

Abstract
Background:
Phosphatidylinositol-3-kinase (PI3K) and androgen receptor pathway activation is common in metastatic castration resistant prostate cancer (mCRPC). Buparlisib is an oral, pan-class I PI3 kinase inhibitor.
Methods:
This was a multisite single arm phase II trial of buparlisib 100 mg ± enzalutamide daily in men with mCRPC whose disease progressed on or who were not candidates for docetaxel. The primary end-point was the rate of radiographic/clinical progression-free survival (PFS) at 6 months.
Results:
Thirty men were accrued: 67% post-docetaxel; median prostate specific antigen (PSA) was 70 ng/dl, 83% had ≥4 prior therapies for mCRPC; 43% received concurrent enzalutamide. The final 6 month PFS rate was estimated to be 10% (95% confidence interval 2.5—23.6%). Median PFS was 1.9 months and was 3.5 months with concurrent enzalutamide. Median overall survival was 10.6 months. Concurrent enzalutamide led to a five-fold reduction in buparlisib concentrations. PSA declines were observed in 23%; no patients achieved a ≥50% decline, and no radiographic responses were observed. Severe adverse events occurred in four men including respiratory infection and multi-organ failure, urinary tract obstruction, confusion and one seizure in the setting of a new central nervous system (CNS) metastasis. Grade III adverse events were seen in 43% of patients; common toxicities included grade I—II weight loss, diarrhoea, nausea, fatigue, anorexia, rash, hyperglycemia and anxiety/mood disorders.
Conclusions:
Buparlisib did not demonstrate significant activity in men with mCRPC, suggesting that PI3K inhibition is not sufficient to reverse resistant mCRPC progression. Future studies of PI3K pathway inhibitors with concurrent enzalutamide should develop optimal dosing and focus on selected patients more likely to benefit.
Keywords: BKM-120, Buparlisib, Prostate cancer, PI3 kinase, Enzalutamide, Metastatic castration, resistant prostate, cancer, Clinical trial
Precis
This was a multisite phase II trial of buparlisib, a panclass I PI3 kinase inhibitor, in men with metastatic castration resistant prostate cancer (mCRPC) who had failed or were not candidates for docetaxel. Buparlisib alone or in combination with enzalutamide did not improve progression-free survival, suggesting that phosphatidylinositol-3-kinase and c-terminal androgen receptor inhibition is not sufficient to reverse resistant mCRPC progression.
1. Introduction
Despite immunotherapy, chemotherapy and novel androgen receptor (AR) directed therapies, men with metastatic castration resistant prostate cancer (mCRPC) develop resistant disease progression within months to a few years [1-6]. Thus, there is an urgent need to develop more effective treatments.
A key oncogenic pathway implicated in CRPC progression is the phosphatidylinositol-3-kinase (PI3K) pathway, which is activated in the majority of metastatic human prostate cancer (PC) samples, often through PTEN loss [7,8]. The PI3K pathway has been shown to promote castration and chemotherapy resistance, stem-ness, cell growth and differentiation, as well as AR signalling, key hallmarks of CRPC lethality and progression [9-12]. Clinical trials of mammalian target of rapamycin (mTOR; TORC1) inhibitors in men with PC have not demonstrated sufficient clinical activity, suggesting alternative pathways such as PI3 kinase and AR signalling may contribute to resistance [13-17].
Buparlisib (BKM-120) is an orally bioavailable panclass I PI3K inhibitor [18]. In preclinical models, growth inhibition was observed in tumours with PIK3CA mutations preferentially, and in PTEN null models [18]. The recommended phase II dose of buparlisib is 100 mg once daily, with significant toxicities including mood disturbances (neuropsychiatric), hyperglycemia, and rash and target inhibition was demonstrated [19]. Pharmacodynamic inhibition of phospho-S6, a downstream biomarker of PI3K/mTOR pathway activity, was observed in 80% of patients at this dose level, along with concurrent increases in insulin and blood glucose, consistent with pathway inhibition. Partial response or stable disease was observed in over 50% of patients, including colorectal and breast carcinoma [19].
Reciprocal feedback inhibition of AR by PI3K signalling in PC has been noted, in which inhibition of PI3K leads to re-repression and activation of AR target genes, and inhibition of AR leads to reciprocal PI3K pathway activation in PC models [20,21]. On the other hand, concomitant suppression of both PI3K and AR pathways led to tumour regressions [20]. Given that PI3K pathway activation is common in men with mCRPC, we conducted a phase II efficacy trial of buparlisib, with and without the potent AR inhibitor enzalutamide [1], in men with progressive mCRPC that progressed after multiple lines of standard therapy. The amendment permitting concurrent AR inhibition with enzalutamide was based on emerging data suggesting that combined PI3K and AR pathway inhibition could overcome reciprocal feedback AR pathway activation by PI3K single agent inhibition [20,21].
2. Methods
2.1. Eligibility
Men with mCRPC were eligible if they were 18 years of age or older, had a Karnofsky performance status ≥70 and a life expectancy of at least 3 months. Patients were required to have histologically confirmed PC, metastatic disease and progressive disease according to Response Evaluation Criteria in Solid Tumours (RECIST 1.1) [22] or Prostate Cancer Working Group 2 (PCWG2) prostate specific antigen (PSA) or radiographic progression (computed tomography [CT]/bone scan) criteria [23]. We amended the study after 17 subjects were enrolled to permit patients who were progressing on enzalutamide continue enzalutamide with the addition of buparlisib therapy (see CONSORT diagram, Supplementary Fig. 1). All men were continued on androgen deprivation therapy in order to maintain a castrate level of testosterone (<50 ng/dl), and bone anti-resorptive therapy if bone metastases were present. Men were required to have had at least one prior systemic taxane-based chemotherapy for PC unless the patient refused or did not tolerate chemotherapy.
Exclusion criteria included untreated brain metastases, chronic liver disease or active infection, significant mood disorders (anxiety, depression, bipolar disorder, psychosis, homicidality or suicidality), or uncontrolled cardiac conditions, diabetes and a fasting blood glucose ≥120, and concurrent use of strong inhibitors/inducers of CYP3A4. Full details are available on clinicaltrials.gov (NCT01385293). All patients provided informed consent under an institutional review board (IRB)-approved consent form.
2.2. Trial design and study treatment
This was a prospective, open-label single arm investigator-initiated trial conducted across 3 sites within the Department of Defence Prostate Cancer Clinical Trials Consortium, with the Duke Cancer Institute as lead site. After meeting eligibility, subjects were treated with buparlisib 100 mg orally once daily. Dose reductions were permitted based on prespecified criteria for toxicity. This study was originally written as a single agent study of buparlisib; however, concurrent buparlisib and enzalutamide at 160 mg once daily was permitted through a protocol amendment in order to test the efficacy of combined pathway inhibition in men with mCRPC progressing on enzalutamide [20,21]; the use of concurrent enzalutamide was not randomised but rather offered to patients progressing on enzalutamide at entry.
Treatment was continued until radiographic or clinical progression as per RECIST 1.1 criteria or physician decision around lack of clinical benefit. Imaging including CT and bone scans was performed at baseline and every 2 months. Cycles were 4 weeks in length with laboratory (bone marrow, kidney and liver function, fasting glucose and lipids, PSA, CTCs, lactate dehydrogenase) toxicity assessments performed on days 1 and 15 in cycle 1 and day 1 of subsequent cycles. Circulating tumour cells (CTCs) were enumerated and analysed by the US Food and Drug Administration approved CELLSEARCH method at baseline and at day 1 of each cycle. Psychiatric assessments were performed using the PHQ-9 scale for depression and the GAD-7 scale for anxiety at screening, days 1 and 15 of cycles 1 and 2, and each cycle thereafter. Adverse events were tracked continuously and reported using National Cancer Institute Common Toxicity Criteria (NCI CTC) version 4.0.
For men treated with concurrent enzalutamide (n = 13) intense pharmacokinetic samples for buparlisib were conducted using previously published methodology [19]. PK samples were drawn on day 1 before dose (0 min) and 30, 60, 120 min, 4 and 8 h, and 24 h after dose (prior to next dose, or trough level). For patients who received single agent buparlisib (n = 17), a single PK sample was drawn before dose on cycle 2 day 1.
2.3. End-points and statistical methods
The primary end-point of this study was progression-free survival (PFS) as determined by the earliest of either radiographic progression based on the PCWG2 criteria (RECIST 1.1 soft tissue progression or confirmed bone scan progression), death, or the onset of a skeletal-related event or symptomatic deterioration or a requirement to start a new systemic therapy. PSA increases did not define progression. The historical 6-month PFS survival rate was 24.5%, based on everolimus-treated patients in men with metastatic che-morefractory CRPC [24] and data reported in the phase III TROPIC trial of cabazitaxel in this setting [4]. The null hypothesis was a 6-month PFS rate of 24.5% and alternative hypothesis was 40%. It was assumed that 60 patients would be accrued over 24 months with 12 months of follow-up, leading to a target sample size of 66 patients assuming withdrawal/ineligibility rate of 10%.
This trial included a futility analysis after 30 patients had been followed for 6 months. Accrual to the trial was suspended while conducting the futility analysis. The futility analysis would be performed when 44% of the PFS events (25 events) had occurred. The probabilities of early termination under the null and alternative hypothesis were 0.62 and 0.12, respectively. The type I error rate after considering the futility boundary was 0.12 and the power was 0.86. The Kaplan-Meier product limit approach was used to estimate the PFS and overall survival distributions for all patients enrolled on the trial, and in men treated with buparlisib alone or with concurrent enzalutamide. The database was locked on 6th May 2015 to perform the interim analysis.
3. Results
From 25th August 2011 through 1st May 2014, 30 patients were enrolled into the phase II trial (see consort diagram, Supplementary Fig. 1). Baseline characteristics are shown in Table 1. Eighty-seven percent of subjects were white, 7% black. The majority of men had prior Gleason 8-10 disease (57%), multiple prior systemic therapies (56% had 6 or more), and 67% had prior docetaxel for mCRPC. Thirteen (43%) had prior enzalutamide and continued on enzalutamide with concurrent buparlisib (following an amendment), while 15 (50%) overall had prior abiraterone acetate (23% of those treated with concurrent enzalutamide, and 27% of those who had no prior/concurrent enzalutamide) and 4 (13%) had prior cabazitaxel chemotherapy. The radiographic pattern of spread of these men was bone predominant ± nodal metastases in 60%, and visceral metastases were observed in 40% of men. The median PSA at entry was 70 ng/dl, and the median CTC count was 15 cells per 7.5 mL. Of those men with evaluable CTCs (n = 22), 11 (50%) had ≥5 CTCs at baseline.
Table 1. Baseline characteristics of the patients.
| Baseline characteristic | Concurrent enzalutamide | Overall (%) | |
|---|---|---|---|
| No (n = 17) | Yes (n = 13) | n = 30 | |
| N (%) | N (%) | N (%) | |
| Age, median and range in years | 66 (48-91) | 68 (53-94) | 67 (48-94) |
| Race | |||
| Black or African American | 1 (6) | 1 (8) | 2 (7) |
| Native American/Alaska Native | 1 (6) | 0 (0) | 1 (3) |
| Native Hawaiian or Other Pacific Islander | 1 (6) | 0 (0) | 1 (3) |
| White non-Hispanic | 14 (82) | 12 (92) | 26 (87) |
| Karnofsky performance status (KPS) | |||
| ≤80 | 8 (47) | 7 (54) | 15 (50) |
| 90-100 | 9 (53) | 5 (38) | 14 (47) |
| Missing | 0 (0) | 1 (8) | 1 (3) |
| Gleason sum | |||
| 6 | 2 (12) | 3 (23) | 5 (17) |
| 7 | 4 (24) | 3 (23) | 7 (23) |
| 8-10 | 11 (65) | 6 (46) | 17 (57) |
| Missing | 0 (0) | 1 (8) | 1 (3) |
| Prior docetaxel | |||
| No | 6 (35) | 4 (31) | 10 (33) |
| Yes | 11 (65) | 9 (69) | 20 (67) |
| #Prior systemic therapies | |||
| 0-2 | 4 (24) | 1 (8) | 5 (17) |
| 3-5 | 5 (29) | 3 (23) | 8 (27) |
| 6-8 | 6 (35) | 7 (54) | 13 (43) |
| ≥9 | 2 (12) | 2 (15) | 4 (13) |
| Metastatic disease | |||
| Bone ± node, no visceral metastases | 10 (59) | 8 (62) | 18 (60) |
| Visceral metastases | 7 (41) | 5 (48) | 12 (40) |
| Opiate use | 9 (53) | 3 (23) | 12 (40) |
| PSA, median (ng/dl) | 82 (0.05-650) | 49 (4.6-5317) | 70 (0.05-5317) |
| Median circulating tumour cells | 4 (0-112) | 21 (1-60) | 15 (0-112) |
| (cells per 7.5 mL), n = 22 evaluable | |||
| Alkaline phosphatase (IU/L) | 87 (25-493) | 88 (26-943) | 88 (25-943) |
| Haemoglobin, g/dl | 11.2 (9.2-14.5) | 11.5 (9.5-13.7) | 11.3 (9.2-14.5) |
| Lactate dehydrogenase (LDH) | 198 (132-1273) | 189 (147-2000) | 195 (132-2000) |
3.1. Safety
Toxicity at least possibly related to study drug(s) as scored by the NCI CTC version 4.0 criteria are reported in Table 2. Three patients (10%) stopped buparlisib due to toxicity. One patient treated with concurrent buparlisib and enzalutamide developed a tonic-clonic seizure in the setting of progressive disease and a new central nervous system (CNS) metastasis, although the seizure was felt to be at least possibly related to enzalutamide and the lower seizure threshold. One patient died of multi-organ failure and pneumonia 3 d after initiating buparlisib (grade V toxicity) but this was felt to be unlikely related to study drug. Mood disorders due to buparlisib were seen in approximately 10% of patients, including anxiety or depression (10% each), agitation (3%) and confusion with hypoactive delirium (7%). Two additional possibly related severe adverse events were observed in one patient each: confusion and urinary tract obstruction due to local progression of disease. Adverse events were generally mild in nature (grade I-II) and included nausea (30%), anorexia (27%), diarrhoea (23%), fatigue (23%), hyperglycemia (13%), pain (13%), constipation (10%), rash with pruritus (10%). More severe adverse events (grade III-IV) included fatigue (13%), hyperglycemia (3%), confusion (3%), myalgia (3%) and urinary tract obstruction (3%).
Table 2. Listing of most commonly observed toxicities at least possibly related to study drugs at a rate of >10% or grades III-V.
| NCI CTC version 4.0 toxicity term |
Grade I-II (%) | Grade III-IV (%) |
|---|---|---|
| Nausea | 35 | 0 |
| 23 | 0 | |
| Fatigue | 29 | 12 |
| 15 | 15 | |
| Anorexia | 29 | 0 |
| 23 | 0 | |
| Diarrhoea | 29 | 0 |
| 15 | 0 | |
| Hyperglycemia | 18 | 6 |
| 8 | 0 | |
| Pruritus | 18 | 0 |
| 0 | 0 | |
| Pain | 12 | 0 |
| 15 | 0 | |
| Anxiety | 6 | 0 |
| 15 | 0 | |
| Oedema limbs | 0 | 0 |
| 15 | 0 | |
| Constipation | 12 | 0 |
| 8 | 0 | |
| ALT increased | 12 | 0 |
| 0 | 0 | |
| AST increased | 12 | 0 |
| 0 | 0 | |
| Depression | 12 | 0 |
| 8 | 0 | |
| Anaemia | 0 | 0 |
| 8 | 8 | |
| Confusion | 0 | 6 |
| 8 | 0 | |
| Myalgia (spasmatic pain) | 0 | 6 |
| 0 | 0 | |
| Urinary tract obstruction | 0 | 6 |
| 0 | 0 | |
Percentages are worst grade reported per patient. No grade V adverse events (AEs) were reported. n = 17 (buparlisib, grey), n = 13 (buparlisib + enza, white).
AST, aspartate transaminase, ALT, alanine transaminase.
While comparisons are limited by sample size, toxicities seen with single agent buparlisib and combination enzalutamide/buparlisib are further described in Table 2. Combination therapy appeared to be associated with a reduced rather than increased toxicity. The most common toxicities observed were nausea, fatigue, anorexia, diarrhoea, hyperglycemia and pruritus, and were typically grade I-II in nature. Hyperglycemia was observed in 4/17 (24%) subjects treated with buparlisib alone as compared to 1/13 (8%) patients treated with concurrent buparlisib/enzalutamide. Mood disorders such as anxiety (6% monotherapy, 15% with combination) and depression (12% monotherapy, 8% combination) were typically mild in nature.
3.2. Pharmacokinetic studies
For men treated with concurrent enzalutami-de-buparlisib and evaluable PK data, median Cmax was 242.5 ng/mL and the median area under the curve (AUC) for buparlisib was 1777 h* ng/ml. For comparison at this pharmacodynamically active dose level in the phase I trial of buparlisib [19] as a single agent, the median Cmax in cycle 1 (single dose) was 1010 ng/ml and median AUC was 8680 h* ng/mL, indicating a significantly lower (~80% lower than expected) concentration of buparlisib when administered with enzalutamide. For men treated with buparlisib alone, the trough cycle 2 PK parameters indicated a median of 614 ng/mL (n = 9, range 36-1550). Intensive PK studies were not performed with buparlisib alone (prior to the amended protocol), limited any comparison with the prior phase I trial. However, patients requiring dose reduction in buparlisib (n = 4) tended to have a greater exposure (trough PK median 805 ng/mL, range 614-1550) than those men who tolerated full dose buparlisib (n = 5, median trough concentration 421 ng/mL, range 36-813). Please see Supplementary Table 1 and Supplementary Fig. 2 for details on PK parameters and within-patient PK heterogeneity for combination patients.
3.3. Efficacy
At the time of the database lock, 27 of 30 patients have progressed before 6 months. The primary end-point of this trial was the rate of composite PFS at 6 months and was 0%. With further follow-up, the 6 month PFS rate was estimated to be 10% (95% confidence interval [CI] 2.5-23.6%) and the median PFS was 1.9 months (95% CI 1.8-3.4 months). The 6 month PFS rate was 11.8% (95% CI 2.0-31.2) among men treated with buparlisib alone and 7.7% (95% CI 0.5-29.2%) among men treated with buparlisib and concurrent enzalutamide (n = 13) (Table 3). The median PFS was 1.9 months (95% CI 1.7-3.0) for buparlisib alone and 3.5 months (95% CI 1.2-5.5) for buparlisib plus concurrent enzalutamide (Fig. 1a). Thus, the trial was stopped at the first stage (n = 30).
Table 3. Efficacy summary.
| Estimate | Concurrent enzalutamide | Total | # Failed | Median, months (95% CI) | 6-month survival (95% CI) | 1-year survival (95% CI) |
|---|---|---|---|---|---|---|
| OS | Total | 30 | 29 | 10.6 (6.5-14.6) | 70.0% (50.3%-83.1%) | 43.3% (25.6%-59.9%) |
| No | 17 | 17 | 10.2 (4.7-14.4) | 70.6% (43.8%-86.6%) | 35.3% (14.5%-57.0%) | |
| Yes | 13 | 12 | 13.6 (3.2-15.4) | 69.2% (37.3%-87.2%) | 53.8% (24.8%-76.0%) | |
| PFS | Total | 30 | 30 | 1.9 (1.8-3.4) | 10.0% (2.5%-23.6%) | 10.0% (2.5%-23.6%) |
| No | 17 | 17 | 1.9 (1.7-3.0) | 11.8% (2.0%-31.2%) | 11.8% (2.0%-31.2%) | |
| Yes | 13 | 13 | 3.5 (1.2-5.5) | 7.7% (0.5%-29.2%) | 7.7% (0.5%-29.2%) | |
Key efficacy end-points, including radiographic progression-free survival (rPFS), overall survival (OS), and 6 and 12 month rates of rPFS and OS overall and according to concurrent enzalutamide use.
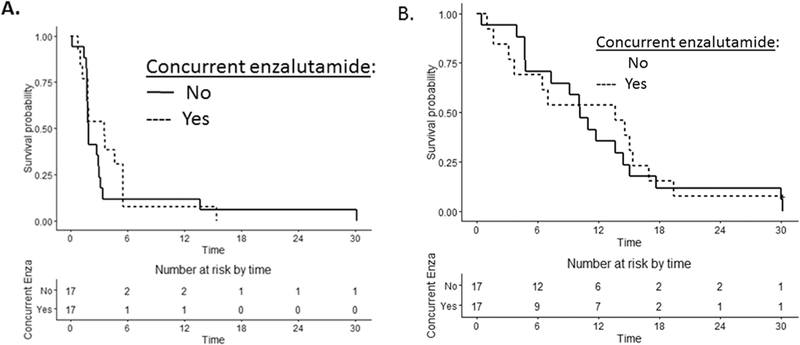
Fig. 1. (A) Kaplan-Meier estimates of progression-free survival (PFS) according to treatment group. (B) Kaplan-Meier estimates of overall survival (OS) according to treatment group.
The median overall survival of all subjects was 10.6 months (95% CI 6.5-14.6) and similar across subgroups of buparlisib alone or buparlisib/enzalutamide (Fig. 1b). The 1-year survival rate was 43.3% (95% CI 5.6-59.9%; Table 3). The 1-year survival rate for buparlisib alone was 35.3% (95% CI 14.5-57.0%) and was 53.8% for buparlisib/enzalutamide (95% CI 24.8-76.0%).
PSA declines were not commonly observed with buparlisib, with only one patient (3%) experiencing a ≥30% PSA decline (see Fig. 2a). The median percent PSA change from baseline was +80% (+104% without concurrent enzalutamide and +45% with concurrent enza-lutamide). Thus, a rise in PSA was the most consistent PSA pattern observed on study, with 6/30 subjects experiencing a more than doubling in PSA over time (20%). There was no discernable difference in the PSA decline rates among men treated with or without enzalutamide. Only 1/13 CTC-evaluable subjects with ≥5 CTCs at baseline had a conversion to <5 CTCs (7%). The best overall radiographic response by RECIST 1.1 was unconfirmed stable disease in 40% of subjects, with confirmed stable disease observed in 17% of patients, and 43% of men had progressive disease as their best response.
Fig. 2.
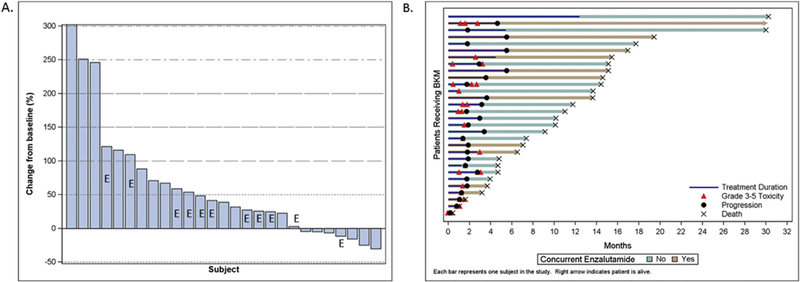
(A) Waterfall plot of best PSA decline from baseline. (B) Swimmer’s plot of the patient experience on buparlisib with or without enzalutamide.
A swimmer’s plot is presented which summarises the individual patient flow including toxicity, progression, drug treatment and survival (Fig. 2b). The median treatment duration was 2.1 months (range 0.2-7.4 months), with the vast majority of patients stopping treatment due to progression within 2-6 months, and only 1 patient remained on study drug for longer than 6 months.
4. Discussion
In this trial, the pan-PI3 kinase inhibitor buparlisib (BKM-120) did not provide sufficient efficacy in men with treatment-refractory mCRPC as a single agent. In addition, we did not observe reversal of resistance or sufficient clinical activity among men with enzalutamide resistant mCRPC treated with continued enzalutamide and the addition of buparlisib. This was accompanied by reduced toxicity related to buparlisib and substantially lower concentrations of buparlisib, suggesting that enzalutamide substantially induces the metabolism and clearance of buparlisib. Enzalutamide is a strong cytochrome P450 CYP2C9 and CYP3A4 enzyme inducer, and buparlisib is a substrate of multiple hepatic cytochrome enzymes, including CYP3A1 andCYP2C11. While the exact mechanism of reduced buparlisib concentration by enzalutamide is not known, it is likely mediated through CYP enzyme induction. The single agent buparlisib data are similar to the modest clinical activity observed in men with mCRPC treated with the TORC1 inhibitors [13-16]. The lack of single agent activity is supported by the limited PSA and CTC declines or radiographic responses, the short time to clinical/radiographic progression.
The reasons for the limited clinical activity of a pan-PI3 kinase inhibitor, despite strong preclinical rationale and clinical evidence of pathway activation in the majority of men with mCRPC, are unclear. Single agent maximum tolerated dose (MTD) dosing of buparlisib at 100 mg once daily has been shown to pharmacodynamically inhibit the PI3K pathway in target tissues in phase I studies [19]. Originally, this study was designed as a single agent buparlisib trial; however, due to limited activity, an amendment was approved which permitted concurrent enzalutamide based on preclinical data demonstrating greater anti-tumour activity with combined AR and PI3K pathway inhibition [20,21]. While drug interactions were anticipated, strikingly insufficient drug levels of buparlisib were observed when dosed concurrently with enzalutamide, which likely limited pharmacodynamic target engagement of the PI3K signalling axis and our ability to assess the efficacy of combination therapy. The lack of pharmacodynamic assessments in this trial is clearly a limitation for determining target engagement. In order to circumvent this, a dose finding study of escalating doses of buparlisib with standard enzalutamide dosing would be needed based on PI3K target engagement assessment and PK assessments as well as dose limiting toxicities (phase I dose optimisation approach). Our present study was not designed originally to investigate such a dose finding PK/PD approach.
Based on evidence for reciprocal feedback regulation of the PI3K and AR pathways in CRPC preclinical models [20,21], we had hypothesised that buparlisib would overcome the acquired or primary resistance observed to enzalutamide, given that the majority of men with CRPC harbour somatic alterations in the PI3K pathway [7,8]. A biomarker-selected population for PI3K pathway inhibition may have led to greater clinical activity, and such approaches should be considered in future trials, given the known molecular heterogeneity between men with mCRPC. Given the high frequency of PI3K pathway alterations in men with mCRPC [7] and the nearly uniform increases in PSA that were observed with buparlisib in this setting, both alone and concurrently with enzalutamide, our data support such a reciprocal feedback mechanism in patients or simply rapid cancer progression and resistance through other pathways including AR activity. The lack of activity of buparlisib may be due to additional mechanisms of CRPC progression including activation of full length AR or AR splice variants (i.e. AR-V7) [25], alternative pathways (such as 4EBP1 abundance [26], RAS/MAPK activation [27], NFkB activation, MYC gain, RB1 loss, TP53 loss, mitochondrial reprogramming [28,29]) or incomplete PI3K/AKT pathway inhibition. Our clinical experience with buparlisib alone suggests that there is limited clinical activity of single agent buparlisib in the patient population examined and highlights the need for improved preclinical models [13,14,24,30]. It also remains biologically plausible that upfront inhibition with combined potent AR and PI3K/Akt inhibition may be more effective than treatment with a PI3K/Akt inhibitor upon the development of AR resistance. While further development of buparlisib in men with CRPC is not planned, further studies of such a combination approach targeting AR and the PI3K/Akt pathway are merited following the determination of safe, effective and pharmacodynami-cally active dosing of a PI3K pathway inhibitor with concurrent AR pathway inhibition, particularly in activated PI3K pathway enriched populations.
Supplementary Material
Acknowledgements
The authors wish to acknowledge the trial coordinators and their staff for their support in the conduct of this trial, and the patients and their families for their dedication to this research. The authors also wish to thank staff psychiatrists Sarah Rivelli MD (Duke) and Anne F. Gross MD (OHSU) for their support and assessments on this study. The authors acknowledge support of the Department of Defence Prostate Cancer Clinical Trials Consortium for the development of the database used for this study and for the infrastructural support needed for this study. This work was supported financially by Novartis.
Funding
This is an investigator-initiated/sponsored trial with funding from Novartis.
AA receives research support from Medivation, Astellas, Janssen, Sanofi-Aventis and Dendreon, is a consultant/advisory board member for Medivation, Bayer and Sanofi-Aventis and a speaker for Dendreon and Sanofi-Aventis. JA receives research support from Novartis and Astellas. TB receives research support from Astellas and Medivation and is a consultant/advisory board member for Astellas. EY receives research support from Astellas, Bayer and Dendreon and is a consultant/ advisory board member for Astellas, Bayer, Dendreon, Ferring, Janssen and Tokai. DG receives research support from Bayer, Dendreon Corporation, Genentech, Innocrin, Janssen, Millenium, Novartis, Pfizer Inc, Sanofi-Aventis and Viamet; is a consultant for Astellas, Bayer, Dendreon Corporation, Genentech, Innocrin, Janssen, Medivation, Novartis, Pfizer Inc, Sanofi-Aventis and Viamet and a speaker for Bayer, Dendreon Corporation, Novartis and Sanofi-Aventis.
Footnotes
Conflict of interest statement
The other authors report no potential conflicts of interest.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.ejca.2017.02.030.
References
- [1].Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014;371:424–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010;363:411–22. [DOI] [PubMed] [Google Scholar]
- [3].Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de SP, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med 2013;368:138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I, et al. Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010; 376:1147–54. [DOI] [PubMed] [Google Scholar]
- [5].Parker C, Nilsson S, Heinrich D, O’Sullivan JM, Fossa SD, Chodacki A, et al. Updated analysis of the phase III, doubleblind, randomized, multinational study of radium-223 chloride in castration-resistant prostate cancer (CRPC) patients with bone metastases (ALSYMPCA). ASCO Meeting Abstracts, vol. 30; [Google Scholar]
- [6].Tannock IF, de WR, Berry WR, Horti J, Pluzanska A, Chi KN, et al. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med 2004;351:1502–12. [DOI] [PubMed] [Google Scholar]
- [7].Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010;18:11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, et al. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res 2010;16: 5692–702. [DOI] [PubMed] [Google Scholar]
- [10].Dubrovska A, Kim S, Salamone RJ, Walker JR, Maira SM, Garcia-Echeverria C, et al. The role of PTEN/Akt/PI3K signaling in the maintenance and viability of prostate cancer stem-like cell populations. Proc Natl Acad Sci U S A 2009;106:268–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Edgar KA, Wallin JJ, Berry M, Lee LB, Prior WW, Sampath D, et al. Isoform-specific phosphoinositide 3-kinase inhibitors exert distinct effects in solid tumors. Cancer Res 2010;70: 1164–72. [DOI] [PubMed] [Google Scholar]
- [12].Schwartz S, Wongvipat J, Trigwell CB, Hancox U, Carver BS, Rodrik-Outmezguine V, et al. Feedback suppression of PI3Kal-pha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell 2015;27:109–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Armstrong AJ, Netto GJ, Rudek MA, Halabi S, Wood DP, Creel PA, et al. A pharmacodynamic study of rapamycin in men with intermediate-to high-risk localized prostate cancer. Clin Cancer Res Off J Am Assoc Cancer Res 2010;16:3057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Armstrong AJ, Shen T, Halabi S, Kemeny G, Bitting RL, Kartcheske P, et al. A phase II trial of temsirolimus in men with castration-resistant metastatic prostate cancer. Clin Genitourin Cancer 2013;11:397–406. [DOI] [PubMed] [Google Scholar]
- [15].Courtney KD, Manola JB, Elfiky AA, Ross R, Oh WK, Yap JT, et al. A phase I study of everolimus and docetaxel in patients with castration-resistant prostate cancer. Clin Genitourin Cancer 2015; 13:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rathkopf DE, Larson SM, Anand A, Morris MJ, Slovin SF, Shaffer DR, et al. Everolimus combined with gefitinib in patients with metastatic castration-resistant prostate cancer: phase 1/2 results and signaling pathway implications. Cancer 2015;121: 3853–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Templeton AJ, Dutoit V, Cathomas R, Rothermundt C, Bartschi D, Droge C, et al. Phase 2 trial of single-agent everolimus in chemotherapy-naive patients with castration-resistant prostate cancer (SAKK 08/08). Eur Urol 2013;64:150–8. [DOI] [PubMed] [Google Scholar]
- [18].Maira SM, Pecchi S, Huang A, Burger M, Knapp M, Sterker D, et al. Identification and characterization of NVP-BKM120, an orally available pan-class I PI3-kinase inhibitor. Mol Cancer Ther 2012;11:317–28. [DOI] [PubMed] [Google Scholar]
- [19].Bendell JC, Rodon J, Burris HA, de JM, VerweijJ, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol 2012;30:282–90. [DOI] [PubMed] [Google Scholar]
- [20].Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011;19:575–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011;19:792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45: 228–47. [DOI] [PubMed] [Google Scholar]
- [23].Scher HI, Halabi S, Tannock I, Morris M, Sternberg CN, Carducci MA, et al. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: recommendations of the Prostate Cancer Clinical Trials Working Group. J Clin Oncol 2008;26:1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].George DAAC P, Morris K, Madden J, Turnbull J, Dewhirst M, Major N, et al. A phase II study of RAD001 in men with hormone-refractory metastatic prostate cancer (HRPC). In: 2008 Genitourinary Cancers Symposium; 2008.abstract 181 [Google Scholar]
- [25].Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med 2014;371:1028–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hsieh AC, Nguyen HG, Wen L, Edlind MP, Carroll PR, Kim W, et al. Cell type-specific abundance of 4EBP1 primes prostate cancer sensitivity or resistance to PI3K pathway inhibitors. Sci Signal 2015;8:ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Will M, Qin AC, Toy W, Yao Z, Rodrik-Outmezguine V, Schneider C, et al. Rapid induction of apoptosis by PI3K inhibitors is dependent upon their transient inhibition of RAS-ERK signaling. Cancer Discov 2014;4:334–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Caino MC, Ghosh JC, Chae YC, Vaira V, Rivadeneira DB, Faversani A, et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc Natl Acad Sci U S A 2015;112:8638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ghosh JC, Siegelin MD, Vaira V, Faversani A, Tavecchio M, Chae YC, et al. Adaptive mitochondrial reprogramming and resistance to PI3K therapy. J Natl Cancer Inst 2015:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nakabayashi M, Werner L, Courtney KD, Buckle G, Oh WK, Bubley GJ, et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU Int 2012;110: 1729–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.