

Summary
Compared to adults, infants suffer higher rates of hospitalization, severe clinical complications, and mortality due to influenza infection. We found that γδ T cells protected neonatal mice against mortality during influenza infection. γδ T cell-deficiency did not alter viral clearance or interferon-γ production. Instead, neonatal influenza infection induced the accumulation of interleukin-17A (IL-17A)-producing γδ T cells, which was associated with IL-33 production by lung epithelial cells. Neonates lacking IL-17A-expressing γδ T cells or Il33 had higher mortality upon influenza infection. γδ T cells and IL-33 promoted lung infiltration of group 2 innate lymphoid cells and regulatory T cells, resulting in increased amphiregulin secretion and tissue repair. In influenza-infected children, IL-17A, IL-33, and amphiregulin expression were correlated, and increased IL-17A levels in nasal aspirates were associated with better clinical outcomes. Our results indicate that γδ T cells are required in influenza-infected neonates to initiate protective immunity and mediate lung homeostasis.
Keywords: γδ T cells, neonatal influenza infection, Type-2 immunity, IL-17A, IL-33, amphiregulin, children
Introduction
Influenza viruses are among the most common and significant pathogens that underlie human respiratory diseases, with high morbidity and mortality (Taubenberger and Kash, 2010; Taubenberger and Morens, 2008). These viruses primarily infect respiratory epithelial cells where, after detection by immune sensors, the host will initiate innate and adaptive immune responses that guide viral clearance and tissue repair (Dash and Thomas, 2014; Iwasaki and Pillai, 2014). Whereas viral clearance is primarily promoted by Type-1 immune responses through effector CD8+ T cells (Cerwenka et al., 1999; Duan and Thomas, 2016; Duan et al., 2015), group 2 innate lymphoid cells (ILC2s) regulated by interleukin (IL)-33-IL-33R signaling promote Type-2 immune responses and are important for lung homeostasis and repair following influenza infection (Chang et al., 2011; Gorski et al., 2013; Guo and Thomas, 2017; Le Goffic et al., 2011; Monticelli et al., 2011). Similarly, regulatory T (Treg) cells and T helper (Th) cells have been implicated in beneficial influenza immune responses mediated by IL-33-driven tissue restoration via production of amphiregulin (Areg) (Arpaia et al., 2015). Although the downstream immune responses and tissue repair process induced by IL-33 have been well defined, the production and upstream regulation of IL-33 is still unclear.
Compared to adults, infants suffer relatively higher rates of hospitalization, severe clinical complications, and mortality as a result of influenza infection (Bhat et al., 2005; Munoz, 2003). The proximate mechanisms guiding these age-dependent differences may also be related to exaggerated Type-2 responses that are characteristic of the infant immune system (Adkins et al., 2004; Dowling and Levy, 2014; Garcia et al., 2000; de Kleer et al., 2016), mediated by the IL-33 pathway (de Kleer et al., 2016; Saluzzo et al., 2017), and may function to prevent tissue damage owed to excessive inflammation. Thus, the pathologies concomitant with influenza infection, and their respective therapeutic targets, likely differ between infants and adults. In adult influenza infection, CD8+ T cells predominate the antiviral response (Cerwenka et al., 1999; Duan and Thomas, 2016; Duan et al., 2015), but in infant influenza infection, CD8+ T cells exhibit relatively reduced functionality (You et al., 2008).
In contrast to CD8+ T cells, γδ T cells are the first T cells to appear in the thymus during fetal development and have the capacity to recognize a wide range of antigens and respond rapidly to infections (Chien et al., 2014; Vantourout and Hayday, 2013) even during infancy; for instance, these cells are known to play important roles in the infant immune response to RSV infection (Huang et al., 2015) and enterocolitis (Weitkamp et al., 2014). In mice, γδ T cells are generally categorized according to their secretion of IFN-γ (Gao et al., 2003), IL-17A (Coffelt et al., 2015; Papotto et al., 2017), or M-CSF (Mamedov et al., 2018). The IL-17A producers, in particular, are known to play a prominent role in lung immunity to bacterial infections, where rapid IL-17A production is critical for the recruitment of protective neutrophil responses (Romagnoli et al., 2016). For the most part, γδ T cells have not been associated with inducing Type-2 immune effectors. Although γδ T cells may likewise act as immune sentinels during neonatal influenza infection, the functions of γδ T cells in influenza-infected infants remain to be elucidated.
In this study, utilizing a neonatal mouse model of influenza infection, we report that γδ T cells played an important role in protecting suckling mice against viral infection. This protection was mediated by augmentation of IL-33 production from the mucosal compartment that was induced by IL-17A secreted from the responding γδ T cells. We further showed that this IL-17A-dependent IL-33 production subsequently generates a local Type-2 immune response with increased accumulation of Areg-producing ILC2s and Treg cells in the lung, thus promoting tissue repair and lung integrity following infection. We also observed correlations between IL-17A, IL-33, and Areg in nasal washes of human influenza-infected infants. Thus, our findings reveal important roles for γδ T cells in directing an axis of IL-17A and IL-33 production, promoting tissue recovery after infection and providing a potential therapeutic target.
Results
γδ T cells protect neonatal mice against influenza infection by promoting tissue restoration independent of viral clearance
In influenza-infected adult mice, cytotoxic CD8+ T cells can express death receptors and produce cytokines and lytic mediators that promote viral clearance (Brincks et al., 2008; Gruta and Turner, 2014; Moskophidis and Kioussis, 1998). However, during infancy the immune system remains developmentally immature, with relatively fewer and less diverse CD8+ T cells compared to adults (You et al., 2008). In contrast, many γδ T cell subsets develop fetally and thus are available to respond in neonates. To investigate if γδ T cells can respond to neonatal influenza infection, we infected 7-day-old wild-type neonates with A/HKx31 (H3N2) influenza virus. After infection, we observed a significant accumulation of γδ T cells, characterized by increases in both frequency and cell number (Figure 1A) although there was no change in mock-infected lungs of the same age (Figure S1). EdU (5-ethynyl-2’-deoxyuridine) cell incorporation assays indicated that the increases in γδ T cells in infected lungs were at least partially due to γδ T cell proliferation (Figure 1B). In order to understand the function of γδ T cells during influenza infection, we next infected a neonatal cohort of wild-type and γδ T cell receptor-deficient (TCRδ−/−) animals with influenza virus. Compared to TCRδ−/− mice, relative weight gains were overall significantly larger in wild-type neonates (Figure 1C). Wild-type neonates also had a significantly increased survival rate compared to TCRδ−/− mice (Figure 1D), yet the two groups demonstrated no detectable difference in tissue viral clearance at any time point after infection (Figure 1E). Consistent with the viral titer, loss of γδ T cells did not alter the levels of IFN-γ at Day 7 after infection (Figure 1F). To determine if γδ T cells also play a role in adult influenza infection, we infected wild-type and TCRδ−/− littermates (8–10 weeks old); no significant differences were observed in the weight loss profile or survival rate (Figures S2A-B) between wild-type and TCRδ−/− adults. These data indicate that in neonatal mice γδ T cells provide protection against influenza infection without influencing antiviral responses.
Figure 1: γδ T cells protect neonatal mice against influenza infection via promotion of lung homeostasis and repair, independent of viral clearance.
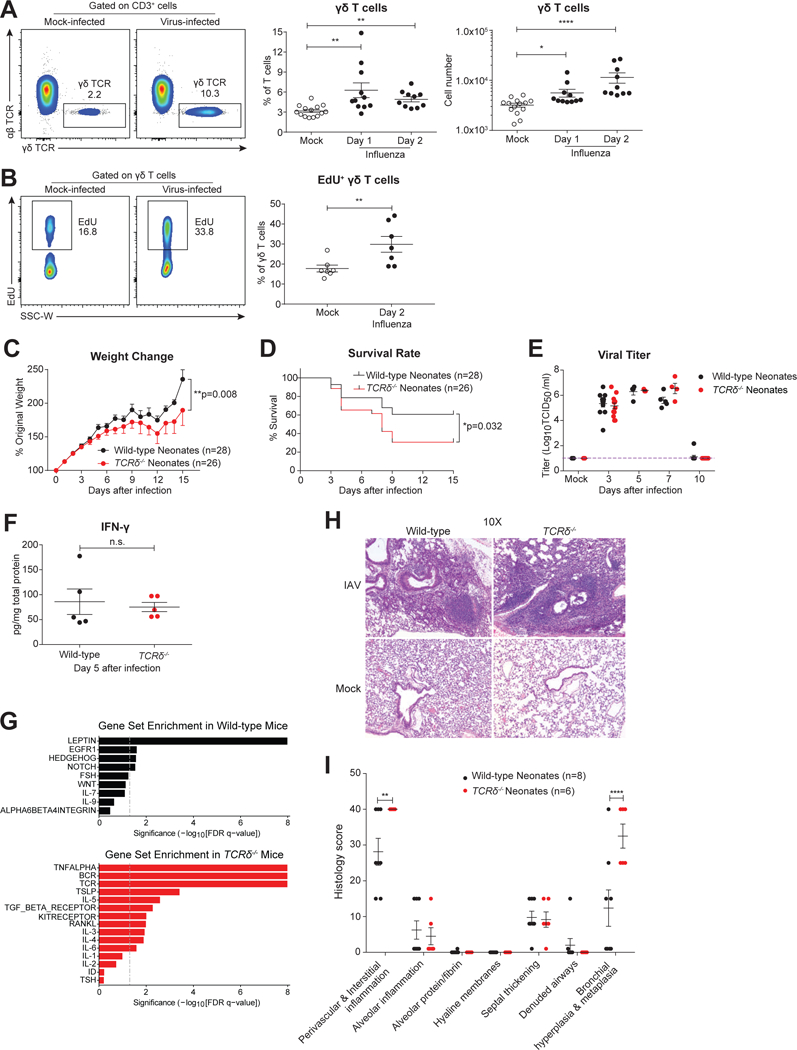
A. Representative flow cytometric plots (left), and frequency and number (right) of γδ T cells in mock- (open circle, n=14) or virus-infected (solid) lungs of wild-type neonates at 1 (n=11) and 2 (n=10) days following intranasal influenza A/x31 virus infection. Data are combined from four independent experiments and presented as mean ± SEM.B. Representative flow cytometric plots (left) and summary frequency plot (right) of EdU+ γδ T cells in mock-infected (n=7) and influenza virus-infected (n=7) lungs of wild-type neonates 2 days after infection. Data are combined from three independent experiments and shown as mean ± SEM.C and D. Body weight profile (% of original weight) (C) and survival rate (D) of wild-type (black, n=28) and TCRδ−/− (red, n=26) neonates following influenza infection. Data are combined from sixteen independent experiments, and weight data are shown as mean ± SEM in change.E. Viral titer (Log10TCID50/ml) of wild-type (black) and TCRδ−/− (red) neonates assessed by plaque assay at days 0, 3, 5, 7 and 10 after influenza infection. Samples are pooled from at least two independent experiments for each time point and data are presented as mean ± SEM.F. Measurement of IFN-γ in the total lung homogenates by ELISA of wild-type (black, n=5) and TCRδ−/− (red, n=5) neonates at 7 days after influenza infection. Samples are pooled from three independent experiments, and data are presented as mean ± SEM.G. Gene Set Enrichment Analysis of whole-lung gene expression, ranked by significance (-Log10[FDR q-value]), from wild-type (black, n=3) and TCRδ−/− (red, n=3) neonates at 8 days after influenza infection.H. Representative images of H&E staining of influenza infected wild-type and TCRδ−/− lungs at 15 days after infection.I. Summary of histological analysis from influenza-infected wild-type (black, n=8) and TCRδ−/− (red, n=6) lungs at 15 days after infection. H-I. Data are combined from two independent experiments and shown as mean ± SEM.*p<0.05, **p<0.01, ****p<0.0001, n.s., not significant.
To determine if γδ T cells function as immune regulators during infection, we performed RNA-Seq using total RNA obtained from whole lungs of 3 wild-type and 3 TCRδ−/− neonates 8 days after infection. Gene Set Enrichment Analysis demonstrated distinct immune pathways in lungs obtained from wild-type and TCRδ−/− mice (Figure 1G). Several pathways relevant to tissue development and regeneration were enriched in wild-type lungs, including the epithelial growth factor receptor (EGFR) pathway (Hall et al., 2016; Monticelli et al., 2011; Zaiss et al., 2015) and the hedgehog pathway (Hogan et al., 2014; Sriperumbudur et al., 2017). The absence of γδ T cells was associated with increased inflammatory pathways, including TNF-α, IL-6, and IL-5. In order to investigate the downstream effects of these γδ T cell-mediated immune responses on infected lungs, we performed H&E staining of lung sections. At 15 days after infection, TCRδ−/− mice were characterized by increased perivascular & interstitial inflammation and bronchiolar hyperplasia & metaplasia (Figure 1H-I), suggesting that γδ T cell deficiency disrupted lung homeostasis and tissue repair. Collectively, these data indicate that γδ T cells expand after influenza infection and provide protection to infants by promoting tissue restoration rather than by enhancing the antiviral response.
γδ T cells rapidly produce IL-17A after neonatal influenza infection
To determine the mechanism by which γδ T cells promoted protection in neonatal animals, we examined their phenotypes after infection by using CD27 and CD44 surface marker expression, which distinguish IL-17A- and IFN-γ- producing γδ T cells (Ribot et al., 2009). We found that γδ T cells were predominantly CD27-CD44hi in virus-infected neonatal lungs, corresponding to IL-17A production, while they were mainly CD27+CD44lo (corresponding to IFN-γ production) after mock infection (Figure 2A). After influenza infection, lung γδ T cells also had higher cell surface expression of CCR6 and Sca-1 (Figure 2B), consistent with the phenotype of IL-17A-producing γδ T cells. To confirm the IL-17A-producing phenotype of lung γδ T cells after influenza infection, we assayed Il17a gene expression using quantitative real-time PCR (qRT-PCR) in sorted γδ T cells at different points. This analysis indicated that Il17a was upregulated transiently 1 day after infection. (Figure 2C). In concordance with these qRT-PCR data, the intracellular staining of IL-17A in γδ T cells showed an enhanced frequency and number of IL-17A-producing γδ T cells in virus-infected lungs, whereas IFN-γ-producing γδ T cells were not significantly different (Figures 2D-E, kinetics shown in Figures S3A-B). This transient IL-17A production by γδ T cells did not significantly alter neutrophil infiltration or accumulation (Figure S3C).
Figure 2. IL-17A-producing γδ T cells rapidly accumulate and respond to influenza infection in neonatal mice.
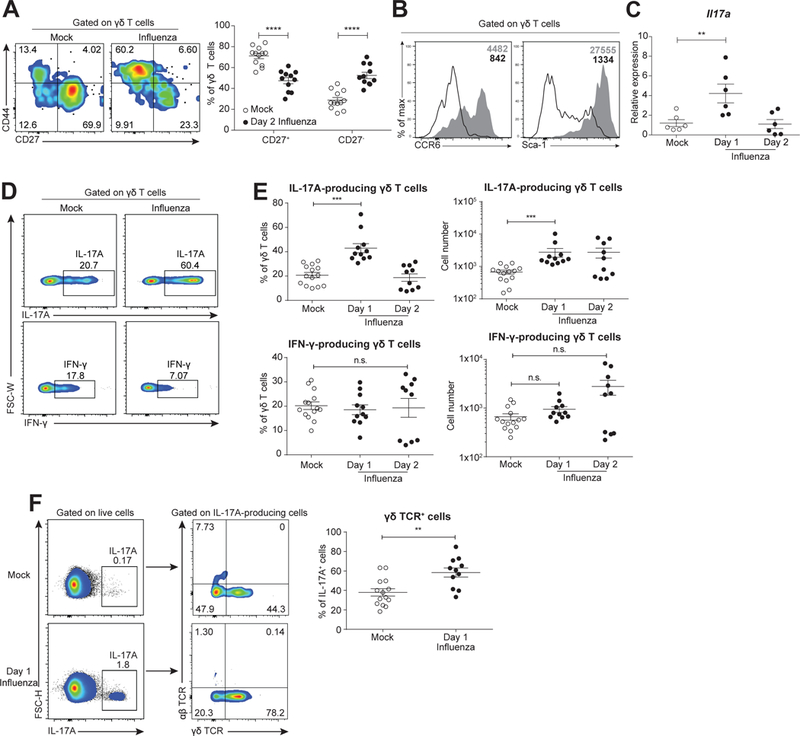
A. Representative flow cytometric plots (left, with CD27 and CD44 expression on x- and y-axis, respectively) and summary frequency plot (right) of γδ T cells from mock- (open, n=12) and influenza virus- (solid, n=11) infected mice at 2 days following infection. Data are combined from four independent experiments and presented as mean ± SEM.B. Representative flow cytometric histogram showing expression of CCR6 (left) and Sca-1 (right) gated on γδ T cells from mock- (open) and virus- (shaded) infected neonates (2 days after infection). Mean fluorescence intensity are shown in the upper right corners.C. Relative expression of Il17a by quantitative real-time PCR in sort-purified γδ T cells from mock (open, n=6) and virus-infected neonates at 1 (n=6) and 2 (n=6) days following infection. Samples are combined from two independent experiments and data are presented as mean ± SEM.D. Representative flow cytometric plots of IL-17A (top) and IFN-γ (bottom) expression in γδ T cells from mock- (left) and virus-infected (right) neonates at 1 day after infection.E. Scatter plot showing frequency and number of IL-17A- (top) and IFN-γ- (bottom) producing γδ T cells from (D). Samples from mock-infected (n=14) neonates were pooled from animals 1 (n=11) and 2 (n=10) days after mock infection.F. Representative flow cytometric plots (left) and summary frequency plots (right) of γδ TCR expression gated on total IL-17A-producing cells from mock- (n=14) and virus- (n=11) infected mice at 1 day following infection. (D-F) PMA/ionomycin was used for stimulation prior to intracellular staining. Data are combined from four independent experiments and presented as mean ± SEM. **p<0.01, ***p<0.001, ****p<0.0001, n.s., not significant.
γδ T cells can be highly clonotypic in different tissues, especially with respect to Vγ chain usage (Carding and Egan, 2002). Given the enrichment in CD27- γδ T cells secreting IL-17A in influenza-infected neonates, we decided to investigate if the T cell receptor repertoires were altered in cells exhibiting this phenotypes. By utilizing single-cell PCR technology (Dash et al., 2011, 2015; Guo et al., 2016), we characterized the TRGV family usage by CD27+ or CD27- neonatal γδ T cells (Kashani et al., 2015) that were mock- or influenza-infected. TRGV family usage varied significantly between CD27+ and CD27- γδ T cells at different time points after infection (Figure S3D & Table S1). Furthermore, we observed shifts in the representation of individual TRGV families in CD27-, but not CD27+, populations. Specifically, TRGV2+ receptors, which were associated with CD27+ cells at baseline, became enriched in the CD27- population.
Upon gating on IL-17A-positive cells, we also found that neonates contained elevated frequencies of γδTCR+ cells among total IL-17A-producing cells in the early stages of influenza infection. Furthermore, γδ T cells became the dominant producers of IL-17A after infection (Figure 2F). However, the expression of IL-17A and IFN-γ by γδ T cells in adult mice showed a distinct pattern from that in neonates: consistent with a previous study (Crowe et al., 2009), adult γδ T cells produced additional IL-17A at a later stage of infection (Figures S2C-E). In conjunction, these data from neonates and adults suggest distinct functionality of γδ T cells across ontogeny, such that neonatal γδ T cells rapidly secrete and are the primary source of IL-17A in response to influenza infection.
IL-17A, primarily secreted by γδ T cells, improves the survival of influenza-infected neonates by enhancing IL-33 production
IL-17A is the canonical cytokine that regulates Type-3 immunity, and its roles in adult influenza infection are still controversial. Some have suggested that IL-17A can be beneficial by modulating B cell responses (Wang et al., 2011), whereas others have proposed that IL-17A signaling may increase immune pathology by promoting neutrophil infiltration in adults (Crowe et al., 2009). Our data suggest that IL-17A might play a protective role in neonates because, in comparison to the lungs of virus-infected TCRδ−/− neonates, IL-17A (mainly produced by γδ T cells) is increased in wild-type lungs one day after influenza infection (Figure 3A). To test this formally in our experimental model, we infected TCRδ−/− neonates concomitant with administration of a low-level of recombinant mouse IL-17A, the dose for which was calculated based on the difference of lung total IL-17A between wild-type and TCRδ−/− neonates, and monitored the survival rate. Low dose IL-17A significantly improved the survival of γδ T cell-deficient mice, indicating that the IL-17A pathway is protective in the neonatal animals (Figure 3B). In contrast, a high dose of IL-17A administered to infected TCRδ−/− mice was detrimental to the host (Figure S4A), as was a low dose of IL-17A administered to infected wild-type neonates (Figures S4B), suggesting that IL-17A fine-tunes the balance between pathogenicity and protection. In order to determine if IL-17A specifically produced by γδ T cells is critical for protection against neonatal influenza infection, we isolated γδ T cells from wild-type or Il17a−/− neonatal lungs, transferred the cells to littermate TCRδ−/− neonates intranasally and infected the recipients with influenza virus (Figure 3C). The weight change (Figure 3D) and survival rate (Figure 3E) profiles suggested that IL-17A production by γδ T cells contributed to the protection γδ T cells provide to infected neonates.
Figure 3. IL-17A, predominantly secreted by γδ T cells, improves the survival of influenza-infected neonates by promoting IL-33 production.
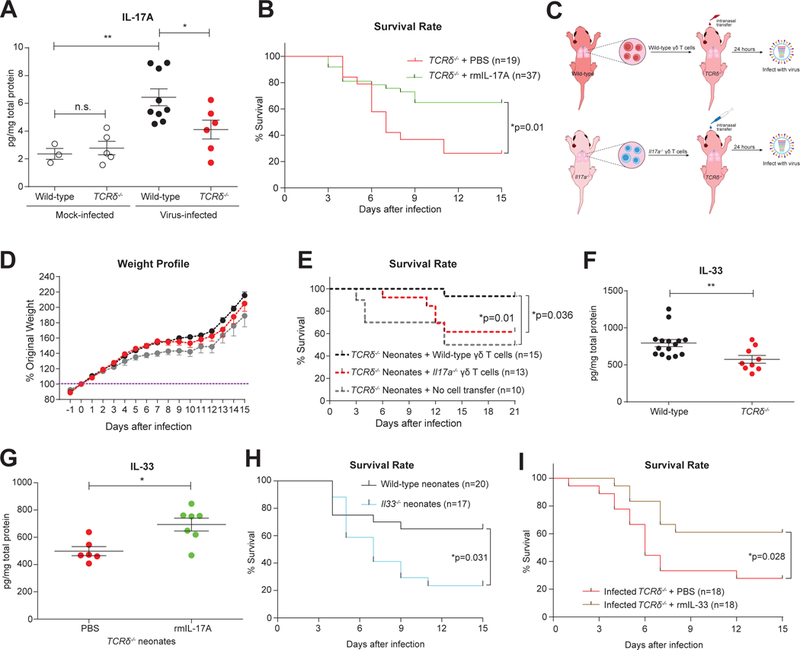
A. Estimation of IL-17A in whole lung homogenates from mock- (open) or influenza virus-(solid)- infected wild-type (black) and TCRδ−/− (red) neonates by ELISA at 1 day after infection. Samples are pooled from at least two independent experiments and data are shown as mean ± SEM (Mann-Whitney test).B. Survival rate of influenza virus-infected TCRδ−/− neonates administered with low levels of recombinant mouse IL-17A (rmIL-17A, green, n=37, 100pg/mouse) or PBS control (red, n=19) at the time of infection. Data are combined from six independent trials, which individually showed the same trend, and data are presented as mean ± SEM.C. Schema outlining wild-type and Il17a−/− γδ T cell transfers to TCRδ−/− neonates and subsequent infection.D and E. Body weight profile (D) and survival rate (E) of influenza-infected TCRδ−/− neonates receiving wild-type (black, n=15) or Il17a−/− (red, n=13) γδ T cells intranasally or no cell transfer (grey, n=10). Data are combined from four independent trials, which individually showed the same trend. Weight profile data are presented as mean ± SEM.F. Protein levels of IL-33 assessed by ELISA in influenza virus-infected wild-type (black, n=15) and TCRδ−/− lungs (red, n=9) at 1 day following infection. Samples are pooled from three independent experiments, and data are shown as mean ± SEM.G. Protein levels of IL-33 detected by ELISA in the lungs of TCRδ−/− neonates that had been infected with influenza virus and simultaneously treated with either a low level of rmIL-17A (green, n=7, 100pg/mouse) or PBS control (red, n=6). Data were collected 1 day after infection/treatment. Samples are pooled from at least two independent experiments, and data are shown as mean ± SEM.H. Survival rate of influenza virus-infected wild-type (black, n=20) and Il33−/− neonates (blue, n=17) with intranasal influenza virus infection. Data are combined from ten independent experiments and shown as mean ± SEM in weight change.I. Survival rate of influenza virus-infected TCRδ−/− neonates administered with recombinant mouse IL-33 (rmIL-33, brown, n=18, 10ng/mouse) or PBS control (red, n=18) at the time of infection. Data are combined from six individual experiments.*p<0.05, **p<0.01, n.s., not significant.
To explore which immune processes may have been disrupted by the loss of IL-17A in TCRδ−/− neonates, we next assessed the concentration of a panel of cytokines in homogenized lungs at day 2 after infection. The majority of cytokines generally associated with responses to influenza infection, including IFN-γ, IL-2, IL-1β, KC, GM-CSF, and IP-10, exhibited no differences between wild-type and TCRδ−/− neonatal lungs at day 2 after infection, suggesting that traditional innate and Type-1 responses are not dependent on γδ T cells (Figure S4C). In contrast, we found that loss of γδ T cells led to diminished levels of IL-33 (Figures 3F & S4D). To confirm this observation, we again assayed IL-33 to determine if γδ T cell-derived IL-17A was sufficient for inducing IL-33 in γδ T cell-deficient mice treated with IL-17A. Compared to virus-only controls, IL-33 levels in whole lung homogenates were elevated in response to low-dose IL-17A (Figure 3G). These data suggest that IL-33 production is upregulated downstream of IL-17A, which is secreted by γδ T cells in influenza virus-infected mice. In order to determine if IL-33 production functions as a component of the protection provided by γδ T cells and IL-17A in influenza-infected neonates, we next infected wild-type and Il33-deficient neonates with influenza virus. Il33−/− neonates exhibited increased mortality compared to wild-type littermates (Figure 3H). In addition, and consistent with our IL-17A results, the administration of IL-33 also improved the survival of infected TCRδ−/− neonate littermates (Figure 3I). Therefore, in neonatal mice, γδ T cells are the primary source of IL-17A, which in turn is necessary for IL-33 production, which itself is protective in these animals.
IL-17A promotes IL-33 production in mouse lung epithelial cells in synergy with influenza virus by elevating STAT3 phosphorylation
One study recently demonstrated that IL-17A promotes the exacerbation of IL-33-induced airway hyperresponsiveness in an allergic asthma model in BALB/c mice (Mizutani et al., 2014). However, the precise relationship between IL-17A and IL-33 production remains unclear. Although IL-33 can be produced by multiple cell types, epithelial cells are thought to be the primary source in mouse lungs (Pichery et al., 2012), and we found sorted lung epithelial cells from wild-type neonates to have the highest Il33 gene expression among other lung stromal cell types at day 2 after influenza infection (Figure S4E). We consequently set out to determine if IL-17A could directly induce IL-33 production in lung epithelial cells. To test this, we stimulated a mouse lung type-1 epithelial cell line (LET1s) (Rosenberger et al., 2014-2) in vitro using influenza virus and recombinant mouse IL-17A (either individually or in combination). After 24 or 48 hours (hrs) of treatment, the cells in each group were collected to assay IL-33 transcripts and protein with qRT-PCR and immunoblot, respectively. Quantification of the relative expression of Il33 mRNA showed that administration of either virus or IL-17A could enhance IL-33 production in LET1 cells, but the effect of the virus and IL-17A combination was especially striking (Figure 4A). Consistent with these findings, substantial IL-33 protein was also detected after treatment with both stimuli by immunoblot (Figure 4B). Interestingly, IL-17A alone was more potent than virus alone in elevating IL-33 mRNA and protein expression.
Figure 4. IL-17A elevates IL-33 production in murine lung epithelial cells via STAT3 phosphorylation.
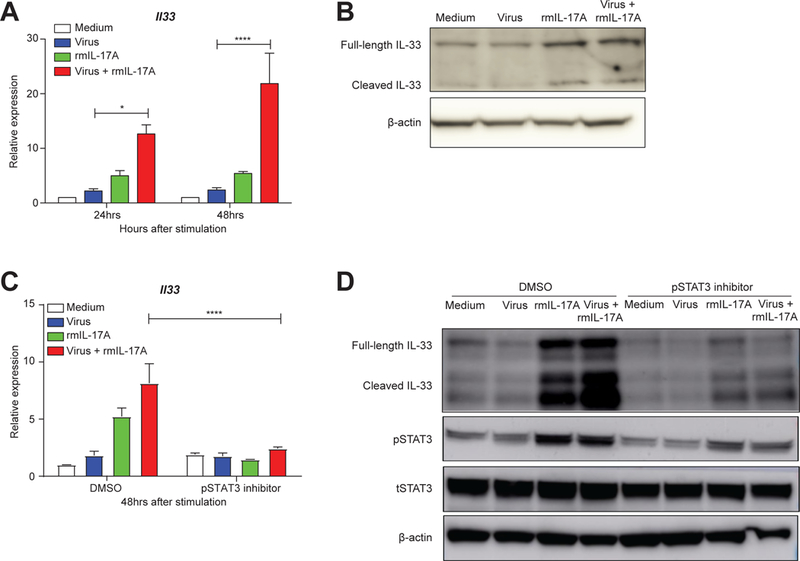
A. Relative expression of Il33 mRNA, as assessed by quantitative real-time PCR, in mouse lung epithelial cells (LET1s) treated with medium (white), A/x31 influenza virus (blue, 3 MOI), rmIL-17A (green, 50ng/ml), or virus+rmIL-17A (red) at 24hrs and 48hrs after treatment. Data are shown as mean ± SEM and combined from three separate experiments that independently showed the same trend.B. Immunoblot assay of IL-33 protein in stimulated LET1s for the same conditions as in (A) at 48 hrs after stimulation.C. Quantitative real-time PCR analysis of Il33 mRNA in LET1 cells treated as in A and B with and without STAT3 phosphorylation inhibitor S3I-201 (100μM in 0.05% DMSO) at 48hrs after treatment. Data are shown as mean ± SEM and combined from two separate experiments that independently showed the same trend.D. Immunoblot analysis of IL-33 and phosphorylated STAT3 (pSTAT3) in the LET1 lysates treated as before (A, B, and C). Total STAT3 (tSTAT3) and β-actin are also shown. Immunoblots were repeated twice showing similar results.*p<0.05, ****p<0.0001
We next examined the signaling pathways induced by influenza virus and IL-17A treatment in LET1 cells. Examination of the transcriptional binding sites in the human IL-33 promoter (ENCODE data in UCSC genome browser, Build hg19) identified Signal Transducer and Activator of Transcription 3 (STAT3) as a potential regulator of Il33 transcription, so we performed a Western blot assay to test whether STAT3 was activated downstream of treatment with either influenza virus or IL-17A. We found that phosphorylated-STAT3 (pSTAT3) was coordinately upregulated with IL-33 (data not shown). To confirm the role of pSTAT3 in IL-33 production, we inhibited the phosphorylation of STAT3 by co-incubating with S3I-201, a well-established small molecule inhibitor that prevents the dimerization and phosphorylation of STAT3 (Siddiquee et al., 2007). Under the same stimulation conditions as above, we compared the relative expression levels of Il33 among IL-17A- and/or virus-stimulated LET1 cells that were co-stimulated with either DMSO or S3I-201 for 48 hrs. The pSTAT3 inhibitor robustly blocked the upregulation of IL-33 induced by IL-17A, even in the presence of influenza infection and IL-17A co-treatment (Figure 4C). Consistent with these transcript assays, IL-33 protein abundance was also attenuated after treatment with S3I-201 (Figure 4D). Thus, our results indicate that IL-17A can directly increase the production of IL-33 in lung epithelial cells downstream of STAT3 phosphorylation.
Deficiency in γδ T cells compromises the accumulation and functions of ILC2s and Treg cells
ILC2s (Monticelli et al., 2011), Treg cells, and Th cells (Arpaia et al., 2015) have been reported to play roles in maintaining tissue homeostasis for influenza-infected adults. Each of these cell types can express the IL-33 receptor (ST2) on their cell surface and can be recruited to the lungs by IL-33. Compared to TCRδ−/− lungs, we observed more ILC2s and total Treg cells in wild-type lungs, but comparable amounts of Th cells were present in both mouse strains at day 5 after infection (Figures 5A-B & S5A). ILC2s and Treg cells have both been implicated in the production of Areg, which is important for tissue repair after influenza infection in adult mice (Arpaia et al., 2015; Monticelli et al., 2011; Zaiss et al., 2015). The assessment of Areg levels in total wild-type lung homogenates showed that the peak of Areg production was at day 5 after infection (Figure S5B). Thus, we measured the amount of Areg-producing ILC2s, Treg cells, and Th cells in infected wild-type and TCRδ−/− neonates at this time point. We found higher frequencies and numbers of Areg-producing ILC2s and Treg cells in wild-type, infected lungs compared to those from TCRδ-deficient mice (Figures 5C-D). In contrast, Th cell responses were equivalent between the two groups at day 5 after infection (Figures 5D & S5C). In addition, neutrophils accumulating in the infected lungs rarely produced Areg (Figure S5D). Consistent with the importance of IL-33 in regulating Type-2 responses, the frequency and number of Areg-producing ILC2s were also higher in wild-type neonates compared to Il33−/− animals (Figures 5E-F). In order to characterize the Areg-producing cells more thoroughly, we isolated ILC2s, ST2+CD4+ cells, and ST2-CD4+ cells from wild-type and TCRδ−/− neonatal lungs at day 5 after infection and performed qRT-PCR assays to measure gene expression of multiple signature genes. Although many factors remained unchanged, cells from wild-type mice exhibited higher levels of Areg and Il10, while cells from TCRδ−/− had elevations in Type-2 response genes, including Il5 and Gata3 (Figures 5G & S5E-F). The consistently lower Areg expression in cells from TCRδ−/− mice strongly suggests that the deficiency in γδ T cells compromises tissue repair after infection (Figure 5G).
Figure 5. Deficiency in γδ T cells results in decreased accumulation and functionality of ILC2s and Treg cells.
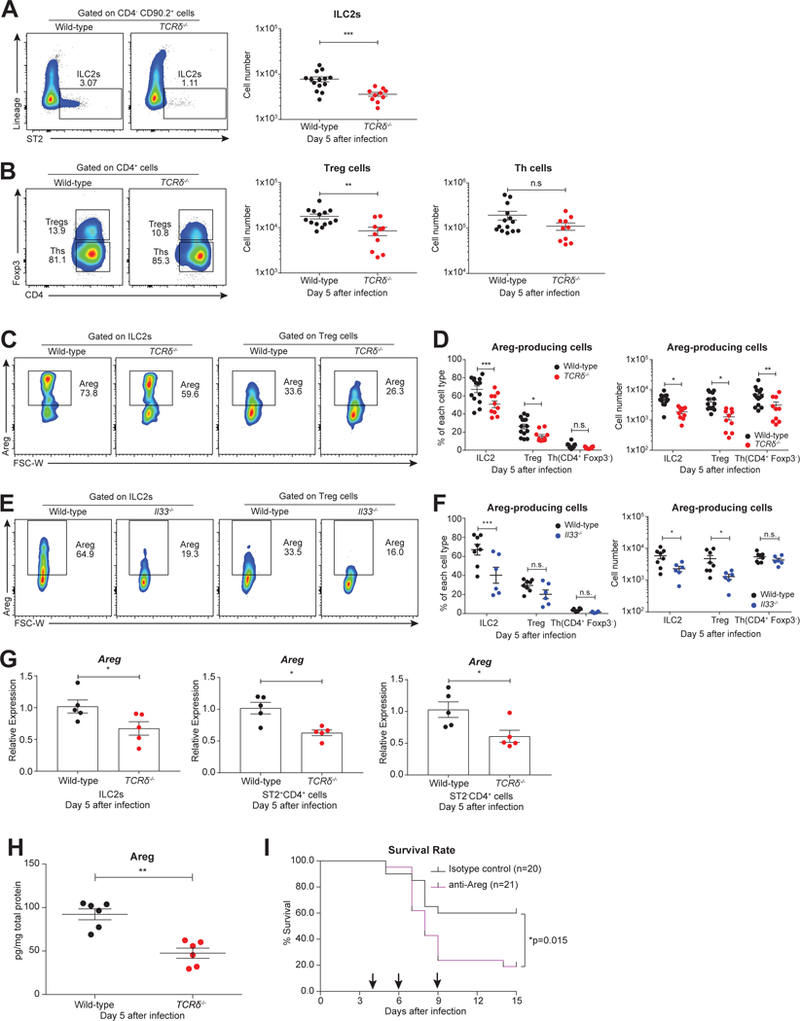
A. Representative flow cytometric plots (left) and cell number (right) of ILC2s (CD4- CD90.2+ Lin- ST2+) in the lungs of infected wild-type (black, n=14) and TCRδ−/− (red, n=10) neonates at day 5 following infection.B. Representative flow cytometric plots (left) and cell number (right) of Treg cells (CD4+ Foxp3+) and Th cells (CD4+ Foxp3-) in the lungs of infected wild-type (black, n=14) and TCRδ−/− (red, n=10) neonates at day 5 following infection.C. Representative flow cytometric plots of Areg-producing cells gated on ILC2s and Treg cells in the lungs of infected wild-type (black, n=14) and TCRδ−/− (red, n=10) neonates at 5 days following infection.D. Frequency (left) and cell number (right) of Areg-producing ILC2s, Treg cells, and Th cells in the lungs of infected wild-type (black, n=14) and TCRδ−/− (red, n=10) neonates at 5 days after infection. (A-D) Data are combined from four independent experiments and shown as mean ± SEM.E. Representative flow cytometric plots of Areg-producing cells gated on ILC2s and Treg cells in the lungs of infected wild-type (black, n=8) and Il33−/− (blue, n=6) neonates at 5 days following infection.F. Frequency (left) and cell number (right) of Areg-producing ILC2s, Treg cells, and Th cells in the lungs of infected wild-type (black, n=8) and Il33−/− (blue, n=6) neonates at 5 days after infection. (E-F) Data are combined from three independent experiments and shown as mean ± SEM.G. Relative expression levels of Areg of ILC2 cells, ST2+CD4+ cells, and ST2-CD4+ cells sorted from wild-type (black, n=5) and TCRδ−/− (red, n=5) neonatal lungs at 5 days after infection measured by quantitative real-time PCR. Samples are pooled from two independent experiments, and data are presented as mean ± SEM.H. Protein levels of Areg in whole-lung lysate from infected wild-type (black, n=6) and TCRδ−/− (red, n=6) neonates at 5 days after infection. Data are combined from two independent experiments and shown as mean ± SEM.I. Survival rate of infected wild-type neonates that were injected intraperitoneally at days 4, 6, and 9 (shown by arrows) after influenza virus infection with Areg neutralizing antibody (purple, α-Areg antibody, 2ug/mouse/timepoint, n=21) or isotype control antibody (black, normal goat IgG control, 2ug/mouse/time point, n=20). Data are combined from three separate trials that individually showed the same trend.*p<0.05, **p<0.01, ***p<0.001, n.s., not significant.
In order to investigate the importance of Areg in the neonatal influenza model, we next assessed the levels of Areg by ELISA. The overall abundance of Areg protein was lower in TCRδ−/− whole-lungs relative to wild-type lungs (Figure 5H), and this deficiency is likely implicated in the reduced survival rate observed in TCRδ−/− mice after infection (Figure 1D). Based on the kinetics of Areg production (Figure S5B), we also delivered Areg neutralizing antibody or isotype-control antibody intraperitoneally to wild-type mice on Days 4, 6, and 9 after infection. After monitoring the survival of these treated mice, we found that Areg is protective during neonatal influenza infection (Figure 5I) in wild-type animals. Taken together, these results suggest that neonatal γδ T cells benefit neonatal mice during influenza infection specifically by mediating Areg production via ILC2s and Treg cells.
IL-17A, IL-33, and Areg are correlated with each other and with improved outcomes in influenza-infected human children
To determine whether the findings in our neonatal influenza mouse model were translatable to humans, we assessed the abundance of IL-17A, IL-33, and Areg in nasal aspirates obtained from children and infants with community-acquired influenza infection. We found that IL-33 levels were significantly and positively correlated with IL-17A levels in the airways of influenza-infected subjects (Figure 6A). However, IL-33 was not correlated with IFN-γ (Figure 6B), suggesting that IL-33 production is not controlled by IFN-γ in this context and that all cytokine levels were not generally correlated. We also found a positive correlation between IL-33 and Areg (Figure 6C). However, IFN-γ and Areg (Figure S4A) were also correlated, possibly indicating the existence of independent pathways associated with Areg-production in human infection, or a non-mechanistic correlation between IFN-γ and Areg kinetics.
Figure 6. The IL-17A/IL-33/Areg cascade in influenza-infected children is associated with robust disease outcome.
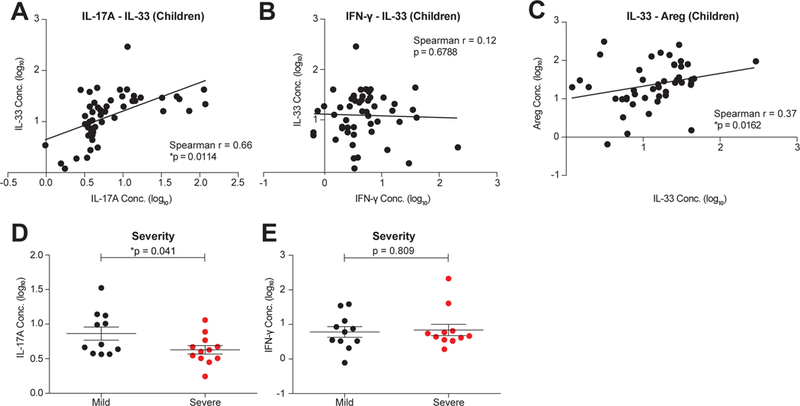
A- C. Correlation between concentrations of human IL-17A and IL-33 (A), IFN-γ and IL-33 (B) and IL-33 and Areg protein levels (C) in the nasal aspirates of influenza-infected children (< 8 years old, n=51).D. Concentration of IL-17A at Day 0 after enrollment in the nasal aspirates of children with mild (black, n=11) and severe (red, n=12) influenza disease outcomes.E. Concentration of IFN-γ at Day 0 after enrollment in the nasal aspirates of children with mild (black, n=11) and severe (red, n=12) disease outcomes.Data are presented as mean ± SEM, and in each case cytokine values (pg/ml) were log10 transformed for visualization only. *p<0.05.
In searching for correlates of disease outcome, we found that IL-17A concentrations at diagnosis (within 96 hours of symptom onset) in nasal aspirates predicted the severity of influenza illness in infants and children, as those with mild disease outcomes had significantly higher levels of IL-17A than those with severe outcomes (Figure 6D). In contrast, IFN-γ was neither correlated with IL-17A (Figure S4B) nor associated with disease severity (Figure 6E).
To confirm our observation in mice that IL-17A can directly induce IL-33 production by lung epithelial cells, we stimulated cells from a human lung epithelial cell line (A549) with virus and/or recombinant human IL-17A together with either DMSO or the STAT3 inhibitor S3I-201 (as above). qRT-PCR analysis of these treatments indicated that, as in mice, human IL-17A can enhance the gene expression of IL33 in lung epithelial cells in a STAT3 phosphorylation-dependent manner during influenza infection (Figures S4C). In summary, IL-17A (rather than IFN-γ) is positively associated with the IL-33-Areg axis and potentially influences the disease outcomes in children with influenza infections.
Discussion
In comparison to older children and young adults, infant influenza infections are associated with higher rates of hospitalization and mortality. In the human FLU09 cohort, we previously reported that viral clearance was not impaired in infants compared to other demographic groups, and yet disease severity was increased (Oshansky et al., 2014). Intriguingly, previous reports have also shown that neonates cannot mount robust Type-1 immune responses, as revealed by the insufficient magnitude and function of neonatal CD8+ T cells (You et al., 2008). Instead, Type-2 immune responses are more dominant in the neonatal immune system (Adkins et al., 2004; Dowling and Levy, 2014; Garcia et al., 2000), perhaps as a mechanism limiting potential inflammatory damage. As a result of the variations in the immune system evident throughout ontogeny, the parameters that determine mild or severe outcomes after influenza infection likely differ between infants and adults.
During the development of the immune system, γδ T cells develop and migrate to mucosal organs prior to αβ T cells, suggesting that the former may play a critical role in immune competence in neonates (Chien et al., 2014; Hayday, 2000; Vantourout and Hayday, 2013). Here, we confirmed that notion by defining a significant role for γδ T cells in maintaining lung homeostasis and tissue integrity during neonatal influenza infection.
The primary function of γδ T cells in this model of influenza infection appears to be the rapid production of IL-17A to initiate a cascade of Type-2 and tissue repair responses, which were characterized by the induction of IL-33, the recruitment of ILC2s and Treg cells, and the production of amphiregulin. However, the ligands for γδ T cell activation in this system are undetermined. This unconventional role for IL-17A diverges significantly from its better characterized contributions to Type-3 immunity, for instance in responses to lung infections by bacteria such as Staphylococcus aureus (Cheng et al., 2012) and Mycobacterium tuberculosis (Lockhart et al., 2006). Thus far, little has been discovered about the potential role of IL-17A in Type-2 immunity, which predominates during allergic reactions and helminth infections rather than bacterial infections (Spellberg and Edwards, 2001). One study has suggested IL-17A-producing γδ T cells can modulate regulatory T cell homeostasis and thermogenesis in adipose tissues by inducing IL-33 production (Kohlgruber et al., 2018). Another study has reported an exacerbation of IL-33 production after IL-17A administration, but this was in a model of allergic asthma, where the effects of IL-17A were primarily pathological (Mizutani et al., 2014). Although IL-17A and influenza virus both induce robust IL-33 production, IL-17A is more potent than virus in upregulating IL-33 expression in lung epithelial cells through STAT3 phosphorylation. Recent studies also found that IL-17A can promote tumor growth (Wang et al., 2009) and angiogenesis (Pan et al., 2015) via STAT3 phosphorylation, suggesting that this may be a common potential target for IL-17A signaling, with effects on tissue repair and remodeling. In contrast, canonical IL-17A signaling proceeds through the nuclear factor-κB (NF-κB) pathway mediated by ACT1 (Gaffen, 2009), which results in the activation of the Type-3 immune response traditionally associated with IL-17A. In LET1 cells we found that NF-κB activation (data not shown) was dispensable for IL-33 induction.
To confirm the specific role of IL-17A produced by γδ T cells in protection, we performed a γδ T cell-transfer experiment from IL-17A wild-type or deficient mice. Our results showed a significant difference in the survival between infected TCRδ−/− neonates receiving wild-type γδ T cells and those receiving Il17a−/− γδ T cells. However, we also observed a delayed, but not significantly different, survival among TCRδ−/− neonates with Il17a−/− γδ T cells, compared to TCRδ−/− neonates receiving a PBS control. Thus, we speculate there may be some small additional potential IL-17A-independent effects of neonatal γδ T cells. Transfer of γδ T cells into mice lacking both IL-33 and γδ T cells would confirm the requirement for an intact IL-33 pathway downstream of γδ T cell activity and will be the subject of future experiments in the lab.
Given the important role of Type-2 immunity in mediating allergy and asthma, as well as the epidemiological linkages between infant infections and future development of these diseases, it seems possible that this IL-17A-IL-33 pathway may be a key link between early infection and atopy. One hypothesis for why this pathway may be more active or essential in neonates than in adults, where the effects of IL-17A appear minimal, is the significantly smaller lung volumes in the very young. Lung damage resulting from infection can, as a proportion of total lung volume, generate a greater impact on lung efficiency. Thus, robust induction of Type-2 immunity and its associated tissue repair function may be necessary to maintain the capacity of the infant lung, albeit at the cost of an increased risk of subsequent pathologies.
Of particular interest, we observed that IL-17A, instead of IFN-γ, is positively correlated with IL-33 in nasal aspirate samples from children with naturally-acquired influenza infection. This correlation also extends to IL-33 and Areg in these samples, consistent with the findings in our mouse model demonstrating an early regulatory role of IL-17A for the subsequent immune response to neonatal influenza infection. Interestingly, this is thus far the only protective association with an increased cytokine concentration that we have observed in this cohort. In contrast, Type-1 immune responses were not associated with IL-33. These data indicate the novel IL-17A-IL-33-Areg axis may serve as a potential therapeutic target during infant influenza infection; further study of these markers across additional human cohorts will be important not only for verifying the importance of this axis in infant influenza infection, but also for fully elucidating the particular conditions underlying this aspect of γδ T cell function in humans in general.
In conclusion, our findings provide evidence that IL-17A, secreted by γδ T cells, has a critical function in protecting infants during influenza infection. IL-17A-producing γδ T cells may provide this protection by inducing IL-33 secretion and Type-2 immune responses which, in turn promote tissue homeostasis and integrity.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Live/Dead Ghost DyeTM Violet 510 | Tonbo Biosciences | Cat.#13–0870 |
| Anti-mouse TCRγ/δ (clone GL3) | Biolegend | Cat.#118108 |
| Anti-mouse TCRβ (clone H57–597) | Biolegend | Cat.#109230 |
| Anti-mouse CD4 (clone RM4–5) | Biolegend | Cat.#100552 |
| Anti-mouse CD8α (clone 53–6.7) | Biolegend | Cat.#100742 |
| Anti-mouse CD3 (clone 17A2) | Biolegend | Cat.#100236 |
| Anti-mouse CD90.2 (clone 30–H12) | Biolegend | Cat.#105328 |
| Anti-mouse CD25 (clone PC61.5) | eBioscience | Cat.#25–0251-82 |
| Anti-mouse CD27 (clone LG.3A10) | Biolegend | Cat.#124216 |
| Anti-mouse CD44 (clone IM7) | Biolegend | Cat.#103049 |
| Anti-mouse CCR6 (clone 29–2L17) | Biolegend | Cat.#129814 |
| Anti-mouse Sca-1 (clone D7) | Biolegend | Cat.#108146 |
| Anti-mouse ST2 (clone DIH9) | Biolegend | Cat.#145304 |
| Anti-mouse Lineage cocktail (clonelin-1) | Biolegend | Cat.#133302 |
| Anti-mouse IL-17A (clone eBio17B7) | eBioscience | Cat.#11–7177–81 |
| Anti-mouse IFN-γ (clone XMG1.2) | Biolegend | Cat.#505826 |
| Anti-mouse Foxp3 (clone FJK-16s) | eBioscience | Cat.#25–5773-82 |
| Anti-mouse Gr-1 (clone RB6–8C5) | Biolegend | Cat.#108406 |
| Anti-mouse CD11b (clone M1/70) | Biolegend | Cat.#101243 |
| Anti-mouse CD11c (clone N418) | Biolegend | Cat.#117339 |
| Anti-mouse CD31 (clone 390) | Biolegend | Cat.#102408 |
| Anti-mouse CD326 (clone G8.8) | Biolegend | Cat.#118214 |
| Anti-mouse Mouse Amphiregulin Biotinylated Antibody |
R&D | Cat.#BAF989 |
| Brilliant Violet 421 Streptavidin | Biolegend | Cat.#405225 |
| Mouse Amphiregulin Antibody (Polyclonal Goat IgG) |
R&D | Cat.#AF989 |
| Normal Goat IgG Control (Polyclonal Goat IgG) |
R&D | Cat.#AB-108-C |
| Mouse IL-33 Goat Polyclonal Antibody | R&D | Cat.#AF3626 |
| Phospho-Stat3 (Tyr705) (D3A7) XP® Rabbit mAb |
Cell Signaling Technology |
Cat.#9145 |
| Stat3 (124H6) Mouse mAb | Cell Signaling Technology |
Cat.#9139 |
| β-Actin (13E5) Rabbit mAb | Cell Signaling Technology |
Cat.#4970 |
| Virus Strains | ||
| Influenza strain X31 (A/HKx31 [H3N2, A/Aichi/2/68 × A/Puerto Rico/8/34 (2 + 6)] |
Lab grown from an 8-plasmid reverse genetic system (Hoffmann et al., 2000) |
N/A |
| Influenza strain PR8 (A/Puerto Rico/8//34, H1N1) |
Lab grown from an 8- plasmid reverse genetic system (Hoffmann et al., 2000) |
N/A |
| Influenza strain Brisbane (A/Brisbane/10/2007, H3N2) |
Lab grown by egg passage |
N/A |
| Biological Samples | ||
| Nasal Aspirates of influenza-infected pediatric patients |
FLU09 cohort (Oshansky et al., 2014) |
N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| S3I-201 (pSTAT3 inhibitor) | Sigma | Cat.#SML0330 |
| Recombinant Mouse IL-17A Protein, CF | R&D | Cat.#421-ML-025/CF |
| Recombinant Mouse IL-33 Protein, CF | R&D | Cat.#3626-ML-010/CF |
| Recombinant Human IL-17A Protein, CF | R&D | Cat.#7955-IL-025/CF |
| Cell Stimulation Cocktail (plus protein transport inhibitors) (500X) |
eBioscience | Cat.#00-4975-03 |
| Protein Transport Inhibitor Cocktail (500X) |
eBioscience | Cat.#00-4980-03 |
| UltraComp eBeads | Invitrogen | Cat.#01-2222-41 |
| ArC Amine Reactive Compensation Bead Kit |
Invitrogen | Cat.# A10346 |
| cOmplete Protease Inhibitor Cocktail | Sigma-Aldrich | Cat.# 11836170001 |
| TruStain fcXTM Rat Anti-Mouse CD16/CD32 (Fc Block) (clone 93) |
Biolegend | Cat.#101320 |
| Critical Commercial Assays | ||
| Click-iT Plus EdU Pacific BlueTM
Flow Cytometry Assay Kit |
Invitrogen | Cat.#C10636 |
| Fixation and Permeabilization Solution Kit with BD GolgiPlugTM |
BD Biosciences | Cat.#555028 |
| Foxp3/Transcription Factor Staining Buffer Set |
eBioscience | Cat.#00-5523-00 |
| MILLIPLEX MAP Mouse Cytokine/Chemokine Magnetic Bead Panel - Premixed 32 Plex |
Millipore | Cat.#MCYTMAG-70K-PX32 |
| Mouse IL-17 DuoSet ELISA | R&D | Cat.#DY421-05 |
| Mouse IL-33 DuoSet ELISA | R&D | Cat.#DY3626-05 |
| Mouse Amphiregulin DuoSet ELISA | R&D | Cat.#DY989 |
| Mouse IFN-gamma DuoSet ELISA | R&D | Cat.#DY485-05 |
| Human IL-17 DuoSet ELISA | R&D | Cat.#DY317-05 |
| Human IL-33 DuoSet ELISA | R&D | Cat.#DY3625B-05 |
| Human Amphiregulin DuoSet ELISA | R&D | Cat.#DY262 |
| MILLIPLEX MAP Human Cytokine/Chemokine Magnetic Bead Panel - Premixed 41 Plex |
Millipore | Cat.# HCYTMAG-60K-PX41 |
| TaqMan Universal PCR Master Mix | Applied Biosystems | Cat.#4304437 |
| SuperScript VILO cDNA Synthesis Kit | Invitrogen | Cat.#11754050 |
| RNeasy Mini Kit | Qiagen | Cat.#74104 |
| TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold |
Illumina | Cat.#20020599 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat.#23225 |
| Deposited Data | ||
| RNA-Seq data | This paper | GEO: GSE99683 |
| Experimental Models: Cell Lines | ||
| LET1 | Laboratory of Alan Aderem (Rosenberger et al., 2014-2) |
N/A |
| A549 | ATCC | Cat.#CCL-185 |
| MDCK | ATCC | Cat.#CCL-34 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory |
Cat.#000664 |
| Mouse: C57BL/6J.129P2-Tcrdtm1Mom/J | The Jackson Laboratory |
Cat.#002120 |
| Mouse: Il33tm1(KOMP)Vlcg | Rederived in C57BL/6J mice by using embryos obtained from KOMP |
Cat.#VG12663 |
| Mouse: Il17a-/- | Rederived in C57BL/6J mice by using embryos donated by Sarah Gaffen and Jay Kolls with the agreement of Yoichiro Iwakura |
N/A |
| Oligonucleotides | ||
| TCR Sequencing Primers, see Table S1 |
This paper | N/A |
| Mouse Il17a Taqman primer | Applied Biosystems | Cat.#Mm00439618_m1 |
| Mouse Il33 Taqman primer | Applied Biosystems | Cat.#Mm00505403_m1 |
| Mouse Areg Taqman primer | Applied Biosystems | Cat.#Mm01354339_m1 |
| Mouse Il5Taqman primer | Applied Biosystems | Cat.#Mm00439646_m1 |
| Mouse Il10 Taqman primer | Applied Biosystems | Cat.#Mm01288386_m1 |
| Mouse Gata3 Taqman primer | Applied Biosystems | Cat.#Mm00484683_m1 |
| Mouse Rorc Taqman primer | Applied Biosystems | Cat.#Mm01261022_m1 |
| Mouse Stat3 Taqman primer | Applied Biosystems | Cat.#Mm01219775_m1 |
| Mouse Tbx21 Taqman primer | Applied Biosystems | Cat.#Mm00450960_m1 |
| Mouse Foxp3 Taqman primer | Applied Biosystems | Cat.#Mm00475162_m1 |
| Mouse Gapdh Taqman primer | Applied Biosystems | Cat.#Mm99999915_g1 |
| Human IL33 Taqman primer | Applied Biosystems | Cat.#Hs04931857_m1 |
| Human GAPDH Taqman primer | Applied Biosystems | Cat.#Hs02786624_g1 |
| Software and Algorithms | ||
| Flowjo V10 | FlowJo, LLC | https://www.flowjo.com/ |
| GraphPad Prism 7.0 | GraphPad Software, Inc |
http://www.graphpad.com/scientific-software/prism/ |
| STAR (2.5.2b) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| edgeR | (Robinson and Oshlack, 2010) | http://bioinf.wehi.edu.au/edgeR/ |
| GSEA (2.2.4) | (Subramanian et al., 2005) | https://software.broadinstitute.org/gsea/index.jsp |
| NetPath | (Merico et al., 2010) | http://www.netpath.org/ |
| Other | ||
| BD Fortessa | BD Biosciences | N/A |
| Sony Biotech Synergy sorter | Sony Biotech | Model SY3200 |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, P.G. Thomas (paul.thomas@stjude.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Strains
C57BL/6J (wild-type) and C57BL/6J.129P2-Tcrdtm1Mom/J (TCRδ−/−) mice were purchased from the Jackson Laboratory. Il33tm1(KOMP)Vlcg (Il33−/−) embryos were obtained from the KnockOut Mouse Project (KOMP; https://www.komp.org/) and rederived using C57BL/6 mice at St. Jude Children’s Research Hospital’s Animal Resource Center. TCRδ+/−, Il33+/− and Il17a+/− mice were bred to generate littermate control as well as homozygous knockout mice. Il17a−/− mice were rederived from mice donated by Sarah Gaffen and Jay Kolls (University of Pittsburgh), with the agreement of Dr. Yoichiro Iwakura at the Tokyo University of Science. Both male and female neonates were infected at the age of 7-day-old, and female adult mice were infected at the age of 8–10 weeks. Infection methods are described below.
An important factor involved in neonatal immunity previously identified in such studies is the potential effect of variation in environment (Cicin-Sain et al., 2012); indeed, in our own experiments we have observed differences in neonates reared by different wild-type and TCRδ−/− dams. These observations suggest that γδ T cells may regulate additional features of immunity via breast milk or some other maternal factor. To avoid such confounding factors in the experiments reported here, all γδ T cell- and IL-33-deficient mouse studies were performed with heterozygous parents and utilized comparisons of littermate controls.
Human Samples
For this study, we used a subset of individuals from the FLU09 cohort which has been previously described (Oshansky et al., 2014). Nasal aspirates were collected at time of enrollment from 25 pediatric patients with laboratory confirmed influenza virus infection with no predisposing conditions (48% female, 88% African American, average age at enrollment = 1.5, age range 0.05 – 7.31, age mean ± STD 1.50 ± 1.47). Patients included in this study were stratified into mild and severe groups using treatment status, with 56% of patients having severe influenza illness outcome.
Ethical Compliance for Mouse and Human Study
Animal study protocols were approved by St. Jude Children’s Research Hospital Committee on Use and Care of Animals. All the mice were bred and maintained at St. Jude Children’s Research Hospital under specific-pathogen free conditions.
For studies involving human samples, inclusion criteria required that participants meet the clinical case definition of influenza virus infection at the time of enrollment. This study was conducted in compliance with 45 CFR 46 and the Declaration of Helsinki. Institutional Review Boards of St. Jude Children’s Research Hospital and the University of Tennessee Health Science Center/Le Bonheur Children’s Hospital approved the study.
METHOD DETAILS
Animal Model of Influenza A Infection
Neonatal mice were anesthetized by isoflurane inhalation and infected intranasally (i.n.) with A/HKx31 [H3N2, A/Aichi/2/68× A/Puerto Rico/8/34 (2 + 6)] in 10μl (0.6×102 EID50/g body weight).
For neonatal EdU experiment, infected neonates were injected intraperitoneal (i.p.) with 50μl EdU (1μg/μl) diluted in PBS at 8 hrs before tissue harvest.
For neonatal influenza infection with rmIL-17A (low-dose: 100pg/mouse, high-dose: 1000pg/mouse) and rmIL-33 (10ng/mouse) administration experiment, TCRδ−/− neonates were intranasally infected with A/HKx31 virus mixed with rmIL-17A (421-ML; R&D) or rmIL-33 (3626-ML; R&D) in 10μl.
For anti-Areg neutralizing experiments, wild-type neonates were infected with A/HKx31 virus in 10μl i.n. followed by intraperitoneal injection of 2ug of anti-Areg (AF989; R&D) or normal goat IgG control antibody (AB-108-C; R&D) in 50μl of PBS on day 4, 6 and 9 after infection.
For adult influenza infection, adult wild-type and TCRδ−/− littermates (8–10 weeks old) were anesthetized by Tribromoethanol (Avertin) i.p. injection and infected i.n. with 1MLD50 A/Puerto Rico/8/34 virus in 30μl.
Quantification of IAV in Infected Lung Tissue
Tissues were homogenized and centrifuged at 10,000 rpm for 15 mins. Supernatant from lung homogenates were measured by traditional 50% Tissue culture Infective Dose assay in Madin-Darby canine kidney (MDCK) cells.
Cytokine Measurement of Mouse Lung Homogenates
Cytokines in the supernatant of lung homogenates were measured by mouse DuoSet ELISA kits (R&D) or Milliplex assays (Millipore) according to manufacturer’s’ instructions. The results were normalized by total protein determined by BCA assay (Thermo Fisher).
Flow Cytometry
Mice were euthanized at described timepoints and lungs were collected for analysis. For analysis of surface markers, cells were stained in PBS containing 2% (wt/vol) BSA, with TCRγ/δ (GL3; Biolegend), TCRβ (H57–597; Biolegend), CD4 (RM4–5; Biolegend), CD8α (53–6.7; Biolegend), CD3 (17A2; Biolegend), CD90.2 (30-H12; Biolegend), CD25 (PC61.5; eBioscience), ST2 (DIH9; Biolegend), CD27 (LG.3A10; Biolegend), CD44 (IM7; Biolegend), CCR6 (29–2L17; Biolegend), Sca-1 (D7; Biolegend), ST2 (DIH9; Biolegend), Lineage cocktail (lin-1; Biolegend), Gr-1 (RB6–8C5; Biolegend), CD11b (M1/70; Biolegend), CD11c (N418; Biolegend), CD31 (390; Biolegend) and CD326 (G8.8; Biolegend). Intracellular IL-17A (eBio17B7, eBioscience), IFN-γ (XMG1.2, Biolegend), Foxp3 (FJK-16s, eBioscience), Amphiregulin Biotinylated Antibody (R&D), and Brilliant Violet 421 Streptavidin (Biolegend) were analyzed by flow cytometry according to the manufacturer’s instructions. For intracellular cytokine staining, T cells were stimulated for 4 hrs with Cell stimulation cocktail plus protein transport inhibitors (eBioscience) before intracellular staining, which is according to the manufacturer’s instructions (eBioscience). Antibody validation profiles are available via 1DegreeBio (http://1degreebio.org/) or CiteAb (http://www.citeab.com). Flow cytometry data were acquired on Fortessa (BD Biosciences) and analyzed using FlowJo software (TreeStar).
RNA-Seq
Genomic DNA-free total RNA was extracted from whole, homogenized lungs using the RNeasy Mini kit (Qiagen). Individually indexed libraries were constructed for each mouse using the Illumina TruSeq Stranded Total RNA kit, and multiplexed libraries were sequenced across two Illumina HiSeq lanes, yielding over 580 million 100bp paired-end reads. Reads were mapped to the mm10 genome using STAR (version 2.5.2b; (Dobin et al., 2013)) with default parameters, and gene counts were obtained with HTSeq (version 0.6.1p1; (Anders et al., 2015)) using Gencode comprehensive chromosomal annotations (vM13). Putative genes, pseudogenes, allosomal genes, and genes with fewer than 5 counts per million (CPM) were excluded from analyses. Gene counts were normalized to trimmed mean of M-values (TMM) using edgeR (Robinson and Oshlack, 2010) and then transformed to log2-CPM and modeled as a function of genotype (i.e., wild-type, TCRδ−/−) using the limma package to determine differential expression across conditions (Ritchie et al., 2015). Results were corrected for multiple testing using the Benjamini and Hochberg method to produce FDR-adjusted p-values. For Gene Set Enrichment Analysis, each gene was assigned a rank by calculating the product of the log2-fold change and the inverse of the FDR-adjusted p-value; this ranked set was then analyzed by GSEA (version 2.2.4; (Subramanian et al., 2005)) using the GseaPreranked function (1,000 permutations, uncollapsed dataset, classic enrichment statistic, 500/15 maximum/minimum set sizes) and the mouse NetPath gene set (Merico et al., 2010). RNA-Seq data were deposited at NCBI GEO under accession GSE99683.
Quantitative Real-time PCR
RNA of sorted γδ T cells, ILC2s, ST2+CD4+ cells and ST2-CD4+ cells was isolated using RNeasy Mini kit (Qiagen) and reverse transcribed into cDNA with the SuperScript Vilo kit® (Invitrogen). Gene expression was assessed using Taqman® Gene Expression Master Mix according to the manufacturer’s instructions (Applied Biosystems). All human and mouse primers and probe sets were purchased from Applied Biosystems. Data were analyzed with the method (Livak and Schmittgen, 2001) and results with Ct value higher than 35 were considered undetected.
In Vitro Stimulation
Murine lung epithelial cells (LET1s) and human lung epithelial cell line (A549) were maintained in our laboratory and cultured in DMEM supplemented with 10% (vol/vol) FBS, 1% (vol/vol) L-Glutamine, and 1% (vol/vol) penicillin-streptomycin. For in vitro stimulation, LET1 cells were plated overnight and treated with 3MOI of A/HKx31, and/or 50ng/ml recombinant mouse IL-17A (421-ML; R&D) for indicated time. For in vitro stimulation of human lung epithelial cells, A549 were plated overnight and treated with 3MOI of A/Brisbane/10/2007 (H3N2), and/or 50ng/ml recombinant human IL-17A (7955-IL-025/CF; R&D) for 48 hrs. For the pSTAT3 inhibition assay, LET1 and A549 cells were stimulated with virus and/or IL-17A with 100μM S3I-201 (SML0330; Sigma), resuspended in DMSO. All samples contained 0.05% DMSO in the culture medium.
Western Blotting
Proteins were extracted from stimulated cells using RIPA lysis buffer supplemented proteinase inhibitors (Roche). Samples were loaded in 12%–15% SDS-PAGE and transferred onto PVDF membranes. Blocking was performed in 5% BSA for 1 hr, and membranes were incubated in primary antibodies overnight at 4°C. Membranes were incubated with HRP-conjugated secondary antibody for 1hr, and proteins were visualized using ECL substrate (Thermo Fisher). The primary antibodies were IL-33 (1:500 dilution, AF3626, R&D), pSTAT3 (1:1,000, 9145, Cell Signaling), STAT3 (1:1,000, 9139, Cell Signaling), and β-actin (1:2,000, 8457, Cell Signaling).
Neonatal γδ T cell Transfer Experiment
Neonatal γδ T cells were harvested and sorted from lungs of naive wild-type and Il17a−/− neonatal littermates. Following sorting, wild-type or Il17a−/− γδ T cells were resuspended in a concentration of 2×104 cells in 10μl of PBS and transferred to TCRδ−/− littermate mice i.n., and a third transfer treatment of plain PBS was used as a control comparison. After 24 hrs, these TCRδ−/− neonates were anesthetized by isoflurane and infected i.n. with A/HKx31 in 10μl (0.6×103 EID50/g body weight). We used the log-rank test to assess survival differences as a function of treatment; after finding a significant effect, we then tested for pairwise differences between: 1) neonates receiving wild-type and Il17a−/− cells, and 2) between neonates receiving wild-type cells and PBS. We verified that there were no effects of experimental batch on survival by independently testing for effects of experiment number.
Single-cell Sorting and Multiplex PCR
Single cell suspension was made from wild-type lungs with mock- or influenza- (day 2 or 6) infections and stained with a live/dead exclusion dye (Live/dead aqua, Tonbo Biosciences), APC-conjugated anti-mouse CD3 (clone: 17A2) and PE-conjugated anti-mouse γδTCR (clone: GL3). The stained cells were resuspended in RNAse inhibitor containing sort buffer (RNAsin, Promega, 200U/μl) and sorted by gating on γδTCR+CD3+ cells directly into a 384-well PCR plate that had been preloaded with 1μl of reverse transcription reaction mix [1X SuperScript VILO Reaction Mix (Thermo Fisher), SuperScript VILO enzyme and 1X0.1% NP40 (Thermo Fisher)] with a sorter (Model SY3200, Sony Biotech Synergy sorter, Sony Biotech, San Jose, CA). The last column of the plate was left empty for use as PCR negative controls. After sorting, plates were, sealed and kept on ice until the sort is over, briefly centrifuged at 500g and stored at −80°C until downstream processing.
Following single-cell sort into a 384 well PCR plate, direct lysis and reverse transcription was performed using SuperScript Vilo (Invitrogen) as described before (Guo et al., 2016). The resultant cDNA was subjected to a first round PCR reaction using a cocktail of TCRVγ region-specific forward and C region-specific reverse primers (Table S1). Using corresponding internal primer pool, the first round PCR products were further subjected to a nested PCR. The PCR products were purified by Exonuclease I/Shrimp Alkaline Phosphatase treatment (Dash et al., 2015) and sequenced as described with a TRGC internal primer using an ABI Big Dye sequencer (Applied Biosystem) at the Hartwell Center, St Jude Children’s Research Hospital. The sequence data were analyzed using a custom-built macro-enabled Microsoft Excel sheet to derive CDR3 nucleotide and amino acid sequences (available upon request). To determine the corresponding TRGV-TRGJ usage, the sequence data derived from single cells were matched against the IMGT database (Lefranc et al., 2009).
Human Samples and Cytokine Measurement
To characterize human cytokines during influenza infection, we utilized nasal lavages obtained as part of the FLU09 study (Oshansky et al., 2014). FLU09 participants provided lavages at varying intervals from the day of enrollment through 12 days after enrollment. For this study, we focused on samples from a subset of infants and children below the age of 8 years (n = 25) who were verifiably infected with influenza A (pH1 or H3). Cells were separated from cell-free/mucus-free supernatants, and samples were stored at −80°C until analysis. Cytokines were measured in supernatants as follows: IL-17A, IL-33, and amphiregulin were assayed by Human IL-17A, IL-33, and amphiregulin DuoSet ELISA kits (R&D), and the level of IFN-γ was detected by Human Milliplex assays (Millipore) according to manufacturer instructions. To determine severity of influenza disease outcome, patients who were admitted to an emergency room or hospital were considered “severe” cases, whereas all other participants were considered “mild” cases.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed with Prism 7.0 software (GraphPad) and presented as mean ± S.E.M. unless otherwise indicated. Experiments were repeated at least twice, as indicated. Statistical significance was determined by Mann-Whitney test between two groups. For comparison across three or more groups, non-parametric one-way ANOVA (Kruskal-Wallis test) was used, and differences between individual groups were estimated using Dunn’s multiple comparisons test. For the comparison of two groups at different time points, two-way ANOVA was used, and differences between individual groups were estimated using Sidak’s multiple comparisons test. To compare relative weight gain between wild-type and TCRδ−/− neonates from 1–15 days after infection, we used a linear mixed-effects model via the “lmer” function in the R package lme4 (v1.1–13) (Bates et al., 2015) and assessed the significance of effects with Wald tests; specifically, genotype was modeled as a function of relative weight gain, with mixed effects allowing intercepts to vary across subjects (in order to control for individual differences among the repeatedly measured mice) and allowing the effect of genotype to vary across time points. The difference of survival rate was determined using the log-rank test. p < 0.05 was considered statistically significant where ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, and n.s., non significant.
For the repertoire analysis, because chi-square tests were not applicable due to the presence of zero-values in contingency table cells, statistical association across treatment conditions was assessed using Fisher’s exact test as implemented in R (Clarkson et al., 1993). Overall comparisons across days-post-infection for a given cell type (i.e., CD27+ or CD27-) were constructed as 3×7 contingency tables representing 3 treatment conditions and 7 possible gene segments. In the case that this overall comparison was significant, the simple effects were resolved by constructing 2×7 contingency tables for each pairwise comparison. P-values were adjusted to control family-wise error rate using Holm’s method (Holm, 1979).
For human cytokine analyses, we initially focused on the putative correlations underlying the proposed IL-17A/IL-33/Areg axis, using generalized linear mixed models with a log-link of the Gaussian distribution, as implemented by the R package MASS (v7.3–47) (Venables and Ripley, 2002), to investigate correlations among the adjacent components of the axis (i.e., IL-33 as a function of IL-17A, and Areg as a function of IL-33). In each case, we also included age, study day, and IFN-γ expression as fixed effects, and patient-ID was included as a random effect in order to account for repeated sampling within patients over the course of the study. We also analyzed IFN-γ expression as a function of axis cytokines, using both the Gamma and Gaussian distributions. To investigate potential correlations between cytokine levels and disease severity, we focused on samples collected on the day at which disease severity was assessed (i.e., study day 0); for these analyses, we used the natural logarithm transformation to obtain normally distributed cytokine measures and ages, and we employed multifactor Analysis of Variance models (R aov function), with age included as a covariate. For significant effects in aov models, we confirmed equal variances between groups using an F-test (R var.test function).
Supplementary Material
Acknowledgements
We would like to thank the St Jude Children’s Research Hospital Animal Resource Center’s staff for their support and excellent animal care. We thank Drs. Greig Lennon and Richard Cross in the St. Jude Flow Cytometry & Cell Sorting Shared Resource for cell sorting. We thank Dr. Stephania Cormier for helpful discussions and instruction in neonatal infections. This work was supported by NIH grant R01 AI121832, The Hartwell Foundation Individual Biomedical Research Award, St. Jude Center of Excellence for Influenza Research and Surveillance HHSN272201400006C, and ALSAC (to P.G.T.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The RNA-seq data from whole lung RNA of wild-type and TCRδ−/− neonates (day 8 after infection) has been deposited in the Gene Expression Omnibus (GEO, NCBI) under accession code GSE99683.
Declaration of Interests
The authors declare no competing interests.
References
- Adkins B, Leclerc C, and Marshall-Clarke S (2004). Neonatal adaptive immunity comes of age. Nat. Rev. Immunol. 4, 553–564. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arpaia N, Green JA, Moltedo B, Arvey A, Hemmers S, Yuan S, Treuting PM, and Rudensky AY (2015). A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162, 1078–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software, Articles 67, 1–48. [Google Scholar]
- Bhat N, Wright JG, Broder KR, Murray EL, Greenberg ME, Glover MJ, Likos AM, Posey DL, Klimov A, Lindstrom SE, et al. (2005). Influenza-Associated Deaths among Children in the United States, 2003–2004. N. Engl. J. Med. 353, 2559–2567. [DOI] [PubMed] [Google Scholar]
- Brincks EL, Katewa A, Kucaba TA, Griffith TS, and Legge KL (2008). CD8 T Cells Utilize TRAIL to Control Influenza Virus Infection. J. Immunol. 181, 4918–4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carding SR, and Egan PJ (2002). Gammadelta T cells: functional plasticity and heterogeneity. Nat. Rev. Immunol. 2, 336–345. [DOI] [PubMed] [Google Scholar]
- Cerwenka A, Morgan TM, and Dutton RW (1999). Naive, Effector, and Memory CD8 T Cells in Protection Against Pulmonary Influenza Virus Infection: Homing Properties Rather Than Initial Frequencies Are Crucial. J. Immunol. 163, 5535–5543. [DOI] [PubMed] [Google Scholar]
- Chang Y-J, Kim HY, Albacker LA, Baumgarth N, McKenzie ANJ, Smith DE, DeKruyff RH, and Umetsu DT (2011). Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat. Immunol. 12, 631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng P, Liu T, Zhou W-Y, Zhuang Y, Peng L-S, Zhang J-Y, Yin Z-N, Mao X-H, Guo G, Shi Y, et al. (2012). Role of gamma-delta T cells in host response against Staphylococcus aureus-induced pneumonia. BMC Immunol. 13, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y-H, Meyer C, and Bonneville M (2014). γδ T Cells: First Line of Defense and Beyond. Annu. Rev. Immunol. 32, 121–155. [DOI] [PubMed] [Google Scholar]
- Cicin-Sain L, Brien JD, Uhrlaub JL, Drabig A, Marandu TF, and Nikolich-Zugich J (2012). Cytomegalovirus Infection Impairs Immune Responses and Accentuates T-cell Pool Changes Observed in Mice with Aging. PLoS Pathog. 8, e1002849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarkson DB, Fan Y-A, and Joe H (1993). A remark on algorithm 643: FEXACT: an algorithm for performing Fisher’s exact test in r x c contingency tables. ACM Trans. Math. Softw. 19, 484–488. [Google Scholar]
- Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau C-S, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, et al. (2015). IL-17-producing γδ T cells and neutrophils conspire to promote breast cancer metastasis. Nature 522, 345–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe CR, Chen K, Pociask DA, Alcorn JF, Krivich C, Enelow RI, Ross TM, Witztum JL, and Kolls JK (2009). Critical Role of IL-17RA in Immunopathology of Influenza Infection. J. Immunol. 183, 5301–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P, and Thomas PG (2014). Host Detection and the Stealthy Phenotype in Influenza Virus Infection In Influenza Pathogenesis and Control - Volume II, M.B.A. Oldstone, and Compans RW, eds. (Springer International Publishing; ), pp. 121–147. [Google Scholar]
- Dash P, McClaren JL, Oguin TH 3rd, Rothwell W, Todd B, Morris MY, Becksfort J, Reynolds C, Brown SA, Doherty PC, et al. (2011). Paired analysis of TCRα and TCRβ chains at the single-cell level in mice. J. Clin. Invest. 121, 288–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash P, Wang GC, and Thomas PG (2015). Single-Cell Analysis of T-Cell Receptor αβ Repertoire. Methods Mol. Biol. 1343, 181–197. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowling DJ, and Levy O (2014). Ontogeny of early life immunity. Trends Immunol. 35, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, and Thomas PG (2016). Balancing Immune Protection and Immune Pathology by CD8+ T-Cell Responses to Influenza Infection. Front. Immunol. 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, Meliopoulos VA, McClaren JL, Guo X-ZJ, Sanders CJ, Smallwood HS, Webby RJ, Schultz-Cherry SL, Doherty PC, and Thomas PG (2015). Diverse Heterologous Primary Infections Radically Alter Immunodominance Hierarchies and Clinical Outcomes Following H7N9 Influenza Challenge in Mice. PLoS Pathog. 11, e1004642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffen SL (2009). Structure and signalling in the IL-17 receptor family. Nat. Rev. Immunol. 9, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Yang W, Pan M, Scully E, Girardi M, Augenlicht LH, Craft J, and Yin Z (2003). Gamma delta T cells provide an early source of interferon gamma in tumor immunity. J. Exp. Med. 198, 433–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AM, Fadel SA, Cao S, and Sarzotti M (2000). T cell immunity in neonates. Immunol. Res. 22, 177–190. [DOI] [PubMed] [Google Scholar]
- Gorski SA, Hahn YS, and Braciale TJ (2013). Group 2 Innate Lymphoid Cell Production of IL-5 Is Regulated by NKT Cells during Influenza Virus Infection. PLoS Pathog. 9, e1003615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruta NLL, and Turner SJ (2014). T cell mediated immunity to influenza: mechanisms of viral control. Trends Immunol. 35, 396–402. [DOI] [PubMed] [Google Scholar]
- Guo X-ZJ, and Thomas PG (2017). New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 39, 541–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X-ZJ, Dash P, Calverley M, Tomchuck S, Dallas MH, and Thomas PG (2016). Rapid cloning, expression, and functional characterization of paired αβ and γδ T-cell receptor chains from single-cell analysis. Mol Ther Methods Clin Dev 3, 15054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall OJ, Limjunyawong N, Vermillion MS, Robinson DP, Wohlgemuth N, Pekosz A, Mitzner W, and Klein SL (2016). Progesterone-Based Therapy Protects Against Influenza by Promoting Lung Repair and Recovery in Females. PLoS Pathog. 12, e1005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayday AC (2000). γδ Cells: A Right Time and a Right Place for a Conserved Third Way of Protection. Annu. Rev. Immunol. 18, 975–1026. [DOI] [PubMed] [Google Scholar]
- Hoffmann E, Neumann G, Kawaoka Y, Hobom G, and Webster RG (2000). A DNA transfection system for generation of influenza A virus from eight plasmids. Proc. Natl. Acad. Sci. U. S. A. 97, 6108–6113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan BLM, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CCW, Niklason L, Calle E, Le A, Randell SH, et al. (2014). Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 15, 123–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm S (1979). A Simple Sequentially Rejective Multiple Test Procedure. Scand. Stat. Theory Appl. 6, 65–70. [Google Scholar]
- Huang H, Saravia J, You D, Shaw AJ, and Cormier SA (2015). Impaired gamma delta T cell-derived IL-17A and inflammasome activation during early respiratory syncytial virus infection in infants. Immunol. Cell Biol. 93, 126–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, and Pillai PS (2014). Innate immunity to influenza virus infection. Nat. Rev. Immunol. 14, 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashani E, Föhse L, Raha S, Sandrock I, Oberdörfer L, Koenecke C, Suerbaum S, Weiss S, and Prinz I (2015). A clonotypic Vγ4Jγ1/Vδ5Dδ2Jδ1 innate γδ T-cell population restricted to the CCR6+CD27− subset. Nat. Commun. 6, 6477. [DOI] [PubMed] [Google Scholar]
- de Kleer IM, Kool M, de Bruijn MJW, Willart M, van Moorleghem J, Schuijs MJ, Plantinga M, Beyaert R, Hams E, Fallon PG, et al. (2016). Perinatal Activation of the Interleukin-33 Pathway Promotes Type 2 Immunity in the Developing Lung. Immunity 45, 1285–1298. [DOI] [PubMed] [Google Scholar]
- Kohlgruber AC, Gal-Oz ST, LaMarche NM, Shimazaki M, Duquette D, Nguyen HN, Mina AI, Paras T, Tavakkoli A, von Andrian U, et al. (2018). γδ T cells producing interleukin-17A regulate adipose regulatory T cell homeostasis and thermogenesis. Nat. Immunol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M-P, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, et al. (2009). IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 37, D1006–D1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goffic R, Arshad MI, Rauch M, L’Helgoualc’h A, Delmas B, Piquet-Pellorce C, and Samson M (2011). Infection with Influenza Virus Induces IL-33 in Murine Lungs. Am. J. Respir. Cell Mol. Biol. 45, 1125–1132. [DOI] [PubMed] [Google Scholar]
- Livak KJ, and Schmittgen TD (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lockhart E, Green AM, and Flynn JL (2006). IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J. Immunol. 177, 4662–4669. [DOI] [PubMed] [Google Scholar]
- Mamedov MR, Scholzen A, Nair RV, Cumnock K, Kenkel JA, Oliveira JHM, Trujillo DL, Saligrama N, Zhang Y, Rubelt F, et al. (2018). A Macrophage Colony-Stimulating-Factor-Producing γδ T Cell Subset Prevents Malarial Parasitemic Recurrence. Immunity 48, 350–363.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, Stueker O, Emili A, and Bader GD (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS One 5, e13984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani N, Nabe T, and Yoshino S (2014). IL-17A Promotes the Exacerbation of IL-33–Induced Airway Hyperresponsiveness by Enhancing Neutrophilic Inflammation via CXCR2 Signaling in Mice. J. Immunol. 192, 1372–1384. [DOI] [PubMed] [Google Scholar]
- Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CGK, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, et al. (2011). Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskophidis D, and Kioussis D (1998). Contribution of Virus-specific CD8+ Cytotoxic T Cells to Virus Clearance or Pathologic Manifestations of Influenza Virus Infection in a T Cell Receptor Transgenic Mouse Model. J. Exp. Med. 188, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz FM (2003). Influenza virus infection in infancy and early childhood. Paediatr. Respir. Rev. 4, 99–104. [PubMed] [Google Scholar]
- Oshansky CM, Gartland AJ, Wong S-S, Jeevan T, Wang D, Roddam PL, Caniza MA, Hertz T, Devincenzo JP, Webby RJ, et al. (2014). Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am. J. Respir. Crit. Care Med. 189, 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan B, Shen J, Cao J, Zhou Y, Shang L, Jin S, Cao S, Che D, Liu F, and Yu Y (2015). Interleukin-17 promotes angiogenesis by stimulating VEGF production of cancer cells via the STAT3/GIV signaling pathway in non-small-cell lung cancer. Sci. Rep. 5, 16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papotto PH, Ribot JC, and Silva-Santos B (2017). IL-17+ γδ T cells as kick-starters of inflammation. Nat. Immunol. 18, 604–611. [DOI] [PubMed] [Google Scholar]
- Pichery M, Mirey E, Mercier P, Lefrancais E, Dujardin A, Ortega N, and Girard J-P (2012). Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 188, 3488–3495. [DOI] [PubMed] [Google Scholar]
- Ribot JC, deBarros A, Pang DJ, Neves JF, Peperzak V, Roberts SJ, Girardi M, Borst J, Hayday AC, Pennington DJ, et al. (2009). CD27 is a thymic determinant of the balance between interferon-γ- and interleukin 17–producing γδ T cell subsets. Nat. Immunol. 10, 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romagnoli PA, Sheridan BS, Pham Q-M, Lefrançois L, and Khanna KM (2016). IL-17A-producing resident memory γδ T cells orchestrate the innate immune response to secondary oral Listeria monocytogenes infection. Proc. Natl. Acad. Sci. U. S. A. 113, 8502–8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberger CM, Podyminogin RL, Askovich PS, Navarro G, Kaiser SM, Sanders CJ, McClaren JL, Tam VC, Dash P, Noonan JG, et al. (2014–2). Characterization of innate responses to influenza virus infection in a novel lung type I epithelial cell model. J. Gen. Virol. 95, 350–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saluzzo S, Gorki A-D, Rana BMJ, Martins R, Scanlon S, Starkl P, Lakovits K, Hladik A, Korosec A, Sharif O, et al. (2017). First-Breath-Induced Type 2 Pathways Shape the Lung Immune Environment. Cell Rep. 18, 1893–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, McLaughlin MM, Lawrence NJ, et al. (2007). Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. U. S. A. 104, 7391–7396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spellberg B, and Edwards JE Jr, (2001). Type 1/Type 2 immunity in infectious diseases. Clin. Infect. Dis. 32, 76–102. [DOI] [PubMed] [Google Scholar]
- Sriperumbudur A, Breitzig M, Lockey R, and Kolliputi N (2017). Hedgehog: the key to maintaining adult lung repair and regeneration. J. Cell Commun. Signal. 11, 95–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, and Kash JC (2010). Influenza Virus Evolution, Host Adaptation, and Pandemic Formation. Cell Host Microbe 7, 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taubenberger JK, and Morens DM (2008). The Pathology of Influenza Virus Infections. Annu. Rev. Pathol.: Mech. Dis. 3, 499–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vantourout P, and Hayday A (2013). Six-of-the-best: unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 13, 88–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, and Ripley BD (2002). Modern Applied Statistics with S: (Springer; New York: ). [Google Scholar]
- Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, and Yu H (2009). IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J. Exp. Med. 206, 1457–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chan CCS, Yang M, Deng J, Poon VKM, Leung VHC, Ko K-H, Zhou J, Yung Yuen K, Zheng B-J, et al. (2011). A critical role of IL-17 in modulating the B-cell response during H5N1 influenza virus infection. Cell. Mol. Immunol. 8, 462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitkamp J-H, Rosen MJ, Zhao Z, Koyama T, Geem D, Denning TL, Rock MT, Moore DJ, Halpern MD, Matta P, et al. (2014). Small intestinal intraepithelial TCRγδ+ T lymphocytes are present in the premature intestine but selectively reduced in surgical necrotizing enterocolitis. PLoS One 9, e99042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You D, Ripple M, Balakrishna S, Troxclair D, Sandquist D, Ding L, Ahlert TA, and Cormier SA (2008). Inchoate CD8+ T Cell Responses in Neonatal Mice Permit Influenza-Induced Persistent Pulmonary Dysfunction. J. Immunol. 181, 3486–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DMW, Gause WC, Osborne LC, and Artis D (2015). Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42, 216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.