

Abstract
Background and Purpose
Homocystinurias are rare genetic defects characterized by altered fluxes of sulfur compounds including homocysteine and cysteine. We explored whether the severely perturbed sulfur amino acid metabolism in patients with homocystinurias affects the metabolism of hydrogen sulfide.
Experimental Approach
We studied 10 treated patients with a block in the conversion of homocysteine to cysteine due to cystathionine β‐synthase deficiency (CBSD) and six treated patients with remethylation defects (RMD) and an enhanced flux of sulfur metabolites via transsulfuration. Control groups for CBSD and RMD patients consisted of 22 patients with phenylketonuria on a low‐protein diet and of 12 healthy controls respectively. Plasma and urine concentrations of selected sulfur compounds were analysed by HPLC and LC–MS/MS.
Key Results
Patients with CBSD exhibited plasma concentrations of monobromobimane‐detected sulfide similar to appropriate controls. Urinary homolanthionine and thiosulfate in CBSD were increased significantly 1.9 and 3 times suggesting higher hydrogen sulfide synthesis by γ‐cystathionase and detoxification respectively. Surprisingly, patients with RMD had significantly lower plasma sulfide levels (53 and 64% of controls) with lower sulfite concentrations, and higher taurine and thiosulfate levels suggesting enhanced cysteine oxidation and hydrogen sulfide catabolism respectively.
Conclusion and Implications
The results from this study suggest that severe inherited defects in sulfur amino acid metabolism may be accompanied by only moderately perturbed hydrogen sulfide metabolism and lends support to the hypothesis that enzymes in the transsulfuration pathway may not be the major contributors to the endogenous hydrogen sulfide pool.
Linked Articles
This article is part of a themed section on Chemical Biology of Reactive Sulfur Species. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.4/issuetoc
Abbreviations
- CARS2
cysteinyl tRNA synthetase 2
- CBS
cystathionine β‐synthase
- CBSD
cystathionine β‐synthase deficiency
- CPERS
cysteine persulfide synthases
- CSE
γ‐cystathionase
- Cys
cysteine
- Cystat
cystathionine
- FE
fractional excretion
- Hcy
homocysteine
- Hlanth
homolanthionine
- Hyp
hypotaurine
- Lanth
lanthionine
- MBB
monobromobimane
- PKU
phenylketonuria
- RMD
remethylation defect
- SDO
sulfur dioxygenase
- tCys
total cysteine
- tHcy
total homocysteine
- Tau
taurine
- TST
thiosulfate transferase
Introduction
Hydrogen sulfide (H2S) has been known for a long time as a toxic gas with a typical smell of rotten eggs. Since the first description of its endogenous production and signalling properties, the number of publications related to the biological functions of H2S increased tremendously, especially in the past few decades. Endogenously‐produced H2S is involved in a number of physiological processes and in the development of human diseases, for example, in neuronal function (Paul and Snyder, 2018), in the cardiovascular system (Pan et al., 2017), in the gastrointestinal mucosa (Wallace et al., 2018) and in kidney disease (Kasinath et al., 2018). Hence, H2S metabolic pathways have been considered as emerging therapeutic targets in various conditions (Yang et al., 2017; Wallace and Wang, 2015; Szabo, 2016). There are many more areas where H2S‐mediated events have been demonstrated to play fundamental roles, and the landmark discoveries relating to this small signalling molecule have recently been reviewed (Szabo, 2018).
Biosynthesis of H2S is linked to the metabolism of sulfur‐containing amino acids, specifically to cysteine (L‐Cys) and homocysteine (Hcy), which are important metabolites in the Hcy transsulfuration pathway (see Figure 1). In human tissues, H2S is widely accepted to be mainly produced from L‐Cys. Contributing pathways using L‐Hcy, and possibly D‐cysteine and methanethiol, were also suggested (Pol et al., 2018). The proposed syntheses of H2S via the transsulfuration pathways use the cytosolic cystathionine β‐synthase (CBS) and γ‐cystathionase (CSE) enzymes or the mitochondrial 3‐mercaptopyruvate sulfurtransferase. Due to the relaxed substrate specificity of CBS and CSE, H2S is synthesized from Cys and Hcy via several alternative reactions (Kabil and Banerjee, 2014), some of which produce thioethers lanthionine (Lanth), cystathionine (Cystat) and homolanthionine (Hlanth). Furthermore, a recent report revealed that a mitochondrial enzyme, cysteinyl tRNA synthetase (CARS2), plays a major role in converting Cys into Cys‐per/polysulfide species, and this involves the catalysis of translational protein Cys polysulfidation (Akaike et al., 2017). In addition, Cys‐per/polysulfides were shown to be efficiently reduced by the thioredoxin and glutathione (GSH) enzyme machineries using NADPH as a source of electrons (Doka et al., 2016). These coupled enzymatic pathways could therefore represent a major source of H2S production in cells. It is important to emphasize that H2S and many of its metabolites (such as inorganic and organic persulfides and polysulfides, thiosulfate or metal‐coordinated sulfur species) are highly reactive molecules, thus forming a dynamic system for H2S storage and release (Nagy et al., 2014). Furthermore, beside the above‐mentioned enzyme‐catalysed reactions, sulfur species can also interconvert via spontaneous fast dynamic equilibria (Bogdandi et al., 2019). Therefore, the nature of the effector molecule in a particular condition is often difficult to assess. In addition to these classical substrates for enzymatic production, other unconventional endogenous and exogenous sources of H2S have recently been proposed (Olson, 2018).
Figure 1.
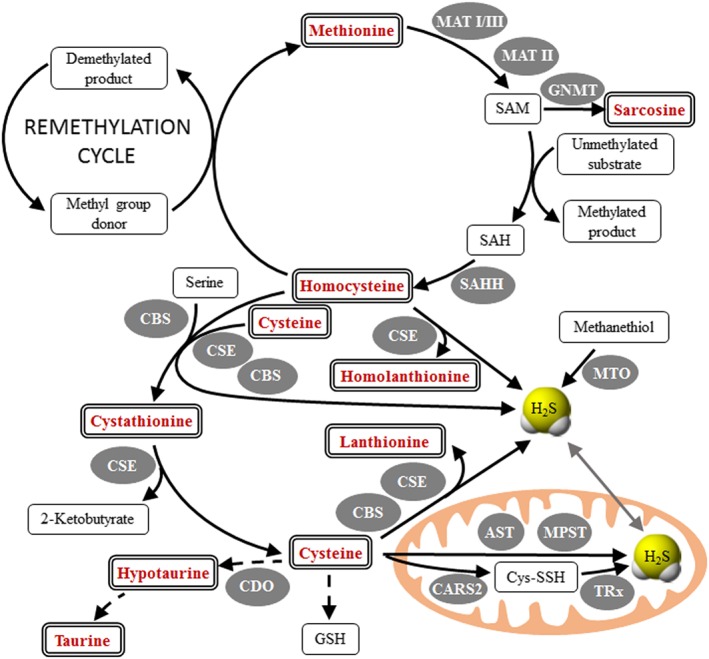
Metabolism of sulfur amino acids and H2S synthesis. The typical intake of the sum of methionine and Cys in adults ranges between 1.8 and 6.8 g·day−1 (Nimni et al., 2007). Methionine is converted by methionine adenosyltransferases (MAT I/III and MATII) to S‐adenosylmethionine (SAM), which donates the methyl group in numerous biologically important methylation reactions. S‐adenosylhomocysteine (SAH) is cleaved to homocysteine (Hcy) and adenosine by S‐adenosylhomocysteine hydrolase (SAHH). Hcy can be metabolized back to methionine by methionine synthase (MTR) with the help of methionine synthase reductase (MTRR) in a folate‐ and cobalamin‐dependent remethylation pathway, or by liver‐dependent betaine‐homocysteine methyltransferase (BHMT) using betaine as a methyl‐group donor. Alternatively, the sulfur of Hcy can be irreversibly metabolized to sulfate by the transsulfuration pathway: Hcy is condensed with serine to form cystathionine (Cystat), which is subsequently cleaved to form Cys, ammonia and α‐ketobutyrate; these reactions are catalysed by CBS and γ‐cystathionase (CSE) respectively. These two cytosolic enzymes can also use Cys and/or Hcy to synthesize H2S. H2S can also be synthesized in mitochondria from Cys by aspartate amino transferase (AST) and mercaptopyruvate sulfurtransferase and from methanethiol by methanethiol oxidase (MTO). Furthermore, cysteinyl t‐RNA synthetase enzymes (e.g. the mitochondrial CARS2 or other CPERS) use Cys to produce Cys‐persulfide (CysSSH) and polysulfide species in a PLP‐dependent manner. These polysulfides are reduced by the thioredoxin (TRx) and GSH systems using NADPH to give H2S and Cys as final products. Excess Cys can be further converted into taurine in a series of reactions – including cysteine dioxygenase (CDO). Mitochondrial oxidation of H2S involves several steps yielding thiosulfate, sulfite and finally sulfate; for details, see Figure 2. In this study, we determined H2S and additional metabolites (shown in bold double framed rectangles). Enzymes catalysing individual reactions are shown in shaded ovals.
Degradation of H2S is located mostly in the mitochondria (see Figure 2) and starts by persulfidation of sulfide : quinone oxidoreductase (SQOR). Two alternative pathways for further oxidation of persulfidated SQR were proposed involving sulfur dioxygenase (SDO), thiosulfate transferase (TST) and sulfite oxidase (SO) with formation of intermediate metabolites sulfite, thiosulfate and thiocyanate, and of the final H2S oxidation product sulfate (Augustyn et al., 2017; Olson, 2018; Kohl et al., 2019).
Figure 2.
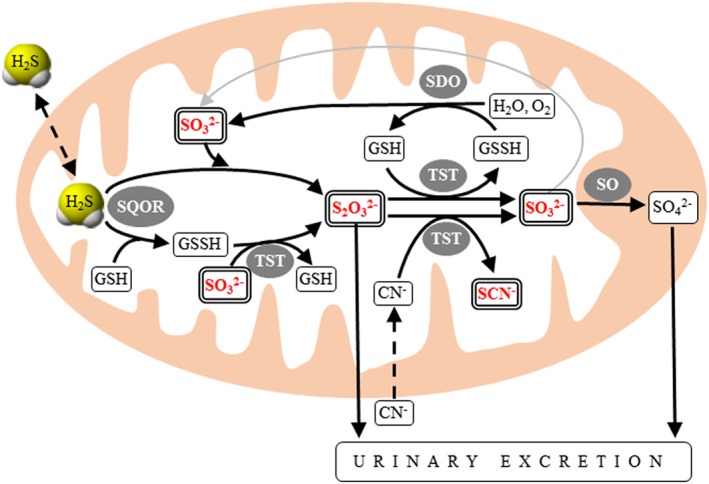
Model of mitochondrial oxidation of H2S. Several models of mitochondrial H2S oxidation have been proposed (Augustyn et al., 2017; Carter and Morton, 2016; Libiad et al., 2018; Melideo et al., 2014; Olson, 2018). This scheme summarizes the alternative pathways of H2S oxidation and interconversions of selected sulfur compounds within mitochondria. H2S is transferred to either GSH and/or sulfite by sulfide : quinone oxidoreductase thus forming GSH persulfide (GSSH) and/or thiosulfate respectively. Thiosulfate transferase (TST; also known as rhodanase) is involved in several bi‐directional reactions that interconvert sulfite and thiosulfate, or GSH and its persulfide. In addition, TST catalyses the detoxification of cyanide to thiocyanate. Persulfidated GSH is oxidized by SDO to sulfite, which is ultimately oxidized by a molybdenum cofactor‐containing enzyme sulfite oxidase to sulfate. In this study, we determined H2S and additional metabolites (shown in bold double framed rectangles). Enzymes catalysing individual reactions are shown in shaded ovals.
Inborn errors of metabolism represent a heterogeneous group of at least 700 rare diseases, which are caused by inherited deficiencies of enzymes, transporters or other gene products (Morava et al., 2015; Zschocke, 2016). Studies of patients with these genetic disorders and extreme biochemical phenotype were instrumental in understanding the physiological function of various metabolic pathways. Homocystinurias are inborn errors of sulfur amino acid metabolism, which are characterized by the accumulation of homocysteine in the body due to a blocked transsulfuration or remethylation pathway; reviews and guidelines on diagnosis and treatment of homocystinurias have been recently published (Huemer et al., 2017; Morris et al., 2017; Kožich et al., 2016). In the present study, we examined the metabolism of sulfur species in patients with two different types of homocystinurias. The first group consisted of patients with the severe form of complete CBS deficiency (CBSD), which is not amenable to the chaperoning effect of pyridoxine, while the second group included patients with remethylation defects (RMD). These disorders manifest by a combination of neuropsychiatric symptoms (cognitive impairment, behavioural problems, seizures and white matter abnormalities), thromboembolism and connective tissue abnormalities (marfanoid features and lens dislocation, only in CBSD). However, biochemical abnormalities in these disorders differ considerably. In CBSD, homocysteine and methionine accumulate while metabolites below the CBS block (i.e. cystathionine and cysteine) are decreased. In contrast, the RMD are characterized by decreased production of Met and S‐adenosylmethionine, an accumulation of Hcy and an increased flux of sulfur compounds through the transsulfuration pathway as indicated by the accumulation of cystathionine. CBSD is managed by administration of pharmacological doses of pyridoxine in the vitamin B6 responsive form of the disease, while patients not responding to pyridoxine require a protein‐restricted diet with amino acid supplements and/or betaine administration. The RMD do not require protein restriction, and patients are treated with betaine, hydroxocobalamin and/or methyltetrahydrofolate; however, the efficacy of this therapy is lower than that in the CBSD.
Despite the large number of studies, the contribution of CBS and CSE into the overall pool of H2S synthesis is only partially understood, and the relative input of Cys and Hcy under physiological and pathological conditions is largely unknown. In patients with CBSD, one of the cytosolic H2S producing enzymes is inactive, and this block in the transsulfuration pathway leads to the accumulation of Hcy and depletion of Cys. In contrast, patients with RMD have preserved activity of CBS and CSE, and the flux through the transsulfuration is enhanced. The differences in enzymatic activities and metabolite fluxes between these two types of homocystinurias may thus help to differentiate the role of CBS and CSE, and of substrates in H2S metabolism. Since the assessment of H2S metabolism in humans using various analytical methods is notoriously intriguing (Nagy et al., 2014; Olson et al., 2014), in a previous study, we measured indirect indices of H2S synthesis, namely, the levels of thioethers (Kozich et al., 2016), and observed, in archived plasma samples of patients, changes compatible with enhanced H2S production. A direct demonstration of disturbed H2S concentrations and of its catabolic products was not possible in archived samples due to pre‐analytical obstacles; thus, in the present prospective study, we analysed sulfur compounds relevant to H2S metabolism in fresh samples obtained from treated patients with CBSD and RMD.
Methods
Patients and controls
In this study, we enrolled a total of 51 subjects from one centre. The patient cohort consisted of 10 cases with classical homocystinuria caused by pyridoxine non‐responsive CBSD (CBSD group) and of six cases with RMD (RMD group) due to cblG (n = 4), cblE (n = 1) and cblJ (n = 1) defects respectively. The primary genetic defect was confirmed by enzymatic and molecular genetic analyses in all patients. The CBSD group involved five males and five females with a median age of 22 years (range 0.5–33 years); all patients received a low‐protein diet (median 8 mg methionine·kg−1·day−1, range 4–15) with amino acid mixtures free of methionine and enriched with cysteine (Cys content in these amino acid mixtures is about two times higher than in mixtures for treatment of PKU); eight patients also received betaine (median 117 mg·kg−1·day−1 p.o., range 41–214); and nine patients also received pyridoxine although they did not meet the criteria of pyridoxine responsiveness. Plasma and urine samples were obtained from 10 and nine CBSD patients respectively. In the RMD group, the sex ratio was 3:3 for males to females and the median age was 5 years (age range 3–24 years); samples of plasma and urine were available from all patients in this group. All patients in the RMD group were on a regular diet without any protein restriction; patients with cblG and cblE defects were treated with betaine (median 157 mg·kg−1·day−1 p.o., range 115–220), hydroxycobalamin (median 48 μg·kg−1·day−1 i.m., range 14–58) and folate and/or calcium folinate p.o.; and one patient with cblJ defect was treated only with hydroxycobalamin (48 μg·kg−1·day−1 i.m.).
Two different control groups were established to match the different dietary treatment of patients with homocystinurias. The size of the control groups was about twice as large as the patient groups. Patients with classical phenylketonuria (C‐PKU control group) served as a control group for patients with CBSD as all PKU patients were on a low‐protein diet (median 11 mg phenylalanine·kg−1·day−1, range 5–43) with amino acid supplementation containing cysteine and methionine, and none of the patients received sapropterine therapy. The PKU control group consisted of 23 patients (15 males and eight females) with median age 31 years (range 1–50 years); plasma and urine samples were available from 22 and 21 patients respectively. Healthy adult controls on a regular diet served as controls (C‐H control group) for patients with RMD; this control group consisted of 13 healthy adults (six males and seven females) with median age of 41 years (range 30–57); one female individual was excluded from the control group due to elevated total Hcy (tHcy).
Plasma samples from patients and controls were collected into lithium‐heparin vacutainer tubes with gel (BD Vacutainer LH PST II) 2–3 h after the last meal and kept on ice/water slush. Blood was processed within 10–30 min after collection, and plasma was separated by centrifugation at 2000× g for 5 min and immediately processed or frozen at −85°C. Unprocessed plasma and urine samples were stored at −85°C for up to 6 weeks prior to analysis. Patient and control samples were collected between October and December 2017, and all study subjects gave a written informed consent. This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Committee of the General University Hospital in Prague (approval No. Grant COST 35/13 and No. AZV 71/15).
Hydrogen sulfide determination
Plasma sulfide levels were determined using two slightly different methods. Both methods utilized monobromobimane (MBB) derivatization followed by HPLC separation and fluorescent quantitation of the sulfide dibimane (SDB) product based on previous publications (Wintner et al., 2010; Shen et al., 2012). All procedures were carried out in the dark to avoid photo‐induced decomposition of MBB derivatives.
In Method A, 10 μL of plasma sample was mixed with 10 μL 50 mmol·L−1 HEPES buffer with pH 8.0 and 10 μL of 10 mmol·L−1 MBB dissolved in acetonitrile. The reaction mixture was placed on an orbital shaker for 10 min, and then 100 μL ethylacetate was added to extract the product SDB and MBB and to quench the reaction. For proper extraction, the microcentrifuge tubes were placed on a tube rotator for 10 min and then centrifuged at 1300× g for 7 min. Fifty microlitres of the organic supernatant were removed and evaporated using a vacuum centrifuge concentrator; 10 μL acetonitrile was added and evaporated again to remove residual ethylacetate; the tubes were stored at −20°C until measurement. Prior to injection, the solid samples were dissolved in 250 μL of acetonitrile.
A Thermo Ultimate 3000 HPLC system equipped with a fluorescent detector was used with an Agilent Zorbax Eclipse XDB‐C18 column (250 × 4.6 mm, 5 μm) for the separation of the analytes. The fluorescence detector was set at 390 nm (excitation wavelength) and 475 nm (emission wavelength). Ten microlitres of the derivatized sample were injected and eluted using a 35‐min‐long gradient profile consisting of 0.1% of formic acid (A) and 0.1% of formic acid in acetonitrile (B) at a flow rate of 1 mL·min−1. The gradient profile begun with 85% A followed by a linear increase to 30% B over 5 min and an isocratic step with 30% B for 10 min. The linear gradient continued with a decrease to 22% B over 3 min, followed by a rapid increase to 70% B over 5 min and a further increase to 90% B over 3 min. The column was then regenerated with 85% A for another 7 min.
In Method B, 25 μL of plasma sample was mixed with 66 μL of the MBB working solution (10 μL of 100 mmol·L−1 MBB in acetonitrile was mixed with 650 μL of 200 mmol·L−1 HEPES buffer, pH 8.2). The derivatization reaction proceeded in the dark at room temperature for exactly 10 min; the derivatization was stopped by adding 5 μL 50% trichloroacetic acid to the samples under vigorous vortexing conditions. Samples were centrifuged (30 000× g for 5 min), and the supernatants were stored in the autosampler in dark vials at 5°C. For this method, we used the same chromatography set‐up; only the gradient was shortened to 21 min. The gradient profile begun with 85% A followed by a linear increase to 30% B over 3 min and an isocratic step with 30% B for 6 min. The linear gradient continued with a decrease to 22% B over 1.8 min, followed by a rapid increase to 70% B over 3 min and further increase to 90% B over 1.8 min. The column was then regenerated with 85% A for another 4.2 min.
Concentrations of H2S were estimated based on calibration curves that were established using derivatized standard sulfide solutions in water together with calibration blanks (we used deionized water instead of the sulfide solution). First, a 50 μmol·L−1 derivatized stock solution was prepared, which was then used to make the standards in the concentration range of 1–5 μmol·L−1 by a serial dilution using the calibration blank solutions.
Determination of organic sulfur compounds
Amino acids methionine and sarcosine, and thioethers Cystat, Hlanth and Lanth were determined by LC–MS/MS using commercially available kit for amino acid analysis (EZ:faast, Phenomenex, Torrance, USA) as described in detail previously (Kozich et al., 2016). The LC–MS/MS analyses were performed on LC–MS/MS system consisting of the Agilent 1100 Series LC System coupled with API 4000 triple quadrupole mass spectrometer.
Plasma and urinary total aminothiols Cys, Hcy, cysteinyl‐glycine, γ‐glutamylcysteine and GSH were determined by reversed‐phase HPLC with fluorescent detection after derivatization with ammonium 7‐fluorobenzo‐2‐oxa‐1,3‐diazole‐4‐sulfonate. The reduction of disulfides and protein bound aminothiols was achieved with tris(2‐carboxyethyl)phosphine as described previously (Krijt et al., 2001).
Taurine (Tau) and hypotaurine (Hyp) were determined by a modification of the LC–MS/MS method published previously (Awwad et al., 2016). The plasma samples (50 μL) were deproteinized with 150 μL of acetonitrile/methanol/formic acid mixture (84.9:15:0.1) containing 20 μmol·L−1 of the isotope labelled internal standards Tau‐d4 and citrulline‐d4 (used as internal standard for Hyp). Urine samples were diluted with water to a creatinine concentration of 1 mmol·L−1, and 50 μL of the diluted sample was mixed with 150 μL of acetonitrile/methanol/formic acid (84.9:15:0.1) containing 20 μmol·L−1 of the isotope‐labelled internal standards as above. The deproteinized urinary or plasma samples were centrifuged at 10 000× g for 5 min, and 2 μL of the supernatants were injected into the LC–MS/MS system. The metabolites were separated on Luna HILIC column (100 × 2 mm, 3 μm particles, Phenomenex, USA) using a gradient elution profile between 0.1% formic acid in acetonitrile (A) and 0.1% formic acid in water (B). The gradient profile begun with 90% A for 1.5 min followed by a linear increase to 60% B over 2 min and an isocratic step with 60% B for 2 min. The column was then regenerated with 90% A for another 6 min. The flow rate was 0.3 mL·min−1. Detection of both analytes was carried out using positive electrospray ionization technique and selected multiple reaction monitoring. The precursor→product mass transitions for Tau, Hyp and their respective internal standards Tau‐d4 and citrulline‐d4 were 126.0 → 108.0, 109.8 → 92 and 130.0 → 112.0, 180 → 163 respectively.
Determination of sulfite, thiosulfate and thiocyanate
Inorganic anions sulfite, thiosulfate and thiocyanate (SO3 2−, S2O3 2−, SCN−) were determined by a modification of the HPLC method published previously (Ji et al., 1995). The method was optimized for determination of all three anions (data not shown) and performed in series containing up to 25 samples as follows: 50 μL of plasma or urine sample was mixed with 60 or 120 μL, respectively, of the MBB working solution (100 μL of 100 mmol·L−1 MBB in acetonitrile was mixed with 5900 μL of buffer containing 14.3 mmol·L−1 EDTA and 14.3 mmol·L−1 ammonium bicarbonate, pH 8.5). The derivatization reaction proceeded in the dark at room temperature for 2 h, and the derivatization was stopped by the addition of 15 μL or 24 μL of 70% sulfosalicylic acid to the plasma or urine sample respectively. After 10 min on ice, samples were centrifuged (10 000× g at 5°C for 10 min) and clear supernatants were stored in the autosampler in dark vials at 5°C; 10 μL of the samples were injected onto the HPLC column.
Calibration samples were prepared by spiking pooled remnants of plasma or urine samples remaining after routine biochemical analyses and processed as described above; two sets of calibration samples were injected at the beginning and the end of each series. For plasma analytes, the concentration ranges were 0.05–2.5 μmol·L−1 for sulfite and thiosulfate and 5–250 μmol·L−1 for thiocyanate; for urinary analytes, we used the addition of 0.05–5 μmol·L−1 for sulfite, 0.5–50 μmol·L−1 for thiosulfate and 2.5–250 μmol·L−1 for thiocyanate respectively. Blank samples for urine was the deionized water used to prepare reagents, while for heparinized plasma, we used water eluates of the empty sampling tubes.
The HPLC system consisted of Shimadzu LC‐20AD system with RF‐20AXs fluorescence detector. Analytes were separated on a Hyperchrome HPLC column (ProntoSIL 120–3‐C18 AQ 3.0 μm, 150 × 3.0 mm, Bischoff chromatography) using a gradient elution profile between 0.1% of formic acid (A) and 0.1% of formic acid in 40% of acetonitrile (B); flow rate was 0.7 mL·min−1. The gradient profile begins with 95% A for 0.5 min followed by a rapid increase to 20% B over 0.5 min and a slow linear increase to 28% B over 6 min. The linear gradient was promptly continued for 5 min up to 55% B, followed by another rapid increase to 100% B over 1 min and an isocratic step with 100% B for 3 min. The column was then regenerated with 95% A for another 5 min. The fluorescence detector was set at 390 nm (excitation wavelength) and 475 nm (emission wavelength).
Assessment of renal handling of metabolites
The fractional excretion (FE) of metabolites was calculated using the standard formula: FEmetabolite [%] = 100 × (Umetabolite × Pcreatinine) / (Pmetabolite × Ucreatinine), where U and P are the concentrations of analytes determined in simultaneously obtained urine and plasma samples respectively. Creatinine in urine and plasma was determined using the automated analyser Hitachi 902 and the commercially available kit ‘Kreatinin liquid 500’ (Jaffe method, Erba Lachema, Czech Republic) and ‘Kreatinin enzymaticky liquid 204’ (enzymatic method, Pliva Diagnostika, Czech Republic) respectively.
Statistical analyses
Samples were processed in a blinded fashion, and each series of analyses contained both control and patient samples. Data were not distributed normally and are shown as medians with the first and third quartiles. To compare the data between the patients and the respective control groups, we used Mann–Whitney U‐test with Benjamini–Hochberg correction for multiple testing; for this exploratory analysis, the data for plasma, urine and FE were considered independent sets of variables. All statistical analyses were performed using Statistica Cz, version 12 (StatSoft, Inc., USA), and the significance of the false discovery rate was set at <0.05.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Concentrations of substrates for H2S synthesis
Compared to appropriate controls, the median concentration of the alternative H2S precursor tHcy was increased significantly 14 and 28 times in plasma and urine of CBSD patients and 5.2 and 5.8 times in RMD patients respectively (see Figure 3). Median levels of the major substrate for H2S synthesis – total Cys (tCys) – were significantly lower in plasma of CBSD and RMD patients, reaching only 72 and 70% of controls, respectively; however the urinary excretion did not differ from controls significantly. The catabolic products of Cys oxidation Tau and Hyp were decreased to 77 and 67% of the median of controls in plasma of CBSD patients, while the urinary excretion did not differ. In contrast, the median Tau and Hyp concentration was 2.1 and 5.5 times higher in the urine and 1.3 and 1.8 times higher in plasma of RMD patients respectively. The data on other related metabolites including sarcosine as a marker of betaine treatment and on aminothiols relating to metabolism of GSH are shown in Supporting Information Tables S1 and S2.
Figure 3.
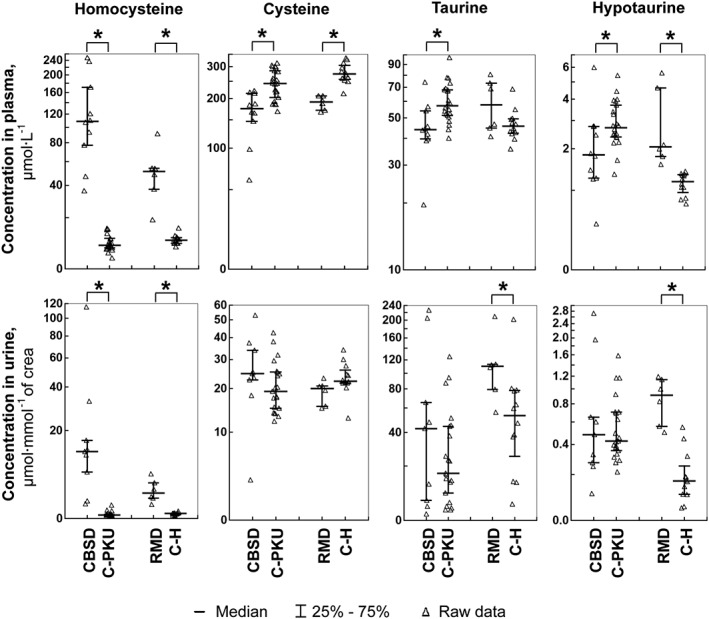
Concentrations of H2S precursors Hcy and Cys, and of Cys oxidation products Tau and Hyp. The left parts of the graphs show the distribution of individual data in the group of pyridoxine non‐responsive patients with CBSD (n = 10) who received a protein‐restricted diet with amino acid supplements enriched with Cys, and the appropriate control group of patients on low protein/amino acid mixture‐supplemented diet suffering from classical phenylketonuria (C‐PKU, n = 22). The right parts of the figures show data from patients with RMD (n = 6) and healthy controls (C‐H, n = 12). Solid bars indicate medians, whiskers represent the first and the third quartile, and asterisks denote significant difference between patients and the respective controls.
Concentrations of thioethers, by‐products of H2S synthesis
Thioethers are produced by CBS and CSE by condensation of serine, Cys or Hcy. H2S is synthesized only if two sulfur‐containing amino acids are condensed: reactions involving serine as one substrate, and Cys or Hcy as the second substrate produce water instead of H2S. The proportion of thioethers originating from reactions producing H2S versus reactions producing water is at present unknown. Cystat is synthesized by both CBS and CSE using serine and Hcy with elimination of water, or Cys and Hcy with production of H2S. As shown in Figure 4, patients with CBSD indeed exhibited significantly lower median plasma and urinary Cystat concentrations of 16 and 22% of controls respectively. In contrast, plasma and urine of patients with RMD showed significantly higher (2.9 and 2.2 times) median Cystat concentrations. Hlanth is synthesized only by CSE from two molecules of Hcy with concomitant release of H2S; this thioether was significantly elevated 1.9 times in both plasma and urine of patients with CBSD and 2.3 times in urine of patients with RMD. Finally, Lanth is a product of CBS‐ or CSE‐catalysed condensation of two Cys molecules with the release of H2S, or of serine and Cys with elimination of water; in patients with CBSD, there was a significant decrease of plasma Lanth to 51% of controls and in patients with RMD 1.6 times increase of urinary Lanth concentration.
Figure 4.
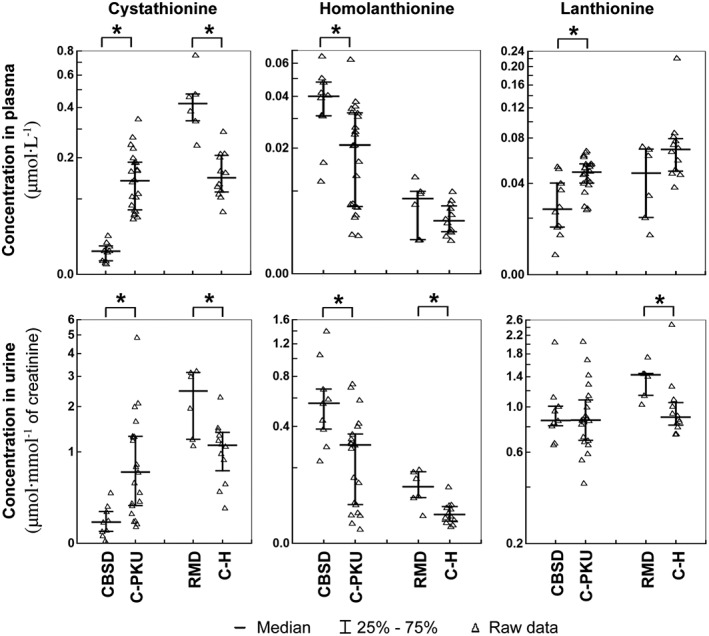
Concentrations of thioethers in plasma and urine. The left parts of the graphs show distribution of individual data in the group of pyridoxine non‐responsive patients with CBSD (n = 10) who received a protein‐restricted diet with amino acid supplements enriched with Cys, and the appropriate control group of patients on low protein/amino acid mixture‐supplemented diet suffering from classical phenylketonuria (C‐PKU, n = 22). The right parts of the figures show data from patients with RMD (n = 6) and healthy controls (C‐H, n = 12). Solid bars indicate medians, whiskers represent the first and the third quartile, and asterisks denote significant difference between patients and the respective controls.
H2S concentrations
We measured H2S levels in patient and control plasma samples using the MBB method (Nagy et al., 2014) under two different assay conditions (see Methods); data are summarized in Figure 5. Both methods indicated that sulfide levels in plasma of CBSD patients did not differ from the PKU control group. Surprisingly, the measured median sulfide concentrations in plasma of patients with RMD was significantly decreased to 53 and 64% of the median in healthy controls as measured by methods A and B respectively. Notice that there is considerable difference in the actual measured sulfide concentrations with the two methods. This is related to the fact that the MBB method uses a derivatization step of sulfide to yield a SDB product in a practically irreversible reaction. Therefore, upon total consumption of free sulfide, the biomolecule bound sulfide pool (likely to represent mostly polysulfides in plasma) slowly starts liberating sulfide due to the shift in the speciation equilibria (Bogdandi et al., 2019; Nagy et al., 2014). This means that the two methods measure different fractions from the bound sulfide pool (mostly due to the difference in the used MBB concentration, which affects the kinetics of sulfide release) beside free sulfide. Note that this shortcoming is not unique to MBB‐based methods. In fact, it is a general problem of all methodologies, which utilize an irreversible binding, evaporation or consumption of free sulfide in any way during its detection, which is in fact the case with most of the available techniques (Nagy et al., 2014) (see also Discussion).
Figure 5.
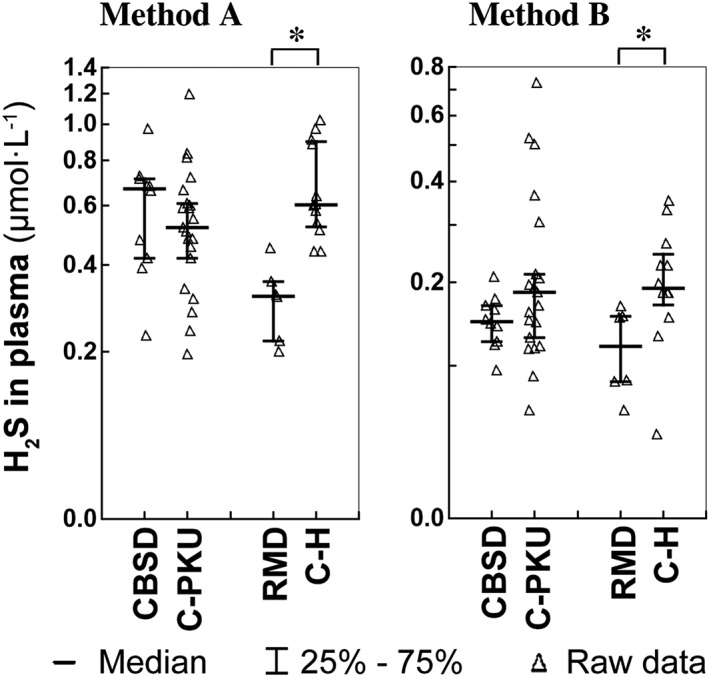
Concentration of H2S in plasma determined by Method A and Method B. The left parts of the graphs show distribution of individual data in the group of pyridoxine non‐responsive patients with CBSD (n = 10) who received a protein‐restricted diet with amino acid supplements enriched with Cys, and the appropriate control group of patients on low protein/amino acid mixture‐supplemented diet suffering from classical phenylketonuria (C‐PKU, n = 22). The right parts of the figures show data from patients with RMD (n = 6) and healthy controls (C‐H, n = 12). Solid bars indicate medians, whiskers represent the first and the third quartile, and asterisks denote significant difference between patients and the respective controls.
Concentrations of inorganic sulfur metabolites in the H2S oxidation pathway
Mitochondrial oxidation of H2S to sulfate produces a number of intermediates. Here, we determined the concentration of sulfite, thiosulfate and thiocyanate (see Figure 6). Sulfite plasma and urine concentrations in patients with CBSD did not differ statistically from PKU controls, while in patients with RMD, the median plasma sulfite was surprisingly decreased to 27% of controls. Median thiosulfate was increased non‐significantly in plasma and significantly three times in urine of patients with CBSD. Thiosulfate was also significantly increased in plasma and urine in patients with RMD (1.7 and 2.4 times compared to controls). Concentration of thiocyanate did not differ from controls in plasma and urine of patients with CBSD. However, median thiocyanate concentration was significantly decreased in plasma to only 18% of control values in patients with RMD.
Figure 6.
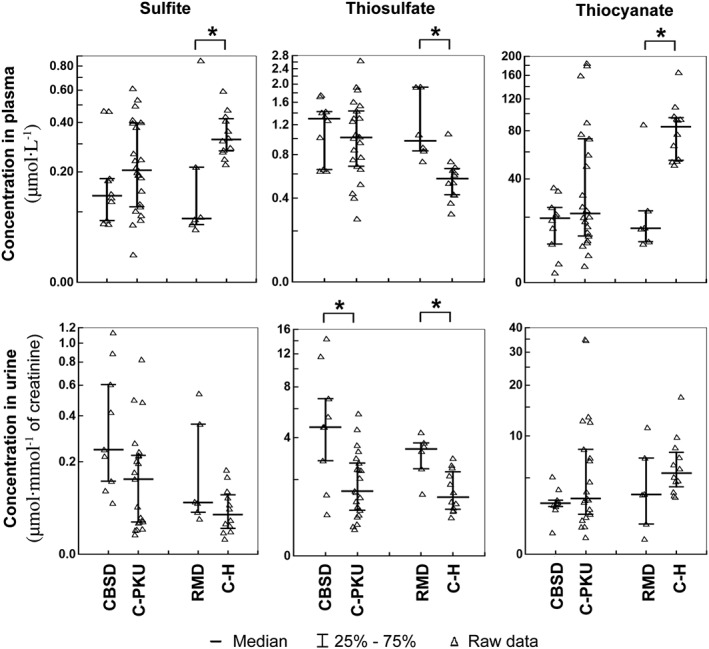
Concentrations of inorganic sulfur compounds. The left parts of the graphs show distribution of individual data in the group of pyridoxine non‐responsive patients with CBSD (n = 10) who received a protein‐restricted diet with amino acid supplements enriched with Cys, and the appropriate control group of patients on low protein/amino acid mixture‐supplemented diet suffering from classical phenylketonuria (C‐PKU, n = 22). The right parts of the figures show data from patients with RMD (n = 6) and healthy controls (C‐H, n = 12). Solid bars indicate medians, whiskers represent the first and the third quartile, and asterisks denote significant difference between patients and the respective controls.
Renal handling of metabolites
Since the study subjects provided both blood and urine, we were able to calculate the FE of analytes to assess the renal handling of metabolites and to compare whether reabsorption or active excretion of sulfur metabolites differs between patients and controls. FE <100% indicates active reabsorption while FE > 100% signifies an active excretion by tubular cells. There were no significant changes in FE of measured analytes between patients and controls indicating that the above described differences between the groups are most likely not caused by changes in renal handling of metabolites. Supporting Information Tables S1 and S2 show that amino acids are effectively reabsorbed in the kidney as demonstrated by the FE ranging between 0.2 and 2.6%. In contrast, the thioethers Cystat and Hlanth appear to be reabsorbed to much lesser extent as indicated by FE between 19.6 and 79.2%, and Lanth appears to be filtered without reabsorption or even actively excreted by the kidney with FE ranging from 96.5 to 184.2%. Surprisingly, sulfite and thiocyanate are also reabsorbed quite efficiently in the kidney while thiosulfate reached FE 10.2 to 20.8%.
Discussion
This study in patients with severe genetic defects in sulfur amino acid metabolism showed surprisingly only moderate changes in steady‐state plasma concentrations of MBB‐detected H2S despite the dramatic alterations in the substrates for H2S synthesis, namely, of Hcy and to a lesser degree of Cys. In patients with CBSD, plasma sulfide levels were similar to the appropriate controls, and paradoxically, a small but significant decrease in patients with RMD was detected. It is difficult to infer from these data whether H2S production is indeed normal or decreased in the tissues of these patients as other indices such as elevation of thioethers and of thiosulfate may indicate higher production of H2S by CBS/CSE and its catabolism by TST respectively. The increased thiosulfate with simultaneously unchanged or decreased plasma sulfide and/or sulfite is intriguing. Sulfite may be formed by SDO from GSH persulfide or by the activity of aspartate aminotransferase from Cys sulfinate, which is synthesized by CDO from Cys (Kohl et al., 2019). Since Cys sulfinate is further converted to Tau, assessment of Tau levels in homocystinurias may help dissecting the changes in the CDO‐related formation of sulfite as discussed below.
We hypothesized that the absent activity of CBS and decreased Cys availability due to blocked transsulfuration will result in lower production of H2S in reactions using Cys as a substrate with concomitant decrease of Cystat and Lanth, while the synthesis of H2S from grossly elevated Hcy will be enhanced at the CSE step. The lack of CBS activity is clearly evidenced by Hcy elevation in plasma and urine and by grossly decreased Cystat in plasma and urine. It is unknown how much Cystat originates from condensation of serine with Hcy versus condensation of Cys with Hcy (with H2S production), mostly by the preserved activity of CSE (Majtan et al., 2018). Cys depletion has been implicated in CBSD as an important mechanism responsible for connective tissue abnormalities (Majors and Pyeritz, 2000); in our study, the Cys depletion is evidenced by decreased concentration of Cys catabolic products Tau and Hyp in plasma and urine of CBSD patients despite the supplementation of the amino acid mixtures enriched with Cys. Congruently, the decreased Cystat and Lanth production in CBSD may in part reflect decreased H2S synthesis from a lowered Cys pool. In contrast, the enhanced formation of H2S from accumulating Hcy is demonstrated by higher production of Hlanth by CSE. In summary, this study showed indirect indices of both decreased synthesis of H2S using Cys as a substrate and increased synthesis using Hcy. The concentration of H2S in plasma reflects not only its production but also the rate of its catabolism. The major finding in the catabolic pathway of H2S is the significant elevation of thiosulfate in urine indicating a possible higher rate of H2S detoxification using sulfite in the TST reaction, which is indirectly supported also by non‐significantly decreased sulfite. Moreover, the slightly lower sulfite may be also due to low production of Cys sulfinate via CDO as evidenced by low Hyp and Tau and/or due to decreased formation of GSH persulfide (Augustyn et al., 2017) resulting from GSH depletion, which was shown in the liver of a CBS‐deficient mouse model (Maclean et al., 2010). To sum up, it is conceivable that patients with CBSD may produce slightly less H2S from Cys, which is compensated by production of H2S from Hcy and that this slightly higher H2S is further metabolized to thiosulfate (Figure 7A). However, the functionally severe knockout of CBS activity did not lead to major perturbations in H2S concentrations and lends support to a hypothesis that CBS may not be the major contributor to the H2S pool and that CARS2/cysteine persulfide synthases (CPERS) or other enzymes can play a more important role in H2S production.
Figure 7.
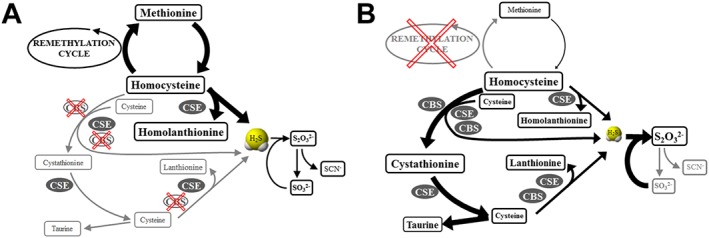
Models of H2S synthesis and catabolism in two types of homocystinuria. (A) Model of disturbed sulfur amino acid metabolism and unchanged H2S production in CBSD. (B) Model of decreased H2S steady‐state levels in patients with RMD due to putatively enhanced conversion to thiosulfate. Changes in steady‐state concentrations and fluxes are indicated by the sizes of the rectangles with metabolites and by the thickness of the arrows respectively.
In patients with RMD, the activity of both CBS and CSE is preserved; moreover, the supply of substrates for H2S synthesis is increased. Thus, we hypothesized that H2S production in RMD will be increased. The enhanced flux of sulfur compounds in RMD is demonstrated by significantly increased concentration of Cystat in plasma and urine. It may appear that patients with RMD exhibit Cys depletion due to observed lower plasma tCys concentrations. However, total plasma Cys is not a suitable marker to assess availability of Cys for subsequent metabolic conversions in patients with homocystinurias (Hargreaves et al., 2002). Plasma proteins harbour only a limited number of free thiol groups for binding of Hcy, Cys and other aminothiols. Thus, the excess of Hcy occupies a large proportion of these sites and consequently less Cys can be bound to plasma proteins leading to decreased concentration of protein‐bound and consequently of tCys. Indeed, a study of patients with severe vitamin B12 deficiency (and functional block in the remethylation pathway similar to the RMD patients reported here) exhibited grossly elevated free and protein‐bound Hcy and proportionally decreased protein‐bound and tCys despite an increased flux through the transsulfuration pathway (Mansoor et al., 1994).
The increased Cys production in RMD is indirectly supported also by higher concentrations of Tau and Hyp, originating from Cys oxidation by the likely up‐regulated CDO expression (Kohl et al., 2019). Despite this increased production of Hcy and Cys, we did not detect any significant changes in Lanth and Hlanth concentrations as by‐products of H2S synthesis in plasma, but we observed marginally elevated concentration in urine. The most surprising finding was the significantly lower detected plasma H2S concentration in patients with RMD compared to healthy controls; the model for these observations is proposed in Figure 7B. In RMD patients, the provision of Cys and Hcy for H2S synthesis is increased and the activity of both CBS and CSE is preserved, suggesting that deficient synthesis of H2S by these two enzymes is quite unlikely reason for low plasma H2S. It is perceivable that increased thiosulfate in plasma and urine may result from enhanced conversion of GSH persulfide and sulfite into thiosulfate via the actions of TST in cellular mitochondria (Kohl et al., 2019), which would entail decreased sulfide levels due to a drop in GSH‐persulfide (a proposed major contributor of the bound sulfide pools). Consistent with this hypothesis, we observed significantly decreased plasma sulfite levels. The markedly decreased sulfite in plasma contrasts with elevated Tau and Hyp, which indicates that the formation of sulfite from Cys sulfinate by aspartate aminotransferase may not be the major route for sulfite production. In summary, we did not find any compelling evidence in support of our hypothesis that H2S production in RMD is increased.
This study has several limitations that should be considered while interpreting data. First, the assessment of H2S homeostasis in body fluids is inherently difficult due to analytical intricacies discussed in the Introduction (Nagy et al., 2014; Olson et al., 2014). We carefully optimized and validated the method for MBB‐detectable H2S using short reaction times and other experimental conditions to minimize the extraction of H2S from the endogenous sulfur pool by MBB (Bogdandi et al., 2019) in a way that the results remain highly reproducible. Importantly, the applied two different conditions provided similar results, and we were able to detect elevated H2S concentrations in patients with ethylmalonic encephalopathy due to mutations in the ETHE1 gene (data not shown). Second, the sample size in this study was limited due to rarity of these diseases, and thus, analysis of additional patients is needed to replicate our findings. It is important to note that archived samples cannot be used for this type of study as H2S and sulfite are unstable. Third, we analysed metabolites in patients with homocystinurias receiving dietary and pharmacological treatment that may have changed the metabolite patterns. Thus, caution should be exercised in interpreting the data as treatment may have alleviated some of the biochemical changes that would be otherwise detectable in untreated patients; indeed, the Supporting Information Figure S1 shows that tHcy and Hlanth in untreated CBSD and RMD patients are substantially higher than in treated patients analysed in the present study. Moreover, dietary low‐protein treatment may introduce changes per se as suggested in slightly higher Hlanth levels in PKU patients than in healthy controls on regular diet. To summarize, it is conceivable that analysis of samples obtained from untreated newly diagnosed patients via international networks such as the E‐HOD consortium may show more pronounced perturbations of H2S metabolism than presented here. Finally, we were able to obtain only plasma and urine samples, and these biological fluids may not reflect appropriately the changes present in tissues as demonstrated previously in CBS‐deficient mice (Majtan et al., 2018); further insight into H2S homeostasis may be obtained by analysing H2S and related metabolites in the tissues of animal models. All these limitations show that the hypothesis of putatively altered H2S metabolism in homocystinurias is not yet fully resolved, and further studies in humans and animal models of these diseases are warranted.
Conclusions
In summary, this study showed grossly abnormal concentrations of many sulfur‐containing metabolites in patients with two types of homocystinurias but only slightly altered H2S levels in plasma. Whether changes in the availability of substrates for H2S synthesis and in the presence or absence of H2S‐synthesizing enzymes alters the homeostasis of this small signalling molecule in patient tissues and possibly contributes to the pathogenesis of homocystinurias remains at present unknown. It is also possible that the role of CBS in producing H2S may be less important than originally thought and that the H2S synthesizing enzymes create a network in which an insufficiency of one enzyme may be compensated for by the other enzymes including CPERS/CARS.
Author contributions
V.K. designed and coordinated the study, interpreted data and wrote the first draft of the manuscript; T.D. and M.K. developed the methods, analysed samples and interpreted data; J.S. analysed samples, interpreted data, performed statistical analyses and prepared figures and tables; J.K. participated in study design, analysed samples and interpreted data; P.J. participated in study design, coordinated sample collection and interpreted data; P.N. participated in study design, interpreted data and contributed an important intellectual input. All authors revised the manuscript and approved its final version.
Conflict of interest
The authors declare no conflict of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Comparison of selected metabolites in plasma in untreated and treated patients with homocystinurias, and in controls. The open and closed symbols show values of analytes in untreated (Kožich et al., 2016) and treated patients (this study), respectively; lines are connecting data of patients that are reported in both studies and clearly indicate that treatment decreases tHcy a homolanthionine in all patients, and cystathionine in RMD patients.
Table S1 Metabolite concentrations in CBSD patients. Table shows plasma and urinary metabolite concentrations, and fractional excretion in a group of pyridoxine non‐responsive patients with CBSD (n = 10) who received a protein restricted diet with amino acid supplements enriched with cysteine, and in the appropriate control group of patients on low protein/amino acid mixture supplemented diet suffering from phenylketonuria (C‐PKU, n = 22). Data are shown as medians with interquartile ranges. Differences in medians were compared using Mann–Whitney U‐test with Benjamini–Hochberg correction for multiple testing.
Table S2 Metabolite concentrations in patients with RMD. Table show plasma and urinary metabolite concentrations, and fractional excretion in a group of patients with RMD (n = 6) on a normal diet and in healthy controls on normal diet (C‐H, n = 12). Data are shown as medians with interquartile ranges. Differences in medians were compared using Mann–Whitney U‐test with Benjamini–Hochberg correction for multiple testing.
Acknowledgements
The authors would like to thank the patients and control subjects for donations of blood and urine. We also want to express our gratitude to Prof. Jiří Zeman, Drs. Eva Hrubá and Eva Košťálová for subject recruitment, to Ms. Šárka Bláhová, BSc, for sample collection, to Ms. Markéta Pavlíková, MSc, for statistical consultation, to Ms. Magda Jandusová, MSc, for aminothiol analyses and to Attila Nagy for assistance in sulfide measurements. This work was supported by the grant 16‐30384A from the Czech Health Research Council to V.K. and grants KH17_126766, K 129286 and K 109843 from the National Research, Development and Innovation Office to P.N.; institutional support to V.K. was provided by the Ministry of Health of the Czech Republic (project RVO‐VFN 64165) and by Charles University (project Progres Q26).
Kožich, V. , Ditrói, T. , Sokolová, J. , Křížková, M. , Krijt, J. , Ješina, P. , and Nagy, P. (2019) Metabolism of sulfur compounds in homocystinurias. British Journal of Pharmacology, 176: 594–606. 10.1111/bph.14523.
Contributor Information
Viktor Kožich, Email: viktor.kozich@lf1.cuni.cz.
Peter Nagy, Email: peter.nagy@oncol.hu.
References
- Akaike T, Ida T, Wei FY, Nishida M, Kumagai Y, Alam MM et al (2017). Cysteinyl‐tRNA synthetase governs cysteine polysulfidation and mitochondrial bioenergetics. Nat Commun 8: 1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustyn KD, Jackson MR, Jorns MS (2017). Use of tissue metabolite analysis and enzyme kinetics to discriminate between alternate pathways for hydrogen sulfide metabolism. Biochemistry 56: 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awwad HM, Geisel J, Obeid R (2016). Determination of trimethylamine, trimethylamine N‐oxide, and taurine in human plasma and urine by UHPLC‐MS/MS technique. J Chromatogr B Analyt Technol Biomed Life Sci 1038: 12–18. [DOI] [PubMed] [Google Scholar]
- Bogdándi V, Ida T, Sutton TR, Bianco C, Ditrói T, Koster G et al (2019). Speciation of reactive sulfur species and their reactions with alkylating agents: have we got a clue of what is inside the cell? Br J Pharmacol 176: 646–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RN, Morton NM (2016). Cysteine and hydrogen sulphide in the regulation of metabolism: insights from genetics and pharmacology. J Pathol 238: 321–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doka E, Pader I, Biro A, Johansson K, Cheng Q, Ballago K et al (2016). A novel persulfide detection method reveals protein persulfide‐ and polysulfide‐reducing functions of thioredoxin and glutathione systems. Sci Adv 2: e1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves IP, Lee PJ, Briddon A (2002). Homocysteine and cysteine – albumin binding in homocystinuria: assessment of cysteine status and implications for glutathione synthesis? Amino Acids 22: 109–118. [DOI] [PubMed] [Google Scholar]
- Huemer M, Diodato D, Schwahn B, Schiff M, Bandeira A, Benoist JF et al (2017). Guidelines for diagnosis and management of the cobalamin‐related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis 40: 21–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji AJ, Savon SR, Jacobsen DW (1995). Determination of total serum sulfite by HPLC with fluorescence detection. Clin Chem 41: 897–903. [PubMed] [Google Scholar]
- Kabil O, Banerjee R (2014). Enzymology of H2S biogenesis, decay and signaling. Antioxid Redox Signal 20: 770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasinath BS, Feliers D, Lee HJ (2018). Hydrogen sulfide as a regulatory factor in kidney health and disease. Biochem Pharmacol 149: 29–41. [DOI] [PubMed] [Google Scholar]
- Kohl JB, Mellis AT, Schwarz G (2019). Homeostatic impact of sulfite and hydrogen sulfide on cysteine catabolism. Br J Pharmacol 176: 554–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich V, Krijt J, Sokolova J, Melenovska P, Jesina P, Vozdek R et al (2016). Thioethers as markers of hydrogen sulfide production in homocystinurias. Biochimie 126: 14–20. [DOI] [PubMed] [Google Scholar]
- Kožich V, Morris AAM, Blom HJ (2016). Disorders of sulfur amino acid metabolism In: Saudubray J‐M, Baumgartner MR, Walter J. (eds). Inborn Metabolic Diseases: Diagnosis and Treatment. Springer Berlin Heidelberg: Berlin, Heidelberg. [Google Scholar]
- Krijt J, Vackova M, Kozich V (2001). Measurement of homocysteine and other aminothiols in plasma: advantages of using tris(2‐carboxyethyl) phosphine as reductant compared with tri‐n‐butylphosphine. Clin Chem 47: 1821–1828. [PubMed] [Google Scholar]
- Libiad M, Motl N, Akey DL, Sakamoto N, Fearon ER, Smith JL et al. (2018). Thiosulfate Sulfurtransferase like Domain Containing 1 Protein Interacts with Thioredoxin. J Biol Chem 293: 2675–2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean KN, Sikora J, Kozich V, Jiang H, Greiner LS, Kraus E et al (2010). Cystathionine beta‐synthase null homocystinuric mice fail to exhibit altered hemostasis or lowering of plasma homocysteine in response to betaine treatment. Mol Genet Metab 101: 163–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majors AK, Pyeritz RE (2000). A deficiency of cysteine impairs fibrillin‐1 deposition: implications for the pathogenesis of cystathionine beta‐synthase deficiency. Mol Genet Metab 70: 252–260. [DOI] [PubMed] [Google Scholar]
- Majtan T, Krijt J, Sokolova J, Krizkova M, Ralat MA, Kent J et al (2018). Biogenesis of hydrogen sulfide and thioethers by cystathionine beta‐synthase. Antioxid Redox Signal 28: 311–323. [DOI] [PubMed] [Google Scholar]
- Mansoor MA, Ueland PM, Svardal AM (1994). Redox status and protein binding of plasma homocysteine and other aminothiols in patients with hyperhomocysteinemia due to cobalamin deficiency. Am J Clin Nutr 59: 631–635. [DOI] [PubMed] [Google Scholar]
- Melideo SL, Jackson MR, Jorns MS (2014). Biosynthesis of a central intermediate in hydrogen sulfide metabolism by a novel human sulfurtransferase and its yeast ortholog. Biochemistry 53: 4739–4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morava E, Rahman S, Peters V, Baumgartner MR, Patterson M, Zschocke J (2015). Quo vadis: the re‐definition of ‘inborn metabolic diseases’. J Inherit Metab Dis 38: 1003–1006. [DOI] [PubMed] [Google Scholar]
- Morris AA, Kozich V, Santra S, Andria G, Ben‐Omran TI, Chakrapani AB et al (2017). Guidelines for the diagnosis and management of cystathionine beta‐synthase deficiency. J Inherit Metab Dis 40: 49–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Palinkas Z, Nagy A, Budai B, Toth I, Vasas A (2014). Chemical aspects of hydrogen sulfide measurements in physiological samples. Biochim Biophys Acta 1840: 876–891. [DOI] [PubMed] [Google Scholar]
- Nimni ME, Han B, Cordoba F (2007). Are we getting enough sulfur in our diet? Nutr Metab (Lond) 4: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson KR (2018). H2S and polysulfide metabolism: conventional and unconventional pathways. Biochem Pharmacol 149: 77–90. [DOI] [PubMed] [Google Scholar]
- Olson KR, Deleon ER, Liu F (2014). Controversies and conundrums in hydrogen sulfide biology. Nitric Oxide 41: 11–26. [DOI] [PubMed] [Google Scholar]
- Pan LL, Qin M, Liu XH, Zhu YZ (2017). The role of hydrogen sulfide on cardiovascular homeostasis: an overview with update on immunomodulation. Front Pharmacol 8: 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem Pharmacol 149: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pol A, Renkema GH, Tangerman A, Winkel EG, Engelke UF, de Brouwer APM et al (2018). Mutations in SELENBP1, encoding a novel human methanethiol oxidase, cause extraoral halitosis. Nat Genet 50: 120–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Peter EA, Bir S, Wang R, Kevil CG (2012). Analytical measurement of discrete hydrogen sulfide pools in biological specimens. Free Radic Biol Med 52: 2276–2283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (2016). Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discov 15: 185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C (2018). A timeline of hydrogen sulfide (H2S) research: from environmental toxin to biological mediator. Biochem Pharmacol 149: 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Motta JP, Buret AG (2018). Hydrogen sulfide: an agent of stability at the microbiome‐mucosa interface. Am J Physiol Gastrointest Liver Physiol 314: G143–G149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace JL, Wang R (2015). Hydrogen sulfide‐based therapeutics: exploiting a unique but ubiquitous gasotransmitter. Nat Rev Drug Discov 14: 329–345. [DOI] [PubMed] [Google Scholar]
- Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P et al. (2010). A monobromobimane‐based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol 160: 941–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CT, Chen L, Xu S, Day JJ, Li X, Xian M (2017). Recent development of hydrogen sulfide releasing/stimulating reagents and their potential applications in cancer and glycometabolic disorders. Front Pharmacol 8: 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zschocke J (2016). SSIEM classification of inborn errors of metabolism In: Blau N, Duran M, Blaskovics ME, Gibson KM. (eds). Physician's Guide to the Diagnosis, Treatment, and Follow‐up of Inherited Metabolic Diseases. Springer: Berlin‐Heidelberg. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Comparison of selected metabolites in plasma in untreated and treated patients with homocystinurias, and in controls. The open and closed symbols show values of analytes in untreated (Kožich et al., 2016) and treated patients (this study), respectively; lines are connecting data of patients that are reported in both studies and clearly indicate that treatment decreases tHcy a homolanthionine in all patients, and cystathionine in RMD patients.
Table S1 Metabolite concentrations in CBSD patients. Table shows plasma and urinary metabolite concentrations, and fractional excretion in a group of pyridoxine non‐responsive patients with CBSD (n = 10) who received a protein restricted diet with amino acid supplements enriched with cysteine, and in the appropriate control group of patients on low protein/amino acid mixture supplemented diet suffering from phenylketonuria (C‐PKU, n = 22). Data are shown as medians with interquartile ranges. Differences in medians were compared using Mann–Whitney U‐test with Benjamini–Hochberg correction for multiple testing.
Table S2 Metabolite concentrations in patients with RMD. Table show plasma and urinary metabolite concentrations, and fractional excretion in a group of patients with RMD (n = 6) on a normal diet and in healthy controls on normal diet (C‐H, n = 12). Data are shown as medians with interquartile ranges. Differences in medians were compared using Mann–Whitney U‐test with Benjamini–Hochberg correction for multiple testing.