelets have specializations that broaden their functional repertoire considerably. These functional capabilities, some of which are recently discovered, include the ability to sense and respond to infectious and immune signals and to act as inflammatory effector cells. Human platelets and platelets from mice and other experimental animals can link the innate and adaptive limbs of the immune system and act across the immune continuum, often also linking immune and hemostatic functions. Traditional and newly recognized facets of the biology of platelets are relevant to defensive, physiological immune responses of the lungs and to inflammatory lung diseases. The emerging view of platelets as blood cells that are much more diverse and versatile than previously thought further predicts that additional features of the biology of platelets and of megakaryocytes, the precursors of platelets, will be discovered and that some of these will also influence pulmonary immune defenses and inflammatory injury.
I. INTRODUCTION
Platelets circulate in the blood and are best known as the chief effector cells of hemostasis, an essential physiological response that is requisite for host defense and repair (451). Because platelets are small in size compared with other blood cells and are anucleate, physicians and investigators have largely considered them to be simple in structure and function and to have limited, albeit essential, activities. A wealth of information proves this impression of simplicity to be wrong (296). Furthermore, the structure and functions of platelets are dynamic, and change dramatically when they transition from a circulating quiescent state under basal conditions to one of activation in response to physiological and pathological signals (454). Recent evidence also indicates that the platelet transcriptome, proteome, and other key phenotypic features change in disease.
Anucleate platelets (2-5 μm diameter, 0.5 μm thickness, 6–10 fl volume) (470) are generated by a nucleated parent cell, the megakaryocyte, in a complex process termed thrombopoiesis (191). An unusual cellular feature of the megakaryocyte is that it is polyploid (191). Polyploid megakaryocytes and anucleate platelets are unique to mammals (241). In other animal species, specialized circulating cells involved in hemostasis and blood coagulation are termed “thrombocytes” and have nuclei. Although it has been suggested that mammalian platelets evolved from primitive multitasking defensive cells with tissue-sealing and antimicrobial capacities similar to those of many modern invertebrates (465), there is no rigorous evidence for this; furthermore, the biologic advantages provided by biogenesis of anucleate platelets from polyploid megakaryocytes are obscure, regardless of how these specializations have evolved (241).
The process of thrombopoiesis in humans yields ∼100 billion platelets a day and 1 × 1012 circulating platelets in healthy adults (451). There is evidence that the lung is a site of active thrombopoiesis, although the magnitude of the contribution of pulmonary megakaryocytes to total thrombopoiesis remains controversial (468). The platelet life span in blood is ∼10 days in humans and 5 days in mice (200). The platelet life span in clots, thrombi, and in inflamed tissues is unknown. It was previously assumed that functional responses of activated platelets are over within minutes, the time required for traditional platelet responses in physiological hemostasis, but it is now known that some can last for many hours, at least in vitro (369).
Although their contributions to physiological and pathological hemostasis remain the best known functions of platelets, they have other activities. Among them is a diverse repertoire of inflammatory and immune capabilities (397, 453, 454, 469). Mounting evidence that platelets are potent and versatile immune and inflammatory effector cells has emerged from recent observations demonstrating that activated platelets are critical links between the hemostatic and immune systems, with the capacity to carry out recognition and signaling functions, transfer biologic information, and orchestrate complex physiological and pathological inflammatory responses in addition to accomplishing specific effector activities (454).
Platelets, like all circulating blood cells, transit the pulmonary circulation, and they have intimate interactions with the pulmonary vasculature in the healthy and diseased lung (46, 165, 319, 468). Maxwell Wintrobe, who is commonly recognized as the architect of modern hematology, noted that “Platelets . . . are present in great numbers in the capillaries . . . of the lungs . . . ” (477). There is evidence that mammalian lungs contain an intravascular pool of “marginated” platelets that can be released into the systemic circulation by ventilatory and pharmacological maneuvers, although the lung marginated pool of platelets is much smaller than is the marginated pool of neutrophils (polymorphonuclear leukocytes, PMNs) (363, 468). It is likely, however, that most platelets pass unimpeded through alveolar capillaries in tidal breathing under physiological conditions because of differences in size of platelets and the vessels (in humans, the mean platelet diameter is ∼2.25 μm; the alveolar capillary diameter is 2–15 μm, with a mean of 7.5 μm) (468). In contrast, activated platelets interact with alveolar capillaries, endothelium in larger pulmonary vessels, with other platelets, and with leukocytes in the pulmonary blood in lung and systemic inflammation and injury, initiating or amplifying lung dysfunction and damage (46, 319, 468).
In this review, we focus on platelets as critical effector cells in pulmonary immune responses and inflammatory lung diseases. We frame these issues in the context of known or emerging activities of platelets in immune and inflammatory responses in other organs and in other physiological and pathological settings. Throughout, we consider “inflammation” (288) to be synonymous with the “immune continuum” (397, 453, 454, 469). In this conceptual framework, the inflammatory and immune continuum encompasses the traditional innate and adaptive limbs of the immune system, the temporal span from acute to chronic inflammation, and a spectrum with defensive, physiological inflammation at one end of the continuum contrasting with injurious, pathological immune responses at the other. There is evidence that platelets have important activities in each setting. It is likely that megakaryocytes also have immune activities, in addition to their requisite functions in generating new platelets, but there has been little investigation of this possibility.
The pivotal role of platelets as critical effector cells in hemostasis is firmly established in physiology, biology, and medicine. The concept that platelets are equally central effector cells in the immune continuum and in inflammatory responses and diseases has taken longer to gain popular recognition but is now evolving with considerable momentum. There is substantial evidence for this, some of the observations dating back more than four decades (311, 333). Much of the current evidence is summarized in more than a dozen reviews published in the last 5 years alone. Many of these recent summaries reference earlier articles that also provide relevant information and commentary. We cite current and earlier reviews on platelets as inflammatory cells, and reviews of individual topics in immune responses and infection, liberally to give interested readers access to comprehensive information and the opportunity to consider evolution of the concept that platelets are versatile effector cells with both inflammatory and hemostatic functions.
II. PLATELETS AS HEMOSTATIC CELLS: PRIMARY HEMOSTASIS, THROMBOSIS, AND THE HEMOSTASIS OF INFLAMMATION
Platelets have critical physiological functions in hemostasis and pathophysiological effector activities in thrombosis (89, 451). Physiological hemostasis is a highly regulated process that seals wounds in injured vessels in a localized fashion by generating clots (platelet-fibrin complexes formed at the sites of vessel injury). Hemostasis also contributes to vessel remodeling after injury and to wound repair (451). Physiological hemostasis requires activation of platelets in parallel with activation of the biochemical coagulation cascade, together with multiple molecular interactions between these two essential hemostatic components (451). Traditionally, platelet-dependent events have been termed “primary” hemostasis and formation of a fibrin clot called “secondary” hemostasis (451). Genetic, acquired, or iatrogenic defects in the coagulation protease cascade or the hemostatic activities of platelets result in bleeding, often severe and life-threatening (324). Impaired hemostasis in the lung is a potential variable in multiple clinical problems including hemoptysis, a variety of alveolar hemorrhage syndromes, trauma and hemothorax, and bleeding after biopsy, pneumonectomy or lobectomy, or other major surgical procedures.
Thrombosis is usually considered to be a pathological process in which clot formation results from inappropriately triggered, dysregulated, or unregulated hemostasis. Thrombotic disorders have been extensively reviewed (89, 265, 451). Although thrombosis has traditionally been considered to be clot (thrombus) formation mediated by the pathways of hemostasis, recent studies indicate that some mechanisms that drive pathological thrombosis may not be essential for physiological hemostasis (136, 282). Thrombosis is a central event in a variety of pulmonary syndromes that range from prothrombotic consequences of hypoxia and altitude exposure to lung involvement in sepsis (10, 444) (Figure 1A). Deep vein thrombosis and pulmonary embolism are extremely common thrombotic problems in clinical medicine (209, 430, 451) that are briefly considered later in this review.
FIGURE 1.

Platelets are effector cells in hemostasis, inflammation, and thrombotic and inflammatory syndromes in the lung and other organs. A: an organized thrombus occupying a medium size pulmonary artery in the lung of a patient who died of ARDS secondary to sepsis is shown. Neutrophils and monocytes are embedded in the thrombus. Primary hemostatic activities of platelets contribute to the formation of clots and thrombi, which are also sites of interaction with myeloid leukocytes and of other inflammatory and immune effector activities of platelets (see Figure 2). Pro-inflammatory responses of platelets, and platelet-leukocyte interactions, can be modeled in vitro (B and C). B: a platelet-fibrin complex formed by thrombin-stimulated human platelets incubated in the presence of extracellular fibrinogen in vitro. Interleukin-1β protein was detected in the activated platelets (yellow staining) and the fibrin mesh (orange staining) by immunocytochemistry. Green staining indicates actin. [From Lindemann et al. (249).] C: aggregates of thrombin-stimulated human platelets and isolated human monocytes that formed in vitro. The dark staining of the monocyte nuclei (arrows) indicates translocation of nuclear factor kappaB (NF-κB) from the leukocyte cytoplasm to the nucleus in response to molecular signals from the activated platelets. [From Weyrich et al. (464).] Signaling of monocytes by activated platelets induces expression of NF-κB-dependent chemokines and other inflammatory mediators by the leukocytes (453, 454, 464).
Multiple platelet activities contribute to hemostasis and thrombosis (43, 89, 282, 374, 451). As a brief overview of traditional hemostatic platelet activities that will be mentioned throughout this discussion, when vessels are damaged, or in pathological conditions such as atherosclerotic plaque rupture, collagens, von Willebrand factor (vWF), and other subendothelial matrix proteins are exposed. Platelets adhere to these proteins using specialized molecular mechanisms that depend in part on local shear forces. The platelet glycoprotein GPlbα-lX-V complex is critical in these adhesive interactions (8). In addition, platelets are activated by soluble agonists that are locally released or generated, including thrombin and other mediators that are recognized by G protein-coupled and other classes of receptors on the platelet surface that are linked to intracellular signal transduction pathways (52). Engagement of these receptors triggers rapid activation-dependent effector functions (Figure 2). Platelet activation is amplified by endogenous synthesis of the eicosanoid thromboxane A2 (TxA2) and by ADP released from intracellular stores.
FIGURE 2.

The functional repertoire of activated platelets includes multiple activities that mediate hemostasis and inflammation. In primary hemostasis, platelets are activated at sites of endothelial injury and exposure of subendothelial matrix. Platelets are also activated in evolving clots and thrombi and can be activated in the circulation in systemic thrombotic and inflammatory conditions. “Traditional” responses of activated platelets that are critical in physiological hemostasis and pathological thrombosis include shape change, inside-out signaling of integrin αIIbβ3 and other integrins, fibrinogen binding, aggregation, synthesis of thromboxane A2, degranulation and release of hemostatic mediators, adhesion strengthening, and clot retraction. Each of these traditional platelet hemostatic activities has documented or proposed inflammatory functions as well (see text and Table 1). Platelets also have additional biologic activities, many of which are relatively newly discovered, that can initiate or amplify inflammatory and immune responses. In many cases, these nontraditional biologic activities can also contribute to physiological or pathological hemostasis. Each response of activated platelets listed in this figure is discussed in the text. PAF, platelet activating factor; ROS, reactive oxygen species; IL-1β, interleukin-1β; PMNs, polymorphonuclear leukocytes; NET, neutrophil extracellular trap.
Key effector responses important in hemostasis that are triggered by activation-dependent signaling pathways in platelets, in addition to TxA2 synthesis, include morphological alteration (“shape change,” spreading), degranulation of soluble and surface factors that mediate or amplify adhesion and have multiple additional functions, and inside-out signaling of platelet surface integrins. Integrin αIIbβ3 (GPIIb-IIIa) is expressed at high density on the surface of activated platelets and is the principal platelet receptor for fibrinogen. Inside-out signaling (a term that is used interchangeably with integrin “activation”) of αIIbβ3 mediates binding of soluble fibrinogen and consequent formation of stable platelet aggregates in which integrin-bound fibrinogen acts as a molecular bridge between adjacent activated platelets. Activation and aggregation occur within seconds to minutes after vessel injury, depending on the specific activation response. Together with the initial matrix adhesion, activation and aggregation of platelets and subsequent molecular events that strengthen and stabilize platelet accumulation lead to formation of a mass of cells that locally seals the injured vessel (often called a platelet “plug”). Recent experimental observations indicate that platelet activation in clotting blood can be triggered by biomechanical stimulation in addition to soluble biochemical signals, that the basal state of platelet activation (“platelet reactivity”) influences their deposition in clots and thrombi, and that in some cases platelet-fibrin clots have an inner core of highly activated platelets and an outer shell of platelets that are more weakly activated and that are recruited by rheological forces (282). In primary physiological hemostasis, this sequence of events is highly regulated and is limited to the site of injury (52). In pathological conditions, platelet adhesion, activation, and aggregation can be unregulated and the extent of platelet deposition can contribute to occlusive thrombus formation (89, 265, 374). In both physiological and pathological hemostasis, changes in the plasma membranes of deposited, spread platelets facilitate steps in the biochemical coagulation cascade, amplifying thrombin generation and conversion of fibrinogen to fibrin (43, 451). Platelets contribute actively to clot evolution and to retraction, a process that may stabilize clots and thrombi (32, 462). Recent in vitro observations demonstrate that human platelets can synthesize tissue factor, a key procoagulant protein (451), and B-cell lymphoma-3 (Bcl-3), an intracellular protein that influences fibrin retraction (367, 391, 462, 463). Platelets also participate in fibrinolysis, a late phase of hemostasis that is required for clot resolution and vessel patency after injury (310, 472).
Hemostasis and inflammation are intimately related and trigger and amplify one another (95, 119, 357, 448). As examples, thrombin, the terminal protease in the coagulation cascade that cleaves fibrinogen to fibrin, is a potent proinflammatory agonist (79, 264, 357), and certain cytokines such as interleukin 1 (IL-1) and tumor necrosis factor-α (TNF-α), which are pleiotropic inflammatory mediators, stimulate tissue factor synthesis and expression of procoagulant activity by monocytes and endothelial cells (348). Myeloid leukocytes (neutrophils; monocytes), which are critical innate immune effector cells, are recruited into clots (Figure 1) and have multiple prothrombotic activities (117). These include tissue factor synthesis, synthesis of IL-1β and TNF-α, and, for PMNs, formation of neutrophil extracellular traps (NETs), which are discussed later. These examples provide only a small sample of mechanisms that link hemostasis and inflammation. A variety of other molecular and cellular events mediate their reciprocal activation, amplification, and, in disease, pathological dysregulation (95, 117, 119, 357, 448). In vivo imaging studies suggest that IL-1 and TNF-α may mobilize vWF to the endothelial surface in inflammation, inducing thrombus formation mediated by platelet GPlbα-lX-V (322). These tightly linked interactions suggest that hemostasis and inflammation should be considered as components of an integrated system of host response to injury and infection rather than as separate physiological systems, and also suggest evolutionary relationships (255, 465).
The intimate linkage between inflammation and hemostasis has generated new terms, including “thromboinflammation” (34) and “immunothrombosis” (117). Immunothrombosis is proposed to be a specific physiological component of innate immune defense that mediates capture and destruction of intravascular pathogens in microvessels but that can also become dysregulated and injurious in bloodstream microbial invasion, culminating in the syndromes of sepsis and septic shock (117). Thromboinflammation and immunothrombosis have also been framed in the context of pathological clotting and inflammatory vasculopathy in atherosclerotic vascular disease and venous thrombosis. In addition, however, coordinate inflammation and hemostasis occurs in a variety of additional pathological settings including experimental and clinical inflammatory lung diseases. For example, deposition of intravascular platelet-fibrin thrombi is a key feature of diffuse alveolar damage, the pathological hallmark of acute respiratory distress syndrome (ARDS) triggered by both noninfectious and infectious insults (289, 440) (Figure 1A) (see sect. V).
Whether the process is called thromboinflammation, immunothrombosis, or by other designations, platelets are key cellular effectors in linked hemostasis and inflammation in multiple physiological and pathological settings (34, 89, 117, 369, 453, 454) (Table 1). Murine experiments suggest that traditional platelet hemostatic factors such as GPlbα-lX-V have both local and systemic effects in inflammation, depending on the model (78, 322). Examples of platelets as effector cells that mediate linked hemostasis and inflammation in the lungs and other organs (Figure 2) will be mentioned throughout this review. Existing and evolving “anti-platelet” drugs, which are primarily aimed at preventing or interrupting thrombosis in atherosclerotic vascular disease and other common thrombotic disorders, may interrupt inflammatory activities of platelets in addition to blocking their hemostatic functions (32, 223, 252, 295).
Table 1.
Human platelet adhesion molecules and receptors trigger pathways that mediate both hemostasis and inflammation
| Adhesion Molecule or Surface Receptor(s) | Receptor or Adhesion Molecule Class | Ligand(s) or Agonist(s) | Hemostatic Activities | Inflammatory Activities |
|---|---|---|---|---|
| Glycoprotein (GP) Ibα | Major ligand binding subunit of GPIb-IX-V complex; leucine-rich repeat family | von Willebrand Factor (vWF); collagen; thrombospondin; P-selectin | Shear-dependent adhesion of platelets to exposed subendothelial matrix; binds thrombin and facilitates thrombin-induced platelet activation; facilitates activation of coagulation cascade; facilitates inside-out signaling of integrin αIIbβ3 | Binds integrin αmβ2 on human myeloid leukocytes, promoting platelet-leukocyte aggregate formation; pathogen recognition functions (mouse models); regulation of systemic inflammation (mouse model of sepsis) |
| Integrin αIIbβ3 (GPIIb-IIIa) | Integrin | Fibrinogen (Fg) (integrin αIIbβ3 also binds other ligands, including fibrin, fibronectin, vWF, vitronectin, and thrombospondin) | Adhesion and outside-in signaling, triggering or contributing to: aggregation; tight platelet adhesion and spreading on extracellular matrix; TxA2 synthesis; fibrin clot stabilization and retraction; generation of platelet procoagulant activity and microparticles; degranulation; synergistic signaling with thrombin, other agonists; signaling to translation control pathways | Degranulation and release of proinflammatory factors; amplification of synthesis IL-1β by platelets; binding of pathogens |
| Protease-activated receptors (PAR) 1 and 4 | G protein-coupled receptors (GPCR) | Thrombin; synthetic thrombin receptor activating peptides (TRAPs); matrix metalloproteinase 1; others | Inside-out signaling of integrin αIIbβ3; fibrinogen binding; aggregation; degranulation; TxA2 synthesis; tissue factor synthesis by platelets; clot retraction | Degranulation and release of chemokines and antibacterial peptides; surface translocation of P-selectin; degranulation of other proinflammatory factors; synthesis of IL-1β; altered surface display of Toll-like receptors; formation of platelet-leukocyte aggregates; triggering of platelet-dependent leukocyte signaling |
| P2Y receptors (P2Y1, P2Y12) | GPCR | ADP | Triggering, amplification of platelet aggregation; stabilization of platelet aggregates; TxA2 synthesis; degranulation | Formation of platelet-leukocyte aggregates; synthesis of IL-1β by platelets |
| TxA2 receptor | GPCR | Thromboxane A2 (TxA2; stable TxA2 mimetics) | αIIbβ3 activation; fibrinogen binding; aggregation; adhesion; potentiation of thrombin signaling | Synthesis of IL-1β by platelets; release of CD40L |
| Platelet-activating factor (PAF) receptor | GPCR | PAF; PAF-like oxidatively modified phospholipids | Weak agonist for aggregation, αIIbβ3 activation; synergistic amplification of platelet activation by thrombin, ADP | Potent agonist for formation of platelet-neutrophil and platelet-monocyte aggregates; platelet IL-1β synthesis |
| GPVI | Immunoreceptor | Collagen (collagen is also recognized by integrin α2β1 on platelets) | Platelet adhesion to collagen; association with GPlb-IX-V; inside-out signaling of αIIbβ3, α2β1; degranulation; release of inorganic polyphosphates (PolyP) with procoagulant actions; maintenance of vascular barrier integrity | Shedding of proinflammatory microparticles; release of PolyP, which also has proinflammatory activities; platelet synthesis of IL-1β induced by collagen (not yet known if this is mediated by GPVI, integrin α2β1, or both); GPVI contributes to platelet modulation of endothelial barrier function in inflammation |
This list of hemostatic and inflammatory activities and responses triggered by engagement of platelet adhesion molecules and surface receptors is illustrative and not comprehensive. See Ref. 73 for an extensive review of platelet receptors and their functions. In some cases, there are differences in humans and mice; for example, mouse platelets express PARs 3 and 4, whereas human platelets express PARs 1 and 4; murine platelets do not express PAF receptors. Additional examples of platelet receptors and adhesion molecules with both hemostatic and inflammatory functions and details of intracellular pathways and mechanisms are included in recent reviews. [Modified from Rondina et al. (369).]
III. PLATELETS AS IMMUNE AND INFLAMMATORY CELLS
In this section we profile some of the key inflammatory and immune activities of platelets. We also emphasize physiological responses and pathological conditions such as ARDS (Figures 1 AND 3) in which inflammatory and immune activities of activated platelets may be key in addition to their hemostatic functions. We include observations made with human platelets and platelets from experimental animals, particularly mice. It should be recognized, however, that human and murine platelets differ in size, circulating number, circulating life span, transcriptome, proteome, and some biologic and immune responses, and that murine models of inflammation and inflammatory and immune diseases often do not precisely recapitulate human conditions that they are developed to reproduce (453).
FIGURE 3.

Activities of activated platelets may be critical in acute inflammatory lung injury in ARDS. Serial chest radiographs demonstrate increased permeability pulmonary edema that progressed over a 48-h interval from initial evaluation in the emergency department (A) until requirement for endotracheal intubation and mechanical ventilation (B) in a previously healthy patient with H1N1 influenza infection. Platelets may be key effector cells in increased alveolar capillary permeability (Figure 4) and alveolar inflammation (Figure 5) in ARDS caused by sterile and infectious triggers, including influenza. Experimental models of influenza indicate that platelets have nontraditional activities (Figure 2) in the lungs in this infection. See text for additional details. [From Matthay et al. (277).]
A. Platelets in Endothelial Barrier Function, Altered Vascular Permeability, and Lymphatic Vessel Integrity
Altered endothelial barrier function and increased vascular permeability are primary responses in inflammation and merit early consideration in a discussion of platelet functions in this context. Increased vascular permeability is the pathophysiological basis for edema formation in inflammation. Increased permeability edema is a cardinal feature of acute and chronic inflammation–the “tumor” of the four classic diagnostic signs: tumor, rubor, calor, and dolor (349, 453). In some organs, particularly the lung (Figure 3) and the brain, inflammatory edema can be lethal (277, 398). Platelets influence endothelial barrier function by mechanisms that appear to be distinct from, but interrelated with, those that mediate primary hemostasis and thrombosis. We focus on these issues in this section. In addition, we outline recent evidence that platelets influence lymphangiogenesis, a specialized developmental function with major immune and inflammatory consequences, and that they contribute to lymphatic vascular integrity in inflammation.
There is evidence that platelets are required for maintenance of endothelial barrier integrity under basal conditions and for preservation of an element of endothelial barrier function in inflammation. Paradoxically, however, they also contribute to disruption of endothelial barriers and vascular leak (176, 369, 468) (Figure 3). The potential mechanisms accounting for these disparate platelet effects are intricate, and there are many open questions.
Thrombocytopenia is reported to lead to increased permeability of systemic and pulmonary vessels in several models and to be associated with vascular leak in clinical conditions (176, 468) (Figure 4, A and B). The lung has been of particular interest in this regard. In vivo models and isolated lung preparations indicate that platelets regulate pulmonary microvascular endothelial barrier function and integrity. In the lung lymph fistula model pioneered by Staub (420), thrombocytopenia was associated with increased pulmonary vascular permeability to protein based on lung lymph-to-plasma protein ratios in chronically instrumented, spontaneously breathing sheep (253). The “leak” of protein was reduced by administration of ovine platelet-rich plasma. In parallel in vitro experiments, isolated human platelets reduced radiolabeled albumin transfer across cultured bovine endothelial monolayers in a fashion that was dependent on platelet number; isolated human red blood cells (RBCs) did not alter albumin permeability. The results were interpreted as demonstrating that platelets are required for maintenance of basal pulmonary vascular barrier function and that an unidentified paracrine mechanism is involved (253). In experiments utilizing isolated sheep lungs, perfusion with thrombocytopenic ovine blood resulted in increased lung lymph flow and a decrease in lymph-to-plasma oncotic pressure ratio compared with measurements in lungs perfused with control whole ovine blood. The study was interpreted as indicating that platelets protect against lung edema but, again, a mechanism was not identified (339). In isolated, perfused rabbit lungs subjected to oxidant injury or ischemia-reperfusion, human platelets reduced lung edema assessed by lung weight and, in some experiments, by alveolar lavage protein concentration (164, 165). Platelet antioxidant enzymes appeared to preserve lung endothelial barrier integrity in these models, and platelet aggregation and degranulation were not thought to be required based on inhibitor studies (165). Other observations from experiments with cultured endothelium and isolated lungs (2, 159, 299, 337, 338) provide evidence that platelets are involved in maintenance of basal pulmonary endothelial barrier integrity and are required for preservation of an element of restricted alveolar capillary barrier permeability to protein in inflammatory or injury states (165, 468) (Figure 4, A AND B).
FIGURE 4.

Platelets are critical in maintenance of endothelial barrier function but also induce increased endothelial permeability in inflammation. A: when platelet numbers are sufficient, semipermeable endothelial barriers that restrict transfer of water and proteins out of systemic and alveolar capillaries are maintained and protected. Release of stabilizing factors by platelets is one mechanism for endothelial barrier maintenance, although others have also been reported or proposed. Vascular endothelial cell cadherin (VE-cadherin) bonds are critical for establishing and maintaining alveolar and systemic endothelial barrier integrity. B: in severe thrombocytopenia, basal endothelial barrier properties are disrupted, leading to leak of water and protein from alveolar and systemic vessels. Large arrows indicate transvascular fluid and RBC escape into the alveolar space, and small arrows indicate leak into the interstitial and lymphatic space. The size of the arrows does not denote the relative magnitudes of the leaks into these compartments. Severe thrombocytopenia also contributes to inflammation-associated hemorrhage in experimental models. Platelet dysfunction, in addition to decreased platelet numbers, may have this effect. C: activated platelets can induce or amplify increased permeability of alveolar and systemic endothelial barriers in inflammation. Several mechanisms have been proposed or demonstrated in experimental models, including release of platelet factors that disrupt endothelial barriers, signaling of endothelial cells, and interaction with PMNs and monocytes, leading to disruption of endothelial bonds and leak of fluid, proteins, and RBC. Large and small arrows indicate escape of fluid and RBC as outlined in B. Increased permeability lung edema is a key feature of ARDS (Figure 3) and occurs in other syndromes of inflammatory lung injury. [Modified from Weyrich and Zimmerman (468).]
In some circumstances, increased vascular barrier permeability without traumatic interruption of endothelial continuity leads to extravascular escape of RBC in addition to leak of protein, water, and solutes. Platelets are critical for preservation of the vascular barrier properties that retard RBC extravasation under basal conditions (312) (Figure 4B). The barrier-preserving function of platelets is particularly important in inflammation and appears to be required to prevent or minimize hemorrhage in inflamed lungs and other organs. Thus platelets contribute to inflammation-mediated thrombosis (see sect. II), but their deficiency is a cause of inflammation-associated hemorrhage, adding to the complexity of their influences in inflammation and immune syndromes.
Thrombocytopenia is associated with nontraumatic bleeding in infected or inflamed lungs and in other tissues in several experimental models (176). In a model of lethal bacterial pneumonia and pneumonia-induced sepsis, mice with severe thrombocytopenia hemorrhaged into the lungs but not in distant organs (92). In mice with lipopolysaccharide (LPS)-induced acute lung injury (ALI), severe thrombocytopenia (<2.5% of baseline platelet numbers) was associated with alveolar hemorrhage whereas animals with the same degree of thrombocytopenia without LPS-induced lung inflammation did not spontaneously bleed into the lungs even though they had prolonged tail bleed times (144). A similar pattern was seen in dermal inflammation and central nervous system injury. Alveolar hemorrhage in thrombocytopenic mice with LPS-induced ALI was sufficient to cause anemia and grossly bloody bronchoalveolar lavage (BAL) fluids (144). In parallel experiments using models of inflammatory dermatitis, mice genetically deficient in vWF, integrin αIIbβ3, GP Ibα, GPVI, or diacylglycerol-regulated guanine nucleotide exchange factor 1, each of which mediates, or regulates, platelet adhesion or aggregation, did not bleed to a greater extent than did wild-type controls, suggesting that platelet factors that are key in primary hemostasis (Table 1) are not required for vascular barrier preservation in inflammation (144). In a different study, however, GPVI and CLEC2, which are respectively members of the immunoreceptor tyrosine activation motif (ITAM) and hemITAM families of platelet receptors (351), and Src-homology leukocyte protein 76 (SLP-76), which is downstream of CLEC2, were required for barrier-preserving activity of platelets in LPS-induced ALI and in immune-mediated skin inflammation; in contrast, G protein-coupled receptor signaling was, unexpectedly, not required (44). Recently published experiments utilizing GPVI-deficient mice provide additional evidence that GPVI is required for prevention of bleeding into inflamed skin, and that GPVI-dependent platelet adhesion, signaling, and secretion are necessary for full vascular stabilization (150). (Platelet degranulation and secretion are discussed later.) The latter studies (44, 150) make a persuasive case for GPVI and CLEC2 as important factors in regulation of vascular barrier integrity and prevention of inflammation-induced hemorrhage by platelets in mice. In additional observations using murine knockouts, other investigators reported triggering receptor expressed on myeloid cells-like (TREM-like) transcript-1 (TLT-1) to also have this function. Mice deficient in TLT-1, an alpha granule protein that is secreted on platelet activation, had increased bleeding when subjected to the localized Schwartzman reaction, a complex model of LPS- and cytokine-induced dermal inflammation (460). The experiments were interpreted as demonstrating that TLT-1 is an autocrine regulator of platelet aggregation that protects against inflammation-associated hemorrhage and dampens inflammation.
Transfusion of platelets from wild-type mice, in very small numbers in some reports, restores endothelial barrier function and reduces inflammation-associated hemorrhage in thrombocytopenic animals or mice deficient in GPVI (44, 92, 144, 150). These experiments have potential clinical relevance since thrombocytopenia is common in critically ill patients with conditions such as sepsis or ARDS (181), in which vascular leak is a central feature of the pathophysiology (10, 277). Decreased platelet counts leading to disrupted alveolar capillary barrier integrity may contribute to alveolar hemorrhage in ARDS, which is frequently observed in histological specimens (278, 289, 440). Nevertheless, patients are usually not as thrombocytopenic as are mice in which platelet numbers are reduced for experimental purposes. This point should be kept in mind when extrapolating findings or mechanisms in animal models to humans with clinical vascular leak syndromes. Furthermore, platelets mediate hemostasis in models of vascular injury or thrombosis in unexpectedly low numbers in mice (304), and also appear to support endothelial barrier function in similarly low numbers in murine experimental models (144). This should also be taken into account in extrapolation of experimental results and design of preclinical models.
How do platelets contribute to preservation of endothelial barrier integrity under basal conditions and in inflammation (Figure 4, A AND B)? Experiments examining pulmonary or systemic endothelium in a variety of models raise several possibilities (176, 468). They include release of soluble molecules that act as signals and stabilizing factors in endothelial barrier maintainance, adhesive or contact-dependent mechanisms that mediate endothelial signaling and preservation of barrier properties, stimulation or enhancement of endothelial cell proliferation, recruitment of endothelial progenitor cells, neutralization or scavenging of agents that disrupt endothelial barrier integrity, and physical obstruction of gaps in the endothelial barrier. The first mechanism has the greatest favor in the field, although others may also be operative depending on the experimental model. In addition, several mechanisms may contribute simultaneously. For example, in a recent study of dermal immune complex-induced inflammation, adherent platelets were reported to seal discrete breaches in the endothelium opened by neutrophils. This physical sealing was supplemented by scavenging of injurious neutrophil elastase, and perhaps by release of platelet barrier-stabilizing factors (150).
Platelets basally express or synthesize multiple candidate factors that alone or in combination may stabilize endothelial permeability and barrier function in physiological conditions and act to preserve barrier integrity in inflammation (176, 312, 468). Much of the evidence for this comes from experiments in which intact platelets reduced permeability or preserved barrier properties of cultured pulmonary or systemic endothelial monolayers, or platelet-rich plasma, conditioned supernatants from platelets, platelet lysates, or isolated platelet factors had similar effects (2, 159, 253, 299, 337, 338, 402). Specific platelet factors that have endothelial barrier enhancing or stabilizing properties include sphingosine-1-phosphate (S1P), angiopoietin-1 (Ang-1), serotonin, epinephrine, adenosine, ATP, and lysophosphatidic acid (176, 468). Serotonin is reported to have both barrier-stabilizing and barrier-disrupting activities (468). There may be differential effects in different vascular beds due to variations in expression of serotonin receptors or other factors (268). In addition to serotonin, S1P and Ang-1 have received substantial recent attention as barrier stabilizers.
S1P has been examined extensively and clearly has endothelial barrier-stabilizing activities (328, 468). Of note, circulating S1P is also a regulator of lymphocyte trafficking (328). Platelets were originally thought to be the principal source of S1P, which is present in plasma under basal conditions, but RBC and endothelial cells also release S1P (60, 315, 328). In vitro experiments demonstrated that human platelets and platelet supernatants enhance transmonolayer electrical resistance of cultured human pulmonary artery endothelial cells and that the effect is dependent on a key receptor for S1P, S1P1, which was formerly called Edg1 (384). Engagement of S1P1 on endothelial cells by S1P induces rearrangement of the actin cytoskeleton and Rac-GTPase-dependent formation of VE-cadherin-containing adherens junctions on endothelial plasma membranes (131, 239, 328). Homophilic VE-cadherin bonds on adjacent endothelial cells are major regulators of adherens junction integrity and endothelial barrier properties (452) (Figure 4A). S1P reduces, or reverses, increased pulmonary vascular permeability and blunts alveolar edema in models of ALI and lung inflammation (60, 340, 378, 431), suggesting that S1P may be a key platelet-derived stabilizing factor for lung endothelium in inflammation as well as under basal conditions (315, 468).
Ang-1 is also released from platelets (244) and reduces vascular leak (134, 439). Decreased levels of Ang-1, correlated with platelet numbers and potentially due to thrombocytopenia, were found in blood samples from patients with dengue hemorrhagic fever and dengue shock syndrome, which are clinical conditions in which endothelial barrier function is destabilized (294). In experimental studies, platelet-rich plasma (PRP), which contains Ang-1, reduced permeability of monolayers of cultured human lung microvascular endothelial cells treated with TNF-α and attenuated disruption of VE-cadherin-containing junctional complexes; PRP also inhibited LPS-induced vascular leak in mouse lungs (271). The barrier-enhancing effect of PRP was abrogated by siRNA knockdown of tunica internal endothelial cell kinase 2 (Tie2), a receptor for Ang-1, and by a soluble inhibitor of Ang-1 interaction with Tie2, suggesting that Ang-1 is a key mediator of barrier stabilization. Platelets were sonicated to release intracellular factors in preparation of the PRP in these experiments (271), so it is unclear if basal release of platelet-derived Ang-1 contributes to pulmonary endothelial barrier maintenance under physiological conditions.
Although the mediators and molecular mechanisms have not been completely characterized, the experimental evidence outlined above, on balance, supports the concepts that platelets enhance pulmonary and systemic endothelial barrier function and that sufficient numbers of circulating platelets are required for maintenance of basal barrier integrity of alveolar capillaries and other capillary beds and for preservation of an element of endothelial barrier integrity in inflammation in vivo (Figure 4, A AND B). S1P, Ang-1, and PRP have been proposed as adjunct therapies for sepsis-induced vascular leak and increased alveolar capillary permeability in ARDS (240, 271, 315), based in part on observations of this nature. It's also possible that platelets stabilize barrier properties of alveolar epithelial cells by releasing barrier-promoting factors that are translocated across the alveolar capillary membrane. For example, transforming growth factor-β (TGF-β), which is a platelet α-granule constituent (126), regulates ion and fluid transport by alveolar epithelial cells (128, 342). Nevertheless, a paradox is that additional experimental evidence indicates that platelets can contribute to increased alveolar-capillary permeability and to accumulation of pulmonary edema fluid in lung inflammation and injury (46, 427, 453, 492) (Figure 4C). Thus platelets may be both protectors and disruptors of alveolar-capillary barrier integrity in disease (46, 468). Similarly, there is evidence that platelets disrupt barrier function of systemic endothelium in pathological vascular inflammation (369). Demonstration that cationic proteins isolated from human platelets can increase vascular permeability was one of the earliest observations identifying platelets as inflammatory cells (311, 313).
Experiments utilizing murine models of systemic noninfectious inflammation (442), acid aspiration (257, 495), sepsis (149, 495), hemorrhagic shock-induced ALI (443), and transfusion-related acute lung injury (TRALI) (257) demonstrate that platelet depletion results in improvement in alveolar barrier function and leak as measured by changes in radiolabeled albumin permeability, BAL total protein, partitioning of labeled dextran, microscopic assessment of interstitial edema, or measurement of extravascular lung water and extravascular plasma equivalents. In some cases the apparent reduction in alveolar capillary leak resulting from platelet depletion was dramatic. In experiments involving systemic vascular beds, platelets were shown to mediate increased vascular permeability in skin and skeletal muscle that was substantial even in mast cell-deficient mice (61). As noted above, platelets release factors that reduce endothelial permeability and preserve barrier function, but they can also release other factors that have the opposite effect. For example, platelet dense granules (see below) contain histamine, which increases vascular permeability (126). Histamine-induced vascular leak is a classic manifestation of acute inflammation (268, 288, 349). Activated platelets also synthesize mediators that increase endothelial permeability, in addition to releasing stored barrier-destabilizing factors (Figure 2). In a mouse model of aspiration-induced ALI, platelet-derived TxA2 was implicated (495). As a second example, human and mouse platelets synthesize IL-1β and export it in solution and in microvesicles (see sect. IIID). IL-1β is a central mediator of increased endothelial permeability in inflammation and infection (179, 348, 499).
The mechanisms that account for barrier-preserving activities of platelets in one condition and barrier disruption in another are unknown (468). One obvious possibility is cellular activation: experiments outlined above usually suggest that unactivated platelets are involved in preservation of basal barrier integrity and restricted endothelial permeability, and platelets are presumably in an unactivated state as they transit the pulmonary and systemic circulations under basal conditions. In contrast, platelet activation, as occurs in inflammation and thrombosis, is required for release of barrier-disrupting factors such as histamine and for synthesis of TxA2 and IL-Iβ (454). Another component appears to be interactions of activated platelets with leukocytes in inflamed vessels, triggering release or generation of barrier-destabilizing factors by platelet-dependent activation of the leukocytes or by reciprocal signaling (46, 427, 454, 468) (Figure 4C). In murine models in which platelet depletion improved indexes of increased alveolar capillary permeability, neutrophil depletion or interruption of platelet-neutrophil interactions also had a similar effect (150, 257, 442, 495). In an early in vitro model, platelet “release products” amplified neutrophil-mediated injury of cultured endothelial cells; serotonin was implicated as the key platelet factor (42). In a recent in vivo model of immune complex dermatitis mentioned previously, activated platelets and neutrophils conspired to mediate inflammation-induced hemorrhage via platelet GPVI; remarkably and paradoxically, parallel GPVI-dependent adhesion and signaling were required for vasculoprotective effects of platelets, which sealed breaches in the endothelial barrier and blocked exposure of subendothelial matrix in this model (150).Thus diverse interactions of platelets and leukocytes can contribute to altered endothelial barrier integrity (468) (Figure 4C).
Additional mechanisms may also be involved in endothelial barrier-stabilizing activity of platelets in one condition versus barrier-destabilizing activity in others. Changes in endothelial receptors (for example, desensitization) for ligands that alter barrier properties, concentration-dependent effects of critical ligands, and time-dependent changes in signaling pathways linked to key receptors and receptor subtypes may result in a switch from barrier stabilization to destabilization in complex inflammatory conditions. Responses to S1P provide important examples (315, 328). While acute administration of S1P or pharmacological SIP1 agonists to endothelial monolayers in vitro resulted in barrier protection, prolonged exposure eliminated this response suggesting altered S1P-S1P1 signaling (401). In parallel, repeated administration of SIP1 agonists increased vascular leak in bleomycin-induced ALI (401). S1P and pharmacological S1P1 agonists have barrier regulatory activates that are highly concentration-dependent in the mouse lung, and can in some cases cause barrier disruption (315, 378). As additional variables, the “cargo” of platelets, and their synthetic output, may change in inflammation and pulmonary immune responses (468). There is now considerable evidence that the platelet transcriptome and proteome are altered in disease, resulting from transcriptional “reprogramming” of megakaryocytes and potentially from synthesis of new proteins by activated platelets in the vasculature (see sect. IIID). Thus the profile of barrier-stabilizing versus barrier-disrupting factors present in circulating and locally deposited platelets or the pattern of their synthesized products may be altered in lung or systemic inflammation, a possibility that is under investigation.
Evolving evidence indicates that platelets also have novel activities in regulating lymphangiogenesis and the integrity of lymphatic vessels, in addition to influencing endothelial barrier properties of pulmonary and systemic blood vessels. S1P and Ang-1 stimulate lymphangiogenesis (428), suggesting the possibility that platelets influence lymphatic development by releasing these mediators. Furthermore, recent studies with genetically modified mice revealed an unexpected mechanism. Binding of CLEC-2 on platelets by podoplanin (PDPN), a ligand on lymphatic endothelial cells (LEC), triggers SLP-76- and SYK-dependent platelet aggregation at dividing zones between embryonic lymphatic vessels and veins (reviewed in Ref. 317). This appears to be critical for separation of lymph and blood in murine lymphatic development. Mice deficient in CLEC-2, PDPN, or SLP-76/SYK signaling function had blood-filled lymphatics during fetal development and died shortly after birth due to impaired lymphatic function. Of note, genetically altered mice deficient in CLEC-2 or PDPN also have other complex lung defects (317). More recent experiments indicate that CLEC-2-dependent platelet activation and integrin αIIbβ3-mediated aggregation and clot formation operate with anatomic barriers provided by lymphovenous valves to preserve separation of lymph and blood in adult mice (171). In additional studies, mice with conditional postnatal deletion of PDPN had impaired integrity of high endothelial venules (HEV), which are specialized vessels in lymph nodes, and spontaneous bleeding in mucosal lymph nodes that appeared to result from lymphocyte trafficking (170). Mice lacking lymphatic PDPN or platelet CLEC-2 had reduced levels of HEV VE-cadherin. CLEC-2 deficiency was rescued by wild-type platelets, and activation of platelet CLEC-2 resulted in release of S1P, which promoted expression of VE-cadherin. The experiments indicated that local S1P release triggered by PDPN/CLEC-2-mediated platelet activation preserves HEV integrity during immune events and lymphocyte trafficking (170). Thus S1P stabilizes lymphatic endothelial barrier properties in addition to promoting pulmonary and systemic endothelial barrier integrity in mice.
Lymphatic function critically influences lymphocyte trafficking and complex immune responses such as tolerance (3, 428, 474). In addition, experimental models demonstrate that lymphangiogenesis is a central response in lung infection and chronic airway inflammation (19, 20, 329). Therefore, it is possible that platelets influence pulmonary immune function in chronic infection or in airway disease by altering lymphangiogenesis in these settings, although this has not been examined. It is not known, however, if the mechanism involving platelet CLEC-2 and PDPN on LEC operates in the lung, or in mammals besides mice. It is also possible that platelets influence alveolar fluid balance and resolution of pulmonary edema by contributing to lung lymphatic integrity (276, 419) or, in chronic inflammatory conditions, lymphangiogenesis (3). Against this, platelet depletion did not alter lung lymph flow in a study of LPS-challenged sheep (410). The degree of thrombocytopenia in these experiments was not profound, however, and the result is somewhat at odds with experiments in the sheep lung lymph fistula model cited earlier (253). Therefore, the influence of platelets on lung lymphatic function in adult animals remains an open question. A further question is whether the platelet-dependent mechanisms of lymphangiogenesis and maintainance of HEV barrier integrity identified in mice mediate reestablishment of lymphatic drainage and function in transplanted lungs, or other transplanted organs.
B. Platelet Receptors, Signaling Cascades, and Effector Mechanisms: Traditional and Newly Recognized Pathways That Mediate Hemostatic, Inflammatory, and Immune Activities
Platelets have a diverse array of receptors and surface molecules that mediate signaling functions (73). Receptors, adhesion molecules, and intracellular signaling pathways originally thought to exclusively have hemostatic effector functions are now known to mediate inflammatory and immune events (283, 454). An abbreviated list of “traditional” hemostatic receptors and adhesion molecules that also have inflammatory activities is shown in Table 1. The signaling cascades that these and other traditional platelet receptors (73) trigger have been reviewed in detail (52). These classic, long-known hemostatic pathways with newly recognized inflammatory and immune functions (89, 369, 454) are now complemented by platelet pathways and molecular checkpoints that have only recently been discovered. As examples, thrombin-induced PAR signaling in human platelets activates mammalian target of rapamycin (mTOR) (14, 17, 462, 463), and platelet collagen receptors signal to events mediated by ADP-ribosylation factor 6 (ARF6-GTP) (206, 213, 380). Thrombin and collagen are also reported to induce intracellular signaling involving nuclear factor kappaB (NF-κB) (130, 251, 270). NF-κB is a well-known transcription factor that regulates inflammatory gene expression in nucleated cells (Figure 1C) but has noncanonical signaling activities in platelets, as do other intracellular factors originally identified as transcription regulators in cells with nuclei (369, 413). In addition to identifying new intracellular signaling mechanisms, experimental observations also continue to reveal previously unrecognized effector activities triggered by platelet receptors and adhesion molecules traditionally known for their primary hemostatic functions. As examples, platelet GPIbα, which is the major ligand-binding glycoprotein of the GPlbα-IX-V complex (Table 1), is key in molecular systems that recognize intravascular pathogens and present them to dendritic cells and specialized hepatic macrophages in murine models of blood-borne infection (449, 478). In addition, GPlbα-IX-V is reported to have systemic immunomodulatory effects in experimental sepsis in mice (78).
Platelets also have a repertoire of receptors and linked intracellular signaling pathways that have immune recognition, or regulation, as their primary or dominant known function. The remainder of this section focuses on some of these nontraditional signaling systems. Of major topical importance, platelets from humans and other mammals express Toll-like receptors (TLRs) (369, 397, 453). TLRs are members of a broader family of pattern recognition receptors that trigger immune pathways and responses when they are engaged by exogenous ligands (including microbial pathogen-associated molecular patterns, or PAMPS, and certain pathogen virulence factors) or endogenous agonists (such as danger-associated molecular patterns, or DAMPs; others) (334). Expression of TLRs is compelling evidence that platelets recognize inflammatory and pathogen-derived signals and are immune effector cells. Megakaryocyte and megakaryocytic cell lines also express TLRs, and there is evidence that megakaryocyte TLR engagement influences thrombopoiesis (25, 73). Megakaryocytes in the marrow (191) or lung (468) may also respond to TLR engagement with proinflammatory activities, although this possibility has not been extensively explored.
Human and mouse platelets express mRNA, protein, or both for multiple TLRs (25, 73, 133, 396, 453). New reports of TLRs and TLR-dependent functions in human platelets continue to appear (5, 228). The majority of studies examining functions of platelet TLRs have focused on TLR4, TLR2, and TLR9 (369). TLR4 has been most extensively examined, largely by studying platelet responses to its defining ligand, LPS (Table 2). In vivo experiments suggest that LPS activates platelets, although complex interactions of platelets with endothelial cells and leukocytes, which also express TLR4, complicate interpretation of some of these reports. Intravenous or intratracheal challenge of rabbits, rats, or mice with LPS causes thrombocytopenia associated with time-dependent deposition of platelets in the lungs, liver, mesenteric vessels, and other sites (6, 12, 72, 116, 195, 215, 219, 330, 404, 418, 497) (Figure 5). In some experiments the magnitude of accumulation of platelets was in part dependent on the bacterial origin of the LPS. Platelet deposition in microvessels or incorporation into thrombi in response to LPS was reduced or eliminated in mice with mutations of TLR4 or of a key intracellular component of TLR signal transduction, myeloid differentiation marker 88 (MyD88) (6, 12, 375, 496). Thrombocytopenia reduced maximal TNF-α release in mice injected with LPS, suggesting that platelet TLR4 modulates TNF-α production (12). In a second study, however, adoptive transfer of platelets from wild-type mice (∼10% of normal circulating platelet number) did not restore plasma levels of TNF-α or IL-1β in TLR4-deficient mice challenged with LPS (418). In humans, administration of LPS under controlled conditions caused thrombocytopenia, formation of circulating platelet-monocyte aggregates, and increased CD40 ligand (CD40L) on the plasma membranes of circulating platelets (203, 246, 476). (Inflammatory and immune significance of platelet-leukocyte aggregates and CD40L are discussed later.) Thus in vivo observations in experimental animals and humans support the notion that platelets have a recognition system for LPS.
Table 2.
Functional responses of platelets induced by TLR agonists in vitro
| TLR | Platelet Preparation | Agonist(s) | Functional Response(s) | Reference Nos. |
|---|---|---|---|---|
| TLR4 | Washed human platelets | LPS (S. minn. R595, 10–250 μg/ml) | Potentiation of serotonin release triggered by IgG aggregates or immune complexes | 141 |
| Human platelet-rich plasma (PRP) | LPS (E. coli 0111:B4, ≤10 ng/ml) | Did not directly induce aggregation or P-selectin translocation or potentiate these responses to ADP, PAF, collagen | 459 | |
| Gel-filtered mouse platelets in buffer with 10% autologous serum | LPS (E. coli 0111:B4, 5 μg/ml) | Did not induce P-selectin translocation (30 min); induced adhesion of platelets to immobilized fibrinogen under flow | 6 | |
| Human PRP or washed human platelets | LPS (Several E. coli LPS types; 1 μg/ml) | Induced PAC-1 binding; CD40L upregulation; binding of fibrinogen; adhesion to cultured microvascular endothelial cells (no adhesion of washed platelets) | 417 | |
| Isolated human platelets in Hanks' balanced salt solution | LPS (E. coli 0111:B4, 5–100 μg/ml) | Did not induce platelet aggregation (∼5 min) or P-selectin expression; induced attachment of platelets to neutrophils immobilized on protein-coated cover slips under flow; induced platelet-dependent neutrophil extracellular trap (NET) formation and neutrophil degranulation | 72 | |
| Human platelets isolated by negative immunoselection; PRP | LPS (E. coli 0111:B4, 100 ng/ml in 0.5% serum or with recombinant CD14+LBP) | Did not directly induce rapid shape change or aggregation; augmented rapid (5 min) ADP-induced aggregation in PRP; induced time-dependent actin polymerization, P-selectin translocation, P-selectin-dependent platelet-neutrophil interaction, CD40L upregulation (1–3 h); time-dependent splicing of IL-1β pre-mRNA, IL-1β protein synthesis | 400 | |
| Washed human platelets or PRP | LPS (several different LPS types; 1–100 μg/ml) | Did not induce rapid aggregation of washed platelets but potentiated aggregation of platelets stimulated with subthreshold concentrations of thrombin, collagen; directly induced ADP release (10 min), P-selectin translocation (30 min) | 496 | |
| Washed mouse platelets | LPS (E. coli 0111:B4, 10 μg/ml) | Potentiated thrombin-induced aggregation, secretion | 496 | |
| Human platelets isolated by negative immunoselection | LPS (E. coli 0111:B4, 10 ng/ml + rCD14, LBP) | Shedding of microparticles; splicing of IL-1β pre-mRNA, synthesis of IL-1β protein; signaling of cultured endothelial cells | 57 | |
| Human platelets isolated by negative immunoselection | LPS (E. coli 0111:B4, 10 ng/ml to 1 μg/ml) | Did not induce P-selectin translocation (60 min); induced splicing of tissue factor (TF) pre-mRNA and generation of TF procoagulant activity (30 min to 4 h) | 367 | |
| Human PRP | LPS (S. typhimurium, 1 μg/ml); histones | LPS did not induce thrombin generation. In contrast, histones triggered platelet aggregation, P-selectin translocation, and thrombin generation that were partially blocked by anti-TLR4 and anti-TLR2 antibodies | 394 | |
| Human washed platelets with 0.1% fetal bovine serum | LPS (E. coli 0III:B4 0.5–10 μg/ml) | Induced degradation of IκBα and phosphorylation of NF-κB p65; synergized with thrombin in inducing IκBα degradation, p65 phosphorylation, aggregation, fibrinogen binding, vWF and ATP release; did not directly induce P-selectin or CD40L translocation, platelet-PMN aggregate formation (5 min) | 362 | |
| TLR2 | Washed human platelets | PAM3CSK4 (1–30 μg/ml) | Induced platelet aggregation, serotonin release, intracellular Ca2+ release, phosphorylation of intracellular proteins | 27 |
| Human PRP | PAM3CSK4 (≤1 μg/ml) | Did not induce aggregation (15 min), P-selectin translocation (1 h), intracellular Ca2+ mobilization; did not activate platelets or potentiate responses to ADP, PAF | 459 | |
| Human platelets isolated by negative selection | PAM3CSK4 (100 μg/ml) | Induced splicing of IL-Iβ PremRNA | 400 | |
| Washed human platelets | PAM3CSK4 (1–10 μg/ml) | Induced aggregation, adhesion to collagen, P-selectin translocation, activation of αIIbβ3, generation of reactive oxygen species; induced aggregation of mouse platelets; triggered formation of platelet-PMN aggregates in whole blood | 34 | |
| Washed human platelets | PAM3CSK4 (10 μg/ml) | Induced intracellular protein phosphorylation, protein-protein interaction; release of α-granules; formation of platelet monocyte aggregates; differential effects compared with thrombin | 360 | |
| Washed human platelets | PAM3CSK4 (1–10 μg/ml), MALP-2 (1–4 μg/ml) | PAM3CSK4 (TLR2/1 agonist) induced aggregation; ATP secretion; TBXA2 production; increased intracellular Ca2+. MALP-2, a TLR 2/6 agonist, did not induce these responses. | 204 | |
| Washed human platelets | PAM3CSK4 (0.5–5 μg/ml) | Induced IKBα degradation; p65 phospholylation; aggregation; fibrinogen binding; vWF & ATP release; P-selectin and CD40L translocation | 362 | |
| TLR7 | Washed human platelets | Synthetic TLR7 agonists | Induced translocation of P-selectin and CD40L; formation of platelet-PMN and platelet-monocyte aggregates; phosphorylation of platelet p38 MAPK and AKT; increased adhesion of human and murine platelets to collagen under flow | 228 |
| TLR9 | Washed human platelets | Synthetic unmethylated type C CpG oligodeoxynucleotides (ODN) | Induced P-selectin translocation and increased surface expression of TLR9; sequestration of ODN by platelets; type IV collagen potentiated platelet responses to ODN | 437 |
| Human gel-filtered platelets, PRP | Carboxy(alkylpyrrole) protein adducts | Induced αIIbβ3 activation; P-selectin translocation; platelet aggregation; IRAK1 and AKT phosphorylation; acted synergistically with traditional platelet agonists | 335 | |
| Mouse gel-filtered platelets, PRP | Carboxy(alkylpyrrole) protein adducts | Platelets from wild type, but not MyD88−/− or TLR9−/− mice responded with activation events similar to those of human platelets (see above) | 335 |
Studies examining TLR4 agonists are listed first because they are most numerous, followed by others focused on TLR2, TLR7, and TLR9. In some reports, blocking antibodies or inhibitors against the TLR were used and/or platelets from genetically altered mice deficient in signaling by the TLR were examined. The table is not comprehensive, and additional relevant articles are cited in reviews mentioned in the text. For example, over a dozen reports of platelet functional responses to LPS, or to LPS motifs such as lipid A, appeared before expression of TLR4 by platelets was demonstrated (369, 453). [Modified from Rondina et al. (369).]
FIGURE 5.

Platelets accumulate in the alveoli of mice in response to intrapulmonary LPS challenge. A: scattered platelets detected by brown immunostaining with an antibody against CD41 (αIIb subunit of integrin αIIbβ3) are present in alveolar vessels of control mice. (Scale bar = 20 μm; zoom of outlined area = original magnification ×60). Some large areas of anti-CD41 staining may indicate megakaryocytes. B: there was dramatic accumulation of CD41-positive platelets in alveoli of mice challenged with intratracheal LPS. Some CD41-positive staining may indicate platelet microparticles or megakaryocytes. In the right-hand enlarged panel, arrows point to CD41 events in the alveoli, many of which demonstrate platelets associated with intra-alveolar leukocytes. Platelets may have novel extravascular inflammatory activities in the alveolar space in addition to intravascular proinflammatory functions. [From Ortiz-Munoz et al. (330), with permission from American Society of Hematology.]
After molecular characterization and cloning of TLR4 (30), its expression on human and murine platelets was documented (6, 73, 74, 459). This provided a molecular explanation for multiple earlier reports of variable responses of platelets to LPS in vitro and evidence supporting the concept that platelets can sense microbial signals (453). Binding of LPS to platelet TLR4, and variable changes in surface expression of TLR4 in response to platelet activation and storage, were reported (12, 72, 74, 133, 395, 417). Multiple subsequent experiments examining platelet responses to LPS and TLR4 signaling in vitro have been published, with results that are at times perplexing, provocative, and contradictory (Table 2) (369). In some cases LPS directly induced responses of platelets that require cellular activation, or potentiated activation triggered by thrombin or other traditional agonists (57, 72, 74, 362, 367, 400, 417, 496). In others, LPS did not directly trigger classic, rapid activation responses of human or murine platelets (surface translocation of P-selectin, shape change, aggregation) and/or did not enhance activation induced by traditional receptor-mediated agonists (6, 72, 362, 367, 375). A current interpretation of these observations, taken together, is that platelet responses to LPS are not stereotyped or as consistent as are responses induced by traditional receptor-mediated pathways, and that LPS- and TLR4-induced activities are highly dependent on the conditions of the experiments. Variables that may influence the “unconventional” (72) or “atypical” (400) activation of platelets by LPS include the bacterial origin and structure of LPS; purity of the platelet preparation; time of stimulation; the presence of plasma factors that facilitate LPS recognition and TLR4 signaling, including CD14 and LPS binding protein (LBP); and whether the platelets are adherent via integrins or are in suspension (212, 369). As examples of the diversity of putative TLR4-mediated responses of human platelets depending on the experimental conditions, different variants of Escherichia coli LPS and a highly purified LPS preparation, Kdo2-Lipid A, had variable potency as direct agonists for ATP secretion by washed platelets and as potentiators of collagen-induced aggregation of platelets in PRP (496); E. coli LPS induced surface expression of P-selectin on isolated platelets at 30 min (496) or over 1–2.5 h (400), whereas traditional agonists such as thrombin trigger P-selectin translocation from alpha granules within minutes; splicing of IL-1β pre-mRNA and posttranscriptional synthesis of IL-1β protein by stringently isolated human platelets in albumin-containing buffer were enhanced by supplementation with recombinant CD14 and LBP (57, 400). (Production of IL-1β and posttranscriptional synthetic pathways of platelets are discussed in sect. IIID.) It is currently unclear exactly how the behavior of platelets in response to LPS in vitro relates to LPS-induced deposition of platelets in the lungs (Figure 5) and other organs in vivo in experimental animals, or to LPS-induced platelet responses in humans in vivo.
Signals delivered via TLR2 also alter platelet effector functions. TLR2 recognizes multiple microbial ligands including lipopeptides and peptidoglycans from gram positive bacteria, and heterodimerizes with TLR1 and TLR6 (25). Human and murine platelets express TLR2, and human platelets express TLR1 and TLR6 (369). Most investigators report that treatment of human platelets with synthetic ligands for TLR2 directly induces activation responses (Table 2). Human platelets have differential responses to synthetic ligands that are putatively selective for TLR 2/1 and 2/6 (204). Functional activities reported to be triggered by engagement of platelet TLR2 include inside-out signaling of integrin αIIbβ3, fibrinogen binding, aggregation, degranulation of alpha granule and dense granule factors, P-selectin translocation, CD40L surface expression, ATP secretion, IL-1β pre-mRNA splicing, and formation of platelet-leukocyte aggregates (27, 34, 204, 360, 362, 400) (Table 2). Triggering of P-selectin translocation and formation of platelet-leukocyte aggregates (see sect. IIIF) via TLR2 likely contributes to inflammatory responses in patients with gram-positive bacteremia (197). Signaling via TLR2, the TxA2 receptor (TP), and purinergic receptors (P2X1, P2Y1, P2Y12) on human platelets appears to occur in concert (204). In vivo challenge with a bacterium that expresses PAMPs recognized by TLR2 resulted in formation of platelet-neutrophil aggregates in the blood of wild-type but not TLR2-deficient mice (34). Thromboinflammatory activities of platelets triggered by TLR2 engagement appear to be differentially regulated by intracellular signaling cascades distinct from those activated by traditional hemostatic receptors (34, 360). Treatment of wild-type mice with a synthetic TLR2 ligand, but not mice genetically deficient in TLR2, increased megakaryocyte maturation and platelet number suggesting, together with in vitro experiments, that TLR2 on megakaryocytes regulates thrombopoiesis in inflammation (25, 26).
Human and murine platelets also express TLR7 and TLR9 and respond to their engagement with effector functions (Table 2). In other cell types, TLR7 and -9 are restricted to intracellular compartments, primarily endosomes (35). Nevertheless, there is evidence from several laboratories that TLR9 is displayed on the plasma membranes of resting platelets and is increased upon cellular activation (369). TLR9 mRNA is expressed by human megakaryocytes and upregulated in proplatelet production (437). Synthetic oligodeoxynucleotide (ODN) ligands for TLR9 increased TLR9 surface display, induced intracellular sequestration of ODNs, and triggered P-selectin translocation by human platelets in vitro (437). Pretreatment of platelets with type IV collagen to induce TLR9 surface expression enhanced ODN-stimulated P-selectin translocation and sequestration of ODN. Together, the experiments indicate that platelets respond to ODNs via TLR9 and that they have a putative system for microbial ODN uptake. Platelet TLR9 is also reported to recognize endogenous DAMPs that may be important in vascular inflammation. Synthetic (ω-carboxy-alkyl) pyrrole protein adducts, which are intended to mimic modified proteins generated in atherosclerosis and oxidant vascular injury, triggered TLR9-dependent platelet aggregation, degranulation, and, in mice, in vivo thrombosis (335). In contrast to TLR9, platelet TLR7 appears to have more parochial functions and to dominantly signal to immune pathways without dramatically influencing hemostasis or thrombosis. A synthetic ligand for TLR7 triggered formation of human and murine platelet-leukocyte aggregates and induced responses that are requisite for survival of experimental viral infection in mice (228) (Table 2). Thus the TLR repertoire on platelets is diverse and complex both in terms of individual TLRs that are represented and effector responses that they are linked to.
The intracellular signaling pathways that link platelet TLRs to effector functions have not been completely dissected. Nevertheless, platelets express MyD88 (29, 335, 496), which mediates downstream signaling by most TLRs. Distal intermediates in the canonical MyD88 pathway (334), including TIR domain-containing adaptor protein, IL-1R-associated kinase 1, IL-1R-associated kinase 4, and TNFR-associated factor 6, are also reported to be present in platelets based on direct or indirect detection methods (29, 57, 369). Additional intracellular signaling activities have also been reported when platelets are stimulated with ligands for TLR2 or TLR4 (360, 362, 496). Mechanisms that integrate signals and control platelet responses when more than one TLR is engaged or when TLRs and traditional receptors are ligated in parallel or in sequence (Figure 2), as likely occurs in complex pathophysiological settings such as pneumonia, ARDS, or sepsis, have not been dissected.
Platelets express diverse receptors with the potential for immune signaling in addition to TLRs. Two laboratories have reported that human platelets express functional IL-1 receptor type 1 (IL-1R1) (24, 58). IL-1R1 was also detected on a human megakaryocyte cell line (24), and functional experiments indicated that IL-1R1 is expressed by cultured murine fetal liver megakaryocytes (321). IL-1R1 recognizes both IL-1α and IL-1β (47). IL-1β is reported to regulate megakaryocyte maturation (24) and IL-1α to regulate thrombopoiesis via a novel mechanism (321). IL-1β signaling amplifies agonist-induced aggregation and adhesion by human platelets (24) and enhances responses to TLR4 engagement via a unique autocrine loop involving endogenous IL-1β (58). In other cell types, IL-1R1 activates an intracellular signal transduction pathway similar to that of many TLRs and that includes MyD88 and other common intermediates (47). Presumably, this pathway also operates in platelets and megakaryocytes.
Of the multiple classes of platelet receptors and signaling pathways (73), a group sometimes termed immunoreceptors (214) has particular relevance to this discussion. Immunoreceptors are characterized by structural sequences known as immunoreceptor tyrosine-based activation motifs (ITAMs), hemITAMs, and immunoreceptor tyrosine-based inhibitory motifs (ITIMs) (214, 351). The subset of ITAM-coupled or hemITAM-containing receptors on human platelets includes GPVI, CLEC-2, GPlb-lX-V, and Fc receptors (FcγRIIa, FcαRI, and FcεRI) (73, 351). Murine platelets express many of the immunoreceptors found on human platelets, but do not express FcγRIIa (214). GPVI has already been introduced, in part because of its parallel hemostatic and inflammatory signaling capacities (Table 1) and intricate activities in endothelial barrier integrity (44, 150), as has CLEC-2 and its unexpected roles in platelet contributions to lymphangiogenesis and lymphatic barrier function (see sect. IIIA). Each receptor has additional activities and signaling mechanisms relevant to inflammatory and immune functions of platelets (351, 453), and will be mentioned again. Platelet Fc receptors recognize immunoglobulins and immune complexes, providing direct mechanisms for immune interaction, signaling, and effector activities (214, 453). In early studies, responses of human platelets to immune complexes and aggregates of IgG were enhanced by LPS, indicating convergence of immune signaling pathways in these cells and providing early evidence for Fc receptor signaling and for an LPS receptor, now known to be TLR4, on platelets (141). Multiple additional Fc receptor-mediated functions of human platelets have been reported. As examples, clustering of FcαRI (CD89) on human platelets induces synthesis of IL-1β and tissue factor in vitro (356), Bacillus anthracis peptidoglycan activates human platelets via FcγRIIa and complement (425), and influenza virus and IgG immunoglobulins form immune complexes that activate platelets via FcγRIIa (38) (see sect. IIIG). FcγRIIa acts cooperatively with integrin αllbβ3 in outside-in signaling of platelets (498). Platelet ITIM-coupled receptors include TLT-1, mentioned earlier, and platelet-endothelial cell adhesion molecule-1 (PECAM-1) (214). PECAM-1 has intricate functions, including positive modification of integrin activity on activated platelets and inhibitory modulation of responses of platelets activated via ITAM-mediated signaling (318).
Small C-type lectin receptors are widely distributed on immune effector cells (73). Dendritic cell-specific intracellular adhesion molecule-3 grabbing nonintegrin (DC-SIGN) is a small C-type lectin receptor that has been shown to mediate interactions of human platelets with viruses, including recognition of human immunodeficiencty virus (HIV) and dengue virus (73, 180, 406). Recent observations demonstrate that dengue activates inflammasome assembly and function in human platelets via DC-SIGN and that DC-SIGN is a route of entry of the virus into platelets, as will be discussed below. Thus DC-SIGN is particularly instructive in this overview of selected platelet receptors since it mediates previously unrecognized pathogen recognition, signaling, and inflammatory effector functions of these cells.
C. Degranulation, Secretion, Release, and Microparticle Formation by Activated Platelets
Activated platelets have diverse mechanisms of intercellular communication and deliver signals to a variety of target cells including other platelets. These cell-cell signaling capacities initiate and amplify responses in hemostasis, inflammation, and injury (453, 469). Rapid secretion of soluble factors with paracrine or, in some cases, autocrine or endocrine activities is the best known of these communication mechanisms and is a traditional platelet functional response (Figure 2). Activated platelets also translocate granule factors that are anchored in, or to, the plasma membrane and can mediate spatially restricted juxtacrine signaling without being released from the cell surface (453). P-selectin, which delivers signals to target monocytes, neutrophils, and other leukocytes in addition to mediating adhesion, is a prime example. Thus secretion of soluble factors, or their release by other mechanisms, is not the only mode of intercellular communication by activated platelets (454, 469).
As with other traditional responses, platelet secretion was originally characterized in the context of its contributions to hemostasis and thrombosis, and is extremely important in this regard (126). Nevertheless, factors released by platelets also mediate important inflammatory and immune responses (126, 454, 469) (Table 3). The platelet secretome contains a remarkable number of soluble factors, many of which are preformed and stored in subcellular compartments and storage units, including alpha granules (α-granules), dense granules, and lysosomes (126). Novel or atypical storage compartments in human platelets have also recently been reported (231, 437).
Table 3.
Human platelet inflammatory, immune-modulating, and antimicrobial factors and mediators
| Class of Mediator or Factor | Factor | Stored/Synthesized | Reported Immune Target Cell(s) |
|---|---|---|---|
| Adhesion and signaling molecule | P-selectin | Stored | PMNs, monocytes, lymphocytes |
| ITIM-containing immunoreceptor | Triggering receptor expressed on myeloid cells–like transcript-1 (TLT-1) | Stored | Platelets |
| Pleiotropic inflammatory modulators | Histamine | Stored | ECs, monocytes, PMNs, NK cells, T and B cells, eosinophils |
| Serotonin (5-HT) | Stored | Monocytes, macrophages, DC | |
| Inflammatory and immunomodulatory lipids | TxA2 | Synthesized | Platelets, T lymphocyte and macrophage subsets |
| PAF | Synthesized | Platelets, PMNs, monocytes, macrophage and lymphocyte subsets | |
| Pleiotropic adaptive immune modulator | CD40L (CD154) | Stored | B cells, T lymphocytes, EC, monocytes, DC subtypes, epithelial cells |
| Growth factors with immune activities | PDGF | Stored | Monocytes, macrophages, T lymphocytes |
| TGF-β | Stored | Monocytes, macrophages, T and B lymphocytes | |
| Chemokines | PF4 (CXCL4) | Stored | PMNs, monocytes, macrophages |
| NAP2 (CXCL7) and related β-TbG variants | Proteolytic cleavage of stored precursors | PMNs | |
| GRO-α (CXCL1) | Stored | PMNs | |
| ENA-78 (CXCL5) | Stored | PMNs | |
| SDF-1 (CXCL12) | Stored | Bone marrow-derived progenitor cells | |
| RANTES (CCL5) | Stored | Monocytes, eosinophils, basophils, NK cells, T lymphocyte and DC subsets | |
| MIP-1α (CCL3) | Stored | Monocytes, eosinophils, basophils, NK cells, lymphocyte and DC subsets | |
| MCP-3 (CCL7) | Stored | Monocytes, basophils, NK cells lymphocyte and DC subsets | |
| Cytokines | IL-1β | Synthesized | Monocytes, DC and macrophage subsets, T-cell lines, EC, vascular smooth muscle cells, synoviocytes |
| IL-1α | Stored (?) | Same as IL-1β | |
| Macrophage migration inhibitory factor (MIF) | Stored | Monocytes, macrophages | |
| HMBG1 | Stored | Macrophages, PMNs, ECs | |
| GM-CSF | Stored | Eosinophils | |
| Antimicrobial peptides | Platelet microbicidal proteins (several classes) | Stored; and in some cases, proteolytically cleaved | No human cellular targets identified; microbicidal for several bacteria and fungi |
| β-Defensin 1 | Stored | PMNs (NET formation) |
The secretome of α-granules has been extensively examined and contains more than 300 factors when evaluated by proteomic analysis, including coagulation modifiers, adhesion molecules with multiple functions (fibrinogen, vWF, thrombospondin), growth factors, angiogenic factors and inhibitors, multiple chemokines of the CXC and CC classes, antimicrobial factors, and immune effector molecules and modulators (126). Proteins in α-granules are synthesized under transcriptional control by megakaryocytes, or are internalized by megakaryocytes or platelets in some cases. Unique subsets of α-granules may subserve specific functions (341, 397). Two key α-granule proteins, platelet factor 4 (PF4; CXCL4) and β-thromboglobulin (βTbg), were the first members of the CXC chemokine family to be identified; PF4 is a marker for megakaryocytes and platelets and has multiple prothrombotic, proinflammatory, and antimimicrobial activities (51, 454). βTbg and related chemokines, including neutrophil activating peptide 2 (NAP-2; CXCL7), are cleavage products of a parent precursor, platelet basic protein. PF4, βTbg, and NAP-2 have differential and overlapping biologic activities (51). βTbg and NAP-2 have been found in BAL samples from patients with ARDS (75, 186).
Regulated upon activation, normal T cell expressed presumed secreted (RANTES; CCL5), a CC chemokine with pleiotropic immune functions, is also a key α-granule protein (454). Heterophilic interaction of PF4 and RANTES promotes monocyte arrest on endothelial cells in in vitro experiments, suggesting an intricate monocyte targeting mechanism (456). More recently, PF4 and RANTES were reported to form heterodimers in LPS-induced ALI in mice. Antibodies against the chemokines, or a peptide that inhibits their heterodimerization, reduced lung neutrophil accumulation and alveolar capillary protein leak (149). The inhibitory peptide also reduced indexes of inflammatory lung injury in acid-induced ALI and the cecal ligation and puncture (CLP) model of sepsis (149). Thus there is evidence for contributions of PF4 and RANTES, which are prototype platelet α-granule chemokines, to acute lung inflammation and injury. In addition, platelets release several other chemokines (121, 424, 454) that may mediate pulmonary inflammatory responses. Additional α-granule constituents with proinflammatory functions also have this potential (126) (Table 3).
CD40 ligand (CD40L; CD154) is rapidly translocated to the plasma membrane upon platelet activation and is reported to be present in α-granules (65, 73, 205), although diffuse cytoplasmic staining was observed in an earlier study (168). Expression of CD40L by megakaryocytes is regulated by nuclear factor of activated T cells (NFAT) in cooperation with early growth response-1 (EGR-1) (82). CD40L is released from the surfaces of activated platelets in a time-dependent fashion by a process that is not completely defined, and is found as a trimer in plasma and in higher concentrations in serum (73, 97). There is evidence that CD40L can engage its signaling partner, CD40, on target cells while it is anchored on the platelet membrane or is in the cleaved, soluble form in solution (73, 166, 454). Thus it can mediate both juxtacrine and paracrine signaling, and the magnitude of CD40L-dependent signaling by activated platelets may not be directly related to its concentration in blood. Human platelets also constitutively express CD40, which triggers their activation when engaged by CD40L and may also have regulatory functions (167, 188). CD40L from platelets has major immunomodulatory activities (36, 97, 111, 454) and is implicated in septic acute lung inflammation (359). Examples of immune signaling by platelet CD40L are considered in later sections; here, we profile it to illustrate complex facets of surface translocation, shedding, and export of a platelet factor with key immune activities.
Surface translocation of a dense granule protein, CD63, is a marker of platelet activation (73, 294). Platelet dense granules also contain histamine, serotonin, polyphosphates, nucleotides (ADP, ATP), glycosidases, and phosphatases (126). Histamine has multiple activities in allergic and inflammatory responses including direct actions on endothelial cells that induce increased permeability (see sect. IIIA) and neutrophil adhesion (260, 285, 288, 349). As outlined previously, serotonin has complex effects on endothelial barrier properties (468), and also has immunomodulatory activities (454). Polyphosphates are proinflammatory and prothrombotic and can induce multistep generation of bradykinin (307, 408), a classic mediator of inflammation that triggers vascular leak and endothelial generation of platelet activating factor (PAF) and prostacyclin (PGI2) (285). ATP also stimulates endothelial synthesis of PAF (285), which is a potent phospholipid inflammatory autacoid (354) that induces lung inflammation, increased pulmonary capillary permeability, and systemic inflammatory responses (60, 490). Given these and other activities of platelet dense granule constituents, degranulation from this compartment could directly or indirectly mediate disruption of alveolar-capillary barrier integrity and induce inflammation in a variety of pulmonary syndromes. Dense granule release has not been specifically explored in this context, however. Similarily, release of factors from platelet lysosomes, which store proteolytic enzymes (elastase, cathepsin G, others) and glucosidases that could attack alveolar structures, has not been examined in lung inflammation and immune syndromes in a focused way.
In addition to releasing soluble molecules from granular compartments, activated platelets release membrane-encapsulated microparticles (192, 320) (Figures 2 AND 6). Other immune and inflammatory cells also release microparticles (303). In vitro studies indicate that microparticles (sometimes termed microvesicles) are budded from the plasma membrane and that activated platelets also release smaller microvesicular exosomes, which are stored in α-granules and released on degranulation (320). Platelet microparticles transport membrane-associated signaling and adhesion molecules and represent a specialized mechanism of intravascular and extravascular cell-cell communication (320, 369, 453). Platelet signaling factors translocated on microparticles include CD40L (414), RANTES (280), IL-1β and IL-1α (37, 57, 179, 249), tissue factor (391), and others (192). Platelet microparticles mediate complex immune signaling functions, outside of the spectrum of traditional intravascular activities of platelets. As an example, microparticles from activated platelets were translocated to the synovial space in mice, inducing chemokine synthesis and inflammation in experimental arthritis (37). Additional examples will be mentioned in later sections. Procoagulant microparticles were found in the alveoli of patients with ARDS (22), but their cellular origin(s) were not identified. Megakaryocytes also shed microparticles based on in vitro experiments (192, 320). Therefore, lung megakaryocytes (468), in addition to platelets, could be sources of alveolar or systemic microparticles in inflammation and thrombosis.
FIGURE 6.

Activated human platelets synthesize IL-1β in response to hemostatic and inflammatory agonists and dengue virus. Human platelets synthesize IL-1β when activated by hemostatic and inflammatory agonists including thrombin, collagen, ADP, PAF, and LPS. In addition, live dengue virus induces synthesis of IL-1β by engaging DC-SIGN. The mechanism of IL-1β synthesis by activated human platelets involves signal-dependent, spliceosome-mediated splicing of the IL-1β pre-mRNA transcript followed by signal-dependent translation of the mature mRNA and generation of pro-IL-1β protein. Newly synthesized pro-IL-1β is processed to IL-1β by NLRP3 inflammasome-dependent caspase-1 activity, as shown in DENV-activated platelets and platelets from DENV-infected patients. Mature, biologically active IL-1β protein is released from activated platelets in solution and in microparticles. IL-1β in microparticles from activated platelets induces increased permeability of human endothelial monolayers and other inflammatory responses of human endothelial cells. Increased vascular permeability in patients with DENV infection correlated with platelet and platelet microparticle IL-1β and with platelet caspase-1 activity. See text for details and references. [Adapted from Hottz et al. (179).]
D. Synthetic Mechanisms of Activated Platelets: Traditional and Newly Identified Pathways
Synthesis of TxA2 is a traditional, “signature” response of activated platelets that is central in hemostasis and thrombosis (89) (Figure 2). TxA2 also has inflammatory and immune activities beyond its known hemostatic functions (300, 434, 454), and it is possible that TxA2 from platelets drives specific pulmonary immune functions. Activated platelets also synthesize PAF (Figure 2), another bioactive lipid with diverse inflammatory activities in the lungs and other organs (354), although it can be difficult to detect its production by activated human platelets without disabling acetylhydrolases that degrade it (127, 331). In addition, activated platelets generate reactive oxygen species (ROS), which have a variety of effector functions (34, 89). In each case, synthesis of these non-protein products is rapidly induced and is catalyzed by enzymes that are present in the platelet when it separates from the megakaryocyte.
In addition to these mechanisms for synthesis of lipid autacoids and oxygen metabolites, recent evidence demonstrates that activated human and murine platelets have posttranscriptional pathways for stimulated synthesis of proteins and peptides. These newly identified synthetic activities were unanticipated because the platelet is anucleate. We termed the general process of synthesis of proteins from endogenous mRNAs by activated platelets signal-dependent translation (463). Signal-dependent translation allows activated platelets to rapidly generate biologically relevant proteins from constitutive or newly spliced mRNAs (see below) and is induced by signals delivered via G protein-coupled receptors with traditional hemostatic functions (PAR1, others), integrin αIIbβ3, TLRs, and immunoreceptors (369, 453). Thus it differs from the low-level basal protein synthesis in platelets that has been recognized for several decades (387, 467). Protein synthesis by platelets was largely dismissed as having little or no biologic importance until recently, but it is now known that new protein synthesis changes the platelet proteome in functionally significant ways (407).
In addition to required translation “machinery,” human and mouse platelets express an unexpectedly rich transcriptome of mRNAs, unspliced pre-mRNAs, and microRNAs. Intracellular signaling pathways link surface receptor engagement to mRNA translation and, in some cases, to splicing of specific pre-mRNAs to translatable mRNA transcripts. In several cases these signal transduction cascades include key molecular components that were not known to be present in platelets until recently. Examples include Cdc2-like kinase1 (Clk1; important in pre-mRNA splicing) and mammalian target of rapamycin (mTOR, which controls translation of a subset of specific mRNA transcripts). Protein products reported to be synthesized by signal-dependent translation in activated human platelets include Bcl-3, IL-Iβ, plasminogen activator inhibitor-1 (PAI-1), tissue factor (TF), cyclooxygenases (COX) 1 and 2, the SVCT transporter, and tissue inhibitor of metalloprotenase-2 (TIMP2) (407). Each of these proteins has important biologic functions relevant to hemostasis, thrombosis, and inflammation and is not simply a “housekeeping” gene product. Although platelets do not synthesize proteins in the same quantities as do other cell types such as monocytes on a per-cell basis, many platelets accumulate in thromboinflammatory conditions and therefore they can be a significant source of new protein products. Furthermore, as noted earlier, signal-dependent translation can occur rapidly, since the time required for transcription is eliminated, and protein synthesis by activated platelets can be prolonged, based on in vitro studies. We have previously reviewed in detail features of the platelet transcriptome, signal-dependent mRNA translation, and protein synthesis by platelets that are outlined in this brief overview (369, 373, 388, 407, 453, 454, 467, 501). In the remainder of this section we will illustrate posttranscriptional synthesis of proteins by activated platelets using IL-1β, a gene product that has been called the gate keeper of inflammation (102) and that contributes to lung and systemic inflammatory diseases (47, 102), as a specific case. Synthesis of IL-1β was the first example of pre-mRNA splicing and linked translation of the mature mRNA, an intricate variation on the general process of signal-dependent translation, to be identified in activated human platelets.
IL-1 was reported to be present in activated platelets over 25 years ago (160, 161). Both IL-1α and IL-1β have been detected in studies involving human or mouse platelets (1, 37, 57, 58, 160, 161, 179, 249, 393, 400, 438). In the original report, IL-1β appeared to be dominant (161). IL-1α and IL-1β are the products of separate genes but are both recognized by IL-1R1, mentioned previously, and are inhibited by a naturally occurring soluble competitive receptor antagonist (IL-1Ra) (47) that is commonly used as a reagent to block IL-1 activity in experimental studies and that has been developed as a therapeutic (102). IL-1β has been detected in inflammatory exudates and tissue samples from humans with a variety of inflammatory and immune syndromes and is thought to mediate paracrine signaling, whereas IL-1α may principally act as an autocrine factor (47). Platelets, in addition to releasing biogically active IL-1 into solution, mediate juxtacrine intercellular signaling using surface-associated IL-1β (211) and by releasing it associated with microparticles (37, 57, 179, 249). Responses mediated by platelet-derived IL-1 include proinflammatory activation of endothelial cells (57, 135, 161, 249), increased endothelial permeability (179) (see below), smooth muscle cell activation (259), proliferation of a T lymphocyte cell line (161), autocrine signaling of platelets activated via TLR4 (58), synoviocyte activation and intra-articular IL-8 release in vivo (37), and immune signaling in experimental malaria (1). Extracellular IL-1β has been detected in platelet-fibrin complexes formed in vitro (249) (Figure 1B) and in murine thrombi in vivo (58), suggesting that clots are reservoirs for this cytokine in thromboinflammatory disorders (369). Increased IL-1β has been detected in platelets, platelet microparticles, or platelet supernates from patients with dengue (179) (see below) and sickle cell disease (90).
Some research articles and reviews indicate that platelets store IL-1α and IL-1β in α-granules or dense bodies (403, 409), in keeping with the traditional capacity of these cells to store and release multiple pre-formed signaling molecules and mediators (126) (Table 3). Nevertheless, we did not find mature IL-1β or its precursor, pro-IL-1β, in resting human platelets by ELISA or immunocytochemistry. We did, however, detect cell and microparticle-associated IL-1β protein and activity when human platelets were stimulated with thrombin, PAF, ADP, or collagen (96, 249). This pattern was similar to that reported in earlier studies (160, 161). Blocking integrin αIIbβ3 reduced maximal accumulation of IL-1β in model platelet-fibrin clots, indicating that engagement of integrin αIIbβ3 transmits outside-in signals that amplify this response (249). Consistent with this, we also found that outside-in signals via integrin αIIbβ3 induce and regulate signal-dependent translation pathways in human platelets (332, 463) and that platelet integrins regulate intracellular distribution of a key translation factor, eukaryotic initiation factor 4E, and its association with mRNA cap structures (250).
Further exploration of the molecular events that govern accumulation and release of IL-1β by activated platelets revealed an unexpectedly complex posttranscriptional pathway for signal-dependent synthesis of the cytokine (96). The transcriptome of resting human platelets includes the unspliced, intron-containing pre-mRNA that encodes pro-IL-1β, as demonstrated by conventional sequencing and in situ polymerase chain reaction (PCR) detection (96) and, more recently, by next generation RNA sequencing (373). In contrast, most of the trancripts in the human and mouse platelets are mature, fully processed mRNAs, although additional unspliced messages are also present (372, 373, 388, 391). In response to activating signals, the IL-1β pre-mRNA is spliced to the mature, translatable mRNA (Figure 6) by the activity of a spliceosomal complex that, like the IL-1β pre-mRNA, is distributed to platelets by parent megakaryocytes during proplatelet formation, based on in vitro human cell modeling (96). Loss of the pre-IL-1β transcript and accumulation of mature IL-1β mRNA were demonstrated by in situ PCR in platelets stimulated by thrombin while adherent to immobilized fibrinogen (96). This observation is consistent with absence of IL-1β mRNA in unclotted whole blood and its accumulation when the blood clots (297), although the IL-1β message is also generated by new transcription and processing in nucleated blood cells such as monocytes under these conditions. In addition to splicing induced by the G protein-coupled receptors that recognize thrombin and other agonists mentioned above, TLR4 (57, 400), TLR2 (400), FcαRI (356), and DC-SIGN (see below) transmit signals that trigger splicing of pre-IL-1β, translation of the mature transcript, and generation of IL-1β protein. Autocrine signaling mediated by newly synthesized IL-1β and IL-1R1 amplifies TLR4-mediated platelet activation and splicing induced by LPS (58). LPS was a more potent agonist for pre-IL-1β splicing than thrombin (400), but these experiments were done in the absence of fibrinogen or fibrin, which would be predicted to influence the relative synthetic outputs because of engagement of integrin αIIbβ3 (see above).
A kinase not previously known to be present in platelets, Clk1, links engagement of surface receptors to activation of the splicing pathway. This was originally discovered in studies of splicing of the pre-mRNA that encodes TF. Pre-TF, like pre-IL-1β, is present in resting human platelets and spliced to the mature transcript in response to cellular activation in a process that requires Clk1 (367, 391, 454). A requirement for Clk1 activity for splicing of pre-IL-1β was subsequently demonstrated (57). Clk1 phosphorylates key spliceosome components, and its inhibition interrupts the platelet splicing pathway (57, 391). The presence of pre-mRNAs and a spliceosome-mediated mechanism of pre-mRNA processing in activated platelets was unanticipated and had not previously been identified outside of the confines of mammalian cell nuclei (96, 291). Nevertheless, splicing of pre-mRNAs by activated platelets has now been observed in additional studies (57, 139, 356, 400) and appears to be a unique posttranscriptional specialization of these anucleate cells.
Translation of the mature, spliced IL-1β mRNA yields the pro-IL-1β precursor protein (∼31 kDa), which must be truncated to yield the smaller (∼17 kDa), biologically active cytokine (Figure 6). A mechanism for processing pro-IL-1β was implied by detection of mature IL-1β in platelets activated in vitro, in their supernatants and microparticles, and in platelets from patients (see above). Accumulation and release of mature IL-1β by activated human platelets occurs over a 1–18 h time course in vitro, depending on agonist and conditions (57, 249). In other cell types, processing of pro-IL-1β to mature IL-1β is accomplished by inflammasomes, which are unique cytosolic multiprotein complexes that mediate capase-1-catalyzed inflammatory responses (94). Inhibition of capase-1 blocked LPS-stimulated accumulation of mature IL-1β in isolated human platelets (57). The presence of a functional platelet inflammasome, suggested by these and other earlier preliminary observations (453), was recently demonstrated. Dengue virus (DENV), which causes clinical thrombocytopenia, vascular leak and shock syndromes, and is a rare cause of alveolar hemorrhage (405), induces synthesis and secretion of IL-1β and shedding of IL-1β-rich platelet microparticles (179) (Figure 6). DENV is recognized by DC-SIGN on human platelets (180, 406) (see sect. IIIB). Components of the nucleotide binding domain leucine-rich repeat containing protein 3 (NLRP3) inflammasome (94) were detected in resting human platelets, and the NLRP3 complex was observed in platelets incubated with DENV in vitro and in platelets from patients infected with dengue (179). In vitro, DENV induced inflammasome activity by a mechanism involving RIP-1/RIP-3 kinases and mitochondrial ROS, resulting in caspase-1-dependent release of soluble and microparticle-associated IL-1β. Microparticles released from DENV-activated platelets induced increased permeability of cultured human microvascular endothelial monolayers that was blocked by IL-IRa (179). The latter findings establish a new mechanism by which dengue infection can induce increased vascular permeability and leak, which are important clinical complications (294, 405). Together, these experiments demonstrated functional inflammasome activity in stimulated human platelets for the first time, broadening the repertoire of molecular mechanisms for immune response in platelets. In other cell types, inflammasome activity is required for cleavage of pro-IL-18 and generation of mature, biologically active IL-18, in addition to production of processed IL-1β (94). In preliminary analysis, IL-18 mRNA is present in the human platelet transcriptome, suggesting that pro-IL-18 and IL-18 may be produced by these cells under some conditions (unpublished data).
Platelet interactions with DENV reveal additional novel and important facets of the capacity for translation and protein synthesis by these cells. After binding to DC-SIGN in vitro, which requires heparin sulfate proteoglycan and is enhanced by thrombin, DENV is internalized by platelets and replicates (406). Viral proteins are then synthesized using the platelet translation machinery and viral RNA as the template. This process, which is blocked by inhibition of platelet protein synthesis, generates complete, infective virus (406). Among the newly synthesized viral proteins is nonstructural protein 1 (NS1), which is present in the blood of patients with clinical dengue and is highly immunogenic (406). Thus “pirating” of platelet translation and protein processing mechanisms by DV may contribute to critical host immune responses to the virus (368, 406).
E. Platelet-Endothelial Cell Interactions
The intricate and important influences of platelets on endothelial barrier function and permeability (Figure 4) were discussed in detail in an earlier section (sect. IIIA). Additional platelet-endothelial cell interactions are also important in physiological conditions, and in pathological inflammation. From the vessel wall side of the equation, endothelial cells release potent factors that inhibit platelet activation and aggregation, including prostacyclin (PGI2) and nitric oxide (NO), and express a surface ectonucleosidase, NTPDase-1 (CD39), that hydrolyzes ADP released from dense granules and prevents it from amplifying platelet responses (23, 89). Platelets amplify local PGI2 synthesis by transferring prostaglandin endoperoxides to endothelium (273, 383).
Endothelial PGI2, NO, and ectonucleosidases prevent platelet adhesion to endothelial cells under physiological conditions (23). Nevertheless, there is evidence that platelet adhesion to endothelium occurs in inflammation and in vascular injury even when the endothelium remains intact, perhaps in part because of alteration or impairment in basal inhibitory mechanisms (66). βTbg released from activated platelets is reported to impair release of PGI2 from endothelial cells, potentially favoring adhesion of platelets, and βTbg and PF4 together appear to inhibit local antithrombotic mechanism (51). Platelets adhere to virally transformed endothelial cells in vitro, in part by mechanisms that are apparently independent of altered PGI release (83). Early in vitro studies indicated that thrombin-stimulated platelets adhere to cultured human umbilical vein endothelial cells (86) and that ovine platelets adhere to activated pulmonary artery endothelial monolayers (210). More recent experiments provide evidence for binding of integrin αIIbβ3 on activated platelets to endothelial intercellular adhesion molecule 1 (ICAM-1) and integrin αvβ3 (40), and for interaction of GPlb-IX-V on platelets with P-selectin and vWF translocated to the surfaces of activated endothelial cells and/or with other endothelial ligands (7, 40, 66, 322, 364).
Activated platelets transmit signals to endothelial cells in injury and inflammation (Figure 7), resulting in endothelial expression of adhesion molecules, chemokines, and other mediators that induce or amplify inflammatory events. As an example, activated platelets induced degranulation of Weibel-Palade bodies and translocation of P-selectin to the surfaces of endothelial cells in mice, resulting in P-selectin-dependent leukocyte rolling (104). In in vitro experiments, platelet-derived IL-1β (see sect. IIID) triggered increased expression of ICAM-1 and vascular endothelial cell adhesion molecule 1 (VCAM-1) by cultured endothelium (57, 135, 160). Platelet IL-1β also stimulated release of IL-6, granulocyte macrophage colony stimulating factor (GM-CSF), IL-8, and monocyte chemotactic protein 1 (MCP) by cultured human endothelial cells (135, 160, 211). Consistent with these responses, IL-1β synthesized and exported by activated platelets induced cultured human endothelium to become adhesive for neutrophils (249), which is dependent on surface display of adhesion molecules and generation of chemokines and other signaling factors by endothelial cells (349). In other observations, platelet CD40L induced expression of E-selectin, ICAM-1, and VCAM-1 and release of IL-8 and MCP-1 by cultured human endothelial cells (166), indicating that activated platelets deliver additional signals for endothelial inflammatory responses. More recently, platelets were reported to induce lung endothelial TF expression in a murine model of acid-induced ALI (115).
FIGURE 7.
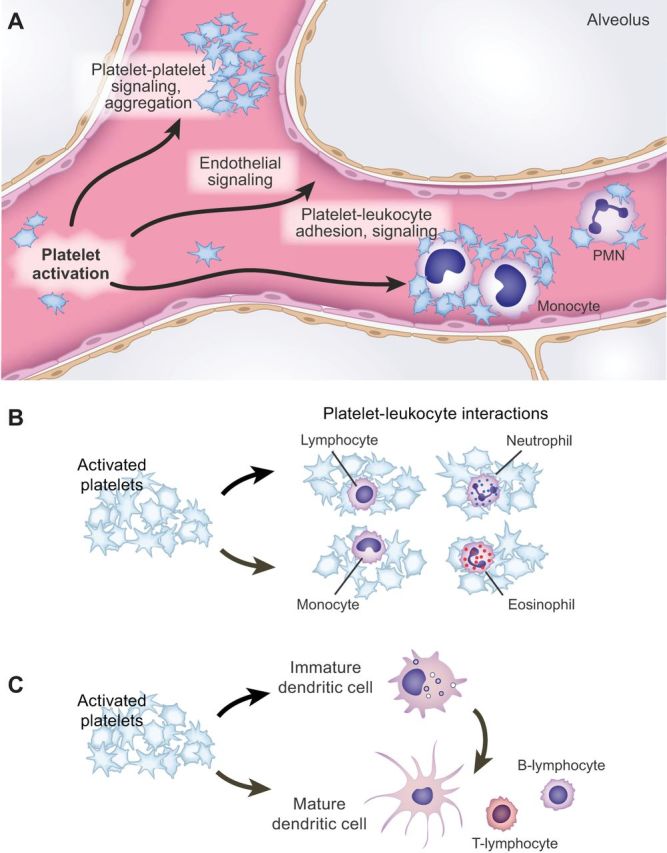
Activated platelets interact with endothelial cells and leukocytes, providing mechanisms for intercellular signaling and induction and amplification of effector responses in inflammation. A: activation of platelets induces platelet-platelet aggregation (see Figure 1B) and intercellular signaling. Platelet aggregation occurs in hemostasis, thrombosis, and inflammation. Platelets and platelet aggregates adhere to and signal endothelial cells in these conditions based on experimental observations. Activated platelets also adhere to leukocytes, forming mixed cell aggregates (“heterotypic aggregates”) that can be retrieved from the blood and detected in vessels and at extravascular sites by histology and intravital microscopy. Platelet-leukocyte aggregates are vehicles for complex signaling and synthesis of inflammatory and hemostatic mediators (see Figure 1C). Microvessels in the lung are sites of platelet-leukocyte aggregate formation and accumulation of platelets and platelet-leukocyte aggregates based on intravital microscopic observations in experimental animals (see FIGURE 5). This may also be an important mechanism in clinical acute lung injury and ARDS. Platelets also interact with leukocytes in clots and thrombi (see Figure 1A). B: activated platelets signal and form aggregates with multiple leukocyte types, including neutrophils (PMNs), eosinophils, monocytes, and lymphocyte subclasses. This is one of several mechanisms that enable platelets to act across the immune continuum, mediate events in the innate and adaptive limbs of the immune system, and link hemostasis and inflammation. C: activated platelets also interact with macrophages (not shown) and dendritic cells, which have multiple regulatory activities in innate and adaptive immune function. Platelets influence dendritic cell differentiation, maturation, and interaction with T- and B-lymphocytes and interact with dendritic cell subsets in the blood. [B and C from Vieira-de-Abreu et al. (454), with permission from Springer Science + Business Media.]
Several studies indicate that platelets can transfer cytosolic mRNAs and microRNAs to endothelial cells or endothelial cell lines in vitro (140, 234, 361). In each case there was evidence for transfer of RNAs in platelet microparticles, and each included evidence for functionality of transferred transcripts. Transfer of argonaute 2 (Ago2)-microRNA complexes with the ability to regulate expression of a reporter gene in endothelial cells was also reported (234). These observations suggest possibilities for complex regulation of endothelial gene expression and phenotype in physiological and inflammatory conditions resulting from transfer of platelet transcripts, made more intricate by the fact that the platelet RNA and microRNA profiles are altered in disease (140, 388).
F. Platelet-Leukocyte Interactions
Activated platelets interact with leukocytes of multiple classes in the circulating blood, in clots and thrombi (Figure 1), and at endothelial surfaces in inflamed vessels (121, 371, 454). Based on evolving observations, platelets also interact with leukocytes at extravascular sites in the lungs and other organs (see below). Circulating myeloid leukocytes (granulocytes, including neutrophils, eosinophils, and basophils; monocytes), lymphocytes of several classes, dendritic cells, and macrophages are all “targets” for activated platelets (454) (Figure 7). Interactions of platelets with circulating myeloid leukocytes are primarily discussed here, and other platelet-leukocyte encounters are considered in a later section. There is evidence that complex platelet-leukocyte interactions, often also involving endothelial cells, occur in both systemic and pulmonary vessels (121, 319, 457).
The cellular and molecular mechanisms that mediate platelet-leukocyte interactions depend on the specific interacting leukocyte type and the experimental or clinical conditions (121, 453, 454). Binding of P-selectin on the surfaces of activated platelets to P-selectin glycoprotein ligand 1 (PSGL-1) on the leukocyte (493) is a critical molecular interaction that has been demonstrated in platelet-leukocyte adhesion involving all circulating leukocyte classes. Additional binding events involving integrins and other adhesion molecules on the activated platelets and on the interacting leukocytes add to critical P-selectin/PSGL-1 tethering in specific cellular interactions (121, 454).
Interactions of activated platelets with neutrophils (PMNs) and monocytes have received considerable attention (454, 494). Recent observations in mice indicate that neutrophils adherent to endothelium in inflamed systemic or pulmonary microvessels locally recruit platelets (173, 195, 196, 281, 415). Capture of platelets by adherent, activated neutrophils has been detected by intravital microscopy in experimental TRALI and LPS-induced ALI (173, 415). Earlier reports in models of antigen-induced and acute sterile inflammation in rabbits also demonstrated neutrophil-mediated accumulation of platelets, including deposition of platelets associated with adherent and emigrating PMNs in the skin and lungs (189, 190, 232). In a recently reported murine model of neutrophil-mediated platelet recruitment, neutrophils were observed to “scan” for activated platelets in the circulation; signals transduced via PSGL-1 (see below) appeared to integrate information required for neutrophil motility and for coordinated interaction with endothelial cells (415). It is unknown if PGI2 or NO generated by locally activated endothelium (see previous section), or ectonucleosidases expressed by endothelium or neutrophils (183), modulate platelet recruitment under these conditions. It is also not clear that this directional cellular interplay is stereotyped in all conditions. In murine models of LPS-induced ALI, septic ALI, and sickle cell injury, platelet-mediated PMN recruitment (457), rather than neutrophil-mediated platelet accumulation, appeared to occur (11, 149, 227, 350, 358). P-selectin-dependent eosinophil recruitment into the lungs by platelets has been observed in murine allergic inflammation (345). In models of leukocyte recruitment in primary hemostasis, platelets adhere to subendothelial proteins, are activated, and recruit neutrophils via mechanisms involving P-selectin/PSGL-1, juxtacrine signaling, and leukocyte β2-integrins (66, 331, 453, 502). Platelet-neutrophil interactions of this nature could occur in inflammation if subendothelium is focally exposed by endothelial retraction or injury (150).
Activated platelets also adhere to leukocytes in circulating blood, forming mixed cell aggregates (sometimes termed heterotypic aggregates) (454). Platelet-leukocyte aggregate formation can be induced in experiments with isolated human cell populations (Figure 1C) or in whole blood samples (453). Formation of platelet-leukocyte conjugates, particularly monocyte-platelet aggregates (MPA), is a particularly useful and durable marker for platelet activation in clinical studies (28). Recent examples of human syndromes in which platelet-leukocyte aggregate formation was detected include aspirin-exacerbated respiratory disease (AERD), HIV infection, influenza, dengue, and severe sepsis and septic shock (146, 178, 235, 365, 366).
The lung is a site of platelet-myeloid leukocyte aggregate formation based on in vivo observations. In mice in which alveolar structures were imagined by two-photon intravital microscopy (256), dynamic formation of neutrophil-platelet aggregates (NPA) was observed in the lung microcirculation in the absence of an inflammatory stimulus (330). Challenge of the animals with LPS resulted in a dramatic increase in the number of NPA and MPA in the pulmonary blood. At later time points, NPA were detected in BAL samples and immobile NPA were observed outside of alveolar vessels by microscopy (Figure 5). The latter findings and additional assays (flow cytometry of BAL samples using an anti-αIIb antibody; BAL TxA2 measurements) indicated that platelets enter alveolar spaces, potentially complexed with leukocytes, in mice with inflamed lungs. In contrast, intra-alveolar platelets and platelet-leukocyte aggregates were not detected in the lungs of control mice (330). Extravascular platelet-leukocyte complexes and platelets have also been reported in the lungs under other conditions (see sect. IV). Accumulation of platelet-leukocyte aggregates and free platelets in inflamed or injured alveoli could have multiple consequences, including fibrin deposition (49, 71, 391) and amplification of inflammation (454). A recent experimental study reported that RANTES (CCL5) released from platelets triggers alveolar macrophage activation, macrophage secretion of IL-8, and neutrophil recruitment to the alveoli in a CLP model (182). Signals to alveolar macrophages by RANTES or other soluble mediators may be delivered by platelets in the alveolar space, by intravascular platelets, or by platelets in both compartments. Platelet-leukocyte aggregates in the peripheral blood, in pulmonary microvessels (Figure 7A), or in the alveolar compartment (Figure 5), have the potential to further amplify and drive inflammation by mediating synthesis of additional mediators (see below).
Aggregation of activated platelets and myeloid leukocytes in the blood also occurs in other models of lung injury. Increased numbers of NPA formed in blood of mice subjected to hydrochloric acid-induced ALI (108, 495). NPA were observed in lung microvessels by electron microscopy in one of these studies, and formation of NPA was dependent on platelet P-selectin (495). Platelet-neutrophil interactions and TxA2 formation were identified as key events in alveolar endothelial injury based on in vitro experiments together with the in vivo observations (495).
In experiments utilizing the acid- or LPS-induced ALI models with relevance to ultimate clinical intervention, aspirin (330, 495), aspirin-induced lipoxins or resolvins (108, 330), or a thromboxane receptor (TP) antagonist (495) reduced NPA formation in vitro and/or in vivo and reduced LPS- or acid-induced ALI. Emerging evidence indicated that lipoxins, resolvins, and related mediators contribute to resolution of acute inflammation in a variety of experimental settings (242). One mechanism for their benefits may be interruption of proinflammatory platelet-leukocyte interactions. Platelet-leukocyte aggregates generate lipoxins (50), however, which potentially have endogenous modulatory effects. Therefore, blocking platelet-leukocyte aggregate formation may not always reduce inflammation in a stereotyped fashion.
Interactions of activated platelets with myeloid leukocytes induce functional responses in addition to simple formation of mixed cell clusters. One functional consequence is activation of leukocyte β2-integrins. This is an important mechanism that can facilitate adhesion and accumulation of PMNs and monocytes in inflamed vessels and tissues (121, 457, 494). Experimental observations cited earlier in this section and a variety of other studies (233, 236, 336) support the conclusion that platelets enhance adhesion and trafficking of myeloid leukocytes under specific conditions. In addition, other functional responses are induced by platelet-myeloid leukocyte interactions. These include transcellular biosynthesis, degranulation and ROS generation by PMNs, fibrin formation, microparticle formation, and vascular remodeling activities (121). Although bidirectional signaling of platelets and leukocytes in whole blood has been detected, most studies have focused on signaling of target leukocytes by activated platelets (454). One important functional outcome is new, or amplified, synthesis of mediators that can drive or modulate inflammatory and immune responses. As an example, NPA, MPA, and eosinophil-platelet aggregates were found in much higher numbers in the blood of patients with AERD than in samples from control subjects, and measurement of a urinary marker of systemic cysteinyl leukotriene (cysLT) formation correlated with the number of NPA (235). Adherent platelets contributed to enhanced leukotriene C4 (LTC4) synthase activity and cysLT production by neutrophils in vitro, and platelets accounted for augmented production of LTC4 by PMNs from patients with AERD compared with cells from control subjects (235). Increased synthesis of LTC4, resulting from such platelet-neutrophil interaction, could further amplify inflammatory and immune responses. For example, LTC4 and related cysLT trigger PAF synthesis and endothelial cell-dependent neutrophil adhesion by human endothelial cells (284). Thus platelet-neutrophil signaling in specific pathophysiological contexts could drive additional key inflammatory events, including augmented neutrophil recruitment and activation.
Platelet-monocyte interactions also result in synthesis of inflammatory proteins by the target leukocytes (reviewed in Refs. 453, 454). This cell-cell dialog has clinical relevance and provides an informative in vitro model of intercellular signaling (Figures 1C AND 7B). Activated human platelets trigger synthesis of chemokines, cytokines, cyclooxygenase-2 (COX-2), metalloproteinases, adhesion molecules, tissue factor, and soluble fms-like kinase (sFlt-1) by isolated human monocytes (269, 454). Early and subsequent experiments demonstrated that engagement of PSGL-1 on human monocytes by platelet P-selectin delivers signals to transcriptional and posttranscriptional pathways that regulate new expression of mRNAs and protein products, and that additional platelet paracrine or juxtacrine factors including RANTES, PAF, and IL-1β can amplify and integrate these signals (103, 153, 266, 454, 461, 464, 466). In some cases, time-dependent multistep mechanisms regulate expression of specific products such as COX-2 (103). Very recent experiments examining platelet-monocyte interactions in dengue indicate that apoptotic platelets utilize P-selectin binding to PSGL-1 and a previously unrecognized mechanism involving exposed phosphatidylserine to differentially trigger synthesis of a specific pattern of chemokines and cytokines by monocytes (178). Other experimental observations also indicate that there are variations in the signaling and output repertoires in platelet-monocyte interactions (109, 151, 423). Studies with anti-platelet and anti-inflammatory drugs suggest that specific molecular checkpoints may be targeted to interrupt synthesis of inflammatory and hemostatic mediators (71, 221, 336, 421, 461), although platelet-monocyte aggregate formation is resistant to conventional anti-platelet therapy under some conditions (486).
Formation of neutrophil extracellular traps (NETs) is a biologic consequence of platelet-neutrophil interactions with much current interest and relevance to lung inflammation. NETs are lattices of extracellular DNA studded with histones, granule enzymes (elastase, cathepsin G, others), and cationic proteins that are extruded by PMNs in infection and inflammation (55, 56, 485). NET formation commonly occurs via a unique cell death pathway often termed “NETosis” that involves nuclear decondensation, nuclear membrane breakdown, and association of histones and granule constituents with the decondensed chromatin in the neutrophil cytoplasm, followed by plasma membrane disruption and extrusion of the NET complexes into the extracellular space (Figure 8). The specific mechanism(s) that mediate NETosis remain to be completely characterized (56, 485). Nonlytic NET formation triggered by certain bacteria and other stimuli has also been described (485). NETs form in pulmonary vessels and in alveoli based on experimental and limited clinical observations (59, 62, 72, 105, 272, 314, 370, 376, 382, 435).
FIGURE 8.

Activated platelets trigger NET formation by neutrophils. Pathogens (bacteria, viruses, fungi, others), chemokines, and other host mediators as well as activated platelets trigger NETosis and NET formation in experimental models. Experimental, and limited clinical, observations indicate that NET formation is a mechanism of extracellular capture, containment, and potential killing of pathogens in vessels and alveoli of the lungs, and in other organs. In addition, however, NET formation can mediate vascular and acute lung injury based on experimental studies. NET-associated factors including histones, neutrophil elastase, and other granule enzymes injure endothelial and alveolar epithelial cells in vitro and in vivo. NETs also induce thrombosis and are components of clots. NET formation may contribute to ARDS and other syndromes of inflammatory injury to the lungs as well as to venous thromboembolism.
NETs capture, immobilize, and in some experimental conditions kill pathogens in vitro, including microbial agents that cause pneumonia and Mycobacterium tuberculosis (56, 485). NETs are proposed to activate lung macrophages in early M. tuberculosis infection (48). Defects in NET formation have been associated with intractable pulmonary fungal infection (33) and neonatal immunodeficiency in humans (489). In a mouse model of Pseudomonas aeruginosa pneumonia, NETs and surfactant protein D coordinately mediated bacterial trapping (105). NETs are proposed to capture and contain pathogens in pulmonary capillaries and other microvessels in bacteremia (72, 281). Thus NET formation may be a key antimicrobial mechanism in pulmonary host defense and in defense of other organs against pathogens (Figure 8), although its relative importance compared with traditional neutrophil mechanisms for intracellular killing of pathogens remains controversial (316, 485). Contributing to both complexity and topical interest, however, there is now considerable experimental evidence that NETs can injure cells and tissues, and that inappropriate or unregulated deployment of NETs by PMNs is a mechanism of inflammatory collateral damage and thrombosis (56, 67, 377, 485). Histones, elastase, myeloperoxidase, and additional NET-associated biochemical agents potently injure several cell types (56, 485). NETs damage alveolar endothelial and epithelial cells in vitro (376), suggesting that NET formation is a mechanism of alveolar capillary membrane injury in vivo (Figure 8). NETs have also been implicated in airway injury in cystic fibrosis (272). Finally, NETs are also prothrombotic (see sect. V). Thus, on the basis of current information in the field, NETs may be effectors of lung injury or of lung defense depending on the experimental or clinical context (67) (Figure 8).
NETosis and NET deployment can be induced by multiple pathogens, by chemokines and inflammatory mediators, and by pharmacological agonists (56, 485). NET formation is also triggered by activated platelets (9, 62, 72, 196, 201, 231, 279, 370) (Figure 8). In vitro, NET formation by human PMNs is induced by platelet signaling factors that include PAF (489), which mediates platelet-dependent neutrophil activation (331); β-defensin-1, an antimicrobial peptide translocated to the surfaces of activated human platelets (231); and high mobility goup box 1 (HMGB1), which is reported to be released from activated human platelets (279). In mice, P-selectin/PSGL-1 signaling (120), and neutrophil activation triggered by platelet RANTES/PF4 heteromers, neutrophil G protein-coupled receptors, and neutrophil β2-integrins (370) induce NET formation. Thus multiple signals from platelets have the potential to induce NET formation based on experimental evidence. In addition, IL-8, which can be generated by intercellular signaling in platelet-monocyte aggregates (461, 466), or when PSGL-1 is engaged on neutrophils (174), is an agonist for NET formation (129) and could induce NET deployment in an inflammatory exudate or thrombus. Extracellular histones associated with NETs can trigger TLR2- and TLR4-dependent platelet activation and aggregation (394), reversing the direction of signal transfer.
NETs are effectors of alveolar damage in mouse models of inflammatory lung injury. In experimental TRALI involving two “hits,” pretreatment with LPS and a subsequent immune trigger, platelets are critical (257) and platelet-dependent NET formation was identified as a key contributor to acute lung dysfunction (62, 435). In one of these studies NET-induced platelet aggregation was found to also be important in the animal model, and NETs were detected in pulmonary vessels in lung tissue from a human subject with fatal TRALI (62). In mice subjected to ventilator-induced lung injury (VILI), which can be considered a pathological consequence of maladaptive inflammation (288) induced by mechanical stress to the lungs, indexes of alveolar damage were dependent on platelets and neutrophils (370). NETs formed in lung microvessels, and circulating NET components were detected in plasma from animals with VILI in this study. In a third model NET formation was platelet dependent and caused inflammatory lung injury in mice with primary graft dysfunction, which is a significant problem in clinical lung transplantation (382). NETs were detected in the lungs by immunofluorescent microscopy in two separate experimental transplant protocols involving ischemia-reperfusion injury, which is thought to be a determinant of primary graft dysfunction (382). In parallel assays, NET components were detected in BAL samples from human lung transplant recipients. In one of the mouse models, involving orthotopic lung transplantation after prolonged cold ischemia, platelet accumulation in lung microvessels was documented by immunohistochemical analysis and was associated with NET formation. Disruption of NETs by intrabronchial administration of DNase improved oxygenation and reduced lung injury as did pretreatment with aspirin before transplant, which also reduced formation of NETs. Together, these surrogate models indicate that NETs have pathophysiological activities in experimental acute lung injury induced by several triggers of deleterious, pathological inflammation: complications of transfusion, mechanical ventilation, and transplantation-induced ischemia and reperfusion.
Experimental models demonstrate that intravascular and extravascular NETs form in endotoxemia, bacteremia, and CLP (72, 77, 281, 290, 429, 475). Platelets are proposed to be important sensors of LPS in bacteremia caused by gram-negative organisms (72, 485), and platelet-induced NET formation is thought to be critical for bacterial containment in the vasculature (281). Nevertheless, NETs can also cause cellular injury as outlined above. Thus NET formation may contribute to septic ALI, a common and lethal form of ARDS (277). Whether NETs are primarily beneficial and adaptive effectors of inflammation that contain and clear pathogens or, conversely, are primarily maladaptive and injurious is an open question in the biology of infection and pathogen-induced lung and systemic injury. The answer is almost certain to be context-specific and will likely depend on whether the model chosen or clinical condition involves live pathogens or toxins such as LPS, whether the pathogens are efficiently cleared by phagocytosis and intracellular killing, and variables such as partitioning of stimuli for NET formation (intravascular versus extravascular) and timing and location of NET deployment. Development of specific inhibitors of NET formation (488) will provide important tools to dissect these issues that may also have therapeutic benefit.
G. Platelets as Innate Sensors and Cellular Effectors in Antimicrobial Host Defense
There is substantial evidence that platelets interact with microbes of a variety of classes (305, 454, 482) (Figure 9). Bacteria and viruses activate platelets directly or indirectly by binding to integrin αIIbβ3, GPIbα, TLRs, Fc receptors, DC-SIGN, and complement receptors (80, 132, 454). Pathogen-dependent activation of platelets may be a key pathophysiological mechanism in host injury in sepsis (93, 247, 369), endocarditis (124), infection of endovascular devices (482), dengue (177), malaria (81, 122, 369), and other infectious syndromes. Platelets were reported to facilitate bacterial dissemination to the lungs and spleen in a model of Streptococcus pyogenes infection (202).
FIGURE 9.

Platelets have sentinel and effector capabilities that allow them to sense and respond to pathogens and microbial products. Platelets utilize a variety of receptors and signal transduction pathways in responses to bacteria, viruses, and other pathogens and have a diverse array of antimicrobial defensive mechanisms. Pathogen-induced platelet activation can also injure the host. Human platelets incubated alone, with Escherichia coli, or with Staphylococcus aureus for 30 min and stained for F-actin and bacterial DNA are shown in this figure. Live E. coli and S. aureus, E. coli LPS, and α-toxin from S. aureus induce a variety of functional responses by platelets in vitro, including signal-dependent protein synthesis (Figure 2). [From Rondina et al. (367).]
While platelets may mediate pathogen-induced injury, there is also evidence that they have a substantial repertoire of antimicrobial activities that benefit the infected host (454, 482) (Table 4). Some of the evidence for platelet defenses against pathogens comes from correlative clinical investigation. For example, patients with idiopathic thrombocytopenia, a syndrome of immune-mediated platelet deficiency, have a substantial risk of infectious complications that is similar to the risk of hemorrhage (323, 352). While in this case the risk of infection may be in part due to complications of treatment, additional clinical data indicate that thrombocytopenia or platelet dysfunction contributes to impaired host defense against infection in a variety of clinical settings (482). Extensive experimental evidence from in vitro and in vivo studies also supports physiologically important antimicrobial activities of platelets (453, 454). As an example, mice with selective genetic deficiency of factor V (FV) in platelets had dramatically increased mortality after group A streptococcal infection as did animals with deficient FV in the plasma compartment (426). The authors of this study concluded that local thrombin generation, fibrin deposition, and platelet-dependent enhancement of these events are key mechanisms that limit dissemination and, perhaps, replication of certain pathogens. As a second example, platelet TLR7 was critical for survival in a murine model of acute viral infection, providing mechanistic experimental evidence indicating that platelets are active in defense against viral pathogens (228). Similarly, platelet depletion in mice infected with lymphocytic choriomeningitis arenavirus, a model of viral hemorrhagic fevers, demonstrated that platelets have complex protective innate and adaptive immune activities in infection with this pathogen (261) (also see sect. IIIH). These and other specific experimental observations, together with clinical correlates, indicate that platelets are innate cellular effectors of antimicrobial host defense (305, 454, 482).
Table 4.
Platelets mediate host responses to microbes
| Biologic Features and Activities of Platelets |
|---|
| Binding, activation by, and in some cases internalization of, certain bacteria and other microbes |
| Expression of multiple functional Toll-like receptors |
| Signaling and enhancement of antimicrobial functions of leukocytes; presentation of bacteria to macrophages and dendritic cells |
| Triggering of NET formation by PMNs |
| Expression and in some cases release of platelet microbicidal proteins, kinocidins, β-defensin-1, and proinflammatory chemokines, cytokines |
| Complex interactions in clinical and experimental bacterial, viral, and protozoal infections |
This table includes a partial list of biologic features and activities of platelets that indicate that they are sensors and cellular effectors in host responses to microbes. Evidence for this comes from studies of both humans and mice. See text and cited references for details and additional information, including examples of platelet activities in complex models of pathogen invasion and pathogen-induced lung injury.
The antimicrobial repertoire of platelets is diverse. One mechanism is enhancement of antimicrobial functions of leukocytes. In earlier sections we mentioned recent experimental studies in which murine platelets present intravascular bacteria to macrophages or dendritic cells for elimination and immune recognition (449, 478) and also outlined recent studies demonstrating that platelets trigger NET formation, a newly recognized innate mechanism of extracellular capture and potential killing of microbes in the lungs and other organs (see sect. IIIF) (Figure 8). In an example in a mouse model, depletion of platelets resulted in increased dissemination of bacteria to the lungs after intraperitoneal E. coli infection, an outcome that was attributed to deficient NET formation and pathogen capture (281). Platelets also enhance a variety of additional antimicrobial activities of neutrophils, monocytes, and lymphocytes (482).
In addition to facilitating control of pathogens by leukocytes, platelets have direct antimicrobial effects. In some cases platelets are reported to phagocytose bacteria and viruses (454). This may, however, not be a particularly robust mechanism, and platelets are inefficient at intracellular killing of at least some bacteria (471). Nevertheless, they store, release, and in some cases retain on their surfaces peptides, proteins, and chemokines with potent antimicrobial activities. Among them are platelet microbicidal proteins (PMPs) and kinocidins, which are ancient immune effector molecules that have antibacterial and antifungal activities and that potentiate the action of some antibiotics (473, 482). PMPs and kinocidins have been identified in platelets from humans and experimental animals and have direct and synergistic activities against bacteria and fungi that cause clinical infections, including pneumonia. Kinocidins are classified in at least five subfamilies, which include chemokines (PF4, NAP-2, RANTES, others) that have antimicrobial activities (482). PF4, for example, kills intraerythrocytic Plasmodium falciparum after binding to the Duffy antigen on infected erythrocytes (287) (see sect. IIIH), and inhibits HIV cellular entry and infection (15). In addition to PMPs and kinocidins, platelets also express β-defensin 1, which inhibits growth of S. aureus in vitro in addition to triggering deployment of NETs (231). Yet another facet of platelet antimicrobial defense is illustrated by release of histidine-rich glycoprotein (HRG), which is synthesized in the liver, internalized by platelets, and stored in α-granules. HRG promoted entrapment of Streptococcus pyogenes in clots in vitro and at infected sites in vivo in a mouse model (399). Clinical and experimental observations of this sort provide evidence that platelets are active contributors to host defense against pathogens, although some microbes can subvert platelet molecular defenses (482).
Evolving evidence demonstrates that platelets are sensors of pathogens and are among the first responders in acute infection (132, 454). Platelets are the earliest and most numerous cells to accumulate at sites of infection involving the endothelium and in infected endovascular devices (482), suggesting rapid sensing of intravascular infection and pathogen-induced vascular injury. Platelets are proposed to be “barometers” of bloodstream invasion by bacteria (72, 281) and to be key cellular effectors in intravascular immune responses to blood-borne pathogens (156) (also see sect. IIIH). As previously noted, platelet TLRs provide molecular pathways for intravascular pathogen sensing, but other platelet receptors also have this function (80). While circulating blood is the principal domain of their microbial sensing, surveillance, and effector functions, platelets also accumulate in infected issues and have the capacity to detect microbes outside of the blood compartment and to influence the natural history of extravascular infections. There is experimental evidence that they may specifically influence outcomes in parenchymal lung infection. In a recent study in mice, an anti-platelet antibody was used to reduce circulating platelet numbers to very low (2–5 × 109/liter over 14–44 h) or low (6–153 × 109/liter over the same interval) levels (92). Mice with very low platelet numbers had dramatically and progressively increased bacterial loads in the lungs after intranasal inoculation with Klebsiella pneumoniae; increased bacterial burdens in blood, spleen, and liver; and increased mortality. Animals with less severe thrombocytopenia had a similar, but less pronounced, pattern of lung and systemic infection. In contrast to results in previously cited models of LPS-induced or sterile inflammation, platelet depletion did not reduce accumulation of neutrophils in the lungs or NET formation in this model of pneumonia and pneumonia-induced sepsis (92). This suggests that innate antimicrobial or signaling activities of platelets were central to pathogen control and to survival of platelet-sufficient animals. Unexpectedly, measurements in thrombocytopenic, infected mice demonstrated enhanced cytokine levels. It is possible that this result was due to impaired inhibitory signaling of macrophages by platelets (479) or differential signaling of monocytes (151) in severely thrombocytopenic animals. The investigators also note that the biologic effects of platelets likely depend on the stimulus and the elapsed time after challenge in models of infection or inflammation, and provide appropriate caveats regarding comparisons of animal models to clinical conditions they are intended to imitate (92).
Examination of platelet activities in experimental and clinical viral syndromes has revealed previously unrecognized pathogen sensing and effector functions. It is now clear that platelets recognize and interact directly with a number of viruses, although in many cases the information is fragmentary and incomplete (454, 482). As with bacteria, plasmodia, and other pathogens that are blood-borne, platelets sense and respond to viruses such as dengue that cause significant clinical viremia (177) (also see sect. IIIF) or are introduced into the bloodstream experimentally (196). In contrast, influenza, a major cause of viral pneumonia and ARDS (277) (Figure 3), induces platelet responses even though the pathogen is dominantly restricted to the alveoli and airways, and is particularly interesting and informative in this regard.
Activated platelets and platelet-monocyte aggregates are present in increased numbers in the blood of critically ill patients with H1N1 influenza A (365). Platelet-monocyte aggregates also form in the blood of subjects undergoing influenza A vaccination, and in vitro experiments suggested that this interaction expands a subset of CD14high CD16+ proinflammatory monocytes (336). Early studies demonstrated that live influenza virus binds to human platelets in vitro, although a specific receptor or binding structure was not identified (433). In addition, infusion of influenza virus into rabbits induced rapid and sustained thrombocytopenia and decreased survival of radiolabeled platelets (433). Recent observations indicate that influenza viruses can activate platelets in a complex fashion. H1N1 influenza A triggered human platelet activation in vitro, detected by translocation of P-selectin, activation of integrin αIIbβ3, microparticle formation, and lipid mediator synthesis (38). The activation mechanism required the presence of serum or plasma and was found to involve formation of virus-IgG immune complexes that are recognized by platelet FcγRIIA. Consistent with this, the pathway could be reproduced in platelets from transgenic mice that express human FcγRIIA (38). Of note, immune complexes containing viral antigen were found in the lungs of patients who died of influenza during the 2009 H1N1 pandemic (301). In addition, FcγRIIA also participates in complex platelet activation by other pathogens (124). The experimental influenza studies further indicated that human platelets have a parallel mechanism by which the virus can induce activation via thrombin formation, presumably mediated by platelet PARs, independent of FcγRIIA and immune complexes (38). This was interpreted as demonstrating that influenza A activates human platelets by both adaptive and innate signaling cascades, in part depending on the immune status of the subject (38). It was proposed that influenza virus is recognized by platelets in systemic blood during viremia, which is detected relatively infrequently in clinical influenza A infection, but may increase in pandemic influenza (70, 248), or in leaky capillaries with increased permeability in the lungs of infected patients (38) (Figure 3). Animal models document increased permeability of the alveolar-capillary membrane in influenza lung infection and indicate that disrupted barrier function and increased capillary permeability can be prolonged (145, 254), consistent with clinical observations (277). If platelets encounter influenza virus in the lung microcirculation as a result of increased microvascular permeability, it will bring a new facet to the functional consequences of altered endothelial barrier integrity in inflammation (see sect. IIIA). There may be alternative mechanisms for platelet activation in severe influenza pneumonia and influenza-induced ARDS. For example, generation of one or more mediators in the lungs or blood that activates megakaryocytes or circulating platelets independent of direct contact with the virus may occur.
Additional recent studies of experimental influenza A infection also support platelet participation. Activated platelets and platelet-leukocyte aggregates were found in the blood of infected mice (238), consistent with observations in humans (365). Platelet activation and formation of platelet-leukocyte aggregates may contribute to thrombocytopenia in influenza, as they do in other viral infections (292). Histopathological studies of influenza A-infected mice demonstrated accumulation of platelets and platelet-leukocyte aggregates in the vascular and extravascular compartments of the lungs (238). In addition, time-dependent accumulation of platelets and serotonin and IL-1β, which could have in part been platelet derived, were detected in BAL samples from these animals (238). Platelets were also found in BAL samples from influenza-infected mice in the previously outlined study (38). Together, these observations add to the idea that platelets are direct or indirect sensors of influenza in vivo and suggest that they can encounter the virus in the alveoli as well as in the blood. There was also evidence that platelets directly contribute to collateral inflammatory lung injury triggered by influenza. Genetic deficiency of integrin αIIbβ3, an αIIbβ3 inhibitor, and pharmacological inhibitors of platelet activation reduced histologic indexes of lung injury and mortality in influenza-infected mice (238).
Whether platelets are protective very early in influenza invasion, as in other experimental infections (see sect. IIIH), is unknown. It is also unknown how platelets influence bacterial pneumonia complicating clinical influenza infection, which is a common and lethal problem (68). As illustrated earlier in this section, platelets have complex activities in bacterial pneumonia that are likely to be compounded by concurrent viral infection. Both bacterial coinfection (68) and thrombocytopenia (258) are reported to be risk factors for increased mortality in clinical influenza infection. Finally, it is unknown how lung megakaryocytes (468) respond to influenza. Both lung megakaryocytes and circulating platelets may sense and respond to influenza and other respiratory viruses.
In addition to bacteria and viruses, platelets interact with fungi, protozoa, spirochetes, and schistosomes (454, 482). Pathogens from each class cause platelet accumulation in lung vessels or pulmonary thrombosis in humans or experimental animals, and in some cases platelet interactions are proposed to be central to lung defense or injury when the host is challenged by these microbes (412, 446). There is little information on platelet sensing or effector responses when the lungs are involved in clinical infections with these agents or in surrogate models, however, leaving gaps in our knowledge and identifying opportunities for future investigation.
Platelet responses to pathogens will also be discussed in the next section, which deals with integrated innate and adaptive immune functions of these cells. Microbial challenge has commonly revealed previously unrecognized immune and inflammatory mechanisms involving a variety of cell types, and has been informative in the immunobiology of platelets in this regard.
H. Platelets as Sensors and Effectors in the Immune Continuum
Platelets clearly function as innate immune cells that detect and respond to infectious and sterile triggers and inducers of inflammation (288) with effector activities (80, 169, 194, 305, 396, 450, 453, 454, 465). Platelets mediate events that are classically associated with innate immune responses, including altered endothelial permeability (369), histamine release (54, 61, 487), complement activation (450), and rapid accumulation of myeloid leukocytes (454) (see earlier sections). Many of the activities of platelets so far discussed in this review can be considered innate immune effector functions, or are parallel hemostatic and innate immune responses (Tables 1–5). Innate immune functions of platelets can clearly be demonstrated in the lungs. As examples, platelets and leukocytes have rapid and coordinate innate immune effector activities in the lungs in experimental models of acute inflammation and immune challenge (46, 189, 190, 442, 487).
As previously introduced, there is evolving evidence that activated platelets also have adaptive immune functions and activities (Table 5) (111, 113, 169, 397, 453, 454, 469). As discussed in section IIIG, in some cases platelet innate and adaptive immune receptors and pathways are triggered in response to the same stimulus (38). In this section we review some of the evidence that platelets operate across the immune continuum and that both adaptive and innate immune activities are in their repertoire.
Table 5.
Platelets act across the immune continuum
| Features of Platelets |
|---|
| Innate immune activities |
| Interaction with pathogens |
| Alterations of endothelial permeability and vascular barrier function |
| Histamine release |
| Complement activation |
| Chemokine, cytokine release |
| Endothelial signaling |
| Targeting, signaling, trafficking of PMNs, monocytes |
| Adaptive immune features and activities |
| CD40L, chemokine, cytokine release; signaling of other immune effector cells |
| T lymphocyte targeting, trafficking, activation, differentiation |
| Alterations in B-cell function, antibody generation, germinal center formation |
| Dendritic cell differentiation, activation, altered function |
| MHCI, costimulatory molecule, and immunoproteasome expression (antigen presentation, MHCI-dependent T-cell activation in one study) |
| Modulation of lymphangiogenesis and lymphatic endothelial barrier integrity |
Some of the reported innate and adaptive activities and phenotypic features of platelets are listed. In many cases, key observations come from studies of mice, although there is also substantial evidence for complex immune activities from studies of human platelets and human cell models. A variety of findings indicate that platelets bridge the innate and adaptive limbs of the immune continuum in specific conditions, thereby mediating information transfer in immune recognition and activation. See text and cited references for additional information and details.
Activated platelets interact with leukocytes that mediate adaptive immune responses, some of which have broad influences at the interface of innate and adaptive immunity. These include lymphocytes and dendritic cells (Figure 7, B AND C), which have protean adaptive immune activities. Activated platelets bind to lymphocytes of several subclasses in vitro and in vivo. As in interactions with PMNs and monocytes, P-selectin on activated platelets mediates both adhesive and signaling functions in platelet-lymphocyte interactions (169, 245, 454). Early and more recent experiments demonstrate that in addition to P-selectin/PSGL-1 binding (87, 302), integrins and CD40L are involved in adhesion of platelets to lymphocyte subsets depending in part on the activation state of the lymphocyte and other conditions (98–101, 411). Circulating activated platelets were found to deliver lymphocytes to HEV and to facilitate lymphocyte homing and immune responses in the skin in mouse models (98, 100), indicating the importance of this platelet-leukocyte interaction. In one of these studies activated platelets enabled lymphocytes to tether to peripheral node addressin, a key adhesion molecule on HEV, in the absence of functional lymphocyte L-selectin, a critical “homing receptor” (98). This mechanism may be important in targeted trafficking of memory T cells, which constitutively express low levels of L-selectin, in nodes (169, 245). There is experimental evidence for platelet-mediated targeting of lymphocytes to the lungs. In a murine model of allergic pulmonary inflammation involving sequential administration of systemic and aerosolized ovalbumin, platelets were required for maximal recruitment of both lymphocytes and eosinophils (345). Platelet P-selectin, PSGL-1, leukocyte integrins, and the formation of platelet-leukocyte complexes were identified as key factors in targeting of the leukocytes to the lungs.
Early and more recent observations indicate that platelet-lymphocyte interactions have functional implications for both cell types (64, 74, 110, 112, 114, 138, 161, 184, 185, 217, 275, 491). These studies also suggest maladaptive, as well as adaptive, immune consequences of interactions between lymphocytes and platelets. As examples, human platelets are reported to initiate T cell-dependent contact hypersensitivity mediated by serotonin release (275), and platelets contributed to cytotoxic T lymphocyte-induced hepatic damage in a mouse model of viral hepatitis (184). Murine platelets present antigens and directly activate naive T cells, activities that may have both injurious and protective immune consequences, in experimental malaria (64) (see also below). Reciprocal interactions between platelets and lymphocytes mediated by CD40L-CD40, adhesion molecules, and paracrine signaling have been reported (88, 245, 491). In one study, T cells were observed to induce RANTES release by platelets which was dependent on CD40L-CD40 recognition, providing a mechanism for immune amplification (88).
Multiple observations, largely in vitro, indicate that platelets influence differentiation of monocytes to dendritic cells (DC) and induce maturation of DC (85, 114, 152, 154, 175, 207, 220, 237, 243, 274) (Figure 7C). DC are central in antigen and pathogen recognition, integration of innate and adaptive immune activities, immune tolerance, and in pathological states, autoimmune disease and allergy (16, 422). P-selectin- and CD40L-dependent mechanisms, and both adhesion-dependent and paracrine signaling, have been reported in platelet-induced DC differentiation and maturation (454). Serotonin induces monocyte differentiation to DC (216) and may have this activity if released by activated platelets in immune events.
In in vitro and in vivo models involving both static and shear conditions, activated human platelets adherent to collagen recruited DC by molecular interactions involving PSGL-1 and integrin αMβ2 on the DC; in mice with injured carotid arteries, DC adhesion to exposed subintima in the vessel was dependent on platelets and was inhibited by soluble GPVI (237). In addition, interaction of human platelets and DC in vitro was associated with phagocytosis of platelets and DC apoptosis (237). In a different study, activated human platelets adhered to immature DC in suspension, and P-selectin/PSGL1 and platelet and DC integrins mediated DC tethering to platelets adherent to collagen; myeloid DC were reported to prune activated platelets from the margins of platelet aggregates under these conditions (267).
Under a variety of experimental conditions, platelets were found to modulate activation and defensive responses of DC including release of proinflammatory cytokines (152, 154, 175, 220, 237). Activated platelets also enhanced lymphocyte proliferation induced by DC in vitro (152, 237). β-Defensins are proposed to link innate and adaptive immunity by activating DC and T cells (480), suggesting that β-defensin 1 on the platelet surface (231) might have this activity. In an in vivo model in mice mentioned in previous sections, platelets recognized Listeria monocytogenes in the bloodstream and shuttled a fraction of bound bacteria to splenic DCs, a mechanism proposed to be important in clearing the blood of pathogens and inducing antibacterial immunity (449). Platelet interactions with DC may also contribute to immune disease. Platelets, apparently activated by immune complexes via FcγRIIA, formed aggregates with plasmacytoid DC and monocytes in the blood of patients with systemic lupus erythematosus (SLE), and activated platelets were observed to trigger interferon-α secretion by plasmacytoid DC (106). It is possible that interaction of activated platelets with plasmacytoid DC and monocytes in this fashion contributes to immune lung injury in SLE (453). Together, these observations demonstrate that activated platelets adhere to and signal DC in vitro, in vivo, and in clinical immune responses.
Macrophages, like DC, have diverse immune and immunoregulatory functions (306, 309). Activated platelets and platelet lysates induced macrophage differentiation in coculture, a response that appeared to be due to one or more platelet membrane lipids (4). PF4 was reported to induce monocyte-to-macrophage differentiation (385), suggesting a different mechanism by which activated platelets may influence this process. In contrast, purified, immobilized P-selectin inhibited monocyte-to-macrophage differentiation in response to macrophage colony stimulating factor (M-CSF) and favored differentiation of a CD14+ CD16+ dendritic cell-like population (243), suggesting that activated platelets retard macrophage development under some conditions and that platelets may differentially regulate the cell fate of monocytes and their evolution to macrophages and DC along divergent pathways. Platelets also interact with and signal mature macrophages based on in vivo and in vitro experiments (182, 195, 392, 478, 479). It is unknown if platelets influence dynamic changes in the functional phenotypes of macrophages (306, 309) by these or other mechanisms. PF4 induces a unique transcriptome signature in cultured macrophages (143) and downregulates macrophage surface receptors (142), suggesting this possibility. Murine peritoneal and bone marrow-derived macrophages treated with LPS express PDPN, which can activate mouse platelets via CLEC2 in vitro (218). This suggests that CLEC2-mediated platelet responses may occur in settings other than lymphangiogenesis (see sect. IIIA).
Platelets influence complex immune responses that mediate innate and adaptive events in vivo, in part by interacting with individual leukocyte types that operate across the immune continuum (circulating monocytes, lymphocytes, DC, macrophages). In this context platelets perform sentinel (detection) and effector functions and appear in specific cases to bridge innate and adaptive limbs of immune recognition and activation. Many of the observations that support this conclusion come from murine models. Platelets control leukocyte recruitment and cytokine generation in the cutaneous Arthus reaction in mice, which is a model of immune complex-mediated inflammation (155). In an early series of experiments utilizing mice challenged with adenoviral vectors after prior immunization, activated platelets induced DC maturation, delivered signals leading to IgM to IgG isotype switching of B cells, and enhanced T-cell effector responses. These events were dependent on platelet CD40L (114). The experimental approaches included adoptive transfer of platelets from wild-type and CD40L-deficient mice and parallel in vitro protocols. The observations indicated that platelets act as sensors of viral antigen and as a cellular communication link between innate and adaptive immune compartments in this model (110, 114, 469). In additional experiments this group also reported that CD40L-mediated signaling by activated platelets or platelet microparticles contributes to lymph node germinal center formation and augmented efficiency of T cell-dependent antibody production (110), and provided evidence that activated platelets can facilitate humoral adaptive immune responses when antigen-specific B- and T-cell numbers are limiting. Here, information transfer to splenic B-cell compartments by CD40L on microparticles released by activated platelets was identified as a key feature of the mechanism (414). These and related observations identified platelet CD40L as a central signal in experimental adaptive immune responses of this nature (111, 113). In parallel studies, platelet CD40L facilitated priming of T cells and protection against experimental Lysteria monocytogenes infection (112), indicating sensing and effector functions of platelets in bacterial infection as well as in challenge with virus and viral antigens. A considerable body of information now indicates that platelets utilize CD40L for both innate and adaptive immune activities (97, 111, 454). Platelet CD40L may be a key determinant of transfusion immunomodulation (36).
Additional examples illustrate complex activities of platelets in innate and adaptive immune responses to infection. Experimental and clinical observations indicate that platelets are sensors of malaria infection and that they are effectors of both host protection and, in severe malarial syndromes, injury (137, 147, 453). Human platelets inhibit P. falciparum growth in vitro (343) and kill intraerythrocytic malarial parasites by a mechanism involving PF4 (262, 286, 287). These observations suggest that platelets defend the host early in malaria infection. Nevertheless, considerable evidence implicates platelets in the injurious vasculopathy of cerebral malaria, one of the most important and lethal malarial complications (81, 122, 453).
Studies in a widely utilized mouse model of severe malaria in which the animals are infected with blood stage P. berghei ANKA (PbA) indicate that platelets have both innate and adaptive immune activities that are defensive or damaging, in part depending on timing after challenge with the pathogen (1). Platelets were activated early after infection of mice with PbA, suggesting that they directly or indirectly sense the infection. Mice in which platelets were depleted prior to infection had increased parasitemia compared with control animals and high mortality, as did mice that received the platelet inhibitor clopidogrel prior to infection. In contrast, mice depleted of platelets or that received the anti-platelet drug beginning 24 h after infection had substantially increased survival. The experiments were interpreted as indicating that platelets have an early protective effect involving innate immune mechanisms but that later in the course of the infection, with ongoing platelet activation, maladaptive platelet effector activities contribute to cerebral malaria and death (1). Early innate protection involved, in part, platelet signaling that enhanced production of acute phase proteins by the liver, based on platelet depletion experiments. Platelet-derived and/or induced IL-1β was key in signaling this acute phase response. In addition, contact-dependent interaction of activated platelets with liver sinusoidal endothelial cells was thought to be important, a conclusion supported indirectly by hepatic accumulation of labeled platelets and microscopic analysis (1). The platelet-induced generation of acute phase proteins limited plasmodium growth and appeared to be critical in protecting mice from cerebral injury. The late injurious effects of platelets, which were blunted by platelet depletion or treatment with clopidogrel after infection, were proposed to result from an inflammatory environment established by circulating activated platelets, based in part on earlier experiments indicating that PF4 can drive inflammation in this model (416). Thus PF4, and platelets as its cellular source, may have both protective (286, 287, 343) and injurious (416) activities in experimental malaria. Platelets also regulate cytokine levels in this model (447). Because PbA infection causes acute lung injury in addition to cerebral involvement, the observations may be relevant to malaria-associated ARDS (432, 446).
Experimental malaria also provides evidence that platelets act across the immune continuum and mediate adaptive responses beyond innate protection involving induction of acute phase proteins and other acute events. Platelets were found to process and present plasmodium-derived antigen and to directly activate naive T cells in a platelet MHC class I (MHCI)-dependent fashion in the P. berghei model (64). In vitro as well as in vivo experiments supported this conclusion. The possibility that platelets not only facilitate acquired immune responses carried out by other immune effector cells (454) but also directly participate in antigen recognition and presentation (64) broadens their potential impact in immunobiology (305). Human platelets express MHCI subunits including HLA class I and β2-microglobulin (13, 208, 372, 379), as do murine platelets (64). Platelets also express T cell costimulatory molecules, an immunoproteasome, and other key components required for antigen processing and presentation (64, 222). In PbA infection, a population of platelets with greatly increased surface levels of MHCI emerges several days after blood stage challenge with the parasite (64). This likely involves changes in the megakaryocyte and/or platelet transcriptome (see sect. IV), although MHCI components are reported to adsorb to the platelet plasma membrane (208). An intriguing possibility from the PbA model is that late interactions of activated platelets and T cells involving antigen presentation may yield immune protection (64) and complement other protective mechanisms such as early acute phase protein synthesis (1) if the infected subject survives acute and subacute complications such as cerebral and lung involvement. The studies also suggest that platelets might be used in cell-based vaccine strategies (64). Such an outcome would expand the roles of platelets in intravascular immunity, which is proposed to result from encounters of immune effector cells with pathogens in the blood (172).
Murine models also suggest additional complex immune functions of platelets. As outlined earlier, platelets facilitate lymphangiogenesis and defend lymph node integrity, which are important in adaptive immune regulation. A model of viral hemorrhagic fever mentioned previously indicates that platelets are also important for splenic integrity and that platelet deficiency alters splenic immune function. In these experiments, platelet numbers were reduced to profoundly low (<2.5% of baseline) or low (<15% of baseline) levels prior to infection with lymphocytic choriomeningitis arenavirus (261). Mice with profoundly low platelet numbers had systemic bleeding with focal hemorrhages in the lungs and other organs and significant mortality, whereas animals with low or normal platelet numbers had no bleeding and survived. Nevertheless, both depletion protocols resulted in impaired clearance of the virus and deficient virus-specific T-cell responses. Platelet-depleted mice had dramatic disruption of splenic architecture, histologic evidence for diffuse viral replication throughout the splenic parenchyma, and reduced numbers of splenic immune effector cells, including DC, macrophages, and T cells, that apparently resulted from cell necrosis. The investigators concluded that platelet-depleted mice were unable to control viral replication due to generalized necrotic depletion of splenic innate and adaptive immune cells, although a specific mechanism was not determined (261). A corollary was that platelet depletion resulted in loss of viral control secondary to accumulated defects in early innate and late adaptive immune responses, and that this occurred even in the animals with only partial platelet depletion. This model, like experimental malaria, provides additional evidence that platelets act across the immune continuum. It is unknown how these experimental murine models translate to human biology and to clinical infectious diseases.
IV. NONTRADITIONAL BIOLOGIC ACTIVITIES OF PLATELETS AND THE PLATELET TRANSCRIPTOME: A SUMMARY
This review has profiled traditional, well-known effector activities of activated platelets–shape change, inside-out signaling of integrin αIIbβ3, aggregation, synthesis of TxA2, degranulation of factors with hemostatic activities, enhanced adhesion, clot retraction–in parallel with an increasing number of important biologic activities that are nontraditional and are newly, or relatively newly, recognized (Figure 2). Within this context we have discussed receptors, signaling cascades, regulatory units such as the inflammasome, and effector functions that have been identified in platelets only in the recent past. Innate and adaptive immune activities are among the “new”, nontraditional biologic capacities of platelets (453, 454, 500), although these functions are likely quite ancient (465), and it has been recognized that platelets are inflammatory cells for a substantial period of time (311, 333). We have also mentioned other recently identified features of platelets that potentially broaden their biologic impact beyond the traditional short list of hemostatic functions, but have not provided a comprehensive review of the new biology of platelets. For example, platelets and platelet precursors can divide and generate progeny after they have been spawned by parent megakaryocytes (390, 436), a recently discovered facet of the new biology of platelets that is not discussed here.
The ability to act outside of the vascular compartment is one of the most interesting nontraditional platelet functions to emerge from recent and earlier studies. As discussed in other sections, this can be accomplished by release of soluble paracrine factors and by shedding of bioactive microparticles by activated platelets. Several examples cited earlier emphasize that platelet microparticles have the potential to mediate diverse physiological and pathophysiological events both within and outside of vessels (37, 57, 140, 179, 234, 249, 361, 391, 414). The ability of platelet microparticles to deliver signals to extravascular cells and drive inflammation in extravascular compartments was demonstrated in lymph nodes (414) and in experimental arthritis (37), but may also extend to the lung. For example, GP VI-mediated shedding of microparticles by platelets adhering to exposed collagen, as occurs in inflamed joints in mice (37), might occur in inflamed or injured lungs and result in microparticle delivery to alveoli.
In addition to the possibility that platelet microparticles or soluble paracrine factors from platelets gain access to the alveolar space, recent studies indicate a third mechanism by which platelets may act in extravascular compartments in the lung: as previously noted, apparently intact platelets, alone or adherent to leukocytes, were found in the alveoli of animals with inflamed or infected lungs (38, 238, 330) (Figure 5) (see sect. III, F and G). Provocatively, the classic electron micrographs of the lungs of humans with ARDS by Bachofen and Weibel include an image showing a “thrombocyte” that appears to be extravasating from an alveolar vessel and another micrograph showing intra-alveolar thrombocytes in the lung of a patient who developed ARDS after multiple trauma and shock (18). Extravasation of platelets was also reported in early studies of bronchial biopsies of patients with asthma (193) and in inflamed airways in experimental and clinical samples (235, 344). In addition, platelets were reported to be present in BAL samples from asthmatic subjects, and in BAL fluid from mice and rabbits in models of allergic lung inflammation (293, 346). Furthermore, platelets were found in inflamed synovial fluid from human subjects in limited clinical reports (453, 481). Several studies report that platelets can migrate in vitro and exit vessels in vivo in experimental models (84, 123, 230, 344). Although active entry of platelets into inflammatory exudates has also been questioned based on experimental modeling (39), together these observations suggest that intact activated platelets can exit vessels in inflamed alveoli, airways, and other tissues under some conditions. In addition, they may be translocated to extravascular compartments of the lung while associated with migrating leukocytes (238, 330). The possibility that extravasated platelets can mediate intercellular signaling or carry out other activities in extravascular compartments of the lungs is provocative, although the functional state of extravascular platelets has not been examined to date. This issue should yield interesting observations in the future. It is also unknown if platelets in inflamed alveoli or at other extravascular sites can reenter the circulation, as has been reported for neutrophils (76).
Among the most significant facets of the newly recognized biology of platelets are the importance of platelet genomics (53), and the size and diversity of the platelet transcriptome (373, 388). Features of the platelet transcriptome and its influence on dynamic changes in the proteome of activated platelets were profiled in section IIID dealing with signal-dependent translation of mRNAs. That discussion does not comprehensively detail the unexpected extent and complexity of the platelet transcriptome as revealed by RNA-seq and other approaches (372, 373, 388), however. Of note, there are differences, in addition to similarities, in the mouse and human platelet transcriptomes (372).
Functional information is emerging rapidly in parallel with charting of the platelet transcriptome. Ongoing analysis of platelet mRNA expression profiles provides insight into the basis for differences in platelet reactivity and function between individuals (53, 388). In a recent study that serves as an example, differences in expression levels of an mRNA, phosphatidylcholine transfer protein (PCTP), and a regulatory microRNA, miR-376c, were found to influence PAR4-mediated platelet reactivity in subjects of different races (107). Platelets express an unexpectedly rich library of microRNAs (355), providing the basis for precise and intricate posttranscriptional fine-tuning of their phenotype and function. As noted earlier, platelets can mediate microRNA and mRNA transfer to leukocytes and endothelial cells in vitro (see sect. IIIE), suggesting that they may influence gene expression in interacting nucleated cells in a transcellular fashion. It is also possible that platelets internalize transcripts originally generated by other cell types. Platelet mRNAs and microRNAs are biomarkers, and there is evolving evidence that their profiles change in thrombotic and inflammatory diseases and that these patterns establish molecular signatures for specific disorders and syndromes (388). An obvious, but critical, point is that megakaryocytes exert dominant influences on the transcriptome profiles of circulating platelets. Human megakaryocytes differentially transfer mRNAs that encode specific proteins to platelets under basal conditions (63). These patterns change based on genetic and environmental variables, including molecular cues generated in thrombosis, inflammation, and infection (388, 453). The extent, nature, and plasticity of the platelet transcriptome and its impact on the biology of platelets and other cells in specific lung syndromes and models of lung disease are only beginning to be explored.
V. PLATELETS IN INFLAMMATORY LUNG SYNDROMES
Platelets are markers and effectors in a variety of inflammatory diseases and syndromes. This spectrum extends from diabetes and other metabolic disorders with inflammatory and thrombotic components to infectious and sterile vasculopathies (369, 409, 453). We have cited observations and reports relevant to pathological lung inflammation and pulmonary immune diseases throughout this review. Here, we focus on a limited group of lung disorders chosen because they illustrate specific aspects of platelet involvement.
There is evidence that platelets are effector cells in ARDS (Figures 1A AND 3) and in less severe inflammatory alveolar involvement in conditions that precipitate this syndrome: sepsis, infectious pneumonia, aspiration, severe trauma, and other triggers (277). Platelets accumulate and are sequestered in the lungs of patients with conditions that predispose to ARDS (18, 41, 162, 386), as they do in experimental animals with inflammatory ALI (163, 330) (Figure 5). Experimental and clinical observations implicate platelets as effectors of diffuse alveolar damage, a pathological constellation that defines the early “exudative” stage of ARDS (18, 278, 289, 440). Animal models intended to mimic inflammatory ALI and ARDS dramatically illustrate the Janus-faced effects of platelets on pulmonary endothelial barrier function and integrity (Figure 4). Multiple experimental observations, many involving platelet interactions with myeloid leukocytes and endothelial cells, demonstrate that platelets mediate increased alveolar-capillary permeability and accumulation of protein-rich alveolar edema fluid in such models; in parallel, experimental thrombocytopenia precipitates alveolar hemorrhage in lung inflammation induced by LPS or live bacteria (reviewed in sect. IIIA). In aggregate, these experimental studies indicate that alterations in platelet number or function can contribute to two key features of diffuse alveolar damage in ARDS: alveolar-capillary leak and alveolar hemorrhage (18, 277, 278, 289, 440). It is not yet known precisely how these experimental findings apply to clinical ARDS (Figure 3). Similarly, platelets facilitate accumulation of myeloid leukocytes in the alveoli and neutrophil-mediated alveolar-capillary membrane injury in specific animal models (see sect. IIIF) (Figure 5 AND 7A), and thus may contribute to these features of diffuse alveolar damage in the lungs of patients with ARDS (277, 278). Platelet-triggered NET formation may be an important mechanism of neutrophil-mediated alveolar injury, although it should be remembered that other host factors, in addition to pathogens, can also induce NET deployment (Figure 8). Platelets have multiple activities that can catalyze deposition of intravascular fibrin or formation of organized thrombi (Figure 1A) in the lungs of patients with ARDS (see sect. II). Platelet-induced NET formation may also be involved in this process (see below). Therefore, activated platelets have key effector function that can initiate or amplify critical events in diffuse alveolar damage and the pathogenesis and inflammatory injury of ARDS (277, 278): increased alveolar-capillary permeability and alveolar flooding with protein-rich edema fluid, alveolar hemorrhage, dysregulated accumulation and activation of myeloid leukocytes, pulmonary microvascular deposition of fibrin, and pulmonary vascular thrombosis. Furthermore, experimental ALI is a setting in which platelets are reported to gain access to the extravascular space (see sect. IV). If this occurs in clinical ARDS, platelets in the alveoli could drive multiple pathways of alveolar inflammation, epithelial injury, and fibrin deposition and might contribute to the formation of hyaline membranes, another key pathological feature of diffuse alveolar damage (289, 440). The presence of platelet degranulation markers in BAL samples from patients with ARDS (75, 186) suggest the possibility that platelets might also have been present at some point, and that these factors may not have been exclusively delivered in paracrine fashion.
In addition to initiating or driving the progression of diffuse alveolar damage, platelets may contribute to complications of supportive care of patients at risk or with established ARDS. Experimental models of VILI demonstrate injurious activities of platelets (370, 484). Key mechanisms included platelet signaling of neutrophils and aberrant platelet-induced changes in endothelial cells in these experiments. VILI triggers NET formation in mice, although the contribution of NETs to alveolar damage depends on the model (370, 483). As noted previously, it is not known how observations in mouse models apply to clinical intensive care. In contrast to VILI, two studies suggest that platelets are not effector cells in pulmonary oxygen toxicity. Thrombocytopenia induced by anti-platelet antibody treatment or genetic deficiency of the thrombopoietin receptor did not protect mice from alveolar injury induced by hyperoxia, although platelets were deposited in the lungs under hyperoxic conditions (21, 257). Platelets contribute to transfusion immunodulation (36) and to the complications of transfusion therapy, as touched on in our earlier discussion of TRALI (62, 257, 283). Pathogen reduction technologies and other features of platelet storage may alter their complex biologic functions (Figure 2) in ways yet to be fully defined (387).
“Anti-platelet” therapy for prevention or treatment of ARDS has been proposed (225, 277). Aspirin was beneficial in short-term animal models including experimental TRALI, acid-induced ALI, LPS-induced ALI, and ischemia-reperfusion lung injury (108, 257, 330, 382, 441), and a P2Yl2 receptor antagonist reduced lung injury in murine CLP-induced sepsis (358). Prehospitalization or preinjury anti-platelet therapy was associated with reduced incidence of acute lung injury in several, but not all, observational clinical studies (118, 157, 224, 325). In prospectively identified patients with ARDS defined by consensus criteria, pre- or in-hospital administration of aspirin was associated with reduced ICU mortality (45). These observations suggest that anti-platelet agents may be beneficial in this syndrome. Additional clinical studies are in progress. A caveat is that drug-induced alterations in platelet function can potentially induce alveolar hemorrhage or worsen leak of water and protein, in addition to interrupting injurious proinflammatory activities (see sect. IIIA) (Figures 3 AND 4). Thus iatrogenic platelet dysfunction resulting from administration of anti-platelet agents could worsen key pathophysiological features of ARDS. As a further issue, inhibition of platelet, or megakaryocyte, functions by anti-platelet drugs could interrupt lung repair pathways (468). These issues await future preclinical and clinical investigation.
Platelets are implicated in chronic and intermittent inflammatory lung syndromes, in addition to the acute alveolar injury of ARDS (453). Some of these disorders include chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF), AERD, and asthma. Thrombocytosis was associated with increased mortality in COPD in one study, and a role for anti-platelet therapy was raised (158). Platelets in blood samples from patients with CF were found to be hyperresponsive to agonists, and there were increased numbers of platelet-leukocyte aggregates (NPA, MPA) in venous blood from CF patients (326). These observations, and a number of earlier reports, suggest that platelets contribute to disregulated lung inflammation in CF and that anti-platelet drugs should be evaluated in therapeutic strategies for this disorder (327). There is also evidence for platelets as inflammatory effector cells in AERD. As previously noted, platelets were found in nasal polyps from patients with this syndrome. Platelet-leukocyte aggregates form in the blood in AERD and mediate transcellular leukotriene synthesis that may drive respiratory inflammation (235) (see sect. IIIF).
Clinical and experimental studies indicate that platelets contribute to key features of the pathophysiology of asthma, including airway hyperresponsiveness, bronchoconstriction, airway inflammation, and airway remodeling (226). Increased numbers of platelet-leukocyte aggregates and other indexes of platelet activation in the blood were detected in studies of patients during asthma attacks or after allergen challenge (91, 148, 229, 293). Platelet-leukocyte aggregates increased in the blood of patients with allergic asthma during late responses to allergen challenge, and also in the blood of sensitized mice in an allergen exposure model (347). Platelet depletion reduced infiltration of leukocytes into the airways of mice after allergen challenge, and adoptive transfer of platelets from allergic mice restored leukocyte accumulation in the airways (347). A variety of additional in vitro and in vivo experiments support the conclusion that platelets facilitate recruitment of eosinophils, lymphocytes, and neutrophils in allergic airway inflammation and provide evidence that platelet P-selectin is critical in these platelet-leukocyte interactions (91, 226, 263, 345).
In additional observations including analysis of cells from asthmatic subjects, activated platelets were thought to facilitate inside-out signaling of β1-integrins on eosinophils via a P-selectin-dependent mechanism and to mediate rapid translocation of platelet-eosinophil aggregates into the lungs (198, 199). In a flow model, platelet P-selectin contributed to enhanced aggregation and adhesion of eosinophils from asthmatic subjects to cultured human endothelial cells (445). Fc receptors that recognize IgE were central in platelet responses in experimental allergic airway inflammation (344) and appear to be important in responses of platelets from patients with allergic asthma (91). Platelets were required for airway remodeling in chronic allergic inflammation in mice and were thought to have activities independent of leukocyte recruitment (346).
The observations profiled here and in other reviews (91, 226) provide substantial evidence that platelets are key effector cells in allergic airway inflammation, asthma, and allergic rhinitis. New links between asthma and hemostasis (91, 298) that may involve critical activities of platelets are emerging. The possibility that platelets influence pathological lymphangiogenesis in chronically inflamed airways was outlined earlier (see sect. III A).
Pulmonary embolism is a common and often deadly clinical event that is precipitated by deep vein thrombosis (430). Traditionally, pathological activation of coagulation proteins or alterations in their levels in blood have been considered to be the primary determinants of risk for venous thromboembolism (VTE) (451). A newer concept is that inflammation and platelet activation are also critical, raising the possibility that anti-platelet therapies that interrupt platelet inflammatory effector activities (223, 295) may be useful in addition to traditional standard treatment focused on inhibiting hemostatic proteins (451). Experimental models of venous thrombosis demonstrate that platelets, neutrophils, and monocytes interact on injured or activated endothelium, generating tissue factor, which drives pathological hemostasis, and cytokines (455). Clots may be reservoirs for IL-1β (Figure 1B) (58, 249) and other cytokines that induce and amplify venous inflammation and thrombosis (458). Formation of NETs, potentially triggered by activated platelets or cytokine stimulation (Figure 8), is a newly recognized feature of clotting that may be a key mechanism in pathological thrombosis (381, 455). Activation of human platelets by extracellular histones, which involves recognition by platelet TLR2 and TLR4, induces a procoagulant phenotype (394). This may reciprocally drive thrombosis when NETs are generated in clots. Thus venous thrombosis is an example of the complex linkage of inflammation and hemostasis (119, 353, 389) that was conceptually introduced at the beginning of this review, and further illustrates that platelets are central effectors that link inflammation and thrombosis. If clots that are formed in venous thrombosis embolize to the lungs, as is frequent (430), platelets may also influence biologic outcomes in the pulmonary vessels. Platelets were reported to retard resolution of pulmonary microemboli in a rodent model (308), although the mechanism for this aberrant activity was not determined.
In clinical studies, activated platelets, platelet microparticles, and platelet-leukocyte aggregates have been found in the blood of patients with VTE (69, 187) and in sepsis (366, 369), which is a risk factor for VTE (209). A single nucleotide polymorphism in GP6, which encodes platelet GPVI, was identified as a gene variant strongly associated with venous thrombosis (31). As outlined earlier in this review, GPVI has hemostatic, inflammatory, and immunomodulatory functions and is unique to platelets. Additional studies may reveal new mechanisms by which GPVI, or other platelet receptors and signaling pathways (Table 1), drive linked inflammation and hemostasis in the pathogenesis of venous thrombosis and its pulmonary complications.
These three examples (ARDS, asthma and inflammatory airway disease, and VTE) demonstrate that platelets have diverse activities in experimental and clinical inflammatory and immune lung syndromes. They also illustrate the potential for platelets to influence pathological inflammatory processes at the alveolar-capillary membrane, in the tracheobronchial tree, and in lung vessels and demonstrate that platelets are active in acute, subacute, and chronic disorders. Thus contributions of platelets in clinical lung inflammation are not restricted to a particular anatomic region or to a specific time interval. Platelets are also likely to have complex activities in many other inflammatory lung diseases besides those profiled in this section, and in inflammatory aspects of lung neoplasia (468). For most of these syndromes, the involvement of platelets has been only superficially examined or is totally unplumbed, however, and will require future inquiry.
GRANTS
Work cited in this article was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants R37HL044525, HL112311, HL066277, HL077671, HL091754, and HL090870. E. A. Middleton is supported by NHLBI Grant T32 HL105321.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
acknowledgments
We thank Kendra Richardson for invaluable efforts in preparation of the manuscript, Diana Lim for creative contributions and preparation of the figures, and our colleagues and students for helpful discussions.
Address for reprint requests and other correspondence: G. A. Zimmerman, Eccles Institute of Human Genetics, 15 N 2030 E, Rm. 4220, Univ. of Utah, Salt Lake City, UT 84112 (e-mail: guy.zimmerman@u2m2.utah.edu).
REFERENCES
- 1.Aggrey AA, Srivastava K, Ture S, Field DJ, Morrell CN. Platelet induction of the acute-phase response is protective in murine experimental cerebral malaria. J Immunol : 4685–4691, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alexander JS, Patton WF, Christman BW, Cuiper LL, Haselton FR. Platelet-derived lysophosphatidic acid decreases endothelial permeability in vitro. Am J Physiol Heart Circ Physiol : H115–H122, 1998. [DOI] [PubMed] [Google Scholar]
- 3.Alitalo K. The lymphatic vasculature in disease. Nature Med : 1371–1380, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Ammon C, Kreutz M, Rehli M, Krause SW, Andreesen R. Platelets induce monocyte differentiation in serum-free coculture. J Leukocyte Biol : 469–476, 1998. [DOI] [PubMed] [Google Scholar]
- 5.Anabel AS, Eduardo PC, Pedro Antonio HC, Carlos SM, Juana NM, Honorio TA, Nicolas VS, Sergio Roberto AR. Human platelets express Toll-like receptor 3 and respond to poly I:C. Hum Immunol : 1244–1251, 2014. [DOI] [PubMed] [Google Scholar]
- 6.Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets express functional Toll-like receptor-4. Blood : 2417–2423, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Andre P, Denis CV, Ware J, Saffaripour S, Hynes RO, Ruggeri ZM, Wagner DD. Platelets adhere to and translocate on von Willebrand factor presented by endothelium in stimulated veins. Blood : 3322–3328, 2000. [PubMed] [Google Scholar]
- 8.Andrews RK, Berndt MC. The GPIb-IX-V complex. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 195–213. [Google Scholar]
- 9.Andrews RK, Arthur JF, Gardiner EE. Neutrophil extracellular traps (NETs) and the role of platelets in infection. Thromb Haemost : 659–665, 2014. [DOI] [PubMed] [Google Scholar]
- 10.Angus DC, van der Poll T. Severe sepsis and septic shock. N Engl J Med : 840–851, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Asaduzzaman M, Lavasani S, Rahman M, Zhang S, Braun OO, Jeppsson B, Thorlacius H. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care Med : 1389–1396, 2009. [DOI] [PubMed] [Google Scholar]
- 12.Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, Ni H, Lazarus AH, Freedman J, Semple JW. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-alpha production in vivo. Blood : 637–641, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Aslam R, Speck ER, Kim M, Freedman J, Semple JW. Transfusion-related immunomodulation by platelets is dependent on their expression of MHC Class I molecules and is independent of white cells. Transfusion : 1778–1786, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Aslan JE, Tormoen GW, Loren CP, Pang J, McCarty OJ. S6K1 and mTOR regulate Rac1-driven platelet activation and aggregation. Blood : 3129–3136, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auerbach DJ, Lin Y, Miao H, Cimbro R, Difiore MJ, Gianolini ME, Furci L, Biswas P, Fauci AS, Lusso P. Identification of the platelet-derived chemokine CXCL4/PF-4 as a broad-spectrum HIV-1 inhibitor. Proc Natl Acad Sci USA : 9569–9574, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol : 669–692, 2009. [DOI] [PubMed] [Google Scholar]
- 17.Babinska A, Markell MS, Salifu MO, Akoad M, Ehrlich YH, Kornecki E. Enhancement of human platelet aggregation and secretion induced by rapamycin. Nephrology Dialysis Transplant : 3153–3159, 1998. [DOI] [PubMed] [Google Scholar]
- 18.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med : 35–56, 1982. [PubMed] [Google Scholar]
- 19.Baluk P, Tammela T, Ator E, Lyubynska N, Achen MG, Hicklin DJ, Jeltsch M, Petrova TV, Pytowski B, Stacker SA, Yla-Herttuala S, Jackson DG, Alitalo K, McDonald DM. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J Clin Invest : 247–257, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Baluk P, Yao LC, Feng J, Romano T, Jung SS, Schreiter JL, Yan L, Shealy DJ, McDonald DM. TNF-alpha drives remodeling of blood vessels and lymphatics in sustained airway inflammation in mice. J Clin Invest : 2954–2964, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barazzone C, Tacchini-Cottier F, Vesin C, Rochat AF, Piguet PF. Hyperoxia induces platelet activation and lung sequestration: an event dependent on tumor necrosis factor-alpha and CD11a. Am J Respir Cell Biol : 107–114, 1996. [DOI] [PubMed] [Google Scholar]
- 22.Bastarache JA, Fremont RD, Kropski JA, Bossert FR, Ware LB. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. Am J Physiol Lung Cell Mol Physiol : L1035–L1041, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beaulieu LM, Freedman JE. Inhibition of platelet function by the endothelium. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 313–342. [Google Scholar]
- 24.Beaulieu LM, Lin E, Mick E, Koupenova M, Weingerg EO, Kramer CD, Genco CA, Tanriverdi K, Larson MG, Benjamin EJ, Freedman JE. Interleukin 1 receptor 1 and interleukin 1beta regulate megakaryocyte maturation, platelet activation, and transcript profile during inflammation in mice and humans. Arterioscler Thromb Vasc Biol : 552–564, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beaulieu LM, Freedman JE. The role of inflammation in regulating platelet production and function: Toll-like receptors in platelets and megakaryocytes. Thromb Res : 205–209, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beaulieu LM, Lin E, Morin KM, Tanriverdi K, Freedman JE. Regulatory effects of TLR2 on megakaryocytic cell function. Blood : 5963–5974, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berg M, Offermanns S, Seifert R, Schultz G. Synthetic lipopeptide Pam3CysSer(Lys)4 is an effective activator of human platelets. Am J Physiol Cell Physiol : C1684–C1691, 1994. [DOI] [PubMed] [Google Scholar]
- 28.Berney-Lang MA, Freilinger AL III, Barnard MR, Michelson AD. Flow cytometry. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 581–602. [Google Scholar]
- 29.Berthet J, Damien P, Hamzeh-Cognasse H, Pozzetto B, Garraud O, Cognasse F. Toll-like receptor 4 signal transduction in platelets: novel pathways. Br J Haematol : 89–92, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Beutler B, Rietschel ET. Innate immune sensing and its roots: the story of endotoxin. Nature Rev Immunol : 169–176, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Bezemer ID, Bare LA, Doggen CJ, Arellano AR, Tong C, Rowland CM, Catanese J, Young BA, Reitsma PH, Devlin JJ, Rosendaal FR. Gene variants associated with deep vein thrombosis. JAMA : 1306–1314, 2008. [DOI] [PubMed] [Google Scholar]
- 32.Bhatt DL, Topol EJ. Scientific and therapeutic advances in antiplatelet therapy. Nature Rev Drug Discovery : 15–28, 2003. [DOI] [PubMed] [Google Scholar]
- 33.Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A, Reichenbach J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood : 2619–2622, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, Hayashi C, Genco CA, Iafrati M, Freedman JE. Stimulation of Toll-like receptor 2 in human platelets induces a thromboinflammatory response through activation of phosphoinositide 3-kinase. Circ Res : 346–354, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity : 305–315, 2010. [DOI] [PubMed] [Google Scholar]
- 36.Blumberg N, Spinelli SL, Francis CW, Taubman MB, Phipps RP. The platelet as an immune cell-CD40 ligand and transfusion immunomodulation. Immunol Res : 251–260, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boilard E, Nigrovic PA, Larabee K, Watts GF, Coblyn JS, Weinblatt ME, Massarotti EM, Remold-O'Donnell E, Farndale RW, Ware J, Lee DM. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science : 580–583, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boilard E, Pare G, Rousseau M, Cloutier N, Dubuc I, Levesque T, Borgeat P, Flamand L. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood : 2854–2863, 2014. [DOI] [PubMed] [Google Scholar]
- 39.Bolam JP, Smith JH. Platelets in inflammatory exudates. J Pharm Pharmacol : 674–676, 1977. [DOI] [PubMed] [Google Scholar]
- 40.Bombeli T, Schwartz BR, Harlan JM. Adhesion of activated platelets to endothelial cells: evidence for a GPIIbIIIa-dependent bridging mechanism and novel roles for endothelial intercellular adhesion molecule 1 (ICAM-1), alphavbeta3 integrin, and GPIbalpha. J Exp Med : 329–339, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bone RC, Francis PB, Pierce AK. Intravascular coagulation associated with the adult respiratory distress syndrome. Am J Med : 585–589, 1976. [DOI] [PubMed] [Google Scholar]
- 42.Boogaerts MA, Yamada O, Jacob HS, Moldow CF. Enhancement of granulocyte-endothelial cell adherence and granulocyte-induced cytotoxicity by platelet release products. Proc Natl Acad Sci USA : 7019–7023, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouchard B, Silveira JR, Tracy PB. Interactions between platelets and the coagulation system. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 425–451. [Google Scholar]
- 44.Boulaftali Y, Hess PR, Getz TM, Cholka A, Stolla M, Mackman N, Owens AP 3rd, Ware J, Kahn ML, Bergmeier W. Platelet ITAM signaling is critical for vascular integrity in inflammation. J Clin Invest : 908–916, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boyle AJ, Di Gangi S, Hamid UI, Mottram LJ, McNamee L, White G, Cross LJ, McNamee JJ, O'Kane CM, McAuley DF. Aspirin therapy in patients with acute respiratory distress syndrome (ARDS) is associated with reduced intensive care unit mortality: a prospective analysis. Critical Care : 109, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bozza FA, Shah AM, Weyrich AS, Zimmerman GA. Amicus or adversary: platelets in lung biology, acute injury, and inflammation. Am J Respir Cell Mol Biol : 123–134, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Braddock M, Quinn A. Targeting IL-1 in inflammatory disease: new opportunities for therapeutic intervention. Nature Rev Drug Discovery : 330–339, 2004. [DOI] [PubMed] [Google Scholar]
- 48.Braian C, Hogea V, Stendahl O. Mycobacterium tuberculosis-induced neutrophil extracellular traps activate human macrophages. J Innate Immun : 591–602, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brambilla M, Camera M, Colnago D, Marenzi G, De Metrio M, Giesen PL, Balduini A, Veglia F, Gertow K, Biglioli P, Tremoli E. Tissue factor in patients with acute coronary syndromes: expression in platelets, leukocytes, and platelet-leukocyte aggregates. Arterioscler Thromb Vasc Biol : 947–953, 2008. [DOI] [PubMed] [Google Scholar]
- 50.Brancaleone V, Gobbetti T, Cenac N, le Faouder P, Colom B, Flower RJ, Vergnolle N, Nourshargh S, Perretti M. A vasculo-protective circuit centered on lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 operative in murine microcirculation. Blood : 608–617, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandt E, Ludwig A, Petersen F, Flad HD. Platelet-derived CXC chemokines: old players in new games. Immunol Rev : 204–216, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Brass LF, Wannermacher KM, Zhu L, Stalker TJ. Signal transduction during platelet plug formation. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 367–398. [Google Scholar]
- 53.Bray PF, Jones CI, Soranzo N, Ouwehand WH. Platelet genomics. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 67–89. [Google Scholar]
- 54.Brindley LL, Sweet JM, Goetzel EJ. Stimulation of histamine release from human basophils by human platelet factor 4. J Clin Invest : 1218–1223, 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science : 1532–1535, 2004. [DOI] [PubMed] [Google Scholar]
- 56.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin? J Cell Biol : 773–783, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J Immunol : 5489–5496, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown GT, Narayanan P, Li W, Silverstein RL, McIntyre TM. Lipopolysaccharide stimulates platelets through an IL-1beta autocrine loop. J Immunol : 5196–5203, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, Jeron A, Latge JP, Brakhage AA, Gunzer M. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathogens : e1000873, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest : 1871–1879, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cara DC, Ebbert KV, McCafferty DM. Mast cell-independent mechanisms of immediate hypersensitivity: a role for platelets. J Immunol : 4964–4971, 2004. [DOI] [PubMed] [Google Scholar]
- 62.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest : 2661–2671, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cecchetti L, Tolley ND, Michetti N, Bury L, Weyrich AS, Gresele P. Megakaryocytes differentially sort mRNAs for matrix metalloproteinases and their inhibitors into platelets: a mechanism for regulating synthetic events. Blood : 1903–1911, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chapman LM, Aggrey AA, Field DJ, Srivastava K, Ture S, Yui K, Topham DJ, Baldwin WM 3rd, Morrell CN. Platelets present antigen in the context of MHC class I. J Immunol : 916–923, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Charafeddine AH, Kim EJ, Maynard DM, Yi H, Weaver TA, Gunay-Aygun M, Russell M, Gahl WA, Kirk AD. Platelet-derived CD154: ultrastructural localization and clinical correlation in organ transplantation. Am J Transplant : 3143–3151, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Lopez JA. Interactions of platelets with subendothelium and endothelium. Microcirculation : 235–246, 2005. [DOI] [PubMed] [Google Scholar]
- 67.Cheng OZ, Palaniyar N. NET balancing: a problem in inflammatory lung diseases. Front Immunol : 1–13, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chertow DS, Memoli MJ. Bacterial coinfection in influenza: a grand rounds review. JAMA : 275–282, 2013. [DOI] [PubMed] [Google Scholar]
- 69.Chirinos JA, Heresi GA, Velasquez H, Jy W, Jimenez JJ, Ahn E, Horstman LL, Soriano AO, Zambrano JP, Ahn YS. Elevation of endothelial microparticles, platelets, and leukocyte activation in patients with venous thromboembolism. J Am Coll Cardiol : 1467–1471, 2005. [DOI] [PubMed] [Google Scholar]
- 70.Choi SM, Xie H, Campbell AP, Kuypers J, Leisenring W, Boudreault AA, Englund JA, Corey L, Boeckh M. Influenza viral RNA detection in blood as a marker to predict disease severity in hematopoietic cell transplant recipients. J Infect Dis : 1872–1877, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christersson C, Johnell M, Siegbahn A. Tissue factor and IL8 production by P-selectin-dependent platelet-monocyte aggregates in whole blood involves phosphorylation of Lyn and is inhibited by IL10. J Thromb Haemost : 986–994, 2008. [DOI] [PubMed] [Google Scholar]
- 72.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nature Med : 463–469, 2007. [DOI] [PubMed] [Google Scholar]
- 73.Clemetson KJ, Clemetson JM. Platelet receptors. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 169–194. [Google Scholar]
- 74.Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-like receptor molecules on human platelets. Immunol Cell Biol : 196–198, 2005. [DOI] [PubMed] [Google Scholar]
- 75.Cohen AB, Stevens MD, Miller EJ, Atkinson MA, Mullenbach G, Maunder RJ, Martin TR, Wiener-Kronish JP, Matthay MA. Neutrophil-activating peptide-2 in patients with pulmonary edema from congestive heart failure or ARDS. Am J Physiol Lung Cell Mol Physiol : L490–L495, 1993. [DOI] [PubMed] [Google Scholar]
- 76.Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, Chavakis T, Brohi K, Imhof BA, Nourshargh S. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity : 1075–1086, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, Bourdeau F, Kubes P, Ferri L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest 3446–3458, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Corken A, Russell S, Dent J, Post SR, Ware J. Platelet glycoprotein Ib-IX as a regulator of systemic inflammation. Arterioscler Thromb Vasc Biol : 996–1001, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost : 1800–1814, 2005. [DOI] [PubMed] [Google Scholar]
- 80.Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost : 1097–1107, 2011. [DOI] [PubMed] [Google Scholar]
- 81.Cox D, McConkey S. The role of platelets in the pathogenesis of cerebral malaria. Cell Mol Life Sci : 557–568, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Crist SA, Elzey BD, Ahmann MT, Ratliff TL. Early growth response-1 (EGR-1) and nuclear factor of activated T cells (NFAT) cooperate to mediate CD40L expression in megakaryocytes and platelets. J Biol Chem : 33985–33996, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Curwen KD, Gimbrone MA Jr, Handin RI. In vitro studies of thromboresistance: the role of prostacyclin (PGI2) in platelet adhesion to cultured normal and virally transformed human vascular endothelial cells. Lab Invest : 366–374, 1980. [PubMed] [Google Scholar]
- 84.Czapiga M, Gao JL, Kirk A, Lekstrom-Himes J. Human platelets exhibit chemotaxis using functional N-formyl peptide receptors. Exp Hematol : 73–84, 2005. [DOI] [PubMed] [Google Scholar]
- 85.Czapiga M, Kirk AD, Lekstrom-Himes J. Platelets deliver costimulatory signals to antigen-presenting cells: a potential bridge between injury and immune activation. Exp Hematol : 135–139, 2004. [DOI] [PubMed] [Google Scholar]
- 86.Czervionke RL, Hoak JC, Fry GL. Effect of aspirin on thrombin-induced adherence of platelets to cultured cells from the blood vessel wall. J Clin Invest : 847–856, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Damle NK, Klussman K, Dietsch MT, Mohagheghpour N, Aruffo A. GMP-140 (P-selectin/CD62) binds to chronically stimulated but not resting CD4+ T lymphocytes and regulates their production of proinflammatory cytokines. Eur J Immunol : 1789–1793, 1992. [DOI] [PubMed] [Google Scholar]
- 88.Danese S, de la Motte C, Reyes BM, Sans M, Levine AD, Fiocchi C. Cutting edge: T cells trigger CD40-dependent platelet activation and granular RANTES release: a novel pathway for immune response amplification. J Immunol 172: 2011–2015, 2004. [DOI] [PubMed] [Google Scholar]
- 89.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med : 2482–2494, 2007. [DOI] [PubMed] [Google Scholar]
- 90.Davila J, Manwani D, Vasovic L, Avanzi M, Uehlinger J, Ireland K, Mitchell WB. A novel inflammatory role for platelets in sickle cell disease. Platelets 1–4, 2014. [DOI] [PubMed]
- 91.De Boer JD, Majoor CJ, van 't Veer C, Bel EH, van der Poll T. Asthma and coagulation. Blood : 3236–3244, 2012. [DOI] [PubMed] [Google Scholar]
- 92.De Stoppelaar SF, van 't Veer C, Claushuis TA, Albersen BJ, Roelofs JJ, van der Poll T. Thrombocytopenia impairs host defense in gram-negative pneumonia-derived sepsis in mice. Blood : 3781–3790, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.De Stoppelaar SF, van 't Veer C, van der Poll T. The role of platelets in sepsis. Thromb Haemost : 666–677, 2014. [DOI] [PubMed] [Google Scholar]
- 94.De Zoete MR, Palm NW, Zhu S, Flavell RA. Inflammasomes. Cold Spring Harbor Perspect Biol : a016287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Delvaeye M, Conway EM. Coagulation and innate immune responses: can we view them separately? Blood : 2367–2374, 2009. [DOI] [PubMed] [Google Scholar]
- 96.Denis MM, Tolley ND, Bunting M, Schwertz H, Jiang H, Lindemann S, Yost CC, Rubner FJ, Albertine KH, Swoboda KJ, Fratto CM, Tolley E, Kraiss LW, McIntyre TM, Zimmerman GA, Weyrich AS. Escaping the nuclear confines: signal-dependent pre-mRNA splicing in anucleate platelets. Cell : 379–391, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dewitte A, Tanga A, Villeneuve J, Lepreux S, Ouattara A, Desmouliere A, Combe C, Ripoche J. New frontiers for platelet CD154. Exp Hematol Oncol : 6, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Diacovo TG, Catalina MD, Siegelman MH, von Andrian UH. Circulating activated platelets reconstitute lymphocyte homing and immunity in l-selectin-deficient mice. J Exp Med : 197–204, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Diacovo TG, deFougerolles AR, Bainton DF, Springer TA. A functional integrin ligand on the surface of platelets: intercellular adhesion molecule-2. J Clin Invest : 1243–1251, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Diacovo TG, Puri KD, Warnock RA, Springer TA, von Andrian UH. Platelet-mediated lymphocyte delivery to high endothelial venules. Science : 252–255, 1996. [DOI] [PubMed] [Google Scholar]
- 101.Diacovo TG, Roth SJ, Morita CT, Rosat JP, Brenner MB, Springer TA. Interactions of human alpha/beta and gamma/delta T lymphocyte subsets in shear flow with E-selectin and P-selectin. J Exp Med : 1193–1203, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dinarello CA. A clinical perspective of IL-1beta as the gatekeeper of inflammation. Eur J Immunol : 1203–1217, 2011. [DOI] [PubMed] [Google Scholar]
- 103.Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, Prescott SM, Zimmerman GA. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest : 2727–2738, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood : 2334–2339, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Douda DN, Jackson R, Grasemann H, Palaniyar N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J Immunol : 1856–1865, 2011. [DOI] [PubMed] [Google Scholar]
- 106.Duffau P, Seneschal J, Nicco C, Richez C, Lazaro E, Douchet I, Bordes C, Viallard JF, Goulvestre C, Pellegrin JL, Weil B, Moreau JF, Batteux F, Blanco P. Platelet CD154 potentiates interferon-alpha secretion by plasmacytoid dendritic cells in systemic lupus erythematosus. Science Transl Med : 47ra63, 2010. [DOI] [PubMed] [Google Scholar]
- 107.Edelstein LC, Simon LM, Montoya RT, Holinstat M, Chen ES, Bergeron A, Kong X, Nagalla S, Mohandas N, Cohen DE, Dong JF, Shaw C, Bray PF. Racial differences in human platelet PAR4 reactivity reflect expression of PCTP and miR-376c. Nature Med : 1609–1616, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eickmeier O, Seki H, Haworth O, Hilberath JN, Gao F, Uddin M, Croze RH, Carlo T, Pfeffer MA, Levy BD. Aspirin-triggered resolvin D1 reduces mucosal inflammation and promotes resolution in a murine model of acute lung injury. Mucosal Immunol : 256–266, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eligini S, Barbieri SS, Arenaz I, Tremoli E, Colli S. Paracrine up-regulation of monocyte cyclooxygenase-2 by platelets: role of transforming growth factor-beta1. Cardiovasc Res : 270–278, 2007. [DOI] [PubMed] [Google Scholar]
- 110.Elzey BD, Grant JF, Sinn HW, Nieswandt B, Waldschmidt TJ, Ratliff TL. Cooperation between platelet-derived CD154 and CD4+ T cells for enhanced germinal center formation. J Leukocyte Biol : 80–84, 2005. [DOI] [PubMed] [Google Scholar]
- 111.Elzey BD, Ratliff TL, Sowa JM, Crist SA. Platelet CD40L at the interface of adaptive immunity. Thromb Res : 180–183, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Elzey BD, Schmidt NW, Crist SA, Kresowik TP, Harty JT, Nieswandt B, Ratliff TL. Platelet-derived CD154 enables T-cell priming and protection against Listeria monocytogenes challenge. Blood : 3684–3691, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Elzey BD, Sprague DL, Ratliff TL. The emerging role of platelets in adaptive immunity. Cell Immunol : 1–9, 2005. [DOI] [PubMed] [Google Scholar]
- 114.Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, Stein CS, Nieswandt B, Wang Y, Davidson BL, Ratliff TL. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune. Immunity : 9–19, 2003. [DOI] [PubMed] [Google Scholar]
- 115.Emin MT, Sun L, Huertas A, Das S, Bhattacharya J, Bhattacharya S. Platelets induce endothelial tissue factor expression in a mouse model of acid-induced lung injury. Am J Physiol Lung Cell Mol Physiol : L1209–L1220, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Endo Y, Shibazaki M, Nakamura M, Takada H. Contrasting effects of lipopolysaccharides (endotoxins) from oral black-pigmented bacteria and Enterobacteriaceae on platelets, a major source of serotonin, and on histamine-forming enzyme in mice. J Infect Dis : 1404–1412, 1997. [DOI] [PubMed] [Google Scholar]
- 117.Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nature Rev Immunol : 34–45, 2013. [DOI] [PubMed] [Google Scholar]
- 118.Erlich JM, Talmor DS, Cartin-Ceba R, Gajic O, Kor DJ. Prehospitalization antiplatelet therapy is associated with a reduced incidence of acute lung injury: a population-based cohort study. Chest : 289–295, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Esmon CT, Xu J, Lupu F. Innate immunity and coagulation. J Thromb Haemost Suppl 1: 182–188, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Etulain J, Martinod K, Wong SL, Cifuni SM, Schattner M, Wagner DD. P-selectin promotes neutrophil extracellular trap formation in mice. Blood : 242–246, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Evangelista V, Smyth SS. Interactions between platelets, leukocytes and the endothelium. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 295–312. [Google Scholar]
- 122.Faille D, El-Assaad F, Alessi MC, Fusai T, Combes V, Grau GE. Platelet-endothelial cell interactions in cerebral malaria: the end of a cordial understanding. Thromb Haemost : 1093–1102, 2009. [DOI] [PubMed] [Google Scholar]
- 123.Feng D, Nagy JA, Pyne K, Dvorak HF, Dvorak AM. Platelets exit venules by a transcellular pathway at sites of F-met peptide-induced acute inflammation in guinea pigs. Int Arch Allergy Immunol : 188–195, 1998. [DOI] [PubMed] [Google Scholar]
- 124.Fitzgerald JR, Foster TJ, Cox D. The interaction of bacterial pathogens with platelets. Nature Rev Microbiol : 445–457, 2006. [DOI] [PubMed] [Google Scholar]
- 126.Flaumenhaft R. Platelet secretion. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 343–366. [Google Scholar]
- 127.Foulks JM, Marathe GK, Michetti N, Stafforini DM, Zimmerman GA, McIntyre TM, Weyrich AS. PAF-acetylhydrolase expressed during megakaryocyte differentiation inactivates PAF-like lipids. Blood : 6699–6706, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Frank JA, Matthay MA. TGF-beta and lung fluid balance in ARDS. Proc Natl Acad Sci USA : 885–886, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, Weinrauch Y, Brinkmann V, Zychlinsky A. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol : 231–241, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gambaryan S, Kobsar A, Rukoyatkina N, Herterich S, Geiger J, Smolenski A, Lohmann SM, Walter U. Thrombin and collagen induce a feedback inhibitory signaling pathway in platelets involving dissociation of the catalytic subunit of protein kinase A from an NFkappaB-IkappaB complex. J Biol Chem : 18352–18363, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Garcia JG, Liu F, Verin AD, Birukova A, Dechert MA, Gerthoffer WT, Bamberg JR, English D. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest : 689–701, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Garraud O, Berthet J, Hamzeh-Cognasse H, Cognasse F. Pathogen sensing, subsequent signalling, and signalosome in human platelets. Thromb Res : 283–286, 2011. [DOI] [PubMed] [Google Scholar]
- 133.Garraud O, Cognasse F. Platelet Toll-like receptor expression: the link between “danger” ligands and inflammation. Inflammation Allergy Drug Targets : 322–333, 2010. [DOI] [PubMed] [Google Scholar]
- 134.Gavard J, Patel V, Gutkind JS. Angiopoietin-1 prevents VEGF-induced endothelial permeability by sequestering Src through mDia. Dev Cell : 25–36, 2008. [DOI] [PubMed] [Google Scholar]
- 135.Gawaz M, Brand K, Dickfeld T, Pogatsa-Murray G, Page S, Bogner C, Koch W, Schomig A, Neumann F. Platelets induce alterations of chemotactic and adhesive properties of endothelial cells mediated through an interleukin-1-dependent mechanism. Implications for atherogenesis. Atherosclerosis : 75–85, 2000. [DOI] [PubMed] [Google Scholar]
- 136.Geddings JE, Mackman N. New players in haemostasis and thrombosis. Thromb Haemost : 570–574, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gerardin P, Rogier C, Ka AS, Jouvencel P, Brousse V, Imbert P. Prognostic value of thrombocytopenia in African children with falciparum malaria. Am J Tropical Med Hygiene : 686–691, 2002. [DOI] [PubMed] [Google Scholar]
- 138.Gerdes N, Zhu L, Ersoy M, Hermansson A, Hjemdahl P, Hu H, Hansson GK, Li N. Platelets regulate CD4(+) T-cell differentiation via multiple chemokines in humans. Thromb Haemost : 353–362, 2011. [DOI] [PubMed] [Google Scholar]
- 139.Gerrits AJ, Koekman CA, van Haeften TW, Akkerman JW. Platelet tissue factor synthesis in type 2 diabetic patients is resistant to inhibition by insulin. Diabetes : 1487–1495, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Gidlof O, van der Brug M, Ohman J, Gilje P, Olde B, Wahlestedt C, Erlinge D. Platelets activated during myocardial infarction release functional miRNA, which can be taken up by endothelial cells and regulate ICAM1 expression. Blood : 3908–3917, s3901–3926, 2013. [DOI] [PubMed] [Google Scholar]
- 141.Ginsberg MH, Henson PM. Enhancement of platelet response to immune complexes and IgG aggregates by lipid A-rich bacterial lipopolysaccharides. J Exp Med : 207–217, 1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gleissner CA, Shaked I, Erbel C, Bockler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res : 203–211, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol : 4810–4818, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Goerge T, Ho-Tin-Noe B, Carbo C, Benarafa C, Remold-O'Donnell E, Zhao BQ, Cifuni SM, Wagner DD. Inflammation induces hemorrhage in thrombocytopenia. Blood : 4958–4964, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gotts JE, Abbott J, Matthay MA. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am J Physiol Lung Cell Mol Physiol : L395–L406, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Green SA, Smith M, Hasley RB, Stephany D, Harned A, Nagashima K, Abdullah S, Pittaluga S, Imamichi T, Qin J, Rupert A, Ober A, Lane HC, Catalfamo M. Activated platelet-T-cell conjugates in peripheral blood of patients with HIV infection: coupling coagulation/inflammation and T cells. AIDS : 1297–1308, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Greenbaum DC, FitzGerald GA. Platelets, pyrexia, plasmodia. N Engl J Med : 526–528, 2009. [DOI] [PubMed] [Google Scholar]
- 148.Gresele P, Dottorini M, Selli ML, Iannacci L, Canino S, Todisco T, Romano S, Crook P, Page CP, Nenci GG. Altered platelet function associated with the bronchial hyperresponsiveness accompanying nocturnal asthma. J Allergy Clin Immunol : 894–902, 1993. [DOI] [PubMed] [Google Scholar]
- 149.Grommes J, Alard JE, Drechsler M, Wantha S, Morgelin M, Kuebler WM, Jacobs M, von Hundelshausen P, Markart P, Wygrecka M, Preissner KT, Hackeng TM, Koenen RR, Weber C, Soehnlein O. Disruption of platelet-derived chemokine heteromers prevents neutrophil extravasation in acute lung injury. Am J Respir Crit Care Med : 628–636, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gros A, Syvannarath V, Lamrani L, Ollivier V, Loyau S, Goerge T, Nieswandt B, Jandrot-Perrus M, Ho-Tin-Noe B. Single platelets seal neutrophil-induced vascular breaches via GPVI during immune complex-mediated inflammation in mice. Blood : 1017–1026, 2015. [DOI] [PubMed] [Google Scholar]
- 151.Gudbrandsdottir S, Hasselbalch HC, Nielsen CH. Activated platelets enhance IL-10 secretion and reduce TNF-alpha secretion by monocytes. J Immunol : 4059–4067, 2013. [DOI] [PubMed] [Google Scholar]
- 152.Hagihara M, Higuchi A, Tamura N, Ueda Y, Hirabayashi K, Ikeda Y, Kato S, Sakamoto S, Hotta T, Handa S, Goto S. Platelets, after exposure to a high shear stress, induce IL-10-producing, mature dendritic cells in vitro. J Immunol : 5297–5303, 2004. [DOI] [PubMed] [Google Scholar]
- 153.Halvorsen B, Smedbakken LM, Michelsen AE, Skjelland M, Bjerkeli V, Sagen EL, Tasken K, Bendz B, Gullestad L, Holm S, Biessen EA, Aukrust P. Activated platelets promote increased monocyte expression of CXCR5 through prostaglandin E2-related mechanisms and enhance the anti-inflammatory effects of CXCL13. Atherosclerosis : 352–359, 2014. [DOI] [PubMed] [Google Scholar]
- 154.Hamzeh-Cognasse H, Cognasse F, Palle S, Chavarin P, Olivier T, Delezay O, Pozzetto B, Garraud O. Direct contact of platelets and their released products exert different effects on human dendritic cell maturation. BMC Immunol : 54, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hara T, Shimizu K, Ogawa F, Yanaba K, Iwata Y, Muroi E, Takenaka M, Komura K, Hasegawa M, Fujimoto M, Sato S. Platelets control leukocyte recruitment in a murine model of cutaneous arthus reaction. Am J Pathol : 259–269, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Harding M, Kubes P. Innate immunity in the vasculature: interactions with pathogenic bacteria. Curr Opin Microbiol : 85–91, 2012. [DOI] [PubMed] [Google Scholar]
- 157.Harr JN, Moore EE, Johnson J, Chin TL, Wohlauer MV, Maier R, Cuschieri J, Sperry J, Banerjee A, Silliman CC, Sauaia A. Antiplatelet therapy is associated with decreased transfusion-associated risk of lung dysfunction, multiple organ failure, and mortality in trauma patients. Crit Care Med : 399–404, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Harrison MT, Short P, Williamson PA, Singanayagam A, Chalmers JD, Schembri S. Thrombocytosis is associated with increased short and long term mortality after exacerbation of chronic obstructive pulmonary disease: a role for antiplatelet therapy? Thorax : 609–615, 2014. [DOI] [PubMed] [Google Scholar]
- 159.Haselton FR, Alexander JS. Platelets and a platelet-released factor enhance endothelial barrier. Am J Physiol Lung Cell Mol Physiol : L670–L678, 1992. [DOI] [PubMed] [Google Scholar]
- 160.Hawrylowicz CM, Howells GL, Feldmann M. Platelet-derived interleukin 1 induces human endothelial adhesion molecule expression and cytokine production. J Exp Med : 785–790, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hawrylowicz CM, Santoro SA, Platt FM, Unanue ER. Activated platelets express IL-1 activity. J Immunol : 4015–4018, 1989. [PubMed] [Google Scholar]
- 162.Hechtman HB, Lonergan EA, Sherpro D. Platelet and leukocyte lung interactions in patients with respiratory failure. Surgery : 155–163, 1978. [PubMed] [Google Scholar]
- 163.Hechtman HB, Lonergan EA, Staunton HPB, Dennis RC, Shepro D. Pulmonary entrapment of platelets during acute respiratory failure. Surgery : 277–283, 1978. [PubMed] [Google Scholar]
- 164.Heffner JE, Cook JA, Halushka PV. Human platelets modulate edema formation in isolated rabbit lungs. J Clin Invest : 757–764, 1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Heffner JK, Repine JE. Platelets. In: The Lung: Scientific Foundations (2nd ed), edited by Crystal RG, West JB, and Weibel ER. Philadelphia, PA: Raven, 1997, p. 947–959. [Google Scholar]
- 166.Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature : 591–594, 1998. [DOI] [PubMed] [Google Scholar]
- 167.Henn V, Steinbach S, Buchner K, Presek P, Kroczek RA. The inflammatory action of CD40 ligand (CD154) expressed on activated human platelets is temporally limited by coexpressed CD40. Blood : 1047–1054, 2001. [DOI] [PubMed] [Google Scholar]
- 168.Hermann A, Rauch BH, Braun M, Schror K, Weber AA. Platelet CD40 ligand (CD40L)–subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets : 74–82, 2001. [DOI] [PubMed] [Google Scholar]
- 169.Herter JM, Rossaint J, Zarbock A. Platelets in inflammation and immunity. J Thromb Haemost : 1764–1775, 2014. [DOI] [PubMed] [Google Scholar]
- 170.Herzog BH, Fu J, Wilson SJ, Hess PR, Sen A, McDaniel JM, Pan Y, Sheng M, Yago T, Silasi-Mansat R, McGee S, May F, Nieswandt B, Morris AJ, Lupu F, Coughlin SR, McEver RP, Chen H, Kahn ML, Xia L. Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature : 105–109, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hess PR, Rawnsley DR, Jakus Z, Yang Y, Sweet DT, Fu J, Herzog B, Lu M, Nieswandt B, Oliver G, Makinen T, Xia L, Kahn ML. Platelets mediate lymphovenous hemostasis to maintain blood-lymphatic separation throughout life. J Clin Invest : 273–284, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hickey MJ, Kubes P. Intravascular immunity: the host-pathogen encounter in blood vessels. Nature Rev Immunol : 364–375, 2009. [DOI] [PubMed] [Google Scholar]
- 173.Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nature Med : 384–391, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J Biol Chem : 28750–28756, 1997. [DOI] [PubMed] [Google Scholar]
- 175.Hilf N, Singh-Jasuja H, Schwarzmaier P, Gouttefangeas C, Rammensee HG, Schild H. Human platelets express heat shock protein receptors and regulate dendritic cell maturation. Blood : 3676–3682, 2002. [DOI] [PubMed] [Google Scholar]
- 176.Ho-Tin-Noe B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost Suppl 1: 56–65, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Hottz E, Tolley ND, Zimmerman GA, Weyrich AS, Bozza FA. Platelets in dengue infection. Drug Discovery Today Dis Mechanisms : e33–38, 2011. [Google Scholar]
- 178.Hottz ED, Medeiros-de-Moraes IM, Vieira-de-Abreu A, de Assis EF, Vals-de-Souza R, Castro-Faria-Neto HC, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT. Platelet activation and apoptosis modulate monocyte inflammatory responses in dengue. J Immunol : 1864–1872, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Hottz ED, Lopes JF, Freitas C, Valls-de-Souza R, Oliveira MF, Bozza MT, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza FA, Bozza PT. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood : 3405–3414, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Hottz ED, Oliveira MF, Nunes PC, Nogueira RM, Valls-de-Souza R, Da Poian AT, Weyrich AS, Zimmerman GA, Bozza PT, Bozza FA. Dengue induces platelet activation, mitochondrial dysfunction and cell death through mechanisms that involve DC-SIGN and caspases. J Thromb Haemost : 951–962, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hui P, Cook DJ, Lim W, Fraser GA, Arnold DM. The frequency and clinical significance of thrombocytopenia complicating critical illness: a systematic review. Chest : 271–278, 2011. [DOI] [PubMed] [Google Scholar]
- 182.Hwaiz R, Rahman M, Syk I, Zhang E, Thorlacius H. Rac1-dependent secretion of platelet-derived CCL5 regulates neutrophil recruitment via activation of alveolar macrophages in septic lung injury. J Leukocyte Biol : 975–984, 2015. [DOI] [PubMed] [Google Scholar]
- 183.Hyman MC, Petrovic-Djergovic D, Visovatti SH, Liao H, Yanamadala S, Bouis D, Su EJ, Lawrence DA, Broekman MJ, Marcus AJ, Pinsky DJ. Self-regulation of inflammatory cell trafficking in mice by the leukocyte surface apyrase CD39. J Clin Invest : 1136–1149, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Iannacone M, Sitia G, Isogawa M, Marchese P, Castro MG, Lowenstein PR, Chisari FV, Ruggeri ZM, Guidotti LG. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nature Med : 1167–1169, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Iannacone M, Sitia G, Isogawa M, Whitmire JK, Marchese P, Chisari FV, Ruggeri ZM, Guidotti LG. Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci USA : 629–634, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Idell S, Maunder R, Fein AM, Switalska HI, Tuszynski GP, McLarty J, Niewiarowski S. Platelet-specific alpha-granule proteins and thrombospondin in bronchoalveolar lavage in the adult respiratory distress syndrome. Chest : 1125–1132, 1989. [DOI] [PubMed] [Google Scholar]
- 187.Inami N, Nomura S, Kikuchi H, Kajiura T, Yamada K, Nakamori H, Takahashi N, Tsuda N, Hikosaka M, Masaki M, Iwasaka T. P-selectin and platelet-derived microparticles associated with monocyte activation markers in patients with pulmonary embolism. Clin Appl Thromb Hemostasis : 309–316, 2003. [DOI] [PubMed] [Google Scholar]
- 188.Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res : 1041–1048, 2003. [DOI] [PubMed] [Google Scholar]
- 189.Issekutz AC, Ripley M. The effect of intravascular neutrophil chemotactic factors on blood neutrophil and platelet kinetics. Am J Hematol : 157–171, 1986. [DOI] [PubMed] [Google Scholar]
- 190.Issekutz AC, Ripley M, Jackson JR. Role of neutrophils in the deposition of platelets during acute inflammation. Lab Invest : 716–724, 1983. [PubMed] [Google Scholar]
- 191.Italiano JE, Hartwig JH. Megakaryocyte development and platelet formation. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 27–49. [Google Scholar]
- 192.Italiano JE Jr, Mairuhu AT, Flaumenhaft R. Clinical relevance of microparticles from platelets and megakaryocytes. Curr Opin Hematol : 578–584, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jeffery PK, Wardlaw AJ, Nelson FC, Collins JV, Kay AB. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am Rev Respir Dis : 1745–1753, 1989. [DOI] [PubMed] [Google Scholar]
- 194.Jenne CN, Urrutia R, Kubes P. Platelets: bridging hemostasis, inflammation, immunity. Int J Lab Hematol : 254–261, 2013. [DOI] [PubMed] [Google Scholar]
- 195.Jenne CN, Wong CH, Petri B, Kubes P. The use of spinning-disk confocal microscopy for the intravital analysis of platelet dynamics in response to systemic and local inflammation. PloS One : e25109, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jenne CN, Wong CH, Zemp FJ, McDonald B, Rahman MM, Forsyth PA, McFadden G, Kubes P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe : 169–180, 2013. [DOI] [PubMed] [Google Scholar]
- 197.Johansson D, Shannon O, Rasmussen M. Platelet and neutrophil responses to Gram positive pathogens in patients with bacteremic infection. PloS One : e26928, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Johansson MW, Han ST, Gunderson KA, Busse WW, Jarjour NN, Mosher DF. Platelet activation, P-selectin, and eosinophil beta1-integrin activation in asthma. Am J Respir Crit Care Med : 498–507, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Johansson MW, Mosher DF. Activation of beta1 integrins on blood eosinophils by P-selectin. Am J Respir Cell Mol Biol : 889–897, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Josefsson EC, Dowling MR, Lebois M, Kile BT. The regulation of platelet life span. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 51–65. [Google Scholar]
- 201.Jung CJ, Yeh CY, Hsu RB, Lee CM, Shun CT, Chia JS. Endocarditis pathogen promotes vegetation formation by inducing intravascular neutrophil extracellular traps through activated platelets. Circulation : 571–581, 2015. [DOI] [PubMed] [Google Scholar]
- 202.Kahn F, Hurley S, Shannon O. Platelets promote bacterial dissemination in a mouse model of streptococcal sepsis. Microbes Infection : 669–676, 2013. [DOI] [PubMed] [Google Scholar]
- 203.Kalsch T, Elmas E, Nguyen XD, Suvajac N, Kluter H, Borggrefe M, Dempfle CE. Endotoxin-induced effects on platelets and monocytes in an in vivo model of inflammation. Basic Res Cardiol : 460–466, 2007. [DOI] [PubMed] [Google Scholar]
- 204.Kalvegren H, Skoglund C, Helldahl C, Lerm M, Grenegard M, Bengtsson T. Toll-like receptor 2 stimulation of platelets is mediated by purinergic P2X1-dependent Ca2+ mobilisation, cyclooxygenase and purinergic P2Y1 and P2Y12 receptor activation. Thromb Haemost : 398–407, 2010. [DOI] [PubMed] [Google Scholar]
- 205.Kamykowski J, Carlton P, Sehgal S, Storrie B. Quantitative immunofluorescence mapping reveals little functional coclustering of proteins within platelet alpha-granules. Blood : 1370–1373, 2011. [DOI] [PubMed] [Google Scholar]
- 206.Kanamarlapudi V, Owens SE, Saha K, Pope RJ, Mundell SJ. ARF6-dependent regulation of P2Y receptor traffic and function in human platelets. PloS One : e43532, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kaneider NC, Kaser A, Tilg H, Ricevuti G, Wiedermann CJ. CD40 ligand-dependent maturation of human monocyte-derived dendritic cells by activated platelets. Int J Immunopathol Pharmacol : 225–231, 2003. [DOI] [PubMed] [Google Scholar]
- 208.Kao KJ, Cook DJ, Scornik JC. Quantitative analysis of platelet surface HLA by W6/32 anti-HLA monoclonal antibody. Blood : 627–632, 1986. [PubMed] [Google Scholar]
- 209.Kaplan D, Casper TC, Elliott CG, Men S, Pendleton RC, Kraiss LW, Weyrich AS, Grissom CK, Zimmerman GA, Rondina MT. VTE Incidence and Risk Factors in Patients with Severe Sepsis and Septic Shock. Chest : 1224–1230, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kaplan JE, Moon DG, Weston LK, Minnear FL, Del Vecchio PJ, Shepard JM, Fenton JW 2nd. Platelets adhere to thrombin-treated endothelial cells in vitro. Am J Physiol Heart Circ Physiol : H423–H433, 1989. [DOI] [PubMed] [Google Scholar]
- 211.Kaplanski G, Porat R, Aiura K, Erban JK, Gelfand JA, Dinarello CA. Activated platelets induce endothelial secretion of interleukin-8 in vitro via an interleukin-1-mediated event. Blood : 2492–2495, 1993. [PubMed] [Google Scholar]
- 212.Kappelmayer J, Beke Debreceni I, Vida A, Antal-Szalmas P, Clemetson KJ, Nagy B Jr.. Distinct effects of Re- and S-forms of LPS on modulating platelet activation. J Thromb Hemostasis : 775–778, 2013. [DOI] [PubMed] [Google Scholar]
- 213.Karim ZA, Choi W, Whiteheart SW. Primary platelet signaling cascades and integrin-mediated signaling control ADP-ribosylation factor (Arf) 6-GTP levels during platelet activation and aggregation. J Biol Chem : 11995–12003, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Kasirer-Friede A, Kahn ML, Shattil SJ. Platelet integrins and immunoreceptors. Immunol Rev : 247–264, 2007. [DOI] [PubMed] [Google Scholar]
- 215.Katayama T, Ikeda Y, Handa M, Tamatani T, Sakamoto S, Ito M, Ishimura Y, Suematsu M. Immunoneutralization of glycoprotein Ibalpha attenuates endotoxin-induced interactions of platelets and leukocytes with rat venular endothelium in vivo. Circ Res : 1031–1037, 2000. [DOI] [PubMed] [Google Scholar]
- 216.Katoh N, Soga F, Nara T, Tamagawa-Mineoka R, Nin M, Kotani H, Masuda K, Kishimoto S. Effect of serotonin on the differentiation of human monocytes into dendritic cells. Clin Exp Immunol : 354–361, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Katz IR, Hoffmann MK, Zucker MB, Bell MK, Thorbecke GJ. A platelet-derived immunoregulatory serum factor with T cell affinity. J Immunol : 3199–3203, 1985. [PubMed] [Google Scholar]
- 218.Kerrigan AM, Navarro-Nunez L, Pyz E, Finney BA, Willment JA, Watson SP, Brown GD. Podoplanin-expressing inflammatory macrophages activate murine platelets via CLEC-2. J Thromb Haemost : 484–486, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kiefmann R, Heckel K, Schenkat S, Dorger M, Goetz AE. Role of p-selectin in platelet sequestration in pulmonary capillaries during endotoxemia. J Vasc Res : 473–481, 2006. [DOI] [PubMed] [Google Scholar]
- 220.Kissel K, Berber S, Nockher A, Santoso S, Bein G, Hackstein H. Human platelets target dendritic cell differentiation and production of proinflammatory cytokines. Transfusion : 818–827, 2006. [DOI] [PubMed] [Google Scholar]
- 221.Klinkhardt U, Bauersachs R, Adams J, Graff J, Lindhoff-Last E, Harder S. Clopidogrel but not aspirin reduces P-selectin expression and formation of platelet-leukocyte aggregates in patients with atherosclerotic vascular disease. Clin Pharmacol Ther : 232–241, 2003. [DOI] [PubMed] [Google Scholar]
- 222.Klockenbusch C, Walsh GM, Brown LM, Hoffman MD, Ignatchenko V, Kislinger T, Kast J. Global proteome analysis identifies active immunoproteasome subunits in human platelets. Mol Cell Proteomics : 3308–3319, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kolandaivelu K, Bhatt D. Novel antiplatelet therapies. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 1185–1213. [Google Scholar]
- 224.Kor DJ, Erlich J, Gong MN, Malinchoc M, Carter RE, Gajic O, Talmor DS. Association of prehospitalization aspirin therapy and acute lung injury: results of a multicenter international observational study of at-risk patients. Crit Care Med : 2393–2400, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kor DJ, Talmor DS, Banner-Goodspeed VM, Carter RE, Hinds R, Park PK, Gajic O, Gong MN. Lung Injury Prevention with Aspirin (LIPS-A): a protocol for a multicentre randomised clinical trial in medical patients at high risk of acute lung injury. BMJ Open : e001606, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kornerup KN, Page CP. The role of platelets in the pathophysiology of asthma. Platelets : 319–328, 2007. [DOI] [PubMed] [Google Scholar]
- 227.Kornerup KN, Salmon GP, Pitchford SC, Liu WL, Page CP. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J Appl Physiol : 758–767, 2010. [DOI] [PubMed] [Google Scholar]
- 228.Koupenova M, Vitseva O, MacKay CR, Beaulieu LM, Benjamin EJ, Mick E, Kurt-Jones EA, Ravid K, Freedman JE. Platelet-TLR7 mediates host survival and platelet count during viral infection in the absence of platelet-dependent thrombosis. Blood : 791–802, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kowal K, Pampuch A, Kowal-Bielecka O, DuBuske LM, Bodzenta-Lukaszyk A. Platelet activation in allergic asthma patients during allergen challenge with Dermatophagoides pteronyssinus. Clin Exp Allergy : 426–432, 2006. [DOI] [PubMed] [Google Scholar]
- 230.Kraemer BF, Borst O, Gehring EM, Schoenberger T, Urban B, Ninci E, Seizer P, Schmidt C, Bigalke B, Koch M, Martinovic I, Daub K, Merz T, Schwanitz L, Stellos K, Fiesel F, Schaller M, Lang F, Gawaz M, Lindemann S. PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). J Mol Med : 1277–1288, 2010. [DOI] [PubMed] [Google Scholar]
- 231.Kraemer BF, Campbell RA, Schwertz H, Cody MJ, Franks Z, Tolley ND, Kahr WH, Lindemann S, Seizer P, Yost CC, Zimmerman GA, Weyrich AS. Novel anti-bacterial activities of beta-defensin 1 in human platelets: suppression of pathogen growth and signaling of neutrophil extracellular trap formation. PLoS Pathogens : e1002355, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Kravis TC, Henson PM. Accumulation of platelets at sites of antigen-antibody-mediated injury: a possible role for IgE antibody and mast cells. J Immunol : 1569–1573, 1977. [PubMed] [Google Scholar]
- 233.Kuckleburg CJ, Yates CM, Kalia N, Zhao Y, Nash GB, Watson SP, Rainger GE. Endothelial cell-borne platelet bridges selectively recruit monocytes in human and mouse models of vascular inflammation. Cardiovasc Res : 134–141, 2011. [DOI] [PubMed] [Google Scholar]
- 234.Laffont B, Corduan A, Ple H, Duchez AC, Cloutier N, Boilard E, Provost P. Activated platelets can deliver mRNA regulatory Ago2*microRNA complexes to endothelial cells via microparticles. Blood : 253–261, 2013. [DOI] [PubMed] [Google Scholar]
- 235.Laidlaw TM, Kidder MS, Bhattacharyya N, Xing W, Shen S, Milne GL, Castells MC, Chhay H, Boyce JA. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood : 3790–3798, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Lam FW, Burns AR, Smith CW, Rumbaut RE. Platelets enhance neutrophil transendothelial migration via P-selectin glycoprotein ligand-1. Am J Physiol Heart Circ Physiol : H468–H475, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Langer HF, Daub K, Braun G, Schonberger T, May AE, Schaller M, Stein GM, Stellos K, Bueltmann A, Siegel-Axel D, Wendel HP, Aebert H, Roecken M, Seizer P, Santoso S, Wesselborg S, Brossart P, Gawaz M. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol : 1463–1470, 2007. [DOI] [PubMed] [Google Scholar]
- 238.Le VB, Schneider JG, Boergeling Y, Berri F, Ducatez M, Guerin JL, Adrian I, Errazuriz-Cerda E, Frasquilho S, Antunes L, Lina B, Bordet JC, Jandrot-Perrus M, Ludwig S, Riteau B. Platelet activation and aggregation promote lung inflammation and influenza virus pathogenesis. Am J Respir Crit Care Med : 804–819, 2015. [DOI] [PubMed] [Google Scholar]
- 239.Lee MJ, Thangada S, Claffey KP, Ancellin N, Liu CH, Kluk M, Volpi M, Sha'afi RI, Hla T. Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell : 301–312, 1999. [DOI] [PubMed] [Google Scholar]
- 240.Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med : 689–691, 2010. [DOI] [PubMed] [Google Scholar]
- 241.Levin J. The evolution of mammalian platelets. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 3–25. [Google Scholar]
- 242.Levy BD, Serhan CN. Resolution of acute inflammation in the lung. Annu Rev Physiol : 467–492, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Li G, Kim YJ, Mantel C, Broxmeyer HE. P-selectin enhances generation of CD14+CD16+ dendritic-like cells and inhibits macrophage maturation from human peripheral blood monocytes. J Immunol : 669–677, 2003. [DOI] [PubMed] [Google Scholar]
- 244.Li JJ, Huang YQ, Basch R, Karpatkin S. Thrombin induces the release of angiopoietin-1 from platelets. Thromb Haemost : 204–206, 2001. [PubMed] [Google Scholar]
- 245.Li N. Platelet-lymphocyte cross-talk. J Leukocyte Biol : 1069–1078, 2008. [DOI] [PubMed] [Google Scholar]
- 246.Li N, Soop A, Sollevi A, Hjemdahl P. Multi-cellular activation in vivo by endotoxin in humans–limited protection by adenosine infusion. Thromb Haemost : 381–387, 2000. [PubMed] [Google Scholar]
- 247.Li Z, Yang F, Dunn S, Gross AK, Smyth SS. Platelets as immune mediators: their role in host defense responses and sepsis. Thromb Res : 184–188, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Likos AM, Kelvin DJ, Cameron CM, Rowe T, Kuehnert MJ, Norris PJ. Influenza viremia and the potential for blood-borne transmission. Transfusion : 1080–1088, 2007. [DOI] [PubMed] [Google Scholar]
- 249.Lindemann S, Tolley ND, Dixon DA, McIntyre TM, Prescott SM, Zimmerman GA, Weyrich AS. Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J Cell Biol : 485–490, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Lindemann S, Tolley ND, Eyre JR, Kraiss LW, Mahoney TM, Weyrich AS. Integrins regulate the intracellular distribution of eukaryotic initiation factor 4E in platelets. A checkpoint for translational control. J Biol Chem : 33947–33951, 2001. [DOI] [PubMed] [Google Scholar]
- 251.Liu F, Morris S, Epps J, Carroll R. Demonstration of an activation regulated NF-kappaB/I-kappaBalpha complex in human platelets. Thromb Res : 199–203, 2002. [DOI] [PubMed] [Google Scholar]
- 252.Liverani E, Kilpatrick LE, Tsygankov AY, Kunapuli SP. The role of P2Y(1)(2) receptor and activated platelets during inflammation. Current Drug Targets : 720–728, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Lo SK, Burhop KE, Kaplan JE, Malik AB. Role of platelets in maintenance of pulmonary vascular permeability to protein. Am J Physiol Heart Circ Physiol : H763–H771, 1988. [DOI] [PubMed] [Google Scholar]
- 254.London NR, Zhu W, Bozza FA, Smith MC, Greif DM, Sorensen LK, Chen L, Kaminoh Y, Chan AC, Passi SF, Day CW, Barnard DL, Zimmerman GA, Krasnow MA, Li DY. Targeting Robo4-dependent Slit signaling to survive the cytokine storm in sepsis and influenza. Science Transl Med : 23ra19, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Loof TG, Morgelin M, Johansson L, Oehmcke S, Olin AI, Dickneite G, Norrby-Teglund A, Theopold U, Herwald H. Coagulation, an ancestral serine protease cascade, exerts a novel function in early immune defense. Blood : 2589–2598, 2011. [DOI] [PubMed] [Google Scholar]
- 256.Looney MR, Bhattacharya J. Live imaging of the lung. Annu Rev Physiol : 431–445, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Looney MR, Nguyen JX, Hu Y, Van Ziffle JA, Lowell CA, Matthay MA. Platelet depletion and aspirin treatment protect mice in a two-event model of transfusion-related acute lung injury. J Clin Invest : 3450–3461, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Lopez-Delgado JC, Rovira A, Esteve F, Rico N, Manez Mendiluce R, Ballus Noguera J, Berrade J. Thrombocytopenia as a mortality risk factor in acute respiratory failure in H1N1 influenza. Swiss Med Weekly : w13788, 2013. [DOI] [PubMed] [Google Scholar]
- 259.Loppnow H, Bil R, Hirt S, Schonbeck U, Herzberg M, Werdan K, Rietschel ET, Brandt E, Flad HD. Platelet-derived interleukin-1 induces cytokine production, but not proliferation of human vascular smooth muscle cells. Blood : 134–141, 1998. [PubMed] [Google Scholar]
- 260.Lorant DE, Patel KD, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Coexpression of GMP-140 and PAF by endothelium stimulated by histamine or thrombin: a juxtacrine system for adhesion and activation of neutrophils. J Cell Biol : 223–234, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Loria GD, Romagnoli PA, Moseley NB, Rucavado A, Altman JD. Platelets support a protective immune response to LCMV by preventing splenic necrosis. Blood : 940–950, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Love MS, Millholland MG, Mishra S, Kulkarni S, Freeman KB, Pan W, Kavash RW, Costanzo MJ, Jo H, Daly TM, Williams DR, Kowalska MA, Bergman LW, Poncz M, DeGrado WF, Sinnis P, Scott RW, Greenbaum DC. Platelet factor 4 activity against P. falciparum and its translation to nonpeptidic mimics as antimalarials. Cell Host Microbe : 815–823, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Lukacs NW, John A, Berlin A, Bullard DC, Knibbs R, Stoolman LM. E- and P-selectins are essential for the development of cockroach allergen-induced airway responses. J Immunol : 2120–2125, 2002. [DOI] [PubMed] [Google Scholar]
- 264.Ma L, Dorling A. The roles of thrombin and protease-activated receptors in inflammation. Semin Immunopathol : 63–72, 2012. [DOI] [PubMed] [Google Scholar]
- 265.Mackman N. Triggers, targets and treatments for thrombosis. Nature : 914–918, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Mahoney TS, Weyrich AS, Dixon DA, McIntyre T, Prescott SM, Zimmerman GA. Cell adhesion regulates gene expression at translational checkpoints in human myeloid leukocytes. Proc Natl Acad Sci USA : 10284–10289, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Maitre B, Mangin PH, Eckly A, Heim V, Cazenave JP, Lanza F, Hanau D, Gachet C. Immature myeloid dendritic cells capture and remove activated platelets from preformed aggregates. J Thromb Haemost : 2262–2272, 2010. [DOI] [PubMed] [Google Scholar]
- 268.Majno G, Palade MD. Studies on inflammation. J Biophys Biochem Cytol : 571–605, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Major HD, Campbell RA, Silver RM, Branch DW, Weyrich AS. Synthesis of sFlt-1 by platelet-monocyte aggregates contributes to the pathogenesis of preeclampsia. Am J Obstet Gynecol : 541–547, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Malaver E, Romaniuk MA, D'Atri LP, Pozner RG, Negrotto S, Benzadon R, Schattner M. NF-kappaB inhibitors impair platelet activation responses. J Thromb Haemost : 1333–1343, 2009. [DOI] [PubMed] [Google Scholar]
- 271.Mammoto T, Jiang A, Jiang E, Mammoto A. Platelet-rich plasma extract prevents pulmonary edema through angiopoietin-Tie2 signaling. Am J Respir Cell Mol Biol : 56–64, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L, Wiedenbauer EM, Krautgartner WD, Stoiber W, Belohradsky BH, Rieber N, Kormann M, Koller B, Roscher A, Roos D, Griese M, Eickelberg O, Doring G, Mall MA, Hartl D. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nature Med : 1018–1023, 2010. [DOI] [PubMed] [Google Scholar]
- 273.Marcus AJ, Weksler BB, Jaffe EA, Broekman MJ. Synthesis of prostacyclin from platelet-derived endoperoxides by cultured human endothelial cells. J Clin Invest : 979–986, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Martinson J, Bae J, Klingemann HG, Tam Y. Activated platelets rapidly up-regulate CD40L expression and can effectively mature and activate autologous ex vivo differentiated DC. Cytotherapy : 487–497, 2004. [DOI] [PubMed] [Google Scholar]
- 275.Matsuda H, Ushio H, Geba GP, Askenase PW. Human platelets can initiate T cell-dependent contact sensitivity through local serotonin release mediated by IgE antibodies. J Immunol : 2891–2897, 1997. [PubMed] [Google Scholar]
- 276.Matthay MA, Folkesson HG, Clerici C. Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev : 569–600, 2002. [DOI] [PubMed] [Google Scholar]
- 277.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest : 2731–2740, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol : 319–327, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D'Angelo A, Bianchi ME, Rovere-Querini P, Manfredi AA. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost : 2074–2088, 2014. [DOI] [PubMed] [Google Scholar]
- 280.Mause SF, von Hundelshausen P, Zernecke A, Koenen RR, Weber C. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arterioscler Thromb Vasc Biol : 1512–1518, 2005. [DOI] [PubMed] [Google Scholar]
- 281.McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe : 324–333, 2012. [DOI] [PubMed] [Google Scholar]
- 282.McFadyen JD, Jackson SP. Differentiating haemostasis from thrombosis for therapeutic benefit. Thromb Haemost : 859–867, 2013. [DOI] [PubMed] [Google Scholar]
- 283.McFadyen JD, Kaplan ZS. Platelets are not just for clots. Transfusion Med Rev : 110–119, 2015. [DOI] [PubMed] [Google Scholar]
- 284.McIntyre TM, Zimmerman GA, Prescott SM. Leukotrienes C4 and D4 stimulate human endothelial cells to synthesize platelet-activating factor and bind neutrophils. Proc Natl Acad Sci USA : 2204–2208, 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.McIntyre TM, Zimmerman GA, Satoh K, Prescott SM. Cultured endothelial cells synthesize both platelet-activating factor and prostacyclin in response to histamine, bradykinin, and adenosine triphosphate. J Clin Invest : 271–280, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.McMorran BJ, Marshall VM, de Graaf C, Drysdale KE, Shabbar M, Smyth GK, Corbin JE, Alexander WS, Foote SJ. Platelets kill intraerythrocytic malarial parasites and mediate survival to infection. Science : 797–800, 2009. [DOI] [PubMed] [Google Scholar]
- 287.McMorran BJ, Wieczorski L, Drysdale KE, Chan JA, Huang HM, Smith C, Mitiku C, Beeson JG, Burgio G, Foote SJ. Platelet factor 4 and Duffy antigen required for platelet killing of Plasmodium falciparum. Science : 1348–1351, 2012. [DOI] [PubMed] [Google Scholar]
- 288.Medzhitov R. Origin and physiological roles of inflammation. Nature : 428–435, 2008. [DOI] [PubMed] [Google Scholar]
- 289.Mendez JL, Hubmayr RD. New insights into the pathology of acute respiratory failure. Curr Opin Crit Care : 29–36, 2005. [DOI] [PubMed] [Google Scholar]
- 290.Meng W, Paunel-Gorgulu A, Flohe S, Hoffmann A, Witte I, MacKenzie C, Baldus SE, Windolf J, Logters TT. Depletion of neutrophil extracellular traps in vivo results in hypersusceptibility to polymicrobial sepsis in mice. Critical Care : R137, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Meshorer E, Misteli T. Splicing misplaced. Cell : 317–318, 2005. [DOI] [PubMed] [Google Scholar]
- 292.Metcalf Pate KA, Lyons CE, Dorsey JL, Shirk EN, Queen SE, Adams RJ, Gama L, Morrell CN, Mankowski JL. Platelet activation and platelet-monocyte aggregate formation contribute to decreased platelet count during acute simian immunodeficiency virus infection in pig-tailed macaques. J Infect Dis : 874–883, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Metzger WJ, Sjoerdsma K, Richerson HB, Moseley P, Zavala D, Monick M, Hunninghake GW. Platelets in bronchoalveolar lavage from asthmatic patients and allergic rabbits with allergen-induced late phase responses. Agents Actions Suppl 21: 151–159, 1987. [DOI] [PubMed] [Google Scholar]
- 294.Michels M, van der Ven AJ, Djamiatun K, Fijnheer R, de Groot PG, Griffioen AW, Sebastian S, Faradz SM, de Mast Q. Imbalance of angiopoietin-1 and angiopoetin-2 in severe dengue and relationship with thrombocytopenia, endothelial activation, and vascular stability. Am J Tropical Med Hygiene : 943–946, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Michelson AD. Antiplatelet therapies for the treatment of cardiovascular disease. Nature Rev Drug Disc : 154–169, 2010. [DOI] [PubMed] [Google Scholar]
- 296.Michelson AD. Platelets (3rd ed.). Amsterdam: Elsevier, 2013, p. 1–1353. [Google Scholar]
- 297.Mileno MD, Margolis NH, Clark BD, Dinarello CA, Burke JF, Gelfand JA. Coagulation of whole blood stimulates interleukin-1 beta gene expression. J Infect Dis : 308–311, 1995. [DOI] [PubMed] [Google Scholar]
- 298.Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, Song LZ, Knight JM, Creighton CJ, Luong A, Kheradmand F, Corry DB. Cleavage of fibrinogen by proteinases elicits allergic responses through Toll-like receptor 4. Science : 792–796, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Minnear FL, Patil S, Bell D, Gainor JP, Morton CA. Platelet lipid(s) bound to albumin increases endothelial electrical resistance: mimicked by LPA. Am J Physiol Lung Cell Mol Physiol : L1337–L1344, 2001. [DOI] [PubMed] [Google Scholar]
- 300.Moalli F, Cupovic J, Thelen F, Halbherr P, Fukui Y, Narumiya S, Ludewig B, Stein JV. Thromboxane A2 acts as tonic immunoregulator by preferential disruption of low-avidity CD4+ T cell-dendritic cell interactions. J Exp Med : 2507–2517, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Monsalvo AC, Batalle JP, Lopez MF, Krause JC, Klemenc J, Hernandez JZ, Maskin B, Bugna J, Rubinstein C, Aguilar L, Dalurzo L, Libster R, Savy V, Baumeister E, Aguilar L, Cabral G, Font J, Solari L, Weller KP, Johnson J, Echavarria M, Edwards KM, Chappell JD, Crowe JE Jr, Williams JV, Melendi GA, Polack FP. Severe pandemic 2009 H1N1 influenza disease due to pathogenic immune complexes. Nature Med : 195–199, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Moore KL, Thompson LF. P-selectin (CD62) binds to subpopulations of human memory T lymphocytes and natural killer cells. Biochem Biophys Res Commun : 173–181, 1992. [DOI] [PubMed] [Google Scholar]
- 303.Morel O, Toti F, Hugel B, Bakouboula B, Camoin-Jau L, Dignat-George F, Freyssinet JM. Procoagulant microparticles: disrupting the vascular homeostasis equation? Arterioscler Thromb Vasc Biol : 2594–2604, 2006. [DOI] [PubMed] [Google Scholar]
- 304.Morowski M, Vogtle T, Kraft P, Kleinschnitz C, Stoll G, Nieswandt B. Only severe thrombocytopenia results in bleeding and defective thrombus formation in mice. Blood : 4938–4947, 2013. [DOI] [PubMed] [Google Scholar]
- 305.Morrell CN, Aggrey AA, Chapman LM, Modjeski KL. Emerging roles for platelets as immune and inflammatory cells. Blood : 2759–2767, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nature Rev Immunol : 958–969, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Muller F, Mutch NJ, Schenk WA, Smith SA, Esterl L, Spronk HM, Schmidbauer S, Gahl WA, Morrissey JH, Renne T. Platelet polyphosphates are proinflammatory and procoagulant mediators in vivo. Cell : 1143–1156, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Murciano JC, Harshaw D, Neschis DG, Koniaris L, Bdeir K, Medinilla S, Fisher AB, Golden MA, Cines DB, Nakada MT, Muzykantov VR. Platelets inhibit the lysis of pulmonary microemboli. Am J Physiol Lung Cell Mol Physiol : L529–L539, 2002. [DOI] [PubMed] [Google Scholar]
- 309.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature Rev Immunol : 723–737, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Mutch NJ. The role of platelets in fibrinolysis. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 469–485. [Google Scholar]
- 311.Nachman RL, Polley M. The platelet as an inflammatory cell. Trans Am Clin Climatol Assoc : 38–43, 1979. [PMC free article] [PubMed] [Google Scholar]
- 312.Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med : 1261–1270, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Nachman RL, Weksler B, Ferris B. Characterization of human platelet vascular permeability-enhancing activity. J Clin Invest : 549–556, 1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Narasaraju T, Yang E, Samy RP, Ng HH, Poh WP, Liew AA, Phoon MC, van Rooijen N, Chow VT. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am J Pathol : 199–210, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Natarajan V, Dudek SM, Jacobson JR, Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L, Bittman R, Weichselbaum R, Berdyshev E, Garcia JG. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol : 6–17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Nauseef WM. Editorial: Nyet to NETs? A pause for healthy skepticism. J Leukocyte Biol : 353–355, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Navarro-Nunez L, Langan SA, Nash GB, Watson SP. The physiological and pathophysiological roles of platelet CLEC-2. Thromb Haemost : 991–998, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Newman PJ, Newman DK. PECAM1. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 283–293. [Google Scholar]
- 319.Nicolls MR, Tamosiuniene R, Babu AN, Voelkel NF. Interactions of pulmonary endothelial cells with immune cells and platelets: Implications for disease pathogenesis. In: The Pulmonary Endothelium: Function in Health and Disease, edited by Voelkel NF, Rounds S. West Sussex, UK: Wiley, 2009, p. 417–436. [Google Scholar]
- 320.Nieuwland R, van der Pol E, Gardiner C, Sturk A. Platelet-derived microparticles. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 453–467. [Google Scholar]
- 321.Nishimura S, Nagasaki M, Kunishima S, Sawaguchi A, Sakata A, Sakaguchi H, Ohmori T, Manabe I, Italiano JE Jr, Ryu T, Takayama N, Komuro I, Kadowaki T, Eto K, Nagai R. IL-1alpha induces thrombopoiesis through megakaryocyte rupture in response to acute platelet needs. J Cell Biol : 453–466, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Nishimura S, Manabe I, Nagasaki M, Kakuta S, Iwakura Y, Takayama N, Ooehara J, Otsu M, Kamiya A, Petrich BG, Urano T, Kadono T, Sato S, Aiba A, Yamashita H, Sugiura S, Kadowaki T, Nakauchi H, Eto K, Nagai R. In vivo imaging visualizes discoid platelet aggregations without endothelium disruption and implicates contribution of inflammatory cytokine and integrin signaling. Blood : e45–56, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Norgaard M, Jensen AO, Engebjerg MC, Farkas DK, Thomsen RW, Cha S, Zhao S, Sorensen HT. Long-term clinical outcomes of patients with primary chronic immune thrombocytopenia: a Danish population-based cohort study. Blood : 3514–3520, 2011. [DOI] [PubMed] [Google Scholar]
- 324.Nurden A, Nurden P. Advances in our understanding of the molecular basis of disorders of platelet function. J Thromb Haemostasis Suppl 1: 76–91, 2011. [DOI] [PubMed] [Google Scholar]
- 325.O'Neal HR Jr, Koyama T, Koehler EA, Siew E, Curtis BR, Fremont RD, May AK, Bernard GR, Ware LB. Prehospital statin and aspirin use and the prevalence of severe sepsis and acute lung injury/acute respiratory distress syndrome. Crit Care Med : 1343–1350, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.O'Sullivan BP, Linden MD, Frelinger AL 3rd, Barnard MR, Spencer-Manzon M, Morris JE, Salem RO, Laposata M, Michelson AD. Platelet activation in cystic fibrosis. Blood : 4635–4641, 2005. [DOI] [PubMed] [Google Scholar]
- 327.O'Sullivan BP, Michelson AD. The inflammatory role of platelets in cystic fibrosis. Am J Respir Crit Care Med : 483–490, 2006. [DOI] [PubMed] [Google Scholar]
- 328.Obinata H, Hla T. Sphingosine 1-phosphate in coagulation and inflammation. Semin Immunopathol : 73–91, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Okazaki T, Ni A, Ayeni OA, Baluk P, Yao LC, Vossmeyer D, Zischinsky G, Zahn G, Knolle J, Christner C, McDonald DM. Alpha5beta1 Integrin blockade inhibits lymphangiogenesis in airway inflammation. Am J Pathol : 2378–2387, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Ortiz-Munoz G, Mallavia B, Bins A, Headley M, Krummel MF, Looney MR. Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood : 2625–2634, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Ostrovsky L, King AJ, Bond S, Mitchell D, Lorant DE, Zimmerman GA, Larsen R, Niu XF, Kubes P. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood : 3028–3036, 1998. [PubMed] [Google Scholar]
- 332.Pabla R, Weyrich AS, Dixon DA, Bray PF, McIntyre TM, Prescott SM, Zimmerman GA. Integrin-dependent control of translation: engagement of integrin alphaIIbbeta3 regulates synthesis of proteins in activated human platelets. J Cell Biol : 175–184, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Packham MA, Nishizawa EE, Mustard JF. Response of platelets to tissue injury. Biochem Pharmacol Suppl: 171–184, 1968. [DOI] [PubMed] [Google Scholar]
- 334.Pandey S, Kawai T, Akira S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harbor Perspect Med : a016246, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Panigrahi S, Ma Y, Hong L, Gao D, West XZ, Salomon RG, Byzova TV, Podrez EA. Engagement of platelet toll-like receptor 9 by novel endogenous ligands promotes platelet hyperreactivity and thrombosis. Circ Res : 103–112, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Passacquale G, Vamadevan P, Pereira L, Hamid C, Corrigall V, Ferro A. Monocyte-platelet interaction induces a pro-inflammatory phenotype in circulating monocytes. PloS One : e25595, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Patil S, Kaplan JE, Minnear FL. Protein, not adenosine or adenine nucleotides, mediates platelet decrease in endothelial permeability. Am J Physiol Heart Circ Physiol : H2304–H2311, 1997. [DOI] [PubMed] [Google Scholar]
- 338.Paty PS, Sherman PF, Shepard JM, Malik AB, Kaplan JE. Role of adenosine in platelet-mediated reduction in pulmonary vascular permeability. Am J Physiol Heart Circ Physiol : H771–H777, 1992. [DOI] [PubMed] [Google Scholar]
- 339.Pearse DB, Brower RG, Adkinson NF Jr, Sylvester JT. Spontaneous injury in isolated sheep lungs: role of perfusate leukocytes and platelets. J Appl Physiol : 1287–1296, 1989. [DOI] [PubMed] [Google Scholar]
- 340.Peng X, Hassoun PM, Sammani S, McVerry BJ, Burne MJ, Rabb H, Pearse D, Tuder RM, Garcia JG. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med : 1245–1251, 2004. [DOI] [PubMed] [Google Scholar]
- 341.Peters CG, Michelson AD, Flaumenhaft R. Granule exocytosis is required for platelet spreading: differential sorting of alpha-granules expressing VAMP-7. Blood : 199–206, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Peters DM, Vadasz I, Wujak L, Wygrecka M, Olschewski A, Becker C, Herold S, Papp R, Mayer K, Rummel S, Brandes RP, Gunther A, Waldegger S, Eickelberg O, Seeger W, Morty RE. TGF-beta directs trafficking of the epithelial sodium channel ENaC which has implications for ion and fluid transport in acute lung injury. Proc Natl Acad Sci USA : E374–383, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Peyron F, Polack B, Lamotte D, Kolodie L, Ambroise-Thomas P. Plasmodium falciparum growth inhibition by human platelets in vitro. Parasitology 99 Pt : 317–322, 1989. [DOI] [PubMed] [Google Scholar]
- 344.Pitchford SC, Momi S, Baglioni S, Casali L, Giannini S, Rossi R, Page CP, Gresele P. Allergen induces the migration of platelets to lung tissue in allergic asthma. Am J Respir Crit Care Med : 604–612, 2008. [DOI] [PubMed] [Google Scholar]
- 345.Pitchford SC, Momi S, Giannini S, Casali L, Spina D, Page CP, Gresele P. Platelet P-selectin is required for pulmonary eosinophil and lymphocyte recruitment in a murine model of allergic inflammation. Blood : 2074–2081, 2005. [DOI] [PubMed] [Google Scholar]
- 346.Pitchford SC, Riffo-Vasquez Y, Sousa A, Momi S, Gresele P, Spina D, Page CP. Platelets are necessary for airway wall remodeling in a murine model of chronic allergic inflammation. Blood : 639–647, 2004. [DOI] [PubMed] [Google Scholar]
- 347.Pitchford SC, Yano H, Lever R, Riffo-Vasquez Y, Ciferri S, Rose MJ, Giannini S, Momi S, Spina D, O'Connor B, Gresele P, Page CP. Platelets are essential for leukocyte recruitment in allergic inflammation. J Allergy Clin Immunol : 109–118, 2003. [DOI] [PubMed] [Google Scholar]
- 348.Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nature Rev Immunol : 803–815, 2007. [DOI] [PubMed] [Google Scholar]
- 349.Pober JS, Sessa WC. Inflammation and the blood microvascular system. Cold Spring Harbor Perspect Biol : a016345, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear AR, Linden J. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler Thromb Vasc Biol : 2392–2399, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Pollitt AY, Hughes CE, Watson SP. GPVI and CLEC-2. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 215–231. [Google Scholar]
- 352.Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood : 2549–2554, 2001. [DOI] [PubMed] [Google Scholar]
- 353.Prescott SM, Weyrich AS, Zimmerman GA. Classification of venous thromboembolism (VTE). The clot is hot: inflammation, myeloid leukocytes, and venous thromboembolism. J Thromb Haemost : 2571–2573, 2005. [DOI] [PubMed] [Google Scholar]
- 354.Prescott SM, Zimmerman GA, Stafforini DM, McIntyre TM. Platelet-activating factor and related lipid mediators. Annu Rev Biochem : 419–445, 2000. [DOI] [PubMed] [Google Scholar]
- 355.Provost P. Platelet microRNAs. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 91–101. [Google Scholar]
- 356.Qian K, Xie F, Gibson AW, Edberg JC, Kimberly RP, Wu J. Functional expression of IgA receptor FcalphaRI on human platelets. J Leukoc Biol : 1492–1500, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Qu Z, Chaikof EL. Interface between hemostasis and adaptive immunity. Curr Opin Immunol : 634–642, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Rahman M, Gustafsson D, Wang Y, Thorlacius H, Braun OO. Ticagrelor reduces neutrophil recruitment and lung damage in abdominal sepsis. Platelets : 257–263, 2014. [DOI] [PubMed] [Google Scholar]
- 359.Rahman M, Roller J, Zhang S, Syk I, Menger MD, Jeppsson B, Thorlacius H. Metalloproteinases regulate CD40L shedding from platelets and pulmonary recruitment of neutrophils in abdominal sepsis. Inflammation Res : 571–579, 2012. [DOI] [PubMed] [Google Scholar]
- 360.Rex S, Beaulieu LM, Perlman DH, Vitseva O, Blair PS, McComb ME, Costello CE, Freedman JE. Immune versus thrombotic stimulation of platelets differentially regulates signalling pathways, intracellular protein-protein interactions, and alpha-granule release. Thromb Haemost : 97–110, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Risitano A, Beaulieu LM, Vitseva O, Freedman JE. Platelets and platelet-like particles mediate intercellular RNA transfer. Blood : 6288–6295, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, Schattner M. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res : 235–243, 2014. [DOI] [PubMed] [Google Scholar]
- 363.Rodrigues RS, Zimmerman GA. Pulmonary endothelial interactions with leukocytes and platelets. In: The Pulmonary Endothelium: Function in Health and Disease, edited by Voelkel NF, Rounds S. West Sussex, UK: Wiley, 2009, p. 143–166. [Google Scholar]
- 364.Romo GM, Dong JF, Schade AJ, Gardiner EE, Kansas GS, Li CQ, McIntire LV, Berndt MC, Lopez JA. The glycoprotein Ib-IX-V complex is a platelet counterreceptor for P-selectin. J Exp Med : 803–814, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Rondina MT, Brewster B, Grissom CK, Zimmerman GA, Kastendieck DH, Harris ES, Weyrich AS. In vivo platelet activation in critically ill patients with primary 2009 influenza A(H1N1). Chest : 1490–1495, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Rondina MT, Carlisle M, Fraughton T, Brown SM, Miller RR 3rd, Harris ES, Weyrich AS, Zimmerman GA, Supiano MA, Grissom CK. Platelet-monocyte aggregate formation and mortality risk in older patients with severe sepsis and septic shock. J Gerontol Ser A Biol Sci Med Sci : 225–231, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, Grissom CK, Weyrich AS, Zimmerman GA. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost : : 748–758, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Rondina MT, Weyrich AS. Dengue virus pirates human platelets. Blood : 286–287, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ Res : 1506–1519, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Rossaint J, Herter JM, Van Aken H, Napirei M, Doring Y, Weber C, Soehnlein O, Zarbock A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood : 2573–2584, 2014. [DOI] [PubMed] [Google Scholar]
- 371.Rossaint J, Zarbock A. Platelets in leucocyte recruitment and function. Cardiovasc Res : 386–395, 2015. [DOI] [PubMed] [Google Scholar]
- 372.Rowley JW, Oler AJ, Tolley ND, Hunter BN, Low EN, Nix DA, Yost CC, Zimmerman GA, Weyrich AS. Genome-wide RNA-seq analysis of human and mouse platelet transcriptomes. Blood : e101–e111, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Rowley JW, Schwertz H, Weyrich AS. Platelet mRNA: the meaning behind the message. Curr Opin Hematol : 385–391, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Ruggeri ZM, Jackson SP. Platelet thrombus formation in flowing blood. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 399–424. [Google Scholar]
- 375.Rumbaut RE, Bellera RV, Randhawa JK, Shrimpton CN, Dasgupta SK, Dong JF, Burns AR. Endotoxin enhances microvascular thrombosis in mouse cremaster venules via a TLR4-dependent, neutrophil-independent mechanism. Am J Physiol Heart Circ Physiol : H1671–H1679, 2006. [DOI] [PubMed] [Google Scholar]
- 376.Saffarzadeh M, Juenemann C, Queisser MA, Lochnit G, Barreto G, Galuska SP, Lohmeyer J, Preissner KT. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PloS One : e32366, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377.Saffarzadeh M, Preissner KT. Fighting against the dark side of neutrophil extracellular traps in disease: manoeuvres for host protection. Curr Opin Hematol : 3–9, 2013. [DOI] [PubMed] [Google Scholar]
- 378.Sammani S, Moreno-Vinasco L, Mirzapoiazova T, Singleton PA, Chiang ET, Evenoski CL, Wang T, Mathew B, Husain A, Moitra J, Sun X, Nunez L, Jacobson JR, Dudek SM, Natarajan V, Garcia JG. Differential effects of sphingosine 1-phosphate receptors on airway and vascular barrier function in the murine lung. Am J Respir Cell Mol Biol : 394–402, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Santoso S, Kalb R, Kiefel V,. and Mueller-Eckhardt C. The presence of messenger RNA for HLA class I in human platelets and its capability for protein biosynthesis. Br J Haematol : 451–456, 1993. [DOI] [PubMed] [Google Scholar]
- 380.Sato H, Suzuki-Inoue K, Inoue O, Ozaki Y. Regulation of adaptor protein GIT1 in platelets, leading to the interaction between GIT1 and integrin alpha(IIb)beta3. Biochem Biophys Res Commun : 157–161, 2008. [DOI] [PubMed] [Google Scholar]
- 381.Savchenko AS, Martinod K, Seidman MA, Wong SL, Borissoff JI, Piazza G, Libby P, Goldhaber SZ, Mitchell RN, Wagner DD. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost : 860–870, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Sayah DM, Mallavia B, Liu F, Ortiz-Munoz G, Caudrillier A, DerHovanessian A, Ross DJ, Lynch JP 3rd, Saggar R, Ardehali A, Ware LB, Christie JD, Belperio JA, Looney MR. Neutrophil extracellular traps are pathogenic in primary graft dysfunction after lung transplantation. Am J Respir Crit Care Med : 455–463, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 383.Schafer AI, Crawford DD, Gimbrone MA Jr. Unidirectional transfer of prostaglandin endoperoxides between platelets and endothelial cells. J Clin Invest : 1105–1112, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Schaphorst KL, Chiang E, Jacobs KN, Zaiman A, Natarajan V, Wigley F, Garcia JG. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol : L258–L267, 2003. [DOI] [PubMed] [Google Scholar]
- 385.Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, Flad HD, Petersen F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood : 1158–1166, 2000. [PubMed] [Google Scholar]
- 386.Schneider RC, Zapol WM, Carvalho AC. Platelet consumption and sequestration in severe acute respiratory failure. Am Rev Respir Dis : 445–451, 1980. [DOI] [PubMed] [Google Scholar]
- 387.Schubert P, Devine DV. De novo protein synthesis in mature platelets: a consideration for transfusion medicine. Vox Sanguinis : 112–122, 2010. [DOI] [PubMed] [Google Scholar]
- 388.Schubert S, Weyrich AS, Rowley JW. A tour through the transcriptional landscape of platelets. Blood : 493–502, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 389.Schulz C, Engelmann B, Massberg S. Crossroads of coagulation and innate immunity: the case of deep vein thrombosis. J Thromb Haemost Suppl 1: 233–241, 2013. [DOI] [PubMed] [Google Scholar]
- 390.Schwertz H, Koster S, Kahr WH, Michetti N, Kraemer BF, Weitz DA, Blaylock RC, Kraiss LW, Greinacher A, Zimmerman GA, Weyrich AS. Anucleate platelets generate progeny. Blood : 3801–3809, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Schwertz H, Tolley ND, Foulks JM, Denis MM, Risenmay BW, Buerke M, Tilley RE, Rondina MT, Harris EM, Kraiss LW, Mackman N, Zimmerman GA, Weyrich AS. Signal-dependent splicing of tissue factor pre-mRNA modulates the thrombogenicity of human platelets. J Exp Med : 2433–2440, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Scull CM, Hays WD, Fischer TH. Macrophage pro-inflammatory cytokine secretion is enhanced following interaction with autologous platelets. J Inflammation : 53, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Sedlmayr P, Blaschitz A, Wilders-Truschnig M, Tiran A, Dohr G. Platelets contain interleukin-1 alpha and beta which are detectable on the cell surface after activation. Scand J Immunol : 209–214, 1995. [DOI] [PubMed] [Google Scholar]
- 394.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: involvement of platelet TLR2 and TLR4. Blood : 1952–1961, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Semple JW, Aslam R, Kim M, Speck ER, Freedman J. Platelet-bound lipopolysaccharide enhances Fc receptor-mediated phagocytosis of IgG-opsonized platelets. Blood : 4803–4805, 2007. [DOI] [PubMed] [Google Scholar]
- 396.Semple JW, Freedman J. Platelets and innate immunity. Cell Mol Life Sci : 499–511, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397.Semple JW, Italiano JE Jr, Freedman J. Platelets and the immune continuum. Nature Rev Immunol : 264–274, 2011. [DOI] [PubMed] [Google Scholar]
- 398.Seydel KB, Kampondeni SD, Valim C, Potchen MJ, Milner DA, Muwalo FW, Birbeck GL, Bradley WG, Fox LL, Glover SJ, Hammond CA, Heyderman RS, Chilingulo CA, Molyneux ME, Taylor TE. Brain swelling and death in children with cerebral malaria. N Engl J Med : 1126–1137, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Shannon O, Rydengard V, Schmidtchen A, Morgelin M, Alm P, Sorensen OE, Bjorck L. Histidine-rich glycoprotein promotes bacterial entrapment in clots and decreases mortality in a mouse model of sepsis. Blood : 2365–2372, 2010. [DOI] [PubMed] [Google Scholar]
- 400.Shashkin PN, Brown GT, Ghosh A, Marathe GK, McIntyre TM. Lipopolysaccharide is a direct agonist for platelet RNA splicing. J Immunol : 3495–3502, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Shea BS, Brooks SF, Fontaine BA, Chun J, Luster AD, Tager AM. Prolonged exposure to sphingosine 1-phosphate receptor-1 agonists exacerbates vascular leak, fibrosis, and mortality after lung injury. Am J Respir Cell Mol Biol : 662–673, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Shepard JM, Moon DG, Sherman PF, Weston LK, Del Vecchio PJ, Minnear FL, Malik AB, Kaplan JE. Platelets decrease albumin permeability of pulmonary artery endothelial cell monolayers. Microvasc Res : 256–266, 1989. [DOI] [PubMed] [Google Scholar]
- 403.Shi G, Morrell CN. Platelets as initiators and mediators of inflammation at the vessel wall. Thromb Res : 387–390, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 404.Shibazaki M, Kawabata Y, Yokochi T, Nishida A, Takada H, Endo Y. Complement-dependent accumulation and degradation of platelets in the lung and liver induced by injection of lipopolysaccharides. Infect Immun : 5186–5191, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Simmons CP, Farrar JJ, Nguyen vV, Wills B. Dengue. N Engl J Med : 1423–1432, 2012. [DOI] [PubMed] [Google Scholar]
- 406.Simon AY, Sutherland MR, Pryzdial EL. Dengue virus binding and replication by platelets. Blood : 378–385, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Smith MC, Schwertz H, Zimmerman GA, Weyrich AS. The platelet proteome. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 103–116. [Google Scholar]
- 408.Smith SA, Choi SH, Collins JN, Travers RJ, Cooley BC, Morrissey JH. Inhibition of polyphosphate as a novel strategy for preventing thrombosis and inflammation. Blood : 5103–5110, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, French PA, Dauerman HL, Becker RC. Platelet functions beyond hemostasis. J Thromb Haemost : 1759–1766, 2009. [DOI] [PubMed] [Google Scholar]
- 410.Snapper JR, Hinson JM Jr, Hutchison AA, Lefferts PL, Ogletree ML, Brigham KL. Effects of platelet depletion on the unanesthetized sheep's pulmonary response to endotoxemia. J Clin Invest : 1782–1791, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Solpov A, Shenkman B, Vitkovsky Y, Brill G, Koltakov A, Farzam N, Varon D, Bank I, Savion N. Platelets enhance CD4+ lymphocyte adhesion to extracellular matrix under flow conditions: role of platelet aggregation, integrins, and non-integrin receptors. Thromb Haemost : 815–821, 2006. [PubMed] [Google Scholar]
- 412.Speth C, Rambach G, Lass-Florl C. Platelet immunology in fungal infections. Thromb Haemost : 632–639, 2014. [DOI] [PubMed] [Google Scholar]
- 413.Spinelli SL, Maggirwar SB, Blumberg N, Phipps RP. Nuclear emancipation: a platelet tour de force. Sci Signal : pe37, 2010. [DOI] [PubMed] [Google Scholar]
- 414.Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood : 5028–5036, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, Nacher M, Pitaval C, Radovanovic I, Fukui Y, McEver RP, Filippi MD, Lizasoain I, Ruiz-Cabello J, Zarbock A, Moro MA, Hidalgo A. Neutrophils scan for activated platelets to initiate inflammation. Science : 1234–1238, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Srivastava K, Cockburn IA, Swaim A, Thompson LE, Tripathi A, Fletcher CA, Shirk EM, Sun H, Kowalska MA, Fox-Talbot K, Sullivan D, Zavala F, Morrell CN. Platelet factor 4 mediates inflammation in experimental cerebral malaria. Cell Host Microbe : 179–187, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Stahl AL, Svensson M, Morgelin M, Svanborg C, Tarr PI, Mooney JC, Watkins SL, Johnson R, Karpman D. Lipopolysaccharide from enterohemorrhagic Escherichia coli binds to platelets through TLR4 and CD62 and is detected on circulating platelets in patients with hemolytic uremic syndrome. Blood : 167–176, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Stark RJ, Aghakasiri N, Rumbaut RE. Platelet-derived Toll-like receptor 4 (Tlr-4) is sufficient to promote microvascular thrombosis in endotoxemia. PloS One : e41254, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Staub NC. Pulmonary edema. Physiol Rev : 678–811, 1974. [DOI] [PubMed] [Google Scholar]
- 420.Staub NC. Pulmonary edema: physiologic approaches to management. Chest : 559–564, 1978. [DOI] [PubMed] [Google Scholar]
- 421.Steiner S, Seidinger D, Huber K, Kaun C, Minar E, Kopp CW. Effect of glycoprotein IIb/IIIa antagonist abciximab on monocyte-platelet aggregates and tissue factor expression. Arterioscler Thromb Vasc Biol : 1697–1702, 2003. [DOI] [PubMed] [Google Scholar]
- 422.Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature : 419–426, 2007. [DOI] [PubMed] [Google Scholar]
- 423.Stephen J, Emerson B, Fox KA, Dransfield I. The uncoupling of monocyte-platelet interactions from the induction of proinflammatory signaling in monocytes. J Immunol : 5677–5683, 2013. [DOI] [PubMed] [Google Scholar]
- 424.Strussmann T, Tillmann S, Wirtz T, Bucala R, von Hundelshausen P, Bernhagen J. Platelets are a previously unrecognised source of MIF. Thromb Haemost : 1004–1013, 2013. [DOI] [PubMed] [Google Scholar]
- 425.Sun D, Popescu NI, Raisley B, Keshari RS, Dale GL, Lupu F, Coggeshall KM. Bacillus anthracis peptidoglycan activates human platelets through FcgammaRII and complement. Blood : 571–579, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Sun H, Wang X, Degen JL, Ginsburg D. Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood : 1358–1364, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Tabuchi A, Kuebler WM. Endothelium-platelet interactions in inflammatory lung disease. Vasc Pharmacol : 141–150, 2008. [DOI] [PubMed] [Google Scholar]
- 428.Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell : 460–476, 2010. [DOI] [PubMed] [Google Scholar]
- 429.Tanaka K, Koike Y, Shimura T, Okigami M, Ide S, Toiyama Y, Okugawa Y, Inoue Y, Araki T, Uchida K, Mohri Y, Mizoguchi A, Kusunoki M. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PloS One : e111888, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Tapson VF. Acute pulmonary embolism. N Engl J Med : 1037–1052, 2008. [DOI] [PubMed] [Google Scholar]
- 431.Tauseef M, Kini V, Knezevic N, Brannan M, Ramchandaran R, Fyrst H, Saba J, Vogel SM, Malik AB, Mehta D. Activation of sphingosine kinase-1 reverses the increase in lung vascular permeability through sphingosine-1-phosphate receptor signaling in endothelial cells. Circ Res : 1164–1172, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Taylor WR, Hanson J, Turner GD, White NJ, Dondorp AM. Respiratory manifestations of malaria. Chest : 492–505, 2012. [DOI] [PubMed] [Google Scholar]
- 433.Terada H, Baldini M, Ebbe S, Madoff MA. Interaction of influenza virus with blood platelets. Blood : 213–228, 1966. [PubMed] [Google Scholar]
- 434.Thomas DW, Rocha PN, Nataraj C, Robinson LA, Spurney RF, Koller BH, Coffman TM. Proinflammatory actions of thromboxane receptors to enhance cellular immune responses. J Immunol : 6389–6395, 2003. [DOI] [PubMed] [Google Scholar]
- 435.Thomas GM, Carbo C, Curtis BR, Martinod K, Mazo IB, Schatzberg D, Cifuni SM, Fuchs TA, von Andrian UH, Hartwig JH, Aster RH, Wagner DD. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood : 6335–6343, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 436.Thon JN, Montalvo A, Patel-Hett S, Devine MT, Richardson JL, Ehrlicher A, Larson MK, Hoffmeister K, Hartwig JH, Italiano JE Jr. Cytoskeletal mechanics of proplatelet maturation and platelet release. J Cell Biol : 861–874, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Thon JN, Peters CG, Machlus KR, Aslam R, Rowley J, Macleod H, Devine MT, Fuchs TA, Weyrich AS, Semple JW, Flaumenhaft R, Italiano JE Jr. T granules in human platelets function in TLR9 organization and signaling. J Cell Biol : 561–574, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Thornton P, McColl BW, Greenhalgh A, Denes A, Allan SM, Rothwell NJ. Platelet interleukin-1alpha drives cerebrovascular inflammation. Blood : 3632–3639, 2010. [DOI] [PubMed] [Google Scholar]
- 439.Thurston G, Rudge JS, Ioffe E, Zhou H, Ross L, Croll SD, Glazer N, Holash J, McDonald DM, Yancopoulos GD. Angiopoietin-1 protects the adult vasculature against plasma leakage. Nature Med : 460–463, 2000. [DOI] [PubMed] [Google Scholar]
- 440.Tomashefski JF., Jr Pulmonary pathology of acute respiratory distress syndrome. Clin Chest Med : 435–466, 2000. [DOI] [PubMed] [Google Scholar]
- 441.Tuinman PR, Muller MC, Jongsma G, Hegeman MA, Juffermans NP. High-dose acetylsalicylic acid is superior to low-dose as well as to clopidogrel in preventing lipopolysaccharide-induced lung injury in mice. Shock : 334–338, 2013. [DOI] [PubMed] [Google Scholar]
- 442.Tvedten HW, Till GO, Ward PA. Mediators of lung injury in mice following systemic activation of complement. Am J Pathol : 92–100, 1985. [PMC free article] [PubMed] [Google Scholar]
- 443.Tweardy DJ, Khoshnevis MR, Yu B, Mastrangelo MA, Hardison EG, Lopez JA. Essential role for platelets in organ injury and inflammation in resuscitated hemorrhagic shock. Shock : 386–390, 2006. [DOI] [PubMed] [Google Scholar]
- 444.Tyagi T, Ahmad S, Gupta N, Sahu A, Ahmad Y, Nair V, Chatterjee T, Bajaj N, Sengupta S, Ganju L, Singh SB, Ashraf MZ. Altered expression of platelet proteins and calpain activity mediate hypoxia-induced prothrombotic phenotype. Blood : 1250–1260, 2014. [DOI] [PubMed] [Google Scholar]
- 445.Ulfman LH, Joosten DP, van Aalst CW, Lammers JW, van de Graaf EA, Koenderman L, Zwaginga JJ. Platelets promote eosinophil adhesion of patients with asthma to endothelium under flow conditions. Am J Respir Cell Mol Biol : 512–519, 2003. [DOI] [PubMed] [Google Scholar]
- 446.Van den Steen PE, Deroost K, Deckers J, Van Herck E, Struyf S, Opdenakker G. Pathogenesis of malaria-associated acute respiratory distress syndrome. Trends Parasitol : 346–358, 2013. [DOI] [PubMed] [Google Scholar]
- 447.Van der Heyde HC, Gramaglia I, Sun G, Woods C. Platelet depletion by anti-CD41 (alphaIIb) mAb injection early but not late in the course of disease protects against Plasmodium berghei pathogenesis by altering the levels of pathogenic cytokines. Blood : 1956–1963, 2005. [DOI] [PubMed] [Google Scholar]
- 448.Van der Poll T, de Boer JD, Levi M. The effect of inflammation on coagulation and vice versa. Curr Opin Infect Dis : 273–278, 2011. [DOI] [PubMed] [Google Scholar]
- 449.Verschoor A, Neuenhahn M, Navarini AA, Graef P, Plaumann A, Seidlmeier A, Nieswandt B, Massberg S, Zinkernagel RM, Hengartner H, Busch DH. A platelet-mediated system for shuttling blood-borne bacteria to CD8alpha+ dendritic cells depends on glycoprotein GPlb and complement C3. Nature Immunol : 1194–1201, 2011. [DOI] [PubMed] [Google Scholar]
- 450.Verschoor A, Langer HF. Crosstalk between platelets and the complement system in immune protection and disease. Thromb Haemost : 910–919, 2013. [DOI] [PubMed] [Google Scholar]
- 451.Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev : 327–358, 2013. [DOI] [PubMed] [Google Scholar]
- 452.Vestweber D, Winderlich M, Cagna G, Nottebaum AF. Cell adhesion dynamics at endothelial junctions: VE-cadherin as a major player. Trends Cell Biol : 8–15, 2009. [DOI] [PubMed] [Google Scholar]
- 453.Vieira-de-Abreu A, Rondina MT, Weyrich AS, Zimmerman GA. Inflammation. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 733–766. [Google Scholar]
- 454.Vieira-de-Abreu A, Campbell RA, Weyrich AS, Zimmerman GA. Platelets: versatile effector cells in hemostasis, inflammation, and the immune continuum. Semin Immunopathol : 5–30, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455.Von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, Khandoga A, Tirniceriu A, Coletti R, Kollnberger M, Byrne RA, Laitinen I, Walch A, Brill A, Pfeiler S, Manukyan D, Braun S, Lange P, Riegger J, Ware J, Eckart A, Haidari S, Rudelius M, Schulz C, Echtler K, Brinkmann V, Schwaiger M, Preissner KT, Wagner DD, Mackman N, Engelmann B, Massberg S. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med : 819–835, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, Hackeng TM, Weber C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood : 924–930, 2005. [DOI] [PubMed] [Google Scholar]
- 457.Von Hundelshausen P, Koenen RR, Weber C. Platelet-mediated enhancement of leukocyte adhesion. Microcirculation : 84–96, 2009. [DOI] [PubMed] [Google Scholar]
- 458.Wakefield TW, Strieter RM, Wilke CA, Kadell AM, Wrobleski SK, Burdick MD, Schmidt R, Kunkel SL, Greenfield LJ. Venous thrombosis-associated inflammation and attenuation with neutralizing antibodies to cytokines and adhesion molecules. Arterioscler Thromb Vasc Biol : 258–268, 1995. [DOI] [PubMed] [Google Scholar]
- 459.Ward JR, Bingle L, Judge HM, Brown SB, Storey RF, Whyte MK, Dower SK, Buttle DJ, Sabroe I. Agonists of toll-like receptor (TLR)2 and TLR4 are unable to modulate platelet activation by adenosine diphosphate and platelet activating factor. Thromb Haemost : 831–838, 2005. [PubMed] [Google Scholar]
- 460.Washington AV, Gibot S, Acevedo I, Gattis J, Quigley L, Feltz R, De La Mota A, Schubert RL, Gomez-Rodriguez J, Cheng J, Dutra A, Pak E, Chertov O, Rivera L, Morales J, Lubkowski J, Hunter R, Schwartzberg PL, McVicar DW. TREM-like transcript-1 protects against inflammation-associated hemorrhage by facilitating platelet aggregation in mice and humans. J Clin Invest : 1489–1501, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 461.Weyrich AS, Denis MM, Kuhlmann-Eyre JR, Spencer ED, Dixon DA, Marathe GK, McIntyre TM, Zimmerman GA, Prescott SM. Dipyridamole selectively inhibits inflammatory gene expression in platelet-monocyte aggregates. Circulation : 633–642, 2005. [DOI] [PubMed] [Google Scholar]
- 462.Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, Spencer E, Kraiss LW, Albertine KH, McIntyre TM, Zimmerman GA. mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood : 1975–1983, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Weyrich AS, Dixon DA, Pabla R, Elstad MR, McIntyre TM, Prescott SM, Zimmerman GA. Signal-dependent translation of a regulatory protein, Bcl-3, in activated human platelets. Proc Natl Acad Sci USA : 5556–5561, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Weyrich AS, Elstad MR, McEver RP, McIntyre TM, Moore KL, Morrissey JH, Prescott SM, Zimmerman GA. Activated platelets signal chemokine synthesis by human monocytes. J Clin Invest : 1525–1534, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Weyrich AS, Lindemann S, Zimmerman GA. The evolving role of platelets in inflammation. J Thromb Haemost : 1897–1905, 2003. [DOI] [PubMed] [Google Scholar]
- 466.Weyrich AS, McIntyre TM, McEver RP, Prescott SM, Zimmerman GA. Monocyte tethering by P-selectin regulates monocyte chemotactic protein-1 and tumor necrosis factor-alpha secretion. Signal integration and NF-kappa B translocation. J Clin Invest : 2297–2303, 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Weyrich AS, Schwertz H, Kraiss LW, Zimmerman GA. Protein synthesis by platelets: historical and new perspectives. J Thromb Haemost : 241–246, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Weyrich AS, Zimmerman GA. Platelets in lung biology. Annu Rev Physiol : 569–591, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol : 489–495, 2004. [DOI] [PubMed] [Google Scholar]
- 470.White J. Platelet structure In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 117–144. [Google Scholar]
- 471.White JG. Why human platelets fail to kill bacteria. Platelets : 191–200, 2006. [DOI] [PubMed] [Google Scholar]
- 472.Whyte CS, Swieringa F, Mastenbroek TG, Lionikiene AS, Lance MD, van der Meijden PE, Heemskerk JW, Mutch NJ. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood : 2568–2578, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Wiesner J, Vilcinskas A. Antimicrobial peptides: the ancient arm of the human immune system. Virulence : 440–464, 2010. [DOI] [PubMed] [Google Scholar]
- 474.Wiig H, Swartz MA. Interstitial fluid and lymph formation and transport: physiological regulation and roles in inflammation and cancer. Physiol Rev : 1005–1060, 2012. [DOI] [PubMed] [Google Scholar]
- 475.Wildhagen KC, Garcia de Frutos P, Reutelingsperger CP, Schrijver R, Areste C, Ortega-Gomez A, Deckers NM, Hemker HC, Soehnlein O, Nicolaes GA. Nonanticoagulant heparin prevents histone-mediated cytotoxicity in vitro and improves survival in sepsis. Blood : 1098–1101, 2014. [DOI] [PubMed] [Google Scholar]
- 476.Wilson M, Blum R, Dandona P, Mousa S. Effects in humans of intravenously administered endotoxin on soluble cell-adhesion molecule and inflammatory markers: a model of human diseases. Clin Exp Pharmacol Physiol : 376–380, 2001. [DOI] [PubMed] [Google Scholar]
- 477.Wintrobe MM. Clinical Hematology (2nd ed). Philadelphia, PA: Lea & Febiger, 1946, p. 194. [Google Scholar]
- 478.Wong CH, Jenne CN, Petri B, Chrobok NL, Kubes P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nature Immunol : 785–792, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Xiang B, Zhang G, Guo L, Li XA, Morris AJ, Daugherty A, Whiteheart SW, Smyth SS, Li Z. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nature Commun : 2657, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Yang D, Chertov O, Bykovskaia SN, Chen Q, Buffo MJ, Shogan J, Anderson M, Schroder JM, Wang JM, Howard OM, Oppenheim JJ. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science : 525–528, 1999. [DOI] [PubMed] [Google Scholar]
- 481.Yaron M, Djaldetti M. Platelets in synovial fluid. Arthritis Rheumatism : 607–608, 1978. [DOI] [PubMed] [Google Scholar]
- 482.Yeaman MR, Bayer AS. Antimicrobial host defense. In: Platelets (3rd ed), edited by Michelson AD. Amsterdam: Elsevier, 2013, p. 767–801. [Google Scholar]
- 483.Yildiz C, Palaniyar N, Otulakowski G, Khan MA, Post M, Kuebler WM, Tanswell K, Belcastro R, Masood A, Engelberts D, Kavanagh BP. Mechanical ventilation induces neutrophil extracellular trap formation. Anesthesiology : 864–875, 2015. [DOI] [PubMed] [Google Scholar]
- 484.Yiming MT, Lederer DJ, Sun L, Huertas A, Issekutz AC, Bhattacharya S. Platelets enhance endothelial adhesiveness in high tidal volume ventilation. Am J Respir Cell Mol Biol : 569–575, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Yipp BG, Kubes P. NETosis: how vital is it? Blood : 2784–2794, 2013. [DOI] [PubMed] [Google Scholar]
- 486.Yong ASC, Pennings GJ, Change M, Hamzah A, Chung T, Qi M, Berieger D, Behnia M, Krilis SA, Ng MKC, Lowe HC, Kritharides L. Intracoronary shear-related up-regulation of platelet P-selectin and platelet-monocyte aggregation despite the use of aspirin and clopidogrel. Blood : 11–20, 2015. [DOI] [PubMed] [Google Scholar]
- 487.Yoshida A, Ohba M, Wu X, Sasano T, Nakamura M, Endo Y. Accumulation of platelets in the lung and liver and their degranulation following antigen-challenge in sensitized mice. Br J Pharmacol : 146–152, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Yost CC. Toward the “ideal” inhibitor of NETs. Blood : 2439–2440, 2014. [DOI] [PubMed] [Google Scholar]
- 489.Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML, Chandler NB, Rodesch CK, Albertine KH, Petti CA, Weyrich AS, Zimmerman GA. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood : 6419–6427, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 490.Yost CC, Weyrich AS, Zimmerman GA. The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie : 692–697, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Zamora C, Canto E, Nieto JC, Ortiz MA, Diaz-Torne C, Diaz-Lopez C, Llobet JM, Juarez C, Vidal S. Functional consequences of platelet binding to T lymphocytes in inflammation. J Leukoc Biol : 521–529, 2013. [DOI] [PubMed] [Google Scholar]
- 492.Zarbock A, Ley K. The role of platelets in acute lung injury (ALI). Front Biosci : 150–158, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood : 6743–6751, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Zarbock A, Polanowska-Grabowska RK, Ley K. Platelet-neutrophil-interactions: linking hemostasis and inflammation. Blood Rev : 99–111, 2007. [DOI] [PubMed] [Google Scholar]
- 495.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest : 3211–3219, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496.Zhang G, Han J, Welch EJ, Ye RD, Voyno-Yasenetskaya TA, Malik AB, Du X, Li Z. Lipopolysaccharide stimulates platelet secretion and potentiates platelet aggregation via TLR4/MyD88 and the cGMP-dependent protein kinase pathway. J Immunol : 7997–8004, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Zhao L, Ohtaki Y, Yamaguchi K, Matsushita M, Fujita T, Yokochi T, Takada H, Endo Y. LPS-induced platelet response and rapid shock in mice: contribution of O-antigen region of LPS and involvement of the lectin pathway of the complement system. Blood : 3233–3239, 2002. [DOI] [PubMed] [Google Scholar]
- 498.Zhi H, Rauova L, Hayes V, Gao C, Boylan B, Newman DK, McKenzie SE, Cooley BC, Poncz M, Newman PJ. Cooperative integrin/ITAM signaling in platelets enhances thrombus formation in vitro and in vivo. Blood : 1858–1867, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499.Zhu W, London NR, Gibson CC, Davis CT, Tong Z, Sorensen LK, Shi DS, Guo J, Smith MC, Grossmann AH, Thomas KR, Li DY. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature : 252–255, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Zimmerman GA, Weyrich AS. Arsonists in rheumatoid arthritis. Science : 528–529, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Zimmerman GA, Weyrich AS. Signal-dependent protein synthesis by activated platelets: new pathways to altered phenotype and function. Arterioscler Thromb Vasc Biol : s17–24, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 502.Zwaginga JJ, Torres HI, Lammers J, Sixma JJ, Koenderman L, Kuijper PH. Minimal platelet deposition and activation in models of injured vessel wall ensure optimal neutrophil adhesion under flow conditions. Arterioscler Thromb Vasc Biol : 1549–1554, 1999. [DOI] [PubMed] [Google Scholar]