

Abstract
Pyridoxine dependent epilepsy (PDE) is a treatable epileptic encephalopathy characterized by a positive response to pharmacologic doses of pyridoxine. Despite seizure control, at least 75% of individuals have intellectual disability and developmental delay. Current treatment paradigms have resulted in improved cognitive outcomes emphasizing the importance of an early diagnosis. As genetic testing is increasingly accepted as first tier testing for epileptic encephalopathies, we aimed to provide a comprehensive overview of ALDH7A1 mutations that cause PDE. The genotypes, ethnic origin and reported gender was collected from 185 subjects with a diagnosis of PDE. The population frequency for the variants in this report and the existing literature were reviewed in the Genome Aggregation Database (gnomAD). Novel variants identified in population databases were also evaluated through in silico prediction software and select variants were over-expressed in an E.coli-based expression system to measure α-aminoadipic semialdehyde dehydrogenase activity and production of α-aminoadipic acid. This study adds 47 novel variants to the literature resulting in a total of 165 reported pathogenic variants. Based on this report, in silico predictions, and general population data, we estimate an incidence of approximately 1:64,352 live births. This report provides a comprehensive overview of known ALDH7A1 mutations that cause PDE, and suggests that PDE may be more common than initially estimated. Due to the relative high frequency of the disease, the likelihood of under-diagnosis given the wide clinical spectrum and limited awareness among clinicians as well as the cognitive improvement noted with early treatment, newborn screening for PDE may be warranted.
Keywords: ALDH7A1, Alpha aminoadipic semialdehyde, Pyridoxine dependent epilepsy, PDE
Introduction:
Pyridoxine dependent epilepsy (PDE; OMIM 266100) is a treatable epileptic encephalopathy characterized by a positive response to pharmacologic doses of pyridoxine (Hunt et al. 1954; Scriver 1960). The classic presentation of PDE is neonatal onset of treatment refractory seizures, with dramatic clinical and electroencephalogram (EEG) improvement following pyridoxine supplementation. The majority of patients are able to achieve adequate seizure control with pyridoxine alone, yet 75% of individuals with PDE have significant intellectual disability (ID) and developmental delay (DD) (Basura et al. 2009; Bok et al. 2012). Current treatment paradigms aim to reduce putative neurotoxic metabolites through a lysine restricted diet and competitive inhibition of lysine transport (van Karnebeek et al. 2012; Coughlin et al. 2015). These therapies have been associated with improved neurologic outcomes, although optimal development is dependent on an early diagnosis and initiation of treatment in the first year of life (Coughlin et al. 2015; Al Teneiji et al. 2017).
PDE results from a deficiency of α-aminoadipic semialdehyde (α-AASA) dehydrogenase (E.C. 1.2.1.3), a key enzyme in lysine metabolism (Mills et al. 2006). α-AASA dehydrogenase deficiency results in the pathognomonic accumulation of α-AASA and the cyclic equivalent Δ-1-piperideine-6 carboxylate (Δ1-P6C). The accumulated Δ1-P6C is reported to sequester the active form of vitamin B6 (pyridoxal 5’-phosphate, PLP) by forming a Knoevenagel condensation product (Mills et al. 2006). The therapeutic strategy of pyridoxine supplementation is to overcome the secondary depletion of PLP. The degree of ID/DD in PDE does not correlate with seizure control or age at which pyridoxine treatment is initiated. Pyridoxine supplementation alone does not treat the underlying defect of lysine oxidation, and treatment with a lysine restricted diet (van Karnebeek et al. 2012) and with a lysine transport inhibitor (arginine) (Mercimek-Mahmutoglu et al. 2014; Coughlin et al. 2015) has resulted in an improved cognitive outcome. Therefore, it is important to differentiate PDE from the other causes of pyridoxine responsive seizures, such as pyridoxamine 5’-phosphate oxidase deficiency and mutations in PROSC, where the recommended treatment is limited to he appropriate vitamin B6 vitamer.
The enzyme α-AASA dehydrogenase is encoded by ALDH7A1 (NM_001182), and the majority of patients with PDE have biallelic mutations consistent with autosomal recessive inheritance (Mills et al. 2006). ALDH7A1 is located at 5q23.2 and contains a transcript of 4,964 base pairs encoding a protein of 539 amino acids. A number of publications have described ALDH7A1 mutations in cohorts of patients with PDE (Plecko et al. 2007; Bennett et al. 2009; Mills et al. 2010; Scharer et al. 2010), although a majority of published mutations are reported in small case series or single case reports. As a result, it is difficult to determine whether each genotype is unique to an individual patient limiting the ability to estimate mutation frequency. The c.1279G>C, p.Glu427Gln (historical name: p.Glu399Gln) mutation is the most commonly reported pathogenic variant in the literature, and is suggested to be present in 30% of patients with European ancestry (Plecko et al. 2007; Mills et al. 2010). A small number of patients were reported to have a single pathogenic variant identified (Plecko et al. 2007; Bennett et al. 2009), and the majority of these patients were later identified to have an intragenic copy number variations (CNVs) within ALDH7A1 (Mefford et al. 2015).
Genetic testing, either through specific gene panels or genomic sequencing, is increasingly accepted as first tier testing for epileptic encephalopathies, even when pathognomonic biomarkers exist. Genetic testing is a powerful diagnostic tool and limits the need for clinical suspicion of PDE and targeted testing. At the same time, the presence of variants of uncertain significance often complicates the interpretation of genetic tests. In order to provide a comprehensive overview of ALDH7A1 mutations that are known to result in PDE, we compiled results from four clinical laboratories, an international patient registry and a review of the literature. Furthermore, we utilized this comprehensive review to estimate the carrier frequency of ALDH7A1 variants in a general population database, which allowed us to estimate the incidence of this treatable disease.
Here we report 369 ALDH7A1 variants in 185 individuals with the clinical diagnosis of PDE. Including the 47 novel variants in this report, there are now 165 pathogenic variants reported in the literature.
Methods:
Literature review:
A systematic literature search was performed with the search criteria of pyridoxine dependent epilepsy, pyridoxine responsive epilepsy, pyridoxine dependent seizures, pyridoxine responsive seizures, antiquitin deficiency, α-amino adipic semialdehyde dehydrogenase deficiency and ALDH7A1. All publications were reviewed for articles that contained clinical data on patients with PDE. PDE due to antiquitin deficiency was defined by either the elevation of α-AASA or Δ1-P6C or biallelic mutations in ALDH7A1. Publications that contained molecular data on patients with PDE were retained and systematically classified.
Subject and variant identification:
Subjects were identified through clinical genetics laboratories that perform clinical testing of ALDH7A1, the international registry for pyridoxine dependent epilepsy (www.pdeonline.org) and the pyridoxine dependent seizures patient registry (Basura et al. 2009). Retrospective data was collected with ethics approval from each main site: University of Colorado, COMIRB 16–1325, COMIRB 16–0434 and Seattle Children’s Research Institute IRB10863 and 15450, and deidentified laboratory data was collected in agreement with the rules of each local ethics committee. Inclusion criteria included any subject with a clinical suspicion of PDE and at least one pathogenic variant identified in ALDH7A1. Genetic testing results and reported gender were collected when available. Sequence variants in both the reported subjects and the literature were compared to the sequence of ALDH7A1 (GenBank NM_001182.4, Ensembl ENST00000409134). Coding nomenclature was corrected using the “A” of the ATG translation initiation codon as position number 1, as opposed to historic nomenclature which had failed to include the mitochondrial leader peptide.
Population analysis:
The minor allele frequency (MAF) was recorded as reported in the Genome Aggregation Database (gnomAD) (last accessed 10 November 2017). The gnomAD browser (gnomad.broadinstitute.org) includes 123,126 exome sequences and 15,496 whole-genome sequences from unrelated individuals. The MAF of all variants identified through the systematic literature review, and reported in this study, were recorded when available. All heterozygote frameshift, canonical splice-acceptor or splice-donor, and nonsense variants were presumed to be pathogenic. All single nucleotide variants (SNVs) were further evaluated to assess pathogenicity. Variants were defined as rare based upon a MAF ≤ 3.34E-04, which is the MAF of the common p.Glu427Gln mutation. Rare SNVs were further evaluated with in silico analysis tools: Protein Variation Effect Analyzer (PROVEAN; http://provean.jcvi.org), Sorting Intolerant From Tolerant (SIFT; http://sift.jcvi.org), and Polymorphism Phenotyping version 2.1.0 (Polyphen-2; http://genetics.bwh.harvard.edu/pph2).
Expression analysis:
An ALDH7A1 gene construct (containing the complete open reading frame of human ALDH7A1) was cloned into the pET15b expression vector as previously described, and was the generous gift of Dr. Coulter-Mackie and colleagues (Coulter-Mackie et al. 2012). In vitro mutagenesis was done with the Qiagen Quikchange II XL kit (Agilent) and the sequence verified by Sanger sequencing. This expression vector was transformed into BL21 Star™ (DE3) E. coli (ThermoFisher). Cultures were grown overnight in LB media at 37oC and antiquitin expression was induced by the addition of 1.0 mM IPTG. Cells were harvested after 3 hours, snap frozen in liquid nitrogen and stored at -80oC. Enzymatic conversion of P6C to AAA was carried out using the previously published antiquitin activity assay using bacterial cell lysate combined with 2.2 mM NAD+ and 0.6 mM P6CH in a total volume of 1.0 mL at 37°C following the production of NADH at 320 nm in a Cary 300 spectrophotometer (Coulter-Mackie et al. 2012, 2014). After 5 minutes, 500 μL of the reaction mixture was combined with 100 μL of glacial acetic acid and analyzed by LC-MS/MS methods.
Results
Literature review:
We retrieved 118 unique variants in ALDH7A1 reported in 62 publications (Supplemental table 1). The frequency of each pathogenic variant was not recorded as the authors could not ensure that a patient was unique to each report. The majority of pathogenic variants (49% of all reported variants) were SNVs that result in single amino acid substitutions (i.e. missense mutations). A high number of missense mutations were identified in exons 15 (25%), 10 (16%), 6 (12%) and 5 (11%) compared to exons 1,2,3,7,8 and 11 where no missense mutations were reported. Other pathogenic variants reported in the literature included variants at the intron/exon boundary (splice site mutations) (21%), insertion-deletion (InDel) mutations (16%), SNVs that result in premature stop codons (nonsense mutations) (7%), CNVs (6%) and SNVs not at the boundary that still result in splicing errors (2%).
Subjects identified from clinical laboratories and the PDE registry:
Molecular genetic results were available for 185 subjects and gender was reported in 139 subjects with 54% of subjects reported to be female. Biallelic mutations were identified in 182 of 185 (98%) subjects (Supplemental table II). All mutation types were identified in this cohort and were similar to those reported in the literature. This study allowed for the evaluation of the frequency of pathogenic variants. Missense mutations were the most frequently identified variant and were present in 57% of all alleles (Supplemental figure 1). The p.Glu427Gln was the most common variant identified and was present in 25.6% of all alleles, and the p.Gly505Arg missense mutation was identified in 3.5% of all alleles. The synonymous variant p.Val278Val (historical nomenclature: c.750G>A, r.748_787del) results in a cryptic splice site (Salomons et al. 2007), and was the second most common recurrent mutation noted in 5.4% of all disease alleles. Despite the presence of these recurrent variants, the majority of missense mutations were private or familial. In fact, 32 of the 49 missense mutations reported in this study were only identified in a single individual. Splice acceptor and splice donor variants were the second most common mutation type present in 18% of all alleles. The c.1566–1G>C variant was identified in 3.5% of all alleles.
Population analysis:
A total of 60 variants were identified in gnomAD which were either previously reported in the literature or reported in this study. These reported variants had an allele frequency of 2.11E-03. There were 23 novel, protein-truncating variants and 9 novel splice acceptor/donor variants, which were assumed to be pathogenic with an allele frequency of 3.18E-04 and 3.66E-05 respectively. A total of 240 rare (MAF ≤ 3.34E-04) novel SNVs were identified; 117 of these SNVs were predicted to be deleterious by at least two in silico programs with an allele frequency of 1.45E-03 (Supplemental table III). Based on the gnomAD data alone, the MAF was calculated at 0.00394. Using the Hardy-Weinberg principle, the estimated carrier frequency of ALDH7A1 mutations is 1:127 and the estimated incidence of α-AASA dehydrogenase deficiency is 1:64,352 births.
Expression analysis:
A total of 10 variants were selected for α-AASADH activity and AAA production. The p.Glu427Gln and p.Cys478Ser variants were selected as positive controls as they have been previously reported to have 0 and 19% of wild type enzyme activity,(Coulter-Mackie et al. 2014) and in our laboratory had 0.3% (SD ±0.2) and 14%; (SD ±1.5) of wild type activity respectively (Figure 1). Four groups of variants were also selected to represent various aspects of this study including variants previously reported in the literature (p.Gly274Glu, p.Ser317Leu) and novel variants reported in this study (p.Arg122Trp in subject PDE-51 and p.Tyr516Cys in subjects PDE-19, 76 and 87). Rare SNVs identified from gnomAD were also selected including variants predicted to be deleterious by in silico tools (p.His339Arg, p.Ala533Thr), and variants not predicted to be deleterious by in silico tools (p.Thr68Asn, p.Phe337Cys). In these 10 variants, the residual α-AASADH activity as measured by NAD reduction rate correlated closely with the amount of AAA production (R2=0.9673), and both provided an equivalent measure of relative enzyme activity. Enzyme activity was quantified for all 10 selected variants and enzyme activity ranged from 0.34% to 89.8% of wild type activity (Figure 1).
Figure 1: AASADH activity of ALDH7A1 variants.
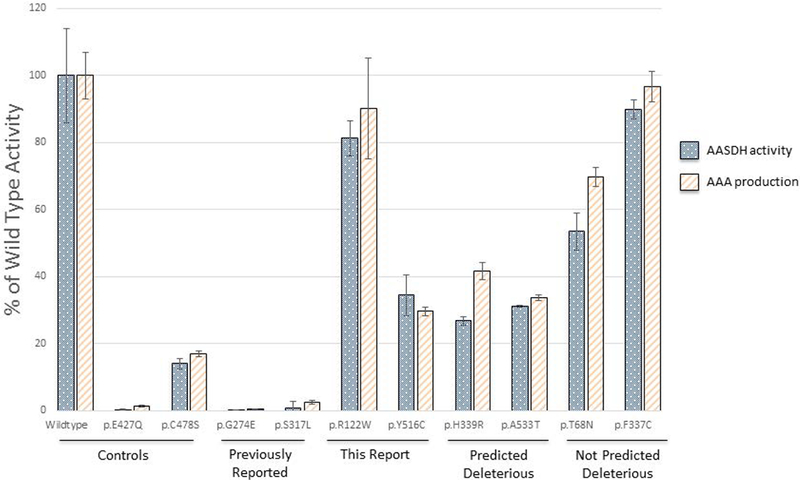
The human ALDH7A1 open reading frame was cloned into an E.coli-based expression vector. The enzyme activity of 10 expressed variants is shown for α-AASA dehydrogenase activity as reduction of NAD (dotted bar) and as AAA production (lined bar), each represented as % of the wild type, with standard deviation provided. The variants expressed included negative and positive controls (Controls), variants previously reported in the literature (Previously Reported), variants identified in subjects within this report (This report), rare variants identified in gnomAD and predicted to be pathogenic (Predicted Deleterious) and rare variants identified in gnomAD and not predicted to be pathogenic (Not Predicted Deleterious).
Discussion:
Herein we report a comprehensive review of ALDH7A1 variants identified in patients with clinical or biochemical suspicion or diagnosis of α-AASA dehydrogenase deficiency. Genetic testing identified biallelic pathogenic variants in 98% of subjects, although biochemical testing using pathognomonic biochemical markers of α-AASA and Δ1-P6C often complements these studies. This result suggests that genetic testing is a reliable method for the diagnosis of α-AASA dehydrogenase deficiency, but eight subjects (4.3%) had at least one CNV present, including one subject that was compound heterozygous for two CNVs. Thus, CNV analysis is important when only one pathogenic variant is identified, or in the case of a high clinical suspicion of PDE, even if no variants are identified through traditional sequencing methods. And, in cases with high clinical suspicion, mRNA studies (Salomons et al. 2007) and testing for pyridoxine dependent seizure disorders should also be considered (Darin et al. 2016; Plecko et al. 2017). This study did not differentiate subjects in whom molecular testing was performed first as opposed to molecular testing performed after a diagnostic biochemical result.
SNVs resulting in single amino acid substitutions were the most common variant type reported in the literature (49% of unique variants reported) and in this cohort (57% of all disease-causing alleles). The p.Glu427Gln variant was the single most common pathogenic variant identified and 32% of all subjects were at least heterozygous for this variant. Ethnicity was not available for the majority of our subjects, but we assume that a significant proportion of our cohort is of European ancestry, where this variant is reported to be present in 30% of patients (Plecko et al. 2007; Mills et al. 2010). Although a number of recurrent variants were identified, twenty-percent of our cohort had at least one private missense mutation. Determining the pathogenic nature of a novel SNV can be difficult, and this report highlights a few strategies for evaluating novel variants in ALDH7A1.
First, the location of the SNV in the gene structure appears relevant. Although pathogenic variants were identified throughout the gene, a significant number of unique variants were identified within exon 15, similar to previous reports that the majority of pathogenic variants occur in exon 15 within the catalytic domain of ALDH7A1 (Figure 2). Missense mutations are equally prominent within exons 5 and 6 within the linker domain and exon 10 within the NAD binding domain of ALDH7A1. The paucity of missense mutations within exons 3,7,8 and 11 is noteworthy.
Figure 2: ALDH7A1 pathogenic variants.
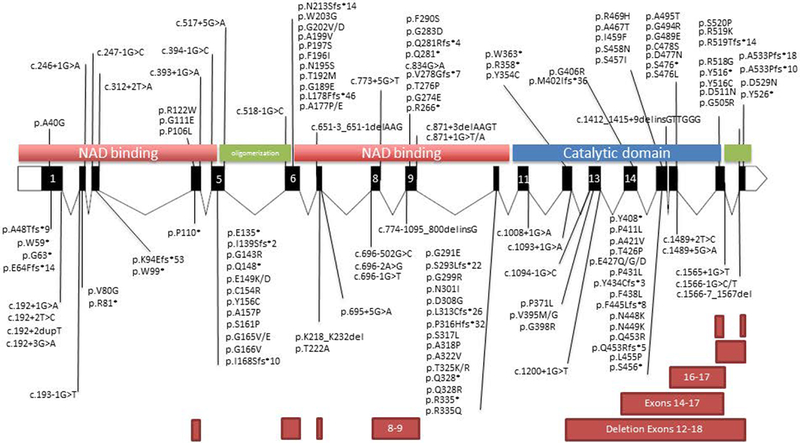
The ALDH7A1 gene is represented as a schematic with exons to scale and introns at 1/10th scale. Placement of predicted protein changes are denoted by solid black line. CNVs are denoted by rectangular boxes.
Functional studies are often utilized to evaluate the effect of novel variants, and a number of ALDH7A1 variants have previously been evaluated in an E.coli based expression system (Table I). In this E.coli based system, decreased AAA production correlated closely with decreased enzyme activity by NAD reduction. In our laboratory, previously reported pathogenic variants had <14% of wild type activity consistent with the previous experiments noted in the literature. Rare variants that were not predicted to be disease causing also had significant residual enzyme activity (>53% of wild type activity) supporting the use of the in silico prediction programs, however in other diseases discrepancies have been reported (Mercimek-Mahmutoglu et al. 2016). Some findings were surprising. For instance, the p.Arg122Trp variant had the second highest enzyme activity of any SNV studied at 81% (SD±5.2) of wild type. This variant was identified in a subject (PDE-51) who was compound heterozygous with the p.Gly63* allele (Supplemental Table II). The p.Arg122Trp is variant is relatively rare with a MAF of 9.02E-05, which is similar to well-reported variants such as p.Gly505Arg. Based on this enzyme activity, more studies are needed to determine whether this SNV is indeed a pathogenic variant.
Table 1:
Variants in ALDH7A1 and in vitro expression analysis
| Codon | Predicted Protein Change | Activity of α-AASADH1 | Expression at 30o,2 (nmol NADH/min/mg ATQ) | Expression at 37o,2 (nmol NADH/min/mg ATQ) | References |
|---|---|---|---|---|---|
| p.Thr68Asn | 53.5% | This report | |||
| c.328C>T | p.Arg110* | 0% | (Mills et al. 2006) | ||
| c.332G>A | p.Gly111Glu | 0% | (Coulter-Mackie et al. 2012) | ||
| p.Arg122Trp | 81.2% | This report | |||
| c.469G>C | p.Ala157Pro | 0% | (Korasick et al. 2017) | ||
| c.494G>T | p.Gln165Val | 0% | (Korasick et al. 2017) | ||
| c.497G>T | p.Gln166Val | 0% | (Korasick et al. 2017) | ||
| c.530C>A | p.Ala177Glu | 0% | (Korasick et al. 2017) | ||
| c.596C>T | p.Ala199Val | 0% | (Mills et al. 2006; Coulter-Mackie et al. 2012) | ||
| c.607T>G | p.Trp203Gly | 0% | (Coulter-Mackie et al. 2012) | ||
| c.821G>A | p.Gly274Glu | 0.22% | This report | ||
| c.848G>A | p.Gly283Asp | 0% | (Korasick et al. 2017) | ||
| c.872G>A | p.Gly291Glu | 0% | (Korasick et al. 2017) | ||
| c.902A>T | p.Asn301Ile | 0% | (Coulter-Mackie et al. 2012) | ||
| c.950C>T | p.Ser317Leu | 0.80% | This report | ||
| c.974C>A | p.Thr325Lys | 0% | (Coulter-Mackie et al. 2012) | ||
| c.974C>G | p.Thr325Arg | 0% | (Coulter-Mackie et al. 2012) | ||
| c.1004G>A | p.Arg335Gln | 0% | (Coulter-Mackie et al. 2012) | ||
| p.Phe337Cys | 89.8% | This report | |||
| p.His339Arg | 26.8% | This report | |||
| c.1184T>G | p.Val395Gly | 64% | 12% | (Coulter-Mackie et al. 2014) | |
| c.1224T>G | p.Tyr408* | 1.8% | (Mills et al. 2006) | ||
| c.1314C>A | p.Phe438Leu | 80% | 13% | (Coulter-Mackie et al. 2014) | |
| c.1279G>C | p.Glu427Gln | 0% | (Mills et al. 2006; Coulter- Mackie et al. 2012) | ||
| c.1280A>G | p.Glu427Gly | 0% | (Coulter-Mackie et al. 2012) | ||
| c.1373G>A | p.Ser458Asn | 0% | (Coulter-Mackie et al. 2012) | ||
| c.1375A>T | p.Ile459Phe | 0% | (Coulter-Mackie et al. 2012) | ||
| c.1432T>A | p.Cys478Ser | 27% | 19% | (Coulter-Mackie et al. 2014) | |
| c.1513G>C | p.Gly505Arg | 0% | (Coulter-Mackie et al. 2012) | ||
| p.Tyr516Cys | 34.4% | This report | |||
| p.Ala533Thr | 31.2% | This report | |||
| c.1596delG | p.Ala533Profs* | 0% | (Mills et al. 2006) |
Represented as a % of wild type activity;
Represented as a % of wild type activity calculated from the mean specific activity.
For the α-AASA enzyme to be functional, the protein must have enzyme activity (which was analyzed in this study) but the protein must also be stable in its natural environment (mitochondria in eukaryotic cells) and must reach its targeted environment (the mitochondrion). Overexpression in E. coli only interrogates a single function, and thus may miss other functions that are equally relevant, and which may well explain the discrepancy between the observed high residual activity of 81% on this study and the proposed pathogenic nature of the mutations. Many variants affect more than one functionality with the ultimate impact being a sum of all required functionalities. This also highlights the difficulty in a genetic-first approach to diagnosis. Fortunately, for PDE caused by α-AASA dehydrogenase deficiency, diagnostic biochemical testing is available in body fluids with the determination of biomarkers such as α-AASA or Δ1-P6C. Although these biomarkers appear excellent, the sensitivity and specificity of biochemical testing still needs to be determined in a sufficiently large study. The combination of clinical findings, molecular genetic testing, and biochemical testing allows clinicians and researchers to be certain of the diagnosis with a high degree of confidence.
Functional studies have been proposed as a primary mechanism to assign pathogenicity of novel variants in general, and high throughput screening of very large numbers of novel genetics variants has been proposed (Starita et al. 2017). However, this study highlights some of the difficulty associated when only one type of functional study is performed. Moreover, a challenge in functional studies is to determine what level of impairment of the functionality will be sufficient for a variant to be pathogenic, which, thus far, has not been sufficiently determined in PDE. Evidence from other inborn errors of metabolism suggests that if a sufficient number of mutations are interrogated, a complex gradation was identified and defining a sharp cutoff was often difficult or even impossible.
Population incidence of PDE:
Previous studies have attempted to utilize population databases, such as the ExAc database, in order to estimate a disease incidence for autosomal recessive and X-linked recessive disorders. This has been most successful when a large number of pathogenic variants are known a priori (Coughlin et al. 2017). We expanded the limited information on pathogenic variants in ALDH7A1 from the literature of case reports and small case series in this large cohort, but the number of variants identified remains limited in comparison to those identified in the gnomeAD database. In order to provide a more comprehensive estimate of disease incidence, we further evaluated rare SNVs noted within gnomAD using computational (in silico) predictive programs as one piece of evidence to evaluate novel variants (Richards et al. 2015). We attempted to apply the same rigor to variants identified in the general population as is used in the context of clinical testing. We first eliminated any SNVs that were either present in homozygous state or had a significantly high prevalence in the general population. We further filtered novel SNVs though in silico prediction programs, resulting in a blend of known, pathogenic variants and predicted pathogenic variants described above. Using this approach, we identified 212 variants within the gnomAD database. Based upon these results and the Hardy-Weinberg principle, we were able to estimate the incidence of α-AASA dehydrogenase deficiency at 1:64,352 births, with incidences in specific subpopulations (Table II)
Table 2:
Estimated incidence of PDE based upon purported pathogenic variants in gnomAD
| Ancestry | East Asian | European (Finish) | African | European Non-(Finish) | Latino | South Asian | Other | Ashkenazi Jewish |
|---|---|---|---|---|---|---|---|---|
| Carrier frequency | 1:66 | 1:123 | 1:128 | 1:134 | 1:143 | 1:147 | 1:160 | 1:1,231 |
| Incidence | 1:16,556 | 1:60,303 | 1:65,474 | 1:71,659 | 1:81,427 | 1:86,299 | 1:101,807 | 1:6,058,365 |
This estimate of disease incidence relies on in silico prediction programs. It does not include the carrier frequency of CNVs, which is a known, but infrequent, disease mechanism, or of non-coding mutations that would impact ALDH7A1 expression. Previous estimates of the incidence of PDE based on clinical diagnosis have varied widely between 1:20,000 in a single center in Germany (Ebinger et al. 1999), 1:396,000 in the Netherlands (Been et al. 2005), and 1:783,000 in the United Kingdom (Baxter 1999). These previous studies only included patients who responded to a pyridoxine trial, often provided as a single dose, as the underlying genetic and biochemical cause had yet to be identified. Since then, multiple limitations of such approaches have been identified including the recognition that EEG improvement following a pyridoxine trial is neither sensitive nor specific (Bok et al. 2010), seizures may not occur until childhood, patients may have delayed or incomplete response to pyridoxine, and patients may present with atypical symptoms (Bass et al. 1996; Mills et al. 2010; van Karnebeek et al. 2016). As a result, if a therapeutic pyridoxine trial is relied upon as the sole diagnostic test, it is likely that a significant number of patients will remain undiagnosed. Indeed, our estimates imply that the incidence is 6 to 12 times higher than that recognized on clinical grounds by a pyridoxine trial. Given this substantial under ascertainment by clinical diagnosis, the treatable nature of PDE, and the improved outcome with early treatment, newborn screening for this disorder should be considered (Jung et al. 2013). Future studies will be required to see if a genomic or a biochemical approach to newborn screening is preferred.
Finally, previous population based studies of PDE have focused on European populations. The ethnicity data present in gnomAD allowed a further preliminary analysis of other populations, even though European subjects remained the largest population present in gnomAD at 44% of all subjects. When we further stratified our findings based on the reported ethnicity in gnomAD, we noted a higher carrier frequency (1:66) and therefore estimated incidence (1:16,556) of PDE in individuals reported to be of East Asian ancestry (Table II). Unfortunately, the number of individuals of East Asian descent in gnomAD is still limited to only 16,556 alleles. Despite this limitation, it may highlight the need for population-targeted testing in populations outside those of European ancestry.
Although it is likely that a significant number of affected patients remain undiagnosed, there may be other reasons for the discrepancy between the estimated incidence of PDE and previous clinical observations. Most notability, the rate of intrauterine demise and neonatal mortality for PDE has not been established. The estimated disease incidence is also limited by current in silico tools used to evaluate many of the variants identified in the general population (Ghosh et al. 2017). In order to overcome this limitation, each variant was assessed based on the MAF and with multiple in silico tools before determining the pathogenic nature of the variant. Population-based studies are required to confirm the incidence of PDE.
Conclusion:
Even in a disorder with well-defined biochemical markers, genetic diagnosis (i.e. sequencing analysis) is common in PDE. A genetic-first approach to inherited disorders is often challenged by determining the pathogenic nature of novel variants. This report provides a comprehensive review of ALDH7A1 variants, which should aid academic and commercial molecular diagnostic laboratories. Using population based genomic sequencing databases we estimated the frequency of α-AASA dehydrogenase deficiency at approximately 1:64,000 live births.
Supplementary Material
Supplemental Table 1: Reported ALDH7A1 mutations in the literature
Supplemental Table 2: Subjects and genotype
Supplemental Table 3: Variants and allele frequency from gnomAD
Supplemental Figure 1: Frequency of ALDH7A1 variants Legend: Pathogenic variants from 185 subjects are presented by mutation type and frequency. All variants are reported by predicted protein consequence and frequency is represented as percentage of all disease alleles.
Synopsis:
We present a comprehensive review of ALDH7A1 mutations that cause PDE
Acknowledgements:
The authors thank Dr. Coulter-Mackie and colleagues for providing the ALDH7A1 gene construct in a pET15b expression vector, colleagues in the International PDE Consortium and Ms. Aisha Ghani and Mr. Sravan Jaggumantri for management of the PDE patient registry. We are grateful to for the support of the ‘Rare Diseases Foundation’ in Vancouver, and the US-based patient organization: ‘Pyridoxine Dependent Epilepsy Foundation.’ This work was supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR001082 and the research endowments of the Division of Neurology, Seattle Children’s Hospital. Contents are the author’s sole responsibility and do not necessarily represent official NIH views.
Details of funding: This work was supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR001082 and the research endowments of the Division of Neurology, Seattle Children’s Hospital
Footnotes
A competing interest statement:C. Coughlin II, M.A. Swanson, E. Spector, NJL Meeks, KE Kronquist, M Aslamy, MF Wempe, CDM van Karnebeek, SM Gospe Jr, VG Aziz, PL Nagy, K Hyland, SJM van Dooren, GS Salomons and JLK Van Hove declare that they have no conflict of interest. H Gao and B Tsai are both employees and stockholders of Fulgent Genetics, a DNA sequencing laboratory. There is no apparent financial gain or loss expected as a result of this study.
Details of ethics approval: Retrospective data was collected with ethics approval from each main site: University of Colorado, COMIRB 16–1325, COMIRB 16–0434 and Seattle Children’s Research Institute IRB10863 and 15450, and deidentified laboratory data was collected in agreement with the rules of each local ethics committee.
A patient consent statement: Individual written informed consent was obtained from patients (or parents/guardians) for registry studies.
Documentation of approval from the Institutional Committee for Care and Use of Laboratory Animals: Not applicable
References:
- Al Teneiji A, Bruun TUJ, Cordeiro D, et al. (2017) Phenotype, biochemical features, genotype and treatment outcome of pyridoxine-dependent epilepsy. Metab Brain Dis 32:443–451. doi: 10.1007/s11011-016-9933-8 [DOI] [PubMed] [Google Scholar]
- Bass NE, Wyllie E, Cohen B, Joseph SA (1996) Pyridoxine-dependent epilepsy: the need for repeated pyridoxine trials and the risk of severe electrocerebral suppression with intravenous pyridoxine infusion. J Child Neurol 11:422–424 [DOI] [PubMed] [Google Scholar]
- Basura GJ, Hagland SP, Wiltse AM, Gospe SM (2009) Clinical features and the management of pyridoxine-dependent and pyridoxine-responsive seizures: review of 63 North American cases submitted to a patient registry. Eur J Pediatr 168:697–704. doi: 10.1007/s00431-008-0823-x [DOI] [PubMed] [Google Scholar]
- Baxter P (1999) Epidemiology of pyridoxine dependent and pyridoxine responsive seizures in the UK. Arch Dis Child 81:431–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Been JV, Bok LA, Andriessen P, Renier WO (2005) Epidemiology of pyridoxine dependent seizures in the Netherlands. Arch Dis Child 90:1293–1296. doi: 10.1136/adc.2005.075069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett CL, Chen Y, Hahn S, et al. (2009) Prevalence of ALDH7A1 mutations in 18 North American pyridoxine-dependent seizure (PDS) patients. Epilepsia 50:1167–1175. doi: 10.1111/j.1528-1167.2008.01816.x [DOI] [PubMed] [Google Scholar]
- Bok LA, Halbertsma FJ, Houterman S, et al. (2012) Long-term outcome in pyridoxine-dependent epilepsy. Dev Med Child Neurol 54:849–854. doi: 10.1111/j.1469-8749.2012.04347.x [DOI] [PubMed] [Google Scholar]
- Bok LA, Maurits NM, Willemsen MA, et al. (2010) The EEG response to pyridoxine-IV neither identifies nor excludes pyridoxine-dependent epilepsy. Epilepsia 51:2406–2411. doi: 10.1111/j.1528-1167.2010.02747.x [DOI] [PubMed] [Google Scholar]
- Coughlin CR, Swanson MA, Kronquist K, et al. (2017) The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med 19:104–111. doi: 10.1038/gim.2016.74 [DOI] [PubMed] [Google Scholar]
- Coughlin CR, van Karnebeek CDM, Al-Hertani W, et al. (2015) Triple therapy with pyridoxine, arginine supplementation and dietary lysine restriction in pyridoxine-dependent epilepsy: Neurodevelopmental outcome. Mol Genet Metab. doi: 10.1016/j.ymgme.2015.05.011 [DOI] [PubMed] [Google Scholar]
- Coulter-Mackie MB, Li A, Lian Q, et al. (2012) Overexpression of human antiquitin in E. coli: enzymatic characterization of twelve ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy. Mol Genet Metab 106:478–481. doi: 10.1016/j.ymgme.2012.06.008 [DOI] [PubMed] [Google Scholar]
- Coulter-Mackie MB, Tiebout S, van Karnebeek C, Stockler S (2014) Overexpression of recombinant human antiquitin in E. coli: partial enzyme activity in selected ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy. Mol Genet Metab 111:462–466. doi: 10.1016/j.ymgme.2014.02.010 [DOI] [PubMed] [Google Scholar]
- Darin N, Reid E, Prunetti L, et al. (2016) Mutations in PROSC Disrupt Cellular Pyridoxal Phosphate Homeostasis and Cause Vitamin-B6-Dependent Epilepsy. Am J Hum Genet 99:1325–1337. doi: 10.1016/j.ajhg.2016.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebinger M, Schultze C, König S (1999) Demographics and diagnosis of pyridoxine-dependent seizures. J Pediatr 134:795–796 [DOI] [PubMed] [Google Scholar]
- Ghosh R, Oak N, Plon SE (2017) Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol 18:225. doi: 10.1186/s13059-017-1353-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt AD, Stokes J, McCRORY WW, Stroud HH (1954) Pyridoxine dependency: report of a case of intractable convulsions in an infant controlled by pyridoxine. Pediatrics 13:140–145 [PubMed] [Google Scholar]
- Jung S, Tran N-TB, Gospe SM, Hahn SH (2013) Preliminary investigation of the use of newborn dried blood spots for screening pyridoxine-dependent epilepsy by LC-MS/MS. Mol Genet Metab 110:237–240. doi: 10.1016/j.ymgme.2013.07.017 [DOI] [PubMed] [Google Scholar]
- Mefford HC, Zemel M, Geraghty E, et al. (2015) Intragenic deletions of ALDH7A1 in pyridoxine-dependent epilepsy caused by Alu-Alu recombination. Neurology 85:756–762. doi: 10.1212/WNL.0000000000001883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, Cordeiro D, Cruz V, et al. (2014) Novel therapy for pyridoxine dependent epilepsy due to ALDH7A1 genetic defect: l-arginine supplementation alternative to lysine-restricted diet. Eur J Paediatr Neurol. doi: 10.1016/j.ejpn.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, Pop A, Kanhai W, et al. (2016) A pilot study to estimate incidence of guanidinoacetate methyltransferase deficiency in newborns by direct sequencing of the GAMT gene. Gene 575:127–131. doi: 10.1016/j.gene.2015.08.045 [DOI] [PubMed] [Google Scholar]
- Mills PB, Footitt EJ, Mills KA, et al. (2010) Genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy (ALDH7A1 deficiency). Brain 133:2148–2159. doi: 10.1093/brain/awq143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills PB, Struys E, Jakobs C, et al. (2006) Mutations in antiquitin in individuals with pyridoxine-dependent seizures. Nat Med 12:307–309. doi: 10.1038/nm1366 [DOI] [PubMed] [Google Scholar]
- Plecko B, Paul K, Paschke E, et al. (2007) Biochemical and molecular characterization of 18 patients with pyridoxine-dependent epilepsy and mutations of the antiquitin (ALDH7A1) gene. Hum Mutat 28:19–26. doi: 10.1002/humu.20433 [DOI] [PubMed] [Google Scholar]
- Plecko B, Zweier M, Begemann A, et al. (2017) Confirmation of mutations inPROSCas a novel cause of vitamin B 6 -dependent epilepsy. J Med Genet 54:809–814. doi: 10.1136/jmedgenet-2017-104521 [DOI] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomons GS, Bok LA, Struys EA, et al. (2007) An intriguing “silent” mutation and a founder effect in antiquitin (ALDH7A1). Ann Neurol 62:414–418. doi: 10.1002/ana.21206 [DOI] [PubMed] [Google Scholar]
- Scharer G, Brocker C, Vasiliou V, et al. (2010) The genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy due to mutations in ALDH7A1. J Inherit Metab Dis 33:571–581. doi: 10.1007/s10545-010-9187-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriver CR (1960) Vitamin B6-dependency and infantile convulsions. Pediatrics 26:62–74 [PubMed] [Google Scholar]
- Starita LM, Ahituv N, Dunham MJ, et al. (2017) Variant Interpretation: Functional Assays to the Rescue. Am J Hum Genet 101:315–325. doi: 10.1016/j.ajhg.2017.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Karnebeek CDM, Hartmann H, Jaggumantri S, et al. (2012) Lysine restricted diet for pyridoxine-dependent epilepsy: first evidence and future trials. Mol Genet Metab 107:335–344. doi: 10.1016/j.ymgme.2012.09.006 [DOI] [PubMed] [Google Scholar]
- van Karnebeek CDM, Tiebout SA, Niermeijer J, et al. (2016) Pyridoxine-Dependent Epilepsy: An Expanding Clinical Spectrum. Pediatr Neurol 59:6–12. doi: 10.1016/j.pediatrneurol.2015.12.013 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1: Reported ALDH7A1 mutations in the literature
Supplemental Table 2: Subjects and genotype
Supplemental Table 3: Variants and allele frequency from gnomAD
Supplemental Figure 1: Frequency of ALDH7A1 variants Legend: Pathogenic variants from 185 subjects are presented by mutation type and frequency. All variants are reported by predicted protein consequence and frequency is represented as percentage of all disease alleles.