

Summary
BLU-667 is a next-generation RET inhibitor that maximizes on-target and minimizes off-target effects. It is an exemplar of genotype-driven drug development followed by multi-histology basket trial validation that is becoming a paradigm for precision oncology. Cancer Discov; 8(7); 797–9. ©2018 AACR.
The identification and successful pharmacologic inhibition of oncogenic, constitutively active cellular kinases has been life-changing for many patients with cancer. Oncogenic kinase alterations occur in multiple ways, including point mutations, insertions/deletions, amplifications, and chromosomal rearrangements. Examples include point mutations in receptor tyrosine kinases (RTK) such as the EGFR point mutations in intracellular kinases such as BRAF, amplifications in RTKs such as HER2, and chromosomal rearrangements in RTKs such as BCR-ABL, anaplastic lymphoma kinase (ALK), and ROS1.
In patients with non-small cell lung cancer (NSCLC), the step-wise refinement of drug development against the oncogenic kinases EGFR and ALK has demonstrated notable clinical benefits. For example, the recently reported FLAURA trial revealed that first-line therapy with the third-generation, mutant-selective EGFR tyrosine kinase inhibitor (TKI) osimertinib more than doubles progression-free survival (PFS) compared with earlier-generation EGFR inhibitors in patients with advanced treatment-naïve EGFR-mutant NSCLC (1). Similarly, the ALEX trial demonstrated that first-line therapy with the “second-generation” ALK inhibitor alectinib more than doubled PFS compared with treatment with the “first-generation” ALK inhibitor crizotinib (25 months vs. 10 months, respectively; ref. 2). Similar drug development paradigms are under way for other targets, including ROS1, NTRK1/2/3, and RET. In this issue of Cancer Discovery, Subbiah and colleagues describe the development of a “next-generation” RET inhibitor, BLU-667, that is entering the analogous field of increasing the specificity of target kinase inhibition in patients with RET-driven malignancies (3).
The RET gene encodes for an oncogenic RTK found in a variety of cancers (4). Constitutive activation of RET can occur through point mutations, either in the extracellular domain or in the kinase domain, or through chromosomal rearrangements. In patients with medullary thyroid cancer (MTC), RET is characteristically altered by activating point mutations, whereas in patients with NSCLC and papillary thyroid cancer (PTC), RET is characteristically altered through chromosomal rearrangements (Fig. 1). Pharmacologic inhibition of RET is currently achieved with multikinase inhibitors (MKI), such as vandetanib and cabozantinib, in patients with RET-driven MTC, PTC, and NSCLC.
Figure 1.
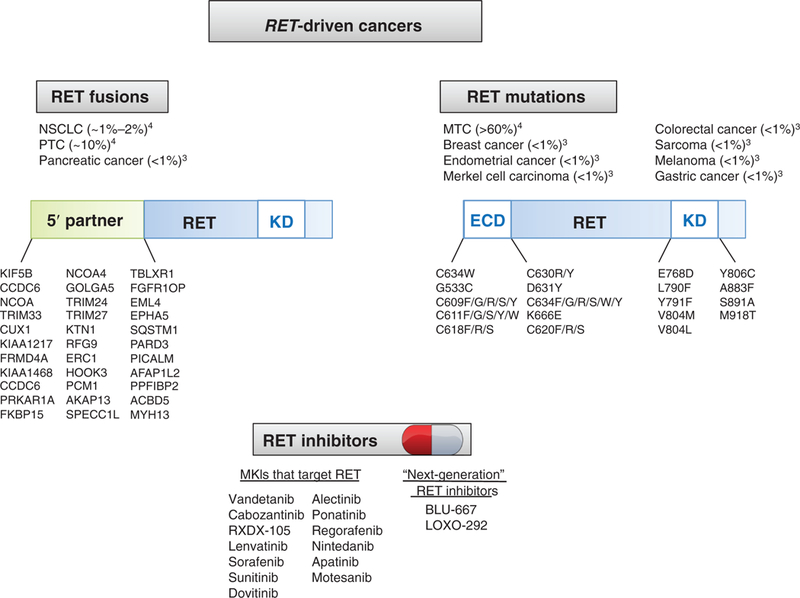
RET alterations by histology and mutation type with current RET Inhibitors. RET-driven cancers tend to harbor either chromosomal rearrangements or point mutations in the RET extracellular domain or tyrosine kinase domain. A variety of multikinase inhibitors have been applied in patients with RET-driven cancer; however, BLU-667 and LOXO-292 are two next-generation RET inhibitors now being broadly applied to patients whose tumors harbor these oncogenic RET alterations.
Subbiah and colleagues describe the strategic development and initial clinical deployment of a novel, next-generation RET-selective inhibitor, BLU-667 (3). Starting with a chemical library screen encompassing >10,000 kinase inhibitors of >60 different chemical scaffolds, the lead compound, BLU-667, was selected to maximize on-target efficacy against wild-type and oncogenic RET while maintaining selectivity toward the target. In subsequent studies in NSCLC and MTC cell lines, BLU-667 exhibited superior potency in vitro against various oncogenic RET alterations compared with the MKIs vandetanib, cabozantinib, and RXDX-105. The authors focused their in vitro toxicity evaluation on inhibition of VEGFR2, as excess VEGFR2 inhibition can result in hypertension, thrombosis, and hemorrhage. BLU-667 achieved greater selectivity for RET over VEGFR2 compared with vandetanib, cabozantinib, and RXDX-105.
Following their in vitro drug potency analyses, the authors show that BLU-667 inhibits MAPK signaling better than vandetanib, cabozantinib, and RXDX-105 in both murine and human cell line models of RET-driven cancer mediated by both chromosomal rearrangements and point mutations, including the common MKI RET resistance mutations V804L and V804M. The authors proceed to quantify the in vivo antitumor efficacy of BLU-667 in allografts and patient-derived xenografts (PDX) of RET-driven cancer mediated by both chromosomal rearrangements and point mutations, again including the common MKI RET resistance mutation V804L and one PDX from a patient with CCDC6-RET-positive colorectal cancer. The authors trace the antitumor effects to on-target MAPK pathway inhibition in on-treatment tumor lysates in the PDX model of KIF5B-RET fusion NSCLC. To evaluate off-target effects, in the KIF5B-RET fusion NSCLC PDX model the authors demonstrate that BLU-667 has significantly less anti-VEGFR2 effect than cabozantinib.
Building on their preclinical studies, the authors describe a phase I “basket” trial (NCT03037385) treating patients with advanced, unresectable RET-altered tumors with BLU-667. The authors describe antitumor responses in 4 patients, including a 47% tumor reduction after 11 months in a patient with TKI-naïve MTC, a 47% tumor reduction after 8 months in a patient with MTC who had progressed after vandetanib, a >30% tumor reduction after 16 weeks in a patient with TKI-naïve KIF5B-RET fusion NSCLC, and a 34% tumor reduction after 16 weeks in a patient with KIF5B-RET fusion NSCLC who had progressed on vandetanib and everolimus. These patients experienced grade 1 toxicities, including nausea, leukopenia, hyperphosphatemia, constipation, dry skin, and rash. In one of the patients with MTC, RET pathway inhibition was demonstrated by decreased DUSP6 and SPRY4 mRNA expression in on-treatment tumor biopsies after 28 days of therapy. We await publication of the full clinical data for a complete efficacy and toxicity evaluation.
This addition to the arsenal for managing RET-altered cancers is noteworthy in several regards. First, the authors describe a compelling story of tailored drug development for an area of need, RET-driven malignancies, with detailed preclinical analysis supporting the compound′s development. Second, the authors document initial clinical activity in patients with RET-driven tumors across histology (MTC, NSCLC) and different types of RET alterations (activating point mutations, rearrangements). Third and most notably, the strategic design of this drug, focusing on minimizing VEGFR2 “off-target” inhibition, is anticipated to decrease the frequency and severity of vascular toxicities (hypertension, thrombosis, hemorrhage) which have been particularly problematic with the MKIs that target both RET and VEGFR2. For example, in one study with vandetanib, grade 3 or higher hypertension affected over half of patients (5). However, it is crucial that this is validated in a larger dataset, because only 4 patients are reported in this article.
These findings also raise interesting questions for ongoing and future studies. First, as “next-generation” RET inhibitors are developed, what is the optimal sequence of RET TKI therapy? In addition to BLU-667, other, more selective inhibitors are being developed, including LOXO-292 (NCT03157128). Will response rates and resistance mechanisms change based on the line of therapy in which a specific RET TKI is received? Will there be a role for earlier-generation MKIs, which target RET and possibly other kinases that allow therapeutic escape?
Second, will the efficacy of BLU-667 and other “next-gen-eration” RET inhibitors cross all histologies of RET-altered tumors? The more common RET-altered tumors were studied here, including MTC and NSCLC; however, RET alterations are found at low frequency across a broad variety of tumor types (Fig. 1). As oncologic care is rapidly integrating site-agnostic therapies, such as the recent FDA approval of pembrolizumab in microsatellite-unstable tumors (6) and breakthrough therapy designation for entrectinib in patients with NTRK-rearranged malignancies (7), it is still critical to consider tumor site of origin. We make particular note of BRAFV600E mutations, which are also found across a variety of tumor types. BRAFV600E inhibitors, such as vemurafenib and dabrafenib, achieve high response rates in BRAF-mutant melanoma (8). In contrast, similar responses are not observed in BRAF-mutant colorectal cancer, where upstream EGFR can provide primary resistance to BRAF inhibitors (9). Time will tell whether tissue site of origin will be critical in mediating the efficacy of RET inhibitors.
Third, will all genomic RET alterations be equally inhibited by BLU-667 and other “next-generation” RET inhibitors? When the clinical context of a target includes multiple genomic alteration types, such as chromosomal rearrangements and point mutations, it is critical to prove drug efficacy in preclinical development in all scenarios before proceeding to broad human testing. Although differences in drug efficacy based on different mutations have been demonstrated in patients with EGFR-mutant NSCLC, differences in drug efficacy based on chromosomal rearrangement fusion partners remain inadequately understood, as demonstrated by the remaining controversy in patients with ALK-rearranged NSCLC (10). The initial studies presented herein suggest that BLU-667 retains broad sensitivity against both RET mutations and RET fusions; however, more nuanced differences regarding degrees of sensitivity across different alterations within this target remain to be defined. It is also likely that mechanisms of acquired resistance to BLU-667 will differ based on the genomic and potentially histologic context.
In conclusion, next-generation TKIs applied in patients with EGFR- and ALK-driven NSCLC have shown impressive improvements in efficacy and acceptable toxicity profiles compared with earlier-generation inhibitors. We remain optimistic that a similar paradigm will play out for next-generation RET inhibitors such as BLU-667 in patients with RET-driven cancers.
Acknowledgments
C.M. Lovly was supported by a Damon Runyon Clinical Investigator Award, a LUNGevity Career Development Award, a V Foundation Scholar-in-Training Award, an AACR-Genentech Career Development Award, a LCFA/IASLC Lori Monroe Scholarship, and by the NIH and NCI R01CA121210, U10CA180864, P01CA129243, and P30CA068485.
Footnotes
Disclosure of Potential Conflicts of Interest
C.M. Lovly reports receiving commercial research support from Novartis, Xcovery, and AstraZeneca and is a consultant/advisory board member for Cepheid, Foundation Medicine, Takeda, Pfizer. Ariad, AstraZeneca, Novartis, Clovis, and Genoptix. No potential conflicts of interest were disclosed by the other author.
References
- 1.Soria JC, Ohe Y, Vansteenkiste J, Reungwetwattana T, Chewaskulyong B, Lee KH, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med 2018;378:113–25. [DOI] [PubMed] [Google Scholar]
- 2.Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med 2017;377:829–38. [DOI] [PubMed] [Google Scholar]
- 3.Subbiah V, Gainor JF, Rahal R, Brubaker JD, Kim JL, Maynard M, et al. Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov 2018;8:836–49. [DOI] [PubMed] [Google Scholar]
- 4.Kato S, Subbiah V, Marchlik E, Elkin SK, Carter JL, Kurzrock R RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin Cancer Res 2017;23:1988–97. [DOI] [PubMed] [Google Scholar]
- 5.Yoh K, Seto T, Satouchi M, Nishio M, Yamamoto N, Murakami H, et al. Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir Med 2017;5:42–50. [DOI] [PubMed] [Google Scholar]
- 6.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drilon A, Siena S, Ou SI, Patel M, Ahn MJ, Lee J, et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: combined results from two phase I trials (ALKA-372–001 and STARTRK-1). Cancer Discov 2017;7:400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Z, Liu W, Gotlieb V. The rapidly evolving therapies for advanced melanoma - towards immunotherapy, molecular targeted therapy, and beyond. Crit Rev Oncol Hematol 2016;99:91–9. [DOI] [PubMed] [Google Scholar]
- 9.Corcoran RB, Andre T, Atreya CE, Schellens JHM, Yoshino T, Bendell JC, et al. Combined BRAF, EGFR, and MEK inhibition in patients with BRAF(V600E)-mutant colorectal cancer. Cancer Discov 2018;8:428–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin JJ, Shaw AT. Differential sensitivity to crizotinib: Does EML4-ALK fusion variant matter? J Clin Oncol 2016;34:3363–5. [DOI] [PubMed] [Google Scholar]