

Summary
Metabolic adaptations accompanying the development of antibiotic resistance in bacteria remain poorly understood. To study this relationship, we profiled the growth of lab-evolved antibiotic-resistant lineages of the opportunistic pathogen Pseudomonas aeruginosa across 190 unique carbon sources. Our data revealed that the evolution of antibiotic resistance resulted in systems-level changes to growth dynamics and metabolic phenotype. A genome-scale metabolic network reconstruction of P. aeruginosa was paired with whole-genome sequencing data to predict genes contributing to observed changes in metabolism. We experimentally validated computational predictions to identify mutations in resistant P. aeruginosa affecting loss of catabolic function. Finally, we found a shared metabolic phenotype between lab-evolved P. aeruginosa and clinical isolates with similar mutational landscapes. Our results build upon previous knowledge of antibiotic-induced metabolic adaptation and provide a framework for the identification of metabolic limitations in antibiotic-resistant pathogens.
Graphical Abstract

eTOC Blurb:
Bacterial metabolism is known to play a role in antibiotic efficacy; however, less is known about how the evolution of antibiotic resistance can affect bacterial metabolism. Dunphy et al. discover systems-level changes in the metabolic functionality and growth dynamics of lab-evolved antibiotic-resistant P. aeruginosa. Through a combined computational and experimental approach, they predict and test the impact of observed genetic mutations on specific metabolic phenotypes.
Introduction
With the threat of a ‘post-antibiotic era’ looming, there is a critical need to develop new strategies to treat bacterial infections (Alanis, 2005; Falagas and Bliziotis, 2007). The design of such approaches could be guided by a better understanding of the relationship between antibiotic resistance and bacterial metabolism. Bacterial metabolism has been shown to be an important factor in the efficacy of certain classes of antibiotics (Lee and Collins, 2011; Lobritz et al., 2015; Martínez and Rojo, 2011; Meylan et al., 2017). For example, mutations to the electron transport chain have been shown to reduce proton motive force (PMF) and limit PMF-dependent influx of aminoglycosides (Martínez and Rojo, 2011; Taber et al., 1987). Such mutations are commonly observed in aminoglycoside-resistant clinical isolates (Schurek et al., 2008; Yen and Papin, 2017). Conversely, quinolone efflux is often PMF-dependent and decreased PMF can result in increased drug susceptibility (Giraud et al., 2000). Links between metabolism and antibiotic resistance have resulted in a variety of proposed treatments to clear infections including drug cycling approaches, which aim to continuously re-sensitize resistant populations, and metabolite supplementation strategies, which jumpstart metabolism to restore drug susceptibility in antibiotic tolerant cells (Imamovic and Sommer, 2013; Meylan et al., 2018; Peng et al., 2015). These tactics do not require new antibiotics, rather they help to prolong the efficacy of existing drugs through the manipulation of metabolism in resistant or tolerant populations.
While it is generally understood that bacterial metabolism affects antibiotic efficacy, it is less understood how the metabolic flexibility and growth dynamics of bacterial pathogens change with the development of antibiotic resistance. The direct impact of sustained antibiotic pressure on metabolic adaptation in clinical isolates is confounded by in vivo pressures, including nutrient stress, oxidative stress, host inflammation, and competition with co-infecting pathogens (Hassett and Cohen, 1989; Hoffman et al., 2010; Singh et al., 2007; Wong et al., 2013). Consequently, reports of metabolic phenotypes of clinical isolates vary across studies and even within the same patient (Hocquet et al., 2016; Jorth et al., 2015; Rau et al., 2010). There has recently been success using adaptive laboratory evolution (ALE) experiments to study the evolution of antibiotic resistance in a controlled in vitro environment (Banerjee et al., 2017; Dragosits and Mattanovich, 2013; Furusawa et al., 2018; Jahn et al., 2017; Toprak et al., 2012; Yen and Papin, 2017; Zampieri et al., 2017). Sequencing and expression profiling of lab-evolved resistant bacteria have revealed genetic mutations responsible for resistance phenotypes (Lázár et al., 2013; Suzuki et al., 2014); however, connecting specific mutations to metabolic phenotypes remains a significant challenge (Palmer and Kishony, 2013; de Visser and Krug, 2014).
Genome-scale metabolic network reconstructions (GENREs) can provide the framework to contextualize genetic and metabolic changes accompanying the development of antibiotic resistance (Bordbar et al., 2014; Oberhardt et al., 2009). A GENRE is a quantitative formalism that captures all known metabolic reactions in an organism (Orth et al., 2010). Within a GENRE, gene-protein-reaction (GPR) rules link annotated metabolic genes to the reactions that their gene products catalyze (Durot et al., 2009; Reed et al., 2003). Among other functions, GENREs can be used to mechanistically evaluate the impact of mutations in single genes on bacterial growth across many environmental conditions (Joyce et al., 2006). This analysis allows for the systematic prediction of the metabolic consequences of individual mutations identified in sequenced drug-evolved lineages. If they can be identified, robust resistant-specific changes in metabolism could be leveraged in the rational design of drug cycling and metabolite supplementation strategies.
To better understand the relationship between antibiotic resistance and bacterial metabolism, we profiled the metabolic phenotypes of previously published lab-evolved antibiotic-resistant lineages of the opportunistic pathogen Pseudomonas aeruginosa. We evaluated growth of piperacillin-, tobramycin-, and ciprofloxacin-resistant P. aeruginosa on 190 unique carbon sources. We also evaluated growth of the starting ancestral lineage and a media-evolved lineage. This effort resulted in the generation of over 2,800 individual growth curves from which we captured the growth dynamics of each evolved lineage. Phenotypic data was integrated with previously collected genomic information on each lineage to probe the importance of reported resistance mutations on the observed metabolic phenotypes (Yen and Papin, 2017). Phenotypic and sequencing data were used in tandem with a recently published GENRE of P. aeruginosa strain UCBPP-PA14 to predict the individual impact of 343 genes deleted in the piperacillin-resistant lineage on loss of catabolic function (Bartell et al., 2017). Finally, a loss of catabolic function observed in the piperacillin-resistant lineage was found to be conserved in clinical isolates with similar large chromosomal deletions (Hocquet et al., 2016).
Altogether, we report that in vitro adaptation to antibiotics resulted in systems-level changes to metabolic function and growth dynamics in P. aeruginosa. We contextualized our data with a computational model to identify genotype-phenotype relationships in the drug-evolved lineages and experimentally interrogated model-driven predictions. By improving our understanding of the metabolic adaptations accompanying the evolution of antibiotic resistance, we aim to help guide the development of novel treatment strategies against bacterial pathogens.
Results
Profiling growth phenotypes of antibiotic-resistant Pseudomonas aeruginosa
P. aeruginosa was previously evolved to lysogeny broth (LB) media through serial passaging for 20 days (Yen and Papin, 2017). In parallel, the same starting ancestral lineage P. aeruginosa was also evolved to each of three antibiotics: ciprofloxacin, piperacillin, and tobramycin (Figure 1A). Whole-genome sequencing was also previously performed on all lineages (Figure 1B) (Yen and Papin, 2017). As formerly reported, the minimal inhibitory concentration (MIC) of each drug measured in its respective drug-evolved lineage increased at least 32-fold while the LB-evolved control lineage had no increase in MIC to any of the drugs relative to the ancestral lineage (Yen and Papin, 2017).
Figure 1. Schematic of phenotypic profiling of antibiotic-resistant Pseudomonas aeruginosa.
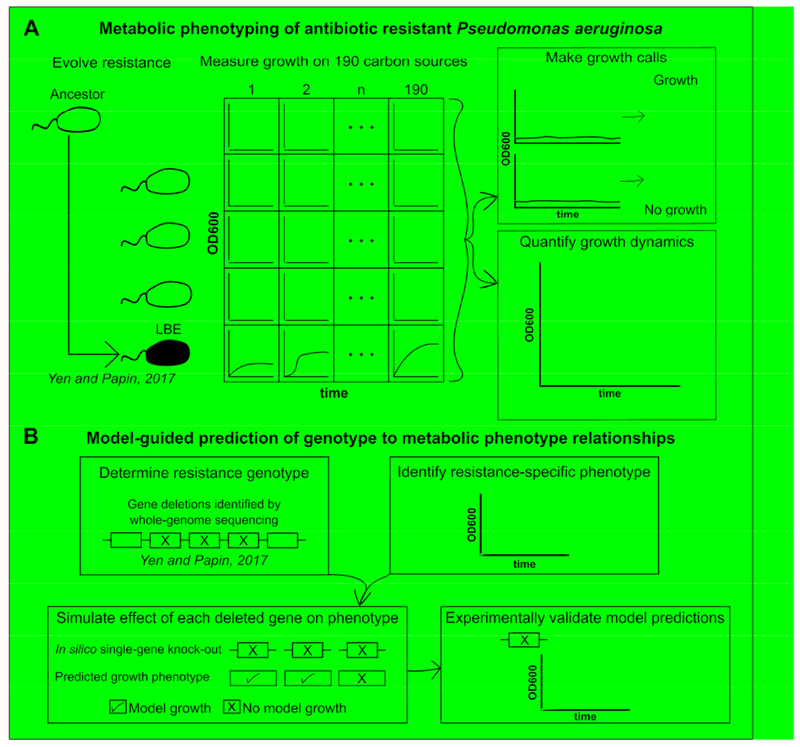
(A) P. aeruginosa strain UCBPP-PA14 was previously adaptively evolved to three antibiotics (ciprofloxacin (CIP), piperacillin (PIP), or tobramycin (TOB)) or to LB media without drug (LBE) for 20 days. Growth of each evolved lineage was profiled by measuring the optical density at 600nm (OD600) of each lineage after inoculation on 190 unique carbon sources for 48-hours. These phenotypic data were used to calculate changes in growth dynamics. (B) Phenotypic data and previously collected whole-genome sequencing data were paired with a genome-scale metabolic network reconstruction of P. aeruginosa UCBPP-PA14 to predict mutations impacting loss of catabolic function in the piperacillin-evolved lineage. In brief, the model was used to predict the individual impact of each gene that was deleted from the piperacillin-evolved lineage on the ability to grow on a given carbon source. If a simulated gene knockout resulted in a loss of model growth on a carbon source where an experimental loss of catabolic function was also observed, then the prediction was experimentally validated with a transposon mutant. This process was repeated for the piperacillin-evolved lineage on 42 carbon sources that could be both experimentally and computationally evaluated.
To characterize phenotypic changes in metabolism that arise with resistance, we evaluated the growth of each lineage on 190 unique carbon sources in the absence of antibiotic-pressure in triplicate (Figure 1A). These growth phenotyping experiments resulted in the generation of 2,850 individual growth curves (Data S1). The median curves were taken for each lineage on each carbon source across three replicate colonies and these 950 median growth curves are referred to for the remainder of the analysis (Figure S1). A lineage was considered to have grown on a given carbon source if the background-subtracted area under the curve (AUC) of the 48-hour growth curve was in the upper quartile of all of the data. This corresponded to a threshold of about 15.8; however, downstream analyses were not qualitatively impacted by changes to this cutoff (Figures 2A and S2, Data S2, and Star Methods). The AUC exceeded this threshold on 25.1% (238/950) of all growth curves, with only 7.4% of all carbon sources (14/190) supporting growth of all five measured lineages (Figure 2B). There were 17 instances in which one or more of the LB- or drug-evolved lineages grew where the ancestral lineage could not, indicating a gain of catabolic function. Losses of catabolic function were more common than gains. A total of 46 carbon sources supported growth of the ancestral lineage while one or more evolved-lineage was unable to grow. Adaptive evolution to LB media and antibiotics resulted in wide-spread loss and occasional gain of catabolic functions.
Figure 2. Summary of phenotypic growth data of antibiotic-resistant P. aeruginosa.
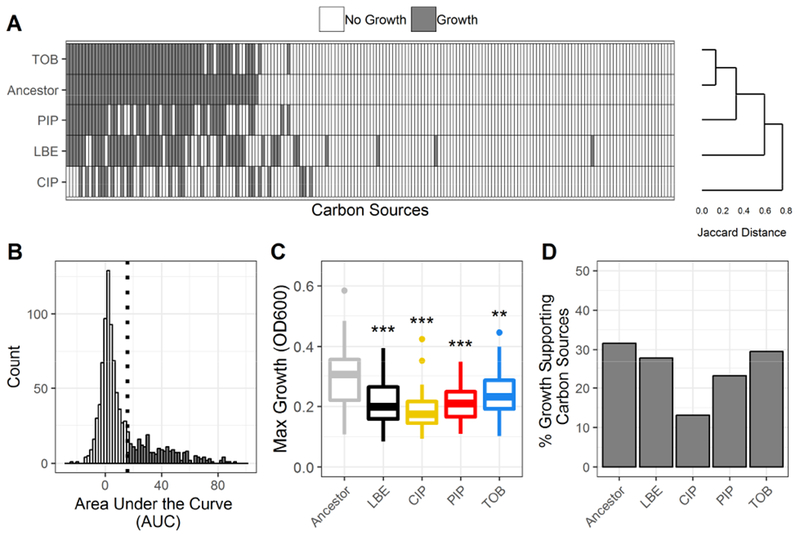
(A) Binary growth calls for each lineage (Ancestor, LBE = LB-Evolved, CIP = ciprofloxacin-resistant, PIP = piperacillin-resistant, TOB = tobramycin-resistant) across 190 unique carbon sources (white = no growth, grey = growth). Lineages are clustered using Jaccard distances and complete linkage. Carbon sources are sorted by growth calls for the ancestral lineage. (B) Histogram of the area under the curve (AUC) for all measured growth curves. A lineage was considered to have grown on a given carbon source if the AUC of the 48-hour growth curve was above the 3rd quartile. The growth/no growth cutoff is indicated by a dotted line. (C) Boxplot summarizes the maximum growth density by lineage across all carbon sources for which each lineage grew. Growth density was measured as the OD600 after subtraction of a negative control. Boxes encompass the 1st to 3rd quartile of each population. Whiskers extend up to 1.5 times the interquartile range from the closest box boundary. Data points that are out of the whisker range are outliers and are denoted by individual points. Significance relative to the ancestor denoted by Wilcoxon rank sum test with Benjamini-Hochberg correction (***P-value < 0.001, **P-value < 0.01, *P-value < 0.05). (D) Summary of the percent of total carbon sources that supported growth of each lineage.
To better understand growth differences across lineages, we measured and summarized the maximum growth densities across growth-supporting carbon sources for each lineage (Figure 2C, Figure S3, and Data S2). The maximum growth density was defined as the maximum background-subtracted optical density measured at 600nm (OD600) of the median of three replicate 48-hour growth curves. Interestingly, all evolved lineages, including the LB-evolved control, exhibited a significantly decreased maximum growth density relative to the ancestral lineage (P-value < 0.05). Each lineage was found to have a different number of growth-supporting carbon sources (Figure 2D). The ancestral lineage grew on 31.6% (60/190) carbon sources. Comparatively, ciprofloxacin- and piperacillin-evolved lineages were the most metabolically limited, only growing on 13.2% (25/190) and 23.2% (44/190) of the tested carbon sources, respectively. The LB-evolved control was only able to grow on 27.9% (53/190) of carbon sources, while tobramycin-evolved lineage grew on 29.5% (56/190) of sources. Overall, adapted lineages were more metabolically limited and had decreased growth relative to the unevolved ancestral lineage.
Altered growth dynamics of antibiotic-resistant P. aeruginosa
To more rigorously examine metabolic differences of our antibiotic-resistant lineages, we quantified the growth dynamics of each individual growth curve across carbon sources that supported growth of all five lineages (Figure 1A and Data S3). For each of the 14 growth-supporting carbon sources, three parameters of bacterial growth were calculated: maximum growth density, doubling time, and time to reach mid-exponential phase (Figure 3A-F). The maximum growth density varied by carbon source and by lineage (Figure 3A). All evolved P. aeruginosa lineages except for the tobramycin-evolved lineage exhibited significant decreases in maximum growth density relative to the ancestral lineage (P-value < 0.05) (Figure 3D). The LB-evolved control had one of the largest decreases in growth density, indicating that media condition impacted the maximum level of growth perhaps more so than antibiotic pressure.
Figure 3. Growth dynamics of antibiotic-resistant P. aeruginosa.
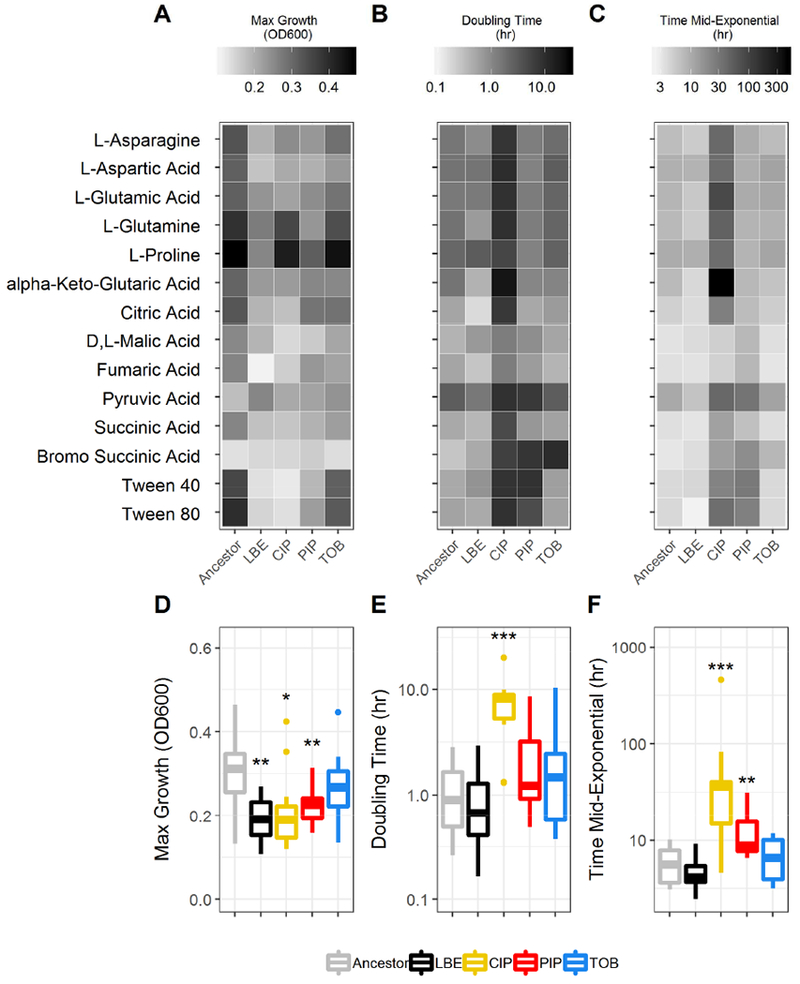
For each carbon source that supported growth on all five studied lineages, growth dynamics, including the maximum growth density (OD600) (A,D), doubling time (hr) (B,E), and time to reach mid-exponential phase (hr) (C,F) were calculated. Boxes encompass the 1st to 3rd quartile of each population. Whiskers extend up to 1.5 times the interquartile range from the closest box boundary. Significance relative to the ancestor determined by Wilcoxon rank sum test with Benjamini-Hochberg correction (***P-value < 0.001; **P-value < 0.01, *P-value < 0.05).
For each growth curve, we also calculated the doubling time and the time taken to reach midexponential phase (Figure 1A). A basic logistic equation was fit to each median growth curve to determine growth dynamics (Sprouffske and Wagner, 2016). Across the 14 universal growth-supporting carbon sources, the ciprofloxacin-evolved lineages had a significantly longer doubling time than the ancestral lineage (P-value < 0.05) (Figures 3B and 3E). The median doubling time of the piperacillin-evolved and tobramycin-evolved lineages also increased and the LB-evolved control slightly decreased; however, these changes were not significant (P-value > 0.05). The ciprofloxacin-evolved and piperacillin-evolved lineages took significantly longer than the ancestral lineage to reach mid-exponential growth (P-value < 0.05) (Figures 3C and 3F). Overall, the ciprofloxacin-evolved lineage exhibited longer lag phases and longer doubling times while the tobramycin-evolved lineage growth curves were relatively similar to the ancestor and LB-evolved control (Figure S1). These findings demonstrate that adaptation to different antibiotics on rich media can heterogeneously impact the growth dynamics of P. aeruginosa across many growth-supporting conditions.
Tobramycin-resistant P. aeruginosa shows enhanced growth on N-acetyl-D-glucosamine
On multiple carbon sources, we observed that an antibiotic-evolved lineage exhibited enhanced growth relative to the ancestral lineage. This phenotype is potentially of greater concern in a clinical setting since a drug treatment that selects for both resistant and metabolically advantaged pathogens could have negative clinical outcomes.
One example of enhanced growth occurred in the tobramycin-evolved lineage, which grew to a higher growth density than the ancestral lineage on the carbon source N-acetyl-D-glucosamine (Figure 4A). N-acetyl-D-glucosamine is a component of human mucin as well as the cell wall of Gram-positive organisms and is thought to be present in sputum of CF patients (Korgaonkar and Whiteley, 2011; Palmer et al., 2007). Consistent with phenotypic profiling data, three additional tobramycin-evolved lineages of P. aeruginosa from the previous study (Yen and Papin, 2017) showed enhanced growth on N-acetyl-D-glucosamine; maximum growth densities were significantly increased in two of these three lineages relative to the ancestral lineage (P < 0.05). This phenotype was not shared with any LB-evolved control lineages, all four of which exhibited significantly decreased maximum growth densities relative to the ancestral lineage (P < 0.05) (Figures 4A and S4). These results indicate that this phenotype of enhanced growth is robust and may be affected by adaptation to tobramycin as opposed to LB media. Growth profiling revealed that the piperacillin-evolved lineage also displayed enhanced growth on N-acetyl-D-glucosamine, suggesting that multiple classes of antibiotics may impact growth on this carbon source (Figure S1A).
Figure 4. Tobramycin-resistant PA14 exhibits enhanced growth on N-Acetyl-D Glucosamine.
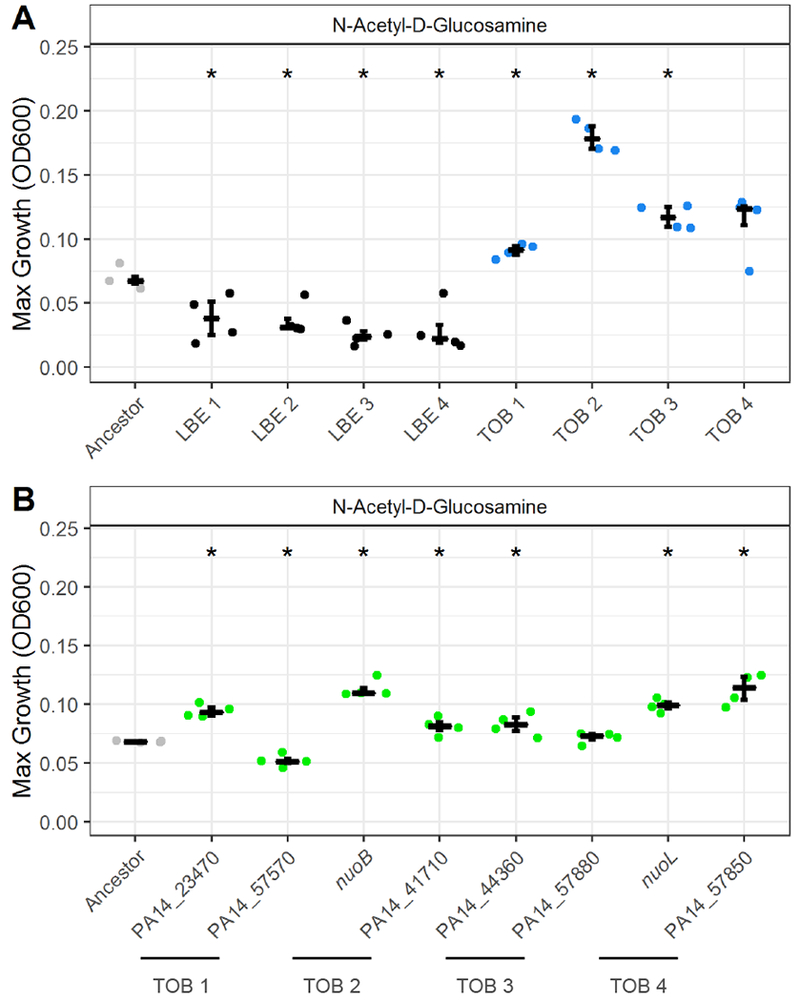
(A) Maximum growth (OD600) of four independent tobramycin-evolved lineages (blue) and four independent LB-evolved lineages (black) on 20mM of N-acetyl-D-glucosamine relative to the unevolved ancestor (grey). Numbers refer to replicate lineages from a previous study (Yen and Papin, 2017). TOB 3 and LBE 2 correspond to TOB and LBE from the phenotypic profiling analysis. (B) Maximum growth (OD600) of eight transposon mutants (green) on 20mM of N-acetyl-D-glucosamine relative to ancestor (grey). Significance relative to the ancestor determined by Wilcoxon rank sum test with Benjamini-Hochberg correction (*P-value < 0.05). Crossbar denotes median and error bars show the interquartile range of four replicate colonies.
Although all four independent tobramycin-evolved lineages exhibited an increase in maximum growth density on N-acetyl-D-glucosamine and some genetic mutations occurred in similar pathways, there were no mutations that were universally shared, indicating that multiple evolutionary paths to tobramycin resistance could be associated with this metabolic phenotype. To explore each path, the effect of two genetic mutations for each lineage on N-acetyl-D-glucosamine utilization were evaluated. In total, eight transposon mutants were selected from the non-redundant PA14 library and grown in N-acetyl-D-glucosamine (Table S1 and Figure S4B) (Liberati et al., 2006). Six out of eight of the mutants had significantly increased maximum growth densities relative to the ancestral lineage: PA14_23470, nuoB, PA14_41710, PA14_44360, nuoL, and PA14_57850 (P < 0.05) (Figure 4B). One mutant, PA14_57570, had significantly decreased growth and another, PA14_57880, was not significantly different from the ancestor. At least one mutated gene for each of the four tobramycin-evolved lineages had enhanced growth on N-acetyl-D-glucosamine (Figure S4B).
Transposon mutants with insertions in the nuoL, nuoB, and PA14_57570 genes exhibited slight increases in resistance to tobramycin relative to wild type P. aeruginosa; however, based on defined clinical breakpoints, the minimal inhibitory concentrations (MIC) were not elevated enough for the mutants to be classified as resistant (Table S1) (EUCAST, 2018). All other mutations had a less than two-fold increase in the MIC of tobramycin. The exact mechanism by which these genes impact growth on N-acetyl-D-glucosamine needs to be resolved with further experimentation. However, these results demonstrate one example of how resistance-associated genetic mutations can unexpectedly and heterogeneously impact metabolism in P. aeruginosa.
Network-guided predictions identify mutations that impact loss of catabolic function in piperacillin-resistant P. aeruginosa
Contrary to other evolved-lineages which contained only a handful of sequenced mutations, the piperacillin-evolved lineage contained a large deletion of 343 genes (Data S4) (Yen and Papin, 2017). This deletion complicated our ability to identify clear genotype-phenotype relationships in the piperacillin-evolved lineage. To contextualize the potential impact of each gene in the large deletion on the multiple losses of catabolic function observed in our metabolic phenotyping data, we simulated single-gene knockout experiments across 42 environments with a genome-scale metabolic network reconstruction of P. aeruginosa PA14 (Figure 1B) (Bartell et al., 2017). Each computationally tested environment contained the components of M9 minimal media and a single carbon source. For a carbon source to be considered for the in silico analysis, it had to meet several criteria. First, it had to have been included in our experimental screen of metabolic phenotypes. Second, the P. aeruginosa metabolic network reconstruction had to contain a transport reaction for the carbon source, representing annotated evidence of a functional transport of the metabolite across the cell membrane. A transport reaction can be thought of as a way for P. aeruginosa to uptake or secrete a particular metabolite. Finally, for a carbon source to be included in the analysis it had to support the production of a non-zero biomass in the complete model, where biomass can be considered a proxy for bacterial growth. For each of the 42 carbon sources that met these requirements, we simulated single-gene knockouts to evaluate the contribution of each gene on model growth and carbon source catabolism. Genes that when knocked out in the model resulted in a total loss of biomass production were predicted to be essential for growth on the tested carbon source.
To predict which genes in the large deletion of the piperacillin-evolved lineage could have impacted observed changes in metabolic phenotype, we looked for overlap between deleted genes identified by whole-genome sequencing and model-predicted essential genes (Figure S5). In total, there were 17 genes shown to be deleted in the piperacillin-evolved lineage that were predicted to be essential for growth on at least one of the 42 experimentally measured carbon sources (Figure 5). For it to be possible to validate an essentiality prediction, the ancestral lineage needed to be able to grow on the carbon source of interest. Otherwise, we could not determine the effect of knocking out a single gene on catabolic function. We were unable to evaluate the effect of eight of the 17 genes because they were predicted to be essential for growth on four carbon sources that were unable to support growth of the ancestral lineage: D-ribose, D-serine, L-serine, and L-phenylalanine. Another two of the 17 genes, bacA and glgA, were predicted to be essential across all environmental conditions, including LB media. While neither of these genes were essential for growth in vitro, they were associated with the ability of the model to produce biomass. Predictions of the knockout of these genes, while perhaps not directly informative for our study of antibiotic resistance, provide useful new information for model curation.
Figure 5. Model-guided prediction of loss of metabolic function in piperacillin-evolved P. aeruginosa.
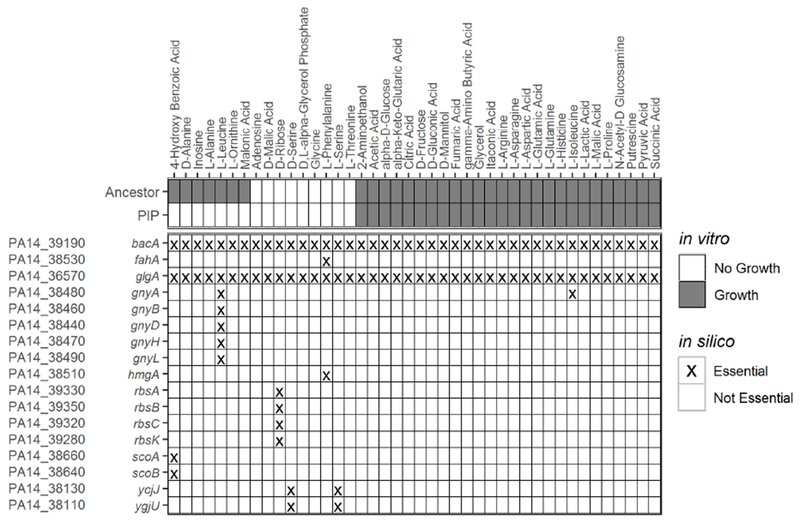
Experimental growth calls of the piperacillin-evolved and ancestral lineages on carbon sources able to support growth on the base iPau1129 model (top). Model predicted genes essential for growth on minimal media with each carbon source (bottom). Only the subset of essential genes that were found to be deleted in the piperacillin-evolved lineage are included (In vitro: white = no growth, grey = growth; in silico: X = essential gene, blank = non-essential gene).
The role of each of the remaining seven predicted essential genes on catabolic function were examined in more detail (Figure 6A-D). Five of the seven remaining genes deleted in the piperacillin-evolved lineage were predicted to be essential for L-leucine utilization: gnyA, gnyB, gnyD, gnyH, gnyL. A cluster of PAO1 genes orthologous to the gnyABDHL cluster has been previously reported to be involved in L-leucine catabolism, specifically in the downstream catabolism of isovaleryl-CoA into the citric acid cycle substrate acetyl-CoA (Figure 6A) (Aguilar et al., 2006). The piperacillin-evolved lineage was found to lack the ability to grow on L-leucine while the ancestral strain was able to grow on this carbon source (Figures 6C and S6A). Based on the predictions of the genome-scale metabolic network reconstruction, we hypothesized that this loss of catabolic function in the piperacillin-evolved lineage was due to the deletion of the five genes in the gny operon. Our hypothesis was supported by the finding that a gnyA transposon mutant was unable to grow in minimal media with L-leucine, demonstrating that gnyA is necessary for L-leucine utilization (Figures 6C and S6A). Our model predicts that deletion of any one of the remaining gny genes would have a similar effect on L-leucine catabolism.
Figure 6. Experimental validation of model-guided gene essentiality predictions.
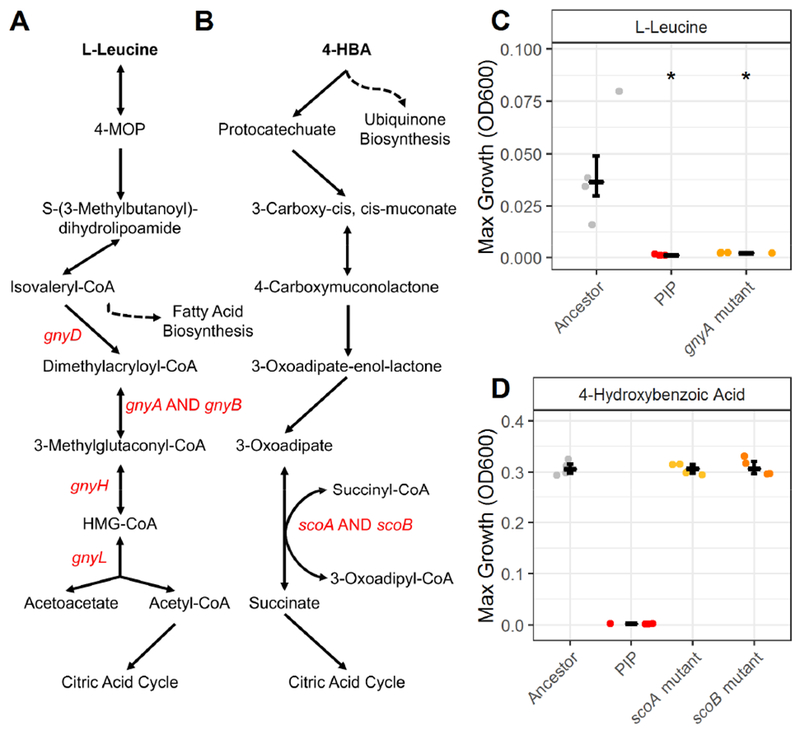
Pathways of L-leucine (A) and 4-hydroxybenzoic acid (B) catabolism according to the GENRE of P. aeruginosa, iPau1129. Genes that were shown to be deleted in the piperacillin-evolved lineage are labelled in red. Dashed lines indicate connections to other pathways in the metabolic network reconstruction. (C) The maximum growth of the ancestral lineage (grey), piperacillin-evolved lineage (red), and a gnyA transposon mutant on L-leucine. (D) The maximum growth of the ancestral lineage (grey), piperacillin-evolved lineage (red), and scoA (solid orange) and scoB (dashed orange) transposon mutants on 4-hydroxybenzoic acid. Significance relative to the ancestor determined by Wilcoxon rank sum test with Benjamini-Hochberg correction (*P-value < 0.05). Crossbar denotes median and error bars show the interquartile range of four replicate colonies.
The remaining three essential gene predictions were experimentally determined to be false positives. The gnyA gene was predicted to be essential for utilization of L-isoleucine; however, both the piperacillin-evolved lineage and gnyA transposon mutant were able to grow on this carbon source (Figures 5 and S6B). The exact reason that gnyA was erroneously predicted as essential remains to be determined and indicates that the current understanding of L-isoleucine utilization in P. aeruginosa as it is represented in our model is incomplete. Two genes, scoA and scoB, were predicted to be essential for the utilization of 4-hydroxybenzoic acid (Figure 5). These genes have been reported to encode for subunits A and B of a CoA-transferase (Winsor et al., 2016). According to the P. aeruginosa GENRE, scoA and scoB use a downstream product of 4-hydroxybenzoic acid degradation to catalyze the conversion of succinyl-CoA to succinate in the citric acid cycle (Figure 6B). We found that both scoA and scoB mutants were able to grow on 4-hydroxybenzoic acid. Growth curves of both mutants matched the growth of the ancestral lineage and while the piperacillin-evolved lineage was unable to grow on 4-hydroxybenzoic acid (Figures 6D and S6C). From this result we conclude that scoA and scoB are not individually necessary for growth on 4-hydroxybenzoic acid. Further evaluation is required to determine why these genes were falsely predicted to be essential as well as why the piperacillin-evolved lineage was unable to grow on 4-hydroxybenzoic acid.
Two independent piperacillin-evolved lineages from a previous study were found to have large deletions partially overlapping with the lineage profiled in this study (Yen and Papin, 2017). Applying the same computational approach to these two piperacillin-resistant lineages, we identified eight additional genes deleted in at least one of these lineages predicted to be essential for utilization of at least one carbon source (Figure S5B-C). We predict that the majority of metabolic phenotypes would be conserved across all three piperacillin-evolved lineages.
Clinical isolates share loss of metabolic function observed in lab-evolved piperacillin-resistant P. aeruginosa
To investigate the clinical relevance of the differential metabolic functionality observed in the drug-evolved lineages, we asked whether clinical isolates with large chromosomal mutations comparable to the piperacillin-resistant lineage would also exhibit an inability to catabolize L-leucine. A previous study characterized four pyomelanogenic (PM) isolates with reduced metabolic flexibility and found that they each had large chromosomal deletions (APM, BPM, CPM, and DPM). For each PM isolate, they also identified ‘parental’ wild type (WT) isolates that were genetically identical to the PM isolates but lacked large deletions, had a significantly faster growth rate on rich media, and did not produce pyomelanin (AWT, BWT, CWT, and DWT) (Hocquet et al., 2016). The large deletions sequenced in the PM isolates were previously found to overlap with the 343 genes deleted in the piperacillin-resistant lineage (Yen and Papin, 2017). To test the hypothesis that the loss of catabolism of L-leucine observed in the piperacillin-resistant lineage and gnyA transposon mutant would be shared with the PM isolates but not the WT isolates, we grew each isolate pair on minimal media with 40mM of L-leucine and measured growth over 48 hours (Figure S6D). The median maximum growth density was measured for each isolate across four replicate colonies. Across all four clinical isolate pairs, the WT isolates reached significantly higher maximum growth densities than the isolates with large chromosomal deletions, which were unable to grow (Figure 7). The DWT and DPM pair, originally isolated from CF sputum, displayed the largest growth difference of the four isolate pairs. This finding suggests that the loss of L-leucine catabolism seen in the piperacillin-resistant lineage and determined to be impacted by the gnyA gene may be a clinically relevant phenotype.
Figure 7. Clinical isolates with large deletions exhibit loss of catabolic function similar to piperacillin-evolved P. aeruginosa.
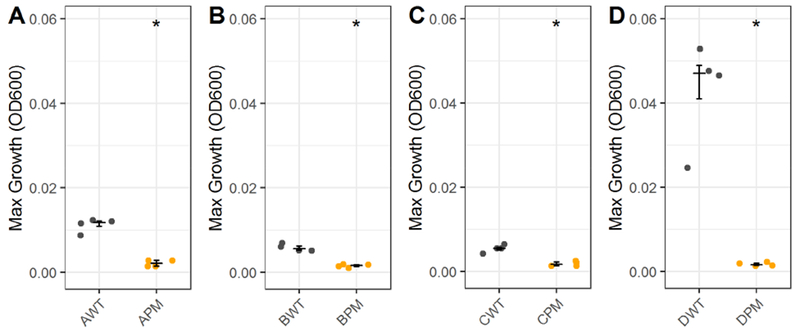
Maximum growth of clinical isolates from (Hocquet et al., 2016) with large chromosomal deletions (PM) and paired parental wild type (WT) isolates without large deletions in L-leucine. Growth was measured on four isolate pairs: (A) AWT and APM, (B) BWT and BPM, (C) CWT and CPM, and (D) DWT and DPM. Significance relative to wild type parent determined by Wilcoxon rank sum test (*P-value < 0.05). Crossbar denotes median and error bars show the interquartile range of four replicate colonies.
Altogether, we paired whole-genome sequencing with a genome-scale metabolic network reconstruction to identify five genes associated with loss of metabolic function in piperacillin-resistant P. aeruginosa. Clinical isolates with similar mutational landscapes to lab-evolved piperacillin-resistant P. aeruginosa were found to share loss of the ability to catabolize L-leucine. Three additional genes predicted to be essential by the model were experimentally determined to be non-essential, highlighting gaps in our current knowledge of metabolism that need to be further investigated. While some of our predictions may be retrospectively obvious, they were all guided by the model, without which we would have been unable to reconcile direct genotype-phenotype relationships in such a complex mutational landscape. A similar approach could be applied to predict metabolic deficits in any bacterial strain with an annotated genome.
Discussion
We have shown that P. aeruginosa experiences systems-level changes to metabolism when exposed to sustained antibiotic pressure. Through the analysis of 950 growth curves on 190 unique carbon sources, we determined that antibiotic-resistant P. aeruginosa exhibit differential growth dynamics from antibiotic-sensitive lineages. We applied a highly curated genome-scale metabolic network reconstruction to predict which mutations in our antibiotic-evolved lineages were impacting loss of metabolic function. Finally, model predictions were validated with mutants with single-gene transposon insertions in genes of interest. Deletions of genes in the gnyABDHL cluster in the piperacillin-evolved lineage were found to result in loss of L-leucine utilization. Our findings emphasize the interconnectivity of antibiotic resistance and metabolism and support future efforts to consider this relationship in the design of new antibiotic regimens.
Consistent with literature, lab-evolved resistant strains displayed mostly decreased metabolic activity with occasional increases in activity across measured environments relative to the unevolved ancestor (Björkman et al., 2000; Fabich et al., 2011; Marvig et al., 2015; Rau et al., 2010). The ciprofloxacin-resistant lineage was found to be the most metabolically limited, growing on the fewest number of carbon sources, exhibiting significantly decreased maximum growth, and taking significantly longer for the population to double and reach mid-exponential phase. The piperacillin-resistant lineage was found to have decreased metabolic flexibility, in part due to a large deletion that removed many metabolic genes and led to the inability to catabolize L-leucine and other carbon sources. We showed that clinical isolates with similar large chromosomal deletions also lacked the ability to catabolize L-leucine and we predict that these isolates would share other metabolic phenotypes with the piperacillin-resistant lineage (Hocquet et al., 2016). Patients from which the isolates were collected had not been recently treated with piperacillin or other beta-lactams, indicating that such large deletions are not necessarily drug-specific, but could be more likely to arise when the cell is under general stress.
The tobramycin-resistant lineage exhibited the fewest metabolic deficits and demonstrated enhanced growth on the carbon source N-acetyl-D-glucosamine. This phenotype was found to be shared with three other independent tobramycin-evolved lineages, and while many of the genetic mutations in these lineages were found to be in similar pathways, there were no universally shared mutations. Mutation of six of these genes, each observed in a single tobramycin-evolved lineage, increased growth on N-acetyl-D-glucosamine. Two of these six mutated genes, nuoL and nuoB, exhibited slight increases in resistance to tobramycin, indicating that resistance-associated mutations had a direct impact on the observed growth phenotype. Mutations in the nuo complex encoding for subunits of the NADH Dehydrogenase I have been previously associated with aminoglycoside tolerance (El’Garch et al., 2007; Schurek et al., 2008; Shan et al., 2015; Yen and Papin, 2017). More generally, disruptions to the electron transport chain have been shown to confer aminoglycoside resistance (Mayer et al., 2015; McCollister et al., 2011; Taber et al., 1987). Consistent with previous literature, mutation of the PA14_57570 gene, encoding for a subunit of the cytochrome c reductase, also resulted in increased resistance to tobramycin (Døtsch et al., 2009); however, this mutation resulted in significantly decreased growth on N-acetyl-D-glucosamine. Our results show that resistance-associated mutations can have heterogeneous impacts on the growth of P. aeruginosa. There remains more experimental work to resolve the exact mechanism by which these genes impact growth on N-acetyl-D-glucosamine, whether through direct catabolism or toxicity (Konopka, 2012).
There were two potential sources of confounding error in this study that were infeasible to resolve. First, all of the growth calls and growth dynamics presented were derived from measurements of optical density over time. Biological processes such as cell widening and pigment production can confound optical measures of population growth (Stevenson et al., 2016); however, we could not have achieved the same level of temporal resolution for as many conditions with other metrics of growth. Second, we observed a significant decrease in the maximum growth density of the LB-evolved control lineage relative to the ancestral lineage, indicating that evolving to media and antibiotic at the same time may have had confounding effects on metabolism. Media condition has been shown to impact evolution of resistance, and evolving to rich media in the absence of drug has been found to result in altered growth phenotypes and deleterious mutations. (Fong et al., 2005; Kram and Finkel, 2015; Westphal et al., 2018; Zampieri et al., 2017). For future adaptive laboratory evolution studies, we recommend that the ancestral lineage be adapted to the base media and then subsequently adapted to the antibiotic of interest to control for the effect of media (Baym et al., 2016). Despite the impact of adaptation to LB media on maximum growth density, we still observed metabolic phenotypes and growth dynamics unique to antibiotic-evolved lineages.
Genome-scale metabolic network reconstructions are becoming increasingly prevalent in the study of antibiotic resistance (Banerjee et al., 2017; Carey et al., 2017; Dunphy and Papin, 2017; Zampieri et al., 2017). Although the P. aeruginosa metabolic network reconstruction does not account for many genes associated with resistance (e.g. regulation of efflux, membrane permeability), the model allowed for the rapid identification of potential mutations impacting growth phenotypes. Erroneous predictions provided useful starting points for future model curation and increased understanding of metabolic functionalities. The majority of the overlap between model genes and mutations uncovered from genome sequencing occurred in the large deletions of the piperacillin-resistant lineages. Due to the limited number of mutations in metabolic genes across the other evolved lineages, we were unable to find specific genetic mechanisms for many of the experimentally observed changes in metabolic phenotype. We speculate that metabolic adaptations that could not be explained by our model simulations may have been affected by resistance mutations that impact transcriptional regulation. We would expect the integration of a transcriptional regulatory network or transcriptomic profiling data with the metabolic network reconstruction to uncover more genotype-phenotype relationships in the resistant lineages (Blazier and Papin, 2012; Carey et al., 2017; Lister et al., 2009; Livermore, 2002; Monk et al., 2017).
While this study shows that there are systems-level metabolic changes following sustained antibiotic pressure, the evolution of resistance is a stochastic process that is sensitive to a wide variety of environmental and host factors not accounted for in our experimental design. As such, observed mutations may have varied with a different selection of antibiotics or media conditions. Nevertheless, many of the mutations we observed were consistent with those in sequenced clinical P. aeruginosa isolates, indicating that the general trends we observed may be robust to nutrient differences between LB media and CF sputum (Hocquet et al., 2016; Mayer et al., 2015). Additionally, through the interrogation of the growth of tobramycin-evolved P. aeruginosa on N-acetyl-D-glucosamine, we have shown that metabolic phenotypes can be shared across resistant lineages with independent evolutionary paths and heterogeneous mutational landscapes. While we have chosen to focus on three antibiotics and one media condition in a single bacterial species, our combined experimental and computational approach can be applied to a large variety of organisms and environmental pressures. Moving forward, we have laid the groundwork for the interrogation of broad metabolic consequences of antibiotic resistance.
Star Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jason Papin (papin@virginia.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains and culture conditions
Pseudomonas aeruginosa strain UCBPP-PA14 was previously evolved to one of three antibiotics (piperacillin, tobramycin, or ciprofloxacin) in lysogeny broth (LB) media for 20 days (Yen and Papin, 2017). As a control, UCBPP-PA14 was also adapted to LB media in the absence of drug for 20 days. One representative lineage from each 20-day evolution described previously (Yen and Papin, 2017) was selected for phenotypic growth profiling in this study. The lineages used were as follows: Day 0 Ancestor, Day 20 Piperacillin Replicate 1, Day 20 Tobramycin Replicate 3, Day 20 Ciprofloxacin Replicate 4, and Day 20 Control Replicate 2. All four Day 20 tobramycin-evolved replicate lineages and control replicate lineages were used in experiments on N-acetyl-D-glucosamine and their numbering was consistent with previous analyses (Figures 4A and S4A) (Yen and Papin, 2017). Day 20 Piperacillin Replicates 2 and 3 were included in expanded computational predictions (Figure S5B-C). Prior to phenotypic screening, frozen stocks of resistant or unevolved sensitive P. aeruginosa were streaked on LB agar plates (1% tryptone, 0.5% yeast extract, 1% NaCl) and incubated for 18–22 hours at 37C. The reported minimal inhibitory concentrations of tobramycin (Sigma) in Table S1 were determined in LB media using the broth microdilution method as previously described (Yen and Papin, 2017).
For growth experiments on the carbon sources N-acetyl-D-glucosamine (>95% (HPLC), Sigma), L-leucine (Sigma), and L-isoleucine (Sigma), frozen stocks of the appropriate P. aeruginosa lineages were streaked on an LB agar plate and incubated for 24 hours at 37C. Single colonies were isolated from the streak plates and grown for 24 hours in 5mL of LB media at 37C and shaken at 120rpm. Overnight cultures were diluted down to a starting OD600 of 0.001 (10Λ6 CFU/mL) in 200uL of M9 minimal media containing the designated carbon source. Prior to inoculation, overnight cultures were washed three times in 1X PBS (6,000rpm for 5 minutes). A concentration of 20mM was used for all carbon sources except L-leucine, which was used at a concentration of 40mM. The OD600 was measured every 10 minutes for 48 hours (Tecan Infinite M200 Pro).
P. aeruginosa UCBPP-PA14 Transposon Mutants
P. aeruginosa mutants with transposon insertions in the nuoL (mutant ID 32173), nuoB (mutant ID 42761), and PA14_41710 (mutant ID 53876) genes were selected from the non-redundant PA14 genome-scale transposon library (PA14NR Set) (Liberati et al., 2006). The mutants were grown on an LB agar plate at 37C. Transposon mutants with insertions in the following genes were grown LB agar plates supplemented with gentamicin and incubated at 37C for 18-22 hours: gnyA (mutant ID 45885), scoA (mutant ID 53819), scoB (mutant ID 34027), PA14_23470 (mutant ID 28941), PA14_57570 (mutant ID 54834), PA14_57880 (mutant ID 35720), PA14_44360 (mutant ID 40719), and PA14_57850 (mutant ID 43223). Single colonies of each mutant were then selected and grown in LB liquid culture at 37C overnight, shaken at 125rpm and were frozen in 25% glycerol. The presence of transposon insertions in the expected genes were verified by PCR (Table S2).
P. aeruginosa Clinical Isolates
Four pairs of clinical isolates were collected by Hocquet et al. as described in their study (Hocquet et al., 2016). Four replicate colonies of each of the eight isolates were grown in 40mM of L-leucine for 48-hours as described above.
METHOD DETAILS
Whole-genome sequencing
Whole-genome sequencing was previously performed on the evolved strains of P. aeruginosa used in this study (NCBI SRA database (www.ncbi.nlm.nih.gov/sra) accession number SRP100674, BioProject number PRJNA376615) (Yen and Papin, 2017). In summary, samples of adaptively evolved strains and wild type ancestor were streaked on LB plates and incubated at 37C before being submitted to Genewiz Incorporation for sequencing (Illumina HiSeq 2500). Sequence reads were aligned to the P. aeruginosa PA14 reference genome (NC_008463.1).
Phenotypic growth profiling via single-carbon source utilization screens
Following incubation on an LB agar plate at 37C for 18-22 hours, bacteria were scraped from a lawn on an LB agar plate and transferred to 1.5mL of IF-0a GN/GP inoculating fluid (Biolog). The sample was vortexed until the liquid culture appeared turbid and contained no visible clumps of bacteria. The culture was then diluted to a starting OD600 of 0.07 in 10-15mL of inoculating fluid. 100uL of liquid culture was transferred to each well of a Phenotype Microarray plate (PM1 or PM2a, Biolog), mixing thoroughly to dissolve lyophilized carbon sources at the bottom of each well, and OD600 was measured every 10 minutes for 48 hours (Tecan Infinite M200 Pro). Three PM1 and three PM2a Phenotype Microarray plates each were used for the ancestral, LB-evolved, piperacillin-evolved, ciprofloxacin-evolved, and tobramycin-evolved lineages. To ensure the homogeneity of each lineage, a different lawn was used for each plate. Each starting lawn was considered a biological replicate. No antibiotics were used in these growth profiling experiments. Plots of all 950 median growth curves can be found in the supplement (Figure S1).
QUANTIFICATION AND STATISTICAL ANALYSIS
Growth cutoff for phenotypic profiling
Each well of the Biolog Microarray plates contained a single carbon source, totaling 190 unique carbon sources and two negative control wells across two, 96-well plates. The median of the negative control wells (median of three colonies for each strain) were subtracted from the median growth curves (across three colonies for each strain). Bacteria were considered to have grown on a given carbon source if the AUC of the growth curve after subtracting the negative control was greater than or equal to a defined threshold. The AUC was chosen to avoid noise associated with single-time point measurements such as the maximum or endpoint optical densities. AUC values correlated well with the maximum optical density across all of the data (Pearson correlation = 0.943, Figure S7). A lineage was considered to have grown on a given carbon source if the AUC was in the upper quartile of data (3rd quartile = 15.8). This threshold was generally consistent with growth calls made by manual inspection of the curves. To rigorously examine the effect of the growth cutoff value on the results of this study, we increased and decreased the cutoff by up to 50% and recalculated the growth dynamics for each lineage on growth-supporting carbon sources. While the carbon sources included in the analyses varied slightly, trends in growth dynamics remained stable across the full range of threshold perturbations (Figure S2).
Calculation of growth dynamics
Three growth parameters were calculated on the background-subtracted median growth curves collected from the Biolog Microarray plates: maximum growth density, doubling time, and time to mid-exponential phase. The maximum growth density was reported as the maximum OD600 value along each growth curve. The doubling time and time to mid-exponential phase were calculated using the growthcurver package in R, which fits a basic logistic equation to growth data (Sprouffske and Wagner, 2016). In brief, the doubling time was taken to be the natural log of 2 divided by the growth rate, where the growth rate was the maximum slope of the natural log of the growth curve. The time to reach mid-exponential phase was defined as the time at which the culture reached half of the maximum stationary phase OD600 or theoretical maximum OD600 if the curve did not reach stationary phase in 48 hours.
The maximum growth densities were calculated for each replicate colony in Figures 4, 6C, 6D, 7, and S4C. Prior to calculating the maximum optical density, the minimum OD600 value of each replicate colony growth curve was subtracted from every time point. The maximum OD600 was taken from each of these minimum-subtracted growth curves and the distributions of maximums were reported.
Genome-scale metabolic network reconstruction of Pseudomonas aeruginosa
The recently published genome-scale metabolic network reconstruction of wild type P. aeruginosa UCBPP-PA14 was used for this study (Bartell et al., 2017). The model accounts for the functions of 1129 genes and 1495 reactions. Previous analysis revealed that the model was 81% accurate at predicting growth phenotypes of wild type P. aeruginosa on 91 experimentally measured carbon sources (Bartell et al., 2017). This accuracy was maintained in the subset of carbon sources examined in this study.
Gene essentiality predictions
75 carbon sources of the 190 measured carbon sources had exchange reactions in the genome-scale metabolic network reconstruction, iPau1129. An exchange reaction represents the ability to import a carbon source into the model system. This is different from a transport reaction, which can be thought of as a way to transport a metabolite between the media and the cell cytosol. We temporarily added exchange reactions for another nine carbon sources that had transport reactions but lacked exchange reactions. The base model supported growth, defined as non-zero biomass, on 42 of these 84 carbon sources. We used iPau11290 to simulate growth of P. aeruginosa in each of these carbon sources with minimal media. Using the singleGeneDeletion function in the COBRA Toolbox (Schellenberger et al., 2011), we simulated single-gene knockouts of each gene in the presence of each unique carbon source. From these simulations, we predicted genes for each media condition that when knocked out, resulted in the model no longer being able to support growth. We refer to genes predicted to be required for growth as essential genes for a given environment. We looked for overlap between essential genes predicted by the model and mutated genes in our evolved resistant strains. This overlap was used to predict resistance-specific essential genes as well as to identify incorrect model predictions.
Statistical tests and clustering
Statistical comparisons between the ancestral lineage and all evolved lineages and transposon mutants were made with the nonparametric two-tailed Wilcoxon rank sum test, which does not require data to be normally distributed. The Benjamini-Hochberg procedure was used to adjust the P-value when there were multiple comparisons (Benjamini and Hochberg, 1995). Tests were performed on distributions of biological replicates or on distributions of medians across biological replicates. A P-value below 0.05 was considered significant (Table S3). Samples were assumed to be independent. The Jaccard distances between lineages were calculated on binary growth calls across all carbon sources using the vegan package in R (Oksanen et al., 2018) and the distance matrix was clustered using the complete-linkage method.
DATA AND SOFTWARE AVAILABILITY
All code and data used to calculate growth dynamics, make gene essentiality predictions, as well as generate figures and supplemental data (excluding Figure 1) can be found at https://github.com/lauradunphy/dunphy_yen_papin_supplement. Complete growth data, binary growth calls, growth dynamics, and computational gene essentiality predictions can be found in the supplement (Data S1–S4).
Supplementary Material
Data S2 Complete binary and numerical growth calls. Related to Figure 2.
Data S3. Growth dynamics. Related to Figure 3.
Data S4. Complete gene essentiality predictions. Related to Figure 5.
Highlights:
Growth profiling of antibiotic-resistant P. aeruginosa on 190 carbon sources
Quantification of growth dynamics reveals altered metabolic functionality
Prediction of metabolic impacts of individual mutations with a computational model
Experimental validation of predicted genotype-phenotype relationships
Acknowledgements
The authors would like to thank lab-mates Anna S. Blazier and Dr. Glynis Kolling for their feedback on the experimental design and manuscript. We would like to give special thanks to Didier Hocquet for providing the clinical isolates used in this study. We would also like to thank Dr. Joanna Goldberg from Emory and Dr. Jason Yang from MIT for their advice on the project. This work was funded by the National Science Foundation Graduate Research Fellowship Program [grant number DDGE-1315231] and the National Institutes of Health [grant number R01-GM088244].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests.
References
- Aguilar JA, Zavala AN, Díaz-Pérez C, Cervantes C, Díaz-Pérez AL, and Campos-García J (2006). The atu and liu Clusters Are Involved in the Catabolic Pathways for Acyclic Monoterpenes and Leucine in Pseudomonas aeruginosa. Appl. Environ. Microbiol 72, 2070–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alanis AJ (2005). Resistance to antibiotics: are we in the post-antibiotic era? Arch. Med. Res 36, 697–705. [DOI] [PubMed] [Google Scholar]
- Banerjee D, Parmar D, Bhattacharya N, Ghanate AD, Panchagnula V, and Raghunathan A (2017). A scalable metabolite supplementation strategy against antibiotic resistant pathogen Chromobacterium violaceum induced by NAD+/NADH+ imbalance. BMC Syst. Biol 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartell JA, Blazier AS, Yen P, Thøgersen JC, Jelsbak L, Goldberg JB, and Papin JA (2017). Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat. Commun 8, ncomms14631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baym M, Lieberman TD, Kelsic ED, Chait R, Gross R, Yelin I, and Kishony R (2016). Spatiotemporal microbial evolution on antibiotic landscapes. Science 353, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol 57, 289–300. [Google Scholar]
- Björkman J, Nagaev I, Berg OG, Hughes D, and Andersson DI (2000). Effects of Environment on Compensatory Mutations to Ameliorate Costs of Antibiotic Resistance. Science 287, 1479–1482. [DOI] [PubMed] [Google Scholar]
- Blazier AS, and Papin JA (2012). Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol 3, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordbar A, Monk JM, King ZA, and Palsson BO (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet 15, 107–120. [DOI] [PubMed] [Google Scholar]
- Carey MA, Papin JA, and Guler JL (2017). Novel Plasmodium falciparum metabolic network reconstruction identifies shifts associated with clinical antimalarial resistance. BMC Genomics 18, 543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Døtsch A, Becker T, Pommerenke C, Magnowska Z, Jänsch L, and Häussler S (2009). Genomewide Identification of Genetic Determinants of Antimicrobial Drug Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 53, 2522–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragosits M, and Mattanovich D (2013). Adaptive laboratory evolution – principles and applications for biotechnology. Microb. Cell Fact 12, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunphy LJ, and Papin JA (2017). Biomedical applications of genome-scale metabolic network reconstructions of human pathogens. Curr. Opin. Biotechnol 51, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durot M, Bourguignon P-Y, and Schachter V (2009). Genome-scale models of bacterial metabolism: reconstruction and applications. FEMS Microbiol. Rev 33, 164–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El’Garch F, Jeannot K, Hocquet D, Llanes-Barakat C, and Plésiat P (2007). Cumulative Effects of Several Nonenzymatic Mechanisms on the Resistance of Pseudomonas aeruginosa to Aminoglycosides. Antimicrob. Agents Chemother 51, 1016–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EUCAST (2018). The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 8.0, 2018 http://www.eucast.org/clinical_breakpoints/.
- Fabich AJ, Leatham MP, Grissom JE, Wiley G, Lai H, Najar F, Roe BA, Cohen PS, and Conway T (2011). Genotype and phenotypes of an intestine-adapted Escherichia coli K-12 mutant selected by animal passage for superior colonization. Infect. Immun 79, 2430–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falagas ME, and Bliziotis IA (2007). Pandrug-resistant Gram-negative bacteria: the dawn of the post-antibiotic era? Int. J. Antimicrob. Agents 29, 630–636. [DOI] [PubMed] [Google Scholar]
- Fong SS, Joyce AR, and Palsson BØ (2005). Parallel adaptive evolution cultures of Escherichia coli lead to convergent growth phenotypes with different gene expression states. Genome Res 15, 1365–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furusawa C, Horinouchi T, and Maeda T (2018). Toward prediction and control of antibiotic-resistance evolution. Curr. Opin. Biotechnol 54, 45–49. [DOI] [PubMed] [Google Scholar]
- Giraud E, Cloeckaert A, Kerboeuf D, and Chaslus-Dancla E (2000). Evidence for Active Efflux as the Primary Mechanism of Resistance to Ciprofloxacin in Salmonella enterica Serovar Typhimurium. Antimicrob. Agents Chemother 44, 1223–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett DJ, and Cohen MS (1989). Bacterial adaptation to oxidative stress: implications for pathogenesis and interaction with phagocytic cells. FASEB J 3, 2574–2582. [DOI] [PubMed] [Google Scholar]
- Hocquet D, Petitjean M, Rohmer L, Valot B, Kulasekara HD, Bedel E, Bertrand X, Plésiat P, Köhler T, Pantel A, et al. (2016). Pyomelanin-producing Pseudomonas aeruginosa selected during chronic infections have a large chromosomal deletion which confers resistance to pyocins. Environ. Microbiol 18, 3482–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LR, Richardson AR, Houston LS, Kulasekara HD, Martens-Habbena W, Klausen M, Burns JL, Stahl DA, Hassett DJ, Fang FC, et al. (2010). Nutrient availability as a mechanism for selection of antibiotic tolerant Pseudomonas aeruginosa within the CF airway. PLoS Pathog 6, e1000712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamovic L, and Sommer MOA (2013). Use of Collateral Sensitivity Networks to Design Drug Cycling Protocols That Avoid Resistance Development. Sci. Transl. Med 5, 204ra132. [DOI] [PubMed] [Google Scholar]
- Jahn LJ, Munck C, Ellabaan MMH, and Sommer MOA (2017). Adaptive Laboratory Evolution of Antibiotic Resistance Using Different Selection Regimes Lead to Similar Phenotypes and Genotypes. Front. Microbiol 8, 816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorth P, Staudinger BJ, Wu X, Hisert KB, Hayden H, Garudathri J, Harding CL, Radey MC, Rezayat A, Bautista G, et al. (2015). Regional Isolation Drives Bacterial Diversification within Cystic Fibrosis Lungs. Cell Host Microbe 18, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce AR, Reed JL, White A, Edwards R, Osterman A, Baba T, Mori H, Lesely SA, Palsson BØ, and Agarwalla S (2006). Experimental and Computational Assessment of Conditionally Essential Genes in Escherichia coli. J. Bacteriol 188, 8259–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka JB (2012). N-acetylglucosamine (GlcNAc) functions in cell signaling. Scientifica 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korgaonkar AK, and Whiteley M (2011). Pseudomonas aeruginosa Enhances Production of an Antimicrobial in Response to N-Acetylglucosamine and Peptidoglycan. J. Bacteriol 193, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kram KE, and Finkel SE (2015). Rich Medium Composition Affects Escherichia coli Survival, Glycation, and Mutation Frequency during Long-Term Batch Culture. Appl Env. Microbiol 81, 4442–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázár V, Singh GP, Spohn R, Nagy I, Horváth B, Hrtyan M, Busa-Fekete R, Bogos B, Méhi O, Csörgő B, et al. (2013). Bacterial evolution of antibiotic hypersensitivity. Mol. Syst. Biol 9, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HH, and Collins JJ (2011). Microbial environments confound antibiotic efficacy. Nat. Chem. Biol 8, 6–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberati NT, Urbach JM, Miyata S, Lee DG, Drenkard E, Wu G, Villanueva J, Wei T, and Ausubel FM (2006). An ordered, nonredundant library of Pseudomonas aeruginosa strain PA14 transposon insertion mutants. Proc. Natl. Acad. Sci 103, 2833–2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister PD, Wolter DJ, and Hanson ND (2009). Antibacterial-Resistant Pseudomonas aeruginosa: Clinical Impact and Complex Regulation of Chromosomally Encoded Resistance Mechanisms. Clin. Microbiol. Rev 22, 582–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livermore DM (2002). Multiple Mechanisms of Antimicrobial Resistance in Pseudomonas aeruginosa: Our Worst Nightmare? Clin. Infect. Dis 34, 634–640. [DOI] [PubMed] [Google Scholar]
- Lobritz MA, Belenky P, Porter CBM, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, and Collins JJ (2015). Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci 112, 8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez JL, and Rojo F (2011). Metabolic regulation of antibiotic resistance. FEMS Microbiol. Rev 35, 768–789. [DOI] [PubMed] [Google Scholar]
- Marvig RL, Dolce D, Sommer LM, Petersen B, Ciofu O, Campana S, Molin S, Taccetti G, and Johansen HK (2015). Within-host microevolution of Pseudomonas aeruginosa in Italian cystic fibrosis patients. BMC Microbiol 15, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer S, Steffen W, Steuber J, and Götz F (2015). The Staphylococcus aureus NuoL-Like Protein MpsA Contributes to the Generation of Membrane Potential. J. Bacteriol 197, 794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollister BD, Hoffman M, Husain M, and Vázquez-Torres A (2011). Nitric oxide protects bacteria from aminoglycosides by blocking the energy-dependent phases of drug uptake. Antimicrob. Agents Chemother 55, 2189–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan S, Porter CBM, Yang JH, Belenky P, Gutierrez A, Lobritz MA, Park J, Kim SH, Moskowitz SM, and Collins JJ (2017). Carbon Sources Tune Antibiotic Susceptibility in Pseudomonas aeruginosa via Tricarboxylic Acid Cycle Control. Cell Chem. Biol 24, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan S, Andrews IW, and Collins JJ (2018). Targeting Antibiotic Tolerance, Pathogen by Pathogen. Cell 172, 1228–1238. [DOI] [PubMed] [Google Scholar]
- Monk JM, Lloyd CJ, Brunk E, Mih N, Sastry A, King Z, Takeuchi R, Nomura W, Zhang Z, Mori H, et al. (2017). iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol 35, 904–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberhardt MA, Palsson BØ, and Papin JA (2009). Applications of genome-scale metabolic reconstructions. Mol. Syst. Biol 5, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, Minchin PR, O’Hara RB, Simpson GL, Solymos P, et al. (2018). vegan: Community Ecology Package. R package version 2.5–2.
- Orth JD, Thiele I, and Palsson B0. (2010). What is flux balance analysis? Nat. Biotechnol 28, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer AC, and Kishony R (2013). Understanding, predicting and manipulating the genotypic evolution of antibiotic resistance. Nat. Rev. Genet 14, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer KL, Aye LM, and Whiteley M (2007). Nutritional Cues Control Pseudomonas aeruginosa Multicellular Behavior in Cystic Fibrosis Sputum. J. Bacteriol 189, 8079–8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng B, Su Y-B, Li H, Han Y, Guo C, Tian Y-M, and Peng X-X (2015). Exogenous alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell Metab 21, 249–261. [DOI] [PubMed] [Google Scholar]
- Rau MH, Hansen SK, Johansen HK, Thomsen LE, Workman CT, Nielsen KF, Jelsbak L, Høiby N, Yang L, and Molin S (2010). Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ. Microbiol 12, 1643–1658. [DOI] [PubMed] [Google Scholar]
- Reed JL, Vo TD, Schilling CH, and Palsson BO (2003). An expanded genome-scale model of Escherichia coli K-12 (iJR904 GSM/GPR). Genome Biol 4, R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schellenberger J, Que R, Fleming RMT, Thiele I, Orth JD, Feist AM, Zielinski DC, Bordbar A, Lewis NE, Rahmanian S, et al. (2011). Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc 6, 1290–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schurek KN, Marr AK, Taylor PK, Wiegand I, Semenec L, Khaira BK, and Hancock REW (2008). Novel Genetic Determinants of Low-Level Aminoglycoside Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 52, 4213–4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan Y, Lazinski D, Rowe S, Camilli A, and Lewis K (2015). Genetic Basis of Persister Tolerance to Aminoglycosides in Escherichia coli. mBio 6, e00078–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Mailloux RJ, Puiseux-Dao S, and Appanna VD (2007). Oxidative Stress Evokes a Metabolic Adaptation That Favors Increased NADPH Synthesis and Decreased NADH Production in Pseudomonas fluorescens. J. Bacteriol 189, 6665–6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprouffske K, and Wagner A (2016). Growthcurver: an R package for obtaining interpretable metrics from microbial growth curves. BMC Bioinformatics 17, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson K, McVey AF, Clark IBN, Swain PS, and Pilizota T (2016). General calibration of microbial growth in microplate readers. Sci. Rep 6, 38828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Horinouchi T, and Furusawa C (2014). Prediction of antibiotic resistance by gene expression profiles. Nat. Commun 5, 5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taber HW, Mueller JP, Miller PF, and Arrow AS (1987). Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev 51, 439–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak E, Veres A, Michel J-B, Chait R, Hartl DL, and Kishony R (2012). Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet 44, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser JAGM, and Krug J (2014). Empirical fitness landscapes and the predictability of evolution. Nat. Rev. Genet 15, 480–490. [DOI] [PubMed] [Google Scholar]
- Westphal LL, Lau J, Negro Z, Moreno IJ, Ismail Mohammed W, Lee H, Tang H, Finkel SE, and Kram KE (2018). Adaptation of Escherichia coli to long-term batch culture in various rich media. Res. Microbiol 169, 145–156. [DOI] [PubMed] [Google Scholar]
- Winsor GL, Griffiths EJ, Lo R, Dhillon BK, Shay JA, and Brinkman FSL (2016). Enhanced annotations and features for comparing thousands of Pseudomonas genomes in the Pseudomonas genome database. Nucleic Acids Res 44, D646–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SM, Bernui M, Shen H, and Akerley BJ (2013). Genome-wide fitness profiling reveals adaptations required by Haemophilus in coinfection with influenza A virus in the murine lung. Proc. Natl. Acad. Sci 110, 15413–15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen P, and Papin JA (2017). History of antibiotic adaptation influences microbial evolutionary dynamics during subsequent treatment. PLOS Biol 15, e2001586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri M, Enke T, Chubukov V, Ricci V, Piddock L, and Sauer U (2017). Metabolic constraints on the evolution of antibiotic resistance. Mol. Syst. Biol 13, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S2 Complete binary and numerical growth calls. Related to Figure 2.
Data S3. Growth dynamics. Related to Figure 3.
Data S4. Complete gene essentiality predictions. Related to Figure 5.
Data Availability Statement
All code and data used to calculate growth dynamics, make gene essentiality predictions, as well as generate figures and supplemental data (excluding Figure 1) can be found at https://github.com/lauradunphy/dunphy_yen_papin_supplement. Complete growth data, binary growth calls, growth dynamics, and computational gene essentiality predictions can be found in the supplement (Data S1–S4).