

Abstract
Cysteine is one of the two key sulfur‐containing amino acids with important functions in redox homeostasis, protein functionality and metabolism. Cysteine is taken up by mammals via their diet and can also be derived from methionine via the transsulfuration pathway. The cellular concentration of cysteine is kept within a narrow range by controlling its synthesis and degradation. There are two pathways for the catabolism of cysteine leading to sulfate, taurine and thiosulfate as terminal products. The oxidative pathway produces taurine and sulfate, while the H2S pathway involves different enzymatic reactions leading to the formation and clearance of H2S, an important signalling molecule in mammals, resulting in thiosulfate and sulfate. Sulfite is a common intermediate in both catabolic pathways. Sulfite is considered as cytotoxic and produces neurotoxic S‐sulfonates. As a result, a deficiency in the terminal steps of cysteine or H2S catabolism leads to severe forms of encephalopathy with the accumulation of sulfite and H2S in the body. This review links the homeostatic regulation of both cysteine catabolic pathways to sulfite and H2S.
Linked Articles
This article is part of a themed section on Chemical Biology of Reactive Sulfur Species. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.4/issuetoc
Abbreviations
- ADO
cysteamine dioxygenase
- AdoMet
S‐adenosylmethionine
- CBS
cystathionine β‐synthase
- CDO
cysteine dioxygenase
- CSA
cysteine sulfinic acid
- CSAD
CSA decarboxylase
- CSE
cystathionine γ‐lyase
- GOT
glutamate oxaloacetate transaminase
- GSH
γ‐glutamylcysteinylglycine
- GSSG
glutathione disulfide
- Moco
molybdenum cofactor
- MPST
3‐mercaptopyruvate sulfurtransferase
- PDO
persulfide dioxygenase
- PLP
pyridoxal phosphate‐dependent
- SO
sulfite oxidase
- SQR
sulfide : quinone oxidoreductase
- SSC
S‐sulfocysteine
- TST
thiosulfate sulfurtransferase
Introduction
Cysteine is one of two sulfur‐containing amino acids with key functions in cellular redox regulation, metabolism, protein synthesis and functionality. Cysteine plays pivotal roles in protein structure due to its ability to form disulfide bridges in an oxidative environment and its functions as active site residues in various families of enzymes. As a result of their high reactivity, cysteine thiols can react with reactive oxygen species forming sulfenic, sulfinic and sulfonic acids, with the latter two being considered as irreversible oxidation products (Poole and Nelson, 2008; Klomsiri et al., 2011). Cysteine is further used as sulfur source for various types of cofactors, such as coenzyme A, biotin or Fe‐S clusters.
Cysteine is able to undergo a large number of different post‐translational modifications (Miseta and Csutora, 2000). For example, S‐palmitoylation is the only reversible form of lipid modification that is found in many neuronal proteins and is catalysed by a large family of palmitoylating enzymes. Other modifications of cysteine residues are related to the action of gasotransmitters NO and H2S forming covalent adducts by S‐nitrosylation and S‐persulfidation (also termed S‐sulfhydration) respectively. Common to all of these modifications is the reactivity and accessibility of a given cysteine residue. In some cases (glyceraldehyde‐3‐phosphate dehydrogenase or parkin), the same residue can undergo nitrosylation and persulfidation, mediating opposing effects on protein function suggesting important roles of those post‐translational modifications as molecular switches (Mustafa et al., 2009; Vandiver et al., 2013).
Cysteine is a semi‐essential amino acid, as it can either be obtained from the diet or enzymatically produced from methionine via the transsulfuration pathway (Figure 1) (Zou and Banerjee, 2005; Banerjee, 2017). Methionine functions as an important methyl group donor in the cell following its conversion to S‐adenosylmethionine. After methyl group transfer, the product S‐adenosylhomocysteine is hydrolysed to adenosine and homocysteine. While a part of the homocysteine generated is regenerated to methionine, a fraction enters the transsulfuration pathway forming cystathionine following the reaction of homocysteine with serine catalysed by cystathionine β‐synthase (CBS). The partitioning of homocysteine to the remethylation cycle to form methionine or the transsulfuration pathway has been described as highly dynamic (Finkelstein, 2000). Cystathionine is subsequently cleaved into cysteine and α‐ketobutyrate by cystathionine γ‐lyase (CSE). Interestingly, both enzymes involved in the transsulfuration pathway are also able to accept cysteine as substrate producing H2S, an important signalling molecule (Figure 1) (Paul and Snyder, 2012; Kabil and Banerjee, 2014).
Figure 1.
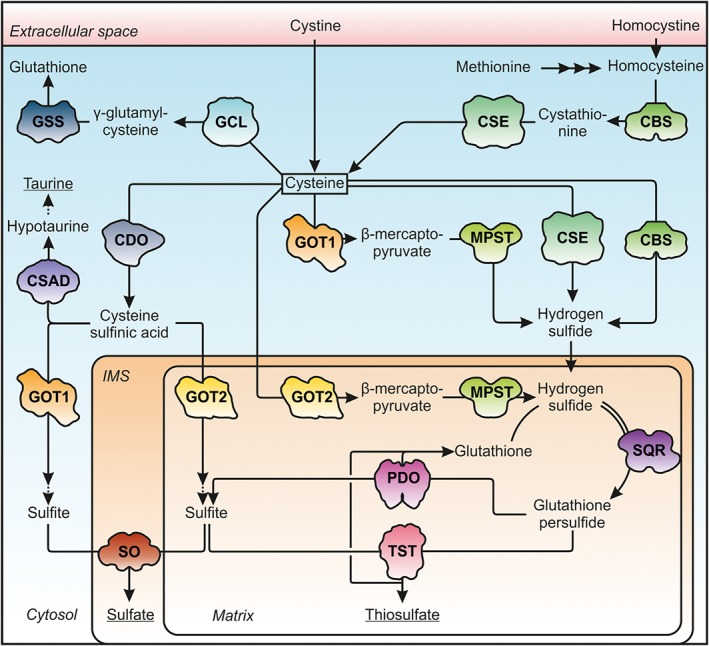
Cysteine anabolism and catabolism pathways. Cysteine is either imported or generated from methionine in the transsulfuration pathway. Cysteine is the rate‐limiting substrate for GSH synthesis. Cysteine degradation proceeds via formation of CSA or H2S to sulfate and taurine or sulfate and thiosulfate respectively. Terminal products are underlined. The shapes of the enzymes involved are depicted according to the crystal structures available. CBS, 1M54; CDO, 2IC1; CSAD, 2JIS; CSE, 2NMP; PDO, 4CHL; GCL, glutamate cysteine ligase, 3LVW; GSS, GSH synthetase, 1M0T; GOT1/2, 3II0/5AX8; IMS, intermembrane space; MPST, 4JGT; SQR, 3H8L; SO, 1SOX; TST, 1RHD.
The vast majority of ‘free’ cysteine in cells is bound in GSH (γ‐glutamylcysteinylglycine), the major antioxidant and most abundant non‐protein thiol in mammals. GSH is synthesized from glutamate, cysteine and glycine, by a two‐step reaction of glutamyl‐cysteine synthetase and GSH synthetase in an ATP‐dependent manner (Figure 1). With 0.5–10 mM cytosolic GSH concentrations (Lu, 2013), GSH is considered as the major source and buffer of cysteine while the cellular concentrations of cysteine are kept within a narrow window in the micromolar range (Lee et al., 2004; Vitvitsky et al., 2004). The availability of cysteine is considered as one of the rate‐limiting steps in GSH synthesis, thereby impacting cellular redox status as well as diverse cellular functions (reviewed in Deponte, 2013; Lu, 2013).
In the extracellular compartment, cysteine is oxidized to cystine, thus representing the major transport form of non‐protein‐bound cysteine (Ueland et al., 1996). Across membranes, cysteine and cystine are transported by different membrane carriers. In the CNS, glial cells mainly import cystine via the cystine‐glutamate antiporter (system xc −) providing the major route for GSH synthesis in the brain (Sato et al., 2005). In its reduced form, cysteine is transported by excitatory amino acid transporters, which are known for their function in glutamate and aspartate clearance. Deficiencies in these transporters have been found to be associated with reduced cellular levels of GSH, oxidative stress and neurodegeneration (Sato et al., 2005; Aoyama et al., 2006).
Alterations in cysteine homeostasis have been associated with various primarily neurodegenerative disorders (see below). Several severe, fast progressing, rare genetic disorders are related to the terminal steps in cysteine degradation and characterized by major alterations in GSH, cysteine/cystine and H2S levels. More complex disorders, such as Huntington's and Alzheimer's disease, have also been linked to altered cysteine homeostasis (McBean et al., 2015). Finally, positive effects of dietary restriction on longevity have been traced back to cysteine‐dependent alterations in the cellular homeostasis of H2S (Hine et al., 2015; Hine and Mitchell, 2015), thus placing the homeostatic control of cysteine metabolism at the heart of cellular signalling and redox regulation. In this review, we discuss the different pathways of cysteine catabolism and provide evidence that sulfite and H2S, as key intermediates, have a major regulatory impact on cysteine homeostasis in health and disease.
Oxidative cysteine catabolism
Cysteine, which is not used for protein or GSH synthesis is further metabolized via two distinct pathways, the oxidative cysteine catabolism and the H2S pathway (Figure 1) (Kabil et al., 2011; Stipanuk and Ueki, 2011). Within the oxidative catabolic pathway, cysteine is first converted to cysteine sulfinic acid (CSA) via oxidation of its thiol group by cysteine dioxygenase (CDO) (Figure 2). CSA is then further processed via two different routes, yielding either taurine or sulfate as end products. Decarboxylation of CSA by CSA decarboxylase (CSAD) releases hypotaurine, which is further oxidized by a non‐enzymatic mechanism to taurine. As the most abundant amino acid derivative in human cells, taurine participates in a broad range of biological processes including bile acid conjugation, antioxidant and anti‐inflammatory actions as well as modulation of the CNS (Ripps and Shen, 2012). Beside taurine synthesis, CSA is deaminated to β‐sulfinylpyruvate, which spontaneously decomposes to pyruvate and sulfite. It has been shown that glutamate oxaloacetate transaminase [GOT; also named aspartate aminotransferase (AST/AAT)] is able to catalyse this reaction in vitro (Singer and Kearney, 1956). Since sulfite is highly toxic, as it inhibits glutamate dehydrogenase (GDH) and malate dehydrogenase (Zhang et al., 2004), fast elimination by oxidation to harmless sulfate is of great importance. This reaction is catalysed by the molybdenum cofactor‐containing mitochondrial enzyme sulfite oxidase (SO) (Schwarz et al., 2009). The individual enzymes involved in oxidative cysteine catabolism will be discussed in the following chapters.
Figure 2.
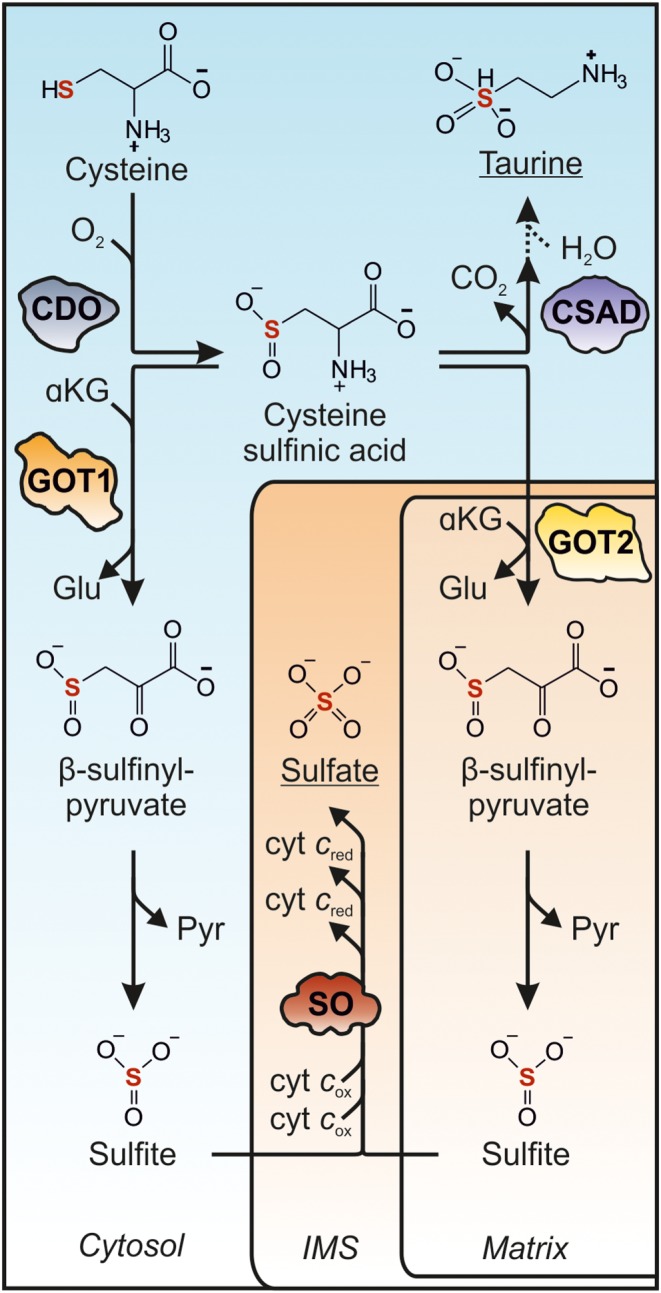
Oxidative cysteine catabolism to taurine and sulfate. Cysteine is converted by CDO (2IC1) to CSA, which is subsequently used for taurine synthesis catalysis by CSAD (2JIS) yielding hypotaurine, which is further converted to taurine. Alternatively, GOT1/2 (3II0/5AX8) catalyse the deamination of CSA to β‐sulfinyl pyruvate, which spontaneously decomposes to sulfite and sulfate. Note that, as yet, it is not known which of the two GOT isoforms functions primarily in oxidative cysteine catabolism. Terminal oxidation of sulfite is catalysed by SO (1SOX) localized to the mitochondrial intermembrane space (IMS). The shapes of the enzymes involved are depicted according to the crystal structures available.
Cysteine dioxygenase
In humans, the amount of dietary cysteine and methionine intake can vary greatly, while intracellular cysteine levels are tightly controlled and maintained within a range of 10–25 μM suggesting a homeostatic control of cysteine‐producing and cysteine‐consuming processes (Lee et al., 2004). It is generally considered that under conditions of cysteine excess, the enzyme CDO plays a key function in the maintenance of cysteine levels (Stipanuk and Ueki, 2011) (Figure 2). Upon increased cysteine availability, CDO concentrations in hepatic and adipose tissue were found to be increased by up to 45‐fold (Dominy et al., 2006). In contrast, in the absence of cysteine, CDO levels are kept low by ubiquitination and subsequent proteasomal degradation of the enzyme. In return, ubiquitination is inhibited in the presence of cysteine, allowing for a rapid adjustment of CDO levels and revealing the physiological importance of the underlying regulatory mechanisms (Dominy et al., 2006).
CDO‐deficient mice are characterized by high postnatal mortality, growth deficiency and connective tissue disease (Ueki et al., 2011). Biochemically, CDO −/− mice display elevated cysteine and severely lowered taurine levels. The remaining production of taurine (~7%) is derived from cysteamine (Ueki et al., 2011), a catabolic cleavage product of coenzyme A. Interestingly, while taurine supplementation prevents the high mortality rate, it does not impact other characteristic phenotypes. Slightly elevated sulfate and H2S levels suggest an increased flux of cysteine through the H2S pathway (see below), which may be connected to the pathology of CDO −/− mice (Ueki et al., 2011; Roman et al., 2013). Interestingly, no deregulation of enzymes involved in the H2S pathway could be detected (Jurkowska et al., 2014).
Catabolism of cysteine sulfinic acid
The distribution of CSA between taurine‐ and sulfate‐generating pathways likely depends on the expression of CSA‐processing enzymes as well as their affinity towards CSA (Figure 2). Interestingly, CSAD and CDO show both high levels in liver and adipose tissue, with only moderate to low levels in other tissues (Stipanuk and Ueki, 2011). The reported K m values of 0.04–0.17 mM are in line with a high specificity of CSAD towards CSA (Guion‐Rain et al., 1975; Oertel et al., 1981). CSAD‐deficient mice resemble CDO −/− mice in terms of taurine levels and mortality rate (Park et al., 2014). Oral administration of taurine is sufficient to restore survival and plasma taurine to wildtype levels.
Deamination of CSA was found to be catalysed by GOT, of which two isoforms are known, one localized in the cytosol (GOT1), the other in the mitochondrial matrix (GOT2) (Recasens et al., 1980) (Figure 2). In contrast to CSAD, both GOT1 and GOT2 display a much broader expression pattern with high levels in the liver, heart, kidney, skeletal muscle and red blood cells (Cechetto et al., 2002). GOT1/2 are important components of the malate–aspartate shuttle, which functions in the transfer of reducing equivalents to mitochondria (Bremer and Davis, 1975). In general, GOT1/2 catalyse the reversible transfer of an amino group from an amino acid donor to an α‐keto acid. They accept three main amino acid donors – aspartate, CSA and cysteine (Nisselbaum and Bodansky, 1964) – while the amino group is usually transferred to α‐ketoglutarate, thereby yielding glutamate.
Based on kinetic data, aspartate is considered to be the main substrate of GOT1/2 (Ubuka et al., 1978; Akagi, 1982). Our own data show a K m of 1.8 mM (GOT1) and 0.4 mM (GOT2) for aspartate, while the K m for CSA was found between 10 and 20 mM for both enzyme isoforms (Schwarz et al., unpublished). In addition, GOT1/2 have also been reported to accept cysteine directly for transamination, thereby participating not only in oxidative cysteine catabolism but also within the H2S pathway (Figure 1) (Ubuka et al., 1978; Akagi, 1982). With K m values for cysteine of around 2 mM for both enzymes, CSA and cysteine represent substrates with similar affinity towards GOT1/2. In contrast to GOT1/2, CSAD exhibits a much lower K m for CSA (0.04–0.17 mM) suggesting a larger flux of CSA towards taurine biosynthesis than towards sulfate formation (Stipanuk and Ueki, 2011).
To this day, it is not known which of the two GOT isoforms represents the primary physiological site for CSA deamination. Kinetic data of CSA deamination by GOT1/2 do not allow any conclusion as to which of the two isoforms is physiologically relevant (Figure 2). Notably, the involvement of GOT2 would require the import of CSA into mitochondria via a yet unidentified transporter. The rate of this import would determine whether or not GOT2 contributes to any significant deamination of CSA. In the future, the individual contribution of each GOT isoform to cysteine catabolism should be clarified by using specific in vivo models. The reaction product of CSA deamination, β‐sulfinyl pyruvate, decomposes into pyruvate and sulfite (Singer and Kearney, 1956); the latter requires immediate oxidation by sulfite oxidase (SO) to avoid cellular damage.
Sulfite oxidase
SO catalyses the two‐electron oxidation of sulfite to sulfate coupled to the reduction of two molecules of cytochrome c (Figures 2 and 3) (Johnson and Rajagopalan, 1980). The structure and reaction mechanism of SO has been extensively reviewed elsewhere (Hille et al., 2014). Vertebrate SO forms homodimers; each monomer harbours an N‐terminal cytochrome b 5‐type haem domain, a catalytic molybdenum cofactor (Moco)‐containing domain and a C‐terminal dimerization domain (Kisker et al., 1997). SO is localized in the mitochondrial intermembrane space where electrons derived from sulfite oxidation are passed to the physiological electron acceptor cytochrome c (Cohen et al., 1972). Until now, the contribution of sulfite to mitochondrial respiration was not considered as a source of any significance. However, we have found that sulfite excess (500 μM) increased oxygen consumption in mitochondria in an SO‐dependent manner by up to 15% (Schwarz et al., unpublished).
Figure 3.
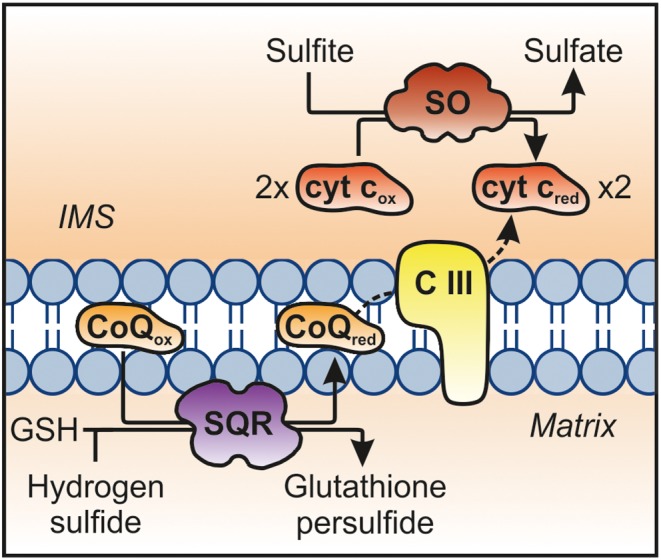
The contribution of SO and sulfide quionone oxidoreductase (SQR) to the mitochondrial electron transport chain. One electron is transferred from H2S to coenzyme Q via SQR (3H8L), which is further transferred to cytochrome c via complex III (CIII). Alternatively, two molecules of cytochrome c are reduced by SO (1SOX)‐catalysed oxidation of one molecule sulfite to sulfate.
SO is highly expressed in liver and kidney, whereas very low levels are detected in the brain (Belaidi et al., 2015). As a mitochondrial enzyme, SO requires a specific translocation and maturation pathway to gain enzymatic function. This process is highly orchestrated and involves targeting and mitochondrial processing of the SO apo‐protein, stepwise integration of Moco and haem cofactors and homodimerization (Klein and Schwarz, 2012).
The catalytic mechanism of SO involves the transfer of two electrons from sulfite to Moco followed by two single‐electron transfer steps via the haem domain to cytochrome c. Electron transfer within SO is dependent on dynamic movements of the haem domain to enable efficient electron transfer (Rapson et al., 2010). These conformational changes have been found to determine a novel, recently identified function of SO: the reduction of nitrite to nitric oxide (Wang et al., 2015). With decreasing intramolecular electron transfer, the rate of nitrite‐dependent nitric oxide synthesis increases.
We have recently generated a mouse model of SO deficiency (isolated SO deficiency – iSOD) (Kohl et al., unpublished data), which resembles in large parts mice that are impaired in the biosynthesis of Moco (Lee et al., 2002). As a result, all Moco‐dependent enzymes are dysfunctional and affected animals die within 10 days of postnatal life. The fact that SO‐deficient mice show a similar survival rate suggests that the lack of SO activity represents the major cause of disease in Moco‐deficient mice.
SO not only serves as terminal enzyme in the oxidative cysteine catabolism. Also via the H2S pathways, significant quantities of sulfite are formed that require the oxidation by SO – if sulfite production from H2S is blocked in rodents, quantities of excreted sulfate are sixfold decreased (Tiranti et al., 2009). Therefore, we will next discuss the generation and catabolism of H2S before linking the contribution of altered sulfite homeostasis to the overall fluxes of S‐containing metabolites.
Enzymatic production of hydrogen sulfide (H2S) from cysteine
While the oxidative cysteine catabolism primarily serves taurine biosynthesis and is stimulated under cysteine excess, the other important branch of cysteine catabolism – the generation of H2S and its subsequent breakdown – has gained increasing attention in recent years. H2S joins the group of physiologically important gasotransmitters and has been found to be involved in multiple cellular functions in cardiovascular systems, neuronal tissues and in the gastrointestinal tract (Abe and Kimura, 1996; Dongó et al., 2017). In the last two decades, H2S has been shown to function as a potent vasodilator with multiple medical applications arising (Caliendo et al., 2010; Beltowski, 2014), for example, ATB‐346, a novel H2S‐releasing non‐steroid anti‐inflammatory drug (Wallace et al., 2010, 2017). H2S is a mediator of tissue inflammation (Li et al., 2011), and early studies provided evidence for its involvement in numerous neurological processes, such as long‐term potentiation in the hippocampus – an important process in memory formation and learning – by enhancing NMDA receptor‐mediated responses (Abe and Kimura, 1996). H2S is closely connected to the post‐translational protein modification of cysteines leading to persulfidation, which has been implicated in mediating a protective function on redox‐active cysteine residues as well as modulating enzymatic activities (Filipovic et al., 2017). Whereas free H2S concentrations in tissues are considered to be rather low (<0.05 μmol·kg−1), concentrations of bound H2S in the blood have been found to be several orders of magnitude higher (Furne et al., 2008; Levitt et al., 2011). Five enzymes in three distinct reaction paths are considered to function in the generation of H2S (Figures 1 and 4). Four additional enzymes are involved in the oxidative degradation of H2S to sulfate and thiosulfate, the latter also involves sulfite derived from the oxidative branch of cysteine catabolism. The high number of enzymes and multitude of pathways involved highlight the physiological importance of these pathways. The contribution of the different enzymes in the formation and breakdown of H2S will be discussed in the following chapters.
Figure 4.
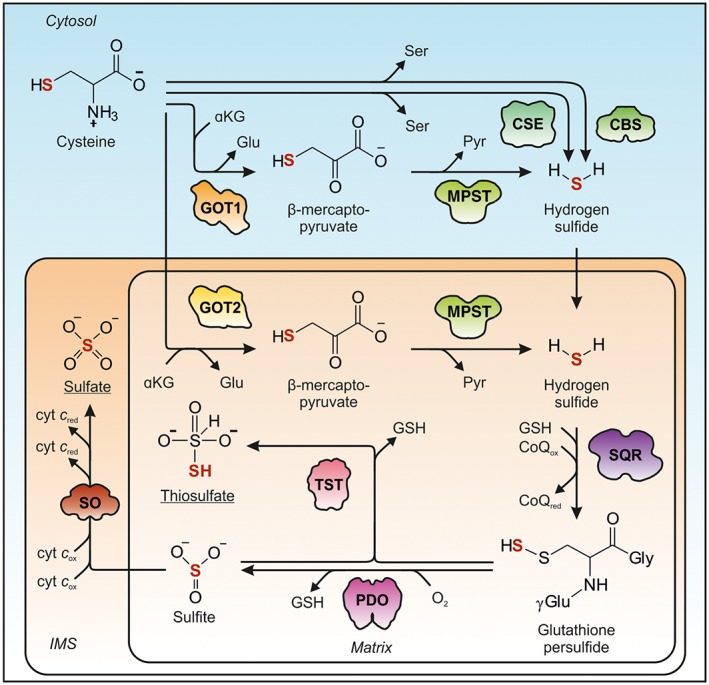
Sulfur flow from cysteine via H2S to sulfate and thiosulfate. Cysteine catabolism via H2S proceeds via three distinct pathways that involve either CBS (1M54), CSE (2NMP), or GOT1/2 (3II0/5AX8) and MPST (4JGT). Within the mitochondrial matrix, H2S is oxidized via SQR (3H8L) to GSH persulfide, which is further converted to either sulfite by PDO (4CHL) or thiosulfate by TST (1RHD). The shapes of the enzymes involved are depicted according to the crystal structures available.
Cystathionine β‐synthase
CBS is the first enzyme in the transsulfuration pathway catalysing the condensation of homocysteine and serine, yielding cystathionine (Figure 1). Initially described as a cysteine‐desulfurating enzyme (Porter et al., 1974), CBS is capable of catalysing multiple reactions towards the generation of H2S (Figure 4). CBS preferentially accepts cysteine as a substrate, which reacts with water releasing serine and H2S or – at a lower rate – with homocysteine and cysteine to form cystathionine and H2S (Chen et al., 2004). Alternatively, cystine may also serve as a substrate to form lanthionine and H2S. Among these alternative H2S‐producing reactions, only the hydrolytic desulfuration of cysteine proceeds with a rate that exhibits physiological relevance (Yadav et al., 2016). CBS is also capable of producing a persulfide by converting cystine to a cysteine persulfide, pyruvate and ammonia. The latter reaction, however, likely only takes place under oxidative stress when intracellular cystine concentrations are elevated (Yadav et al., 2016).
CBS is localized in the cytosol and expressed in several tissues, including neuronal, specifically in glial cells and astrocytes (Enokido et al., 2005). Although CBS is strongly expressed in, that is, hepatic tissues, its relative activity towards H2S production is considered to be tissue‐specific and relatively low in non‐neuronal tissues (Yang et al., 2008) (see below). A recent study suggests that hepatic H2S production by CBS is strongly dependent on the concentration of S‐adenosylmethionine (AdoMet), an allosteric activator of CBS, and serine, a substrate of CBS (Majtan et al., 2018). CBS regulation of both the transcriptional and post‐translational level is complex. CBS is a pyridoxal phosphate‐dependent (PLP) haem enzyme and responds to an oxidative environment via its haem domain, which directly influences the reactivity of the phosphate of PLP, inducing an increase in activity (Banerjee et al., 2003). Several dedicated CBS‐deficient mouse models have been generated and replicate the human disease phenotype and biomarker level abnormalities further described below (Watanabe et al., 1995; Gupta et al., 2009; Maclean et al., 2010). It is noteworthy that homozygous Cbs‐deficient pups display high pre‐ and neonatal mortality due to liver failure, highlighting the importance of CBS in the hepatic transsulphuration pathway (Watanabe et al., 1995). The translational aspects of CBS‐deficient mice to the human disease phenotype have been reviewed extensively elsewhere (Kruger, 2017). Neither sulfate nor thiosulfate levels have been reported in these mice; however, since CBS is the major H2S‐generating enzyme in neuronal tissues, it is reasonable to assume that H2S synthesis and downstream pathways are affected.
Cystathionine γ‐lyase
CSE primarily functions in the trans‐sulfuration pathway by catalysing the downstream reaction of CBS, the hydrolytic cleavage of cystathionine into cysteine, β‐ketobutyrate and ammonia (Figure 1). Additionally, CSE is considered as an important H2S‐producing enzyme (Kabil et al., 2011) catalysing five different reactions all leading to the generation of H2S (Figure 4). However, only two of these reactions are likely relevant under physiological cysteine and homocysteine concentrations: (i) the CBS‐like reaction of cysteine and water to H2S and serine has been estimated to account for up to 70% of CSE‐derived H2S, and (ii) the remaining 30% are covered by the reaction of homocysteine to H2S and α‐ketobutyrate (Yadav et al., 2016), while all other reactions produce only insignificant amounts of H2S. In addition, CSE is able to produce cysteine and homocysteine persulfides, with a rate again considered to be marginal (Yadav et al., 2016; Filipovic et al., 2017).
CSE is a PLP cytosolic enzyme strongly expressed in endothelial tissues of blood vessels (Morikawa et al., 2012). While in the past, the contribution of CSE to neuronal H2S synthesis received little attention, recent studies have found a significant impact of CSE‐dependent H2S production in murine brains under both healthy and disease conditions (Paul et al., 2014). The basal expression levels of CSE are controlled by a specificity protein 1, a ubiquitously expressed transcription factor mainly involved in cellular development (Ishii et al., 2004). Under conditions of cysteine deprivation and ER stress, increased presence of the activating transcription factor 4 increases expression of CSE drastically, which is believed to provide sufficient supply of cysteine for GSH biosynthesis (Dickhout et al., 2012).
To study the function of CSE in a physiological context, two mouse models have been developed and investigated (Yang et al., 2008; Ishii et al., 2010). In general, only mild neurological phenotypes have been reported for both models. CSE‐deficient mice require dietary cysteine supplementation (provided as bioavailable N‐acetyl‐cysteine) to avoid the development of a lethal myopathy and excessive oxidative damage (Ishii et al., 2010; Yamada et al., 2012). They also display increased serum cystathionine and homocysteine levels and decreased taurine levels, which can be correlated to an impaired cysteine biosynthesis, ultimately reducing the flux of cysteine towards the oxidative pathway. Sulfate and thiosulfate levels have not been reported for these mouse models, but are likely to be decreased, as the production of the precursor to both metabolites – H2S – in the heart and endothelial tissues is impaired (Yang et al., 2008). Additionally, protein persulfidation levels in the lung, heart, kidney and brain are significantly decreased under CSE deficiency (Wedmann et al., 2016), in addition to the inability of these mice to produce cysteine from methionine.
Mercaptopyruvate sulfurtransferase
A third pathway capable of producing H2S is dependent on several enzymatic activities. First, a transamination reaction by GOT1/2 converts cysteine and α‐ketoglutarate to 3‐mercaptopyruvate and glutamate. Under which conditions both enzymes are able to accept cysteine as substrate remains unknown, given that the K M values for cysteine are in the low millimolar range (Schwarz et al., unpublished data). In a subsequent reaction, 3‐mercaptopyruvate sulfurtransferase (MPST) catalyses the reductive desulfuration of 3‐mercaptopyruvate, a reaction that also requires thioredoxin, yielding pyruvate and H2S (Nandi et al., 2000). As described above, human GOT1 and GOT2 show similar kinetic properties, whereas rat liver mitochondrial MPST activity (5.2 U·mg−1) was found to be threefold higher than that of the cytosolic fraction (1.7 U·mg−1) (Nagahara et al., 1998). The fractional contribution of cytosolic and mitochondrial MPST to the overall H2S production remains to be elucidated.
MPST expression can be ubiquitously detected, with highest levels in the liver, large intestine and kidney (Shibuya et al., 2013), coinciding with GOT expression. Recently, another source for β‐mercaptopyruvate has been reported: the conversion of D‐cysteine to β‐mercaptopyruvate, hydrogen peroxide and ammonia via D‐amino acid oxidase, a peroxisomal enzyme (Shibuya et al., 2013). D‐cysteine is mostly taken up via the diet, and administration of D‐cysteine via an oral diet to rats has previously been found to lead to an increase in sulfate levels (Krijgsheld et al., 1981). MPST contains two rhodanese‐like domains, and, as such, MPST shares the scope of its substrates with other rhodaneses. MPST transfers the sulfur atom of β‐mercaptopyruvate to a cysteine residue on its C‐terminal rhodanese domain, forming a persulfide. This persulfide can be transferred to cyanide yielding thiocyanide or – more likely – the persulfide is transferred to thioredoxin, releasing H2S through auto‐oxidation (Yadav et al., 2013). Thioredoxin has also been shown to be the primary persulfidation‐resolving enzyme in cells (Dóka et al., 2016; Wedmann et al., 2016).
MPST‐deficient mice have been generated and display increased anxiety‐related behaviour, with no observed physical abnormalities (Nagahara et al., 2013); a comparison to the phenotype of the rare cases of human mercaptolactate‐cysteine disulfiduria is difficult to establish due to the low number of cases reported. Mental retardation, however, seems to be a common phenotype for this disease and was related to the anxiety phenotype of MPST‐deficient mice (Nagahara et al., 2013). While primary neurons derived from these mice display decreased H2S and polysulfide (H2Sn) production in response to β‐mercaptopyruvate, urinary or blood plasma thiosulfate and sulfate levels have not yet been reported (Kimura et al., 2015).
In the light of the three routes of H2S formation, the majority of H2S is formed within the cytosol and only a minor fraction is produced in mitochondria. In contrast, the major H2S‐dependent post‐translational modification, protein persulfidation, has been mainly detected in mitochondria (Wedmann et al., 2016). Although all three H2S‐producing enzymes are capable of H2S generation with significant rates under substrate saturation conditions, their relative contribution to the free and bound H2S pools vary between different tissues and is expected to be highly dependent on the local concentration of substrate and the expression levels of CBS, CSE and MPST in the respective cell type (Kabil et al., 2011). One reason that the action of H2S is highly controlled in a special and temporal manner resides in its efficient oxidation, which will be discussed in the following chapters.
Mitochondrial oxidation of hydrogen sulfide
The biogenesis of H2S takes place primarily in the cytosol, yet its degradation is restricted to the mitochondrial matrix and the intermembrane space (Figures 1 and 4). H2S is exceptionally adept at diffusing through biological membranes and therefore should easily reach the mitochondrial matrix (Riahi and Rowley, 2014). There are several enzymes involved in H2S oxidation resulting in the formation of sulfate and thiosulfate as terminal products (Kabil and Banerjee, 2014). While different routes of H2S oxidation have been proposed, only a few are expected to take place under substrate concentrations that are physiologically relevant. While sulfate is essential for several cellular processes and present in high micromolar concentrations in mammalian tissues (Markovich, 2001), thiosulfate has numerous medical implications, including being an antidote to cyanide poisoning. Furthermore, thiosulfate has been shown to be nontoxic to neurons, although causing nausea and headache when rapidly infused intravenously (Baskin et al., 1992; Kumar et al., 2017).
Sulfide : quinone oxidoreductase
The key enzyme in H2S oxidation is sulfide : quinone oxidoreductase (SQR), which is associated with the matrix side of the inner mitochondrial membrane (Figure 4) and harbours a flavine adenine dinucleotide (FAD) cofactor. It oxidizes H2S to a zero‐valent sulfur by generating a protein‐bound persulfide, which is further transferred to an acceptor. In a two‐step reaction, the electron from the sulfur atom is first transferred to the FAD cofactor and then to coenzyme Q located in the mitochondrial inner membrane, thus feeding into the electron transport chain of mitochondria (Marcia et al., 2010). Therefore, both cysteine degradation intermediates (sulfite and H2S) represent inorganic substrates for the electron transport chain in vertebrates. The final acceptors of the SQR‐bound persulfide are controversial (Filipovic et al., 2017), but GSH and sulfite have been proposed as the most likely candidates, while other acceptors discussed in the field (Libiad et al., 2014) exhibit mitochondrial concentrations that makes their physiological relevance questionable. Due to the high concentration of GSH (in contrast to low sulfite in healthy individuals), one can expect GSH as primary acceptor for the SQR persulfide, thus producing GSH persulfide. However, it is worth mentioning that the k cat and K M determined for the sulfite‐dependent reactivities of SQR and SO are nearly identical. Therefore, under conditions of low mitochondrial GSH levels, SQR could take over a significant share of sulfite clearance from SO (Jackson et al., 2012).
Persulfide dioxygenase
The first path to oxidize the terminal sulfur of the GSH persulfide is dependent on persulfide dioxygenase (PDO), also known as the ethylmalonic encephalopathy 1 (ETHE1) protein. Consistent with the localization of its substrate, PDO is located within the mitochondrial matrix (Figure 4) and appears to be most strongly expressed in liver and muscle tissues (Hildebrandt et al., 2013). For catalysis, the GSH persulfide is coordinated to a mononuclear iron in the active site of PDO, followed by stepwise oxidation of the terminal sulfur atom and an H2O‐dependent displacement of the generated sulfur dioxide, converting it to sulfite. PDO is only capable of oxidizing the persulfide of GSH, but not of cysteine or homocysteine (Kabil and Banerjee, 2012). It was also proposed that PDO may convert thiosulfate, oxygen and water to two sulfite moieties (Kabil and Banerjee, 2012); however, in vitro studies could not provide evidence for such a reaction under physiological pH (Kabil and Banerjee, 2012). The reaction of PDO holds interesting implications for the oxygen‐dependence of the H2S oxidation, and its enzymatic reaction may very well be restricted under hypoxic conditions.
ETHE1 −/− mice have been reported to largely mimic the human disease phenotype, displaying increased C4‐ and C5‐acylcarnitine levels as well as strongly increased tissue, serum and urinary thiosulfate levels (Tiranti et al., 2009). H2S levels have also been found to be increased, with mice showing signs of H2S‐dependent inhibition of cytochrome c oxidase (Tiranti et al., 2009). Interestingly, urinary sulfate concentrations have been demonstrated to be approximately sixfold decreased in these mice (Tiranti et al., 2009), providing evidence for a quantitative flux of sulfite towards thiosulfate via rhodanese.
Thiosulfate sulfurtransferase: rhodanese (TST)
The second path to further metabolize GSH persulfide produced by SQR is catalysed by rhodanese (also labelled TST), the second member of the sulfurtransferase superfamily present in this pathway. Rhodanese is most prominently expressed in hepatic and gastrointestinal tissues and localizes to the mitochondrial matrix. The TST reaction mechanism has been described as largely similar to MPST, with the exception that rhodanese does not rely on thioredoxin to resolve its enzyme‐bound persulfide (Huang and Yu, 2016). Rhodanese is capable of catalysing the transfer of a sulfur atom from a GSH persulfide to sulfite, thereby generating thiosulfate and recovering GSH (Figure 4), as well as the reverse reaction (Hildebrandt and Grieshaber, 2008). Kinetic studies of the enzyme support the view that, under physiological concentrations of substrates, rhodanese preferentially runs in the forward reaction producing thiosulfate (Libiad et al., 2015). However, in vitro, it was shown that rhodanese is able to generate H2S and oxidized GSH in the presence of GSH persulfide and reduced GSH, which may play a significant role in hepatic H2S production (Libiad et al., 2015).
Deficiencies in cysteine catabolism
In the past, different approaches have been followed to investigate the in vivo regulation of cysteine catabolism. Hepatocytes isolated from rats fed with diets of varying sulfur amino acid content were used to estimate the cysteine‐flux via the different catabolic pathways. Under the conditions of a basal diet, the majority of metabolized cysteine primarily enters the H2S pathway. However, with increasing sulfur amino acid intake, the amount of cysteine catabolized via the oxidative pathway increases, as demonstrated by an increased CDO activity. Within the oxidative pathway, it is considered that about 60–70% of CSA is converted into taurine (Bagley and Stipanuk, 1995). These correlations were mainly reported for the liver, the most prominent site of cysteine catabolism; however, cysteine flux might differ in other tissues. Besides feeding studies, major insights into the homeostatic regulation of cysteine catabolism were obtained from specific deficiencies in cysteine catabolism leading to rare and severe inborn errors of metabolism (Bagley and Stipanuk, 1995).
Alterations and deficiencies in the oxidative cysteine catabolism
For most enzymes participating in the oxidative cysteine catabolism, there are no human mutations reported, indicating that the respective enzymes might be essential. This hypothesis is supported by different mouse models; for example, CDO, CSAD and SO knockout mice display a high mortality rate (Ueki et al., 2011; Park et al., 2014). For GOT1/2, a mouse model is lacking and the only known human mutation is a heterozygous single nucleotide polymorphism in the GOT1 gene, leading to a decrease in plasma GOT activity (Shen et al., 2011). Remarkably, no association between the mutation and metabolic traits including alterations in other sub‐clinical markers could be identified. CDO, GOT2 and CSAD have not been associated with any genetic alterations in humans.
Within the oxidative pathway of cysteine catabolism, the only enzyme for which a well‐known human disease has been reported is SO (Shih et al., 1977; Schwarz, 2016). SO deficiency can either be caused by mutations in the SUOX gene (iSOD) or by mutations in genes (MOCS1, MOCS2, MOCS3, GPHN) required for the biosynthesis of Moco (Moco deficiency – MoCD), the active site cofactor of four Mo‐containing enzymes. Aside from SO, other Moco‐dependent enzymes in mammals include xanthine oxidase, aldehyde oxidase and the mitochondrial amidoxime reducing components 1 and 2 (Schwarz and Belaidi, 2013). On a biochemical level, MoCD and iSOD patients are only distinguishable through accumulation of xanthine and hypoxanthine, as well as the absence of uric acid in MoCD, but not iSOD patients (Sass et al., 2010; Schwahn et al., 2015). MoCD and iSOD patients present in their neonatal period with feeding difficulties, intractable seizures with an exaggerated startle reaction followed by severe neurological abnormalities, lens dislocation and head dismorphism. More than 150 MoCD cases and approximately 40 cases of iSOD have been reported (Schwarz, 2016). Disease progression is accompanied by psychomotor retardation due to progressive cerebral atrophy and ventricular dilatation. Patients that survive the acute neonatal period lack neuronal development and usually die within their first years of life (Schwarz and Veldman, 2014). As most of the symptoms of MoCD are mirrored in iSOD, SO is considered as the most important Moco‐dependent enzyme in humans. Therefore, sulfite accumulation presents the primary cause of neurodegeneration in both disorders, although the cytosolic or mitochondrial origin of sulfite remains to be elucidated.
An accumulation of sulfite (0.1–1 mM) has been shown to impair important cellular functions like inhibition of GDH and malate dehydrogenase, decreasing NADH concentrations and mitochondrial membrane potential, and ultimately ATP production (Vincent et al., 2004; Zhang et al., 2004). Sulfite appears to act predominantly on mitochondria, as it was demonstrated that sulfite diminishes mitochondrial respiration, membrane potential and Ca2+ retention capacity, presumably by damaging the electron transport chain (Zhang et al., 2004). In line with this, the administration of sulfite leads to increased ROS levels in different neuronal and kidney cell lines (Vincent et al., 2004; Zhang et al., 2004). Furthermore, GDH, a central metabolic enzyme, is inhibited by sulfite in a dose‐dependent manner. It has been speculated that inhibition of GDH leads to a decreased flux of the tricarboxylic acid cycle, which might further contribute to the reduced ATP production (Zhang et al., 2004). Within the extracellular space, sulfite can reduce disulfide‐bridges of proteins, thus affecting protein folding, stability and activity. Sulfite‐dependent cleavage of disulfide‐bridges produces S‐sulfonated species, such as S‐sulfocysteine (SSC) or S‐sulfonated transthyretin (Kishikawa et al., 2002).
Given the high plasma concentrations of cystine, the reaction of sulfite with cystine to SSC can be seen as the first scavenging mechanism for sulfite (Figure 5). SSC accumulation is inversely associated with a dramatic reduction in cysteine levels in iSOD and MoCD patients (Belaidi et al., 2012; Schwarz and Belaidi, 2013). While these processes likely take place throughout the body, the local damage in the brain suggests a specific sensitivity of neurons towards those metabolic changes. As a result of low cystine, GSH levels may also be reduced, which particularly impacts neuronal redox homeostasis. While no patient data regarding GSH levels under disease conditions exist, in vitro studies show that glutathione disulfide (GSSG), but not GSH, levels were found to be reduced when rat hepatocytes were acutely incubated with high dosages of sulfite (Niknahad and O'Brien, 2008). Furthermore, cystine depletion and glutamate excitotoxicity were found to decrease cellular GSH concentrations (Kato et al., 1992).
Figure 5.
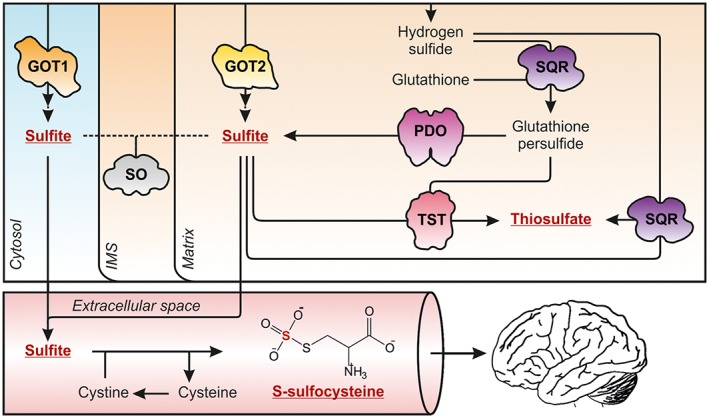
Sulfite excess facilitates the production of S‐sulfonates and causes brain damage. Missing SO activity results in the accumulation of cytotoxic sulfite, which diffuses out of the cell into the blood stream where it reduces disulfide bridges to form S‐sulfonates. S‐sulfocysteine (SSC) is the product of sulfite‐dependent cleavage of cystine. Within the brain, SSC is – like its structural homologue glutamate – excitotoxic. To what extent SSC can cross the blood–brain‐barrier or is formed in the brain is currently unknown. Under sulfite excess, an alternative reaction of SQR with H2S and sulfite leads to the formation thiosulfate. GOT1/2, 3II0/5AX8; PDO, 4CHL; SQR, 3H8L; SO, 1SOX; TST, 1RHD.
SSC is structurally similar to glutamate and – once accumulated in the brain – able to bind to NMDA receptors, likely in the same manner as other amino acids previously discovered to be excitotoxic, including homocysteic acid (Olney et al., 1975). In this context, we have studied SSC‐mediated neuronal cell death and found an NMDA receptor‐mediated calcium influx followed by calpain activation and proteolytic cleavage of synaptic proteins. Therefore, the accumulation of neuronal SSC represents a major contribution to neurodegeneration in iSOD and MoCD, triggering excitotoxicity (Kumar et al., 2017). Since, under physiological conditions, SO expression and activity in the brain is extremely low compared to the kidney or liver (Belaidi et al., 2015), it remains to be elucidated to what extent SSC formed in the periphery is able to cross the blood–brain barrier or if sulfite derived from the periphery or from the brain reacts with neuronal cysteine (Figure 5).
Besides reduced plasma cysteine/cystine levels under MoCD conditions, a reduction of homocysteine further underlines the severe homeostatic dysregulation caused by sulfite accumulation (Sass et al., 2003). Finally, the observed accumulation of thiosulfate suggests an increased sulfur flux via the H2S pathway (Figures 1 and 5), which we will discuss next.
Metabolic disorders in H2S metabolism
In humans, several inherited enzyme deficiencies have been associated with H2S metabolism, which vary in both their presentation and severity (Schwarz and Veldman, 2014). While some known syndromes, such as CSE deficiency, have little to mild symptoms, others, such as ETHE1 deficiency or MoCD and iSOD, are severe disorders with a mostly lethal outcome. The severity of a given disease is closely related to the respective metabolite(s) that primarily accumulate in the absence of the defective enzyme, while the lack of downstream products is less harmful.
A deficiency of CSE leads to cystathioninuria, sometimes also termed cystathioninaemia, which is described as a benign biochemical anomaly rather than a disease (Mudd et al., 2001). CSE deficiency has no or very diffuse clinical symptoms under normal conditions and is characterized by high plasma levels and urinary excretion of cystathionine. In contrast, an impairment of CBS activity may lead to a more pronounced effect in classical homocystinuria including an increased risk of thrombosis, embolism, skeletal abnormalities, developmental delay and hepatic steatosis. Patients may display drastically increased serum methionine and S‐adenosylmethionine as well as homocysteine levels; the severity of the disease is often correlated with the increase in plasma homocysteine levels (Barić and Fowler, 2014). CBS deficiency, however, has been shown to be symptomatically diverse, with differences between pyridoxine‐responsive and non‐responsive patients, which also influences biomarker levels and has been reviewed elsewhere (Kery et al., 1994; Huemer et al., 2015). For MPST, only very few deficiency or insufficiency cases have been reported. The increased presence of a specific, eponymous biomarker is the primary method for the identification of human mercaptolactate‐cysteine disulfidura (Crawhall et al., 1971), with patients displaying either severe phenotypes related to mental retardation and facial abnormalities (Crawhall et al., 1968; Ampola et al., 1969), or displaying essentially no phenotype (Niederwieser et al., 1973). The degree of severity is likely related to the level of insufficiency. Whether the phenotype is a result of the presence of this specific metabolite or due to the reduced production of H2S during the prenatal period is currently unclear (Nagahara et al., 2013). Furthermore, no symptoms or cases concerning a rhodanese deficiency have been reported, likely due to the fact that the most prominent reaction catalysed by rhodanese – the production of thiosulfate – can also be performed, albeit to a lesser degree, by SQR. The detoxification of cyanide to thiocyanide may also be performed by other rhodanese‐domain‐containing enzymes like MPST, for which this capacity has indeed been reported (Yadav et al., 2013). Interestingly, rhodanese has been identified as a candidate in obesity resistance, linking sulfur catabolism to other metabolic processes (Morton et al., 2016).
More severe disorders of cysteine catabolism impair reactions related to the detoxification of H2S. The toxic properties of sulfite and related metabolites have been discussed already. Again, the role of H2S in disease pathology is complex. On the one hand, several beneficial physiological functions have been described for H2S including up‐regulation of antioxidant systems, anti‐inflammatory and cytoprotective pathways as well as modulation of the nervous system by enhancement of NMDA‐receptor‐mediated responses (reviewed in Tiranti and Zeviani, 2013). On the other hand, when present in high micromolar concentrations, H2S has also been reported to interfere with a variety of cellular functions; impaired mitochondrial respiration via inhibition of cytochrome c oxidase is among the most pronounced phenotypes associated with H2S toxicity (Tiranti et al., 2009). Exposure to gaseous H2S can lead to anoxic brain injury, pulmonary oedema and ultimately death in humans (Yalamanchili and Smith, 2008). While brain hypoxia and acute respiratory syndrome are the most likely causes of death after acute H2S exposure, chronic exposure to low levels leads to irritant effects including upper airway inflammation (Tiranti and Zeviani, 2013).
The most prominent disease associated with an increase in H2S levels is ethylmalonic encephalopathy (ETHE1 deficiency) or PDO deficiency. The disease is invariably fatal and characterized by early‐onset encephalopathy, chronic diarrhoea, microangiopathy and excretion of ethylmalonic acid in urine (Tiranti et al., 2004). Another molecular hallmark of the disease is H2S‐mediated inhibition of cytochrome c oxidase, which represents a cause of the accumulation of C4‐ and C5‐acylcarnitines. Interestingly, while urinary and tissue thiosulfate levels are increased in ETHE1‐deficient mice, urinary sulfate levels are decreased sixfold (Tiranti et al., 2009) underscoring a major flux of sulfur via the H2S pathway. The strong accumulation of thiosulfate suggests an increased flux of sulfur via the thiosulphate sulfurtransferase (TST) enzyme due to the accumulation of persulfidated GSH.
More recently, coenzyme Q deficiency, a classical mitochondrial disorder, has been added to the list H2S‐related disorders, as it was found to negatively affect the activity of the H2S‐metabolizing enzyme SQR (Ziosi et al., 2016; Luna‐Sánchez et al., 2017). While no direct deficiency of SQR has been described for humans, an impaired production of coenzyme Q, the cofactor of SQR, appears to correlate with low SQR protein levels (Kühl et al., 2017). Coenzyme Q deficiency causes a variety of severe symptoms including encephalomyopathy, severe infantile multi‐systemic disease, cerebellar ataxia, isolated myopathy and steroid‐resistant nephrotic syndrome (Desbats et al., 2015). The pathogenesis is complex, which is likely related to the different functions of coenzyme Q. Consistent with the reduced enzymatic capacity of SQR, kidney H2S levels are increased whereas urinary thiosulfate levels are decreased (Ziosi et al., 2016). Interestingly, kidney levels of enzymes downstream of SQR were also reduced, suggesting a regulatory mechanism that controls expression of H2S‐catabolizing enzymes in general (Ziosi et al., 2016).
The homeostatic link between oxidative and H2S‐dependent cysteine catabolism in health and disease
Both the oxidative cysteine catabolism and the H2S pathway converge in the formation of sulfite as a terminal intermediate leading to the formation of sulfate and thiosulfate. Therefore, the accumulation of sulfite, caused by iSOD/MoCD, impacts on metabolite and protein levels of both branches of cysteine catabolism in multiple ways.
The scavenging reaction of sulfite with cystine results in a depletion of cystine. As a result, cellular cysteine is reduced as found under conditions where the cystine/glutamate transporter xCT if deficient, causing a reduction in plasma GSH concentrations (Sato et al., 2005). To what extent GSH levels are reduced in patients of iSOD and MoCD remains to be determined. Besides the multiple functions in redox signalling, mitochondrial GSH levels also control the rate of H2S catabolism. Therefore, a sulfite‐dependent depletion of GSH should ultimately limit the capacity of mitochondria to oxidize H2S. In addition to sulfite‐dependent inhibition of mitochondrial respiration, it is reasonable to assume that increasing H2S levels further exaggerates metabolic deficits in iSOD and MoCD.
Mitochondria play a central and critical role in cysteine metabolism in health and disease. On the one hand, reducing equivalents derived from oxidative reactions in both H2S and sulfite oxidation contribute to mitochondrial respiration (Figure 3). On the other hand, dysfunctions in sulfite and H2S oxidation severely impact mitochondrial function and thereby contribute to mitochondria‐driven signalling and cell death (Zhang et al., 2004; Tiranti et al., 2009). Sulfite‐induced mitochondrial damage might be associated with increased taurine levels in iSOD/MoCD patients (Belaidi and Schwarz, 2013). The primary source of taurine biosynthesis under healthy conditions is CSA, which is predominantly produced under cysteine excess. However, since iSOD/MoCD patients display very low cystine/cysteine levels, an alternative pathway for taurine must be up‐regulated. Hypotaurine can also be produced by cysteamine dioxygenase (ADO) using cysteamine as a substrate (Dominy et al., 2007). However, studies using CDO −/− or CSAD −/− mice show that ADO only accounts for about 10% of overall taurine biosynthesis under normal conditions (Ueki et al., 2011; Park et al., 2014). One possibility is that the degradation of coenzyme A to cysteamine is increased under mitochondrial stress (Robishaw and Neely, 1985) given that mitochondrial function has been shown to be impaired under sulfite stress (Salman et al., 2002; Zhang et al., 2004). As coenzyme A concentrations are in the low millimolar range, an increased degradation of this important coenzyme, which involves the release of cysteamine and its further deamination by ADO, might represent a source of increased taurine production in iSOD/MoCD patients.
Another aspect of iSOD is the accumulation of thiosulfate, suggesting an increased flux of accumulated sulfite via rhodanese towards thiosulfate (Touati et al., 2000). As previously discussed, the production of thiosulfate may be accomplished by both SQR and rhodanese. One line of argument is that an increased thiosulfate production might also deplete persulfidated GSH, the product of the SQR‐dependent oxidation of H2S and a possible substrate for rhodanese (Hildebrandt and Grieshaber, 2008). The other is that increased thiosulfate excretion could indicate an increased H2S flux, which is used in the SQR‐dependent production of thiosulfate (Jackson et al., 2012; Libiad et al., 2014). Both hypotheses suggest that iSOD might cause a reduction in H2S levels, and therefore, some aspects of the disease symptoms may be associated with reduced H2S production. The SQR‐dependent thiosulfate production may gain further importance in iSOD when considering the reduced plasma cystine levels in iSOD patients (Touati et al., 2000; Rocha et al., 2014), since cysteine/cystine levels regulate GSH homeostasis (Yu and Long, 2016). With a K M for sulfite similar to SO (Jackson et al., 2012), a reduction in its primary substrate GSH would likely cause a switch from the production of persulfidated GSH to the production of thiosulfate (Figure 5), which further supports the view of a depletion of H2S in iSOD/MoCD.
An alternative scenario could also explain high thiosulfate levels: the low concentration of cystine in iSOD patients (Touati et al., 2000; Rocha et al., 2014) may trigger an up‐regulation of the transsulfuration pathway via CSE (Yu and Long, 2016; Paul et al., 2018), which is an important factor in H2S biosynthesis. As a result, high H2S levels would contribute to increased thiosulfate production under sulfite excess. In conclusion, the complex and fast progressing neurodegenerative phenotype in iSOD and MoCD needs an out‐of‐box analysis of the entire cysteine metabolism to understand the hierarchical order of degenerative events. To what extent H2S accumulation or depletion contributes to iSOD/MoCD pathophysiology needs further investigation using SO‐deficient animal models.
Thiosulfate accumulation is also a hallmark of ETHE1 deficiency. Loss of PDO activity in these patients leaves TST as the sole GSH persulfide degrading enzyme. The observed accumulation of thiosulfate demonstrates an increased flux of H2S towards thiosulfate, accompanied by a loss in sulfate excretion (Tiranti et al., 2009), suggesting that sulfite is primarily incorporated into thiosulfate. The fact that H2S accumulates in ETHE1 deficiency suggest that either TST activity or sulfite availability limits GSH persulfide turnover. Given that N‐acetylcysteine supplementation partially suppresses the phenotype, an increased availability of GSH would allow more H2S to be scavenged (Viscomi et al., 2010). An alternative explanation might be that increased cysteine levels down‐regulate CSE activity, thus producing less H2S and therefore less thiosulfate.
Treatment options for sulfite and H2S toxicity disorders
iSOD is currently a non‐treatable disorder. In some cases, dietary restriction in the intake of methionine and cysteine showed therapeutic effectiveness in patients with mild phenotypes. For example, a patient harbouring a mutation in the SO N‐terminal haem domain (H143N) presented at the age of 12 months with acute left hemiparesis, generalized mild hypotonia and developmental delay. Biomarkers (cystine, SSC and taurine) were typical for iSOD. The introduction of a low‐protein diet with a sixfold reduction in methionine and cysteine intake triggered a sustained reduction of pathological biomarkers (SSC, taurine), followed by clinical improvement, partial shrinking of lesions in the basal ganglia and achievements of additional motor milestones (Del Rizzo et al., 2013). Given the fact that the H143A mutation – affecting one of the two haem‐coordinating histidines in SO – does not affect the catalytic Moco domain, one can speculate that in the absence of a functional haem domain, the catalytic cycle of SO is closed by the use of O2 as alternative electron acceptor (Belaidi et al., 2015), thus retaining a partial capacity to reduce excess sulfite. The accompanied formation of ROS might however accumulate over time and cause other deficits.
MoCD represents another form of iSOD, accompanied by the loss of all Mo‐enzymes (Schwarz et al., 2009). Both disease symptoms and progression are highly similar suggesting iSOD as major disease‐causing mechanism in MoCD, which was also mirrored by SO‐deficient mice. For a subgroup of MoCD patients that carry mutations in the first biosynthetic step of Moco (MOCS1, MoCD type A), a substitution therapy with the Moco‐intermediated cyclic pyranopterin monophosphate (cPMP) has been established in mice (Schwarz et al., 2004) and translated to a first patient (Veldman et al., 2010). In the following years, more than 20 MoCD patients have been administered cPMP showing that initiation of treatment before the manifestation of neurological symptoms has been a prerequisite for treatment success and near normal development (Schwahn et al., 2015). Therefore, patient diagnosis and symptomatic treatment of SSC‐evoked seizures by use of NMDA receptor blockers is considered as key to extend the window of treatability to improve clinical outcome for these patients (Kumar et al., 2017).
Beside dietary restriction of methionine and cysteine in mild forms of iSOD and MoCD, no other treatment options are available today. Based on the homeostatic impact of sulfite and H2S on cysteine catabolism discussed here, additional treatment strategies need to be considered. We suggested earlier that an enzyme substitution therapy with SO might be an option; however, the production of H2O2 as a side product of SO‐mediated sulfite oxidation in the presence of oxygen is challenging (Belaidi et al., 2015). Alternatively, to reduce sulfite accumulation and cysteine depletion, blocking CDO activity or GOT1/2 activity might shift the flux of sulfur into H2S pathways. Considering the scenarios mentioned above, this approach would work, if H2S depletion would accompany iSOD. Crossing CDO and SO‐deficient mice to create double‐knockout animals may answer this question, if additional taurine supplementation can be warranted. Alternatively, the scavenging of excess sulfite would restore cystine/cysteine and regenerate the intracellular GSH pool as a primary antioxidative barrier.
Similar to iSOD/MoCD, thiosulfate is also accumulated in ETHE1 (PDO) deficiency. As discussed above, N‐acetylcysteine treatment diminished the disease phenotype, suggesting a decrease in H2S formation. An alternative approach towards the productive elimination of excess H2S in ETHE1 deficiency might be an increase in sulfite availability by reducing SO activity. Therefore, crossing SO −/− and ETHE1 −/− mice, if viable, could answer this question. In the case of a positive outcome, further increased thiosulfate formation would be expected. From a treatment point of view, the administration of tungstate is well known to inhibit SO activity in a dose‐dependent manner (Kumar et al., 2017).
Concluding remarks
Altered sulfite and H2S metabolism is associated with various types of neurodegenerative disorders ranging from rare inborn errors in metabolism to complex neurodegenerative disorders such as Huntington's, Parkinson's and Alzheimer's disease (McBean et al., 2015; Salmina et al., 2015). The recognized dramatic impact of sulfite, the common intermediate in oxidative cysteine and H2S catabolism on the overall homeostatic balance of cysteine, GSH, homocysteine and thiosulfate, collectively suggests an important effect of sulfite on H2S levels and H2S‐related signalling such as protein persulfidation. Therefore, future studies focusing on metabolic fluxes under different nutrient conditions and enzymatic activities will help to develop a better understanding of which thiol‐species contribute to a particular aspect of a given type of neurodegenerative disorder. Studying rare, mostly severe, metabolic disorders will allow us to extrapolate common principle from extreme cases towards multifactorial disorders. In this context, the beneficial effect of dietary restriction in general and low sulfur diet more specifically suggests the key importance of a tight control of cysteine, H2S, sulfite and GSH persulfide levels for the homeostatic balance in cell signalling and survival.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a, 2017b).
Conflict of interest
G.S. is inventor on a patent for the treatment of molybdenum cofactor deficiency type A using cPMP and CEO of Colbourne Pharmaceuticals GmbH that consults Alexion Pharma in the clinical development of cPMP therapy.
Acknowledgements
Continues support for our research (G.S.) by the German Science Foundation (DFG) is gratefully acknowledged. Current projects on cysteine catabolism are supported by DFG grants SFB1218‐B08 (to G.S.) and Center for Molecular Medicine Cologne grant C13 (to G.S.).
Kohl, J. B. , Mellis, A.‐T. , and Schwarz, G. (2019) Homeostatic impact of sulfite and hydrogen sulfide on cysteine catabolism. British Journal of Pharmacology, 176: 554–570. 10.1111/bph.14464.
References
- Abe K, Kimura H (1996). The possible role of hydrogen sulfide as an endogenous neuromodulator. J Neurosci 16: 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akagi R (1982). Purification and characterization of cysteine aminotransferase from rat liver cytosol. Acta Med Okayama 36: 187–197. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ampola MG, Efron ML, Bixby EM, Meshorer E (1969). Mental deficiency and a new aminoaciduria. Am J Dis Child 117: 66–70. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Sang WS, Hamby AM, Liu J, Wai YC, Chen Y et al (2006). Neuronal glutathione deficiency and age‐dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci 9: 119–126. [DOI] [PubMed] [Google Scholar]
- Bagley PJ, Stipanuk MH (1995). Rats fed a low protein diet supplemented with sulfur amino acids have increased cysteine dioxygenase activity and increased taurine production in hepatocytes. J Nutr 125: 933–940. [DOI] [PubMed] [Google Scholar]
- Banerjee R (2017). Catalytic promiscuity and heme‐dependent redox regulation of H2S synthesis. Curr Opin Chem Biol 37: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Evande R, Kabil Ö, Ojha S, Taoka S (2003). Reaction mechanism and regulation of cystathionine β‐synthase. Biochim Biophys Acta – Proteins Proteomics 1647: 30–35. [DOI] [PubMed] [Google Scholar]
- Barić I, Fowler B (2014). Sulphur amino acids BT In: Blau N, Duran M, Gibson KM, Dionisi Vici C. (eds). Physician's Guide to the Diagnosis, Treatment, and Follow‐Up of Inherited Metabolic Diseases. Springer Berlin Heidelberg: Berlin, Heidelberg, pp. 33–46. [Google Scholar]
- Baskin SI, Horowitz AM, Nealley EW (1992). The antidotal action of sodium nitrite and sodium thiosulfate against cyanide poisoning. J Clin Pharmacol 32: 368–375. [DOI] [PubMed] [Google Scholar]
- Belaidi AA, Arjune S, Santamaria‐Araujo JA, Sass JO, Schwarz G (2012). Molybdenum cofactor deficiency: a new HPLC method for fast quantification of s‐sulfocysteine in urine and serum. JIMD Rep 5: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belaidi AA, Röper J, Arjune S, Krizowski S, Trifunovic A, Schwarz G (2015). Oxygen reactivity of mammalian sulfite oxidase provides a concept for the treatment of sulfite oxidase deficiency. Biochem J 469: 211–221. [DOI] [PubMed] [Google Scholar]
- Belaidi AA, Schwarz G (2013). Molybdenum cofactor deficiency: metabolic link between taurine and S‐sulfocysteine BT In: El Idrissi A, L'Amoreaux WJ. (eds). Taurine 8. Springer New York: New York, NY, pp. 13–19. [DOI] [PubMed] [Google Scholar]
- Beltowski J (2014). Hydrogen sulfide in pharmacology and medicine – an update. Pharmacol Rep 67: 647–658. [DOI] [PubMed] [Google Scholar]
- Bremer J, Davis EJ (1975). Studies on the active transfer of reducing equivalents into mitochondria via the malate‐aspartate shuttle. Biochim Biophys Acta – Bioenerg 376: 387–397. [DOI] [PubMed] [Google Scholar]
- Caliendo G, Cirino G, Santagada V, Wallace JL (2010). Synthesis and biological effects of hydrogen sulfide (H2S): development of H2S‐releasing drugs as pharmaceuticals. J Med Chem 53: 6275–6286. [DOI] [PubMed] [Google Scholar]
- Cechetto J, Sadacharan S, Berk P, Gupta R (2002). Immunogold localization of mitochondrial aspartate aminotransferase in mitochondria and on the cell surface in normal rat tissues. [DOI] [PubMed]
- Chen X, Jhee KH, Kruger WD (2004). Production of the neuromodulator H2S by cystathionine beta‐synthase via the condensation of cysteine and homocysteine. J Biol Chem 279: 52082–52086. [DOI] [PubMed] [Google Scholar]
- Cohen HJ, Betcher‐Lange S, Kessler DL, Rajagopalan KV (1972). Hepatic sulfite oxidase. J Biol Chem 247: 7759–7766. [PubMed] [Google Scholar]
- Crawhall JC, Bir K, Purkiss P, Stanbury JB (1971). Sulfur amino acids as precursors of β‐mercaptolactate‐cysteine disulfide in human subjects. Biochem Med 5: 109–115. [DOI] [PubMed] [Google Scholar]
- Crawhall JC, Parker R, Sneddon W, Young EP, Ampola MG, Efron ML et al (1968). Beta mercaptolactate‐cysteine disulfide: analog of cystine in the urine of a mentally retarded patient. Science (80‐) 160: 419–420. [DOI] [PubMed] [Google Scholar]
- Deponte M (2013). Glutathione catalysis and the reaction mechanisms of glutathione‐dependent enzymes. Biochim Biophys Acta ‐General Subj 1830: 3217–3266. [DOI] [PubMed] [Google Scholar]
- Del Rizzo M, Burlina AP, Sass JO, Beermann F, Zanco C, Cazzorla C et al (2013). Metabolic stroke in a late-onset form of isolated sulfite oxidase deficiency. Mol Genet Metab 108: 263–266. [DOI] [PubMed] [Google Scholar]
- Desbats MA, Vetro A, Limongelli I, Lunardi G, Casarin A, Doimo M et al (2015). Primary coenzyme Q 10 deficiency presenting as fatal neonatal multiorgan failure. Eur J Hum Genet 23: 1254–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickhout JG, Carlisle RE, Jerome DE, Mohammed‐Ali Z, Jiang H, Yang G et al (2012). Integrated stress response modulates cellular redox state via induction of cystathionine γ‐lyase: cross‐talk between integrated stress response and thiol metabolism. J Biol Chem 287: 7603–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dóka É, Pader I, Bíró A, Johansson K, Cheng Q, Ballagó K et al (2016). A novel persulfide detection method reveals protein persulfide‐ and polysulfide‐reducing functions of thioredoxin and glutathione systems. Sci Adv 2: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy JE, Hirschberger LL, Coloso RM, Stipanuk MH (2006). Regulation of cysteine dioxygenase degradation is mediated by intracellular cysteine levels and the ubiquitin–26 S proteasome system in the living rat. Biochem J 394: 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominy JE, Simmons CR, Hirschberger LL, Hwang J, Coloso RM, Stipanuk MH (2007). Discovery and characterization of a second mammalian thiol dioxygenase, cysteamine dioxygenase. J Biol Chem 282: 25189–25198. [DOI] [PubMed] [Google Scholar]
- Dongó E, Beliczai‐Marosi G, Dybvig AS, Kiss L (2017). The mechanism of action and role of hydrogen sulfide in the control of vascular tone. Nitric Oxide – Biol Chem : 1–13. [DOI] [PubMed] [Google Scholar]
- Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H (2005). Cystathionine β‐synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J 19: 1854–1856. [DOI] [PubMed] [Google Scholar]
- Filipovic MR, Zivanovic J, Alvarez B, Banerjee R (2017). Chemical biology of H2S signaling through persulfidation. Chem Rev 118: 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein JD (2000). Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost 26: 219–225. [DOI] [PubMed] [Google Scholar]
- Furne J, Saeed A, Levitt MD (2008). Whole tissue hydrogen sulfide concentrations are orders of magnitude lower than presently accepted values. AJP Regul Integr Comp Physiol 295: R1479–R1485. [DOI] [PubMed] [Google Scholar]
- Guion‐Rain MC, Portemer C, Chatagner F (1975). Rat liver cysteine sulfinate decarboxylase: purification, new appraisal of the molecular weight and determination of catalytic properties. BBA – Enzymol 384: 265–276. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kühnisch J, Mustafa A, Lhotak S, Schlachterman A, Slifker MJ et al (2009). Mouse models of cystathionine beta‐synthase deficiency reveal significant threshold effects of hyperhomocysteinemia. FASEB J 23: 883–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrandt TM, Grieshaber MK (2008). Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J 275: 3352–3361. [DOI] [PubMed] [Google Scholar]
- Hildebrandt TM, Di Meo I, Zeviani M, Viscomi C, Braun H‐P (2013). Proteome adaptations in Ethe1‐deficient mice indicate a role in lipid catabolism and cytoskeleton organization via post‐translational protein modifications. Biosci Rep 33: 575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille R, Hall J, Basu P (2014). The mononuclear molybdenum enzymes. Chem Rev 114: 3963–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L et al (2015). Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Mitchell JR (2015). Calorie restriction and methionine restriction in control of endogenous hydrogen sulfide production by the transsulfuration pathway. Exp Gerontol 68: 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GT, Yu JSK (2016). Enzyme catalysis that paves the way for S‐sulfhydration via sulfur atom transfer. J Phys Chem B 120: 4608–4615. [DOI] [PubMed] [Google Scholar]
- Huemer M, Kožich V, Rinaldo P, Baumgartner MR, Merinero B, Pasquini E et al (2015). Newborn screening for homocystinurias and methylation disorders: systematic review and proposed guidelines. J Inherit Metab Dis 38: 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M (2010). Cystathionine gamma‐lyase‐deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285: 26358–26368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii I, Akahoshi N, Yu X‐N, Kobayashi Y, Namekata K, Komaki G et al (2004). Murine cystathionine gamma‐lyase: complete cDNA and genomic sequences, promoter activity, tissue distribution and developmental expression. Biochem J 381: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MR, Melideo SL, Jorns MS (2012). Human sulfide: quinone oxidoreductase catalyzes the first step in hydrogen sulfide metabolism and produces a sulfane sulfur metabolite. Biochemistry 51: 6804–6815. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Rajagopalan KV (1980). The oxidation of sulphite in animal systems. Ciba Foundation Symposium 72 ‐ Sulphur Biology 72: 119–133. [DOI] [PubMed] [Google Scholar]
- Jurkowska H, Roman HB, Hirschberger LL, Sasakura K, Nagano T, Hanaoka K et al (2014). Primary hepatocytes from mice lacking cysteine dioxygenase show increased cysteine concentrations and higher rates of metabolism of cysteine to hydrogen sulfide and thiosulfate. Amino Acids 46: 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Banerjee R (2012). Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J Biol Chem 287: 44561–44567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Banerjee R (2014). Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal 20: 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Vitvitsky V, Xie P, Banerjee R (2011). The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal 15: 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato S, Negishi K, Mawatari K, Kuo CH (1992). A mechanism for glutamate toxicity in the C6 glioma cells involving inhibition of cystine uptake leading to glutathione depletion. Neuroscience 48: 903–914. [DOI] [PubMed] [Google Scholar]
- Kery V, Bukovska G, Kraus JP (1994). Transsulfuration depends on heme in addition to pyridoxal 5′‐phosphate: cystathionine beta‐synthase is a heme protein. J Biol Chem 269: 25283–25288. [PubMed] [Google Scholar]
- Kimura Y, Toyofuku Y, Koike S, Shibuya N, Nagahara N, Lefer D et al (2015). Identification of H2S3 and H2S produced by 3‐mercaptopyruvate sulfurtransferase in the brain. Sci Rep 5: 14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishikawa M, Sass JO, Sakura N, Nakanishi T, Shimizu A, Yoshioka M (2002). The peak height ratio of S‐sulfonated transthyretin and other oxidized isoforms as a marker for molybdenum cofactor deficiency, measured by electrospray ionization mass spectrometry. Biochim Biophys Acta – Mol Basis Dis 1588: 135–138. [DOI] [PubMed] [Google Scholar]
- Kisker C, Schindelin H, Pacheco A, Wehbi WA, Garrett RM, Rajagopalan KV et al (1997). Molecular basis of sulfite oxidase deficiency from the structure of sulfite oxidase. Cell 91: 973–983. [DOI] [PubMed] [Google Scholar]
- Klein JM, Schwarz G (2012). Cofactor‐dependent maturation of mammalian sulfite oxidase links two mitochondrial import pathways. J Cell Sci 125: 4876–4885. [DOI] [PubMed] [Google Scholar]
- Klomsiri C, Karplus PA, Poole LB (2011). Cysteine‐based redox switches in enzymes. Antioxid Redox Signal 14: 1065–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijgsheld KR, Glazenburg EJ, Scholtens E, Mulder GJ (1981). The oxidation of L‐ and D‐cysteine to inorganic sulfate and taurine in the rat. Biochim Biophys Acta 677: 7–12. [DOI] [PubMed] [Google Scholar]
- Kruger WD (2017). Cystathionine β‐synthase deficiency: of mice and men. Mol Genet Metab 121: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühl I, Miranda M, Atanassov I, Kuznetsova I, Hinze Y, Mourier A et al (2017). Transcriptomic and proteomic landscape of mitochondrial dysfunction reveals secondary coenzyme Q deficiency in mammals. Elife 6: e30952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Dejanovic B, Hetsch F, Semtner M, Fusca D, Arjune S et al (2017). S‐sulfocysteine/NMDA receptor‐dependent signaling underlies neurodegeneration in molybdenum cofactor deficiency. J Clin Invest 127: 4365–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H‐J, Adham IM, Schwarz G, Kneussel M, Sass JO, Engel W et al (2002). Molybdenum cofactor‐deficient mice resemble the phenotype of human patients. Hum Mol Genet 11: 3309–3317. [DOI] [PubMed] [Google Scholar]
- Lee JI, Londono M, Hirschberger LL, Stipanuk MH (2004). Regulation of cysteine dioxygenase and gamma‐glutamylcysteine synthetase is associated with hepatic cysteine level. J Nutr Biochem 15: 112–122. [DOI] [PubMed] [Google Scholar]
- Levitt MD, Abdel‐Rehim MS, Furne J (2011). Free and acid‐labile hydrogen sulfide concentrations in mouse tissues: anomalously high free hydrogen sulfide in aortic tissue. Antioxid Redox Signal 15: 373–378. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P, Moore PK (2011). Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol 51: 169–187. [DOI] [PubMed] [Google Scholar]
- Libiad M, Sriraman A, Banerjee R (2015). Polymorphic variants of human rhodanese exhibit differences in thermal stability and sulfur transfer kinetics. J Biol Chem 290: 23579–23588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R (2014). Organization of the human mitochondrial hydrogen sulfide oxidation pathway. J Biol Chem 289: 30901–30910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC (2013). Glutathione synthesis. Biochim Biophys Acta 1830: 3143–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna‐Sánchez M, Hidalgo‐Gutiérrez A, Hildebrandt TM, Chaves‐Serrano J, Barriocanal‐Casado E, Santos‐Fandila Á et al (2017). CoQ deficiency causes disruption of mitochondrial sulfide oxidation, a new pathomechanism associated with this syndrome. EMBO Mol Med 9: 78–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean KN, Sikora J, Kožich V, Jiang H, Greiner LS, Kraus E et al (2010). A novel transgenic mouse model of CBS‐deficient homocystinuria does not incur hepatic steatosis or fibrosis and exhibits a hypercoagulative phenotype that is ameliorated by betaine treatment. Mol Genet Metab 101: 153–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majtan T, Krijt J, Sokolová J, Křížková M, Ralat MA, Kent J et al (2018). Biogenesis of hydrogen sulfide and thioethers by cystathionine beta‐synthase. Antioxid Redox Signal 28: 311–323. [DOI] [PubMed] [Google Scholar]
- Marcia M, Ermler U, Peng G, Michel H (2010). A new structure‐based classification of sulfide: quinone oxidoreductases. Proteins Struct Funct Bioinforma 78: 1073–1083. [DOI] [PubMed] [Google Scholar]
- Markovich D (2001). Physiological roles and regulation of mammalian sulfate transporters. Physiol Rev 81: 1499–1533. [DOI] [PubMed] [Google Scholar]
- McBean GJ, Aslan M, Griffiths HR, Torrão RC (2015). Thiol redox homeostasis in neurodegenerative disease. Redox Biol 5: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miseta A, Csutora P (2000). Relationship between the occurrence of cysteine in proteins and the complexity of organisms. Mol Biol Evol 17: 1232–1239. [DOI] [PubMed] [Google Scholar]
- Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y et al (2012). Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide‐sensitive hydrogen sulfide pathway. Proc Natl Acad Sci 109: 1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NM, Beltram J, Carter RN, Michailidou Z, Gorjanc G, McFadden C et al (2016). Genetic identification of thiosulfate sulfurtransferase as an adipocyte‐expressed antidiabetic target in mice selected for leanness. Nat Med 22: 771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudd SH, Cerone R, Schiaffino MC, Fantasia AR, Minniti G, Caruso U et al (2001). Glycine N‐methyltransferase deficiency: a novel inborn error causing persistent isolated hypermethioninaemia. J Inherit Metab Dis 24: 448–464. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK et al (2009). H2S signals through protein S‐sulfhydration. Sci Signal 2: ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara N, Ito T, Kitamura H, Nishino T (1998). Tissue and subcellular distribution of mercaptopyruvate sulfurtransferase in the rat: confocal laser fluorescence and immunoelectron microscopic studies combined with biochemical analysis. Histochem Cell Biol 110: 243–250. [DOI] [PubMed] [Google Scholar]
- Nagahara N, Nagano M, Ito T, Shimamura K, Akimoto T, Suzuki H (2013). Antioxidant enzyme, 3‐mercaptopyruvate sulfurtransferase‐knockout mice exhibit increased anxiety‐like behaviors: a model for human mercaptolactate‐cysteine disulfiduria. Sci Rep 3: 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi DL, Horowitz PM, Westley J (2000). Rhodanese as a thioredoxin oxidase. Int J Biochem Cell Biol 32: 465–473. [DOI] [PubMed] [Google Scholar]
- Niederwieser A, Giliberti P, Baerlocher K (1973). β‐mercaptolactate cysteine disulfiduria in two normal sisters. Isolation and characterization of β‐mercaptolactate cysteine disulfide. Clin. Chim. Acta Theriol 43: 405–416. [DOI] [PubMed] [Google Scholar]
- Niknahad H, O'Brien PJ (2008). Mechanism of sulfite cytotoxicity in isolated rat hepatocytes. Chem Biol Interact 174: 147–154. [DOI] [PubMed] [Google Scholar]
- Nisselbaum JS, Bodansky O (1964). Immunochemical and kinetic cationic from human properties of anionic and transaminases separated and human liver *. J Biol Chem 239: 4232–4236. [PubMed] [Google Scholar]
- Oertel WH, Schmechel DE, Weise VK, Ransom DH, Tappaz ML, Krutzsch HC et al (1981). Comparison of cysteine sulphinic acid decarboxylase isoenzymes and glutamic acid decarboxylase in rat liver and brain. Neuroscience 6: 2701–2714. [DOI] [PubMed] [Google Scholar]
- Olney JW, Misra CH, De Gubareff T, De Gubareff T (1975). Cysteine‐S‐sulfate: brain damaging metabolite in sulfite oxidase deficiency1. J Neuropathol Exp Neurol 34: 167–177. [DOI] [PubMed] [Google Scholar]
- Park E, Park SY, Dobkin C, Schuller‐Levis G (2014). Development of a novel cysteine sulfinic Acid decarboxylase knockout mouse: dietary taurine reduces neonatal mortality. J Amino Acids 2014: 346809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Sbodio JI, Snyder SH (2018). Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol Sci 39: 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM et al (2014). Cystathionine γ‐lyase deficiency mediates neurodegeneration in Huntington's disease. Nature 509: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2012). H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol 13: 499–507. [DOI] [PubMed] [Google Scholar]
- Poole LB, Nelson KJ (2008). Discovering mechanisms of signaling‐mediated cysteine oxidation. Curr Opin Chem Biol 12: 18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter PN, Grishaver MS, Jones OW (1974). Characterization of human cystathionine beta‐synthase. Biochim Biophys Acta 364: 128–139. [DOI] [PubMed] [Google Scholar]
- Rapson TD, Astashkin AV, Johnson‐Winters K, Bernhardt PV, Kappler U, Raitsimring AM et al (2010). Pulsed EPR investigations of the Mo (V) centers of the R55Q and R55M variants of sulfite dehydrogenase from Starkeya novella. J Biol Inorg Chem 15: 505–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recasens M, Benezra R, Basset P, Mandel P (1980). Cysteine sulfinate aminotransferase and aspartate aminotransferase isoenzymes of rat brain. Purification, characterization, and further evidence for identity. Biochemistry 19: 4583–4589. [DOI] [PubMed] [Google Scholar]
- Riahi S, Rowley CN (2014). Why can hydrogen sulfide permeate cell membranes? J Am Chem Soc 136: 15111–15113. [DOI] [PubMed] [Google Scholar]
- Ripps H, Shen W (2012). Review: taurine: a ‘very essential’ amino acid. Mol Vis 18: 2673–2686. [PMC free article] [PubMed] [Google Scholar]
- Robishaw JD, Neely JR (1985). Coenzyme A metabolism. Am J Physiol 248: E1–E9. [DOI] [PubMed] [Google Scholar]
- Rocha S, Ferreira AC, Dias AI, Vieira JP, Sequeira S (2014). Sulfite oxidase deficiency – an unusual late and mild presentation. Brain Dev 36: 176–179. [DOI] [PubMed] [Google Scholar]
- Roman HB, Hirschberger LL, Krijt J, Valli A, Kožich V, Stipanuk MH (2013). The cysteine dioxgenase knockout mouse: altered cysteine metabolism in nonhepatic tissues leads to excess H2S/HS − production and evidence of pancreatic and lung toxicity. Antioxid Redox Signal 19: 1321–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salman MS, Ackerley C, Senger C, Becker L (2002). New insights into the neuropathogenesis of molybdenum cofactor deficiency. Can J Neurol Sci 29: 91–96. [DOI] [PubMed] [Google Scholar]
- Salmina AB, Komleva YK, Szijártó IA, Gorina YV, Lopatina OL, Gertsog GE et al (2015). H2S‐ and NO‐signaling pathways in Alzheimer's amyloid vasculopathy: Synergism or antagonism? Front Physiol 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sass JO, Gunduz A, Araujo Rodrigues Funayama C, Korkmaz B, Dantas Pinto KG, Tuysuz B et al (2010). Functional deficiencies of sulfite oxidase: differential diagnoses in neonates presenting with intractable seizures and cystic encephalomalacia. Brain Dev 32: 544–549. [DOI] [PubMed] [Google Scholar]
- Sass JO, Kishikawa M, Puttinger R, Reiss J, Erwa W, Shimizu A et al (2003). Hypohomocysteinaemia and highly increased proportion of S‐sulfonated plasma transthyretin in molybdenum cofactor deficiency. J Inherit Metab Dis 26: 80–82. [DOI] [PubMed] [Google Scholar]
- Sato H, Shiiya A, Kimata M, Maebara K, Tamba M, Sakakura Y et al (2005). Redox imbalance in cystine/glutamate transporter‐deficient mice. J Biol Chem 280: 37423–37429. [DOI] [PubMed] [Google Scholar]
- Schwahn BC, Van Spronsen FJ, Belaidi AA, Bowhay S, Christodoulou J, Derks TG et al (2015). Efficacy and safety of cyclic pyranopterin monophosphate substitution in severe molybdenum cofactor deficiency type A: a prospective cohort study. Lancet 6736: 1–9. [DOI] [PubMed] [Google Scholar]
- Schwarz G (2016). Molybdenum cofactor and human disease. Curr Opin Chem Biol 31: 179–187. [DOI] [PubMed] [Google Scholar]
- Schwarz G, Belaidi AA (2013). Molybdenum in human health and disease. Met Ions Life Sci 13: 415–450. [DOI] [PubMed] [Google Scholar]
- Schwarz G, Mendel RR, Ribbe MW (2009). Molybdenum cofactors, enzymes and pathways. Nature 460: 839–847. [DOI] [PubMed] [Google Scholar]
- Schwarz G, Santamaria‐Araujo JA, Wolf S, Lee HJ, Adham IM, Gröne HJ et al (2004). Rescue of lethal molybdenum cofactor deficiency by a biosynthetic precursor from Escherichia coli. Hum Mol Genet 13: 1249–1255. [DOI] [PubMed] [Google Scholar]
- Schwarz G, Veldman A (2014). Molybdenum cofactor disorders BT In: Blau N, Duran M, Gibson KM, Dionisi Vici C. (eds). Physician's Guide to the Diagnosis, Treatment, and Follow‐Up of Inherited Metabolic Diseases. Springer Berlin Heidelberg: Berlin, Heidelberg, pp. 191–203. [Google Scholar]
- Shen H, Damcott C, Shuldiner SR, Chai S, Yang R, Hu H et al (2011). Genome‐wide association study identifies genetic variants in GOT1 determining serum aspartate aminotransferase levels. J Hum Genet 56: 801–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Koike S, Tanaka M, Ishigami‐Yuasa M, Kimura Y, Ogasawara Y et al (2013). A novel pathway for the production of hydrogen sulfide from D‐cysteine in mammalian cells. Nat Commun 4: 1366–1367. [DOI] [PubMed] [Google Scholar]
- Shih VE, Abroms IF, Johnson JL, Carney M, Roseann B, Robb RM et al (1977). Sulfite oxidase deficiency. Biochemical and clinical investigations of a hereditary metabolic disorder in sulfur metabolism. N Engl J Med . [DOI] [PubMed] [Google Scholar]
- Singer TP, Kearney EB (1956). Intermediary metabolism of L‐cysteinesulfinic in animal tissues' acid originates from cysteine and that it is on the main pathway of the oxi‐ isotope studies (5), and its central role in the conversion of sulfur amino tion to alanine and SO2 in the pre.
- Stipanuk MH, Ueki I (2011). Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 34: 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, D'Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E et al (2004). Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am J Hum Genet 74: 239–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C et al (2009). Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat Med 15: 200–205. [DOI] [PubMed] [Google Scholar]
- Tiranti V, Zeviani M (2013). Altered sulfide (H(2)S) metabolism in ethylmalonic encephalopathy. Cold Spring Harb Perspect Biol 5: a011437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touati G, Rusthoven E, Depondt E, Dorche C, Duran M, Heron B et al (2000). Dietary therapy in two patients with a mild form of sulphite oxidase deeficiency. Evidence for clinical and biological improvement. J Inherit Metab Dis 23: 45–53. [DOI] [PubMed] [Google Scholar]
- Ubuka T, Umemura S, Yuasa S, Kinuta M, Watanabe K (1978). Purification and characterization of mitochondrial cysteine aminotransferase from rat liver. Physiol Chem Phys 10: 483–500. [PubMed] [Google Scholar]
- Ueki I, Roman HB, Valli A, Fieselmann K, Lam J, Peters R et al (2011). Knockout of the murine cysteine dioxygenase gene results in severe impairment in ability to synthesize taurine and an increased catabolism of cysteine to hydrogen sulfide. Am J Physiol Endocrinol Metab 301: E668–E684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueland PM, Mansoor MA, Guttormsen AB, Müller F, Aukrust P, Refsum H et al (1996). Reduced, oxidized and protein‐bound forms of homocysteine and other aminothiols in plasma comprise the redox thiol status – a possible element of the extracellular antioxidant defense system. J Nutr 126: 1281–1284. [DOI] [PubMed] [Google Scholar]
- Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM et al (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat Commun 4: 1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldman A, Santamaria‐Araujo JA, Sollazzo S, Pitt J, Gianello R, Yaplito‐Lee J et al (2010). Successful treatment of molybdenum cofactor deficiency type A with cPMP. Pediatrics 125: e1249–e1254. [DOI] [PubMed] [Google Scholar]
- Vincent AS, Lim BG, Tan J, Whiteman M, Cheung NS, Halliwell B et al (2004). Sulfite‐mediated oxidative stress in kidney cells. Kidney Int 65: 393–402. [DOI] [PubMed] [Google Scholar]
- Viscomi C, Burlina AB, Dweikat I, Savoiardo M, Lamperti C, Hildebrandt T et al (2010). Combined treatment with oral metronidazole and N‐acetylcysteine is effective in ethylmalonic encephalopathy. Nat Med 16: 869–871. [DOI] [PubMed] [Google Scholar]
- Vitvitsky V, Dayal S, Stabler S, Zhou Y, Wang H, Lentz SR et al (2004). Perturbations in homocysteine‐linked redox homeostasis in a murine model for hyperhomocysteinemia. Am J Physiol Regul Integr Comp Physiol 287: R39–R46. [DOI] [PubMed] [Google Scholar]
- Wallace JL, Caliendo G, Santagada V, Cirino G (2010). Markedly reduced toxicity of a hydrogen sulphide‐releasing derivative of naproxen (ATB‐346). Br J Pharmacol 159: 1236–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Vaughan D, Dicay M, MacNaughton WK, de Nucci G (2017). Hydrogen sulfide‐releasing therapeutics: translation to the clinic. Antioxid Redox Signal 28 ars.2017.7068. [DOI] [PubMed] [Google Scholar]
- Wang J, Krizowski S, Fischer‐Schrader K, Niks D, Tejero J, Sparacino‐Watkins C et al (2015). Sulfite oxidase catalyzes single‐electron transfer at molybdenum domain to reduce nitrite to nitric oxide. Antioxid Redox Signal 23: 283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR et al (1995). Mice deficient in cystathionine beta‐synthase: animal models for mild and severe homocyst (e)inemia. Proc Natl Acad Sci U S A 92: 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedmann R, Onderka C, Wei S, András Szijártó I, Miljkovic J, Femic A et al (2016). Improved tag‐switch method reveals that thioredoxin acts as depersulfidase and controls the intracellular levels of protein persulfidation. Chem Sci 7: 3414–3426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav PK, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR et al (2016). Biosynthesis and reactivity of cysteine persulfides in signaling. J Am Chem Soc 138: 289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav PK, Yamada K, Chiku T, Koutmos M, Banerjee R (2013). Structure and kinetic analysis of H2S production by human mercaptopyruvate sulfurtransferase. J Biol Chem 288: 20002–20013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalamanchili C, Smith MD (2008). Acute hydrogen sulfide toxicity due to sewer gas exposure. Am J Emerg Med 26: 518.e5–518.e7. [DOI] [PubMed] [Google Scholar]
- Yamada H, Akahoshi N, Kamata S, Hagiya Y, Hishiki T, Nagahata Y et al (2012). Methionine excess in diet induces acute lethal hepatitis in mice lacking cystathionine γ‐lyase, an animal model of cystathioninuria. Free Radic Biol Med 52: 1716–1726. [DOI] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ‐lyase. Science (80‐.) 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Long YC (2016). Crosstalk between cystine and glutathione is critical for the regulation of amino acid signaling pathways and ferroptosis. Sci Rep 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Vincent AS, Halliwell B, Wong KP (2004). A mechanism of sulfite neurotoxicity: direct inhibition of glutamate dehydrogenase. J Biol Chem 279: 43035–43045. [DOI] [PubMed] [Google Scholar]
- Ziosi M, Di Meo I, Kleiner G, Gao X‐H, Barca E, Sanchez‐Quintero MJ et al (2016). Coenzyme Q deficiency causes impairment of the sulfide oxidation pathway. EMBO Mol Med 199: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou C‐G, Banerjee R (2005). Homocysteine and redox signaling. Antioxid Redox Signal 7: 547–559. [DOI] [PubMed] [Google Scholar]