

Abstract
Nearly all infectious agents contain DNA or RNA genomes, making sequencing an attractive approach for pathogen detection. The cost of high-throughput or next-generation sequencing has been reduced by several orders of magnitude since its advent in 2004, and it has emerged as an enabling technological platform for the detection and taxonomic characterization of microorganisms in clinical samples from patients. This review focuses on the application of untargeted metagenomic next-generation sequencing to the clinical diagnosis of infectious diseases, particularly in areas in which conventional diagnostic approaches have limitations. The review covers (a) next-generation sequencing technologies and common platforms, (b) next-generation sequencing assay workflows in the clinical microbiology laboratory, (c) bioinformatics analysis of metagenomic next-generation sequencing data, (d) validation and use of metagenomic next-generation sequencing for diagnosing infectious diseases, and (e) significant case reports and studies in this area. Next-generation sequencing is a new technology that has the promise to enhance our ability to diagnose, interrogate, and track infectious diseases.
Keywords: clinical diagnostics, metagenomics, next-generation sequencing, pathogen detection, infectious disease, nanopore sequencing
INTRODUCTION
Overview of Next-Generation Sequencing
Next-generation sequencing (NGS), also termed high-throughput or massively parallel sequencing, is a genre of technologies that allows for thousands to billions of DNA fragments to be simultaneously and independently sequenced. The applications of NGS in clinical microbiological testing are manifold and include metagenomic NGS (mNGS), which allows for an unbiased approach to the detection of pathogens. This review focuses on using mNGS methods to identify pathogens directly from clinical samples from patients (1–13). Untargeted mNGS approaches use what is known as shotgun sequencing of clinical samples or pure microbial cultures in which random samples of analyte DNA or RNA are surveyed en masse, in contrast to targeted approaches that utilize singleplex or multiplex polymerase chain reaction (PCR), primer extension, or bait probe enrichment methods, thus restricting detection to a list of specific targets. Whole-genome sequencing of cultured microbial isolates using NGS for organism typing, epidemiology, susceptibility prediction, and virulence factor determination are not discussed in this review, but several excellent descriptions of these applications are available (14–16). Other applications of NGS to infectious diseases include lineage tracing (17), drug-resistance testing of viruses or culture isolates (18–20), and microbiome studies (21). Previous reviews of NGS technologies (22, 23) and the application of clinical NGS to infectious diseases exist (17, 19, 24), but this review includes the latest advances in a rapidly evolving field.
Sequencing Platforms
Illumina (San Diego, CA) offers a popular series of sequencing platforms (iSeq, MiSeq, MiniSeq, NextSeq, HiSeq, and NovaSeq) that are used by the majority of published series. All of these platforms use a strategy of bridge amplification, whereby single molecules of DNA are first attached to a flow cell and then amplified locally into a clonal cluster, analogous to how a single bacterium grows into a colony on a media plate (25). This is followed by sequencing by synthesis, which builds the complementary DNA one nucleotide per cycle, and an optical readout of fluorescently labeled nucleotides then determines its identity (A, G, T, or C). Illumina sequencers have the highest throughput of all sequencers on the market, but it is important to note that this technology has the disadvantage of barcode index switching (26), in which high-frequency barcodes, or indices, that are designed to uniquely identify multiplexed samples may be misassigned during scanning of the flow cell. For mNGS, this can lead to microbial reads from one sample containing a high-titer pathogen cross-contaminating other samples on the same run, thus generating false-positive detections. This problem is exacerbated in the higher throughput HiSeq 3000, −4000, and -X, and NovaSeq sequencers due to the new techniques of exclusion amplification chemistry on a patterned flow cell.
Thermo Fisher Scientific (Waltham, MA) offers the Ion Torrent platform, which clones single DNA molecules on a bead within an emulsion (27). The beads are then placed onto a semiconductor chip containing a matrix of individual pH sensors. As the DNA clones undergo sequencing by synthesis, a localized pH change identifies the sequenced nucleotide.
BGI (Cambridge, MA) offers the BGISEQ platform, which clones single DNA molecules locally on a flow cell through a DNA origami strategy that produces clonal DNA nanoballs (28). The nanoballs then undergo sequencing by synthesis, and there is a fluorescent readout similar to that used by the Illumina platform. While this platform has been used for infectious disease sequencing of clinical samples (10, 29), it is not yet commercially available in the United States.
Oxford Nanopore Technologies (Oxford, United Kingdom) offers portable sequencers under the names MinION, GridION, and PromethION (8). This technology guides single-stranded DNA through a grid of protein nanopores that gathers the DNA sequence through electrical current disruptions. This genre of technology is a significant departure from the previous strategies, and there are implications for performance characteristics. Notably, for infectious disease diagnostics, nanopore DNA sequencing is orders of magnitude faster than other strategies that use sequence-by-synthesis methods. Nanopore sequencing also does not require prior PCR amplification, although often this is still performed due to the high baseline sample input requirement (>500 ng). However, the nanopore approach currently has more sequencing errors, lower throughput, and higher per-read costs than other NGS platforms, which may limit its utility for certain applications.
Advantages of Metagenomic Next-Generation Sequencing for Pathogen Detection
The etiology of suspected infections in acutely ill hospitalized patients often remains undiagnosed, resulting in delayed or inadequate treatment, prolonged stays, readmissions, and increased mortality and morbidity (30, 31). Frequently, these patients are immunocompromised due to cancer, hereditary syndromes, or transplantation, especially if they are in tertiary care medical centers, making them extremely vulnerable to infections. In this setting, the causative agent can include a number of both common and uncommon pathogens, ranging from viruses to bacteria, fungi, and parasites. Organism recovery from routine culture (i.e., growth in media) is limited due to the early administration of broad-spectrum or prophylactic antimicrobial drugs, as well as organisms that are fastidious or slow growing. Hypothesis-driven molecular testing such as PCR can involve numerous individual tests for specifically targeted organisms but may still miss a rare pathogen or use primers containing mismatches to the microbial strain involved, which decreases the sensitivity of detection (1). A hypothesis-free diagnostic approach that has the potential to detect nearly any organism would lead to a dramatic paradigm shift in microbial diagnostic testing. The various diagnostic testing methods used in clinical microbiology have distinct advantages and drawbacks, as described in Table 1. However, a common concern with conventional testing methods is the limitation in the breadth of pathogens detected, and clinicians are often left with negative results and the nagging question of whether the acute illness was actually caused by an infection for which testing was not done.
Table 1.
Comparison of testing methods for diagnosing infectious diseases
| Diagnostic test | Advantages | Disadvantages |
|---|---|---|
| Direct PCR | Simple Rapid Inexpensive Potential for quantitative PCR |
Depends on hypothesis Requires primers that may not always work Limited to a very small portion of genome |
| Multiplex PCR | Rapid Able to detect multiple organisms |
Low specificity and false positives for many organisms due to difficulty in quantitation Often requires more than one amplification Limited to a small portion of genome Requires primers that may not always work |
| Targeted universal multiplex PCR (e.g., 16S, ITS) for Sanger sequencing | Can differentiate multiple species within one pathogen type | Requires primers that may not always work Limited to a very small portion of genome |
| Targeted universal multiplex PCR (e.g., 16S, ITS) for NGS | Can differentiate multiple species within one pathogen type Multiplexing capability Potential for quantitation |
Requires primers that may not always work Expensive and time consuming Often requires more than one amplification Limited to a very small portion of genome |
| Targeted NGS | Sensitive detection for selected organism types Potential for quantitation Potential to be combined with 16S NGS (see above) |
Sequencing library preparation more complex, typically with more than one amplification Limited to a small portion of genome Expensive and time consuming Prone to contamination with environmental species |
| Metagenomic NGS | Hypothesis-free, or unbiased, testing Discovery of new or unexpected organisms Potential for quantitation Ability to detect any portion of genome |
Must also sequence human host background Expensive Time consuming Not all genomes are available Prone to contamination with environmental species |
| Serology | Potential for diagnosis after acute infection Inexpensive |
May be negative during early infection False-negatives in humoral immune deficiencies False-positives |
| Microscopy and staining (e.g., Gram stain, auramine–rhodamine, calcofluor-white) | Rapid Inexpensive |
Low sensitivity unless there is a high burden of disease Low specificity |
| Culture | Able to accommodate large sample volumes Inexpensive Well studied |
Sensitivity limited by use of antibiotics and antifungals Sensitivity limited for fastidious organisms Limited use in viral testing Long time to result, especially in acid-fast and fungal cultures |
| Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry | High specificity Rapid after culture |
Requires culture-positive isolate |
Abbreviations: ITS, internal transcribed spacer; NGS, next-generation sequencing; PCR, polymerase chain reaction.
In comparison to other diagnostic technologies, mNGS offers numerous advantages, but as with other tests, it also has drawbacks (Table 1). A chief advantage of mNGS is unbiased sampling, which enables broad identification of known as well as unexpected pathogens or even the discovery of new organisms (32). mNGS can also be coupled to targeted approaches, such as the use of primers from conserved 16S ribosomal RNA (rRNA) and internal transcribed spacer sequences for, respectively, universal bacterial and fungal detection (33, 34), which can allow for species-level identification of these organisms. Another advantage of mNGS is that it can provide the auxiliary genomic information necessary for evolutionary tracing (35), strain identification (36, 37), and prediction of drug resistance (20). NGS can provide quantitative or semiquantitative data regarding the concentration of organisms in the sample via the counting of sequenced reads, which is useful for polymicrobial samples or in cases in which more than one pathogen has been implicated in the disease process (34).
Limitations of Metagenomic Next-Generation Sequencing for Pathogen Detection, and Potential Solutions
A key disadvantage inherent to mNGS, given its shotgun sequencing approach, is that microbial nucleic acids from most patients’ samples are dominated by human host background. The vast majority of reads, generally >99%, derive from the human host, thus limiting the overall analytical sensitivity of the approach for pathogen detection, given the relative scarcity of microbial nonhuman reads that are sequenced. This disadvantage, which is inherent to unbiased mNGS, is partly mitigated either by targeted sequencing or host depletion methods (7, 38). If only bacterial sequences are of interest, then targeted sequencing of the 16S rRNA gene would be able to distinguish most species while, not incidentally, sequencing human host background (39, 40). Thus, targeted sequencing in combination with mNGS may be particularly useful for nonsterile specimens, such as those from bronchoalveolar lavage, stool, or polymicrobial abscesses.
Host depletion methods use a different approach than targeted sequencing. Instead of leveraging a known pathogen target such as the 16S rRNA gene, host depletion methods aim to decrease the relative proportion of human host background sequences in mNGS data. This approach retains the advantage of unbiased metagenomic sequencing in that it is fully agnostic to the pathogen that one is seeking. For RNA sequencing libraries, most of the host background typically corresponds to human rRNA or mitochondrial RNA sequences, and depletion of these human host sequences would indirectly boost the proportion of nonhuman microbial reads and thus improve the analytic sensitivity for pathogen detection. Methods that have been developed for host RNA depletion include using capture probes for subtractive hybridization (41, 42), ribonuclease (RNase) H–based depletion methods (43), or CRISPR-Cas9 cleavage of targeted sequences (7). These methods are generally effective for RNA libraries, which may contain a high proportion of noncoding rRNA sequences, but they are much less useful for DNA libraries, given that it is impractical due to cost and efficiency considerations to target the entire human host DNA genome.
Alternative methods exist for the depletion of human background DNA during the preanalytical phase, and these are based on differential physical characteristics between the signal (pathogen) and background (human host). One approach is to selectively lyse human white blood cells using saponin or other chemical reagents, followed by treating the released human genomic content with deoxyribonuclease (DNase), thereby enriching for microbial DNA that is protected within viral capsids or microbial cell walls (38, 44). A caveat to this approach is that the enrichment may also indiscriminately increase the relative prevalence of the microbial background that may result from microbial contamination of the reagents used for depletion (45). A different approach is to target low-molecular-weight cell-free DNA or RNA and remove high-molecular-weight genetic content that is often associated with human genomic material. This is accomplished by physically separating the cellular and cell-free compartments of clinical samples using methods such as centrifugation. Although there is the risk of decreased microbial reads after the removal of intact or intracellular microorganisms (e.g., human T cell lymphotropic virus, Listeria monocytogenes), several studies have demonstrated a relative enrichment of pathogen as compared with human reads using this procedure (46).
Another potential drawback of mNGS is the detection of microbial contaminants present in the sample, reagents used for processing, or laboratory environment, which can complicate the analysis and interpretation of results. Even biopsies of presumably sterile sites in the body can be inadvertently contaminated during the routine collection of clinical samples, and this may include contamination from skin flora during fine needle aspiration or oral flora during bronchoalveolar lavage procedures. Therefore, stringent adherence to reagent and workflow quality control procedures are needed to maintain a testing environment that is as sterile and nucleic acid–free as possible. The use of negative controls, reagent assessments, and periodic swipe tests are needed to ensure that laboratory and sample cross-contamination are not generating false-positive results. Additionally, the laboratory must be familiar with the commonly encountered microbial flora present in a range of clinical samples for each specimen type to be tested (13, 24, 47).
METAGENOMIC NEXT-GENERATION SEQUENCING ANALYSIS
In the microbiology (wet) laboratory, mNGS analysis involves a series of clinical sample processing, library preparation, and sequencing steps. This series is followed by bioinformatics analysis and the interpretation of mNGS data in the computational (dry) lab (Figure 1). Here, we discuss the individual steps in detail and the controls that are used during each step in the process for quality assurance.
Figure 1.
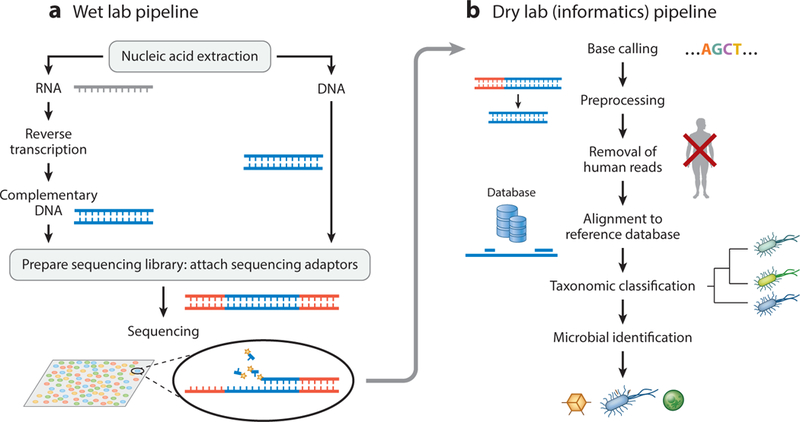
Schematic of the generalized workflow of metagenomic next-generation sequencing for diagnostic clinical use. The workflow has two components: (a) a wet lab protocol in which samples are collected, processed, extracted for nucleic acids, prepared into a sequencing library, and sequenced; (b) a dry lab computational pipeline that includes microbial identification, statistical analysis, and interpretation. The sequencing library may be targeted, undergo DNA amplification, or both.
Sample Collection and Transport
mNGS is generally flexible as to the sample source and nucleic acid quantity. Potential samples for mNGS analysis include tissue, body fluids, swabs, and environmental samples. Input DNA and RNA concentrations amenable to mNGS can be <100 pg, as is often the case for cerebrospinal or vitreous fluids (1, 2), or up to 6 orders of magnitude higher, as is often found in purulent fluids or abscesses. Sample stability is an especially important consideration for the sequencing of RNA, which is labile and vulnerable to degradation by host and environmental RNase enzymes, but stability is also a factor for DNA as well. To minimize the possibility of nucleic acid degradation, the use of chemical DNA or RNA stabilizers at the time of sample collection may be considered. Formalin-fixed paraffin-embedded (FFPE) samples are also associated with nucleic acid degradation when they are allowed to stay unfixed for prolonged periods, and degradation is also enhanced by age and formalin-associated chemical modifications of RNA (48). When frozen, DNA and RNA remain relatively intact; however, multiple freeze–thaw steps during sample aliquoting and processing may result in nucleic acid degradation that is partly due to the release of endogenous nucleases (49).
Nucleic Acid Extraction
The type of nucleic acid extraction used for mNGS is highly dependent on the sample type and whether DNA, RNA, or both, will be sequenced. The raw input varies based on where the sample was taken from and the type of sample and by the method of preprocessing, such as fresh tissue or fluids versus FFPE samples, or cellular versus cell-free nucleic acids. Accordingly, a single commercial vendor will often have a number of different kits for manual extraction or liquid-handling robots for automated extraction.
Library Preparation
Library preparation is the wet lab process of extracting RNA or DNA from samples and preparing it so that it is ready to be sequenced. Library preparation can be thought of in computer terminology as a required process for the compression and conversion of biological data. The amount of biological DNA data encoded in samples is several orders of magnitude higher than what can be practically sequenced even by the latest high-throughput sequencers (nearly 5 × 1014 base pairs are present in just 1 μg of DNA). In contrast, a sequencing run on a dual flow cell Illumina HiSeq 2500 in rapid-run mode yields about 1011 base pairs, or 0.02% of the original data content. Thus, all library preparations significantly subsample the original DNA and RNA content and are subject to representation biases introduced by even small modifications in the library generation process, such as the number of PCR cycles (50, 51).
Shotgun mNGS is perhaps the most unbiased approach to library preparation. The original DNA is randomly subsampled, providing a library that uniformly covers all genomes in the sample on the basis of their prevalence, but sequencing depth is minimized. Library preparation is performed by DNA recombination (tagmentation) of sequencing adapters to DNA (e.g., Illumina’s Nextera preparation) (52) or ligation of the adapter to sheared or fragmented DNA (e.g., Illumina’s TruSeq preparation) (25). For RNA, one common approach is reverse transcription using random primers, followed by second-strand synthesis into complementary DNA, which can then be prepared in a similar fashion to DNA. In contrast to untargeted shotgun sequencing, small panels of targeted sequencing will instead cover narrow areas of all possible positions in the present genomes, but they will cover each area deeply and often completely.
Nearly all DNA and RNA content in most clinical samples is host (human) derived, whereas the nucleic acids of interest for mNGS are microbial (or nonhuman). This poses a significant needle-in-a-haystack challenge for detecting pathogens from metagenomic data. Computational human host subtraction can be performed during the bioinformatics analysis step, but it would be more economical to remove unwanted human DNA or RNA earlier in the mNGS process—that is, during library preparation—as this would avoid the sequencing of irrelevant human background reads that are not used for mNGS. Numerous methods for host depletion have been demonstrated, and these include using saponin lysis to selectively lyse human cells and then to degrade all DNA, assuming that pathogen DNA is protected within its native enclosure, which is either a cell wall for bacteria or fungi or a protein capsid for viruses (38). For RNA, the removal of abundant human ribosomal or mitochondrial RNA can be performed by hybridization using capture probes (53) or by using the Cas9 nuclease to selectively target and deplete stereotypic background human RNA sequences (7).
The proportion of human DNA and RNA that is sequenced can also be reduced by targeting one or more genomic loci specific to pathogens for amplification by PCR using conserved primers, followed by library preparation. In some instances, the conserved primers are linked to the adapters used for sequencing; thus PCR amplification and adapter ligation are combined for single-step library preparation. One common application of this technique is PCR amplification of 16S rRNA using conserved primers and targeting the hypervariable regions (V1–V9) of this gene, followed by either Sanger sequencing or NGS of the resulting amplicon. Sequencing of the hypervariable regions then permits genus- and even species-level identification of the bacteria present in the sample (33, 34). This approach is routinely used for microbiome and metagenomic analyses, and it has also been used to diagnose complex bacterial infections in polymicrobial clinical samples (34, 39, 40). However, it may be less useful for organisms such as viruses, which exhibit high sequence diversity, or for fungi and parasites, which have eukaryotic ribosomes that are similar to human ribosomes, thus leading to nonspecific amplification.
Bioinformatics Analysis
Computational pipelines for metagenomic analyses of mNGS data have unique challenges and requirements that are distinct from other NGS pipelines designed for finding human germline and somatic mutations. Multiple open-source and private software packages for detecting and characterizing microbial sequences from mNGS data now exist, including SURPI (sequence-based ultrarapid pathogen identification) (54), Kraken (55), Taxonomer (56), and private pipelines (2, 3, 6, 11, 57). These informatics pipelines typically (a) preprocess sequencing reads to remove sequenced adapters and low-quality and low-complexity regions (58); (b) optionally, align to the human genome to remove human reads (computational host subtraction); (c) align the processed, nonhuman sequencing reads to a curated pathogen database and assign a taxonomic classification to each sequence read; and (d) perform organizational and statistical analyses on the resultant data with optional visualization, often in a graphical user interface (Figure 1).
The pathogen database can be built from the top down by starting with a comprehensive database such as GenBank, the sequence database maintained by the National Center for Biotechnology Information (~240 gigabases as of 2017; ftp://ftp.ncbi.nlm.nih.gov/genbank/release.notes), and making important adjustments, such as excluding sequences corresponding to the human host genome and low-complexity regions (54–56). Alternatively, the database can be built from the bottom up by aggregating individually curated genomes from a large selection of pathogens. Yet another approach is to curate the reference database for regions that are specific at a given taxonomic level, such as species or genus. It is important to note that not all pathogen genomes may be available, especially when the organism is rare (6). In these cases, de novo assembly may be attempted if the pathogen sequence data are readily abundant in the specimen or if an isolate is obtainable (59, 60). It would be rare to have sufficient read coverage for organisms other than viruses to assemble de novo a full genome that is not already in the reference database. However, de novo assembly of reads into longer contiguous sequences (referred to as contigs) may improve the sensitivity and specificity of database alignments.
Interpretation and Reporting
There is no standard method for interpreting mNGS results. A few competing constraints should be considered: (a) the reference database is incomplete for rare pathogens or emerging strains of pathogens; (b) the reference database is biased toward certain organisms; (c) certain pathogens that are important to distinguish may be similar genetically (e.g., species of mycobacteria); and (d) contamination with normal flora and reagents is a common occurrence that can limit specificity (61–63). To provide the most accurate results, reporting algorithms may need to take into account the quality of the test process and sample, the rarity of the pathogen in other samples on the current run and historical runs, both the relative and absolute abundance of the organism, whether there is a more abundant presence of a genetically similar organism, and the genomic coverage of the organism. Workflows for reporting results need to have preestablished metrics for quality control and interpreting findings, and these may include expert review for all cases or a subset of cases meeting defined criteria or with unusual findings (24).
QUALITY ASSURANCE FOR METAGENOMIC NEXT-GENERATION SEQUENCING
Multiple clinical samples are typically prepared and sequenced together on one mNGS run, and these are distinguished by unique nucleotide barcodes assigned to each sample. This format offers the opportunity to run controls together with many samples throughout the entire process.
Internal Controls
Spiking-in mock organisms, their genomic content, or uniquely identifiable genomic nucleic acids to every sample can help quantify the original sample and serve as a quality check on the entire process. Internal spike-in controls may consist of whole organisms, extracted nucleic acids, or synthetic DNA and RNA sequences, and these can be added to the original specimen or at later time points in the wet lab pipeline. One example of appropriate synthetic RNA internal controls (ICs) is the use of External RNA Controls Consortium spike-ins, which are not only commercially available but also were originally developed in conjunction with the National Institute of Standards and Technology for quality control of RNA gene expression measurements (64).
Consideration should also be given to any potential pathogenic contaminant in the IC, as this spike-in will be present in all samples. For example, if a phage or plasmid spike-in IC is manufactured using Escherichia coli, a human pathogen, then all samples may potentially be contaminated by E. coli, complicating the detection of this organism in clinical samples.
Multiple ICs can be used and at different steps in the process as long as these controls are readily distinguishable, as in the case of spike-in oligonucleotides with unique sequences. For example, if two uniquely identifiable ICs are spiked into the sample just prior to and after the extraction process, then the efficiency of the extraction process can be precisely monitored. If the ICs are at certain sizes, methods can also be used prior to sequencing to assess whether the ICs are present. For example, if an IC is known to be exactly 100 base pairs in length, then it would be possible to quantify it as a marker on capillary electrophoresis. If the IC has a foreign sequence that does not overlap with the sample’s likely genomic content (DNA or RNA, either human or microbial), then quantitative PCR assays can be designed to quantify the IC. Monitoring the recovery of IC material can also be used to assess an individual patient’s results and the performance of the assay over time (65).
External Controls
Both positive and negative external controls should be present in each sequencing run and treated as separate samples using the same lot of reagents and procedural workflow. Ideally, the positive and negative controls should be based on a matrix that simulates the characteristics of the sample matrix. Appropriate matrix substitutes, such as synthetic cerebrospinal fluid (CSF) matrix (Golden West Biologicals, Temecula, CA), can be used if human sample matrices (e.g., CSF) would be impractical to obtain.
Negative controls serve to monitor for external or reagent microbial contamination and cross-sample contamination. Positive controls are useful for detecting performance failures in nucleic acid extraction, the library preparation process, or informatics. Typically, positive controls consist of negative control matrix with quantitated spike-ins of representative pathogens, including a minimum of one of each microorganism type to be detected (e.g., virus, bacteria, fungus, and parasite). Nonpathogenic representative organisms can be selected to reduce the risk of cross-contamination by a pathogenic organism leading to a false-positive detection.
Process Controls
Process controls and checkpoints can be used at multiple points during mNGS to ensure the quality of materials prior to moving on to the next step in analysis. This is particularly important before starting a sequencing run since the reagents used are relatively expensive and repeat sequencing would add significant costs.
Quantification of the amount of DNA in a sequencing library is accomplished using a variety of methods, including spectrometry (e.g., a NanoDrop spectrophotometer, Thermo Fisher Scientific, Waltham, MA) and quantitative PCR. The amount of nucleic acid must be adequate in the final library preparation, and the relative amounts for each library within a sequencing pool have to be considered and normalized during mixing in order to ensure adequate representation for each library in the pool. While methods such as using SPRI (solid-phase reversible immobilization) or AMPure (Beckman Coulter, Beverly, MA) beads can be used to enrich or exclude DNA fragment sizes within or outside a desired range, an assessment of the library size profile can also be performed to confirm the average length and distribution of DNA within the library prior to sequencing. This can be done using gel electrophoresis, such as through Bioanalyzer analysis (Agilent, Santa Clara, CA) or by using Fragment Analyzer (Agilent) to ensure library quality on the basis of the length distribution.
Contamination Control
Contaminating microbes are ubiquitous and may be present in reagents or labware (61, 62), the environment, or normal human flora (63). Due to the sensitivity of mNGS, even minute amounts of outside contamination can be present in the sequencing data. Contamination can be introduced at every step of the process. Samples should be handled not only in a sterile manner but also to minimize contamination from exogenous nucleic acids. All reagents and disposables used during mNGS potentially may be contaminated. Therefore, documenting lot numbers and replacing materials promptly in the event of contamination are critical tasks. Negative controls should also be checked on each run. Similar to other molecular testing methods involving exponential amplification, such as PCR, it is critical to maintain a unidirectional workflow and strict physical separation between preamplification and postamplification processes.
A second form of contamination comes from bleed-through, or cross talk, from other samples in the same run or from library preparation that has high pathogen loads. This can occur via different mechanisms: index-hopping during Illumina sequencing, barcode contamination during primer or adapter synthesis, or cross-contamination during any part of the mNGS process. Index-hopping is a phenomenon that may occur when using Illumina sequencers, whereby DNA molecules assigned to one sample and its corresponding unique index barcode instead present themselves under an index barcode assigned to a different sample (26). This problem has been exacerbated by recent exclusion amplification Illumina technology that uses patterned flow cells, and it is an integral part of the higher throughput and more economic sequencers: HiSeq 3000, HiSeq 4000, HiSeq X Ten, and NovaSeq. The extent of exclusion amplification cross-contamination can be up to 10%, resulting in appreciable false-positive detections for sensitive applications, such as metagenomic sequencing.
Dual index barcoding, in which unique index barcodes are placed on either side of the DNA insert, is able to mitigate but not entirely eliminate contamination from index-hopping. Regardless of the mechanism, the interpretation of metagenomic data should always include evaluation for possible cross-contamination, especially when a sequencing run contains a sample with high levels of a pathogen.
Another source of contamination is barcoded primers or adapters that are contaminated during the synthesis process (66). When oligonucleotides are ordered en masse, the typical process is to synthesize the primers in parallel, which significantly increases the risk of cross-contamination. Purifying oligonucleotides through the same column, as occurs in high-performance liquid chromatography, may exacerbate cross-contamination. The evaluation of new oligonucleotide primers should include a method of evaluating the degree of cross-contamination, especially in adjacent wells of a plate. Thus, in addition to practicing standard reagent quality control to avoid contamination, it may be useful to prepare a high-titer sample with each new lot of primer barcodes and check whether reads from that microorganism are seen in other barcodes when they are analyzed in parallel.
Database Quality Control
Given the exponential growth of GenBank, databases are continually updated. The genomes of common pathogens are available; however, for rare pathogens, often only specific genes or limited regions of the genome have been sequenced, severely limiting sensitivity for detection on the basis of the alignment of reads to these reference sequences. Thus, regular updates to the reference database are important, as the addition of new reference sequences of organisms will improve the sensitivity of metagenomic testing. However, there is also a risk for increased false-positives if these new reference sequences are inadvertently contaminated by sequences corresponding to other species (for example, bacterial reads that are misannotated as a eukaryotic genome). In addition, even minor updates to the reference database require version control and will likely require re-validation of the bioinformatics pipeline, as they may impact the accuracy of the results from mNGS analyses.
Bioinformatics Quality Control
Similarly to documenting reagent lots and adhering to standard operating procedures, the computational pipeline requires version control, with clear tracking of software packages, the reference database, and the input parameters used. However, unlike wet lab analyses, bioinformatics runs can be performed using the same original data without requiring additional sample processing. Thus, it is not uncommon for the same mNGS data to be reanalyzed multiple times as the bioinformatics pipeline evolves and is optimized. Changes to the dry lab pipeline may include changes to software versions, input parameters, the algorithms used to calculate results, and the reference databases, as mentioned previously. It is often useful to maintain standardized mNGS data sets that can be used to benchmark changes in the existing bioinformatics pipeline or to compare the performance of different pipelines.
Recently, the Association for Molecular Pathology and the College of American Pathologists made a joint recommendation for NGS bioinformatics pipelines (67). While the recommendations are based on human germline and somatic mutation sequencing analyses, nearly all of them are also applicable to NGS bioinformatics for metagenomic sequencing. The recommendations advise that the assessment of the sequencing data should start with metrics, such as the number of sequencing reads, depth per sample, and quality of reads. Low-quality reads may be explained by instrument failure, poor library construction, or degraded input samples. A low number of sequencing reads in a given sample may be explained by uneven pooling of sequencing libraries, nonuniform instrument clustering (if run on an Illumina sequencer), or inaccurately quantified input. After sequencing, bioinformatics analyses can provide several metrics for quality assessment, including sequence quality and complexity. For individual sequencing runs, the results corresponding to the input controls (spiked internal control, external negative control, and external positive control) are used to provide quality metrics for the run.
Proficiency Testing
Like any clinical laboratory test, mNGS analysis is subject to proficiency testing (PT) require-ments. Since there are no commercial providers of material intended for PT of mNGS, alternative means of assessment are needed. Most commonly, excess clinical samples with and without prior metagenomics detections (i.e., positive or negative for an infectious agent) are reanalyzed in a blinded fashion for intralaboratory PT documentation. As more laboratories offer mNGS testing on more sample types, interlaboratory exchanges of excess or standardized reference materials, or both, will be useful to assess the variability of results generated by different laboratories.
It may also be useful to confirm mNGS results regarding the presence or absence of organisms using orthogonal detection methods, such as PCR, when identifying samples for PT assessment. Because the spectrum of organisms potentially detected using mNGS is extremely broad, PT materials should contain various organism types, at least on a rotating basis. Independent confirmation of new organisms detected during routine testing is also advisable, and this can serve to broaden the list of species known to be detectable by the mNGS assay over time.
VALIDATING METAGENOMIC NEXT-GENERATION SEQUENCING
mNGS test validation can be divided into wet- and dry-lab components, and both are important to ensure the safety and accuracy of the NGS test. It is possible to validate or revalidate one component, but often, optimizing the design of the test requires simultaneous changes to both components. For example, if controls are changed, then the wet lab change would be swapping the physical control while the dry lab change would be ensuring that the new internal controls are detectable by the bioinformatics pipeline and modifying the reporting algorithm if it depends on the control (e.g., normalization using the external negative no-template control). If, however, only the dry lab component is changed, prior sequencing data or pre=generated standardized mNGS data sets can be used to revalidate the bioinformatics pipeline. The topic of mNGS validation in the clinical laboratory has been previously discussed (24); here we briefly summarize the key points.
Accuracy: Wet Lab
Since mNGS testing is a discovery-based method rather than a hypothesis-driven method, as in the case of targeted testing, it is impractical to use mNGS to test for all possible organisms in the reference database. Instead we recommend an NGS methods–based approach, as previously suggested by the College of American Pathologists (68, 69). A limited set of organisms representing each type of infectious agent (e.g., DNA virus, RNA virus, gram-positive bacteria, gram-negative bacteria, yeast, molds, parasite) can be tested to assess accuracy. The preferred specimen type is residual samples from infected patients that contain the organism to be assayed for, but spiking-in analyte (whole organisms or purified nucleic acid) is also valid if representative samples are not easily available.
Accuracy: Dry Lab
While the entirety of the reference sequences in GenBank is enormous, genome sequences for many pathogens are still missing, and the quality of assembled reference genomes can be variable and often contain contaminating nucleic acids from other organisms or shared plasmids. Notably, the FDA is generating a standardized Database for Reference Grade Microbial Sequences (FDA-ARGOS) (https://www.ncbi.nlm.nih.gov/bioproject/231221). The initial sequences deposited in FDA-ARGOS were generated using a rigorous approach, including independent sequencing on two different platforms [e.g., Illumina and PacBio (Menlo Park, CA)] and genome assembly using multiple algorithms. The development and use of standardized, representative data sets can be used to benchmark the performance of different pipelines and changes to reference databases.
Precision
The precision and reproducibility of mNGS analyses are determined through the generation of replicate sequencing runs, and they are similar to other laboratory tests in approach. In addition to reproducibility of the final result, metrics such as library and sequence data quality, and the assessment of background flora can be useful to monitor the performance of the mNGS assays over time.
Reportable Range
The spectrum of organisms defined as reportable by the mNGS assay should be defined, and organisms determined to be background contaminants or clinically insignificant should be described. Similar to finding new variants of undetermined significance in oncologic molecular testing, the detection of atypical organisms needs to be critically assessed for their potential clinical significance, and uncertainties in clinical applicability should be communicated through the result report. Orthogonal confirmation of new or unusual results using an independent testing method, such as PCR, can be used to expand the types of reportable organisms over time. In these instances, laboratories can perform additional follow-up testing or involve public health laboratories to perform these confirmatory analyses, or both.
Reference Range
The reference range for molecular infectious disease tests is typically “not detected,” but for unbiased mNGS assays, some organisms may be detected as part of normal flora, even in specimens from sterile sites. Some of these organisms may be attributed to background contamination (e.g., human papillomaviruses), while others are true infections with circulating viremia but no known direct clinical significance (e.g., anelloviruses and GB virus C) (11). Others are simply database errors or misannotations (e.g., a stealth virus, which aligns to bacterial sequences). For clinical mNGS, reporting should include organisms with known or at least suspected pathogenic potential, without calling attention to nonpathogenic agents that could be inappropriately considered to be causing pathologic infection and, thus, lead to inappropriate treatment.
Several agents are opportunistic pathogens, and their presence needs to be evaluated in the context of the patient’s presentation to determine their clinical significance. In chronically infected individuals, human herpesviruses are integrated into the genome or present episomally, and their detection may simply indicate the presence of latently infected white blood cells in the sample (70). However, these viruses can be clinically significant pathogens, so reporting these may be appropriate, along with describing the possibility that their detection represents latent infection rather than active infection.
Limits of Detection
The lowest concentration of organismal nucleic acid detectable with 95% confidence is typically determined through replicate testing of diluted control material and probit analysis. Since each organism type may have different sequence recovery rates based on extraction efficiency and genome fragmentation, these studies are performed using representative organisms, and the results are extrapolated to similar organism types. The limits of detection may be compared with other diagnostic test methods, such as culture or PCR, to determine the sensitivity of mNGS relative to these methods.
The sensitivity of mNGS for microbe detection depends on the assay’s ability to efficiently extract and prepare libraries from the genomic material present in clinical samples. Therefore, similar types of organisms are expected to behave similarly in terms of the proportion of mNGS reads produced, enabling a representative-organism approach to be taken. However, when certain organisms are seen as part of the background of the mNGS assay, distinguishing a truly positive sample from the background may require a higher organism concentration to be present. For example, reads mapping to Cutibacterium (Propionibacterium) acnes are commonly seen in mNGS libraries due to contamination of the background sample or reagent. Normalizing the detection threshold to background levels can be useful to establish sample positivity and avoid false-positive calls. One way to accomplish this is to divide the number of sequence reads seen in the sample by the number present in negative or no-template controls and then use a threshold for organism detection based on this ratio.
Interfering Substances
Similar to other molecular methods, substances known to inhibit nucleic acid extraction or enzymatic activity should be added to known positive material to determine their potential for generating false-negative results. Commonly tested interfering substances include heme, protein (at high levels), and bilirubin. For mNGS analysis, human genomic DNA and RNA can be considered as interfering substances, with excess levels in samples essentially masking the presence of organismal nucleic acid. Therefore, spike-in experiments using exogenous DNA, RNA, or cellular material can be used to determine mNGS assay performance for samples with high levels of human host nucleic acid. As described earlier, the spiked-in internal control can be used to effectively determine when an individual sample has high levels of host nucleic acids that might decrease the sensitivity for organism detection.
Contamination
Both a positive and a negative control should be included in every sequencing run to monitor for microbial contamination during mNGS testing. This is especially important when new lots of commercial reagents and consumables are used. The nucleic acid content of the negative control matrix should be lower than in patients’ specimens to maximize the sensitivity for detecting contaminants. Contamination should be tracked over time and monitored, with investigation of potential sources of contamination and, once identified, efforts to eliminate or minimize contamination, especially if it arises from clinically significant organisms. As described above, an approach to normalize for the background present in mNGS results from negative samples can help to avoid false-positives.
Stability
Proper storage and transport conditions should be determined for the original patient sample, as well as for intermediate assay material, such as extracted nucleic acid and the sequencing library. The stability of various types of organisms after refrigeration and freezing and after multiple freeze–thaw cycles can be assessed using known control material to determine adequate recovery under various conditions.
CLINICAL UTILITY OF METAGENOMIC NEXT-GENERATION SEQUENCING
Central Nervous System Infections
The etiologies of meningitis and encephalitis are often unclear, but they may be rooted in infectious causes that often go undiagnosed. The list of potential pathogens is quite broad, and the causative pathogens in some cases are missed despite extensive diagnostic testing. Unbiased mNGS has been used to identify the etiological agent in several cases, with diagnostic confirmation using conventional tests, and some of these infections have been shown to respond to treatment (1, 71). There are now multiple case reports in which viruses (4, 5, 13, 72, 73), bacteria (1), fungi (7, 13), and parasites (6, 7, 13) have been identified from mNGS of CSF and brain tissue.
CSF is a particularly interesting body fluid because it is localized to the central nervous system (CNS) yet flows from deep sites in the CNS that are nontrivial to biopsy: areas adjacent to the lateral ventricles, third ventricle, and fourth ventricle. Interestingly, recent studies have also shown that CSF from lumbar punctures can yield tumor-specific DNA mutations from CNS tumors located upstream of the CSF flow and abutting the CSF space (74, 75). The evidence suggests the potential to diagnose the cause of CNS masses using minimally invasive procedures, such as lumbar puncture.
Bloodstream Infections
Multiple case reports and preliminary studies show that circulating cell-free pathogen DNA or RNA in blood from either circulating or noncirculating pathogens can be associated with an infection (9–11, 12, 57, 76–79). Sequencing can detect pathogen DNA in high-risk patients who are on antimicrobial therapy, raising the possibility that mNGS testing can be used for diagnosing infections in patients with culture-negative sepsis. However, bacterial nucleic acids have also been reported in healthy volunteers assessed by NGS, raising the issue of potential contamination and the question of the clinical significance of DNA detected in plasma (77).
Respiratory Infections
Pneumonia is a common infection that often lacks a diagnosis. Many patients are on antibiotic therapy, which limits the yield of culture-based testing. Complicating the analysis of mNGS is the presence of commensal oral flora organisms, which in some cases may also be pathogens. Therefore, quantitative or semiquantitative statistical analyses may help to distinguish infection from colonization. mNGS (3, 80, 81) and 16S NGS (33) have both identified pathogens using quantitative approaches. Clinical assessment is needed to determine the significance of organisms detected by mNGS, especially for cases not confirmed with conventional testing.
Gastrointestinal Infections
While multiple studies have analyzed the stool microbiome, only a few have attempted to diagnose associated clinical diseases, such as diarrhea, using mNGS techniques (82). Unbiased mNGS has been used to identify a predominance of potential pathogens in patients with acute cholecystitis (83). The detection of extended-spectrum β-lactamase genes was correlated with the susceptibility profiles of cultured isolates, indicating that NGS could be used to provide information about the relevant pathogen’s antimicrobial resistance.
Ocular Infections
Patients with known and unknown ocular infections have been diagnosed using mNGS, including a case of chronic intraocular rubella infection (2, 84). The application of mNGS to limited volume eye specimens may enable a larger list of pathogens to be tested for than is possible with conventional methods.
SUMMARY
mNGS is a revolutionary technology that has disrupted traditional clinical diagnostics on several fronts. This review demonstrates how this new technology and its associated tools can be used for meaningful clinical diagnostics in microbiology.
While the emergence of these new mNGS technologies is exciting, their rapid evolution often outpaces clinical test validation and the comprehensive collection of clinical evidence. Similar to other types of clinical testing, the application of these new diagnostic testing methods should be accompanied by rigorous clinical studies that (a) demonstrate clinical utility, (b) guide usage, and (c) uncover potential areas of misinterpretation. As with any new technology, the clinical adoption of mNGS testing will take time as providers become familiar with it and new guidelines are developed.
Acknowledgments
C.Y.C. is the director of the UCSF–Abbott Viral Diagnostics and Discovery Center (VDCC) and receives research support from Abbott Laboratories, Inc. C.Y.C. and S.M. are inventors of a patent application on algorithms related to SURPI and bioinformatics software titled, Pathogen detection using next-generation sequencing (PCT/US/16/52912).
LITERATURE CITED
- 1.Wilson MR, Naccache SN, Samayoa E, Biagtan M, Bashir H, et al. 2014. Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N. Engl. J. Med 370(25):2408–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doan T, Wilson MR, Crawford ED, Chow ED, Khan LM, et al. 2016. Illuminating uveitis: metagenomic deep sequencing identifies common and rare pathogens. Genome Med 8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Langelier C, Zinter MS, Kalantar K, Yanik GA, Christenson S, et al. 2017. Metagenomic sequencing detects respiratory pathogens in hematopoietic cellular transplant patients. Am. J. Respir. Crit. Care Med 197(4):524–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson MR, Suan D, Duggins A, Schubert RD, Khan LM, et al. 2017. A novel cause of chronic viral meningoencephalitis: Cache Valley virus. Ann. Neurol 82(1):105–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilson MR, Zimmermann LL, Crawford ED, Sample HA, Soni PR, et al. 2017. Acute West Nile virus meningoencephalitis diagnosed via metagenomic deep sequencing of cerebrospinal fluid in a renal transplant patient. Am. J. Transplant 17(3):803–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson MR, Shanbhag NM, Reid MJ, Singhal NS, Gelfand JM, et al. 2015. Diagnosing Balamuthia mandrillaris encephalitis with metagenomic deep sequencing. Ann. Neurol 78(5):722–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gu W, Crawford ED, O’Donovan BD, Wilson MR, Chow ED, et al. 2016. Depletion of abundant sequences by hybridization (DASH): using Cas9 to remove unwanted high-abundance species in sequencing libraries and molecular counting applications. Genome Biol 17:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Greninger AL, Naccache SN, Federman S, Yu G, Mbala P, et al. 2015. Rapid metagenomic identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med 7:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grumaz S, Stevens P, Grumaz C, Decker SO, Weigand MA, et al. 2016. Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med 8:73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long Y, Zhang Y, Gong Y, Sun R, Su L, et al. 2016. Diagnosis of sepsis with cell-free DNA by next-generation sequencing technology in ICU patients. Arch. Med. Res 47(5):365–71 [DOI] [PubMed] [Google Scholar]
- 11.De Vlaminck I, Khush KK, Strehl C, Kohli B, Luikart H, et al. 2013. Temporal response of the human virome to immunosuppression and antiviral therapy. Cell 155(5):1178–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlaminck ID, Martin L, Kertesz M, Patel K, Kowarsky M, et al. 2015. Noninvasive monitoring of infection and rejection after lung transplantation. PNAS 112(43):13336–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilson MR, O’Donovan BD, Gelfand JM, Sample HA, Chow FC, et al. 2018. Chronic meningitis investigated via metagenomic next-generation sequencing. JAMA Neurol 75(8):947–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rossen JWA, Friedrich AW, Moran-Gilad J, ESCMID Study Group for Genomic and Molecular Diagnostics (ESGMD). 2017. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin. Microbiol. Infect 24(4):355–60 [DOI] [PubMed] [Google Scholar]
- 15.Popovich KJ, Snitkin ES. 2017. Whole genome sequencing—implications for infection prevention and outbreak investigations. Curr. Infect. Dis. Rep 19(4):15. [DOI] [PubMed] [Google Scholar]
- 16.Ellington MJ, Ekelund O, Aarestrup FM, Canton R, Doumith M, et al. 2017. The role of whole genome sequencing in antimicrobial susceptibility testing of bacteria: report from the EUCAST subcommittee. Clin. Microbiol. Infect 23(1):2–22 [DOI] [PubMed] [Google Scholar]
- 17.Gardy JL, Loman NJ. 2018. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet 19:9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang C, Mitsuya Y, Gharizadeh B, Ronaghi M, Shafer RW. 2007. Characterization of mutation spectra with ultra-deep pyrosequencing: application to HIV-1 drug resistance. Genome Res 17(8):1195–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefterova MI, Suarez CJ, Banaei N, Pinsky BA. 2015. Next-generation sequencing for infectious disease diagnosis and management: a report of the Association for Molecular Pathology. J. Mol. Diagn 17(6):623–34 [DOI] [PubMed] [Google Scholar]
- 20.Sahoo MK, Lefterova MI, Yamamoto F, Waggoner JJ, Chou S, et al. 2013. Detection of cytomegalovirus drug resistance mutations by next-generation sequencing. J. Clin. Microbiol 51(11):3700–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinstock GM. 2012. Genomic approaches to studying the human microbiota. Nature 489(7415):250–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu C, Miller S. 2016. Next-generation sequencing. In Molecular Microbiology: Diagnostic Principles and Practice, ed. Persing DH, Tenover FC, Hayden RT, Ieven M, Miller MB, et al. , pp. 68–79. Washington, DC: ASM [Google Scholar]
- 23.Goodwin S, McPherson JD, McCombie WR. 2016. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet 17(6):333–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlaberg R, Chiu CY, Miller S, Procop GW, Weinstock G. 2017. Validation of metagenomic next-generation sequencing tests for universal pathogen detection. Arch. Pathol. Lab. Med 141(6):776–86 [DOI] [PubMed] [Google Scholar]
- 25.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456(7218):53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha R, Stanley G, Gulati GS, Ezran C, Travaglini KJ, et al. 2017. Index switching causes “spreading-of-signal” among multiplexed samples in Illumina HiSeq 4000 DNA sequencing. bioRxiv 125724 10.1101/125724 [DOI]
- 27.Rothberg JM, Hinz W, Rearick TM, Schultz J, Mileski W, et al. 2011. An integrated semiconductor device enabling non-optical genome sequencing. Nature 475(7356):348–52 [DOI] [PubMed] [Google Scholar]
- 28.Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327(5961):78–81 [DOI] [PubMed] [Google Scholar]
- 29.Fang C, Zhong H, Lin Y, Chen B, Han M, et al. 2018. Assessment of the cPAS-based BGISEQ-500 platform for metagenomic sequencing. GigaScience 7(3):gix133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bleeker-Rovers CP, Vos FJ, de Kleijn EMHA, Mudde AH, Dofferhoff TSM, et al. 2007. A prospective multicenter study on fever of unknown origin: the yield of a structured diagnostic protocol. Medicine 86(1):26–38 [DOI] [PubMed] [Google Scholar]
- 31.Ewig S, Torres A, Angeles Marcos M, Angrill J, Rañó A, et al. 2002. Factors associated with unknown aetiology in patients with community-acquired pneumonia. Eur. Respir. J 20(5):1254–62 [DOI] [PubMed] [Google Scholar]
- 32.Chiu CY. 2013. Viral pathogen discovery. Curr. Opin. Microbiol 16(4):468–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cummings LA, Kurosawa K, Hoogestraat DR, SenGupta DJ, Candra F, et al. 2016. Clinical next generation sequencing outperforms standard microbiological culture for characterizing polymicrobial samples. Clin. Chem 62(11):1465–73 [DOI] [PubMed] [Google Scholar]
- 34.Salipante SJ, Hoogestraat DR, Abbott AN, SenGupta DJ, Cummings LA, et al. 2014. Coinfection of Fusobacterium nucleatum and Actinomyces israelii in mastoiditis diagnosed by next-generation DNA sequencing. Clin. Microbiol 52(5):1789–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gire SK, Goba A, Andersen KG, Sealfon RSG, Park DJ, et al. 2014. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345(6202):1369–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salipante SJ, SenGupta DJ, Cummings LA, Land TA, Hoogestraat DR, Cookson BT. 2015. Application of whole-genome sequencing for bacterial strain typing in molecular epidemiology. J. Clin. Microbiol 53(4):1072–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deurenberg RH, Bathoorn E, Chlebowicz MA, Couto N, Ferdous M, et al. 2017. Application of next generation sequencing in clinical microbiology and infection prevention. J. Biotechnol 243:16–24 [DOI] [PubMed] [Google Scholar]
- 38.Hasan MR, Rawat A, Tang P, Jithesh PV, Thomas E, et al. 2016. Depletion of human DNA in spiked clinical specimens to improve the sensitivity of pathogen detection by next generation sequencing. J. Clin. Microbiol 54(4):919–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cummings LA, Kurosawa K, Hoogestraat DR, SenGupta DJ, Candra F, et al. 2016. Clinical next generation sequencing outperforms standard microbiological culture for characterizing polymicrobial samples. Clin. Chem 62(11):1465–73 [DOI] [PubMed] [Google Scholar]
- 40.Salipante SJ, Sengupta DJ, Rosenthal C, Costa G, Spangler J, et al. 2013. Rapid 16S rRNA next-generation sequencing of polymicrobial clinical samples for diagnosis of complex bacterial infections. PLOS ONE 8(5):e65226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.He S, Wurtzel O, Singh K, Froula JL, Yilmaz S, et al. 2010. Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nat. Methods 7(10):807–12 [DOI] [PubMed] [Google Scholar]
- 42.Giannoukos G, Ciulla DM, Huang K, Haas BJ, Izard J, et al. 2012. Efficient and robust RNA-seq process for cultured bacteria and complex community transcriptomes. Genome Biol 13:r23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adiconis X, Borges-Rivera D, Satija R, DeLuca DS, Busby MA, et al. 2013. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10(7):623–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marotz CA, Sanders JG, Zuniga C, Zaramela LS, Knight R, Zengler K. 2018. Improving saliva shotgun metagenomics by chemical host DNA depletion. Microbiome 6:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strong MJ, Xu G, Morici L, Bon-Durant SS, Baddoo M, et al. 2014. Microbial contamination in next generation sequencing: implications for sequence-based analysis of clinical samples. PLOS Pathog 10(11):e1004437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu W, Lee M, Arevalo S, Federman S, Whitman J, et al. 2017. Pathogen detection by metagenomic next generation sequencing of purulent body fluids. J. Mol. Diagn 19(6):943–1067 [Google Scholar]
- 47.Bukowska-Ośko I, Perlejewski K, Nakamura S, Motooka D, Stokowy T, et al. 2016. Sensitivity of next-generation sequencing metagenomic analysis for detection of RNA and DNA viruses in cerebrospinal fluid: the confounding effect of background contamination. Adv. Exp. Med. Biol In press. 10.1007/978-3-319-44488-8 [DOI] [PubMed]
- 48.Masuda N, Ohnishi T, Kawamoto S, Monden M, Okubo K. 1999. Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res 27(22):4436–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chirgwin JM, Przybyla AE, MacDonald RJ, Rutter WJ. 1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry 18(24):5294–99 [DOI] [PubMed] [Google Scholar]
- 50.Dohm JC, Lottaz C, Borodina T, Himmelbauer H. 2008. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res 36(16):e105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kozarewa I, Ning Z, Quail MA, Sanders MJ, Berriman M, Turner DJ. 2009. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of GC-biased genomes. Nat. Methods 6(4):291–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adey A, Morrison HG, Asan Xun X, Kitzman JO, et al. 2010. Rapid, low-input, low-bias construction of shotgun fragment libraries by high-density in vitro transposition. Genome Biol 11:R119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM. 2014. Comparison of RNA-seq by poly(A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genom 15:419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Naccache SN, Federman S, Veeeraraghavan N, Zaharia M, Lee D, et al. 2014. A cloud-compatible bioinformatics pipeline for ultrarapid pathogen identification from next-generation sequencing of clinical samples. Genome Res 24(7):1180–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wood DE, Salzberg SL. 2014. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol 15:R46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Flygare S, Simmon K, Miller C, Qiao Y, Kennedy B, et al. 2016. Taxonomer: an interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome Biol 17:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan W, Ngo TTM, Camunas-Soler J, Song C-X, Kowarsky M, et al. 2017. Simultaneously monitoring immune response and microbial infections during pregnancy through plasma cfRNA sequencing. Clin. Chem 63(11):1695–704 [DOI] [PubMed] [Google Scholar]
- 58.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ruby JG, Bellare P, Derisi JL. 2013. PRICE: software for the targeted assembly of components of (meta) genomic sequence data. G3 3(5):865–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng X, Naccache SN, Ng T, Federman S, Li L, et al. 2015. An ensemble strategy that significantly improves de novo assembly of microbial genomes from metagenomic next-generation sequencing data. Nucleic Acids Res 43(7):e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Naccache SN, Greninger AL, Lee D, Coffey LL, Phan T, et al. 2013. The perils of pathogen discovery: origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol 87(22):11966–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, et al. 2014. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12:87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mollerup S, Friis-Nielsen J, Vinner L, Hansen TA, Richter SR, et al. 2016. Propionibacterium acnes: disease-causing agent or common contaminant? Detection in diverse patient samples by next-generation sequencing. J. Clin. Microbiol 54(4):980–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Munro SA, Lund SP, Pine PS, Binder H, Clevert D-A, et al. 2014. Assessing technical performance in differential gene expression experiments with external spike-in RNA control ratio mixtures. Nat. Commun 5:5125. [DOI] [PubMed] [Google Scholar]
- 65.Hardwick SA, Deveson IW, Mercer TR. 2017. Reference standards for next-generation sequencing. Nat. Rev. Genet 18:473–84 [DOI] [PubMed] [Google Scholar]
- 66.Quail MA, Smith M, Jackson D, Leonard S, Skelly T, et al. 2014. SASI-Seq: sample assurance spike-ins, and highly differentiating 384 barcoding for Illumina sequencing. BMC Genom 15:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roy S, Coldren C, Karunamurthy A, Kip NS, Klee EW, et al. 2018. Standards and Guidelines for Validating Next-Generation Sequencing Bioinformatics Pipelines: a joint recommendation of the Association for Molecular Pathology and the College of American Pathologists. J. Mol. Diagn 20(1):4–27 [DOI] [PubMed] [Google Scholar]
- 68.Schrijver I, Aziz N, Jennings LJ, Richards CS, Voelkerding KV, Weck KE. 2014. Methods-based proficiency testing in molecular genetic pathology. J. Mol. Diagn 16(3):283–87 [DOI] [PubMed] [Google Scholar]
- 69.Aziz N, Zhao Q, Bry L, Driscoll DK, Funke B, et al. 2014. College of American Pathologists’ laboratory standards for next-generation sequencing clinical tests. Arch. Pathol. Lab. Med 139(4):481–93 [DOI] [PubMed] [Google Scholar]
- 70.Gentile G, Micozzi A. 2016. Speculations on the clinical significance of asymptomatic viral infections. Clin. Microbiol. Infect 22(7):585–88 [DOI] [PubMed] [Google Scholar]
- 71.Frémond M-L, Pérot P, Muth E, Cros G, Dumarest M, et al. 2015. Next-generation sequencing for diagnosis and tailored therapy: a case report of astrovirus-associated progressive encephalitis. J. Pediatr. Infect. Dis. Soc 4(3):e53–57 [DOI] [PubMed] [Google Scholar]
- 72.Chiu CY, Coffey LL, Murkey J, Symmes K, Sample HA, et al. 2017. Diagnosis of fatal human case of St. Louis Encephalitis virus infection by metagenomic sequencing, California, 2016. Emerg. Infect. Dis 23(10):1964–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Murkey JA, Chew KW, Carlson M, Shannon CL, Sirohi D, et al. 2017. Hepatitis E virus–associated meningoencephalitis in a lung transplant recipient diagnosed by clinical metagenomic sequencing. Open Forum Infect. Dis 4(3):ofx121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pan W, Gu W, Nagpal S, Gephart MH, Quake SR. 2015. Brain tumor mutations detected in cerebral spinal fluid. Clin. Chem 61(3):514–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, et al. 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med 6(224):224ra24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vlaminck ID, Valantine HA, Snyder TM, Strehl C, Cohen G, et al. 2014. Circulating cell-free DNA enables noninvasive diagnosis of heart transplant rejection. Sci. Transl. Med 6(241):241ra77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gosiewski T, Ludwig-Galezowska AH, Huminska K, Sroka-Oleksiak A, Radkowski P, et al. 2017. Com-prehensive detection and identification of bacterial DNA in the blood of patients with sepsis and healthy volunteers using next-generation sequencing method—the observation of DNAemia. Eur. J. Clin. Microbiol. Infect. Dis 36(2):329–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wylie KM, Mihindukulasuriya KA, Sodergren E, Weinstock GM, Storch GA. 2012. Sequence analysis of the human virome in febrile and afebrile children. PLOS ONE 7(6):e27735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abril MK, Barnett AS, Wegermann K, Fountain E, Strand A, et al. 2016. Diagnosis of Capnocytophaga canimorsus sepsis by whole-genome next-generation sequencing. Open Forum Infect. Dis 3(3):ofw144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pendleton KM, Erb-Downward JR, Bao Y, Branton WR, Falkowski NR, et al. 2017. Rapid pathogen identification in bacterial pneumonia using real-time metagenomics. Am. J. Respir. Crit. Care Med 196(12):1610–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Graf EH, Simmon KE, Tardif KD, Hymas W, Flygare S, et al. 2016. Unbiased detection of respiratory viruses by use of RNA sequencing-based metagenomics: a systematic comparison to a commercial PCR panel. J. Clin. Microbiol 54(4):1000–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou Y, Wylie KM, El Feghaly RE, Mihindukulasuriya KA, Elward A, et al. 2016. Metagenomic approach for identification of the pathogens associated with diarrhea in stool specimens. J. Clin. Microbiol 54(2):368–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kujiraoka M, Kuroda M, Asai K, Sekizuka T, Kato K, et al. 2017. Comprehensive diagnosis of bacterial infection associated with acute cholecystitis using metagenomic approach. Front. Microbiol 8:685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Doan T, Acharya NR, Pinsky BA, Sahoo MK, Chow ED, et al. 2017. Metagenomic DNA sequencing for the diagnosis of intraocular infections. Ophthalmology 124(8):1247–48 [DOI] [PMC free article] [PubMed] [Google Scholar]