

Abstract
Parkinson´s disease (PD) is a disorder with highly variable clinical phenotype. The identification of genetic variants modifying age at onset and other traits is of great interest, since it may provide insight into disease mechanisms and potential therapeutic targets. A variant in the DNM3 gene (rs2421947) has been reported as a genetic modifier of age at onset in LRRK2-associated PD. To test the possible effect of genetic variation in DNM3 on age at onset in idiopathic PD, we examined rs2421947 in a total of 5918 PD patients from seven datasets. We also assessed the potential effect of all common variants in the DNM3 locus. There was no significant association between rs2421947 and age at onset in any of the individual studies. Meta-analysis of the seven studies was non-significant and the between-study heterogeneity was minimal. No other common variants within the DNM3 locus affected age at onset. In conclusion, we find no evidence of an association between DNM3 variants and age at onset in idiopathic PD.
Keywords: Parkinson’s disease, age at onset, DNM3
1. Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder with a complex etiology. A small proportion of PD patients have a monogenic form of the disease with highly penetrant mutations following an autosomal dominant or recessive inheritance pattern. However, the majority of PD cases are idiopathic, presumably caused by the combined action of multiple genetic variants in interplay with epigenetic, environmental and stochastic factors (Lill, 2016). To date, genome-wide association studies (GWASs) have linked more than 40 risk loci to PD susceptibility (Chang et al., 2017).
In addition to affecting the risk of disease development, genetic variants may also affect the clinical phenotype once the disease has manifested. The clinical heterogeneity of PD is characterized by a marked variation in the pattern and progression of motor-, cognitive- and other non-motor symptoms. Both rare monogenic mutations and common genetic variants have been shown to contribute to the clinical heterogeneity of PD (Pihlstrom et al., 2016; Puschmann, 2013; Winder- Rhodes et al., 2013).
There is a broad range of age at onset in PD, varying between debut in early adulthood to patients reaching the 8th and 9th decade of life before onset of motor symptoms. Several studies have investigated the effect of PD risk loci on onset age. Cumulative genetic risk scores calculated across PD risk loci have been shown to have a small, but consistent, effect on age at onset (Escott-Price et al., 2015; Lill et al., 2015; Nalls et al., 2015; Pihlstrom and Toft, 2015). In addition, risk loci having the greatest effect in PD GWAS meta-analysis (GBA, SNCA, MAPT and TMEM175) (Nalls et al., 2014) are reported to individually be associated with age at onset (Brockmann et al., 2013; Davis et al., 2016; Lill et al., 2015; Nalls et al., 2015).
Linking common variation to age at onset represents an interesting step toward a better understanding of how genetics affect PD phenotype. In a recent genome-wide study of genetic modifiers of age at onset in leucine-rich repeat kinase 2 (LRRK2) p.G2019S carriers, a DNM3 haplotype tagged by rs2421947 was identified (Trinh et al., 2016). This LRRK2 mutation is the most frequent genetic cause of PD in many populations, estimated to have a frequency of 1% in white North American and as high as 39% in North African Arab patients (Healy et al., 2008).
LRRK2 mutations cause an autosomal dominant form of PD often segregating in families, while GWASs provide consistent evidence that common variation at this locus also modulates disease risk. Since LRRK2 is part of the genetic background for idiopathic PD, variants that modulate age at onset in LRRK2 parkinsonism may also exert an effect in a much wider group of patients. Herein we report analyses of data from seven studies of PD from Europe and North America to determine associations between the DNM3 rs2421947 variant and age at onset of idiopathic PD. To study the potential effect of other DNM3 variants, we also performed a complete assessment of common variation in the gene locus.
2. Methods
2.1. Study populations
We analyzed individual-level genotypes from seven different datasets. Samples originated from genetic studies of PD from Oslo University Hospital and Mayo Clinic Jacksonville. The remaining five datasets were publicly available and selected due to available individual genotype information in PD patients with a reported age at onset. The following four datasets were accessed from dbGaP: 1) CIDR: Genome Wide Association Study in Familial Parkinson Disease (Accession number: phs000126.v1.p1), 2) Mayo-Perlegen LEAPS (Linked Efforts to Accelerate Parkinson’s Solutions) Collaboration (Accession number: phs000048.v1.p1), 3) National Institute of Neurological Disorders and Stroke (NINDS) Genome-Wide genotyping in Parkinson’s Disease (Accession number: phs000089.v3.p2), 4) Genome-Wide Association Study of Parkinson Disease: Genes and Environment performed by The NeuroGenetics Research Consortium (NGRC) (Accession number: phs000196.v2.p1). The last dataset is made available by the Parkinson’s Progression Markers Initiative (PPMI, http://www.ppmi-info.org).
All patients have been examined by a neurologist. The Oslo and Mayo Clinic patients were diagnosed according to the revised UKPDSBB criteria. A detailed description of inclusion criteria for the other studies have previously been described (Hamza et al., 2010; Maraganore et al., 2005; Nalls et al., 2016; Pankratz et al., 2009; Simon-Sanchez et al., 2009). Age at onset is either reported as age at symptom onset or age at diagnosis. In the current analysis, PD patients reporting other than Caucasian non-Hispanic ethnicity have been excluded along with LRRK2 p.G2019S carriers that were identified by imputation or had previously been genotyped. A large subset of the Oslo and Mayo Clinic patients was sequenced for genes causing Mendelian forms of PD and mutation carriers were excluded from the analysis. Also, no known carriers of Mendelian PD mutations in the publicly available datasets were included in the analysis. Demographic characteristics are summarized in Table 1. All participants gave written, informed consent. Sample and data collection at each study site was approved by local ethics committees. The study was approved by the Regional Committee for Medical Research Ethics (Oslo, Norway).
Table 1.
Demographic characteristics of study samples
| Study | Population | PD patients | % Male | Age at onset ± SD | Genotyping method |
|---|---|---|---|---|---|
| Oslo | Norway | 472 | 64 | 55.9 ± 11.2 | Illumina Infinium OmniExpress v.1.1. |
| Mayo | USA | 987 | 64 | 65.5 ± 11.8 | Taqman assay |
| CIDR | North America, Europe and Australia | 823 | 59 | 62.0 ± 10.7 | Illumina HumanCNV370 BeadChip |
| LEAPS | USA | 439 | 62 | 60.9 ± 11.1 | Perlegen DNA chip (85k SNP markers) |
| NINDS | USA | 912 | 60 | 58.4 ± 13.2 | Illumina HumanHap550 BeadChip |
| NGRC | USA | 1971 | 68 | 58.4 ± 11.9 | Illumina HumanOmni1_Quad |
| PPMI | USA and Europe | 314 | 68 | 59.7 ± 9.8 | Illumina NeuroX |
CIDR, Center for Inherited Disease Research; LEAPS, Linked Efforts to Accelerate Parkinson’s Solutions; NINDS, National Institute of Neurological Disorders and Stroke; NGRC, NeuroGenetics Research Consortium; PPMI, Parkinson’s Progression Markers Initiative; PD, Parkinson’s disease; SD, Standard Deviation.
2.2. Genotyping and quality control
Mayo Clinic patients were genotyped for rs2421947 using a Taqman assay. A subset of genotypes was validated by Sanger sequencing with complete concordance. For the other studies genome-wide genotypes were available. Oslo samples were genotyped using the Illumina Infinium OmniExpress v.1.1 array. Pre-imputation quality filtering included filtering out variants with genotype rate < 0.95 or Hardy-Weinberg equilibrium p<10−6 and removal of individuals with call rate < 0.95, excess heterozygosity > 4 standard deviations (SD) from mean, evidence of cryptic relatedness or sex-check failure. Population outliers were excluded from analysis after inspection of principal component analysis plot (> 2.5 SD from mean). Details of genotyping methods and data quality assessments for the publicly available GWASs and the PPMI study are described in previous publications (Hamza et al., 2010; Maraganore et al., 2005; Nalls et al., 2016; Pankratz et al., 2009; Simon-Sanchez et al., 2009).
Common, pruned, genotyped variants (minor allele frequency > 0.05 and r2 < 0.5) were used to calculate principal components for each of the six genome-wide datasets. For all genome-wide datasets, imputation was performed using the Michigan imputation server (Das et al., 2016) with reference data from the Haplotype Reference Consortium (McCarthy et al., 2016) setting a quality cutoff of r2>0.3 for variants included in the analysis. A set of common, pruned variants from each imputed dataset was merged to assess cryptic relatedness across studies. Duplicates and related samples were removed. The final sample sets included in analysis after quality control comprise a total of 5918 PD patients (Table 1).
2.3. Statistical analyzes
First we tested all seven studies individually for association between the DNM3 rs2421947 variant and age at onset under an additive linear regression model. In an alternative binary analysis, age at onset was dichotomized by the median onset calculated across all seven datasets (61 years of age) and logistic regression was used to test for association with the DNM3 rs2421947 variant within each dataset. In both regression analyses, sex and the first five principal components were used as covariates in the genome-wide datasets, while sex was the single covariate in analysis of the Mayo Clinic study. Association analyses were performed in PLINK (https://www.cog-genomics.org/plink/1.9/) (Chang et al., 2015). Inverse-variance, fixed-effects meta-analysis of the seven studies was conducted using GWAMA (Magi and Morris, 2010). Between-study heterogeneity was assessed using Cochran’s Q test and Higgins’s I statistic. Due to the increased burden of recessive disease-causing mutations in early-onset PD (Puschmann, 2013), we repeated the linear regression analysis excluding all patients with an age at onset < 40 years of age.
Next, common variation within DNM3 as well as 100kb upstream and downstream of the gene was analyzed by linear regression in the six genome-wide datasets. Variants with a minor allele frequency below 0.01 in each dataset were excluded from analysis. Association analysis, both within the individual datasets and meta-analysis were performed as described for DNM3 rs2421947. To estimate the degree of multiple testing, we generated a combined, pruned dataset using a cutoff of LD > r2=0.5, leaving 226 independent variants. Adjusting for 226 independent tests by Bonferroni correction, a p-value < 0.0002 was considered significant. Power calculations were performed with the function pwr.f2.test in the R package pwr (version 1.2–1; https://cran.r-project.org/web/packages/pwr/index.html).
3. Results
We found no significant association between rs2421947 and age at onset in any of the individual studies, both when analyzed as a quantitative trait and in the alternative binary analysis. The frequency of the alternative allele G of rs2421947 was similar in all the seven studies, varying between 54% and 56%. Meta-analysis of the seven studies analyzed as a quantitative trait was non-significant (p = 0.55) and the between-study heterogeneity was minimal (I2= 0%, p=0.77). Results from linear regression analysis of the individual studies and meta-analysis are shown in Figure 1. We obtained similar results when we excluded patients with an age at onset < 40 years from the analysis. Meta-analysis of age at onset as a binary trait was also nonsignificant (OR=1.03, 95% CI=0.96 – 1.11, p=0.44) and between-study heterogeneity was minimal (I2=0%, p=0.85).
Figure 1. Study-specific and meta-analysis results for the DNM3 variant rs2421947.
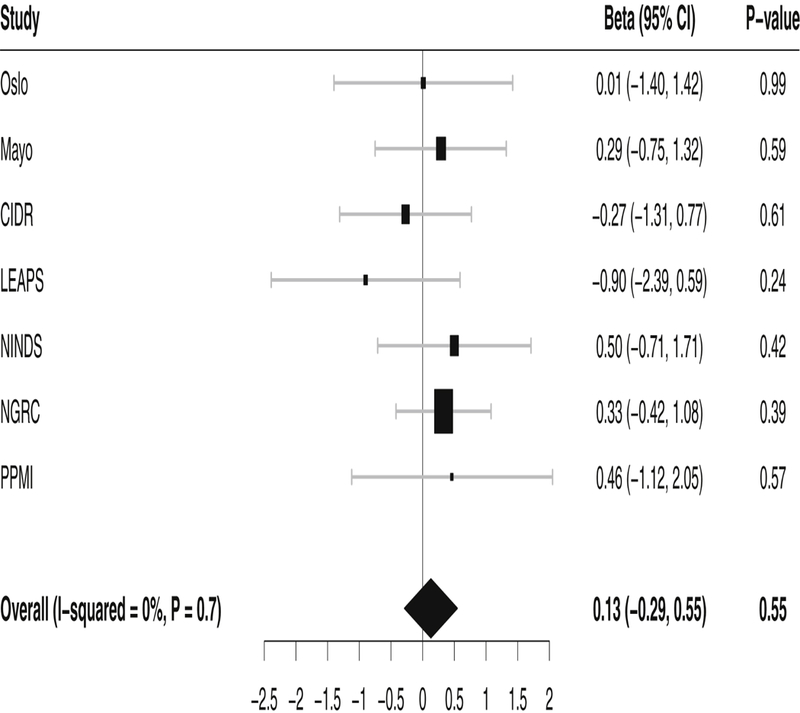
Forest plot showing the effect of rs2421947 on age at onset in idiopathic Parkinson’s disease in individual studies and meta-analysis. The effect size of the G allele is given as a beta estimate with a 95% confidence interval (CI). The size of the squares indicates the size of the datasets. CIDR, Center for Inherited Disease Research; LEAPS, Linked Efforts to Accelerate Parkinson’s Solutions; NINDS, National Institute of Neurological Disorders and Stroke; NGRC, NeuroGenetics Research Consortium; PPMI, Parkinson’s Progression Markers Initiative.
Next, we analyzed all common variants within the DNM3 gene and the flanking genomic region. Variants covered in at least four out of the six analyzed genome-wide datasets were included in the meta-analysis, constituting 1932 variants. None of the meta-analyzed variants had a significant association with age at onset when corrected for multiple testing. Results from the extended DNM3 analysis are provided in Supplementary Table 1. Tests for statistical heterogeneity indicated that heterogeneity was low (I2< 50%) for the vast majority (97%) of the meta-analyzed variants. Studies of the combined impact of PD risk loci on age at onset report a phenotypic variance explained by the calculated genetic risk score of 0.6% and 0.7%, with the signal mostly being driven by two individually age at onset-associated variants (Lill et al., 2015; Nalls et al., 2015). Assuming a variance explained of ~ 0.5% for the tested variant, we have a power of over 99% for the primary analysis of rs2421947 (N=5918) and a power of 89.5% to achieve a p-value of 0.0002 (N=4931) in the extended DNM3 analysis.
The effect of rs2421947 on age at onset was also assessed in the LRRK2 p.G2019S carriers (N=39) that had been excluded from the aforementioned analyses, although this test was limited by a small sample size. Association with age at onset analyzed as a quantitative trait, with sex and dataset as covariables, was nonsignificant for rs2421947, with the trend for direction of effect reversed relative to the original report by Trinh et al. (Trinh et al., 2016) (Effect allele=G, Beta=1.79, 95% CI=−3.31–6.89, p=0.50). Trinh et al. report a correlation between rs2421947 genotype and DNM3 mRNA levels in striatal brain tissue (Trinh et al., 2016). We explored the Genotype-Tissue Expression (GTEx) Portal (version 7; https://www.gtexportal.org/home/) and found that significant expression quantitative trait loci (eQTLs) for DNM3 are reported in cerebellar hemisphere and cerebellum, but not in any of the other brain regions examined by the GTEx project. Interestingly, rs2421947 and variants in high LD are not reported as significant eQTLs for DNM3 in any brain tissue.
4. Discussion
In this study, we found no evidence for a modifying effect of rs2421947 or other common DNM3 variants on age at onset in idiopathic PD. A DNM3 haplotype tagged by rs2421947 was identified by Trinh et al. as a modifier of age at onset in LRRK2 p.G2019S carriers. They reported that the median age at onset of DNM3 GG homozygotes was 12.5 years younger than that of CC homozygotes. This is a large difference in onset age compared to the effect of other variants associated with age at onset in PD, and could be meaningful in the clinical setting. As shown by the 95% confidence interval of our primary analysis, it is highly unlikely that we did not detect an effect altering the onset of PD more than a few months per G allele.
We performed a meta-analysis including a total of 5918 PD patients, and the high number of analyzed individuals is a strength of our study. However, by including cases from different study sites with variations in study design, heterogeneity may be introduced in meta-analysis of the genetic data. We assessed this and found low heterogeneity. We accounted for population substructure within the individual studies by including five eigenvectors in the regression model. The meta-analyzed studies use mostly self-reported symptom onset. Age at onset is subjective and may be prone to recall bias. Nevertheless, the reliability of self- and family-reported age at onset compared to medical records is high and all three methods have been regarded as valid (Reider et al., 2003).
A recent study of LRRK2 p.G2019S carriers in the Spanish population did not find and association between DNM3 rs2421947 and age at onset of PD (Fernandez- Santiago et al., 2018). Linkage patterns vary among populations, and the possibility that disease relevant variation could be tagged by different genetic markers in Europeans/North Americans as compared to the Arab-Berber population studied by Trinh et al. prompted us to extend our analysis to all imputed common variants across the DNM3 locus. The genomic region flanking the DNM3 gene is included in our analysis to cover potential regulatory variants, although this increases the multiple testing burden. On the other hand, regulatory variants affecting DNM3 expression may reside in an even more distal part of the genome not covered in our analysis. eQTL data could be used to identify potential regulatory variants and reduce the number of tests to adjust for. However, the currently available databases are incomplete since eQTLs in addition to depending on tissue and cell-type, also may vary between different physiological conditions (Albert and Kruglyak, 2015).
Despite recent efforts to elucidate the genetic architecture behind age at onset and other clinical characteristics of PD, the vast majority of genetic variation affecting PD phenotypes remains unexplained. Many studies have limited their analysis to known risk loci of PD, while attempts at identifying novel genetic modifiers of age at onset have proven challenging. Genetic modifiers of age at onset may be limited to sub-groups of patients carrying specific mutations or susceptibility variants. Variations in the MAPT gene have been found to be associated with age at onset in PD patients carrying a LRRK2 mutation (Gan-Or et al., 2012; Golub et al., 2009). A recent GWAS of age at onset analyzed PD patients with and without a family history of the disease separately. No significant association was found in those without a family history of PD, while two signals were detected in individuals reporting to have a first or second-degree relative with PD. Both these signals mapped to gene regions that are not known to affect PD risk (Hill-Burns et al., 2016).
Discovering genetic modifiers of phenotype in PD is important, since it may provide insight into disease mechanisms and help in identifying potential therapeutic targets. DNM3 has previously not been identified by GWASs of disease risk or onset age (Chang et al., 2017; Hill-Burns et al., 2016; Latourelle et al., 2009; Nalls et al., 2014). We found no association between common DNM3 variants and age at onset in idiopathic PD, but the possible contribution of rare variants within this genetic locus cannot be excluded. Variability within the DNM3 locus may be a specific modifier of LRRK2 parkinsonism, although this has yet to be replicated in independent studies. DNM3 encodes the protein dynamin-3 which is highly expressed in neurons (Raimondi et al., 2011). The LRRK2 protein has been shown to interact with dynamin-3 and other members of the dynamin GTPase superfamily that regulate membrane dynamics important for endocytosis and mitochondrial morphology (Stafa et al., 2014). Trinh et al. report a correlation between rs2421947 genotype and DNM3 mRNA levels in striatal tissue, but such an association is not observed in the GTEx Portal. There are several methodological differences between these analyses and additional evidence is needed before conclusions regarding rs2421947 as an eQTL for DNM3 in the brain can be drawn.
Further insight into disease mechanisms may be gained by examining complex genetic interactions. A novel epistatic interaction between two genetic variants was reported by a recent study incorporating genetic, molecular and clinical data into models to predict motor progression in PD (Latourelle et al., 2017). Larger patient cohorts with comprehensive characterization of disease phenotype will benefit future studies of how genetics affect clinical heterogeneity.
Supplementary Material
Highlights.
No association between rs2421947 and age at onset in idiopathic Parkinson’s disease
Common genetic variation in the DNM3 locus has no modifying effect on age at onset
Genetic modifier of LRRK2-parkinsonism is not transferable to idiopathic disease
Acknowledgements
The authors thank all study participants, investigators and funding partners for their contribution to the data analyzed in the current publication. The funding organizations had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Victoria Berge-Seidl and Mathias Toft are funded by a grant from the South-Eastern Norway Regional Health Authority. Mathias Toft is also supported by the Research Council of Norway. Lasse Pihlstrøm is supported by the Norwegian Health Association. Mayo Clinic is a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P50 #NS072187 to OR and ZKW), Zbigniew K. Wszolek is also funded by Mayo Clinic Center for Individualized Medicine, Mayo Clinic Neuroscience Focused Research Team (Cecilia and Dan Carmichael Family Foundation, and the James C. and Sarah K. Kennedy Fund for Neurodegenerative Disease Research at Mayo Clinic in Florida), the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch, The Sol Goldman Charitable Trust, and Donald G. and Jodi P. Heeringa. Owen Ross is supported by NINDS R01 NS078086, Department of Defense award W81XWH-16-PRMRP-IIRA, Michael J. Fox Foundation. The analyses presented in the current publication are based on the use of study data downloaded from the dbGaP web site, under dbGaP accession phs000126.v1.p1, phs000048.v1.p1, phs000089.v3.p2 and phs000196.v2.p1. Data was also obtained from the Parkinson’s Progression Markers Initiative (PPMI) database (www.ppmi-info.org/data). For up-to-date information on the study, visit www.ppmi-info.org. PPMI - a public-private partnership - is funded by the Michael J. Fox Foundation for Parkinson’s Research and funding partners including AbbVie, Avid Radiopharmaceuticals, Biogen, BioLegend, Bristol-Myers Squibb, GE Healthcare, Genentech, GlaxoSmithKline, Lilly, Lundbeck, Merck, Meso Scale Discovery, Pfizer, Piramal, Roche, Sanofi Genzyme, Servier, Takeda, Teva and UCB.
Footnotes
Disclosure statement: The authors report no conflicts of interest concerning the research related to the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Albert FW, Kruglyak L, 2015. The role of regulatory variation in complex traits and disease. Nature reviews. Genetics 16(4), 197–212. [DOI] [PubMed] [Google Scholar]
- Brockmann K, Schulte C, Hauser AK, Lichtner P, Huber H, Maetzler W, Berg D, Gasser T, 2013. SNCA: major genetic modifier of age at onset of Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 28(9), 1217–1221. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ, 2015. Second- generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR, 2017. A meta-analysis of genome- wide association studies identifies 17 new Parkinson’s disease risk loci. Nature genetics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, Schlessinger D, Stambolian D, Loh PR, Iacono WG, Swaroop A, Scott LJ, Cucca F, Kronenberg F, Boehnke M, Abecasis GR, Fuchsberger C, 2016. Next-generation genotype imputation service and methods. Nature genetics 48(10), 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AA, Andruska KM, Benitez BA, Racette BA, Perlmutter JS, Cruchaga C, 2016. Variants in GBA, SNCA, and MAPT influence Parkinson disease risk, age at onset, and progression. Neurobiology of aging 37, 209e201–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escott-Price V, Nalls MA, Morris HR, Lubbe S, Brice A, Gasser T, Heutink P, Wood NW, Hardy J, Singleton AB, Williams NM, 2015. Polygenic risk of Parkinson disease is correlated with disease age at onset. Annals of neurology 77(4), 582–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Santiago R, Garrido A, Infante J, Gonzalez-Aramburu I, Sierra M, Fernandez M, Valldeoriola F, Munoz E, Compta Y, Marti MJ, Rios J, Tolosa E, Ezquerra M, 2018. alpha-synuclein (SNCA) but not dynamin 3 (DNM3) influences age at onset of leucine-rich repeat kinase 2 (LRRK2) Parkinson’s disease in Spain. Movement disorders : official journal of the Movement Disorder Society 33(4), 637–641. [DOI] [PubMed] [Google Scholar]
- Gan-Or Z, Bar-Shira A, Mirelman A, Gurevich T, Giladi N, Orr-Urtreger A, 2012. The age at motor symptoms onset in LRRK2-associated Parkinson’s disease is affected by a variation in the MAPT locus: a possible interaction. Journal of molecular neuroscience : MN 46(3), 541–544. [DOI] [PubMed] [Google Scholar]
- Golub Y, Berg D, Calne DB, Pfeiffer RF, Uitti RJ, Stoessl AJ, Wszolek ZK, Farrer MJ, Mueller JC, Gasser T, Fuchs J, 2009. Genetic factors influencing age at onset in LRRK2-linked Parkinson disease. Parkinsonism & related disorders 15(7), 539–541. [DOI] [PubMed] [Google Scholar]
- Hamza TH, Zabetian CP, Tenesa A, Laederach A, Montimurro J, Yearout D, Kay DM, Doheny KF, Paschall J, Pugh E, Kusel VI, Collura R, Roberts J, Griffith A, Samii A, Scott WK, Nutt J, Factor SA, Payami H, 2010. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nature genetics 42(9), 781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S, Brice A, Aasly J, Zabetian CP, Goldwurm S, Ferreira JJ, Tolosa E, Kay DM, Klein C, Williams DR, Marras C, Lang AE, Wszolek ZK, Berciano J, Schapira AH, Lynch T, Bhatia KP, Gasser T, Lees AJ, Wood NW, 2008. Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. The Lancet. Neurology 7(7), 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill-Burns EM, Ross OA, Wissemann WT, Soto-Ortolaza AI, Zareparsi S, Siuda J, Lynch T, Wszolek ZK, Silburn PA, Mellick GD, Ritz B, Scherzer CR, Zabetian CP, Factor SA, Breheny PJ, Payami H, 2016. Identification of genetic modifiers of age-at-onset for familial Parkinson’s disease. Human molecular genetics 25(17), 3849–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latourelle JC, Beste MT, Hadzi TC, Miller RE, Oppenheim JN, Valko MP, Wuest DM, Church BW, Khalil IG, Hayete B, Venuto CS, 2017. Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: a longitudinal cohort study and validation. The Lancet. Neurology 16(11), 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latourelle JC, Pankratz N, Dumitriu A, Wilk JB, Goldwurm S, Pezzoli G, Mariani CB, DeStefano AL, Halter C, Gusella JF, Nichols WC, Myers RH, Foroud T, 2009. Genomewide association study for onset age in Parkinson disease. BMC medical genetics 10, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lill CM, 2016. Genetics of Parkinson’s disease. Molecular and cellular probes 30(6), 386–396. [DOI] [PubMed] [Google Scholar]
- Lill CM, Hansen J, Olsen JH, Binder H, Ritz B, Bertram L, 2015. Impact of Parkinson’s disease risk loci on age at onset. Movement disorders : official journal of the Movement Disorder Society 30(6), 847–850. [DOI] [PubMed] [Google Scholar]
- Magi R, Morris AP, 2010. GWAMA: software for genome-wide association metaanalysis. BMC bioinformatics 11, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PV, Frazer KA, Cox DR, Ballinger DG, 2005. High-resolution whole- genome association study of Parkinson disease. Am J Hum Genet 77(5), 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, Luo Y, Sidore C, Kwong A, Timpson N, Koskinen S, Vrieze S, Scott LJ, Zhang H, Mahajan A, Veldink J, Peters U, Pato C, van Duijn CM, Gillies CE, Gandin I, Mezzavilla M, Gilly A, Cocca M, Traglia M, Angius A, Barrett JC, Boomsma D, Branham K, Breen G, Brummett CM, Busonero F, Campbell H, Chan A, Chen S, Chew E, Collins FS, Corbin LJ, Smith GD, Dedoussis G, Dorr M, Farmaki AE, Ferrucci L, Forer L, Fraser RM, Gabriel S, Levy S, Groop L, Harrison T, Hattersley A, Holmen OL, Hveem K, Kretzler M, Lee JC, McGue M, Meitinger T, Melzer D, Min JL, Mohlke KL, Vincent JB, Nauck M, Nickerson D, Palotie A, Pato M, Pirastu N, McInnis M, Richards JB, Sala C, Salomaa V, Schlessinger D, Schoenherr S, Slagboom PE, Small K, Spector T, Stambolian D, Tuke M, Tuomilehto J, Van den Berg LH, Van Rheenen W, Volker U, Wijmenga C, Toniolo D, Zeggini E, Gasparini P, Sampson MG, Wilson JF, Frayling T, de Bakker PI, Swertz MA, McCarroll S, Kooperberg C, Dekker A, Altshuler D, Willer C, Iacono W, Ripatti S, Soranzo N, Walter K, Swaroop A, Cucca F, Anderson CA, Myers RM, Boehnke M, McCarthy MI, Durbin R, 2016. A reference panel of 64,976 haplotypes for genotype imputation. Nature genetics 48(10), 1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Escott-Price V, Williams NM, Lubbe S, Keller MF, Morris HR, Singleton AB, 2015. Genetic risk and age in Parkinson’s disease: Continuum not stratum. Movement disorders : official journal of the Movement Disorder Society 30(6), 850–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Keller MF, Hernandez DG, Chen L, Stone DJ, Singleton AB, 2016. Baseline genetic associations in the Parkinson’s Progression Markers Initiative (PPMI). Movement disorders : official journal of the Movement Disorder Society 31(1), 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M, DeStefano AL, Kara E, Bras J, Sharma M, Schulte C, Keller MF, Arepalli S, Letson C, Edsall C, Stefansson H, Liu X, Pliner H, Lee JH, Cheng R, Ikram MA, Ioannidis JP, Hadjigeorgiou GM, Bis JC, Martinez M, Perlmutter JS, Goate A, Marder K, Fiske B, Sutherland M, Xiromerisiou G, Myers RH, Clark LN, Stefansson K, Hardy JA, Heutink P, Chen H, Wood NW, Houlden H, Payami H, Brice A, Scott WK, Gasser T, Bertram L, Eriksson N, Foroud T, Singleton AB, 2014. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nature genetics 46(9), 989–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankratz N, Wilk JB, Latourelle JC, DeStefano AL, Halter C, Pugh EW, Doheny KF, Gusella JF, Nichols WC, Foroud T, Myers RH, 2009. Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124(6), 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pihlstrom L, Morset KR, Grimstad E, Vitelli V, Toft M, 2016. A cumulative genetic risk score predicts progression in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 31(4), 487–490. [DOI] [PubMed] [Google Scholar]
- Pihlstrom L, Toft M, 2015. Cumulative genetic risk and age at onset in Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 30(12), 1712–1713. [DOI] [PubMed] [Google Scholar]
- Puschmann A, 2013. Monogenic Parkinson’s disease and parkinsonism: clinical phenotypes and frequencies of known mutations. Parkinsonism & related disorders 19(4), 407–415. [DOI] [PubMed] [Google Scholar]
- Raimondi A, Ferguson SM, Lou X, Armbruster M, Paradise S, Giovedi S, Messa M, Kono N, Takasaki J, Cappello V, O’Toole E, Ryan TA, De Camilli P, 2011. Overlapping role of dynamin isoforms in synaptic vesicle endocytosis. Neuron 70(6), 1100–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reider CR, Halter CA, Castelluccio PF, Oakes D, Nichols WC, Foroud T, 2003. Reliability of reported age at onset for Parkinson’s disease. Movement disorders : official journal of the Movement Disorder Society 18(3), 275–279. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, Schulte C, Bras JM, Sharma M, Gibbs JR, Berg D, Paisan-Ruiz C, Lichtner P, Scholz SW, Hernandez DG, Kruger R, Federoff M, Klein C, Goate A, Perlmutter J, Bonin M, Nalls MA, Illig T, Gieger C, Houlden H, Steffens M, Okun MS, Racette BA, Cookson MR, Foote KD, Fernandez HH, Traynor BJ, Schreiber S, Arepalli S, Zonozi R, Gwinn K, van der Brug M, Lopez G, Chanock SJ, Schatzkin A, Park Y, Hollenbeck A, Gao J, Huang X, Wood NW, Lorenz D, Deuschl G, Chen H, Riess O, Hardy JA, Singleton AB, Gasser T, 2009. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nature genetics 41(12), 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafa K, Tsika E, Moser R, Musso A, Glauser L, Jones A, Biskup S, Xiong Y, Bandopadhyay R, Dawson VL, Dawson TM, Moore DJ, 2014. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Human molecular genetics 23(8), 2055–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Gustavsson EK, Vilarino-Guell C, Bortnick S, Latourelle J, McKenzie MB, Tu CS, Nosova E, Khinda J, Milnerwood A, Lesage S, Brice A, Tazir M, Aasly JO, Parkkinen L, Haytural H, Foroud T, Myers RH, Sassi SB, Hentati E, Nabli F, Farhat E, Amouri R, Hentati F, Farrer MJ, 2016. DNM3 and genetic modifiers of age of onset in LRRK2 Gly2019Ser parkinsonism: a genome- wide linkage and association study. The Lancet. Neurology 15(12), 1248–1256. [DOI] [PubMed] [Google Scholar]
- Winder-Rhodes SE, Evans JR, Ban M, Mason SL, Williams-Gray CH, Foltynie T, Duran R, Mencacci NE, Sawcer SJ, Barker RA, 2013. Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain : a journal of neurology 136(Pt 2), 392–399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.