

SUMMARY
Salmonella enterica serovar (S.) Typhi is an extraintestinal pathogen that evolved from Salmonella serovars causing gastrointestinal disease. Compared to non-typhoidal Salmonella serovars, the genomes of typhoidal serovars contain various loss-of-function mutations. However, the contribution of these genetic differences to this shift in pathogen ecology remains unknown. We show that the ydiQRSTD operon, which is deleted in S. Typhi, enables S. Typhimurium to utilize microbiota-derived butyrate during gastrointestinal disease. Unexpectedly, genetic ablation of butyrate utilization reduces S. Typhimurium epithelial invasion and attenuates intestinal inflammation. Deletion of ydiD renders S. Typhimurium sensitive to butyrate-mediated repression of invasion gene expression. Combined with the gain of virulence-associated (Vi) capsular polysaccharide and loss of very-long O antigen chains, two features characteristic of S. Typhi, genetic ablation of butyrate utilization abrogates S. Typhimurium-induced intestinal inflammation. Thus, the transition from a gastrointestinal to extraintestinal pathogen involved discrete genetic changes, providing insights into pathogen evolution and emergence.
Graphical Abstract
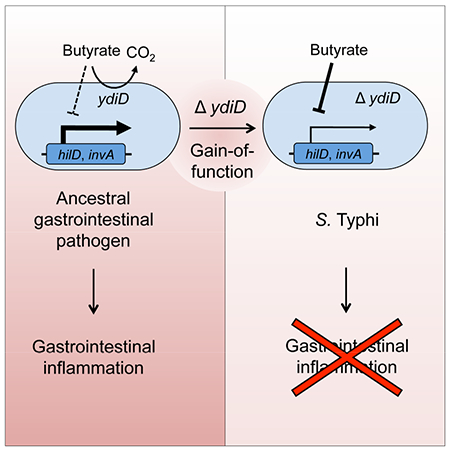
eTOC blurb
Bronner et al. show that genes enabling Salmonella enterica serovar (S.) Typhimurium to utilize microbiota-derived butyrate are deleted in the genome of the closely related S. Typhi, thereby moderating intestinal inflammation induced by the pathogen. Thus, gene loss can aid in the transition from gastrointestinal to extraintestinal pathogens.
INTRODUCTION
From the standpoint of human disease, the genus Salmonella is traditionally divided into zoonotic non-typhoidal Salmonella serovars and human-adapted typhoidal Salmonella serovars. After an incubation period of less than 24 hours, non-typhoidal Salmonella serovars cause a localized gastrointestinal disease in patients with an intact immune system (Glynn and Palmer, 1992). In contrast, typhoidal Salmonella serovars cause an extraintestinal disease after an average incubation period of two weeks (Olsen et al., 2003). This extraintestinal disease is referred to as typhoid fever, when associated with Salmonella enterica serovar (S.) Typhi, or as paratyphoid fever, when caused by S. Paratyphi A (Crump et al., 2004; Kirk et al., 2015). The long incubation period of typhoid fever suggests that unlike non-typhoidal Salmonella serovars, S. Typhi limits severe intestinal inflammation early after infection. One genetic factor implicated in moderating intestinal inflammation during typhoid fever is the virulence-associated (Vi) capsular polysaccharide, which was acquired by S. Typhi through horizontal gene transfer of the viaB locus (Haneda et al., 2009; Wilson et al., 2008; Wilson et al., 2011). However, the viaB locus is not present in S. Paratyphi A, suggesting that additional genetic changes curbing a rapid onset of intestinal inflammation remain to be identified in typhoidal Salmonella serovars.
A virulence factor crucial for inducing severe intestinal inflammation during infection with the non-typhoidal S. Typhimurium is the invasion-associated type III secretion system (T3SS-1) (Tsolis et al., 1999). Expression of genes encoding T3SS-1 can be modulated by short-chain fatty acids (SCFAs) both in vitro and in the gastrointestinal tract (Garner et al., 2009; Lawhon et al., 2002). SCFAs are fermentation end products of the gut microbiota that can trigger acid stress responses as well as modulate virulence factor expression in enteric pathogens (Altier, 2005). Butyrate and propionate have been shown to reduce expression of Salmonella invasion genes, whereas acetate enhances invasion gene expression (Gantois et al., 2006; Lawhon et al., 2002). Mixtures representing colonic SCFA concentrations, which contain higher proportions of butyrate and propionate, exhibit a greater inhibitory effect than mixtures representing ileal SCFA concentrations, which contain a higher proportion of acetate (Altier, 2005). Since typhoidal and non-typhoidal Salmonella serovars encounter similar SCFA concentrations during infection, this regulatory mechanism is not a likely candidate for explaining why only the latter pathogens cause severe intestinal inflammation after an incubation period of less than 24 hours.
An overt genetic difference between typhoidal and non-typhoidal Salmonella serovars is the presence in the former genomes of significantly larger numbers of loss-of-function mutations (McClelland et al., 2004; Parkhill et al., 2001). The majority of functions lost by deletion or gene disruption (pseudogene formation) in genomes of typhoidal Salmonella serovars are involved in central anaerobic metabolism, which is required for expansion of non-typhoidal Salmonella serovars in the gut lumen during gastrointestinal disease (Nuccio and Baumler, 2014). However, in many cases the predicted functions of metabolic pathways lost in typhoidal Salmonella serovars remain to be established experimentally using an animal model. For example, both the S. Typhi and S. Paratyphi A genomes carry a deletion of the ydiQRSTD operon, but this deletion is not present in other Salmonella genomes (Nuccio and Baumler, 2014). Sequence homology predicts that the S. Typhimurium ydiQRSTD operon encodes a pathway for β-oxidation of fatty acids in the presence of nitrate (Campbell et al., 2003), an electron acceptor becoming available in the intestinal lumen during gastrointestinal disease (Lopez et al., 2015; Lopez et al., 2012). This predicted role suggests S. Typhi and S. Paratyphi A lost the ydiQRSTD operon because they do not cause gastrointestinal disease and hence no longer need metabolic pathways that enhance bacterial growth in the inflamed intestine. Here we characterize the role of the ydiQRSTD operon during infection to illuminate its role in the evolutionary path from a gastrointestinal to an extraintestinal pathogen.
RESULTS
S. Typhimurium utilizes microbiota-derived butyrate using anaerobic β-oxidation
To investigate whether the anaerobic pathway for β-oxidation of fatty acids conferred a fitness advantage during growth in the intestinal lumen, we compared the fitness of the S. Typhimurium wild type (IR715) and a ydiD mutant (DNB4) by infecting genetically resistant (CBA) mice with a 1:1 mixture of both strains. The S. Typhimurium wild type was recovered in significantly higher numbers (P < 0.001) from colon contents than the ydiD mutant (Fig. 1A), suggesting that anaerobic β-oxidation conferred a fitness advantage during pathogen growth in the lumen. Interestingly, in germ-free (Swiss Webster) mice, wild type and ydiD mutant were recovered in similar numbers from colon contents; however, when pre-colonized with a community of 17 human Clostridia isolates, anaerobic β-oxidation conferred a significant fitness advantage (P < 0.001) (Fig. 1B). Clostridia are the main producers in gut-associated microbial communities of the SCFA butyrate (Louis and Flint, 2009; Vital et al., 2014). When germ-free mice infected with a 1:1 mixture of the S. Typhimurium wild type and a ydiD mutant received butyrate supplementation, anaerobic β-oxidation conferred a significant fitness advantage (P < 0.01) (Fig. 1B). The finding that deletion of ydiD generated a phenotype in germ-free mice only after supplementation with Clostridia or with butyrate supported the idea that the anaerobic β-oxidation pathway is required in vivo for utilization of microbiota-derived butyrate, but not for utilization of host or diet derived fatty acids. Consistent with the idea that the anaerobic β-oxidation pathway is required for butyrate utilization, the S. Typhimurium wild type consumed butyrate under anaerobic conditions in vitro, while butyrate consumption was not observed with a ydiD mutant (Fig. 1C). Butyrate consumption in vitro could be restored in a ydiD mutant by introducing the cloned ydiD gene on a plasmid (pYDID).
Figure 1: S. Typhimurium uses anaerobic β-oxidation to utilize microbiota-derived butyrate.
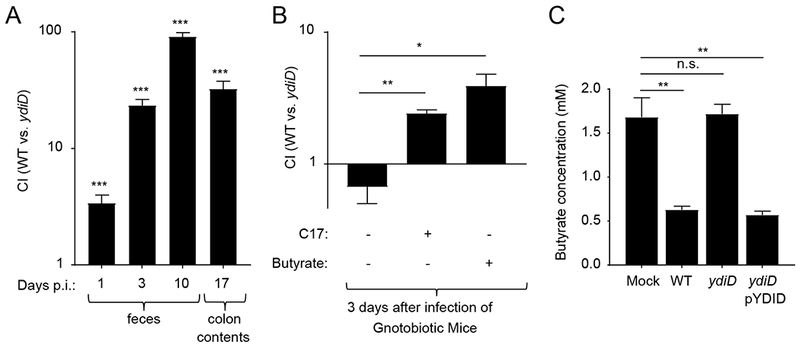
(A) CBA mice (n = 6) were intragastrically infected with an equal mixture of S. Typhimurium IR715 wild type (WT) and a ydiD mutant. The competitive Index (CI) was determined on days 1, 3, 10, and 17 after infection (p.i.) in fecal and colon contents. (B) Swiss-Webster germ-free mice (n = 6) were mock treated, supplemented with 17 human Clostridia isolates or with butyrate (150 mM in drinking water) and then infected intragastrically with a 1:1 mixture of the S. Typhimurium wild type (WT) and a ydiD mutant. (C) Medium containing 3mM butyrate was inoculated with the indicated bacterial strains, incubated anaerobically for 8 hours and butyrate concentrations were measured using liquid chromatography-mass spectrometry. Error bars represent geometric mean ± SE. *, ** and *** represents p-value <0.01, 0.001 and <0.0001, respectively.
Genetic ablation of butyrate utilization attenuates the severity of colitis
Next, we wanted to further characterize the ydiD mutant by infecting mice with individual bacterial strains. In genetically resistant (CBA) mice, S. Typhimurium causes severe colitis within 10 days after infection, as indicated by measuring the inflammatory marker lipocalin-2 in feces by ELISA (Rivera-Chavez et al., 2016). Remarkably, deletion of ydiD attenuated the ability of S. Typhimurium to trigger intestinal inflammation, as indicated by significantly (P < 0.0001) lower lipocalin-2 levels in feces of CBA mice infected with a ydiD mutant compared to mice infected with S. Typhimurium wild type (Fig. 2A). This result was unexpected, because genetic ablation of other metabolic pathways required for bacterial growth in the intestinal lumen, such as ethanolamine utilization (Thiennimitr et al., 2011) or nitrate respiration (Lopez et al., 2015), does not moderate intestinal inflammation. Introducing the cloned ydiD gene on a plasmid (pYDID) restored fecal lipocalin-2 levels elicited by the ydiD mutant to levels observed in the S. Typhimurium wild type, suggesting that the effect was not due to unlinked mutations. Similarly, analysis of histopathological changes revealed that a ydiD mutant caused significantly reduced intestinal inflammation compared to the S. Typhimurium wild type or a complemented ydiD mutant (Fig. 2B). Transcript levels of Il17a and Kc, encoding two cytokines (IL-17A and KC) involved in neutrophil recruitment, were significantly (P < 0.0001) higher in cecal tissue of mice infected with the S. Typhimurium wild type or complemented ydiD mutant compared to mice infected with a ydiD mutant (Fig. 2C). T3SS-1-mediated neutrophil recruitment into the intestinal lumen is associated with a depletion of Clostridia from the gut-associated microbial community (Gill et al., 2012; Sekirov et al., 2010). Quantification of Clostridia by real-time PCR using class-specific primers revealed that the abundance of this taxon in the gut-associated microbial community was markedly reduced in mice infected with S. Typhimurium wild type or the complemented ydiD mutant compared to mice infected with a ydiD mutant (Fig. 2D), which correlated with reduced cecal butyrate concentrations (Fig. 2E). These data suggested that, surprisingly, genetic ablation of butyrate utilization markedly blunted the severity of intestinal inflammation during S. Typhimurium infection.
Figure 2: Genetic ablation of anaerobic β-oxidation moderates intestinal inflammation.
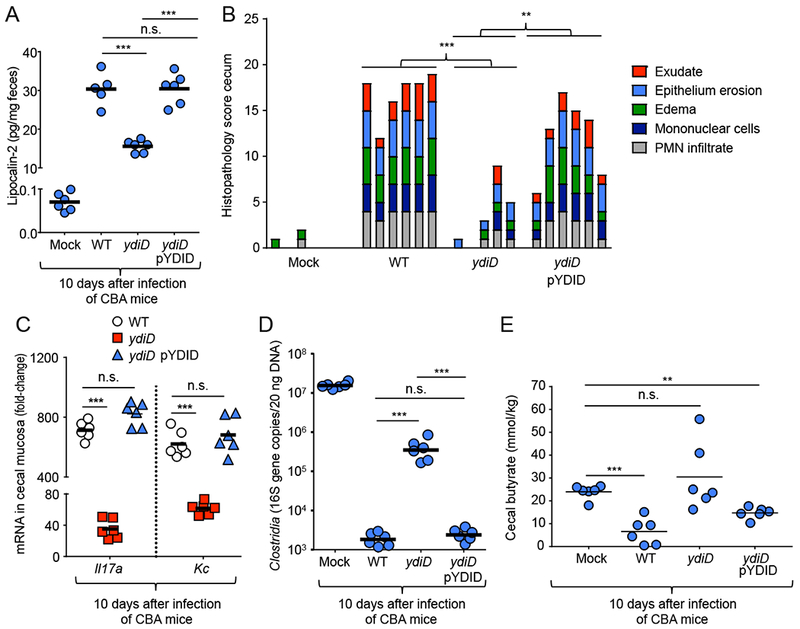
(A) Lipocalin-2 levels were determined by ELISA in colon contents of mice 10 days after infection with the indicated S. Typhimurium IR715 strains. (B) Histopathological changes were scored in blinded sections of the cecum. Each bar represents the combined scoring results for one individual animal. (C) Expression levels of Il17a mRNA and Kc mRNA isolated from the cecal tissue 10 days after infection in CBA mice. Transcript levels were normalized to actin levels and are shown as fold-change over trsanscript levels in mock-infected animals. (D) Clostridia 16S rRNA gene copy numbers present in 20 ng of total bacterial DNA were determined at 10 days post infection in CBA mice infected with the indicated S. Typhimurium strains. (E) The butyrate concentration was determined in cecal contents using liquid chromatography-mass spectrometry 10 days after infection of mice with the indicated S. Typhimurium strains. For (A – D) each circle and square represents data from an individual animal. *** represents p-value <0.0001, n.s. = not significant.
Butyrate utilization is essential for robust T3SS-1 gene expression and epithelial invasion
Next, we wanted to determine the mechanism by which deletion of ydiD attenuated gastrointestinal disease. Anaerobic growth of the ydiD mutant was impaired in the presence of 3 mM butyrate, but not in the absence of butyrate (Fig. S1A and S1B). However, this growth defect alone was not a likely explanation for attenuated gastrointestinal disease, because reduced bacterial growth in vivo caused by genetic ablation of ethanolamine utilization or nitrate respiration does not moderate intestinal inflammation (Lopez et al., 2015; Thiennimitr et al., 2011). A functional T3SS-1 is required for gastrointestinal disease caused by S. Typhimurium in calves (Tsolis et al., 1999) and for eliciting colitis in mice (Barthel et al., 2003). Butyrate inhibits expression of hilD, encoding a positive regulator of T3SS-1 gene expression, through an unknown mechanism (Gantois et al., 2006). Since S. Typhi lacks the anaerobic β-oxidation pathway encoded by ydiQRSTD, we explored whether S. Typhi would exhibit reduced invasion gene expression in the presence of butyrate. No differences in expression of invA, encoding a component of the T3SS-1 apparatus, were observed between S. Typhi and S. Typhimurium when bacteria were cultured anaerobically in the presence of acetate (Fig. S1C). However, in the presence of 3 mM butyrate (mimicking butyrate levels encountered in the small intestine), transcript levels of invA were significantly lower in S. Typhi and in a S. Typhimurium ydiD mutant compared to the S. Typhimurium wild type (Fig. 3A). While S. Typhi, S. Typhimurium and a S. Typhimurium ydiD mutant were equally invasive for T84 cells in a standard gentamicin protection assay, invasion of S. Typhi and the S. Typhimurium ydiD mutant was significantly reduced compared to the S. Typhimurium wild type when the assay was performed under hypoxic (0.8% O2) conditions with SCFA concentrations resembling those in the small intestine (12 mM acetate, 5 mM propionate and 3 mM butyrate) (Fig. 3B).
Figure 3: Genetic ablation of anaerobic β-oxidation decreases invasion in the presence of SCFAs.
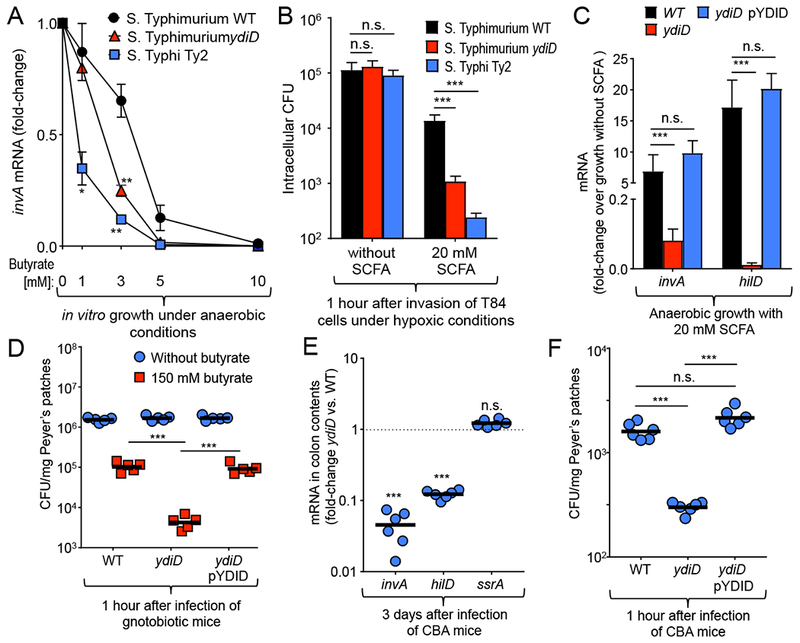
(A) S. Typhimurium wild type (WT), a S. Typhimurium ydiD mutant, and S. Typhi strain Ty2 were grown under anaerobic conditions in rich broth supplemented with the indicated concentrations of butyrate. Transcript levels of invA were quantified by real-time polymerase chain reaction (PCR), normalized to 16S ribosomal RNA (rRNA) levels and shown as fold-change over transcript levels detected in medium without SCFA supplementation. (B) T84 cells were infected for 1 hour with the indicated bacterial strains under hypoxic conditions (0.8% O2). Bacterial numbers were recovered after 90 minutes of gentamicin treatment (n = 3). (C) The S. Typhimurium IR715 wild type (WT), a ydiD mutant, and a complemented ydiD mutant (ydiD pYDID) were grown under anaerobic conditions in rich broth supplemented with 20 mM SCFAs (12 mM acetate, 5 mM propionate, and 3 mM butyrate) and bacterial RNA was collected. (D) Swiss-Webster gnotobiotic mice (n = 6) were infected with the S. Typhimurium IR715 wild type, a ydiD mutant, or a complemented ydiD mutant (109 CFU). To measure invasion, mice were euthanized one hour after infection to enumerate intracellular bacteria in Peyer’s patches. (E) Bacterial RNA was isolated from colon contents of S. Typhimurium-infected mice. (C and E) Transcript levels of invA, hilD, and ssrA were quantified by real-time polymerase chain reaction (PCR), normalized to 16S ribosomal RNA (rRNA) levels and shown as fold-change over transcript levels detected in medium without SCFA supplementation. (F) CBA mice (n = 6) were infected as described in (D). Error bars in (A-C) represent mean ± SD of n≥3 independent experiments. For (B – D) each circle and square represents data from an individual animal. ** and *** represents p-values <0.001 and <0.0001, n.s. = not significant.
Butyrate utilization maintains T3SS-1 gene expression in the small intestine
Seeing that invA expression was repressed in the ydiD mutant, we explored whether hilD gene expression and invasion efficacy were defective in the presence of SCFAs in vitro. Anaerobic growth in medium mimicking SCFA levels encountered in the small intestine (12 mM acetate, 5 mM propionate, and 3 mM butyrate) resulted in significantly (P < 0.0001) reduced expression of hilD in a ydiD mutant compared to the S. Typhimurium wild type (Fig. 3C). Introducing the cloned ydiD gene on a plasmid (pYDID) restored invasion gene expression in the ydiD mutant in the presence of SCFAs. When grown in the presence of 12 mM acetate, no significant differences in invA or hilD gene expression were observed between the S. Typhimurium wild type, a ydiD mutant or the complemented ydiD mutant (Fig. S2A). However, supplementation with 3 mM butyrate significantly (P < 0.0001) reduced invA and hilD expression in the ydiD mutant when compared to wild type or the complemented ydiD mutant (Fig. S2B). The results of an electrophoretic mobility shift assay (EMSA) suggested that reduced hilD expression was not caused by butyrate inhibiting HilD to bind its own promoter (Fig. S2C).
Next, we investigated whether inactivation of butyrate utilization would reduce bacterial invasion of Peyer’s patches in the small intestine in vivo. To monitor invasion in the absence of microbiota-derived SCFAs, germ-free mice were infected intragastrically with the S. Typhimurium wild type, a ydiD mutant or the complemented ydiD mutant, ileal Peyer’s patches were collected one hour later and treated with gentamicin to kill extracellular bacteria. No differences in invasiveness between bacterial strains were observed in the absence of butyrate; however, when germ-free mice received drinking water supplemented with butyrate, the ydiD mutant was recovered in significantly (P < 0.0001) lower numbers from Peyer’s patches than the wild type or the complemented ydiD mutant (Fig. 3D).
Finally, we examined the importance of butyrate utilization for invasion gene expression and Peyer’s patch invasion in the presence of a normal gut microbiota. The invA and hilD genes were expressed in significantly lower levels in the cecal contents of CBA mice three days after infection with the ydiD mutant compared to mice infected with S. Typhimurium wild type (Fig. 3E). As a control, we determined expression of ssrA, a positive regulator of the second S. Typhimurium type III secretion system (T3SS-2), which remained unchanged in the S. Typhimurium wild type compared to a ydiD mutant (Fig. 3E). Recovery of gentamicin-resistant bacteria from Peyer’s patches revealed that the ydiD mutant was significantly (P < 0.0001) less invasive in mice with a normal microbiota than the wild type or the complemented ydiD mutant (Fig. 3F). Collectively, these data suggested that genetic ablation of butyrate utilization rendered S. Typhimurium more sensitive to a butyrate-mediated repression of T3SS-1 invasion gene expression, thereby diminishing epithelial invasion.
Three genetic changes in S. Typhi cooperate to moderate gastrointestinal disease
Two genetic changes have been implicated in moderating intestinal inflammation during typhoid fever. One is the viaB locus, a DNA region containing genes for the synthesis of the virulence-associated (Vi) capsular polysaccharide, which was acquired by S. Typhi through horizontal gene transfer (Haneda et al., 2009; Wilson et al., 2008; Wilson et al., 2011). Introduction of the S. Typhi viaB locus into S. Typhimurium enhances proliferation of the pathogen at extraintestinal sites, such as the spleen (Jansen et al., 2011) and moderates the severity of intestinal inflammation in a mouse model (Haneda et al., 2009). The second genetic change is a loss-of-function mutation in fepE, encoding the length regulator of very-long O-antigen chains (Crawford et al., 2013). Since the ydiQRSTD operon is absent in S. Typhi, we wanted to investigate whether genetic ablation of ydiD could further reduce intestinal inflammation caused by a S. Typhimurium phoN::viaB fepE mutant.
One hour after intragastric infection of CBA mice, both the S. Typhimurium phoN::viaB fepE mutant and S. Typhimurium ydiD mutant exhibited significantly reduced invasion of Peyer’s patches compared to S. Typhimurium wild type (Fig. 4A). Importantly, the S. Typhimurium phoN::viaB fepE ydiD mutant was recovered in significantly lower numbers than either of the aforementioned strains (Fig. 4A). Fecal lipocalin-2 levels were significantly lower 10 days after infection of mice with a S. Typhimurium phoN::viaB fepE ydiD mutant compared to mice infected with a S. Typhimurium phoN::viaB fepE mutant or a S. Typhimurium ydiD mutant (Fig. 4B). Furthermore, inflammatory changes induced by infection with S. Typhimurium wild type were significantly reduced in mice infected with either a S. Typhimurium phoN::viaB fepE mutant or a S. Typhimurium ydiD mutant. Importantly, inflammation was abrogated in mice infected with a S. Typhimurium phoN::viaB fepE ydiD mutant (Fig. 4C) These data suggested that deletion of ydiD cooperates with inactivation of fepE and acquisition of viaB in moderating intestinal inflammation during typhoid fever. Bacterial recovery from systemic sites (spleen and liver) suggested that enhanced dissemination to or survival in extraintestinal tissue was mediated mainly by the phoN::viaB fepE mutations (Fig. 4D and S3A–S3C). Collectively, our data suggests just three genetic changes in S. Typhi cooperated to moderate intestinal inflammation during the initial stages of typhoid fever. Interestingly, two of these genetic changes, formation of a pseudogene (fepE) and a chromosomal deletion (ydiQRSTD), are loss-of-function mutations.
Figure 4: Deletion of ydiD cooperates with acquisition of viaB and pseudogene formation in fepE to moderate intestinal inflammation.
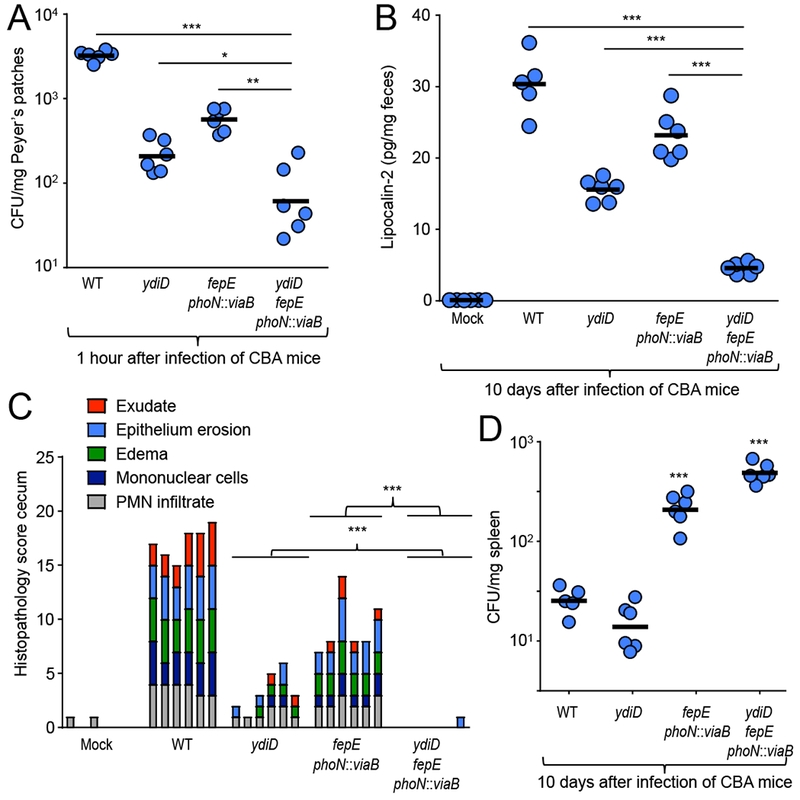
(A) CBA mice (n = 6) were infected with the S. Typhimurium IR715 wild type (WT), a ydiD mutant, a fepE phoN::viaB mutant or a fepE phoN::viaB ydiD mutant. To measure invasion, mice were euthanized one hour after infection and Peyer’s patches collected to enumerate intracellular bacteria. (B) Lipocalin-2 levels were determined by ELISA from colon contents in mice infected with the indicated S. Typhimurium strains 10 days after infection. (C) Histopathological changes were scored in blinded sections of the cecum. Each bar represents the combined scoring results for one individual animal. (D) Spleen CFU of S. Typhimurium in CBA mice (n = 6) 10 days after infection. For (A – C) each circle represents data from an individual animal. *, **, and *** represents p-values <0.01, <0.001, and <0.0001.
DISCUSSION
Generally, loss-of-function mutations reduce bacterial fitness. Contrary, our results suggest that deletion of ydiQRSTD bestowed the ability upon S. Typhi and S. Paratyphi A to moderate intestinal inflammation (Fig. 2). Acquisition of the viaB locus and pseudogene formation in fepE further decreased intestinal inflammation (Crawford et al., 2013; Haneda et al., 2009), while increasing dissemination to and/or survival in the liver and spleen (Fig. 4D), by enabling S. Typhi to avert the phagocyte respiratory burst (Miller et al., 1972). S. Paratyphi A, a typhoidal Salmonella serovar lacking the viaB locus, acquired the ability to avert the respiratory burst of phagocytes through pseudogene formation in rfbE, an example of convergent evolution (Hiyoshi et al., 2018). The result was a gain-of-function leading to the transition from a gastrointestinal to an extraintestinal infection profile. The idea that deletion of ydiQRSTD was involved in the transition from a gastrointestinal to an extraintestinal pathogen is further supported by previous genome comparisons. The most parsimonious explanation for the presence of a ydiQRSTD deletion in both S. Typhi and S. Paratyphi A, but not in genomes of non-typhoidal Salmonella serovars, is horizontal exchange of this DNA region between typhoidal Salmonella serovars. This is consistent with observations made by genome comparison, which suggests that S. Typhi exchanged a remarkable 23% of its genome with S. Paratyphi A through horizontal gene transfer, an event that is postulated to mark the origin of typhoid and paratyphoid fever (Didelot et al., 2007; Holt et al., 2008). Our results suggest that this horizontal transfer event included exchange of the ydiQRSTD deletion between S. Typhi and S. Paratyphi A, which resulted in a moderation of intestinal inflammation, a characteristic that distinguishes typhoidal from non-typhoidal Salmonella serovars.
Subsequent to this large-scale horizontal exchange, the S. Typhi and S. Paratyphi A lineages began to accumulate loss-of-function mutations at an accelerated rate, a process that is still in progress (Holt et al., 2008). Many pseudogenes acquired during the phase of accelerated genomic decay resulted in loss of pathways involved in central anaerobic metabolism, which are required for expansion of non-typhoidal Salmonella serovars, such as S. Typhimurium, in the gut lumen during gastrointestinal disease (Nuccio and Baumler, 2014). Our results support a model in which accelerated genome decay was preceded by acquisition of genetic changes (i.e. a deletion of ydiQRSTD, acquisition of viaB and pseudogen formation in fepE) that moderated intestinal inflammation (Fig. 5), suggesting that this phenotypic change was an evolutionary driver of accelerated genome decay in typhoidal Salmonella serovars, because it made functions obsolete that are only needed for taking advantage of intestinal inflammation.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andreas J. Bäumler (ajbaumler@ucdavis.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions.
The 17 human Clostridia isolates were kindly provided by K. Honda (Atarashi et al., 2011; Narushima et al., 2014) and were cultured individually as described previously (Atarashi et al., 2013). Unless indicated otherwise, S. Typhimurium and E. coli strains (Key Resources Table) were routinely grown aerobically at 37°C in LB broth (BD Biosciences, cat. # 244620) or on LB agar plates. If appropriate, antibiotics were added to the media at the following concentrations: 0.03 mg/ml chloramphenicol, 0.1 mg/ml carbenicillin, 0.05 mg/ml kanamycin, and 0.05 mg/ml nalidixic acid. For growth under anaerobic conditions, reduced rich broth (tryptone, 10 g/L, NaCl, 5 g/L, and yeast extract 1 g/L, 0.1M MOPS pH 7.8, 40 mM sodium fumarate) was inoculated with the indicated strain and incubated at 37°C in an anaero be chamber (0% oxygen). When necessary SCFAs were added at a concentration of 20 mM (12 mM acetate, 5 mM propionate, and 3 mM butyrate). Six hours after inoculation, cultures were harvested and stored at −80°C for subsequent RNA extraction.
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Ty2 | S. Typhi isolate, Vi+ | ATCC 19430 |
| IR715 | Nalidixic acid-resistant derivative of ATCC 14028s | (Stojiljkovic et al., 1995) |
| FF176 | IR715 phoN::Tn10d-Cam | (Faber et al., 2016) |
| AJB715 | IR715 phoN::KmR | (Kingsley et al., 2003) |
| FF357 | IR715 ΔydiD | This study |
| DNB4 | IR715 phoN::KmR ΔydiD | This study |
| RC60 | IR715 phoN::viaB fepE::pGP704 | (Crawford et al., 2013) |
| DNB3 | IR715 phoN::viaB fepE::pGP704 ΔydiD | This study |
| Salmonella Typhi Ty2 | ATCC | ATCC 19430 |
| Salmonella Typhimurium IR715 | Nalidixic acid-resistant derivative of ATCC 14028s | (Stojiljkovic et al., 1995) |
| DH5α λpir | F− endA1 hsdR17 (r−m+) supE44 thi-1 recA1 gyrA relA1 Δ(lacZYA-argF)U189 ϕ80lac ΔM15 λpir | (Pal et al., 2005) |
| S17-1 λpir | C600::RP4 2-(Tet::Mu) (Kan::Tn7) λpir recA1 thi pro hsdR (r−m+) | (Simon et al., 1983) |
| BL21 (DE3) | F- ompT hsdSB (rB-mB-) gal dcm (DE3) | (Studier and Moffatt, 1986) |
| Plasmids used in this study | ||
| pRDH10 | ori(R6K) mobRP4 sacRB TetR CmR | (Kingsley et al., 1999) |
| pWSK29 | ori(pSC101) CarbR | (Wang and Kushner, 1991) |
| pYDID | pWSK29 carrying ydiD gene transcribed from its native promoter | This study |
| pET28a | Expression vector with a His6-tag, KanR | Novagen |
| pWJ19 | pET28a carrying hilD gene transcribed from T7 promoter | This study |
| Critical Commercial Assays | ||
| TRI-reagent | Molecular Research Center | cat#: RT 111 |
| DNA-free kit | Applied Biosystems | cat#: AM1906 |
| Aurum Total RNA kit | BioRad | cat#: 7326820 |
| PowerSoil DNA Isolation Kit | Mo-Bio | cat#: 12888 |
| Bradford assay | BioRad | cat#:5000006 |
| Gel extraction kit | Qiagen | cat#: 28706 |
| EMSA kit | Invitrogen | cat#: E33075 |
| Lipocalin-2 ELISA | R&D systems | cat#: DY1857 |
| Experimental Models: Cell Lines | ||
| T84 colonic carcinoma cell | ATCC | cat#: CCL-248 RRID: CVCL_0555 |
| Experimental Models: Organisms/Strains | ||
| Mus musculus CBA/J | Jackson Labs | cat#: 000656 RRID: IMSD JAX:000656 |
| Mus musculus Gnotobiotic Swiss Webster | Bred in-house; originally acquired from Taconic | cat#: SW-F and SW-M |
| Oligonucleotides | ||
| Primers used in this study, see Table S1 | This paper | N/A |
| Software and Algorithms | ||
| Prims v7.0a | GraphPad | N/A |
Cell Culture Systems.
The colonic carcinoma cell line T84 was obtained from the American Type Culture Collection. T84 cells were routinely maintained in DMEM-F12 medium (1.2 g/l sodium bicarbonate, 2.5 mM L-glutamine, 15 mM HEPES, 0.5 mM sodium pyruvate (Invitrogen), and 10% fetal bovine serum (FBS; Invitrogen).
Animal Experiments.
All experiments in this study were approved by the Institutional Animal Care and Use Committee at the University of California at Davis. Female CBA mice, aged 8 weeks, were obtained from The Jackson Laboratory. Germ-free Swiss-Webster mice were bred in house. Mice were housed in ventilated cage racks on corn bedding, providing water and mouse chow ad libidum. Mice were monitored twice daily and cage bedding changed every two weeks.
CBA mice were infected with either 0.1 ml of LB broth (mock-infected) or S. Typhimurium in LB broth. For single infections, mice were inoculated with 1 × 109 CFU of the indicated S. Typhimurium strains. Peyer’s patches were collected 1 hour after infection for quantifying Salmonella invasion. To assess intestinal inflammation, mice were euthanized at 10 days after infection, cecal contents, colon contents, spleen, and liver were collected for enumeration of bacterial numbers and the proximal colon and cecal tip were collected for histopathology. Bacterial numbers were determined by plating serial ten-fold dilutions onto LB agar containing the appropriate antibiotics.
For competitive infections, conventional mice were inoculated with 1 × 109 CFU of a 1:1 mixture of the indicated strains. Salmonella-infected germ-free mice were inoculated with 17 human Clostridia isolates by oral gavage or given 150 mM sodium butyrate in the drinking water then inoculated with 1 × 109 CFU of a 1:1 mixture of the indicated strains. Fecal pellets were collected at the indicated time points to monitor colonization over time.
METHOD DETAILS
Invasion assay.
T84 cells were seeded 2.5 χ 105 cells/well and infected with indicated strains at a multiplicity of infection (MOI) of 10. The bacteria and cells were exposed to hypoxic conditions in a humidified hypoxia chamber (0.8% O2, Coy Laboratory products) while being incubated for 1 h at 37°C in DMEM-F12 medium contai ning 20 mM SCFA mix (12 mM acetate, 5 mM propionate, and 3 mM butyrate) during invasion. Each well was washed three times with sterile PBS (KCl at 2.7 mM, KH2PO4 at 1.8 mM, NaCl at 140 mM, Na2HPO4 at 10 mM, pH 7.4) to remove extracellular bacteria, and medium containing gentamicin at a concentration of 0.1 mg/ml was added for a 90-min incubation in conditions stated above. After three washes with PBS, the cells were lysed with 1 ml of 1% Triton X-100 and the lysates were transferred to sterile tubes. Tenfold serial dilutions were plated to enumerate intracellular bacteria.
ELISA assay.
Colon contents were used to assess lipocalin-2 levels. Lipocalin-2 levels were determined by sandwich enzyme-linked immunosorbent assay (ELISA) according to the manufacturer’s instructions. A minimum of 6 biological replicates were used for each experimental group.
Quantitative real-time PCR analysis.
After euthanasia for murine RNA isolation, cecal tissue sections were homogenized in a Mini-Beadbeater (BioSpec Products, Bartlesville, OK) and RNA was isolated by the TRI-Reagent method following the manufacturer’s protocol. Contaminating DNA was removed using the DNA-free kit and RNA was stored at −80°C.
To determine invasion gene expression during in vitro growth in the presence of SCFA, RNA from frozen bacterial pellets was extracted using the Aurum Total RNA mini Kit (BioRad, cat. #: 7326820). DNA remaining in the RNA isolation portion was removed using the DNA-free kit (Applied Biosystems, cat. #: AM1906). Target gene transcription was normalized to the levels of β-actin mRNA for murine gene expression. Target gene transcription was normalized to the levels of S. Typhimurium 16S rRNA gene mRNA for bacterial gene expression. As a control, bacterial target gene expression was normalized to the S. Typhimurium rpoA gene message, which produced similar results as normalization with the 16S rRNA gene message (data not shown). Fold change in mRNA levels was determined using the comparative threshold cycle (CT) method and conditions were compared to either S. Typhimurium or S. Typhi in the absence of SCFAs for bacterial gene expression (invA, hilD) or mock-infected cecal tissue for murine gene expression experiments (Il17a, Kc).
DNA from the cecal contents was extracted using the PowerSoil DNA Isolation kit according to the manufacturer’s protocol. PCR mix and the appropriate primer sets (Supplementary table 1) at a final concentration of 0.25 mM. Absolute values were calculated using a plasmid carrying the cloned gene to generate a standard curve ranging from 108 to 101 copies/ml diluted in a 0.02 mg/ml yeast RNA (Sigma-Aldrich, cat. # R6750) solution.
Construction of Salmonella Typhimurium mutants.
All strains, plasmids and primers used in this study are listed in Supplementary tables 1 or the Key Resources Table. PCR products were confirmed by sequencing. The suicide plasmid pRDH10 was propagated in E. coli DH5a λpir.
To generate a ydiD mutant, regions upstream and downstream of ydiD were PCR amplified from the S. Typhimurium wild-type strain IR715 using primers ydiD-P1,2,3 and 4 (Supplementary table 1). The PCR fragments were gel purified and cloned into BamHI digested pRDH10 using Gibson Assembly Master Mix (NEB) yielding plasmid pXY. Plasmid pXY was conjugated into S. Typhimurium IR715 (wild type) and AJB715 (phoN::KmR) using E. coli S17-1λpir as a donor strain. Exconjugants were plated onto LB+Nal+Cm to select for clones that had integrated the suicide plasmid. Sucrose counter-selection was performed as published previously (Lawes and Maloy, 1995). Strains that were sucrose resistant and CmS were verified by PCR to carry the ydiD deletion. The resulting ydiD mutants were designated FF357 (ydiD) and DNB4 (phoN::KmR ydiD).
To generate the fepE::pGP704 phoN::viaB ydiD mutant, fepE::pGP704 and phoN::viaB were subsequently transduced into FF357 (ydiD) by generalized P22 HT int-105 phage transduction using strain RC60 as donor strain. Transductants were cleaned from phage contaminations on Evans blue-Uranine (EBU) plates and tested for phage sensitivity by crossstreaking against P22 H5.
For complementation of FF357, the ydiD gene together with its native promoter region was PCR amplified using primers ydiDc-P1, P2, P3 and P4. The PCR fragments were gel purified and cloned into BamHI digested pWSK29 using Gibson Assembly Master Mix (NEB). The resulting plasmid (pYDID) was transformed into strain FF357 for complementation.
Plasmid construction.
For the construction of pWJ19 producing His6-HilD (HilD protein tagged with six histidines at its N-terminus) under T7 promoter, the DNA containing the Salmonella Typhimurium 14028s hilD gene was amplified by PCR using primers of HilD-His-F and HilD-His-R, and the PCR products were introduced between NdeI and EcoRI sites of pET28a (Novagen, cat. # 69864).
Expression and purification of His6-HilD.
E. coli BL21 (DE3) (Novagen, 69450) containing pWJ19 expressing His6-HilD was grown in 100 ml of LB media at 37°C in a shaken incubator at 220 rpm. At an optical density of 0.5, the expression of His6-HilD was induced by adding 1 mM isopropyl-β-D-thiogalactopyranoside and the bacterial culture was allowed to grow for another 16 hours at 18°C in a shaken incubator at 180 rpm. The His6-HilD proteins were purified using nikel-nitrilotriacetic acid (Ni-NTA) resin (Qiagen, cat. # 30410) according to the manufacturer’s instructions, and the bound proteins were eluted with elution buffer [20 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 250 mM imidazole]. The eluted proteins were concentrated by using a VivaSpin 20 instrument (3,000-molecular-weight cutoff [MWCO] polyethersulfone; GE Healthcare, 28-9323-58), and the elution buffer was replaced with storage buffer [20 mM Tris-HCl (pH 8.0), 300 mM NaCl, and 50% glycerol] using a PD MidiTrap G-25 column (GE Healthcare, 28-9180-08). Protein concentration was determined using the Bradford assay with BSA as the standard.
EMSA.
EMSA experiments were performed as described previously (Campbell et al., 2008; Martinez et al., 2014). DNA fragments containing the hilD promoter (the −130/+75 region of hilD) were amplified by PCR using primers hilD-EMSA-F and hilD-EMSA-R with wild-type Salmonella Typhimurium 14028s chromosomal DNA as a template. The amplified PCR products were purified from agarose gels using a gel extraction kit (Qiagen, cat. # 28706). Binding reactions were performed by mixing 0.8 pmol of PCR products with increasing concentrations of purified His6-HilD in binding buffer containing 10 mM Tris-HCl (pH 8.0), 0.5 mM EDTA (pH 8.0), 1 mM DTT, 50 mM KCl, 10 μg/ml BSA, and 10 μg/ml Poly dI∙dC, in a total volume of 20 μl. Protein-DNA binding reaction mixtures were incubated at room temperature for 30 minutes and then electrophoretically separated in 6% nondenaturing poly-acrylamide gels in 0.5X Tris-borate-EDTA buffer. The DNA fragments were stained by using EMSA Kit according to the manufacturer’s instructions and visualized with a Gel Doc™ EZ (Bio-Rad, cat. # 1708270).
Measurements of short-chain fatty acid concentrations.
Samples of cecal and colon contents were diluted with nanopore water (10 μl/mg) and gently agitated overnight at 4°C. The homogenized samples were centrifuged at 21,000 × g for 5 min. 100 μl of the supernatants were transferred centrifuged at 21,000 × g again for 20 min. For each sample, 20 μl of the supernatant was mixed with 20 μl of 100 mM N-(3-Dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (1-EDC HCl) (Sigma-Aldrich, cat. # E7750) in 5% pyridine (Sigma-Aldrich cat. # 270407) and 40 uL of 200 mM 2-Nitrophenylhydrazine (2-NPH) (Sigma-Aldrich, cat. # N21588) in 80% acetonitrile (ACN) (Sigma-Aldrich, cat. # BJAH015-4) with 50 mM HCl. The mixture was incubated at 40°C for 30 min. After reacting, 420 μl of 10% ACN was added to the solution. Then 1 μl the solution was injected into an Agilent 6490 triple quadruple mass spectrometer for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are expressed as the mean and standard error of the mean. Group sizes of animals were determined by power calculation. The number of animals (n) for each group is provided in the figure legends. Ratios were converted logarithmically prior to statistical analysis to ensure data are normally distributed. A Student’s t-test was used for statistical analyses of all measurements. A P value of < 0.05 was considered significant.
Supplementary Material
Highlights.
Anaerobic β-oxidation enables S. Typhimurium to utilize microbiota-derived butyrate
Loss of anaerobic β-oxidation renders invasion gene expression sensitive to butyrate
S. Typhi gained the ability to moderate inflammation by losing anaerobic β-oxidation
Gene loss drove the transition from a gastrointestinal to an extraintestinal pathogen
ACKNOWLEDGEMENTS
E.E.O. was supported by Public Health Service Grant TR001861 (E.E.O.). Work in A.J.B.’s laboratory is supported by Public Health Service Grants AI044170, AI096528, AI112445 and AI112949.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Altier C (2005). Genetic and environmental control of salmonella invasion. J Microbiol 43 Spec No, 85–92. [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, et al. (2013). Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, et al. (2011). Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthel M, Hapfelmeier S, Quintanilla-Martinez L, Kremer M, Rohde M, Hogardt M, Pfeffer K, Russmann H, and Hardt WD (2003). Pretreatment of mice with streptomycin provides a Salmonella enterica serovar Typhimurium colitis model that allows analysis of both pathogen and host. Infect Immun 71, 2839–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JW, Morgan-Kiss RM, and Cronan JE Jr. (2003). A new Escherichia coli metabolic competency: growth on fatty acids by a novel anaerobic beta-oxidation pathway. Mol Microbiol 47, 793–805. [DOI] [PubMed] [Google Scholar]
- Campbell LA, Faivre EJ, Show MD, Ingraham JG, Flinders J, Gross JD, and Ingraham HA (2008). Decreased recognition of SUMO-sensitive target genes following modification of SF-1 (NR5A1). Mol Cell Biol 28, 7476–7486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford RW, Wangdi T, Spees AM, Xavier MN, Tsolis RM, and Baumler AJ (2013). Loss of very-long O-antigen chains optimizes capsule-mediated immune evasion by Salmonella enterica serovar Typhi. mBio 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crump JA, Luby SP, and Mintz ED (2004). The global burden of typhoid fever. Bulletin of the World Health Organization 82, 346–353. [PMC free article] [PubMed] [Google Scholar]
- Didelot X, Achtman M, Parkhill J, Thomson NR, and Falush D (2007). A bimodal pattern of relatedness between the Salmonella Paratyphi A and Typhi genomes: convergence or divergence by homologous recombination? Genome research 17, 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber F, Tran L, Byndloss MX, Lopez CA, Velazquez EM, Kerrinnes T, Nuccio SP, Wangdi T, Fiehn O, Tsolis RM, et al. (2016). Host-mediated sugar oxidation promotes postantibiotic pathogen expansion. Nature 534, 697–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantois I, Ducatelle R, Pasmans F, Haesebrouck F, Hautefort I, Thompson A, Hinton JC, and Van Immerseel F (2006). Butyrate specifically down-regulates salmonella pathogenicity island 1 gene expression. Appl Environ Microbiol 72, 946–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner CD, Antonopoulos DA, Wagner B, Duhamel GE, Keresztes I, Ross DA, Young VB, and Altier C (2009). Perturbation of the small intestine microbial ecology by streptomycin alters pathology in a Salmonella enterica serovar typhimurium murine model of infection. Infect Immun 77, 2691–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill N, Ferreira RB, Antunes LC, Willing BP, Sekirov I, Al-Zahrani F, Hartmann M, and Finlay BB (2012). Neutrophil elastase alters the murine gut microbiota resulting in enhanced Salmonella colonization. PloS one 7, e49646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glynn JR, and Palmer SR (1992). Incubation period, severity of disease, and infecting dose: evidence from a Salmonella outbreak. Am J Epidemiol 136, 1369–1377. [DOI] [PubMed] [Google Scholar]
- Haneda T, Winter SE, Butler BP, Wilson RP, Tukel C, Winter MG, Godinez I, Tsolis RM, and Baumler AJ (2009). The capsule-encoding viaB locus reduces intestinal inflammation by a Salmonella pathogenicity island 1-independent mechanism. Infect Immun 77, 2932–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiyoshi H, Wangdi T, Lock G, Saechao C, Raffatellu M, Cobb BA, and Bäumler AJ (2018). Mechanisms to evade the phagocyte respiratory burst arose by convergent evolution in typhoidal Salmonella serovars. Cell Reports, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt KE, Parkhill J, Mazzoni CJ, Roumagnac P, Weill FX, Goodhead I, Rance R, Baker S, Maskell DJ, Wain J, et al. (2008). High-throughput sequencing provides insights into genome variation and evolution in Salmonella Typhi. Nature genetics 40, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen AM, Hall LJ, Clare S, Goulding D, Holt KE, Grant AJ, Mastroeni P, Dougan G, and Kingsley RA (2011). A Salmonella Typhimurium-Typhi Genomic Chimera: A Model to Study Vi Polysaccharide Capsule Function In Vivo. PLoS Pathog 7, e1002131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley RA, Humphries AD, Weening EH, De Zoete MR, Winter S, Papaconstantinopoulou A, Dougan G, and Baumler AJ (2003). Molecular and phenotypic analysis of the CS54 island of Salmonella enterica serotype typhimurium: identification of intestinal colonization and persistence determinants. Infect Immun 71, 629–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley RA, Reissbrodt R, Rabsch W, Ketley JM, Tsolis RM, Everest P, Dougan G, Baumler AJ, Roberts M, and Williams PH (1999). Ferrioxamine-mediated Iron(III) utilization by Salmonella enterica. Appl Environ Microbiol 65, 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk MD, Pires SM, Black RE, Caipo M, Crump JA, Devleesschauwer B, Dopfer D, Fazil A, Fischer-Walker CL, Hald T, et al. (2015). World Health Organization Estimates of the Global and Regional Disease Burden of 22 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med 12, e1001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawes M, and Maloy S (1995). MudSacI, a transposon with strong selectable and counterselectable markers: use for rapid mapping of chromosomal mutations in Salmonella typhimurium. J Bacteriol 177, 1383–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawhon SD, Maurer R, Suyemoto M, and Altier C (2002). Intestinal short-chain fatty acids alter Salmonella typhimurium invasion gene expression and virulence through BarA/SirA. Mol Microbiol 46, 1451–1464. [DOI] [PubMed] [Google Scholar]
- Lopez CA, Rivera-Chavez F, Byndloss MX, and Baumler AJ (2015). The Periplasmic Nitrate Reductase NapABC Supports Luminal Growth of Salmonella enterica Serovar Typhimurium during Colitis. Infect Immun 83, 3470–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez CA, Winter SE, Rivera-Chavez F, Xavier MN, Poon V, Nuccio SP, Tsolis RM, and Baumler AJ (2012). Phage-mediated acquisition of a type III secreted effector protein boosts growth of salmonella by nitrate respiration. mBio 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis P, and Flint HJ (2009). Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS microbiology letters 294, 1–8. [DOI] [PubMed] [Google Scholar]
- Martinez LC, Banda MM, Fernandez-Mora M, Santana FJ, and Bustamante VH (2014). HilD induces expression of Salmonella pathogenicity island 2 genes by displacing the global negative regulator H-NS from ssrAB. J Bacteriol 196, 3746–3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Clifton SW, Latreille P, Porwollik S, Sabo A, Meyer R, Bieri T, Ozersky P, McLellan M, et al. (2004). Comparison of genome degradation in Paratyphi A and Typhi, human-restricted serovars of Salmonella enterica that cause typhoid. Nat Genet 36, 1268–1274. [DOI] [PubMed] [Google Scholar]
- Miller RM, Garbus J, and Hornick RB (1972). Lack of enhanced oxygen consumption by polymorphonuclear leukocytes on phagocytosis of virulent Salmonella typhi. Science 175, 1010–1011. [DOI] [PubMed] [Google Scholar]
- Narushima S, Sugiura Y, Oshima K, Atarashi K, Hattori M, Suematsu M, and Honda K (2014). Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut microbes 5, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccio SP, and Baumler AJ (2014). Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. mBio 5, e00929–00914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen SJ, Bleasdale SC, Magnano AR, Landrigan C, Holland BH, Tauxe RV, Mintz ED, and Luby S (2003). Outbreaks of typhoid fever in the United States, 1960-99. Epidemiol Infect 130, 13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal D, Venkova-Canova T, Srivastava P, and Chattoraj DK (2005). Multipartite regulation of rctB, the replication initiator gene of Vibrio cholerae chromosome II. J Bacteriol 187, 7167–7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhill J, Dougan G, James KD, Thomson NR, Pickard D, Wain J, Churcher C, Mungall KL, Bentley SD, Holden MT, et al. (2001). Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 413, 848–852. [DOI] [PubMed] [Google Scholar]
- Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, et al. (2016). Depletion of Butyrate-Producing Clostridia from the Gut Microbiota Drives an Aerobic Luminal Expansion of Salmonella. Cell Host Microbe 19, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekirov I, Gill N, Jogova M, Tam N, Robertson M, de Llanos R, Li Y, and Finlay BB (2010). Salmonella SPI-1-mediated neutrophil recruitment during enteric colitis is associated with reduction and alteration in intestinal microbiota. Gut microbes 1, 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon R, Priefer U, and Puhler A (1983). A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Bio/Technology 1, 784–791. [Google Scholar]
- Stojiljkovic I, Baumler AJ, and Heffron F (1995). Ethanolamine utilization in Salmonella typhimurium: nucleotide sequence, protein expression, and mutational analysis of the cchA cchB eutE eutJ eutG eutH gene cluster. J Bacteriol 177, 1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, and Moffatt BA (1986). Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. J Mol Biol 189, 113–130. [DOI] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, and Baumler AJ (2011). Intestinal inflammation allows Salmonella to utilize ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsolis RM, Adams LG, Ficht TA, and Baumler AJ (1999). Contribution of Salmonella typhimurium virulence factors to diarrheal disease in calves. Infect Immun 67, 4879–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vital M, Howe AC, and Tiedje JM (2014). Revealing the bacterial butyrate synthesis pathways by analyzing (meta)genomic data. mBio 5, e00889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang RF, and Kushner SR (1991). Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199. [PubMed] [Google Scholar]
- Wilson RP, Raffatellu M, Chessa D, Winter SE, Tukel C, and Baumler AJ (2008). The Vi-capsule prevents Toll-like receptor 4 recognition of Salmonella. Cell Microbiol 10, 876–890. [DOI] [PubMed] [Google Scholar]
- Wilson RP, Winter SE, Spees AM, Winter MG, Nishimori JH, Sanchez JF, Nuccio SP, Crawford RW, Tukel C, and Baumler AJ (2011). The Vi capsular polysaccharide prevents complement receptor 3-mediated clearance of Salmonella enterica serotype Typhi. Infection and immunity 79, 830–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.