

Abstract
Pancreatic ductal adenocarcinoma (PDA) is an aggressive cancer that is highly refractory to the current standards of care. The difficulty in treating this disease is due to a number of different factors, including altered metabolism. In PDA, the metabolic rewiring favors anabolic reactions which supply the cancer cell with necessary cellular building blocks for unconstrained growth. Furthermore, PDA cells display high levels of basal autophagy and macropinocytosis. KRAS is the driving oncogene in PDA and many of the metabolic changes are downstream of its activation. Together, these unique pathways for nutrient utilization and acquisition result in metabolic plasticity enabling cells to rapidly adapt to nutrient and oxygen fluctuations. This remarkable adaptability has been implicated as a cause of the intense therapeutic resistance. In this review, we discuss metabolic pathways in PDA tumors and highlight and how they contribute to the pathogenesis and treatment of the disease.
Keywords: Pancreatic cancer, Autophagy, metabolism, Microenvironment, macropinocytosis
Introduction:
PDA Biology
Within the last 30 years considerable progress has been made in cancer detection and treatment, leading to a significant increase in survival rates of many cancer types. Despite these recent advances, the 5 year survival of pancreatic cancer remains dismal at ~8% [1]. In the United States there were 53,670 estimated new cases and 43,090 estimated deaths for 2017, making pancreatic cancer one of the deadliest cancers [1]. Despite only representing < 3% all cancer diagnoses, it is predicted to become the second leading cause of cancer deaths by 2020 [2]. The poor prognosis and aggressive nature of pancreatic cancer is linked to late onset of disease presentation, cancer cell intrinsic alterations, and microenvironmental factors.
The most common subtype of pancreatic cancer, accounting for about 90% of all cases, is pancreatic ductal adenocarcinoma (PDA) and is the focus of the review. The majority of PDA cases arise from pancreatic intraepithelial neoplasms (PanIN) which are microscopic lesions of dysplasia. The progression of PanIN to invasive carcinoma is a gradation brought on by accumulation of genetic alterations and development of the distinctive microenvironment [3]. The driving oncogene in PDA results from an activating mutation in the KRAS gene which is found in > 90% of tumors [4]. KRAS mutations are found in low grade PanIN lesions (PanIN-1A) and are likely an early event in malignant transformation. There is, however, discordance between the prevalence of benign precursor lesions and rates of cancer incidence, suggesting other genetic and environmental factors cooperate in the development of PDA [3]. Consistent with this observation, mouse models of pancreatic cancer driven by a pancreas-specific Cre-deleter and a latent knock-in allele of oncogenic Kras (LSL-KrasG12D;PdxCre) have long latency or require the loss of tumor suppressor genes (TP53, CDKN2A, SMAD4, and others) to develop a tumor [5, 6].
Despite the complexity of PDA initiation, it has been established that the KRAS mutation is a key driver of tumor progression and multiple studies report that tumors are highly dependent on this oncogene for tumor maintenance [7, 8]. The KRAS oncogene encodes for a small GTPase which acts as a molecular switch bridging signals from membrane bound receptors to central cellular signaling pathways. The activity of KRAS is regulated by GTPase activating proteins (GAPs) and guanine exchange factors (GEFs) which toggle the protein between inactive (GDP bound) and active (GTP bound) states [9]. In PDA, KRAS is most commonly mutated at the G12 residue preventing the interaction with GAPs which results in KRAS bound to GTP and is therefore constitutively active [10]. This results in aberrant downstream signaling through pathways such as mitogen-activated protein kinase (MAPK) and phosphoinsotide-3 kinase (PI3K). These signaling pathways are responsible for regulating a number of key cellular functions including growth and survival. Unchecked KRAS signaling results in increased proliferation, decreased apoptosis, and an invasive phenotype [9]. While tumors require KRAS for growth, thus making it an attractive drug target, disrupting this oncogene pharmaceutically has proven problematic [11]. Recently, however, the development of inhibitors targeting specific KRAS mutations (G12C) have provided evidence that some mutations lead to vulnerabilities that can be exploited for therapeutic gain [12].
Appreciable effort is also underway to identify other frequently mutated genes in PDA as potential therapeutic targets. Whole exosome sequencing of pancreatic tumors revealed reproducible alterations in TP53, SMAD4, and CDK2NA, but have yet to discover targetable recurrent mutations [4, 10]. PDA tumors are characterized as heterogeneous and genetically complex with a number of chromosomal rearrangements which likely contributes to treatment resistance of the disease [13]. Sequencing of primary tumors has been challenging because of the lack of neoplastic cellularity, considering the bulk of the tumor is composed of stromal components [14]. Utilization of techniques such as laser-captured microdissection have aided in isolating malignant populations of cells for analysis [15].
One of the defining characteristics of PDA tumors is excessive desmoplasia which is comprised of a heterogeneous mixture of extracellular matrix (ECM) proteins and a multitude of cell types [16]. Stellate cells, a specialized type of fibroblast found in the pancreas, constitute a major cell type in the tumor stroma and have multifaceted roles within the tumor microenvironment (TME) such as the production of ECM [17]. Other cells included within the stromal reaction are endothelial cells, immune cells and neurons. The contribution of these cells to pancreatic cancer progression is an active area of investigation. The PDA microenvironment is understood to be immunosuppressive and accordingly, immune checkpoint inhibitors have been largely unsuccessful as a monotherapy [18]. These results highlight the complexity of immunological reactions within the TME and suggest that successfully treating PDA will require a multipronged approach [19, 20]. The switch of pancreatic stellate cells from a quiescent to an activated state results in deposition of the collagenous components of the ECM. This leads to extensive fibrosis which impinges on vasculature causing increased hydrostatic pressure, lower nutrient influx, and hypoxia [21]. The deficient vascular network in pancreatic tumors has been shown to impede drug delivery, further contributing to the highly refractory nature of these tumors [22]. Consistent with the lack of functional vasculature, PDA are likely nutrient depleted when compared to benign adjacent tissue [23]. To survive in such austere conditions, PDA tumors rely on a unique combination of adaptive metabolic networks for nutrient acquisition and utilization which are described in this review.
Tumor Metabolism:
In order to support the ability to proliferate in an unconstrained manner, tumor cells have adapted their metabolism in many different ways. Perhaps the most well-known example comes from the observation that cancer cells have increased rates of aerobic glycolysis, known as the Warburg effect [24]. This leads to decreased glucose oxidation and more flux through the anabolic side branches of glycolysis, such as the pentose phosphate pathway (PPP). Indeed, a major consequence of many of the metabolic changes seen in tumors is to shift fuel sources into anabolic pathways in order to provide the cells with the necessary substrates to increase biomass. Interestingly, it has been recently appreciated that many of the oncogenes and tumor suppressor genes important in tumor pathogenesis can actually rewire metabolism to favor a more anabolic state and therefore support tumor growth. This includes oncogenes such as KRAS and MYC as well as the TP53 tumor suppressor. Given that many of the metabolic adaptations occur downstream of these genes selectively in cancers, they may provide opportunities for therapeutic intervention. This review will focus on the metabolic alterations seen in PDA, with a particular emphasis on the metabolic plasticity that is characteristic of these tumors.
Nutrient Scavenging:
Autophagy
Macroautophagy is a highly conserved catabolic process that results in the lysosomal degradation of intracellular components and functions to maintain metabolic and cellular homeostasis through recycling of cytoplasmic materials to basic cellular building blocks (amino acids, fatty acids, nucleotides). This process involves sequestration of cytoplasmic material such as proteins or organelles into double membrane structures known as autophagosomes. The fusion of the autophagosome and the lysosome results in degradation of the cargo and release of recycled components back into the cytosol [25, 26]. There are three different types of autophagy; macroautophagy, microautophagy and chaperone mediated autophagy which each utilize different mechanisms in delivering cargo to the lysosome [27]. This review will focus on the role of macroautophagy (hereafter autophagy) which plays a prominent role in the metabolism and progression of PDA.
Nearly all tissues have low levels of basal autophagy which generally serves as a protective mechanism, ridding the cells of damaged organelles and protein aggregates which have the potential to become cytotoxic. Indeed, dysregulation of autophagy has been implicated in a number of different pathological conditions, including neurodegenerative diseases, inflammation and cancer [28]. The major function of autophagy is understood to be pro-survival, as has been shown in the initial mouse studies where autophagy genes were deleted embryonically and resulted in death soon after weaning [29]. Consistently, autophagy flux is increased under cellular stress (nutrient deprivation, hypoxia, and chemotherapeutics).
Autophagy can be broken down into discreet steps beginning with initiation of the autophagosome. One of the most potent activators of autophagy is nutrient deprivation, which is tightly regulated by the MTOR pathway [30]. Through degradation of cytosolic components, autophagy can supply nutrients to essential metabolic pathways and promote survival during starvation. The canonical process is tightly controlled through a complex series of steps that involve more than 30 autophagy related genes (ATG) and the process has been extensively reviewed elsewhere [27]. In brief, under nutrient replete conditions MTOR is active, and MTORC1 mediated signaling suppresses autophagy activation. In contrast, starvation stimulates autophagy through modulation of MTORC1 activity as well as AMPK signaling. Upon induction of autophagy, machinery involved in the nucleation of the phagophore is recruited. Following initiation is autophagosome elongation/maturation and lysosomal fusion. The maturation phase involves a concert of ATG proteins to form the autophagosome membrane. During this process, cytosolic components become engulfed by the forming autophagosome which are subsequently degraded by the lysosome following fusion. In addition to bulk autophagy, where cytosolic contents are degraded non-selectively, there are multiple forms of selective autophagy whereby cargo is selectively targeted to the autophagosome by autophagy receptors [31]. Importantly, perturbation at multiple steps of this process results in defective autophagosomes and diminished autophagic flux. Autophagy can also be blocked at the level of the lysosome with drugs such as hydroxychloroquine which inhibits lysosomal acidification.
In PDA, autophagy levels are basally high even when grown in complete media suggestive of alternative or supplemental means of autophagy activation. Indeed, it has been shown that the MiT/TFE family of transcription factors can stimulate high levels of basal autophagy independent of MTOR activity. These transcription factors are constitutively activated in PDA in a nutrient-independent manner and conversely, knockdown of MiT/TFE factors impairs PDA lysosomal function and autophagic flux [32]. Additionally, the identification of an ULK1 (ATG1) phosphatase (PP2A-B55alpha) that stimulates autophagy has also been shown to be required for high basal levels of autophagy in PDA [33].
Utilizing genetically engineered mouse models of cancer harboring mutations or deletions in essential autophagy genes, the function of autophagy in cancer progression and initiation have been studied. Autophagy has dichotomous roles in cancer in that it can have both pro and anti-tumor properties. Indeed, its function during tumorigenesis can be dependent on biological factors such as the tumor type, tumor mutations, as well as technical aspects such as the model system and assays used [34]. By removing damaged organelles and aggregated proteins, autophagy reduces levels of reactive oxygen species (ROS) and mitigates tissue damage, thereby creating a barrier to malignant transformation. In established tumors, however, the ability of autophagy to fuel metabolism from recycled intracellular material results in a pro-tumorigenic effect. Consistently, tumor cells in hypoxic and nutrient devoid areas of tumors would benefit from having high levels of autophagy. Indeed, autophagy is essential for tumor growth in multiple different cancers both in vitro and in vivo, including PDA, breast, lung, prostate, and brain tumors [35]. Expectedly, genetic ablation of autophagy using ATG5 or ATG7 conditional null mouse models results in attenuated growth and progression of KRAS-driven PDA tumors [36, 37]. Pharmacologically targeting autophagy with hydroxychloroquine (HCQ), to inhibit lysosomal acidification, has similar results in tumor xenografts [37, 38]. One of the proposed mechanisms by which autophagy promotes PDAC growth is through its role in metabolism. Given, the diversity of substrates that can be degraded by autophagy, there is the potential to fuel nearly every pathway in central carbon metabolism. This has the effect of providing metabolic plasticity to these tumors and can give them a survival advantage in the austere TME that was described above. Indeed, inhibition of autophagy in PDA cells results in impaired mitochondrial function with decreased oxidative phosphorylation and a subsequent drop in ATP levels [37, 38]. While not performed in PDA, work in KRAS driven lung cancer models has also shown an important metabolic role of autophagy in these tumors. In this setting, autophagy deficiency results in substantial defects in nucleotide pool sizes and similar to PDA, a decrease in energy charge [39]. Taken together, these results suggest prominent cancer cell autonomous roles for autophagy in PDA (and likely multiple other tumor types) metabolism and survival.
In addition to the cancer cell intrinsic roles for autophagy, emerging evidence indicates that there are non-cell-autonomous autophagic mechanisms that support tumor cells. A recent publication from our group identified an autophagy-dependent tumor-stroma metabolic crosstalk through secretion of alanine by stellate cells to support PDA growth. Stellate cells are abundant in the pancreatic TME and, by releasing an unknown factor, PDA cells can stimulate them to produce alanine through an autophagic-dependent mechanism [40]. These findings have major implications in the nutrient deprived TME since the alanine is used by the PDA cells to fuel the TCA cycle. Use of this “alternative” fuel source allows glucose carbon to be shunted into biosynthetic pathways, such as serine synthesis, to support tumor growth. Impairing autophagy selectively in the stellate cell compartment results in decreased tumor development in co-transplantation experiments. These results suggest that autophagy inhibitors will act on both the stellate cell and tumor cell compartment, effectively decreasing a major exogenous carbon source for PDA cells while also compromising intracellular metabolism. Due to the lack of a potent and specific autophagy inhibitor, our group recently developed a tetracycline inducible ATG4B dominant negative (ATG4B DN) mouse model to recapitulate acute autophagy inhibition in fully formed PDA tumors, analogous to the therapeutic situation [41]. Indeed, autophagy inhibition resulted in prolonged survival and even caused tumor regressions. Using this model, we were able to show the effect of autophagy inhibition was likely due to its impact on both stroma and tumor cells. Another interesting finding from this study revealed that tumor regression in the context of autophagy inhibition was, in part, mediated by macrophages. Further examination of the whole body effects of autophagy inhibition will begin to elucidate contributions of different cellular compartments to tumor progression and may help identify novel therapeutic targets.
Macropinocytosis
While autophagy is clearly an important process in PDA metabolic scavenging, allowing them to survive in the nutrient poor TME, there are many unresolved questions. Importantly, while autophagy can produce diverse nutrients, it cannot create a net increase in biomass due to the fact that these cells are degrading themselves. Interestingly, PDA cells also rely on another lysosomal-dependent pathway to fuel their elevated metabolic demand. Macropinocytosis is the non-specific bulk internalization of large portions of extracellular fluid. The engulfed cargo consists of protein, lipid, virus and bacteria. To internalize extracellular material, macropinocytosis relies on polymerization of actin filaments to create characteristic membrane ruffling. The extended plasma membrane can then fold inwards fusing with the basal membrane giving rise to endocytic vesicles termed macropinosomes. Through a series incompletely resolved steps, macropinosomes mature as they traffic in the cytosol where they are recycled to the cell membrane or fused with the lysosome for degradation. The latter process results in the release of digested cargo, which includes amino acids and lipids, into the cytosol [42]. Nutrients acquired through macropinocytosis have been shown to play an important role PDA metabolism. In contrast to autophagy, these internalized nutrients can lead to increased biomass.
Oncogenic RAS has been shown to increase levels of macropinocytosis [43]. While it has been known for some time that RAS promotes macropinocytosis, the implications of this finding have only recently been explored in the context of cancer. In PDA, uptake of extracellular isotopically labeled protein via macropinocytosis contributes to the central carbon metabolism. Consistent with this, providing cells with exogenous albumin can rescue cell death from glutamine deprivation. This effect was abrogated by treatment with EIPA, an agent known to inhibit macropinocytosis. Indeed, treatment of mice with PDA xenografts with EIPA significantly inhibited tumor growth [44]. Recent reports also provide evidence of macropinocytosis occurring in human PDA tumors as well as exogenous protein contributing to the amino pools of PDA tumors in vivo [23, 45]. Recently, PDA were shown to take up collagen from the ECM, at least in part through macropinocytosis. The collagen-derived proline contributed to central carbon metabolism and promoted PDA survival under nutrient deprived conditions [46]. Lastly, it has been shown that RAS transformed cells can also scavenge extracellular lipid [47]. While it is not clear that the uptake is due to macropinocytosis, it is yet another form of extracellular nutrient scavenging.
Taken together, these studies define a critical role for recycling and scavenging mechanisms in PDA metabolism. These methods of nutrient acquisition provide malignant cells with a means to grow in an exceptionally harsh microenvironment. Autophagy, in addition to providing metabolites for growth, also functions in therapeutic resistance as it can promote survival during periods of stress (chemotherapy, radiation). While autophagy is a potent and adaptive survival mechanism, cells are ultimately limited by intracellular content. In contrast, macropinocytosis is a means of generating a diverse array of essential building blocks from an exogenous source which can contribute to cellular biomass. Given the prominent roles of autophagy and macropinocytosis in PDA metabolism, targeting these pathways therapeutically may result in meaningful clinical responses. Considering both of these pathways converge at the lysosome it is tempting to speculate that lysosomal perturbation would be especially toxic to PDA tumors. A number of clinical trials are underway to test HCQ treatment in PDA and other tumor types. Early results point to possible improved outcomes when HCQ is added to chemotherapy in localized PDA [48].
Fuel Sources:
Glucose:
Glucose fuels cancer cell proliferation by providing carbon for anabolic reactions that generate biosynthetic intermediates and through the production of ATP (fermentation or oxidation of pyruvate) [49]. To meet the elevated demand for glucose required by PDA, oncogenic KRAS confers a number of changes within the glycolytic pathway. The generation of a genetically engineered mouse model of PDA with expression of Kras G12D under the control of a doxycycline inducible promoter helped elucidate many of the changes observed in PDA glucose metabolism [8]. Employing this model, it has been reported that KRAS expression results in the transcriptional upregulation of the glucose transporter GLUT1 as well as the enzymes HK1, HK2, PFK1, and LDHA. Upregulation of GLUT1 leads to an augmented capacity for glucose uptake, fueling downstream pathways, while the increase in glycolytic enzymes dictates the fate of glucose carbon. Expectedly, silencing of oncogenic Kras expression in tumors markedly diminishes glucose uptake and flux through the glycolytic pathway and associated anabolic branches (described in detail below) [8]. While this model allowed the assessment of Kras-dependent metabolic changes, it is important to remember that these are all occurring in the context of TP53 loss, a critical genetic alteration in PDA (~70% of cases) [10]. Indeed, TP53 deletion or mutation itself has been linked to a number of metabolic changes. It has been shown that TP53 regulates glycolysis through a variety of mechanisms including transcriptional repression of glucose transporters (e.g. GLUT1) [50]. Additionally, TP53 has also been shown to regulate the PPP [50].
In PDA, as in many other types of cancers, glucose-derived pyruvate is preferentially converted to lactate instead of undergoing oxidative phosphorylation. The prevalence for this mechanism of pyruvate metabolism in PDA is driven in part by KRAS-mediated upregulation of LDHA [8]. Further control is exerted on LDHA through posttranslational modifications of the enzyme. Decreased acetylation of the lysine 5 residue leads to increased stability of the enzyme in PDA [51]. LDHA plays another important function in the regeneration of NAD+, a necessary cofactor for efficient glycolytic metabolism. The ratio of NAD+/NADH governs glycolytic flux and the presence of LDHA ensures the ratio remains high to allow for heightened glycolysis [52].
Additionally, pancreatic tumors are hypoxic resulting in the stabilization of HIF1which leads to transcription of glycolytic enzymes and decreased activity of pyruvate dehydrogenase (PDH) [53, 54]. These changes serve to preserve high glycolytic flux and lactate production. Taken together, these data provide some insight into maintenance of the glycolytic phenotype found in PDA tumors. In agreement with the proclivity of PDA to utilize pyruvate fermentation over oxidative phosphorylation, in vivo tracing of labeled glucose carbon makes very little contribution to TCA cycle metabolites in PDA [55]. It has been reported, however, that lactate excreted by hypoxic pancreatic cancer cells can provide an alternative fuel source for those PDA cells that are well oxygenated [56]. Interestingly, labeled lactate carbon contributes to the labeling of TCA cycle metabolites in PDA as well as in other types of cancer [55, 57]. Once thought to be a waste product of glycolysis, it is now becoming clear that lactate excretion by PDA cells has various roles within the TME, including providing nutrients to neighboring cells and contributing to an immunosuppressive phenotype [58].
A key feature of the elevated rate of glycolysis is that it allows PDA tumors to shuttle glucose carbon to various anabolic pathways to support growth. One such anabolic branch that is fed by glycolysis is the pentose phosphate pathway (PPP). This pathway is important for the generation of intermediates for nucleotide biosynthesis and can be broken down into two distinct arms: the oxidative arm and the non-oxidative arm. In the oxidative arm of the PPP, glucose-6-phospate is oxidized to ribose 5-phosphate and, in the process, generates 2 molecules of NADPH. These reducing equivalents are in turn utilized for such processes as redox homeostasis and fatty acid synthesis. The non-oxidative arm consists of a series of reversible reactions that can generate ribose 5-phosphate for nucleotide synthesis or provide intermediates to fuel glycolysis. It is important to note that these two branches of the PPP can be decoupled and the non-oxidative arm does not produce NADPH. Interestingly, it has been shown that oncogenic KRAS in PDA tumors increases glucose carbon flux exclusively through the non-oxidative arm of the PPP [8]. KRAS mediates the upregulation of rate limiting enzymes RPE and RPIA which leads to increased flux into the non-oxidative PPP. Indeed, inhibition of these enzymes with RNAi decreases clonogenicity of cells and growth of xenografts, highlighting the importance of the non-oxidative PPP in PDA tumor growth [8]. The synthesis of nucleotide precursors through the non-oxidative PPP implies that PDA tumors have another means of generating required pools of NADPH (discussed below). A recent report, however, highlights a role of the oxidative PPP in PDA metastasis [59].
Another major anabolic pathway through which PDA cells increase glucose flux is the hexosamine biosynthesis pathway (HBP). This pathway results in the synthesis of uridine diphosphate-N-acetylglucosamine (UDP-GlcNAc) which executes a number of functions, including intracellular signaling and post-translational modifications (PTM). The committed and rate limiting step in this pathway is through the enzyme GFPT1 which is transcriptionally increased in PDA. Extinction of Kras using the doxycycline inducible mouse model results in decreased expression of Gfpt1 as well as decreased glycosylation, indicating upregulation is downstream KRAS signaling [8]. The addition of O-GlcNAc to proteins (glycosylation) is analogous to phosphorylation and has been shown to rewire metabolism in cancer [60]. The enzyme OGT is responsible for the addition of O-GlcNAc to proteins. Indeed, PDA was found to have increased levels of OGT and hyper-O-GlcNAacylation which activates NF-kB signaling resulting in an anti-apoptotic phenotype. Knockdown of GFPT1 or OGT results in decreased proliferation of PDA in vitro and in vivo [61].
Considering anabolic glucose metabolism plays such a prominent role in PDA progression, targeting these pathways therapeutically may lead to clinical response. Currently, there are no clinical grade inhibitors against GFPT1, RPIA, or RPE. These enzymes, however, are upregulated due to KRAS signaling. While a specific and clinical-grade pan-KRAS inhibitor does not yet exist, progress has been made in targeting a specific KRAS mutation - G12C (unfortunately a rare occurrence in PDA). Indirectly targeting glucose metabolism through downstream components of KRAS signaling (MEK/ERK) is possible through MEK inhibition (MEKi). Expectedly, treatment with MEKi results in the decrease of many of the glycolytic enzymes under the transcriptional control of KRAS [8]. Unfortunately, MEKi PDA trials were largely unsuccessful with multiple reasons put forth for the lack of success, including signaling redundancy and a multitude of resistance mechanisms being identified [62]. Indeed, it has been shown that ERK reactivation is a major cause of treatment resistance to upstream inhibitors (RAF, MEK) in RAS mutant cancers. A report by Hayes and colleagues suggests that targeting ERK directly may be a more effective treatment strategy as prolonged ERK inhibition results in MYC degradation and attenuates PDA growth [63].
Glutamine:
The nonessential amino acid glutamine is the most abundant amino acid in the blood and has versatile roles in cancer cell metabolism. Although mammalian cells can synthesize glutamine, it is considered conditionally essential for the growth of many types of cancer cells. Continuous replenishment of the TCA cycle with glutamine-derived carbon (anapleurosis) is important for generating reducing equivalents as well precursors for biosynthesis. Canonical glutamine metabolism takes glutamine-derived glutamate and, through glutamate dehydrogenase (GLUD1), converts glutamate to α-ketoglutarate, thereby fueling the TCA cycle. This process generates NADH as well as metabolites required for de novo synthesis of macromolecules and lipids. Glutamine is also an important source of reduced nitrogen required for the biosynthesis of both purines and pyrimidines [64, 65].
In addition to aforementioned roles of glutamine, we identified a non-canonical glutamine metabolism pathway in PDA which is critical to maintain redox homeostasis. KRAS mutant PDA cells suppress levels of GLUD1 and increase levels of cytosolic aspartate aminotransferase (GOT1), resulting in reprogrammed glutamine metabolism. In this situation, the mitochondrial aspartate aminotransferase (GOT2) produces aspartate. This aspartate is released into the cytoplasm where it is metabolized by GOT1, followed by a cascade of reactions by malic dehydrogenase (MDH1) and malic enzyme (ME1). The end result is increased reducing potential of the cell in the form of NADPH [66]. In a related manner, it has been recently reported that cell survival in the acidic TME imparted by lactate production from elevated glycolysis relies on increased expression of GOT1 and the aforementioned non-canonical glutamine pathway [67]. Considering that KRAS shunts glucose carbon from glycolysis away from the oxidative, NADPH generating branch of the PPP, the GOT2-GOT1-ME1 pathway becomes a major source of cytosolic NADPH production. Indeed perturbation at multiple steps of this pathway (GLS, GOT1, ME1) results in redox imbalance and decreased cellular proliferation [66].
The putative reliance of cancer cells on glutamine metabolism accompanied by the observation that glutaminolysis is dispensable for nonmalignant cells makes therapeutically targeting this pathway appealing. Indeed multiple inhibitors directed against glutaminase (GLS), the initiating enzyme in PDA glutamine metabolism, are being evaluated for efficacy in a number of different cancers. One such example is second generation GLS inhibitor CB-839 [68]. We have recently shown, however, that sensitivity to glutamine metabolism inhibition in vitro may not translate to efficacy in vivo [69]. To this end, GLS inhibition potently suppresses PDA cell growth in short term assays in vitro, yet there is no significant change in tumor growth rates in multiple different mouse models of PDA. Interestingly, PDA cells treated for longer durations in culture rapidly rewire their metabolism through a coordinated change in the expression of multiple key metabolic enzymes. This results in altered metabolic fluxes, replenishment of critical metabolite pools, and ultimately a restoration of proliferative capacity [69]. In contrast, other cancers such as Keap1 mutant lung cancer have sustained responses to GLS inhibition both in vitro and in vivo, thus identifying a genetic biomarker for targeting glutamine metabolism [70].
It has also been reported that glutamine metabolism is influenced by extracellular nutrient content and when cells are grown in media that may mimic in vivo conditions, the contribution of glutamine to central carbon metabolism is markedly reduced in lung cancer [71]. While these are intriguing data, in vivo tracing experiments reveal that pancreatic tumors consume significant amounts of glutamine which outcompetes glucose and lactate for TCA cycle labeling [55]. The importance of glutamine metabolism in vivo is further supported by concomitant treatment of PDA tumors with CB-839 and ROS generating agents. Combinatorial targeting of GLS and redox metabolism (BSO, β-lapachone) leads to a synergistic response in xenograft models, suggesting that glutamine metabolism is necessary for redox maintenance in vivo [69, 72]. The lack of response exhibited by PDA tumors when treated with CB-839 as a single agent lends credence to the metabolically plastic nature of these tumors. Given that GLS is so proximal in the pathways utilizing glutamine carbon, it is likely that these tumors can rapidly rewire their metabolism to generate alternative sources of glutamate as a mechanism of resistance [69]. Taken together these data suggest that targeting enzymes further downstream in glutamine metabolism may lead to a more potent and sustainable response. Furthermore, as there were multiple compensatory metabolic pathways that were upregulated in response to GLSi it is unlikely that targeting a single pathway will lead to sustained therapeutic efficacy. By understanding a priori these compensations, combinatorial therapeutic strategies can be identified. Lastly, it also advocates that longer term cell culture assays may be more predictive of in vivo efficacy than standard short-term growth assays when evaluating metabolic inhibitors since these assays better recapitulate temporal exposure to the compound seen in vivo.
Lipids and Fatty Acids:
Lipid and fatty acid metabolism are expanding areas of research in cancer metabolism. Lipid metabolism is essential for production of cellular membranes (especially during cell division) and production of ATP through beta-oxidation [73]. Altered signaling pathways in malignant cells result in aberrant lipid metabolism in the form of both scavenging and de novo synthesis of lipids. To this end, human pancreatic ductal epithelial cells (HPNE) transformed with the KRAS oncogene increase lipid uptake from the environment [47]. Additionally, Fatty acid synthase (FASN), a key enzyme in fatty acid synthesis (FAS), is upregulated by EGFR/ERK signaling and inhibition of this enzyme is detrimental to PDA cells [74]. The augmented capacity of PDA cells to either uptake or synthesize lipids and fatty acids (FAs) seems to play an important role in cancer progression. Indeed, high transcriptional levels of FASN or LDLR (cholesterol uptake) are associated with poor overall survival or reoccurrence in PDA respectively [75].
The pancreatic TME is considered deficient in levels of lipids and fatty acids when compared to benign pancreas [76]. The interpretation of this data, as in any steady state measurements, can either indicate scarcity of lipids or excess utilization. Interestingly, it has been reported that PDA cells have increased propensity to uptake exogenous cholesterol through elevated levels of low density lipoprotein receptor (LDLR) [77]. Cholesterol synthesis requires a large contribution from oxygen and given the hypoxic microenvironment of PDA, it is tempting to speculate that uptake is the preferred means of cholesterol acquisition. There is also evidence that high fat diets in mouse models of PDA or treatment of cells with certain species of fatty acids can increase PDA cell growth [78, 79]. Taken together these data support the idea that exogenous lipids are beneficial for PDA tumors.
It has also been reported that lipid synthesis accounts for 75–90% of the cellular palmitate pools in PDA and other cancer types [47]. The biology of lipids and FAs is complex and this observation does not exclude the possibility that cancer cells also rely on exogenous lipids, but does make a case that FAS is an essential contributor to lipid metabolism. The synthesis of fatty acids requires cytosolic pools of acetyl-CoA and to keep up with elevated rates of FAS in many cancers, there are multiple sources from which these pools can be generated. One mechanism of acetyl-CoA production is through metabolism of citrate. In this case, citrate is exported from the mitochondria via tricarboxylate transporter SLC25A1 where it is metabolized by ACLY to generate acetyl-CoA and oxaloacetate. Another mechanism of generating acetyl-coA comes from the metabolism of acetate derived from dietary or other exogenous sources. The role of acetate in cancer metabolism and progression has been thoroughly discussed by Schug and colleagues[80]. Interestingly, it has been proposed that monocarboxylate carriers (MCT1/4), which normally function in the transport of lactate and pyruvate, can transport acetate into the cell. Furthermore, these transporters are overexpressed in PDA, potentially contributing to de novo lipid synthesis and the overall metabolic plasticity of these tumors [81]. Indeed it has been shown that pancreatic cancer cells consume acetate from the environment and the rate of uptake is dictated by expression of ACSS2 [82]. The next steps in FAS involves the enzyme acetyl-CoA carboxylase (ACC) converting acetyl-CoA into malonyl-CoA which generates C16 palmitate through successive condensation reactions catalyzed by FASN. The palmitate generated through de novo synthesis has a number of intracellular functions, including regulation of signaling networks in PDA and perturbations at multiple steps (ACC, FASN) attenuates PDA growth [83]. It has been recently reported that high FASN expression correlates with gemcitabine resistance in PDA and combination treatment of Orlistat (FASN inhibitor) and gemcitabine results in a synergistic response [84].
To add to the complexity of lipid metabolism in PDA, some lipids and FAs have a clear role in supporting PDA tumor growth while others have a purported anti-tumor effects. Although dietary factors and obesity are established risk factors for PDA [85], some types of FAs are reported to decrease tumor cell proliferation. Polyunsaturated fatty acids (PUFAs) of the n-6 variety were reported to stimulate growth of PDA cells, while n-3 PUFAs decreased proliferation [86]. Indeed, further elucidating mechanisms involved in the regulation and response to different FAs may aid in identifying novel therapeutics and treatment options for patients. Currently, a number of inhibitors targeting lipid metabolism are being evaluated for efficacy in clinical trials in PDA and other cancers [75].
Hypoxia and HIF1
While most solid tumors have some degree of hypoxia, human PDA tumors were found to have the lowest level of oxygen when compared to other tumor types [87]. This is probably not surprising given the fibrotic and poorly functioning vasculature of the PDA microenvironment. The lack of available oxygen has a number of consequences on the metabolism and clinical response of PDA tumors. Orchestrating much of the hypoxic response is the transcription factor HIF1, a heterodimer consisting of HIF1α and HIF1β subunits. At high oxygen tensions, HIF1α is ubiquitinated and subsequently degraded by the proteasome. This degradation is dependent on the hydroxylation of proline residues by prolyl-hydroxylase domain protein 2 (PDH2). Under hypoxic conditions PDH2 activity is reduced resulting in the stabilization of HIF1α which now can dimerize with the HIF1β resulting in successive downstream signaling [88].
Perhaps the best known function of HIF1 signaling is the metabolic rewiring to favor anaerobic conditions. As mentioned previously, HIF1 increases transcription of glycolytic enzymes and decreases glucose carbon entry into the TCA cycle. This is a beneficial adaptation under hypoxia as it allows cells to produce ATP regardless of oxygen availability [89]. Besides energy production, it has been reported that hypoxia influences flux into the HBP in PDA [56]. The consequences of increased O-GlcNAcylation under hypoxia are not well established, but further investigation will be imperative to understanding PDA signaling and metabolism under low oxygen conditions.
Elevated expression of HIF1 is associated with poor survival in PDA patients [90]. This is likely due to a number of different factors including the observation that hypoxic cells are resistant to radiotherapy [91]. A recent finding by Shukla and colleagues adds to the understanding of the poor clinical response by demonstrating a HIF1 dependent metabolic adaption that results in resistance to gemcitabine, a standard of care in PDA. Through increased pyrimidine biosynthesis imparted by MUC-1/HIF1 crosstalk, PDA tumors develop resistance to gemcitabine [92]. These results demonstrate another instance where the plasticity of PDA metabolism leads to treatment resistance, yet highlights potential therapeutic targets that may improve clinical outcome.
ROS in PDA initiation and Progression
Reactive oxygen species (ROS) are by products of cellular metabolism and have versatile roles within cancer cells. At high levels, ROS are detrimental, causing oxidative damage to proteins, lipids and DNA. Sustained ROS and irreparable damage will lead to cell death. On the other hand, ROS are important signaling molecules that can regulate cellular metabolism through alterations of signaling networks [93]. One such example of ROS acting in a pro-tumorigenic capacity is through modulation of tumor suppressors. The exposed thiols on the catalytic site of tumor suppressor PTEN are susceptible to ROS attack, which results in inhibited function. Inactivation of PTEN causes aberrant downstream signaling through PI3K/AKT and activation of pro-survival genes contributing to oncogenesis [94]. The elevated levels of ROS found in most cancers are important not only for tumor maintenance but have been shown to play a critical role in tumor initiation. To this end, treating KC (LSL-KrasG12D; p48Cre) mice with mitochondrial ROS scavenger MitoQ leads to a marked reduction in the PanIN lesions [95]. Additionally, it has been reported that non-mitochondrial generated superoxide may play an important role in PDA growth. Indeed, scavenging of superoxides through increased expression of SOD or pharmaceutical treatment resulted in decreased tumor growth in PDA in vivo [96].
There exists a delicate balance between the levels of ROS that are tumor promoting and cytotoxic. In order to maintain this critical equilibrium, PDA tumors have evolved a number of different mechanisms to fine tune the levels of intracellular ROS. One such mechanism is through non-canonical glutamine metabolism which produces cytosolic NADPH (discussed above). The reducing equivalent NADPH is important for maintaining levels of reduced glutathione, an essential molecule for cellular detoxification. Another mechanism in PDA to counteract excessive ROS accumulation is through upregulation of the transcription factor NRF2 by oncogenic KRAS. Activation of NRF2 results in the transcription of antioxidant proteins such as NQO1 and thioredoxin. Indeed, it has been shown that NRF2 plays a role in oncogenesis of KRAS mutant PDA tumors and deletion results in significantly decreased tumors in KPC mice [97]. In addition to its role in PDA development, it has been shown that NRF2 is necessary for tumor maintenance. The loss of NRF2 in PDA in vitro and in vivo leads to the oxidation of proteins involved in mRNA translation resulting in decreased tumor growth. These results could be recapitulated by combined targeting of AKT and glutathione metabolism [98]. Taken together, these data present a fundamental role for ROS in both tumor initiation and progression and disturbing the ROS equilibrium identifies a potential therapeutic opportunity for PDA prevention and treatment.
Conclusion:
PDA, like many tumors, have altered their metabolism to meet the needs of unconstrained proliferation. The unique TME in which these tumor cells reside poses particular challenges for the cells to meet their basic metabolic demands. These tumors exhibit profound hypoxia, markedly impaired perfusion, and low nutrient availability. To counter this, PDA have developed a series of adaptations that allow them to not only grow, but to thrive under these conditions. While these seminal discoveries have propelled the field forward, many challenges still exist in defining how nutrients are acquired and used in tumors. The complexity of the TME in shaping metabolism and contribution of non-malignant cells remains to be explored. For example, it is not clear how autophagy (recycling), macropinocytosis (extracellular scavenging), and nutrient cross-talk circuits are all integrated. It is tempting to speculate that autophagy may provide the ability of tumor cells to survive rapid oscillations of nutrients and oxygen in the TME. Through cooperation with macropinocytosis and nutrient sharing with other cell-types in the TME, a tumor cell could restore biomass and resume proliferation. The difficulty in answering such fundamental questions is due, in part, by our limited capability of studying tumor metabolism in vivo. Analyses are confounded by the heterogeneous populations of cells found in the TME making it difficult to ascertain relative contributions to a metabolite profile. While in vivo labeling studies have certainly contributed to our understanding of tumor fuel source utilization in mice and now even in people, it cannot discern the contribution of the non-neoplastic component of the tumor and therefore only gives the aggregate of all cell types. Utilizing in vitro culture systems has certainly contributed a great deal of our understanding of PDA metabolism, but growth in vitro is clearly distinct from that in vivo. To this end, all in vitro metabolic dependencies require extensive in vivo validation in relevant autochthonous models with an intact immune systems and relevant stroma. While this is certainly achievable, it is relatively low throughput. One potential way of circumventing this problem is through the use of genetic techniques such as pooled CRISPR screens performed in vivo. Mapping metabolism through loss of function screens would identify required metabolic enzymes for growth in vivo. Through syngeneic transplantation models, these screens could be performed in the presence of functioning immune system further elucidating the roles different cellular compartments in cancer progression. The contributions of the TME and requirements for PDA cells to grow in vivo will add to our understanding of PDA metabolism and potentially identify promising therapeutic targets.
In the end, the translation of PDA metabolic dependencies in vivo to therapeutic approaches will require an extensive understanding of how these tumors adapt or rewire their metabolism in response to such perturbations. Extensive pre-clinical studies will allow for the development of effective combinatorial approaches that can be tested in patients. The preliminary success of HCQ in PDA patients provides much excitement and rational that targeting metabolism in PDA can lead to improved outcomes in the future.
Figure 1.
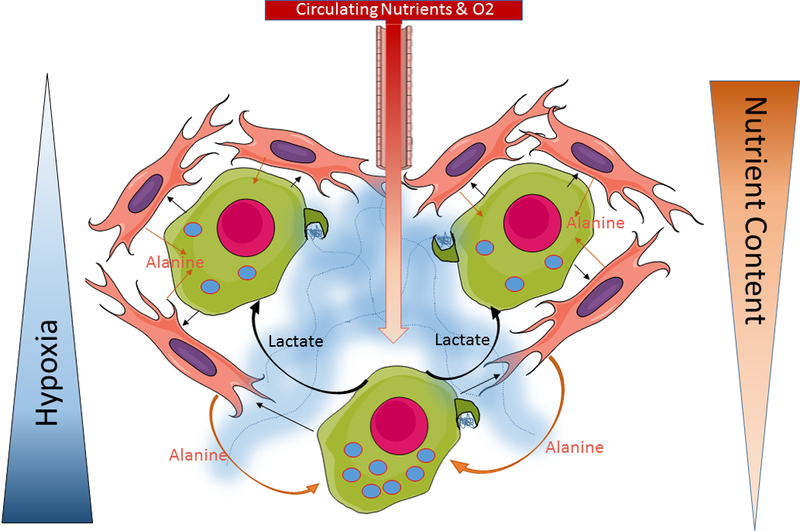
Representative image of metabolic cross-talk in the PDA microenvironment. Moving further from the vasculature results in the decrease in nutrient content and increase in hypoxia. PDA Cells (green cell, magenta nucleus) in the nutrient devoid and hypoxic areas of the tumor display increased autophagy (represented by blue dots with red outlines) and lactate secretion (curved black arrows). Secreted lactate serves as a fuel source for neighboring PDA cells. Stellate cells (orange cell, purple nucleus) secrete alanine into the microenvironment (orange arrows) which is rapidly consumed by PDA cells. The process of autophagy-dependent alanine secretion is stimulated by PDA cells through release of an unknown factor (straight black arrow). Collagen (blue area) is deposited by activated stellate cells into the TME. Through macropinocytosis (dark green membrane protrusions on tumors cell), cell can uptake extracellular protein as well as components of the ECM (like collagen) as an external fuel source for central carbon metabolism.
Figure 2.
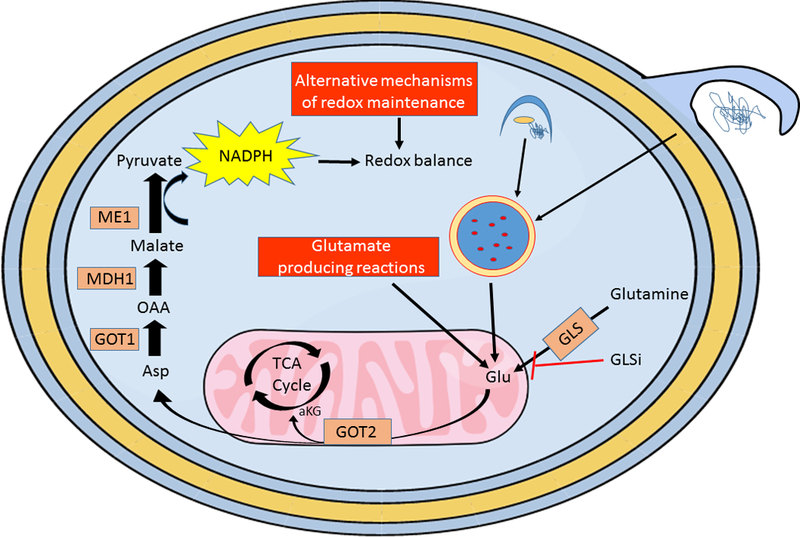
PDA cells have a remarkable ability to adapt to nutrient fluctuations and targeted therapies. For example, PDA cells can rapidly adapt to GLS inhibition (GLSi) through multiple mechanisms of metabolic rewiring. Blocking the conversion of glutamine to glutamate perturbs redox balance in PDA cells by disrupting the initial steps of the non-canonical glutamine pathway that generates NADPH. One means by which PDA overcomes GLSi is by upregulating cellular reactions that generate glutamate bypassing the need for GLS activity (lower red box). Scavenging by macropinocytosis (blue membrane protrusion) and recycling by autophagy (dark blue forming phagophore) of macromolecules through the lysosome (dark blue circle, red dots) can also generate glutamate to fuel PDA metabolism downstream of GLS. Another means of adaptation is by directly upregulating metabolic reactions that restore reducing potential of the cell (upper red box). The plasticity of PDA metabolism allows for adaption at multiple levels making the cells exceptionally resistant to microenvironmental or therapeutic induced changes in nutrients.
Acknowledgements
We apologize for the omission of any primary citations. Figures created in the Mind the Graph platform-www.mindthegraph.com. This work was supported by National Cancer Institute Grants R01CA157490, R01CA188048 and P01CA117969; ACS Research Scholar Grant RSG-13–298-01-TBG; NIH grant R01GM095567; and the Lustgarten Foundation to A.C.K.
Footnotes
Disclosure of Potential Conflicts of Interest: ACK has financial interests in Vescor Therapeutics, LLC. A.C.K. is an inventor on patents pertaining to Kras regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K is on the SAB of Cornerstone/Rafael Pharmaceuticals.
References
- 1.Siegel RL, Miller KD, and Jemal A, Cancer statistics, 2018. CA Cancer J Clin, 2018. 68(1): p. 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Rahib L, et al. , Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res, 2014. 74(11): p. 2913–21. [DOI] [PubMed] [Google Scholar]
- 3.Ying H, et al. , Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev, 2016. 30(4): p. 355–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waddell N, et al. , Whole genomes redefine the mutational landscape of pancreatic cancer. Nature, 2015. 518(7540): p. 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aguirre AJ, et al. , Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev, 2003. 17(24): p. 3112–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tuveson DA, et al. , Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell, 2004. 5(4): p. 375–87. [DOI] [PubMed] [Google Scholar]
- 7.Collins MA, et al. , Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest, 2012. 122(2): p. 639–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ying H, et al. , Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell, 2012. 149(3): p. 656–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Waters AM and Der CJ, KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb Perspect Med, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Raphael BJ, et al. , Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell, 2017. 32(2): p. 185-203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox AD, et al. , Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov, 2014. 13(11): p. 828–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hobbs GA, Wittinghofer A, and Der CJ, Selective Targeting of the KRAS G12C Mutant: Kicking KRAS When It’s Down. Cancer Cell, 2016. 29(3): p. 251–3. [DOI] [PubMed] [Google Scholar]
- 13.Kimmelman AC, et al. , Genomic alterations link Rho family of GTPases to the highly invasive phenotype of pancreas cancer. Proc Natl Acad Sci U S A, 2008. 105(49): p. 19372–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ansari D, et al. , Pancreatic cancer stroma: controversies and current insights. Scand J Gastroenterol, 2017. 52(6–7): p. 641–646. [DOI] [PubMed] [Google Scholar]
- 15.Witkiewicz AK, et al. , Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun, 2015. 6: p. 6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feig C, et al. , The pancreas cancer microenvironment. Clin Cancer Res, 2012. 18(16): p. 4266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Omary MB, et al. , The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest, 2007. 117(1): p. 50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng M, et al. , PD-1/PD-L1 and immunotherapy for pancreatic cancer. Cancer Lett, 2017. 407: p. 57–65. [DOI] [PubMed] [Google Scholar]
- 19.Bayne LJ, et al. , Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell, 2012. 21(6): p. 822–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pylayeva-Gupta Y, et al. , Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell, 2012. 21(6): p. 836–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DuFort CC, DelGiorno KE, and Hingorani SR, Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology, 2016. 150(7): p. 1545–1557.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olive KP, et al. , Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science, 2009. 324(5933): p. 1457–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamphorst JJ, et al. , Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res, 2015. 75(3): p. 544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vander Heiden MG, Cantley LC, and Thompson CB, Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science, 2009. 324(5930): p. 1029–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glick D, Barth S, and Macleod KF, Autophagy: cellular and molecular mechanisms. J Pathol, 2010. 221(1): p. 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur J and Debnath J, Autophagy at the crossroads of catabolism and anabolism. Nature Reviews Molecular Cell Biology, 2015. 16: p. 461. [DOI] [PubMed] [Google Scholar]
- 27.Levine B and Klionsky DJ, Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell, 2004. 6(4): p. 463–77. [DOI] [PubMed] [Google Scholar]
- 28.Levine B and Kroemer G, Autophagy in the pathogenesis of disease. Cell, 2008. 132(1): p. 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yonekawa T and Thorburn A, Autophagy and cell death. Essays Biochem, 2013. 55: p. 105–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saxton RA and Sabatini DM, mTOR Signaling in Growth, Metabolism, and Disease. Cell, 2017. 169(2): p. 361–371. [DOI] [PubMed] [Google Scholar]
- 31.Mancias JD and Kimmelman AC, Mechanisms of Selective Autophagy in Normal Physiology and Cancer. J Mol Biol, 2016. 428(9 Pt A): p. 1659–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perera RM, et al. , Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature, 2015. 524(7565): p. 361–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong PM, et al. , Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat Commun, 2015. 6: p. 8048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White E, The role for autophagy in cancer. The Journal of Clinical Investigation, 2015. 125(1): p. 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levy JMM, Towers CG, and Thorburn A, Targeting autophagy in cancer. Nature Reviews Cancer, 2017. 17: p. 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rosenfeldt MT, et al. , p53 status determines the role of autophagy in pancreatic tumour development. Nature, 2013. 504(7479): p. 296–300. [DOI] [PubMed] [Google Scholar]
- 37.Yang A, et al. , Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov, 2014. 4(8): p. 905–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang S, et al. , Pancreatic cancers require autophagy for tumor growth. Genes Dev, 2011. 25(7): p. 717–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guo JY, et al. , Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev, 2016. 30(15): p. 1704–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sousa CM, et al. , Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature, 2016. 536(7617): p. 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang A, et al. , Autophagy sustains pancreatic cancer growth through both cell autonomous and non-autonomous mechanisms. Cancer Discov, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim JP and Gleeson PA, Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol, 2011. 89(8): p. 836–43. [DOI] [PubMed] [Google Scholar]
- 43.Bar-Sagi D and Feramisco JR, Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science, 1986. 233(4768): p. 1061–8. [DOI] [PubMed] [Google Scholar]
- 44.Commisso C, et al. , Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature, 2013. 497(7451): p. 633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davidson SM, et al. , Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med, 2017. 23(2): p. 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olivares O, et al. , Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun, 2017. 8: p. 16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamphorst JJ, et al. , Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A, 2013. 110(22): p. 8882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boone BA, et al. , Safety and Biologic Response of Pre-operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann Surg Oncol, 2015. 22(13): p. 4402–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeBerardinis RJ and Chandel NS, Fundamentals of cancer metabolism. Sci Adv, 2016. 2(5): p. e1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vousden KH and Ryan KM, p53 and metabolism. Nat Rev Cancer, 2009. 9(10): p. 691–700. [DOI] [PubMed] [Google Scholar]
- 51.Zhao D, et al. , Lysine-5 acetylation negatively regulates lactate dehydrogenase A and is decreased in pancreatic cancer. Cancer Cell, 2013. 23(4): p. 464–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Canto C, Menzies KJ, and Auwerx J, NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab, 2015. 22(1): p. 31–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robin ED, Murphy BJ, and Theodore J, Coordinate regulation of glycolysis by hypoxia in mammalian cells. J Cell Physiol, 1984. 118(3): p. 287–90. [DOI] [PubMed] [Google Scholar]
- 54.Seagroves TN, et al. , Transcription factor HIF-1 is a necessary mediator of the pasteur effect in mammalian cells. Mol Cell Biol, 2001. 21(10): p. 3436–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hui S, et al. , Glucose feeds the TCA cycle via circulating lactate. Nature, 2017. 551(7678): p. 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Guillaumond F, et al. , Strengthened glycolysis under hypoxia supports tumor symbiosis and hexosamine biosynthesis in pancreatic adenocarcinoma. Proc Natl Acad Sci U S A, 2013. 110(10): p. 3919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Faubert B, et al. , Lactate Metabolism in Human Lung Tumors. Cell, 2017. 171(2): p. 358–371.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romero-Garcia S, et al. , Lactate Contribution to the Tumor Microenvironment: Mechanisms, Effects on Immune Cells and Therapeutic Relevance. Front Immunol, 2016. 7: p. 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McDonald OG, et al. , Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet, 2017. 49(3): p. 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrer CM, Sodi VL, and Reginato MJ, O-GlcNAcylation in Cancer Biology: Linking Metabolism and Signaling. J Mol Biol, 2016. 428(16): p. 3282–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma Z, Vocadlo DJ, and Vosseller K, Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem, 2013. 288(21): p. 15121–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cutsem EV, et al. , Phase II randomized trial of MEK inhibitor pimasertib or placebo combined with gemcitabine in the first-line treatment of metastatic pancreatic cancer. Journal of Clinical Oncology, 2015. 33(3_suppl): p. 344–344. [Google Scholar]
- 63.Hayes TK, et al. , Long-Term ERK Inhibition in KRAS-Mutant Pancreatic Cancer Is Associated with MYC Degradation and Senescence-like Growth Suppression. Cancer Cell, 2016. 29(1): p. 75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.DeBerardinis RJ and Cheng T, Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene, 2010. 29(3): p. 313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Pavlova NN, and Thompson CB, Cancer cell metabolism: the essential role of the nonessential amino acid, glutamine. Embo j, 2017. 36(10): p. 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Son J, et al. , Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature, 2013. 496(7443): p. 101–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abrego J, et al. , GOT1-mediated anaplerotic glutamine metabolism regulates chronic acidosis stress in pancreatic cancer cells. Cancer Lett, 2017. 400: p. 37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gross MI, et al. , Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther, 2014. 13(4): p. 890–901. [DOI] [PubMed] [Google Scholar]
- 69.Biancur DE, et al. , Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun, 2017. 8: p. 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sayin VI, et al. , Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muir A, et al. , Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chakrabarti G, et al. , Targeting glutamine metabolism sensitizes pancreatic cancer to PARP-driven metabolic catastrophe induced by ss-lapachone. Cancer Metab, 2015. 3: p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Beloribi-Djefaflia S, Vasseur S, and Guillaumond F, Lipid metabolic reprogramming in cancer cells. Oncogenesis, 2016. 5: p. e189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bian Y, et al. , Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem Biophys Res Commun, 2015. 463(4): p. 612–7. [DOI] [PubMed] [Google Scholar]
- 75.Sunami Y, Rebelo A, and Kleeff J, Lipid Metabolism and Lipid Droplets in Pancreatic Cancer and Stellate Cells. Cancers (Basel), 2017. 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ma X, et al. , The metabolic features of normal pancreas and pancreatic adenocarcinoma: preliminary result of in vivo proton magnetic resonance spectroscopy at 3.0 T. J Comput Assist Tomogr, 2011. 35(5): p. 539–43. [DOI] [PubMed] [Google Scholar]
- 77.Guillaumond F, et al. , Cholesterol uptake disruption, in association with chemotherapy, is a promising combined metabolic therapy for pancreatic adenocarcinoma. Proc Natl Acad Sci U S A, 2015. 112(8): p. 2473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Philip B, et al. , A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology, 2013. 145(6): p. 1449–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang F, et al. , Increased lipid metabolism and cell turnover of MiaPaCa2 cells induced by high-fat diet in an orthotopic system. Metabolism, 2009. 58(8): p. 1131–6. [DOI] [PubMed] [Google Scholar]
- 80.Schug ZT, Vande Voorde J, and Gottlieb E, The metabolic fate of acetate in cancer. Nat Rev Cancer, 2016. 16(11): p. 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Parks SK, Chiche J, and Pouyssegur J, Disrupting proton dynamics and energy metabolism for cancer therapy. Nat Rev Cancer, 2013. 13(9): p. 611–23. [DOI] [PubMed] [Google Scholar]
- 82.Bulusu V, et al. , Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep, 2017. 18(3): p. 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nishi K, et al. , Inhibition of Fatty Acid Synthesis Induces Apoptosis of Human Pancreatic Cancer Cells. Anticancer Res, 2016. 36(9): p. 4655–60. [DOI] [PubMed] [Google Scholar]
- 84.Tadros S, et al. , De novo Lipid Synthesis Facilitates Gemcitabine Resistance through Endoplasmic Reticulum Stress in Pancreatic Cancer. Cancer Res, 2017. 77(20): p. 5503–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bracci PM, Obesity and pancreatic cancer: overview of epidemiologic evidence and biologic mechanisms. Mol Carcinog, 2012. 51(1): p. 53–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mohammed A, et al. , Endogenous n-3 polyunsaturated fatty acids delay progression of pancreatic ductal adenocarcinoma in Fat-1-p48(Cre/+)-LSL-Kras(G12D/+) mice. Neoplasia, 2012. 14(12): p. 1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vaupel P, Hockel M, and Mayer A, Detection and characterization of tumor hypoxia using pO2 histography. Antioxid Redox Signal, 2007. 9(8): p. 1221–35. [DOI] [PubMed] [Google Scholar]
- 88.Masoud GN and Li W, HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B, 2015. 5(5): p. 378–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eales KL, Hollinshead KE, and Tennant DA, Hypoxia and metabolic adaptation of cancer cells. Oncogenesis, 2016. 5: p. e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hoffmann AC, et al. , High expression of HIF1a is a predictor of clinical outcome in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA, VEGF, and bFGF. Neoplasia, 2008. 10(7): p. 674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brown JM, Tumor hypoxia in cancer therapy. Methods Enzymol, 2007. 435: p. 297–321. [DOI] [PubMed] [Google Scholar]
- 92.Shukla SK, et al. , MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell, 2017. 32(3): p. 392. [DOI] [PubMed] [Google Scholar]
- 93.Reczek CR and Chandel NS, The Two Faces of Reactive Oxygen Species in Cancer. Annual Review of Cancer Biology, 2017. 1(1): p. 79–98. [Google Scholar]
- 94.Kitagishi Y and Matsuda S, Redox regulation of tumor suppressor PTEN in cancer and aging (Review). Int J Mol Med, 2013. 31(3): p. 511–5. [DOI] [PubMed] [Google Scholar]
- 95.Liou GY, et al. , Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep, 2016. 14(10): p. 2325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Du J, et al. , Regulation of pancreatic cancer growth by superoxide. Mol Carcinog, 2013. 52(7): p. 555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.DeNicola GM, et al. , Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature, 2011. 475(7354): p. 106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chio IIC, et al. , NRF2 Promotes Tumor Maintenance by Modulating mRNA Translation in Pancreatic Cancer. Cell, 2016. 166(4): p. 963–976. [DOI] [PMC free article] [PubMed] [Google Scholar]