

Abstract
Hyperpolarization-activated cyclic nucleotide-gated (HCN) channels mediate rhythmic electrical activity of cardiac pacemaker cells, and in neurons play important roles in setting resting membrane potentials, dendritic integration, neuronal pacemaking, and establishing action potential threshold. Block of HCN channels slows the heart rate and is currently used to treat angina. However, HCN block also provides a promising approach to the treatment of neuronal disorders including epilepsy and neuropathic pain. While several molecules that block HCN channels have been identified, including clonidine and its derivative alinidine, lidocaine, mepivacaine, bupivacaine, ZD7288, ivabradine, zatebradine, and cilobradine, their low affinity and lack of specificity prevents wide-spread use. Different studies suggest that the binding sites of these inhibitors are located in the inner vestibule of HCN channels, but the molecular details of their binding remain unknown. We used computational docking experiments to assess the binding sites and mode of binding of these inhibitors against the recently solved atomic structure of human HCN1 channels, and a homology model of the open pore derived from a closely related CNG channel. We identify a possible hydrophobic groove in the pore cavity that plays an important role in conformationally restricting the location and orientation of drugs bound to the inner vestibule. Our results also help explain the molecular basis of the low-affinity binding of these inhibitors, paving the way for the development of higher affinity molecules.
Introduction
Hyperpolarization-activated cyclic-nucleotide gated (HCN) channels are the molecular correlate of the currents If or Ih in sinoatrial node (SAN) cells and neurons. Four mammalian isoforms have been identified (HCN1-4) with 60% sequence identity among them. Topologically, HCN channels resemble voltage-gated potassium (Kv) channels, however, functionally they are spectacularly different. HCN channels are formed by homo- or hetero-tetrameric assembly of subunits1. Each subunit contains 6 transmembrane α-helices (S1–S6), a re-entrant loop between the S5 and S6 helices that forms the selectivity filter and a C-terminal cyclic-nucleotide binding domain (CNBD) attached to the S6 via an 80 amino acid C-linker. Like other voltage-gated channels, HCN channels contain a positively charged S4 helix that functions as a voltage sensor that moves with the same directionality as voltage sensors of other channels2,3. However, HCN channels slowly activate at very negative (hyperpolarized) membrane potentials in which other voltage-gated cation channels close. Electrophysiological recordings have characteristic properties, including activation upon membrane hyperpolarization, a lack of voltage-dependent inactivation, conduction of Na+ and K+, a shift in the activation curve due to direct interaction with cAMP and cGMP, and inhibition by external Cs+4. The rates of opening and closing differ for each mammalian HCN isoform. HCN1 channels activate in less than 300 ms, while HCN4 channels require seconds to open. Moreover, the half-maximal voltage for activation (V1/2) for HCN1 and HCN3 are significantly depolarized compared to HCN2 and HCN4. HCN isoforms also differ from one another in their response to cyclic nucleotides. cAMP shifts the V1/2 in HCN2 and HCN4 by +15 mV, while HCN1 and HCN3 are only weakly modulated, with cAMP inducing shifts in V1/2 of less than +5mV5–8.
HCN1 and HCN2 channels are widely expressed in the central and peripheral nervous systems where they are open at sub-threshold potentials and play roles in setting resting membrane potentials, dendritic integration, neuronal pacemaking, and establishing action potential threshold. HCN1 knockout mice have impaired motor learning9,10 and enhance susceptibility to seizures11. HCN2 knockout mice present symptoms of absence epilepsy and tremoring12, and do not demonstrate neuropathic pain in response to mechanical or thermal stimuli13. The gain of function and loss of function mutations in HCN1 and 2 are linked to various genetic epilepsies in humans14–18. Altered HCN-cAMP signaling in prefrontal cortex networks also appears to contribute to the working memory deficits in schizophrenia and stress19–21. Mutations in the scaffolding protein SHANK3 may predispose people to autism by inducing an Ih channelopathy with increased neuronal input resistance, enhanced neuronal excitability and reduced synaptic transmission22. Additionally, HCN4 is the principal component of Ih in all mammalian sinoatrial node (SAN) and other cardiac conduction tissue5,23–26. HCN4−/− resulted in embryonic death in mice due to a failure to generate mature pacemaking cells12,27 while HCN4 conditionally deficient mice have a 70–80% reduction in SAN Ih28. Genetic variants in HCN channels have been linked to cardiac disorders including sinus node dysfunction, atrial fibrillation29–39, ventricular tachycardia40–42, atrio-ventricular block43, Brugada syndrome38,44, sudden infant death syndrome45,46, and sudden unexpected death in epilepsy47.
Since neuronal HCN channels are open at sub-threshold potentials, and make the cell membrane less responsive to incoming inputs, they are excellent targets for fine-tuning of intrinsic neuronal excitability. Inhibition of cardiac Ih by bradycardic agents such as ivabradine have been found useful in reducing the incidence of cardiovascular mortality and hospitalisation for some subclasses of heart failure48. Moreover, since HCN channel expression is largely limited to the heart and nervous system, and HCNs are not found in vascular tissue, targeted inhibition of HCN channels has strong therapeutic potential for several cardiac and neuronal disorders, without inducing any adverse effects on pulmonary and vascular smooth muscle tone. While several molecules that target HCN channels have been identified, including ZD728849–53, zatebradine54,55, cilobradine54,55, and ivabradine56–58, their low affinity and lack of isoform specificity prevents wide-spread use of these current HCN inhibitors. Additionally, HCN channels have been shown to interact with molecules such as clonidine59 and its derivative alinidine60, bupivacaine, lidocaine, and mepivacaine61. However, these molecules are not HCN specific and interact with numerous other channels and receptors as well.
In light of the recent high-resolution structures of human HCN162 and the related eukaryotic CNG channel63 we set out to characterize the interactions of 9 known HCN inhibitors within the pore using a computational docking approach. Our data provide insights into the drug binding modes and potentially explain the molecular bases for their low affinities.
Results
Docking to the closed HCN1 pore
500 docking attempts were made for each ligand to the closed human HCN1 pore extracted from the cryo-EM structure (PDB: 5U6O)62. For all ligands, 0 of the 500 docking conformations resided in the central cavity (Fig. 1). Similarly, ligands were not observed to bind within the pore cavity in any of the 500 docking attempts to the cAMP-bound HCN1 pore (PDB: 5U6P) (Supp. Fig. 3). We estimate the diameter of the central cavity measured at Y386 of opposite subunits to be 7.7 Å, enabling little more than 2–5 waters to occupy this space. Given that ligands such as ivabradine and ZD7288 are much larger and have been suggested to be trapped in the closed state56–58,64,65, our data suggests that the ligand-trapped closed state must differ from the apo-closed state that was observed in the cryo-EM structures. We therefore generated an open state model of human HCN1, based on the structure of the closely related eukaryotic CNG channel TAX-463.
Figure 1.

Docking of HCN blockers to the apo-closed HCN1 pore. The results from 500 attempts to dock clonidine (A) alinidine (B) lidocaine (C) ZD7288 (D) and ivabradine (E) to the closed pore of the cryo-EM structure (PDB: 5U6O). Despite these ligands being known to block or be “trapped” in the closed state, none of the 500 docked poses for any of these inhibitors were observed in the pore cavity. These data indicate that the apo-closed pore conformation must differ from the ligand-trapped closed pore.
Clonidine and alinidine binding to the open HCN1 pore
Clonidine, a well-known α2-adrenergic receptor agonist, also inhibits HCN2 and HCN4 with IC50’s of ~3–10 µM, shifting their voltage dependence to more hyperpolarized potentials by 10–20 mV, and is 4–10 fold less potent in HCN159. Docking of clonidine to the open HCN1 pore lead to the identification of two putative clusters within the pore cavity that cannot be distinguished based on binding energy alone. The autodock algorithm estimates a binding energy per non-hydrogen atom of −0.357 +/− 0.01 kcal/mol. Both of these clusters are well defined by position and orientation, with RMSDs of 0.02 Å and 0.17 Å from the reference pose within each clusters. The first cluster (Fig. 2A blue) contained 95% of the docked poses, and fit like a “lock and key” into a groove in the internal cavity formed by residues C358, Y386, and A387. In this conformation, a persistent hydrogen bond could be observed between the imidazole hydrogen of clonidine, and the hydroxyl oxygen of Y386. Another stabilizing hydrogen bond is observed between the N1 nitrogen and the hydroxyl hydrogen of the same Y386. In the second cluster, containing only 5% of the poses (Fig. 2B red), the clonidine is stabilized a single hydrogen bond between the imidazole hydrogen and the backbone carbonyl oxygen of A383 and reciprocating anion- π interactions between clonidine and the Y386 aromatic rings.
Figure 2.
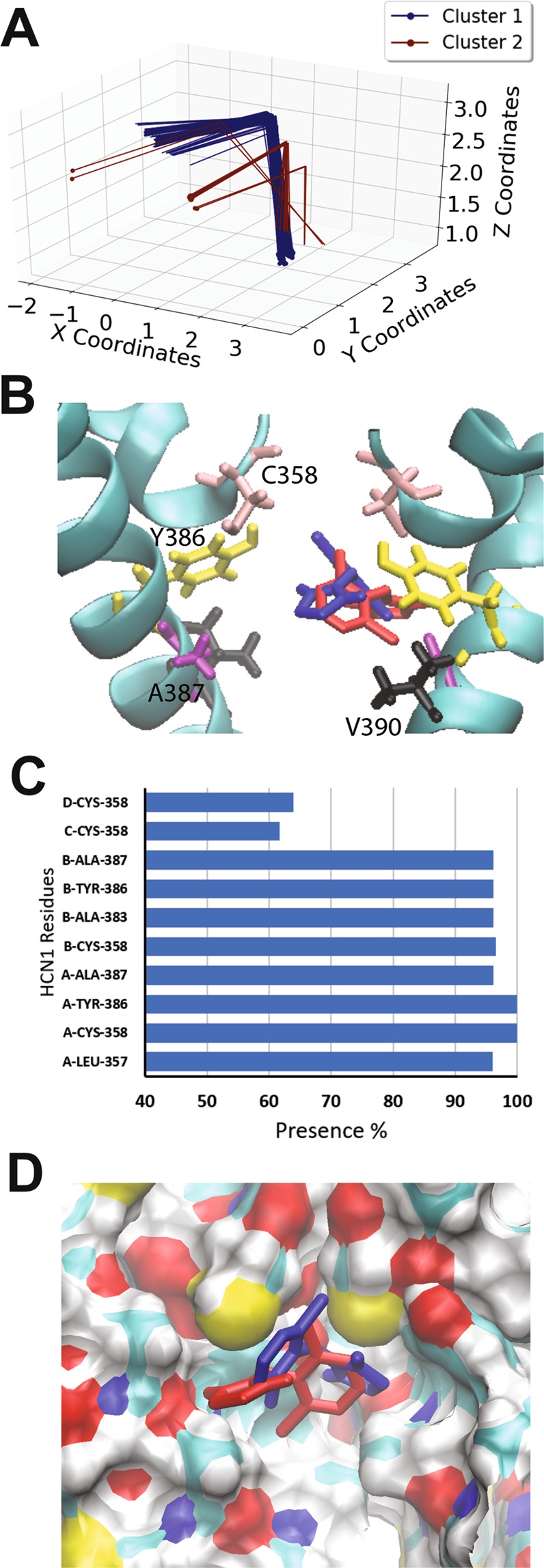
Docking of clonidine to the open HCN1 pore. (A) Clustering analysis of the 500 docking attempts. Docks can be placed into 2 clusters according to our automated algorithm. (B) Representative poses from cluster 1 (95% of poses; blue) and cluster 2 (5% of poses; red) are shown. Residues C358 (pink), Y386 (yellow), A387 (purple) and V390 (black) are highlighted. (C) The frequency of residues within 4 Å of the ligand for each pose was assessed and is indicated as a percentage. (D) A comparison of clonidine poses in cluster 1 (blue) and cluster 2 (red). The ligand fits into a hydrophobic groove lined by residues C358, Y386, and A387.
Alinidine (N-allyl-clonidine) is a clonidine derivative with differing pharmacological effects, including its bradycardic66–68 and analgesic properties69. Alinidine induces the slowing of spontaneous activity of rabbit SAN accompanied by a 10% prolongation of the action potential duration60 and by reducing the steepness of diastolic depolarization of Purkinje fibres with limited side effects on action potential duration and inotropic state70. Alinidine shifts the voltage-dependence of Ih activation to more negative voltages and reduced maximal conductance by 27% with no use or frequency dependence, indicating that alinidine binds equally well to HCN open and closed channel states. Alindine docking to the human HCN1 open pore resulted in 3 clusters (Fig. 3A), however, the third cluster (red) was populated by only 4 poses and bound with higher energy. Common to each of the two highest occupied and lowest energy clusters, the α-butylene group of alinidine extends into the S4 ion binding site of the selectivity filter, and would be expected to displace an ion in this site (Fig. 3C). Similar to clonidine, in these two clusters, alinidine fit like a “lock and key” into a groove in the internal cavity formed by residues C358, Y386, and A387. The binding energy per non-hydrogen atom was estimated to be −0.320 +/− 0.001 kcal/mol, −0.332 +/− 0.001 kcal/mol for clusters 1 and 2 respectively, which are indistinguishable according to Autodock. 319 docked poses (64%) reside in cluster 1, where a persistent hydrogen bond between the imidazole hydrogen of clonidine, and the hydroxyl oxygen of Y386 is observed. In addition, in this cluster, alinidine is stabilized by an anion-π or parallel-displaced π-stacking interaction between its aromatic group and the Y386 aromatic ring of another subunit. Poses in cluster 2 (177 of 500 docks) bind in the similar region with a different orientation, such that hydrogen bonding between the imidazole ring and the Y386 hydroxyl groups can still occur, but only parallel-displaced π-stacking between the alinidine and neighbouring Y386 aromatic rings can occur, and not an anion-π interaction.
Figure 3.
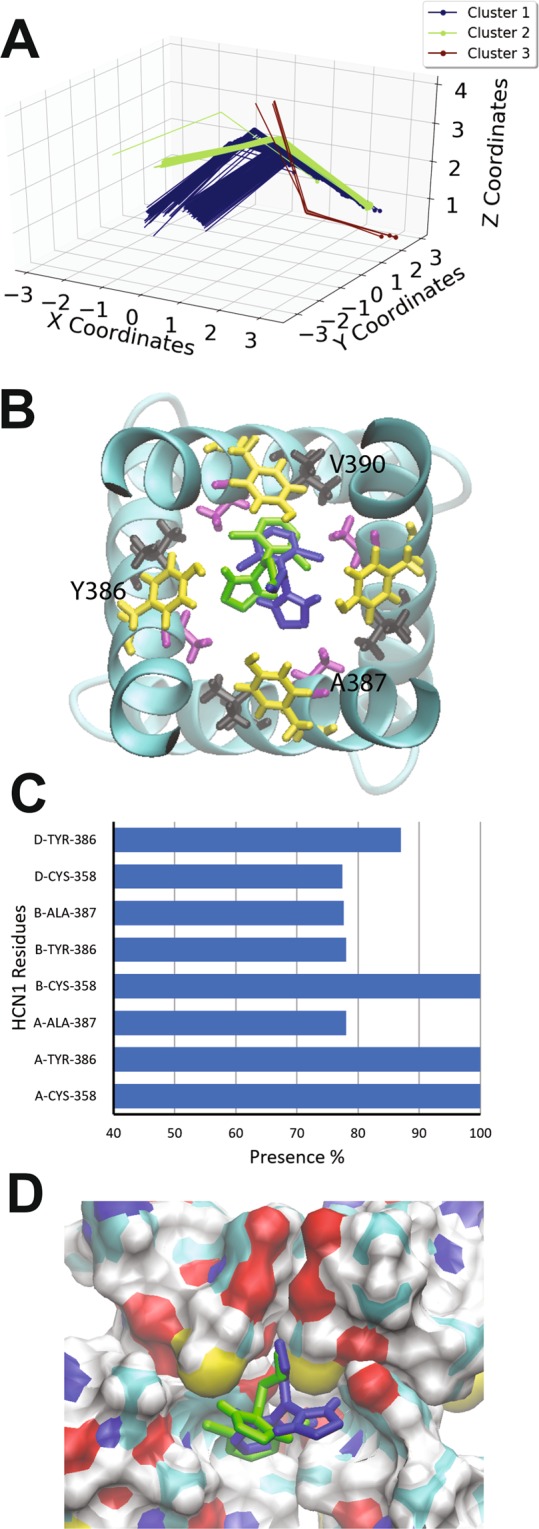
Docking of alinidine to the open HCN1 pore. (A) Clustering analysis of the 500 docking attempts. Docks can be placed into 3 clusters according to our automated algorithm. Cluster 1 (blue), cluster 2 (green) and cluster 3 (red) contained 319, 177 and 4 poses respectively. (B) Top view of the HCN1 pore with representative poses from cluster 1 (blue) and cluster 2 (green) are shown with residues C358 (pink), Y386 (yellow), A387 (purple) and V390 (black) highlighted (C) The frequency of residues within 4 Å of the ligand for each pose was assessed and is indicated as a percentage. (D) A comparison of clonidine poses in cluster 1 (blue) and cluster 2 (green). The ligand fits into a hydrophobic groove lined by residues C358, Y386, and A387.
The overall examination of clonidine and alinidine binding to the HCN1 pore indicate that a hydrophobic groove generated by residues C358, Y386, and A387 can assist in conformationally restraining the ligand. Moreover, Y386 of neighbouring subunits co-ordinate these ligands through a combination of hydrogen bonding and π-bonding interactions.
Binding of lidocaine to the open HCN1 pore
Lidocaine, mepivacaine and bupivacaine are local anesthetics known to inhibit voltage-gated sodium (Nav) channels71. Recently, these molecules have been shown to also inhibit HCN currents in DRG neurons with similar potencies61, suggesting clinical relevance. Since HCN blockade by lidocaine appears to occur from the inside of the cell72, we performed docking experiments of lidocaine to our model of the open human HCN1 pore. 5 clusters of docking poses were identified, with the binding sites lined by residues L357, C358, A383, Y386, A387, and V390 (Fig. 4). Similar to what we observed for clonindine and alinidine, the most occupied cluster and with low energy (cluster 2; occupied by 306 poses), is stabilized by π-stacking interactions of the aromatic rings of the ligand and Y386. This enables the ligand to sit deeper in the hydrophobic groove than clonidine and alinidine (Fig. 4D) and close enough to interact with V390. A hydrogen bond network is also formed between the amide hydrogen of lidocaine and the hydroxyl oxygen of the same Y386 that π-stacks with the ligand, as well as the sulfhydryl sulfur atom of C358. Cluster 1 (occupied by 136 poses) is rotated such that the aromatic ring can no longer π-stack, and the amide bond is rotated such that the hydrogen bond network is now formed by the oxygen of the amide bond in lidocaine, and the hydrogen atoms of the Y386 and C358 sidechains. In general, the aromatic of ring in most lidocaine poses is constrained in the hydrophobic groove, independent of whether or not π-stacking can occur, while the amine portion points toward the central pore axis. Notably, the binding energies per non-hydrogen atom are comparable to clonidine and alinidine, with a mean of −0.307 kcal/mol and a maximum of −0.333 kcal/mol. The highest energy poses occupied a cluster (cluster 4; orange) with only 4 out of 500 poses with the ligand displaced against one of the S6 pore helices and may represent a local minimum, and not the physiologically relevant binding pose.
Figure 4.

Docking of lidocaine and derivatives to the open HCN1 pore. (A) Clustering analysis of the 500 docking attempts. Docks can be placed into 5 clusters according to our automated algorithm. (B) Top view of the HCN1 pore with representative poses from cluster 1 (blue) cluster 2 (cyan) and cluster 3 (green) are shown (C) The frequency of residues within 4 Å of the ligand for each pose was assessed and is indicated as a percentage. (D) Side view of representative poses from cluster 1 (blue) cluster 2 (cyan) and cluster 3 (green) are shown. The ligand aromatic ring of lidocaine fits into a hydrophobic groove lined by residues C358, Y386, and A387, while the amine portion of the ligand points toward the central pore axis. (E) Overlay of lidocaine (black) and mepivacaine (blue) docked in the pore of HCN1 channels. Mepivacaine docks with both rings in hydrophobic groove formed by neighbouring subunits, while lidocaine’s amine points toward the pore axis. (F) Mepivacaine (red) and bupivacaine (blue) dock similarly, however, the butyl group on the aromatic ring extends toward the central axis of the pore cavity likely enhancing the occlusion of ions.
Bupivacaine and mepivacaine are lidocaine derivatives with ringed R-groups replacing the ethylamine. Bupivacaine and mepivacaine had comparable binding energies per non-hydrogen atom to lidocaine (−0.320 kcal/mol and −0.332 kcal/mol respectively). However, we found mepivacaine to dock with greater complementarity into the pore grove than lidocaine. Poses can be arranged into 2 clusters (Supp. Fig. 3) with the ligands simply rotated by approximately 180° around the center of the molecule. Notably, while the aromatic ring of lidocaine resides in the hydrophobic groove, its amine does not arrange itself into the portion of the hydrophobic groove formed by the neighboring subunit, but rather points toward the central pore axis (Fig. 4E). On the other hand, both ring structures of mepivacaine can arrange themselves into both portions of the hydrophobic groove formed by neighbouring subunits. Interestingly, bupivacaine largely appears to dock into the same positions as mepivacaine, with the butyl group extended toward the central axis of the pore cavity (Fig. 4F), similar to the amine group of lidocaine. Considering that lidocaine, bupivacaine and mepivacine have increasing IC50’s73, our results suggest that while the nitrogen containing ring is entropically favourable to help orient the “caines” into the hydrophobic groove, there may be an important role in occluding permeant ions by having a greater portion of the ligand occupying the central axis of the pore cavity.
ZD7288 binding to the open HCN1 pore
ZD7288 is an open-state blocker of HCN channels51,65,74,75 with bradycardic and antianginal activity in animal models49,76. ZD7288 induces at ~15 mV hyperpolarizing shift in voltage-dependent Ih activation and reduces maximal activity by more than 50%49. This drug also reduces the generation of hippocampal epileptic discharges in rabbits77 and reverse pain behavior and spontaneous discharges in injured rat nerve fibers78–80. The 500 docking attempts for ZD7288 against the human HCN1 open pore resulted in 24 clusters (Fig. 5A), with 271 poses (54%) in one cluster, interacting with residues L357, C358, A383, Y386, A387, V390 and G381 of 3 subunits within the tetramer (Fig. 5B,C). Poses in this cluster are largely stabilized by hydrogen bonds between the ligand’s nitrogen atoms and the hydroxyl groups of Y386. The remaining clusters reside on the same plane, and interact with the same residues, however, are not rotationally constrained, thereby reducing the number of hydrogen bonds formed. However, the binding energies per non-hydrogen atom between the best occupied cluster (cluster 1; −0.305 +/− 0.003 kcal/mol) and the other clusters (all ranging between −0.29 and −0.31) are indistinguishable. This suggests that binding of ZD7288 in HCN channels does not likely occur with a preferred orientation, and that the ligand is relatively free to rotate within the pore cavity. Moreover, unlike the binding of clonidine, alinidine and lidocaine, π-stacking interactions between the aromatic portions of the ligand, and the aromatic sidechain of Y386 do not appear to contribute to the mechanism of binding of ZD7288.
Figure 5.
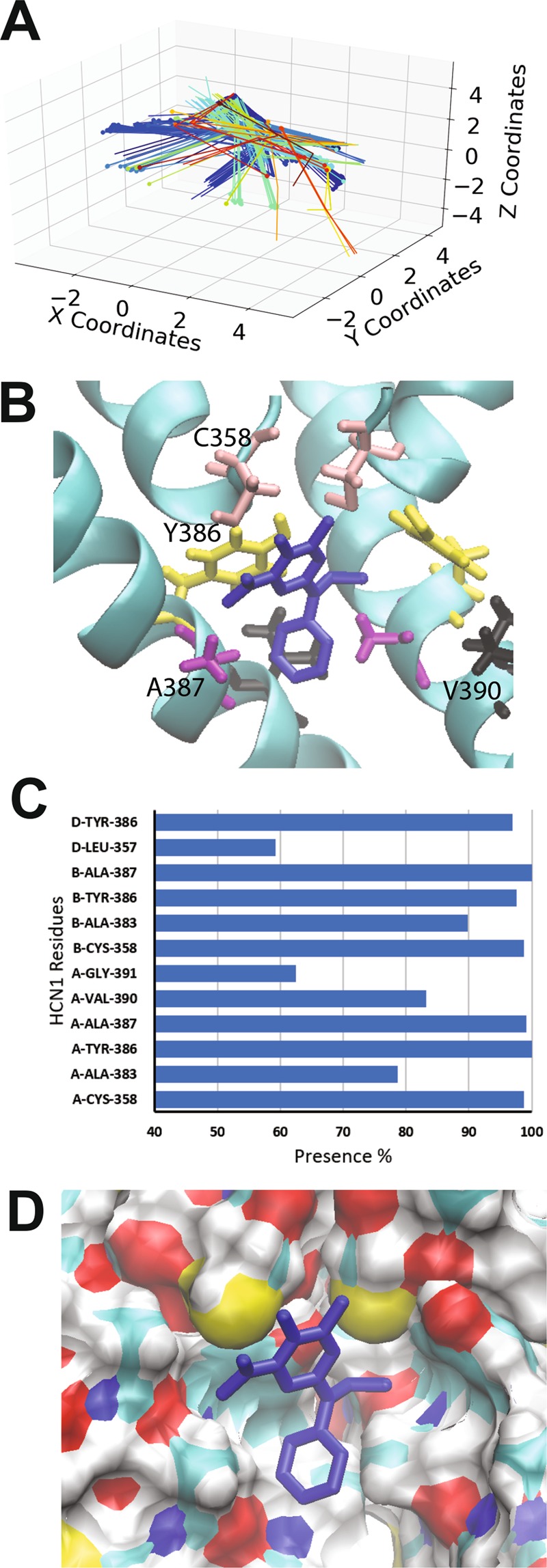
Docking of ZD7288 to the open HCN1 pore. (A) Clustering analysis of the 500 docking attempts. Docks can be placed into 24 clusters according to our automated algorithm, indicating that ZD7288 does not bind in a preferred orientation. (B) A representative pose of ZD7288 with residues C358 (pink), Y386 (yellow), A387 (purple) and V390 (black) highlighted. (C) The frequency of residues within 4 Å of the ligand for each pose was assessed and is indicated as a percentage. (D) ZD7288 does not fit nicely into the hydrophobic groove and the aromatic sidechain of Y386 does not appear to contribute to the mechanism of binding as it does for other ligands.
Ivabradine binding to the open HCN1 pore
Ivabradine was the first clinically approved molecule that specifically targeted HCN channels for the treatment of heart failure81–83. Ivabradine block of HCN4 can occur only from the intracellular side58 when the channels are opened by hyperpolarization with enhanced binding upon frequent changes in the direction of ion flow56–58. In contrast, HCN1 channels can also be inhibited from the closed state58. Intriguingly, the 500 docking attempts of ivabradine to the human HCN1 open pore could not be clustered using our algorithm. The ligand binds in the pore cavity in a U-shaped configuration (Fig. 6B), without positional or orientational restriction (Fig. 6A). Consequently, ivabradine can interact with residues C358, A383, Y386, A387, V390, G391, and T394 of all 4 subunits. The binding energy per non-hydrogen atom is −0.185 +/− 0.018 kcal/mol with a maximum binding energy for the lowest energy pose of −0.258 kcal/mol. Binding of ivabradine to the human HCN1 open pore appears largely driven by van der Waals and hydrophobic interactions, with no consistent hydrogen bonding, anion-π or π-stacking interactions observed. Notably, no part of the ivabradine molecule fits into the hydrophobic groove generated by residues C358, Y386, and A387. Consistent with this observation, it was previously shown that the equivalent mutation to A387V in HCN4 channels (A507V) did not affect ivabradine block of HCN channels64. Interactions with these residues are weaker and less specific than for ligands such as clonidine, alinidine and lidocaine. Similar results were observed for zatebradine and cilobradine (Supp. Figs 5 and 6). Our findings are consistent with the model that ivabradine requires an open channel to access the binding site, and that outward current favours blocker binding. Our data also indicates that the “ligand trapped” closed conformation is not the same as the apo-closed conformation observed in the cryo-EM structure, since the apo-closed pore cavity lacks the volume necessary to accommodate any of the inhibitors we examined.
Figure 6.
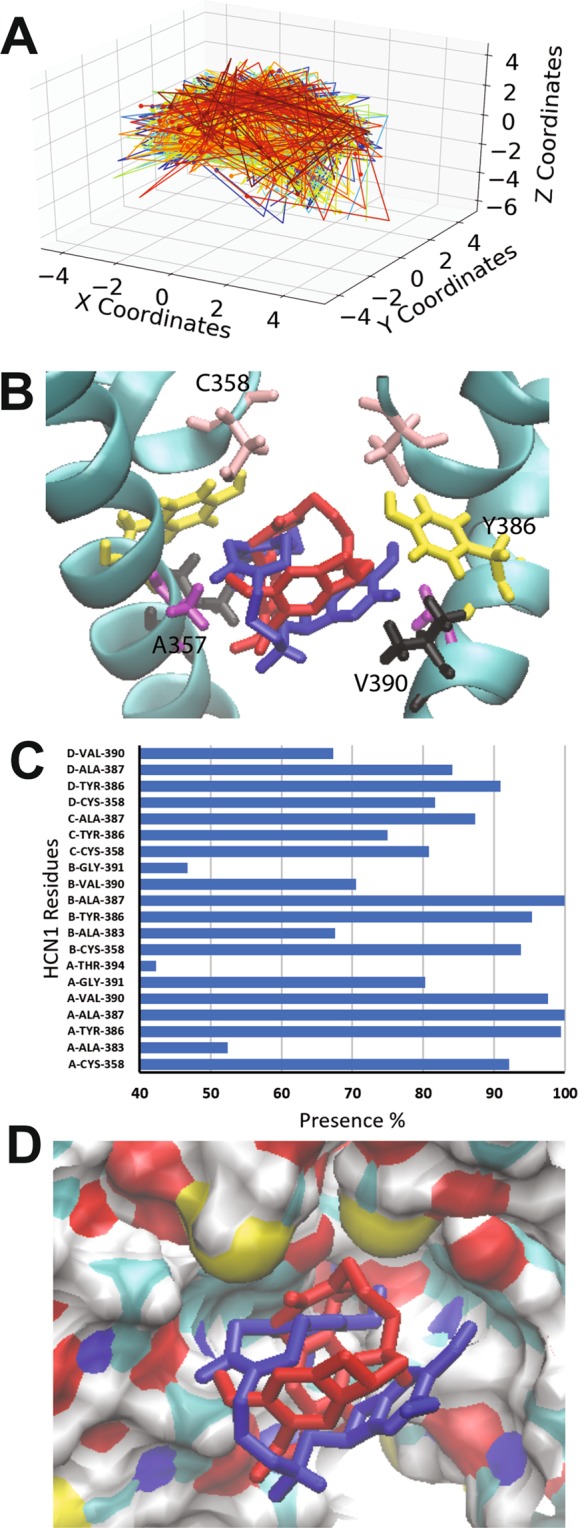
Docking of ivabradine to the open HCN1 pore. (A) Clustering analysis of the 500 docking attempts. Docks cannot be clustered according to our automated algorithm, indicating that ivabradine does not bind in a preferred orientation. (B) Two poses of ivabradine are shown with residues C358 (pink), Y386 (yellow), A387 (purple) and V390 (black) highlighted. Ivabradine in all poses folds into a U-shape, however, is free to rotate 360° in the pore cavity. (C) The frequency of residues within 4 Å of the ligand for each pose was assessed and is indicated as a percentage. (D) None of the ivabradine fit nicely into the hydrophobic groove which is largely prevented by the presence of the methoyx groups on the aromatic ring.
Discussion
Targeted inhibition of HCN channels has strong therapeutic potential as anti-convulsants, analgesics, anti-depressants, anti-psychotics, and bradycardic agents without adverse effects on pulmonary and vascular smooth muscle tone. However, the lack of channel or isoform specificity and low affinity for most currently available inhibitors hinders their use. Thus, to aid in the development of higher affinity molecules, a detailed understanding of drug-channel interactions are required. We therefore sought to investigate molecular details of block of 9 known inhibitors of HCN channels (with differing scaffolds) and identify residues involved in their binding.
Drug binding in the central cavity
The first intriguing result obtained from our docking experiments is that for each of the ligands examined, 0 of 500 docks to the closed human HCN1 pore contained ligands bound in the cavity. Our estimates using the CHARMM-GUI webserver suggest that the cavity can accommodate only 2–5 waters in this space with a diameter measured at Y386 of opposite subunits of 7.7 Å. Given that ligands such as ivabradine and ZD7288 are much larger and have been suggested to bind in the open state and be trapped in the closed state57,65, our data suggests that the ligand-trapped closed state differs from the apo-closed state that was observed in the cryo-EM structure.
Docking of these ligands to the open pore model of human HCN1 was more successful, with 100% of docking poses for each drug residing in the pore cavity. The residues that appear to be most critical for ligand binding of these pore blockers are C358, A383, Y386, A387, V390. Residues C358, Y386, and A387 appear to generate a hydrophobic groove that helps to conformationally restrict the ligands such as clonidine and alinidine, whose rings can fit. Binding of these ligands appears to be driven by a hydrogen bonds, anion-π and π-stacking interactions within this hydrophobic groove. On the other hand, “bradine” inhibitors appear to bind to the HCN1 open pore in a geometrically unconstrained manner via primarily van der Waals and hydrophobic interactions, and are sterically hindered from similar coordinated binding to the hydrophobic groove used for clonidine and alinidine binding. This lack of strong interactions and high rotational freedom within the pore cavity may provide a molecular basis for the low (2–10 µM) affinity of “bradines” to HCN channels. This may present a region that may be exploited for the development of higher affinity pore-blocking ligands. This idea is supported by the examination of binding of ZD7288 and “caine” inhibitors (such as lidocaine). The “caine” inhibitors can form parallel displaced pi interactions with Y386 and fit sterically into the pore groove, and thus dock in a relatively small number of clusters (≤5; Fig. 4). ZD7288 can form H-bond interactions, however, does not have a portion that can fit within this pore groove and form π-stacking interactions, and thus is relatively unconstrained in its binding (Fig. 5). Unfortunately, this lack of conformational restriction in the pore for ligands such as ZD7288 and ivabradine may present serious challenges for solving ligand bound structures using x-ray crystallography or cryo-EM approaches.
Comparison with Previous Studies
Previous studies have attempted to examine the binding of ivabradine and ZD7288 to HCN channels using computational docking approaches51,64,84,85. Unfortunately, these studies required the use of homology model generated using prokaryotic KcsA51,64,85 and MthK84,85 channel as templates for the closed and open conformations, as an atomic resolution structure of HCNs or more closely related channels were not yet available at the time. Our study extracted the closed pore-domain from the the high-resolution structure of human HCN1 generated by cryo-EM experiments (PDB: 5U6O) to dock against. Structural alignment of the closed model derived from KcsA (Fig. 1A; red) and the closed pore extracted from the cryo-EM structure (Fig. 1A; cyan) indicate that there is a large difference between the structures. Firstly, the pore cavity in our models are lined by S6 residues C358, A383, Y386, A387, V390, G391, T394, A395, and Q398 in the closed and open states. This is largely in agreement with previous reports50,51,64, however differs for 2 key residues. L357 and F389 do not line the pore cavity. In the cryo-EM structure, the sidechain of F389 is faces S5 residues I298, I302, and M304 of the same chain and T384 of the neighbouring S6. This seems more reasonable than previously suggested, since the energetic cost of hydrating a phenylalanine group within the pore would be greater than if the residue was buried amongst non-polar residues. Similarly, in the cryo-EM structure, L357 packs as part of the interface between the pore-helix of one subunit and the N-terminal region of the neighbouring S6. These differences may play an important role in drug binding models, since many of the HCN inhibitors (such as bradines and ZD7288) largely interact with the channel through hydrophobic interactions and van der Waals forces, rather than through electrostatic interactions and hydrogen bonds. Most notably, the cryo-EM structure differs from previous models in the position of residue Y386 relative to the pore helix (Supp. Fig. 3). The distance between C358 and Y386 is 9.2 Å in the KcsA derived model, versus only 4.8 Å in the high-resolution structure. It is this difference that enables ivabradine to bind above residue Y386 in previous studies64, while it is sterically restricted from this space in our study. This difference also has a carry-over effect on the generation of an open model. Our open pore model was derived using the open pore observed in the cryo-EM structure of the closely related CNG channel TAX-463. In the KcsA-based model of HCN4, I510 (equivalent to residue 390 in human HCN1) does not interact with ivabradine directly, but was proposed to stabilize the conformation of Y506 (equivalent to Y386 in HCN1) which does interact with the ligand64. However, our model, derived from the atomic resolution structures of HCN1 and TAX-4 CNG channels, indicates that ivabradine and ZD7288 bind below Y386 and can interact directly with V390 of HCN1 channels (Figs 5 and 6).
Conclusions
Overall, our study provide novel insights into the mode of binding for 9 known inhibitors of HCN channels, and indicate that the inhibitor bound-closed pore state of HCN1 must differ from the apo-closed pore state observed in the high resolution structure. We also identify a hydrophobic groove within the pore cavity lined by residues C358, Y386, and A387 that appears to conformationally restrict ligands and has the potential to be exploited for the development of higher affinity molecules. Our results also help explain the molecular basis of the low-affinity binding of these inhibitors.
Methods
Model Building and Docking Calculations
The closed pore domain (S5-P-loop-S6) of human HCN1 (residues 296–402) was extracted from the PDB (5U6O or 5U6P)62 and processed through the CHARMM-GUI PDB Reader86 which enabled the corrections of any missing atoms and/or side-chains. In the absence of the crystal structure of an human HCN1 channel with an open pore domain, we turned to homology modelling using the template of the eukaryotic CNG channel, TAX-4 (PDB 5H3O)63. Sequences of the S5-pore-S6 residues were aligned using ClustalΩ87 (Supp. Fig. 1) and used to generate a homology model using ICM-Pro88 (Molsoft LLC, LaJolla). The model was processed through CHARMM-GUI PDB Reader86 to assist with repairing any missing atoms, repair any improper bond lengths or angles, and provide the appropriate PDB format for used for docking.
Preparation of the ligands and HCN1 channel pore domains in the open or closed states for docking experiments was performed using AutodockTools4 (ADT4)89. Grid parameter files and grid maps were generated by AutoGrid 4.2 within ADT with a grid spacing of 0.375 Å and positioned to exclude extracellular binding of the ligands. Each ligand was independently docked 500 times using Lamarckian GA docking algorithm90 with the maximum number of energy evaluations set to 2 500 000. Docking was performed on single processors of the supercomputer cluster Briaree (Compute Canada/CalculQuébec). Prior knowledge that these ligands are pore blockers enabled us to remove any docked poses outside of the pore cavity.
Clustering Analysis
To account for the four-fold symmetry of the HCN pore, docked ligand poses were rotated into the same quadrant. Following this, ligands were clustered by (i) position in the pore and (ii) the orientation of functional groups. This was achieved by simplifying each ligand to a vector generated from the centroid of atoms selected in different regions of the molecule, as demonstrated for ivabradine (Supp. Fig. 1A). Table 1 lists the atoms used to determine each of the centroids determined for each ligand.
Table 1.
Simplified ligand vectors used for clustering were generated by calculating 3–4 centroids within each ligand. The atoms used to calculate each centroid is listed.
| Ligands | Centroid 1 | Centroid 2 | Centroid 3 | Centroid 4 |
|---|---|---|---|---|
| Clonidine | C7, C8, & C9 | N1 | C1 & C4 | |
| Alinidine | C4 & C7 | N1 | C10, C11, & C12 | |
| Lidocaine | C8 & C11 | C6 | N2 | |
| Mepivacaine | N2 | C13 & C10 | N1 | C6 & C3 |
| Bupivacaine | N2 | C5 & C8 | N1 | C12 & C15 |
| ZD7288 | C3 & C6 | C9 & N2 | N4 | |
| Ivabradine | C20 & C21 | C2 | C7 & C12 | |
| Zatebradine | C19 & C22 | C2 | C7 & C12 | |
| Cilobradine | C3 & C6 | C11 & C14 | C19 & C24 |
To cluster, using our simplified molecules, we generated a variant of Silhouette clustering91 to which we added a scoring function in order to find a suitable number of clusters without user bias. A “reference dock” is selected at random to generate the first cluster. The distance of every other dock is then compared to this reference position via the sum of linear distance between each of the centroids from our selections in Table 1 according to:
where D is the sum of the distances between each centroid, n the number of centroids we selected as best representative of the whole molecule, and a and b represent the docks being are compared.
If D lies within a defined threshold distance it is merged to the cluster, otherwise a new cluster is created. 4 Å (equivalent to a weak hydrogen bond) was selected as the starting threshold.
Since the number of clusters calculated is dependent on the threshold set, we added a scoring function (S) to each cluster to choose the optimal number.
with Inside Distance defined as:
With c being the cluster’s global centroid and x the centroid for each individual dock inside the cluster.
The Outside Distance is defined as:
with y being the centroid for every dock from the nearest cluster.
Ideally, S should be <1 for all clusters calculated, which indicates every cluster is justified and the number of clusters is well determined. If the S determined for any cluster >1, the threshold is automatically lowered by 0.1 Å for each iteration until every S falls below 1. If S remains >1 for every iteration until 0, the algorithm selects for the threshold yielding the lowest maximum S and the number of clusters is determined accordingly.
Supplementary information
Acknowledgements
This work was supported by a grant from Natural Sciences and Engineering Research Council to ND (DG-435649-2013). ND was a junior research fellow of the Fonds de recherche du Québec – Santé.
Author Contributions
N.D. designed the study and contributed to data analysis. J.T. performed the docking experiments. K.M.C. and J.T. performed data analysis. N.D. and J.T. wrote the manuscript.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-37116-2.
References
- 1.Ulens C, Tytgat J. Functional heteromerization of HCN1 and HCN2 pacemaker channels. J Biol Chem. 2001;276:6069–6072. doi: 10.1074/jbc.C000738200. [DOI] [PubMed] [Google Scholar]
- 2.Mannikko R, Elinder F, Larsson HP. Voltage-sensing mechanism is conserved among ion channels gated by opposite voltages. Nature. 2002;419:837–841. doi: 10.1038/nature01038. [DOI] [PubMed] [Google Scholar]
- 3.Vemana S, Pandey S, Larsson HP. S4 movement in a mammalian HCN channel. J Gen Physiol. 2004;123:21–32. doi: 10.1085/jgp.200308916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiological reviews. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- 5.Ishii TM, Takano M, Xie LH, Noma A, Ohmori H. Molecular characterization of the hyperpolarization-activated cation channel in rabbit heart sinoatrial node. J Biol Chem. 1999;274:12835–12839. doi: 10.1074/jbc.274.18.12835. [DOI] [PubMed] [Google Scholar]
- 6.Ludwig A, et al. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO J. 1999;18:2323–2329. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mistrik P, et al. The murine HCN3 gene encodes a hyperpolarization-activated cation channel with slow kinetics and unique response to cyclic nucleotides. J Biol Chem. 2005;280:27056–27061. doi: 10.1074/jbc.M502696200. [DOI] [PubMed] [Google Scholar]
- 8.Seifert R, et al. Molecular characterization of a slowly gating human hyperpolarization-activated channel predominantly expressed in thalamus, heart, and testis. Proc Natl Acad Sci USA. 1999;96:9391–9396. doi: 10.1073/pnas.96.16.9391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nolan MF, et al. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 10.Nolan MF, et al. The hyperpolarization-activated HCN1 channel is important for motor learning and neuronal integration by cerebellar Purkinje cells. Cell. 2003;115:551–564. doi: 10.1016/S0092-8674(03)00884-5. [DOI] [PubMed] [Google Scholar]
- 11.Huang Z, Walker MC, Shah MM. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009;29:10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludwig A, et al. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science. 2011;333:1462–1466. doi: 10.1126/science.1206243. [DOI] [PubMed] [Google Scholar]
- 14.Dibbens LM, et al. Augmented currents of an HCN2 variant in patients with febrile seizure syndromes. Ann Neurol. 2010;67:542–546. doi: 10.1002/ana.21909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiFrancesco JC, et al. Recessive loss-of-function mutation in the pacemaker HCN2 channel causing increased neuronal excitability in a patient with idiopathic generalized epilepsy. J Neurosci. 2011;31:17327–17337. doi: 10.1523/JNEUROSCI.3727-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura Y, et al. Novel HCN2 mutation contributes to febrile seizures by shifting the channel’s kinetics in a temperature-dependent manner. PLoS One. 2013;8:e80376. doi: 10.1371/journal.pone.0080376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nava C, et al. De novo mutations in HCN1 cause early infantile epileptic encephalopathy. Nat Genet. 2014;46:640–645. doi: 10.1038/ng.2952. [DOI] [PubMed] [Google Scholar]
- 18.Tang B, Sander T, Craven KB, Hempelmann A, Escayg A. Mutation analysis of the hyperpolarization-activated cyclic nucleotide-gated channels HCN1 and HCN2 in idiopathic generalized epilepsy. Neurobiol Dis. 2008;29:59–70. doi: 10.1016/j.nbd.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnsten AF. Prefrontal cortical network connections: key site of vulnerability in stress and schizophrenia. Int J Dev Neurosci. 2011;29:215–223. doi: 10.1016/j.ijdevneu.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamo NJ, et al. Stress Impairs Prefrontal Cortical Function via D1 Dopamine Receptor Interactions With Hyperpolarization-Activated Cyclic Nucleotide-Gated Channels. Biol Psychiatry. 2015;78:860–870. doi: 10.1016/j.biopsych.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 2013;23:1643–1654. doi: 10.1093/cercor/bhs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yi F, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352:aaf2669. doi: 10.1126/science.aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brioschi C, et al. Distribution of the pacemaker HCN4 channel mRNA and protein in the rabbit sinoatrial node. J Mol Cell Cardiol. 2009;47:221–227. doi: 10.1016/j.yjmcc.2009.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Moosmang S, et al. Cellular expression and functional characterization of four hyperpolarization-activated pacemaker channels in cardiac and neuronal tissues. Eur J Biochem. 2001;268:1646–1652. doi: 10.1046/j.1432-1327.2001.02036.x. [DOI] [PubMed] [Google Scholar]
- 25.Shi W, et al. Distribution and prevalence of hyperpolarization-activated cation channel (HCN) mRNA expression in cardiac tissues. Circ Res. 1999;85:e1–6. doi: 10.1161/01.RES.85.1.1. [DOI] [PubMed] [Google Scholar]
- 26.Tellez JO, et al. Differential expression of ion channel transcripts in atrial muscle and sinoatrial node in rabbit. Circulation research. 2006;99:1384–1393. doi: 10.1161/01.RES.0000251717.98379.69. [DOI] [PubMed] [Google Scholar]
- 27.Harzheim D, et al. Cardiac pacemaker function of HCN4 channels in mice is confined to embryonic development and requires cyclic AMP. EMBO J. 2008;27:692–703. doi: 10.1038/emboj.2008.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herrmann S, Stieber J, Stockl G, Hofmann F, Ludwig A. HCN4 provides a ‘depolarization reserve’ and is not required for heart rate acceleration in mice. The EMBO journal. 2007;26:4423–4432. doi: 10.1038/sj.emboj.7601868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duhme N, et al. Altered HCN4 channel C-linker interaction is associated with familial tachycardia-bradycardia syndrome and atrial fibrillation. Eur Heart J. 2013;34:2768–2775. doi: 10.1093/eurheartj/ehs391. [DOI] [PubMed] [Google Scholar]
- 30.Laish-Farkash A, et al. A novel mutation in the HCN4 gene causes symptomatic sinus bradycardia in Moroccan Jews. J Cardiovasc Electrophysiol. 2010;21:1365–1372. doi: 10.1111/j.1540-8167.2010.01844.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macri V, et al. A novel trafficking-defective HCN4 mutation is associated with early-onset atrial fibrillation. Heart Rhythm. 2014;11:1055–1062. doi: 10.1016/j.hrthm.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milanesi R, Baruscotti M, Gnecchi-Ruscone T, DiFrancesco D. Familial sinus bradycardia associated with a mutation in the cardiac pacemaker channel. The New England journal of medicine. 2006;354:151–157. doi: 10.1056/NEJMoa052475. [DOI] [PubMed] [Google Scholar]
- 33.Milano A, et al. HCN4 mutations in multiple families with bradycardia and left ventricular noncompaction cardiomyopathy. J Am Coll Cardiol. 2014;64:745–756. doi: 10.1016/j.jacc.2014.05.045. [DOI] [PubMed] [Google Scholar]
- 34.Nof E, et al. Point mutation in the HCN4 cardiac ion channel pore affecting synthesis, trafficking, and functional expression is associated with familial asymptomatic sinus bradycardia. Circulation. 2007;116:463–470. doi: 10.1161/CIRCULATIONAHA.107.706887. [DOI] [PubMed] [Google Scholar]
- 35.Schulze-Bahr E, et al. Pacemaker channel dysfunction in a patient with sinus node disease. The Journal of clinical investigation. 2003;111:1537–1545. doi: 10.1172/JCI16387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schweizer PA, et al. cAMP sensitivity of HCN pacemaker channels determines basal heart rate but is not critical for autonomic rate control. Circ Arrhythm Electrophysiol. 2010;3:542–552. doi: 10.1161/CIRCEP.110.949768. [DOI] [PubMed] [Google Scholar]
- 37.Schweizer PA, et al. The symptom complex of familial sinus node dysfunction and myocardial noncompaction is associated with mutations in the HCN4 channel. J Am Coll Cardiol. 2014;64:757–767. doi: 10.1016/j.jacc.2014.06.1155. [DOI] [PubMed] [Google Scholar]
- 38.Ueda K, et al. Functional characterization of a trafficking-defective HCN4 mutation, D553N, associated with cardiac arrhythmia. J Biol Chem. 2004;279:27194–27198. doi: 10.1074/jbc.M311953200. [DOI] [PubMed] [Google Scholar]
- 39.Zhou J, et al. A novel HCN4 mutation, G1097W, is associated with atrioventricular block. Circ J. 2014;78:938–942. doi: 10.1253/circj.CJ-13-0996. [DOI] [PubMed] [Google Scholar]
- 40.Kuwabara Y, et al. Increased expression of HCN channels in the ventricular myocardium contributes to enhanced arrhythmicity in mouse failing hearts. J Am Heart Assoc. 2013;2:e000150. doi: 10.1161/JAHA.113.000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oshita K, et al. Ectopic automaticity induced in ventricular myocytes by transgenic overexpression of HCN2. J Mol Cell Cardiol. 2015;80C:81–89. doi: 10.1016/j.yjmcc.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 42.Ueda K, et al. Role of HCN4 channel in preventing ventricular arrhythmia. J Hum Genet. 2009;54:115–121. doi: 10.1038/jhg.2008.16. [DOI] [PubMed] [Google Scholar]
- 43.Baruscotti M, et al. Deep bradycardia and heart block caused by inducible cardiac-specific knockout of the pacemaker channel gene Hcn4. Proc Natl Acad Sci USA. 2011;108:1705–1710. doi: 10.1073/pnas.1010122108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Crotti L, et al. Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J Am Coll Cardiol. 2012;60:1410–1418. doi: 10.1016/j.jacc.2012.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans A, Bagnall RD, Duflou J, Semsarian C. Postmortem review and genetic analysis in sudden infant death syndrome: an 11-year review. Hum Pathol. 2013;44:1730–1736. doi: 10.1016/j.humpath.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 46.Tester DJ, Ackerman MJ. Sudden infant death syndrome: how significant are the cardiac channelopathies? Cardiovasc Res. 2005;67:388–396. doi: 10.1016/j.cardiores.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 47.Tu E, Waterhouse L, Duflou J, Bagnall RD, Semsarian C. Genetic analysis of hyperpolarization-activated cyclic nucleotide-gated cation channels in sudden unexpected death in epilepsy cases. Brain Pathol. 2011;21:692–698. doi: 10.1111/j.1750-3639.2011.00500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Swedberg K, et al. Ivabradine and outcomes in chronic heart failure (SHIFT): a randomised placebo-controlled study. Lancet. 2010;376:875–885. doi: 10.1016/S0140-6736(10)61198-1. [DOI] [PubMed] [Google Scholar]
- 49.BoSmith RE, Briggs I, Sturgess NC. Inhibitory actions of ZENECA ZD7288 on whole-cell hyperpolarization activated inward current (If) in guinea-pig dissociated sinoatrial node cells. Br J Pharmacol. 1993;110:343–349. doi: 10.1111/j.1476-5381.1993.tb13815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan YC, et al. Probing the bradycardic drug binding receptor of HCN-encoded pacemaker channels. Pflugers Arch. 2009;459:25–38. doi: 10.1007/s00424-009-0719-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cheng L, Kinard K, Rajamani R, Sanguinetti MC. Molecular mapping of the binding site for a blocker of hyperpolarization-activated, cyclic nucleotide-modulated pacemaker channels. J Pharmacol Exp Ther. 2007;322:931–939. doi: 10.1124/jpet.107.121467. [DOI] [PubMed] [Google Scholar]
- 52.Gasparini S, DiFrancesco D. Action of the hyperpolarization-activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflugers Arch. 1997;435:99–106. doi: 10.1007/s004240050488. [DOI] [PubMed] [Google Scholar]
- 53.Harris NC, Constanti A. Mechanism of block by ZD 7288 of the hyperpolarization-activated inward rectifying current in guinea pig substantia nigra neurons in vitro. J Neurophysiol. 1995;74:2366–2378. doi: 10.1152/jn.1995.74.6.2366. [DOI] [PubMed] [Google Scholar]
- 54.DiFrancesco D. Some properties of the UL-FS 49 block of the hyperpolarization-activated current (i(f)) in sino-atrial node myocytes. Pflugers Arch. 1994;427:64–70. doi: 10.1007/BF00585943. [DOI] [PubMed] [Google Scholar]
- 55.Goethals M, Raes A, van Bogaert PP. Use-dependent block of the pacemaker current I(f) in rabbit sinoatrial node cells by zatebradine (UL-FS 49). On the mode of action of sinus node inhibitors. Circulation. 1993;88:2389–2401. doi: 10.1161/01.CIR.88.5.2389. [DOI] [PubMed] [Google Scholar]
- 56.Bois P, Bescond J, Renaudon B, Lenfant J. Mode of action of bradycardic agent, S 16257, on ionic currents of rabbit sinoatrial node cells. Br J Pharmacol. 1996;118:1051–1057. doi: 10.1111/j.1476-5381.1996.tb15505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bucchi A, Baruscotti M, DiFrancesco D. Current-dependent block of rabbit sino-atrial node I(f) channels by ivabradine. J Gen Physiol. 2002;120:1–13. doi: 10.1085/jgp.20028593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bucchi A, Tognati A, Milanesi R, Baruscotti M, DiFrancesco D. Properties of ivabradine-induced block of HCN1 and HCN4 pacemaker channels. J Physiol. 2006;572:335–346. doi: 10.1113/jphysiol.2005.100776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knaus A, et al. Direct inhibition of cardiac hyperpolarization-activated cyclic nucleotide-gated pacemaker channels by clonidine. Circulation. 2007;115:872–880. doi: 10.1161/CIRCULATIONAHA.106.667675. [DOI] [PubMed] [Google Scholar]
- 60.Satoh H, Hashimoto K. Electrophysiological study of alinidine in voltage clamped rabbit sino-atrial node cells. Eur J Pharmacol. 1986;121:211–219. doi: 10.1016/0014-2999(86)90492-9. [DOI] [PubMed] [Google Scholar]
- 61.Bischoff U, Brau ME, Vogel W, Hempelmann G, Olschewski A. Local anaesthetics block hyperpolarization-activated inward current in rat small dorsal root ganglion neurones. Br J Pharmacol. 2003;139:1273–1280. doi: 10.1038/sj.bjp.0705363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee CH, MacKinnon R. Structures of the Human HCN1 Hyperpolarization-Activated Channel. Cell. 2017;168:111–120 e111. doi: 10.1016/j.cell.2016.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li M, et al. Structure of a eukaryotic cyclic-nucleotide-gated channel. Nature. 2017;542:60–65. doi: 10.1038/nature20819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bucchi A, et al. Identification of the molecular site of ivabradine binding to HCN4 channels. PLoS One. 2013;8:e53132. doi: 10.1371/journal.pone.0053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin KS, Rothberg BS, Yellen G. Blocker state dependence and trapping in hyperpolarization-activated cation channels: evidence for an intracellular activation gate. J Gen Physiol. 2001;117:91–101. doi: 10.1085/jgp.117.2.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kobinger W, Lillie C, Pichler L. Cardiovascular actions of N-allyl-clonidine (ST 567), a substance with specific bradycardic action. Eur J Pharmacol. 1979;58:141–150. doi: 10.1016/0014-2999(79)90005-0. [DOI] [PubMed] [Google Scholar]
- 67.Lillie C, Kobinger W. Comparison of the bradycardic effects of alinidine (St 567), AQ-A 39 and verapamil on guinea-pig sinoatrial node superfused with different Ca2+ and NaCl solutions. Eur J Pharmacol. 1983;87:25–33. doi: 10.1016/0014-2999(83)90046-8. [DOI] [PubMed] [Google Scholar]
- 68.Traunecker W, Walland A. Haemodynamic and electrophysiologic actions of alinidine in the dog. Arch Int Pharmacodyn Ther. 1980;244:58–72. [PubMed] [Google Scholar]
- 69.Stockhaus, K. In Problems of Drug Dependence 355-366 (Cambridge: National Academy of Sciences 1977).
- 70.Snyders DJ, Van Bogaert PP. Alinidine modifies the pacemaker current in sheep Purkinje fibers. Pflugers Arch. 1987;410:83–91. doi: 10.1007/BF00581900. [DOI] [PubMed] [Google Scholar]
- 71.Brau ME, Vogel W, Hempelmann G. Fundamental properties of local anesthetics: half-maximal blocking concentrations for tonic block of Na+ and K+ channels in peripheral nerve. Anesth Analg. 1998;87:885–889. doi: 10.1097/00000539-199810000-00026. [DOI] [PubMed] [Google Scholar]
- 72.Putrenko I, Yip R, Schwarz SKW, Accili EA. Cation and voltage dependence of lidocaine inhibition of the hyperpolarization-activated cyclic nucleotide-gated HCN1 channel. Sci Rep. 2017;7:1281. doi: 10.1038/s41598-017-01253-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romanelli MN, et al. HCN Channels Modulators: The Need for Selectivity. Curr Top Med Chem. 2016;16:1764–1791. doi: 10.2174/1568026616999160315130832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benetos A, Rudnichi A, Thomas F, Safar M, Guize L. Influence of heart rate on mortality in a French population: role of age, gender, and blood pressure. Hypertension. 1999;33:44–52. doi: 10.1161/01.HYP.33.1.44. [DOI] [PubMed] [Google Scholar]
- 75.Wu S, et al. Inner activation gate in S6 contributes to the state-dependent binding of cAMP in full-length HCN2 channel. J Gen Physiol. 2012;140:29–39. doi: 10.1085/jgp.201110749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Briggs I, BoSmith RE, Heapy CG. Effects of Zeneca ZD7288 in comparison with alinidine and UL-FS 49 on guinea pig sinoatrial node and ventricular action potentials. J Cardiovasc Pharmacol. 1994;24:380–387. doi: 10.1097/00005344-199409000-00005. [DOI] [PubMed] [Google Scholar]
- 77.Kitayama M, et al. Ih blockers have a potential of antiepileptic effects. Epilepsia. 2003;44:20–24. doi: 10.1046/j.1528-1157.2003.22702.x. [DOI] [PubMed] [Google Scholar]
- 78.Chaplan SR, et al. Neuronal hyperpolarization-activated pacemaker channels drive neuropathic pain. J Neurosci. 2003;23:1169–1178. doi: 10.1523/JNEUROSCI.23-04-01169.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee DH, Chang L, Sorkin LS, Chaplan SR. Hyperpolarization-activated, cation-nonselective, cyclic nucleotide-modulated channel blockade alleviates mechanical allodynia and suppresses ectopic discharge in spinal nerve ligated rats. J Pain. 2005;6:417–424. doi: 10.1016/j.jpain.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 80.Sun Q, Xing GG, Tu HY, Han JS, Wan Y. Inhibition of hyperpolarization-activated current by ZD7288 suppresses ectopic discharges of injured dorsal root ganglion neurons in a rat model of neuropathic pain. Brain Res. 2005;1032:63–69. doi: 10.1016/j.brainres.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 81.Bohm M, et al. Heart rate as a risk factor in chronic heart failure (SHIFT): the association between heart rate and outcomes in a randomised placebo-controlled trial. Lancet. 2010;376:886–894. doi: 10.1016/S0140-6736(10)61259-7. [DOI] [PubMed] [Google Scholar]
- 82.Fox K, et al. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:807–816. doi: 10.1016/S0140-6736(08)61170-8. [DOI] [PubMed] [Google Scholar]
- 83.Fox K, et al. Heart rate as a prognostic risk factor in patients with coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): a subgroup analysis of a randomised controlled trial. Lancet. 2008;372:817–821. doi: 10.1016/S0140-6736(08)61171-X. [DOI] [PubMed] [Google Scholar]
- 84.Giorgetti A, Carloni P, Mistrik P, Torre V. A homology model of the pore region of HCN channels. Biophys J. 2005;89:932–944. doi: 10.1529/biophysj.104.045286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Griguoli M, et al. Nicotine blocks the hyperpolarization-activated current Ih and severely impairs the oscillatory behavior of oriens-lacunosum moleculare interneurons. J Neurosci. 2010;30:10773–10783. doi: 10.1523/JNEUROSCI.2446-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jo S, Kim T, Iyer VG, Im W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–1865. doi: 10.1002/jcc.20945. [DOI] [PubMed] [Google Scholar]
- 87.Sievers F, Higgins DG. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018;27:135–145. doi: 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins. 1995;23:403–414. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- 89.Morris, G. M. et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem19, 1639–1662, 10.1002/(Sici)1096-987x(19981115)19:14<1639::Aid-Jcc10>3.0.Co;2-B (1998).
- 90.Morris GM, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rousseeuw PJ. Silhouettes - a Graphical Aid to the Interpretation and Validation of Cluster-Analysis. J Comput Appl Math. 1987;20:53–65. doi: 10.1016/0377-0427(87)90125-7. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.