

Abstract
Purpose
We assessed the usefulness of real-time molecular profiling through next-generation sequencing (NGS) in predicting the tumor biology of advanced pancreatic neuroendocrine tumors (panNETs) and in characterizing genomic evolution.
Methods
Patients with metastatic panNETs were recruited in the routine clinical practice setting (between May 2014 and March 2017) for prospective NGS of their tumors as well as for germline analysis using the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) sequencing platform. When possible, NGS was performed at multiple time points.
Results
NGS was performed in 96 tumor samples from 80 patients. Somatic alterations were identified in 76 of 80 patients (95%). The most commonly altered genes were MEN1 (56%), DAXX (40%), ATRX (25%), and TSC2 (25%). Alterations could be defined in pathways that included chromatin remodeling factors, histone methyltransferases, and mammalian target of rapamycin pathway genes. Somatic loss of heterozygosity was particularly prevalent (55 of 95 tested samples [58%]), and the presence of loss of heterozygosity resulted in improved overall survival (P = .06). Sequencing of pre- and post-treatment samples revealed tumor-grade progression; clonal evolution patterns were also seen (molecular resistance mechanisms and chemotherapy-associated mutagenesis). Germline genetic analysis identified clinically actionable pathogenic or likely pathogenic variants in 14 of 88 patients (16%), including mutations in high-penetrance cancer susceptibility genes (MEN1, TSC2, and VHL).
Conclusion
A clinical NGS platform reveals pertubations of biologic pathways in metastatic panNETs that may inform prognosis and direct therapies. Repeat sequencing at disease progression reveals increasing tumor grade and genetic evolution, demonstrating that panNETs adopt a more aggressive behavior through time and therapies. In addition to frequent somatic mutations in MEN1 and TSC2, germline mutations in these same genes underlie susceptibility to panNETs and highlight the need to re-evaluate whether germline genetic analysis should be performed for all patients with panNETs.
INTRODUCTION
Pancreatic neuroendocrine tumors (panNETs) represent 1% to 2% of all pancreatic neoplasms, but they have a rising incidence and prevalence.1-4 Whole-exome and whole-genome sequencing, largely of resected panNETs, has defined genomic events and improved our understanding of disease pathogenesis.5,6 These efforts have identified changes in chromatin remodeling, telomere maintenance, and mammalian target of rapamycin (mTOR) pathway genes. In addition to the well-characterized hereditary syndromes associated with panNET development (multiple endocrine neoplasia 1, Von Hippel-Lindau syndrome, neurofibromatosis type 1), recent data suggest the presence of germline alterations in DNA damage repair genes in some patients.6,7
Treatments for unresectable panNETs include targeted and cytotoxic therapies, as well as liver-directed approaches.8-13 No data exist to guide therapy for panNETs with defined genomic alterations. In addition, disease heterogeneity poses a significant challenge in defining the selection and sequencing of therapy.
To that end, using an institutional matched tumor-normal platform, we prospectively performed next-generation sequencing (NGS) of tumors in patients with metastatic well-differentiated (WD) panNETs. We also performed germline analysis for 76 cancer susceptibility genes to detect inherited pathogenic alleles. We hypothesized that NGS could help predict panNET biology and aggressiveness. We also hypothesized that panNETs evolve over time with a more aggressive behavior that could be characterized biologically through NGS of repeat biopsies after treatment.
METHODS
Study Population
Written informed consent was obtained from patients with a diagnosis of WD panNET using a prospective, institutional review board–approved protocol (ClinicalTrials.gov identifier: NCT01775072). NGS was performed on patients accrued between May 2014 and March 2017. Pathologic features of the tumor samples (differentiation and grade) were determined according to the 2017 WHO classification, which recognizes the existence of a grade 3 (G3) category within WD panNETs; historical values for differentiation and grade were collected through review of the electronic medical records. Electronic medical records were also reviewed for patient data on demographics, treatments, and survival.
Sequencing of Tumor Samples
NGS was performed using Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT), a matched tumor-normal sequencing platform approved for clinical use by the New York State Department of Health, to detect base substitutions, small indels, copy number changes, and selected gene rearrangements.14-17 Updated assay versions were released between 2014 and 2017, and NGS of tumor tissue consisted of deep sequencing of all exons and selected introns of 275 (preclinical pilot test), 341 (version 1), 410 (version 2), or 468 (version 3) cancer-related genes. Genotyping was performed using the pilot test for two of 96 samples (2%), version 2 for 16 of 96 samples (17%), version 3 for 57 of 96 samples (59%), and version 4 for 21 of 96 samples (22%). Actionable genetic alterations were defined as changes that could guide therapy, including standard of care and investigational treatments (annotated according to OncoKB).18
Germline Sequencing
Germline variants from the .bam file of the normal DNA sequence were called using the MuTect and GATK haplotype caller as described previously; all variants with < 1% population frequency in the Exome Aggregation Consortium database were interpreted.19,20 Germline analysis included 76 genes known to be associated with hereditary cancer predisposition, including all cancer predisposition genes identified in the American College of Medical Genetics and Genomics guidelines.19,21,22 Variants were interpreted on the basis of American College of Medical Genetics and Genomics criteria.
Loss of Heterozygosity
Loss of heterozygosity (LOH) was determined through analysis of total, allele-specific, and integer DNA copy number genome-wide using FACETS.23
Statistical Analysis
Descriptive statistics were used to characterize the study population and the somatic and germline genetic results. Overall survival (OS), defined as the time from the date of first tissue acquisition (from which NGS was completed) until death or last known follow-up, was estimated by Kaplan-Meier methods. An association between somatic alterations and OS, as well as between LOH status and OS, was computed using a permuted log-rank test.24 All statistical analyses were performed using R version 3.3.2 (R Foundation for Statistical Computing, Vienna, Austria). Fisher’s exact test was used to examine the association between somatic alterations and tumor grade. All P values were two sided, and P < .05 was considered to indicate statistical significance.
RESULTS
Clinical Characteristics
As of March 2017, NGS had been performed on 96 tumor samples from 80 patients (eight patients with two samples analyzed; four patients with three samples analyzed). The median time from pathologic diagnosis to tumor profiling was 27 months (range, 0 to 218 months). The median age was 54 years; 56% were female. All patients had metastatic disease and had been treated heavily with systemic and liver-directed therapies. In 29 patients, the primary tumor was sequenced, and in 58 patients, a metastatic tumor was sequenced; in seven patients, both primary and metastases were sequenced (in one instance, sequencing took place synchronously). Patient and tumor characteristics are summarized in Table 1.
Table 1.
Baseline Patient Characteristics

Genomic Data
In the 96 samples, the median depth of sequencing was 665.5X (range, 75X to 1,255X); the median somatic mutational burden (mutations/megabyte) was 2.95 (range, 0 to 201.76). Recurrent somatic alterations occurred in key pathways and functional groups: chromatin remodeling factors (CRFs), histone methyltransferases (HMTs), and mTOR pathway genes.
Somatic alterations in CRFs (DAXX, ATRX, PBRM1, SMARCB1, ARID5B, ARID2, ARID1B, and ARID1A) were observed in 52 of 80 patients (65%). Frequently altered CRFs included DAXX (32 of 80 [40%]), ATRX (20 of 80 [25%]), and ARID1A (nine of 80 [11%]). Somatic alterations in HMTs (MEN1, SETD2, KMT2A, KMT2C, and KMT2D) were observed in 53 of 80 patients (66%). Frequently altered HMTs included MEN1 (45 of 80 [56%]), SETD2 (14 of 80 [18%]), and KMT2C (seven of 80 [9%]). Somatic alterations in the mTOR pathway (IGF1R, IGF2, MTOR, TSC1, TSC2, AKT2, AKT3, PIK3R3, PIK3C3, PIK3CG, PIK3CB, PIK3CD, PIK3C2G, PTEN, PIK3CA, RHEB, IRS1, and IRS2) were observed in 34 of 80 patients (43%). mTOR pathway alterations were largely seen in TSC2 (20 of 80 [25%]) and PTEN (10 of 80 [13%]). Somatic alterations in cell cycle signaling genes were seen in 31 of 80 patients (39%). TP53 alterations were observed in 10 of 80 patients (13%); all were characterized as likely oncogenic.
Somatic LOH in ≥ 50% of the genome was highly prevalent; it was identified in 55 of 95 tested samples (58%). A significant association (q value < 0.05) was noted between LOH and the presence of MEN1, DAXX, PTEN, or TSC2 alterations. LOH was observed recurrently in chromosomes 1, 2, 3, 6, 8, 10, 11, 15, 16, 21, and 22 (Appendix Fig A1).
NGS results for the first sequenced sample in all patients are summarized in Figure 1. In the 96 sequenced samples, average mutations/megabyte were as follows: 2.74 in grade 1 (G1) tumors, 4.20 in grade 2 (G2) tumors, and 23.64 in G3 tumors. In the recurrently altered genes, there were no statistical differences in genetic spectrum on the basis of G1 versus G2 tumors, but a trend was noted of G2 and G3 tumors harboring TP53, SETD2, and BRAF alterations.
Fig 1.

Genomic landscape of pancreatic neuroendocrine tumors, including the first sequenced sample for each patient (n = 80), as identified by the Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets. LOH, loss of heterozygosity; Mb, megabyte; mTOR, mammalian target of rapamycin.
Novel Recurrently Altered Genes
In this metastatic cohort, we observed a much higher frequency of SETD2 alterations (14 of 80 patients [18%]) in comparison with previous investigations, and a novel finding of BRAF alterations (six of 80 patients [8%]). Many of the somatic alterations in SETD2 and BRAF were first noted after treatment (SETD2, four patients; BRAF, two patients).
SETD2
Seventeen tumor samples (collected from 14 patients) harbored SETD2 alterations; one G1 (6%), 12 G2 (71%), and four G3 (24%). The mean Ki-67 proliferation index (available for 15 of 17 samples) was 20% (range, 1% to 70%). The clinical behavior of these tumors, including the single G1 tumor, was more aggressive.
BRAF
Ten tumor samples (collected from six patients) harbored BRAF alterations: two of 10 G1 (20%), three of 10 G2 (30%), and five of 10 G3 (50%). In addition to two tumors with V600E mutations, several non-V600 BRAF alterations were seen (K601E, T599K, and T310I), and one tumor expressed both BRAF G596D and E451K mutations.
Survival
With a median follow-up for survivors of 27.6 months, we observed 14 deaths; median OS was 118.9 months (95% CI, 99.6%-not achieved). Significantly longer 1-year OS was observed in MEN1-altered tumors compared with wild-type (WT) tumors (98% [95% CI, 84% to 99%] versus 82% [95% CI, 63% to 92%], P = .01) and in DAXX- or ATRX-altered tumors compared with WT tumors (97% [95% CI, 83% to 99%] versus 83% [95% CI, 64% to 93%], P < .01). Significantly inferior 1-year OS was observed in BRAF-altered tumors compared with WT tumors (75% [95% CI, 13% to 96%] versus 95% [95% CI, 87% to 98%], P < .01; Fig 2). No significant OS differences were noted with mTOR pathway or TP53 alterations.
Fig 2.

Significant associations between somatic alterations and overall survival.
Genetic Testing Before and After Treatment
Twelve patients underwent NGS of tumor collected before and after treatment.
Molecular mechanisms of resistance.
For patients treated with targeted agents, NGS before and after treatment allowed for the evaluation of potential resistance mechanisms. Pre- and post-treatment samples were available for three patients receiving the mTOR inhibitor everolimus and for one patient receiving the BRAF V600E inhibitor vemurafenib.
In all three patients receiving everolimus, mTOR pathway alterations were identified at progression. Patient 1 enrolled in a clinical trial of everolimus with BYL719 (PI3K inhibitor). On disease progression, a biopsy specimen revealed an oncogenic PTEN mutation (Q298*), and the sampled areas of tumor progressed from G1 to G3 (Appendix Figure A2). This PTEN mutation was seen previously at a low allele frequency when the original pancreas tumor was sequenced, reflecting the possible emergence of this clone through targeted therapy.
Patient 2 received everolimus for 8 weeks, at which point treatment was held for infectious complications. A liver lesion increased in size and a biopsy was performed; NGS identified a hotspot mutation in RHEB (Y35S).25 Subsequent sampling and NGS of this same liver lesion after 16 months of observation demonstrated the RHEB alteration at a higher allele frequency. In patient 2, the sampled areas increased from G1 to G3 (Appendix Figure A3). The MSK-IMPACT version used to sequence the pretreatment sample did not interrogate RHEB, so the allele fraction of the RHEB mutation is unknown in this sample; however, it increased notably after treatment with andprogression on everolimus.
Patient 3 received everolimus for > 9 months before disease progression. NGS of the pancreatic tumor before starting everolimus demonstrated an IGF2 mutation (G226S). At disease progression, NGS of a growing liver lesion demonstrated loss of this IGF2 alteration, acquisition of a TSC2 splice site mutation (X161_splice), and an AKT2 mutation (G16D; Appendix Figure A4). Surprisingly, no shared mutations were noted in sequencing of the two samples; clinically, however, the disease was consistent with a single primary, suggesting the emergence of a previously unrecognized clone through treatment, although the possibility of a second primary cannot be excluded.
Patient 4, whose tumor harbored a BRAF V600E mutation, enrolled in a basket clinical trial with vemurafenib and developed an NRAS hotspot mutation (Q61R; Appendix Figure A5).26
Chemotherapy-associated mutagenesis.
Three patients underwent NGS of their tumor tissue before and after cytotoxic chemotherapy. Although all patients had a confirmed partial response to chemotherapy before progression, post-treatment NGS noted an acquired mutational burden of > 100 alterations (117, 165, and 267 additional mutations). Two patients received alkylating agents, and the third received oxaliplatin. A review of the tumors exposed to alkylating drugs showed that the variants shared by both samples were observed at a higher allele fraction compared with those unique to the individual samples, and the evolutionary pattern demonstrated a predominant C>T signature consistent with alkylating-agent–associated mutagenesis; in addition, consistent with previous observations, acquired alterations were observed in the mismatch repair genes for both tumors (Fig 3).27-29 The sampled tumor in both patients increased from G1 to G3.
Fig 3.

Post-treatment samples with high mutational burden after exposure to alkylating agents, as well as acquisition of mutations in the mismatch repair genes. (A) Patient received dacarbazine. (B) Patient received temozolomide.
Germline Genetic Testing
Germline testing for 76 cancer-associated genes was performed in 88 patients, 78 in the original prospective cohort and an additional 10 with surgically resected panNETs. Fourteen likely pathogenic germline alterations were identified in known cancer susceptibility genes (16%): three in established high-penetrance genes (MEN1, VHL, and TSC2 [3%]), four in moderate-penetrance genes (CHEK2 in four patients [5%]), and seven in low-penetrance genes (monoallelic MUTYH in four patients and APC I1307K in three patients [8%]). Notably, we identified a germline TSC2 mutation in a 32-year-old patient who did not exhibit the classic clinical features of tuberous sclerosis complex (TSC) and did not meet the criteria for TSC genetic testing; magnetic resonance imaging of the brain identified tubers and radial bands, typical of TSC (Appendix Figure A6). In addition, three pathogenic or likely pathogenic germline alterations were identified in genes with uncertain clinical actionability (RAD50) or in recessive alleles (RECQL4 and FANCC; Table 2).
Table 2.
Pathogenic or Likely Pathogenic Germline Alterations in 76 Cancer Predisposition–Associated Genes in Well-Differentiated Pancreatic Neuroendocrine Tumors

Somatic Actionable Alterations
NGS results were classified according to the OncoKB actionability scale (Table 3). Tumor from 34 patients had mTOR pathway alterations, and 12 of 34 patients (35%) received everolimus. Among these patients, as best therapy response, two of 12 (17%) had tumor regression, six of 12 (50%) had stable disease, and one of 12 (8%) had progressive disease; three of 12 (25%) are still receiving treatment. In the remaining 46 patients without somatic mTOR pathway alterations, 17 of 46 (37%) received everolimus; five of 17 (29%) had tumor regression, seven of 17 (41%) had stable disease, four of 17 (24%) had progressive disease, and there was an unknown response in one patient (lost to follow-up). Results from NGS led to the enrollment of three patients into clinical trials, but with little response noted.
Table 3.
Actionable Alterations Identified in Well-Differentiated Pancreatic Neuroendocrine Tumors by MSK-IMPACT (as classified according to OncoKB)

DISCUSSION
To our knowledge, this is the first study to evaluate the real-time clinical usefulness of NGS for patients with advanced, metastatic panNETs. This study, like others, suggests that the presence of MEN1, DAXX, or ATRX alterations is associated with improved OS in the metastatic setting.5,30 Confirmation of these findings is important; unlike previous investigations, this study included metastatic patients only, and previous studies have demonstrated shortened survival with loss of DAXX or ATRX expression in the nonmetastatic setting.30-32 In addition, we observed that BRAF alterations (found predominantly in post-treatment biopsy specimens) were significantly associated with poor survival; it is possible that the more advanced, progressive, metastatic state of our cohort could explain our identification of BRAF mutations, a well-established marker of aggressive disease in other GI cancers.33 We identified SETD2 alterations with aggressive histopathologic features predominantly in post-treatment biopsy specimens; most of the SETD2 alterations were truncating (gene loss of function). SETD2 alterations have been implicated in the pathogenesis of many cancers (breast, lung, acute lymphoblastic leukemia, renal cell carcinoma, and gliomas), supporting a role for SETD2 as a tumor suppressor across many diseases.34-41 Given our findings, one might consider closer monitoring and/or cytotoxic therapies in patients with SETD2- or BRAF-altered tumors and a higher burden of disease earlier in the treatment course.
Our study identifies pertubations of biologic pathways in metastatic panNETs that were found in previous whole-exome and whole-genome studies evaluating mostly localized panNETs.5,6 We observed alterations largely along three pathways: CRFs, HMTs, and mTOR. Our study clarifies panNET genetics exclusively in the metastatic cohort, the group which gains immediate clinical value from real-time sequencing as we make steps toward precision medicine.
In addition, we found that 16% of patients harbored a germline pathogenic or likely pathogenic alteration in high-, moderate-, or low-penetrance cancer predisposition genes. In looking at those patients found to have mutations in low- or moderate-penetrance genes, it is notable that both CHEK2 and monoallaleic MUTYH were noted in previous germline assessments of patients with panNETs.6 Notably, the patient in our series with a TSC2 germline mutation did not meet the clinical criteria for TSC genetic testing; the identification of this mutation was incidental. As our group and others have shown, TSC2 is an important somatic driver in panNETs, and the occurrence of panNETs has been reported previously in TSC germline mutations.42 However, our report suggests that the burden of TSC2 germline mutations in panNETs may be underestimated because of an absence of classic phenotypic TSC features. Because mutations in MEN1 and TSC2 are two of the most common somatic findings in panNETs, our finding of germline mutations within these two high-penetrance genes demonstrates the importance of these genes in panNET pathogenesis. Although the contribution of the other germline alterations to panNET susceptibility has yet to be elucidated, our data suggest that there is likely a larger-than-anticipated germline contribution in this disease. Given the lack of predictable phenotypic findings in most high- and moderate-risk mutation carriers, combined with the relative rarity of this tumor subtype, consideration of genetic risk assessment for all patients with panNETs is not unreasonable. Such germline findings would have a profound impact on long-term cancer surveillance and cascade genetic testing of at-risk family members.
In a small number of patients, somatic mutational signatures guided therapy. We identified somatic mTOR alterations in a much greater proportion of patients (43%) than described previously, but the presence or absence of an mTOR pathway mutation was not predictive of response or resistance to treatment.8 Our numbers are small, however, so additional work is needed to clarify the role of mTOR pathway alterations in predicting response and/or resistance to targeted mTOR pathway inhibitors.
Our series included only patients with WD tumors, and consistent with previous findings, RB1 alterations were essentially absent and were observed in only one post-treatment sample exposed to chemotherapy.43,44 WD panNETs are often thought of as cancer in slower motion, and we have used our sequencing efforts to help clarify panNET evolutionary patterns over time and through therapy. In the 12 patients who underwent NGS of tumor tissue at multiple time points, we noted increased tumor grade in eight patients (67%), associated with marked genetic evolution after treatment. Most notably, in patients treated with alkylating agents, profound hypermutation was seen, consistent with a mutational signature described previously in malignant gliomas. To our knowledge, we are the first to observe this evolutionary pattern in panNETs.27-29
In conclusion, comprehensive molecular profiling of panNET samples using archival tissue is feasible. In a disease in which the natural history is thought to be indolent, we have demonstrated that the cancer transforms with grade and genetic progression over time and as therapy progresses. The practical usefulness of this analysis remains limited at this time; our data suggest a higher-than-expected germline mutation rate, and consideration of genetic risk assessment for all patients with panNETs is reasonable. In addition, given the heterogeneity of the disease, rapid, readily available, and clinically feasible NGS should be considered in clinical trials to help stratify these patients. Although our efforts represent the largest cohort of panNETs prospectively undergoing NGS, it is important to recognize the limitations of our investigation. Specifically, our numbers are too small to clarify the genetic predictors of response or resistance to commonly used therapies in panNETs, the prognostic effects of the identified genetic alterations, or the patterns of genetic evolution (because only 12 patients underwent sequencing at multiple time points). To clarify these issues, our future efforts will expand to study panNETs genetically at multiple time points during treatment using noninvasive technologies to define the biology of panNET evolution and to potentially guide future treatments for our patients.
ACKNOWLEDGMENT
The authors thank the patients who kindly agreed to participate in this research effort.
Appendix
Fig A1.
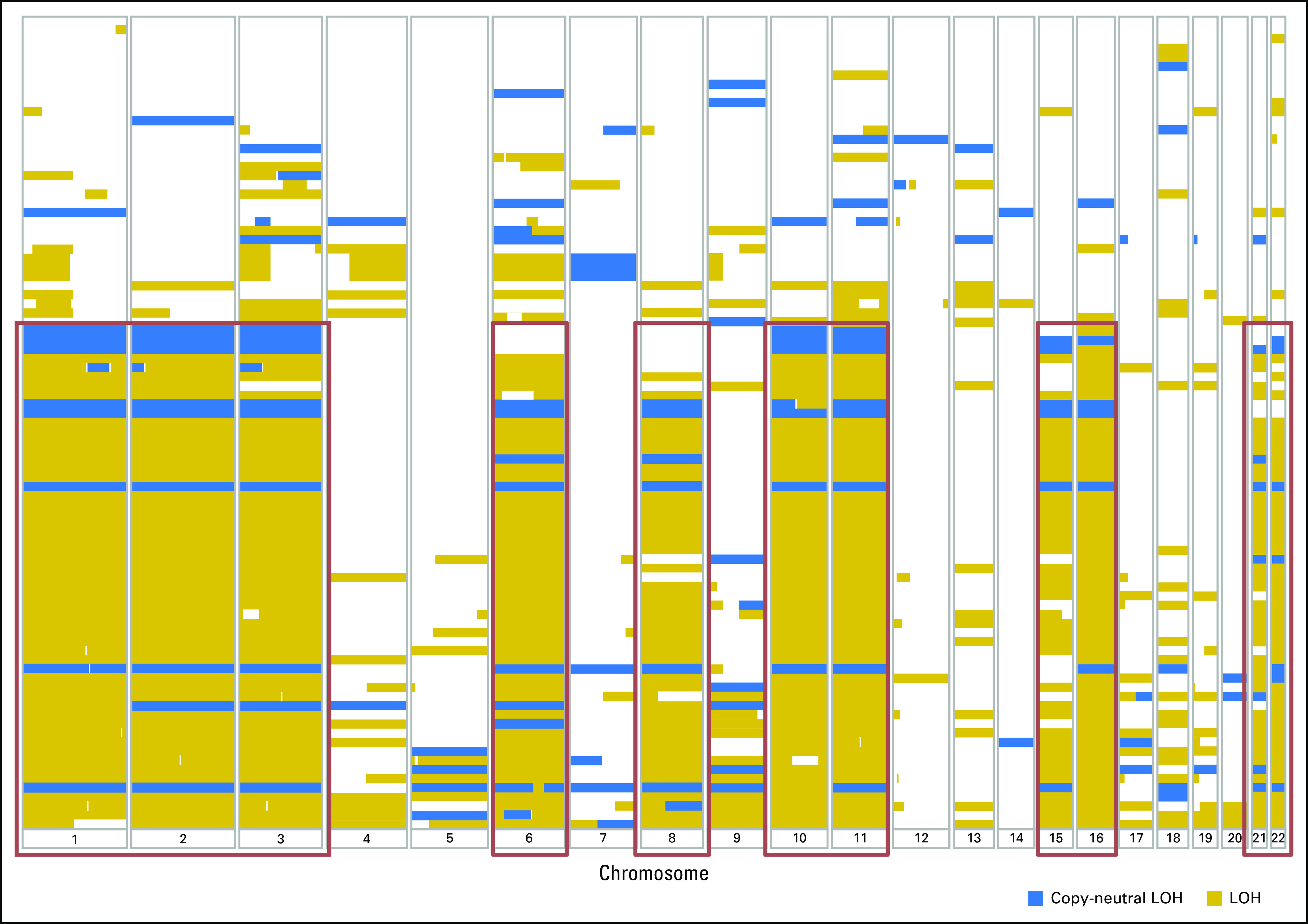
Loss of heterozygosity (LOH) and copy-neutral LOH across all autosomes in well-differentiated pancreatic neuroendocrine tumors. LOH was observed recurrently in chromosomes 1, 2, 3, 6, 8, 10, 11, 15, 16, 21, and 22.
Fig A2.
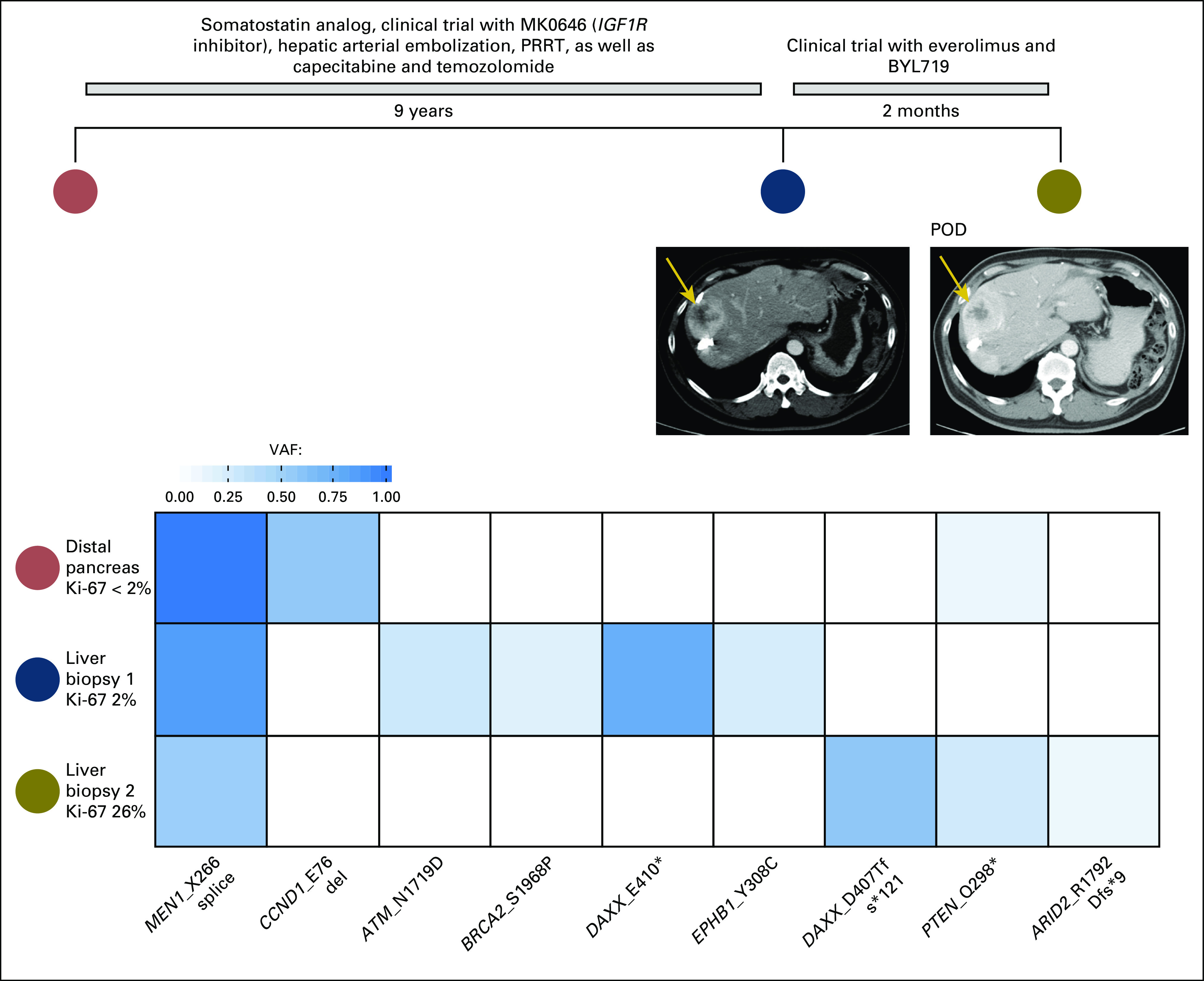
Acquisition of PTEN mutation (Q298*) after treatment with everolimus. POD, progression of disease; PRRT, peptide receptor radionuclide therapy; VAF, variant allele frequency.
Fig A3.
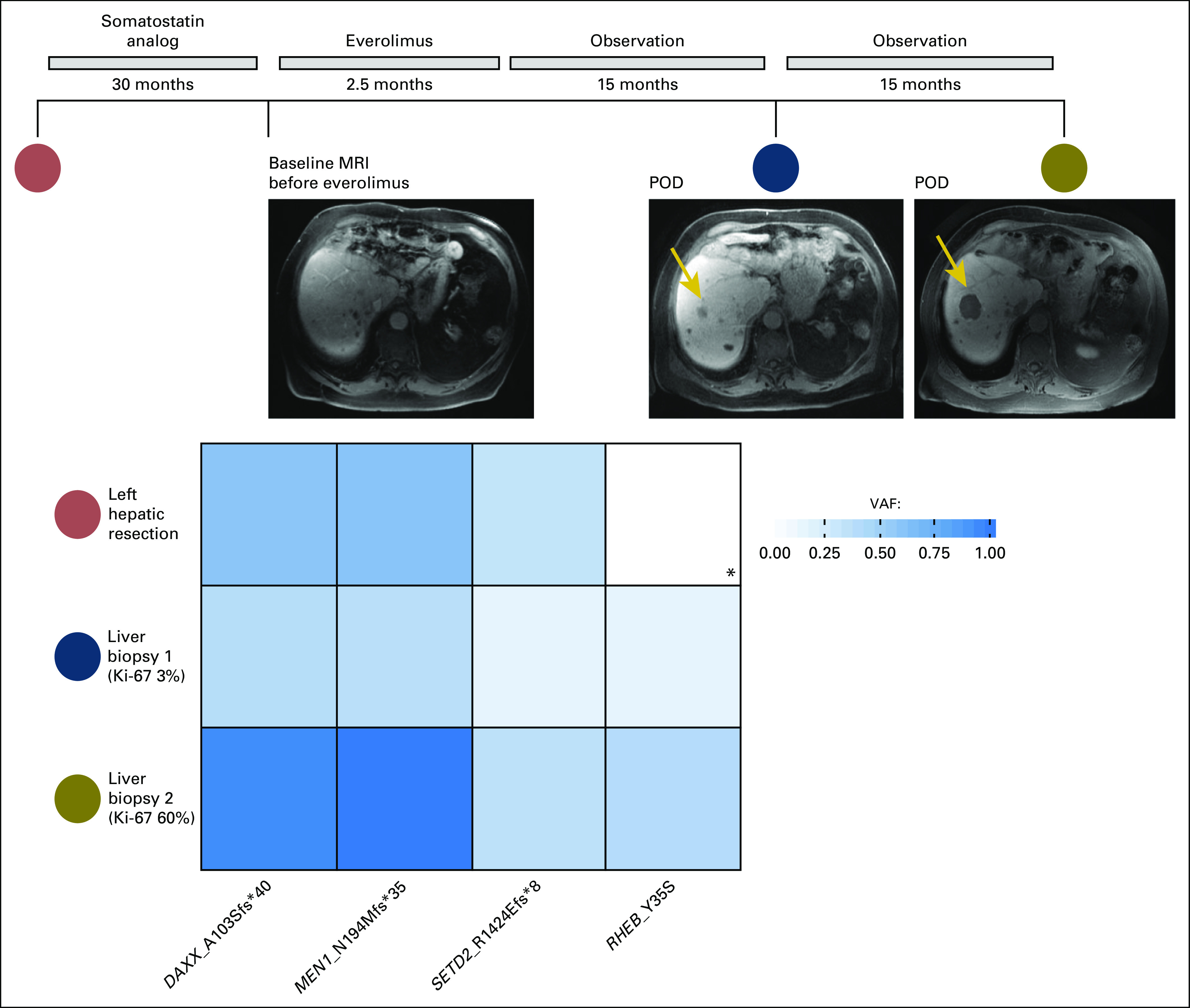
Acquisition of RHEB mutation (Y35S) after treatment with everolimus. (*) Gene was not interrogated in the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets version used for next-generation sequencing of the tumor sample. MRI, magnetic resonance imaging; POD, progression of disease; VAF, variant allele frequency.
Fig A4.

After treatment with everolimus, loss of previously seen IGF2 alteration, and acquisition of TSC2 and AKT2 mutations. MRI, magnetic resonance imaging; POD, progression of disease; VAF, variant allele frequency.
Fig A5.

Acquisition of NRAS mutation after treatment with the BRAF V600E inhibitor vemurafenib. (*) Gene was not interrogated in the Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets version used for next-generation sequencing of the tumor sample. POD, progression of disease; PR, partial response; VAF, variant allele frequency.
Fig A6.

Magnetic resonance imaging of the brain demonstrating bilateral lesions consistent with tubers and radial bands, characteristic of tuberous sclerosis complex.
Footnotes
Presented, in part, as oral abstract presentations at the 2015 North American Neuroendocrine Tumor Society annual meeting (Austin, TX, October 15-17, 2015) and 2016 North American Neuroendocrine Tumor Society annual meeting (Jackson, WY, September 30-October 1, 2016). Presented, in part, as poster presentations at the 2015 Gastrointestinal Cancers Symposium (San Francisco, CA, January 15-17, 2015) and 2016 Gastrointestinal Cancers Symposium (San Francisco, CA, January 21-23, 2016). Presented, in part, in abstract publication format at the 2016 American Society of Clinical Oncology Annual Meeting (Chicago, IL, June 3-7, 2016).
Supported by the Conquer Cancer Foundation of the American Society of Clinical Oncology (2015 Young Investigator Award to N.R.); Cycle for Survival (D.R.-L.); the National Cancer Institute Memorial Sloan Kettering Cancer Core Grant (P30-CA008748); and the Center for Molecular Oncology of Memorial Sloan Kettering Cancer Center.
AUTHOR CONTRIBUTIONS
Conception and design: Nitya Raj, Leonard B. Saltz, David S. Klimstra, Diane Reidy-Lagunes
Financial support: Nitya Raj, Diane Reidy-Lagunes
Administrative support: Nitya Raj, Joanne Chou, Virginia Kelly, Diane Reidy-Lagunes
Provision of study material or patients: Nitya Raj, Leonard B. Saltz, Marc Ladanyi, David S. Klimstra, Diane Reidy-Lagunes
Collection and assembly of data: Nitya Raj, Ronak Shah, Janet Li, Virginia Kelly, Leonard B. Saltz, Marc Ladanyi, Michael F. Berger, David S. Klimstra, Diane Reidy-Lagunes
Data analysis and interpretation: Nitya Raj, Ronak Shah, Zsofia Stadler, Semanti Mukherjee, Joanne Chou, Brian Untch, Janet Li, Muyinat Osoba, Leonard B. Saltz, Diana Mandelker, Marc Ladanyi, Michael F. Berger, David S. Klimstra, Diane Reidy-Lagunes
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Nitya Raj
Research Funding: Novartis (Inst)
Ronak Shah
No relationship to disclose
Zsofia Stadler
Consulting or Advisory Role: Allergan (I); Genentech (I), Regeneron Pharmaceuticals (I), Optos (I), Adverum (I)
Semanti Mukherjee
Employment: Regeneron Pharmaceuticals
Stock and Other Ownership Interests: Regeneron Pharmaceuticals
Research Funding: Regeneron Pharmaceuticals
Joanne Chou
No relationship to disclose
Brian Untch
No relationship to disclose
Janet Li
Research Funding: Eisai (Inst)
Virginia Kelly
No relationship to disclose
Muyinat Osoba
No relationship to disclose
Leonard B. Saltz
Consulting or Advisory Role: McNeil PPC (I)
Research Funding: Taiho Pharmaceutical
Diana Mandelker
No relationship to disclose
Marc Ladanyi
Honoraria: Merck (I)
Consulting or Advisory Role: NCCN/Boehringer Ingelheim Afatinib Targeted Therapy Advisory Committee, National Comprehensive Cancer Network/AstraZeneca Tagrisso Request for Proposal Advisory Committee
Research Funding: Loxo (Inst)
Michael F. Berger
Research Funding: Illumina
David S. Klimstra
Stock and Other Ownership Interests: PAIGE.AI
Consulting or Advisory Role: Wren Laboratories, Ipsen
Diane Reidy-Lagunes
Honoraria: Novartis
Consulting or Advisory Role: Ipsen, Pfizer, Novartis
Research Funding: Novartis
REFERENCES
- 1.Dumlu EG, Karakoç D, Özdemir A: Nonfunctional pancreatic neuroendocrine tumors: Advances in diagnosis, management, and controversies. Int Surg 100:1089-1097, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Metz DC, Jensen RT: Gastrointestinal neuroendocrine tumors: Pancreatic endocrine tumors. Gastroenterology 135:1469-1492, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dasari A, Shen C, Halperin D, et al. : Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol 3:1335-1342, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao JC, Hassan M, Phan A, et al. : One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 26:3063-3072, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Jiao Y, Shi C, Edil BH, et al. : DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331:1199-1203, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scarpa A, Chang DK, Nones K, et al. : Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543:65-71, 2017 [DOI] [PubMed] [Google Scholar]
- 7.Jensen RT, Berna MJ, Bingham DB, et al. : Inherited pancreatic endocrine tumor syndromes: Advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 113:1807-1843, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao JC, Shah MH, Ito T, et al. : Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 364:514-523, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raymond E, Dahan L, Raoul JL, et al. : Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med 364:501-513, 2011 [DOI] [PubMed] [Google Scholar]
- 10.Kulke MH, Lenz HJ, Meropol NJ, et al. : Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 26:3403-3410, 2008 [DOI] [PubMed] [Google Scholar]
- 11.Yao JC, Phan AT, Chang DZ, et al. : Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: Results of a phase II study. J Clin Oncol 26:4311-4318, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yao JC, Lombard-Bohas C, Baudin E, et al. : Daily oral everolimus activity in patients with metastatic pancreatic neuroendocrine tumors after failure of cytotoxic chemotherapy: A phase II trial. J Clin Oncol 28:69-76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strosberg JR, Fine RL, Choi J, et al. : First-line chemotherapy with capecitabine and temozolomide in patients with metastatic pancreatic endocrine carcinomas. Cancer 117:268-275, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cheng DT, Mitchell TN, Zehir A, et al. : Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn 17:251-264, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gnirke A, Melnikov A, Maguire J, et al. : Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27:182-189, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagle N, Emery C, Berger MF, et al. : Dissecting therapeutic resistance to RAF inhibition in melanoma by tumor genomic profiling. J Clin Oncol 29:3085-3096, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wagle N, Berger MF, Davis MJ, et al. : High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov 2:82-93, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakravarty D, Gao J, Phillips SM, et al. : OncoKB: A precision oncology knowledge base. JCO Precis Oncol 2017:1-16, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng DT, Prasad M, Chekaluk Y, et al. : Comprehensive detection of germline variants by MSK-IMPACT, a clinical diagnostic platform for solid tumor molecular oncology and concurrent cancer predisposition testing. BMC Med Genomics 10:33, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandelker D, Zhang L, Kemel Y, et al. : Mutation detection in patients with advanced cancer by universal sequencing of cancer-related genes in tumor and normal DNA vs guideline-based germline testing. JAMA 318:825-835, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rahman N: Realizing the promise of cancer predisposition genes. Nature 505:302-308, 2014 [Erratum: Nature 510:175, 2014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kalia SS, Adelman K, Bale SJ, et al. : Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet Med 19:249-255, 2017 [DOI] [PubMed] [Google Scholar]
- 23.Shen R, Seshan VE: FACETS: Allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res 44:e131, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heller G, Venkatraman ES: Resampling procedures to compare two survival distributions in the presence of right-censored data. Biometrics 52:1204-1213, 1996 [Google Scholar]
- 25.Chang MT, Asthana S, Gao SP, et al. : Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 34:155-163, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hyman DM, Puzanov I, Subbiah V, et al. : Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 373:726-736, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Johnson BE, Mazor T, Hong C, et al. : Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343:189-193, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosales RA, Drummond RD, Valieris R, et al. : signeR: An empirical Bayesian approach to mutational signature discovery. Bioinformatics 33:8-16, 2017 [DOI] [PubMed] [Google Scholar]
- 29.van Thuijl HF, Mazor T, Johnson BE, et al. : Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol 129:597-607, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JY, Brosnan-Cashman JA, An S, et al. : Alternative lengthening of telomeres in primary pancreatic neuroendocrine tumors is associated with aggressive clinical behavior and poor survival. Clin Cancer Res 23:1598-1606, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singhi AD, Liu TC, Roncaioli JL, et al. : Alternative lengthening of telomeres and loss of DAXX/ATRX expression predicts metastatic disease and poor survival in patients with pancreatic neuroendocrine tumors. Clin Cancer Res 23:600-609, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marinoni I, Kurrer AS, Vassella E, et al. : Loss of DAXX and ATRX are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology 146:453-460, 2014 [DOI] [PubMed] [Google Scholar]
- 33.Yaeger R, Saltz L: BRAF mutations in colorectal cancer: Clinical relevance and role in targeted therapy. J Natl Compr Canc Netw 10:1456-1458, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piva F, Santoni M, Matrana MR, et al. : BAP1, PBRM1 and SETD2 in clear-cell renal cell carcinoma: Molecular diagnostics and possible targets for personalized therapies. Expert Rev Mol Diagn 15:1201-1210, 2015 [DOI] [PubMed] [Google Scholar]
- 35.Fontebasso AM, Schwartzentruber J, Khuong-Quang DA, et al. : Mutations in SETD2 and genes affecting histone H3K36 methylation target hemispheric high-grade gliomas. Acta Neuropathol 125:659-669, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu L, Guo R, Zhang X, et al. : Loss of SETD2, but not H3K36me3, correlates with aggressive clinicopathological features of clear cell renal cell carcinoma patients. Biosci Trends 11:214-220, 2017 [DOI] [PubMed] [Google Scholar]
- 37.Mar BG, Bullinger LB, McLean KM, et al. : Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun 5:3469, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Al Sarakbi W, Sasi W, Jiang WG, et al. : The mRNA expression of SETD2 in human breast cancer: Correlation with clinico-pathological parameters. BMC Cancer 9:290, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfister SX, Ahrabi S, Zalmas LP, et al. : SETD2-dependent histone H3K36 trimethylation is required for homologous recombination repair and genome stability. Cell Reports 7:2006-2018, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walter DM, Venancio OS, Buza EL, et al. : Systematic in vivo inactivation of chromatin-regulating enzymes identifies Setd2 as a potent tumor suppressor in lung adenocarcinoma. Cancer Res 77:1719-1729, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hakimi AA, Ostrovnaya I, Reva B, et al. : Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD2: A report by MSKCC and the KIRC TCGA research network. Clin Cancer Res 19:3259-3267, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larson AM, Hedgire SS, Deshpande V, et al. : Pancreatic neuroendocrine tumors in patients with tuberous sclerosis complex. Clin Genet 82:558-563, 2012 [DOI] [PubMed] [Google Scholar]
- 43.Tang LH, Basturk O, Sue JJ, et al. : A practical approach to the classification of WHO grade 3 (G3) well-differentiated neuroendocrine tumor (WD-NET) and poorly differentiated neuroendocrine carcinoma (PD-NEC) of the pancreas. Am J Surg Pathol 40:1192-1202, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yachida S, Vakiani E, White CM, et al. : Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol 36:173-184, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]