

Abstract
The transsulfuration pathway is a metabolic pathway where transfer of sulfur from homocysteine to cysteine occurs. The pathway leads to the generation of several sulfur metabolites, which include cysteine, GSH and the gaseous signalling molecule hydrogen sulfide (H2S). Precise control of this pathway is critical for maintenance of optimal cellular function and, therefore, the key enzymes of the pathway, cystathionine β‐synthase and cystathionine γ‐lyase, are regulated at multiple levels. Disruption of the transsulfuration pathway contributes to the pathology of several conditions such as vascular dysfunction, Huntington's disease and during ageing. Treatment with donors of hydrogen sulfide and/or stimulation of this pathway have proved beneficial in several of these disorders. In this review, we focus on the regulation of the transsulfuration pathway pertaining to cysteine and H2S, which could be targeted to develop novel therapeutics.
Linked Articles
This article is part of a themed section on Chemical Biology of Reactive Sulfur Species. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.4/issuetoc
Abbreviations
- MPST
3‐mercaptopyruvate sulfurtransferase
- ATF4
activating transcription factor 4
- CDO
cysteine dioxygenase
- CSE
cystathionine γ‐lyase
- FXR
farnesoid X receptor
- GH
growth hormone
- GPABR1
GPCR for secondary bile acids
- H2S
hydrogen sulfide
- HD
Huntington's disease
- HO2
haem oxygenase 2
- Keap1
kelch‐like ECH‐associated protein 1
- mHtt
mutant huntingtin
- NMDA
N‐methyl D‐aspartate
- Nrf2
nuclear factor (erythroid‐derived 2)‐like 2
- PLP
pyridoxal 5′‐phosphate
- SAM
S‐adenosyl‐methionine
- SP1
specificity protein 1
Introduction
The transsulfuration pathway plays a central role in sulfur metabolism and redox regulation in cells. In mammals, the pathway involves the transfer of sulfur from homocysteine to cysteine via cystathionine and is the only route for biosynthesis of cysteine (Figure 1). Homocysteine, which is derived from dietary methionine, is converted to cystathionine by cystathionine β‐synthase (CBS), which is acted on by cystathionine γ‐lyase (CSE) to generate cysteine. In prokaryotes, fungi and plants, the pathway can operate in the opposite direction to synthesize methionine from cysteine and was initially referred to as the transsulfuration pathway, whereas in mammals, it was designated as the reverse transsulfuration pathway (Steegborn et al., 1999; Wang, 2012). For the sake of clarity, this review will refer to the pathway in mammals as the transsulfuration pathway as the conversion from cysteine to methionine does not occur here.
Figure 1.
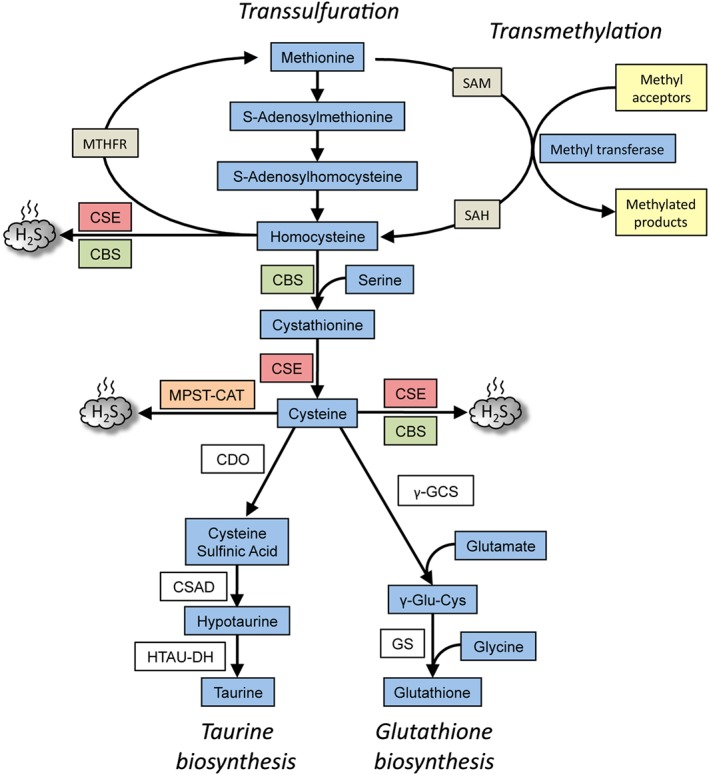
Overview of the transsulfuration pathway. The pathway results in the generation of cysteine from homocysteine, which in turn, is derived from dietary methionine in mammals. CBS condenses homocysteine with serine to generate cystathionine, the substrate for CSE, to generate cysteine. CSE can generate H2S from either cysteine or homocysteine. While CSE can utilize cysteine to generate H2S, CBS uses a combination of cysteine and homocysteine to generate H2S. A third H2S‐generating enzyme, MPST, in conjunction with cysteine amino acid transferase (CAT) utilizes cysteine to generate H2S. The transsulfuration pathway intersects with the transmethylation pathway at homocysteine, which can be remethylated back to methionine by N5,N10‐methylenetetrahydrofolate reductase (MTHFR). The cysteine generated by the pathway can be chanelled into GSH synthesis by the action of the enzymes, γ‐glutamyl cysteine synthetase (γ‐GCS) and glutathione synthetase (GS) or converted to other sulfur‐containing molecules such as taurine. Taurine is generated by the action of three enzymes, CDO, cysteine sulfinic acid decarboxylase (CSAD) and hypotaurine dehydrogenase (HTAU‐DH).
In addition to its essential role in protein synthesis, cysteine is also a component of the major antioxidant GSH and a potent antioxidant itself. Disruption of cysteine and GSH metabolism has been frequently linked to aberrant redox homeostasis and neurodegeneration (McBean et al., 2015; Paul et al., 2018). Cysteine and GSH play central roles in maintaining thiol redox balance in the brain with the transsulfuration pathway being a major source of cysteine in astrocytes (McBean, 2012; McBean, 2017). Both CSE and CBS play important roles in the regulation of redox balance. It has been reported that approximately 50% of the cysteine generated by the transsulfuration pathway is utilized for GSH biosynthesis in hepatic cells (Mosharov et al., 2000; Banerjee and Zou, 2005). In the mouse brain, the activity of the pathway is lower as compared to the liver, but the flux can be increased by oxidative stress (Vitvitsky et al., 2006; Diwakar and Ravindranath, 2007). Cysteine is also the precursor of the gaseous signalling molecule hydrogen sulfide (H2S) and other sulfur metabolites (Paul and Snyder, 2012; Paul and Snyder, 2015a; Paul and Snyder, 2015b). Besides GSH and H2S, cysteine is converted to the sulfur containing molecule taurine by the action of the enzyme cysteine dioxygenase (CDO) to form cysteinesulfinic acid, which can then be decarboxylated to hypotaurine by cysteinesulfinic acid decarboxylase, and the hypotaurine generated, oxidized to taurine (Stipanuk and Ueki, 2011) (Figure 1). Since CDO acts directly on cysteine, it can modulate H2S production by influencing substrate availability. Mice lacking CDO have elevated cysteine and H2S production capacity (Jurkowska et al., 2014; Rose et al., 2017). Taurine plays a role in osmoregulation, immunomodulation, neuromodulation, Ca2+ homeostasis, ocular function and possesses antioxidant and anti‐inflammatory effects (Schaffer and Kim, 2018). An intact taurine biosynthetic pathway responsive to hypertonic conditions has been demonstrated in neurons, consistent with its role in osmoregulation (Vitvitsky et al., 2011).
This pathway is intimately linked to the transmethylation pathway via homocysteine, which can be remethylated to generate methionine or be irreversibly converted to cysteine (Figure 1). This article will focus on the regulation of the transsulfuration pathway pertaining to cysteine and H2S metabolism and its role during normal and pathological conditions.
Enzymes of the transsulfuration pathway
CSE and CBS, the key enzymes regulating the flux through the transsulfuration pathway, are regulated at multiple levels. CBS (also called l‐serine hydrolyase) catalyses the first committed step of transsulfuration by condensing serine with homocysteine to generate cystathionine in a β‐replacement reaction. CBS is believed to function predominantly in the nervous system, although it is active in the periphery in the liver and kidneys. CBS is a cytosolic homotetrametic enzyme of about 63 kDa subunits that binds two cofactors, pyridoxal 5′‐phosphate (PLP) and haem (Meier et al., 2001). The enzyme is activated allosterically by S‐adenosyl‐methionine (SAM) and also by a cleavage at its carboxyl terminal at R413 to generate a 45 kDa monomer, which is not only twice as active as the full length form but also refractory to SAM‐mediated activation (Kery et al., 1998; Majtan et al., 2014). CBS can also utilize cysteine instead of serine to generate H2S and cystathionine. In addition, CBS can act on two molecules of cysteine to produce H2S. However, the preferred substrates for generation of H2S by CBS are cysteine and homocysteine. CSE can use homocysteine alone to generate H2S and is responsible for the clearance of homocysteine under conditions of hyperhomocysteinemia, unlike CBS which is unresponsive under these conditions (Chen et al., 2004; Singh et al., 2009). Over 150 mutations in the CBS protein have been reported, of which several, especially those causing misfolding, are linked to enzyme activity (Kozich et al., 2010). CBS mice rarely live past two weeks and exhibit a variety of abnormalities, which include growth retardation, severe hepatopathy, vascular abnormalities, dislocation of the eye‐lens and skeletal deformities (Watanabe et al., 1995; Kruger, 2017). Human subjects with CBS mutations exhibit several neurological deficits including anxiety, depression, obsessive–compulsive behaviour and psychosis (Abbott et al., 1987).
CSE, encoded by the gene Cth, also known as γ‐cystathionase, cysteine lyase, cysteine desulfhydrase, cystathionase, cystathioninase, cystine desulfhydrase, homoserine deaminase or homoserine dehydratase, is the second enzyme in the pathway. CSE utilizes cystathionine generated by CBS to generate cysteine, a semi‐essential amino acid. As CSE is the only enzyme that can directly generate cysteine de novo in mammals, its depletion is deleterious (Ishii et al., 2010; Mani et al., 2011). In AIDS patients, the very low levels of CSE render cysteine an essential amino acid (Martin et al., 2001). In addition to loss of CSE function, loss of CBS also leads to lowered cysteine synthesis in cells (Gupta and Kruger, 2011). CSE is predominantly expressed in the periphery, although it is now recognized to be functional in the brain with neuroprotective roles (Vitvitsky et al., 2006; Diwakar and Ravindranath, 2007; Paul and Snyder, 2012; Paul et al., 2014; Paul and Snyder, 2018). The crystal structure of human CSE revealed that the enzyme is a homotetramer with each subunit being 45 kDa, and with the cofactor PLP bound to each subunit (Sun et al., 2009). PLP interacts with the carboxylate of Asp187 of CSE via hydrogen bonding and mutation of this residue to Ala abolishes catalytic activity, as assessed by H2S production. Unlike CBS, which is constitutively expressed, CSE is inducible by a wide variety of stimuli ranging from oxidative and endoplasmic reticulum stress to nutrient deprivation. CSE, like CBS, can also generate H2S. In the liver, CSE is the dominant enzyme for generation of H2S although CBS is also expressed at high levels (Kabil et al., 2011b). Loss of CSE can lead to oxidative stress, aberrant stress responses, vascular deficits and hyperhomocysteinemia (Yang et al., 2008; Sbodio et al., 2016; Sbodio et al., 2018). The third enzyme that produces H2S, 3‐mercaptopyruvate sulfurtransferase (MPST), is part of the cysteine catabolic pathway, generating the gaseous signalling molecule in concert with cysteine amino acid transferase (Shibuya et al., 2009).
Signalling by H2S
H2S exerts its effects on cellular physiology in several ways. Similar to NO, H2S can modify reactive cysteine residues on proteins, a post‐translational modification termed sulfhydration or persulfidation, analogous to nitrosylation mediated by NO. In the case of persulfidation, –SH groups of reactive cysteine residues are converted to persulfide or –SSH groups (Mustafa et al., 2009; Paul and Snyder, 2015a; Filipovic et al., 2018). Sulfhydration is a physiological modification that is reversible by endogenous reductants, such as the thioredoxin system (Krishnan et al., 2011; Doka et al., 2016). Sulfhydration is more prevalent than nitrosylation with about 50% of hepatic proteins being sulfhydrated (Mustafa et al., 2009). In several instances, sulfhydration and nitrosylation can target the same cysteine residue but have opposite effects. For example, the glycolytic enzyme GAPDH is activated by persulfidation but inhibited by nitrosylation (Hara et al., 2005; Mustafa et al., 2009). Thus, mice lacking CSE have reduced levels of sulfydration and lower GAPDH activity. Sulfhydration can modulate several physiological processes including responses to inflammatory stimuli, oxidative stress, neuronal signalling pathways and vasodilatation (Mustafa et al., 2011; Paul and Snyder, 2012; Sen et al., 2012; Vandiver et al., 2013; Yang et al., 2013).
H2S also has roles independent of sulfhydration, for example, in cellular bioenergetics. At lower concentrations (0.1–1 μM), the H2S donor NaHS can stimulate electron transport in rat isolated liver mitochondria, whereas higher concentrations (3–30 μM) suppress mitochondrial activity (Modis et al., 2013). H2S can directly donate electrons to the mitochondrial electron transport chain (ETC) at complex II and increase mitochondrial cAMP, whereas at higher concentrations, it disrupts the ETC by inhibiting mitochondrial cytochrome c oxidase (Szabo et al., 2014). Another mode by which H2S acts is by binding to metal centres of metalloenzymes (Pietri et al., 2011). One of the several haem proteins acted on by H2S is GC C, where a reduction in the ferric (Fe3+) haem to the ferrous (Fe2+) form enhances its interaction with NO, which activates the enzyme (Zhou et al., 2016). H2S can also interface with NO signalling by forming nitrosothiols, which act as signalling molecules (Whiteman et al., 2006; Filipovic et al., 2012).
Regulation of the transsulfuration pathway at the post‐translational level
As H2S and cysteine participate in a wide variety of physiological processes, precise control of their production is critical for the maintenance of optimal cellular function. Thus, it is not surprising that the transsulfuration pathway is subject to regulatory controls at multiple levels. Both CBS and CSE undergo several post‐translational modifications that can alter their enzymatic activity or sub‐cellular localizations (Figure 2). One of the less studied aspects of the transsulfuration pathway is the elucidation of conditions leading to cysteine synthesis by CSE as opposed to the production of H2S, GSH or taurine. The haem in CBS plays a key role in switching the transsulfuration pathway from cysteine production to H2S generation (Kabil et al., 2016). When haem is bound by endogenous ligands, the production of cystathionine occurs due to the higher intracellular levels of serine and its greater affinity for CBS as compared to cysteine. Similarly, the affinity of cystathionine for CSE is greater than that of cysteine, favouring the generation of cysteine as opposed to H2S. During conditions of stress, when NO or carbon monoxide (CO) levels rise, causing inhibition of CBS due to the formation of nitrosyl or ferrous carbonyl CBS, homocysteine accumulates, leading to the production of H2S by CSE (Banerjee, 2017). Enzymes which catabolize cysteine also have an effect on the production of H2S.
Figure 2.
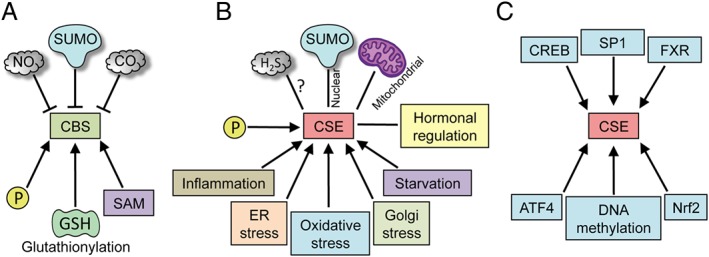
Regulation of CSE and CBS. CBS and CSE are regulated at multiple levels. (A) Post‐translational regulation of CBS. CBS is a constitutively expressed enzyme and can be regulated by several post‐translational modifications (PTMs) such as sumoylation, phosphorylation and glutathionylation. It is allosterically regulated by SAM, which activates it and increases its stability. The haem centre in CBS can bind NO and CO. (B) CSE is a highly inducible protein, which is regulated by a wide variety of stimuli such as inflammation, ER stress (which can cause translocation to the mitochondria), oxidative stress and Golgi stress, starvation and by hormones. CSE is also modified by PTMs such as phosphorylation, sulfhydration and sumoylation (which may cause translocation to the nucleus). (C) Unlike CBS, whose activity in the cell is regulated predominantly by PTMs, the effects of CSE are exerted mainly by regulation of its expression at the transcriptional level. Several transcription factors, such as the cAMP‐response element binding protein (CREB), SP1, FXR, activating transcription factor 4 and Nrf2, have binding sites on the CSE promoter. In addition, the expression of CSE is also regulated by methylation of its promoter.
Regulation of CBS
The haem cofactor in the N‐terminal domain of CBS can bind CO or NO, which can inhibit its activity (Singh et al., 2007). This inhibition has relevance in the regulation of cerebral microvasculature. During normoxia, CO produced by the O2‐dependent enzyme haem oxygenase 2 (HO2) binds to the haem of CBS and keeps the enzyme suppressed. HO2 is localized to the endothelial cells of the cerebral vasculature, which is in close proximity to the astrocytes where CBS is localized (Enokido et al., 2005; Morikawa et al., 2012). During hypoxia, when O2 becomes limiting, activity of HO2 and CO production drops, relieving the inhibition on CBS, which can then produce H2S to mediate arteriolar vasodilation (Morikawa et al., 2012). Although it is not known whether the CO mediating CBS inhibition originates from the neuronal or endothelial pools HO2, it is evident that two gaseous signalling molecules interact to modulate a major cerebral function. Another mode by which CBS can be modulated is by phosphorylation. For instance, phosphorylation of Ser227 of human CBS in human urothelium occurs in a cGMP/PKG‐dependent manner to increase H2S production (d'Emmanuele di Villa Bianca et al., 2016). The activity of CBS can also be modulated allosterically by SAM, a component of the transmethylation pathway (Ereno‐Orbea et al., 2014). In addition, SAM stabilizes CBS. Methionine restriction leads to a significant decrease in CBS protein levels due to decrease in SAM concentrations and destabilization of CBS (Prudova et al., 2006). CBS has also been reported to be a substrate for sumoylation by the E3 SUMO ligase, human polycomb group protein 2, which decreases its activity (Agrawal and Banerjee, 2008). However, glutathionylation stimulates the activity of human CBS (Niu et al., 2015). CBS activity increases the flux via the transsulfuration pathway, which leads to GSH production, which becomes oxidized and depleted during oxidative stress. Thus, it seems reasonable that CBS would be modified by GSH to stimulate production of cystathionine, the precursor for cysteine, whose availability governs the rate of GSH production (Figure 2A).
Regulation of CSE
Like CBS, CSE can also be regulated by post‐translational modifications. Phosphorylation sites have been reported on CSE, which modulates its activity. In response to bile acid receptor activation, CSE is phosphorylated via the Akt pathway, which increases its catalytic activity (Renga et al., 2015). CSE has been reported to be sumoylated, which has been speculated to be responsible for its nuclear localization (Agrawal and Banerjee, 2008). Apart from induction, translocation of CSE from the cytosol to other cellular compartments such as mitochondria in response to stress has been observed, adding an additional layer of regulation to the regulation of H2S production (Fu et al., 2012). Sulfhydration of CSE itself has been reported by our laboratory; however, the significance of the modification is yet to be explored (Mustafa et al., 2009) (Figure 2B).
Regulation of the transsulfuration pathway at the transcriptional level
Between the two enzymes, CSE and CBS, CSE is highly inducible and is regulated in response to a wide variety of extrinsic and intrinsic signals. CSE is induced by oxidative stress, ER stress, Golgi stress, mitochondrial stress, inflammation and starvation, among several others (Harding et al., 2003; Kandil et al., 2010; Sen et al., 2012; Sbodio et al., 2016; Sbodio et al., 2018) (Figure 2B). Expression of CSE is regulated by the specificity protein 1 (SP1) under basal conditions (Yang et al., 2011; Zhang et al., 2011).
CSE is a highly inducible protein and can be regulated by several factors depending on the cell type and tissue (Figure 2C). In the liver, the bile‐acid‐activated farnesoid X receptor (FXR) regulates CSE by binding to the CSE promoter thereby regulating H2S production and hepatic microcirculation (Renga et al., 2009). FXR binds the sequence, AGTTCAgTGTACCT, with two inverted repeats separated by one nucleotide and increases CSE expression. These studies showed that mice lacking FXR display decreased levels of CSE and H2S in the systemic circulation. The GPCR for secondary bile acids, GPABR1, activates the cAMP/PKA pathway, which results in the phosphorylation of the cAMP response element binding protein and binding to the CSE promoter and increased transcription of CSE. Agonists of GPABR1 reverse the vasoconstriction induced by noradrenaline and methoxamine on isolated rat livers via H2S production. Inhibiting CSE activity with propargylglycine reverses these effects. CSE is also regulated during inflammation, for example, during LPS stimulation of macrophages, which leads to increased association of SP1 with the CSE promoter. This leads to elevated H2S production and sulfhydration of the p65 subunit of the transcription factor NF‐κB and increases its recruitment to promoters of anti‐apoptotic genes (Sen et al., 2012). Apart from inflammation, CSE is inducible by oxidative stress and the CSE promoter harbours a binding site for the master regulator of oxidative stress response, nuclear factor (erythroid‐derived 2)‐like 2 (Nrf2) (Martin et al., 2007; Hassan et al., 2012; Guo et al., 2014). In addition, Nrf2 function is also regulated by H2S produced by CSE. Treatment of cells with NaHS, the H2S donor, improves the stability of Nrf2 and also sulfhydrates Kelch‐like ECH‐associated protein 1 (Keap1), the repressor of Nrf2 (Hourihan et al., 2013). An independent study also reported sulfhydration of Keap1 leading to Nrf2 activation (Yang et al., 2013). Nrf2 has been reported to have binding sites, which function as antioxidant response elements, with the sequences, 5′‐GTGATCTAGCA‐3′ and 5′‐ATGAGGCAGCT‐3′, for the upstream regions of CBS and CSE respectively (Hassan et al., 2012; Hourihan et al., 2013). Thus, Nrf2 can elevate the expression of both CSE and CBS. The induction of CSE by oxidative stress is not surprising as it is a central part of a pathway that leads to the synthesis of two major antioxidants, cysteine and GSH, in addition to H2S, which can mediate the antioxidant stress response via sulfhydration of proteins involved in this response (Yang et al., 2013).
Another stress stimulus that up‐regulates CSE expression and H2S production is nutrient restriction and amino acid starvation. The transcription factor involved in response to amino acid starvation is activating transcription factor 4 (ATF4), which binds to the CSE promoter in addition to promoters of other amino acid biosynthetic and transport genes (Harding et al., 2003).
The expression of CSE can also be modulated by several hormones. In the long‐lived Ames dwarf mice, which have reduced growth hormone (GH) and thyroid‐stimulating hormone signalling, as well as in mice lacking GH receptors, CSE and CBS levels are elevated and H2S production is increased. GH and TH negatively regulate hepatic H2S production through distinct mechanisms involving the hypothalamic–pituitary axis (Hine et al., 2017).
In addition to regulation by various transcription factors, the CSE promoter has been found to be controlled by DNA methylation wherein cytosine is modified to 5‐methylcytosine (Giannakopoulou et al., 2017) (Figure 2C). Several CpG islands (regions rich in cytosine and guanosine residues) have been identified in the CSE promoter, which are hypermethylated in patients with coronary artery disease. Interestingly, there was a gender‐specific difference in methylation status, with hypermethylation being observed only in males. These patients exhibited a decrease in H2S production and increase in cytosine methylation of the Cth promoter.
Dysregulation of the transsulfuration pathways and therapeutic opportunities
As the transsulfuration modulates several physiological processes, its dysregulation can lead to deleterious effects. The levels of H2S are highly regulated during normal cellular processes with controls operating at multiple levels as discussed above. Too much and too little H2S production can have damaging consequences for cells. Thus, the dose–response of H2S can explain its variable effects reported in literature. During ageing and other pathophysiological conditions, the fine balance of H2S can be upset. A few of the conditions associated with a dysregulated transsulfuration pathway are described below.
Vascular abnormalities
Like NO, H2S plays an important role in the vascular system with effects on BP, vasorelaxation and angiogenesis (Szabo, 2017; Gheibi et al., 2018). Lower H2S production has been associated with myocardial ischaemia, angina and cardiovascular disease (Polhemus et al., 2014). Depletion of CSE and H2S causes a range of vascular deficits ranging from hypertension and impaired vasorelaxation to impaired angiogenesis (Yang et al., 2008; Coletta et al., 2012; Cindrova‐Davies et al., 2013). The cGMP signalling pathway, which plays a central role in vasodilatation and smooth muscle relaxation, is disrupted in mice lacking CSE. H2S can not only inhibit PDEs which degrade cGMP but also stimulate GCs which synthesize cGMP. Several of the PDE inhibitors, such as sildenafil citrate and tadalafil, which are currently marketed to treat erectile dysfunction and cardiovascular deficits, stimulate H2S production (Salloum et al., 2009; Fusco et al., 2012). In addition to the generation of H2S, CSE and CBS can also be considered to be homocysteine‐clearing enzymes. Depletion of both CBS and CSE causes hyperhomocystenaemia, which is an independent risk factor for developing cardiovascular deficits and neurodegenerative disorders such as Alzheimer's disease (Mattson and Shea, 2003; Smith et al., 2018). Thus, in addition to generating cysteine, the transsulfuration pathway utilizes homocysteine, whose accumulation can be harmful, by activating the NMDA receptors causing excitotoxicity and elevated oxidative stress (Lipton et al., 1997).
Huntington's disease
We have recently shown that the transsulfuration pathway is disrupted in the autosomal dominant neurodegenerative disorder, Huntington's disease (HD) leading to lowered cysteine and H2S levels (Paul et al., 2014; Paul and Snyder, 2014). HD is triggered by the expansion of polyglutamine repeats in the protein huntingtin, leading to motor, cognitive and psychiatric disturbances (Group, 1993). At the molecular level, mutant huntingtin (mHtt) aggregates and affects multiple cellular processes including transcriptional regulation, response to stress and autophagy (Bates et al., 2015). In cell cultures and mouse models of HD as well as in patient samples, the levels of CSE are drastically diminished, due to the sequestration of SP1 by mHtt (Paul et al., 2014). In addition to impaired SP1 function, transcriptional regulation of CSE in response to amino acid starvation is also affected in HD (Sbodio et al., 2016) as demonstrated in striatal progenitor cells, STHdh Q7/Q7 and STHdh Q111/Q111 (referred to as Q7 and Q111 cells: Figure 3). The transporters for cystine and cysteine are also dysregulated in HD (Li et al., 2010; Frederick et al., 2014). Together, the lack of cysteine and GSH cause an elevation of ROS, which prevents the stress response elicited by ATF4 in a vicious cycle, which mediates cell death. As a result of these abnormalities, the Q7 cells are unable to grow under cysteine‐deprived conditions. Accordingly, stimulating the transsulfuration pathway affords therapeutic benefits. Recently, we showed that the Golgi stress response can be harnessed to up‐regulate cysteine and H2S biosynthesis. Similar to ER stress and oxidative stress responses, the Golgi stress response can elicit the induction of CSE (Sbodio et al., 2018). Golgi stressors, such as the ionophores monensin and nigericin, can activate the integrated stress response by activating the phosphorylation of PKR‐like ERK, which then leads to the induction of ATF4 and CSE. Thus, pretreatment of Q111 cells with monensin promotes growth in cysteine‐free conditions via induction of CSE (Figure 3).
Figure 3.
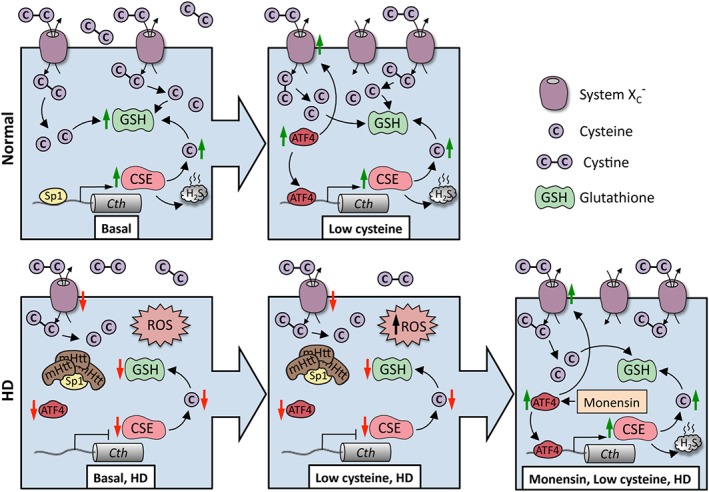
Huntington's disease (HD) as an example of disrupted cysteine metabolism. In normal striatal cells, during basal conditions, the expression of CSE is controlled by the transcription factor SP1, which has binding sites in the CSE (Cth) promoter, resulting in cysteine (denoted as C) and GSH production. When cysteine becomes limiting, ATF4 is induced leading to an elevated expression of CSE as well the transporters for cystine (denoted as C–C) (System Xc −). System Xc − imports cystine, the oxidized form of cysteine, which is subsequently reduced to cysteine inside cells. Both the basal as well as ATF4‐mediated induction of CSE in response to cysteine deprivation are compromised in HD. SP1 is sequestered by mHtt, leading to diminished expression of CSE, causing an increase in ROS. Initially, ATF4 is functional, but during disease progression, a further increase in ROS prevents this response, leading to a decline in its induction. Stimulating the transsulfuration pathway via the Golgi stress response (as shown in the case of monensin, an ionophore and Golgi stressor) can protect HD cells and prolong survival.
Ageing
It has been shown that increased flux via the transsulfuration pathway delays ageing and increases lifespan. In the long‐lived Ames dwarf mice, an enhanced expression and activity of CSE was observed, which correlated with increased GSH levels (Uthus and Brown‐Borg, 2003). The positive impact of the pathway on longevity has also been observed in the fruit fly, Drosophila melanogaster (Kabil et al., 2011a). More recently, it was demonstrated that H2S was responsible for increased longevity afforded by caloric restriction in yeast, C. elegans, Drosophila and mice (Hine et al., 2015). In Werner's syndrome, an autosomal recessive disease characterized by accelerated ageing is caused by a mutation in the Werner protein, which leads to defects in genomic instability and aberrant DNA repair; CSE and CBS are down‐regulated and this may contribute to the elevated oxidative stress associated with the disease (Murano, 1995). The administration of H2S donors has proved beneficial in a cell culture model of Werner's syndrome leading to reversal of abnormal morphology and protein aggregation associated with the disease (Talaei et al., 2013).
Strategies to combat dysregulated transsulfuration
As discussed in the preceding sections, aberrant redox homeostasis occurs in a wide variety of diseases and during normal ageing. In cases where elevated CBS or CSE expression occur, use of specific inhibitors for these enzymes may be beneficial as in the case of Down syndrome, where trisomy of chromosome 21, where Cbs is located, causes excess H2S production (Kamoun et al., 2003). However, specific inhibitors of CBS are currently unavailable (Asimakopoulou et al., 2013). In conditions where impaired transsulfuration occurs and causes redox imbalance due to suboptimal expression of CSE, several strategies can be followed to mitigate abnormalities. A case in point is HD, where decreased CSE expression causes decreased cysteine and H2S production, elevated oxidative stress and associated abnormalities. Several approaches can be taken to counteract the disrupted redox balance in HD (Figure 4). Supplements of cysteine or its precursors mitigated symptoms and delayed disease progression in mouse models of HD (Paul et al., 2014). Another approach is to mitigate oxidative stress by administration of antioxidants such as ascorbate and N‐acetyl cysteine. In HD, the response of cytoprotective pathways decline due to oxidative stress and the administration of antioxidants have proved beneficial (Wright et al., 2015; Sbodio et al., 2016). Stimulating the transsulfuration pathway as a whole by inducing mild stress such as Golgi stress can induce CSE expression to increase flux through the pathway and precondition cells to withstand future insults (Sbodio et al., 2018). Another as yet unexplored option is to induce the expression of CSE and/or CBS by modulating epigenetic changes such as DNA methylation or histone modifications (Figure 4).
Figure 4.
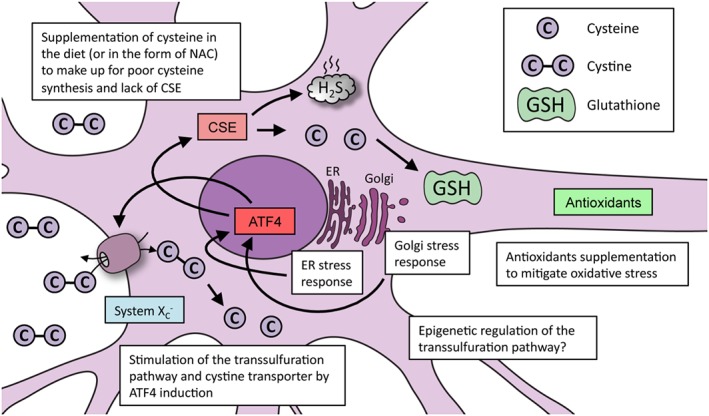
Strategies used to promote redox balance in cells via cysteine metabolism: Huntington's disease as an example. In conditions involving elevated oxidative stress caused by cysteine imbalance (as exemplified by Huntington's disease), several approaches can be followed to improve cell survival. Supplementation of cysteine or N‐acetyl cysteine (NAC) via the diet can reverse abnormalities in cells. Mitigating oxidative stress, which improves stress‐response pathways, can promote optimal functioning of the transsulfuration pathway. Up‐regulating the expression of CSE via the stress‐responsive transcription factor ATF4 can correct abnormalities associated with cysteine deprivation. Mild forms of ER and Golgi stress can elicit cytoprotective responses, which may provoke cellular adaptations that can help protect against future damaging insults. In addition to these strategies, altering the epigenetic state of the CSE and CBS promoters may also induce their expression.
Summary
It is increasingly evident that the transsulfuration pathway plays a central role in the maintenance of redox homeostasis and integration of stress responses. Both cysteine and H2S participate in a plethora of signalling processes. In order to better understand the molecular mechanisms underlying the action of these versatile sulfur‐containing molecules, precise measurement of their levels is necessary. Thus, the development of more specific and sensitive reagents to detect these signalling molecules is necessary. Confusion in the field is especially caused by the lack of methods to accurately estimate endogenous concentrations of H2S. In addition to these aspects, the generation of tissue‐specific knockout models of CSE, CBS and MPST would prove invaluable to assess the roles of the three enzymes in normal signalling pathways. In order to identify pathways regulated by H2S, proteomic approaches in conjunction with metabolomics would yield information on nodes for therapeutic intervention. Screening for compounds, both endogenous and exogenous, that stimulate the transsulfuration pathway would lead to a better understanding of the homeostatic regulation of processes controlled by H2S and the development of novel precision therapeutics.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b,c,d).
Conflict of interest
The authors declare no conflicts of interest.
Sbodio, J. I. , Snyder, S. H. , and Paul, B. D. (2019) Regulators of the transsulfuration pathway. British Journal of Pharmacology, 176: 583–593. 10.1111/bph.14446.
References
- Abbott MH, Folstein SE, Abbey H, Pyeritz RE (1987). Psychiatric manifestations of homocystinuria due to cystathionine β‐synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B6‐responsiveness. Am J Med Genet 26: 959–969. [DOI] [PubMed] [Google Scholar]
- Agrawal N, Banerjee R (2008). Human polycomb 2 protein is a SUMO E3 ligase and alleviates substrate‐induced inhibition of cystathionine beta‐synthase sumoylation. PLoS One 3: e4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Cidlowski JA, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Nuclear hormone receptors. Br J Pharmacol 174: S208–S224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017c). The Concise Guide to PHARMACOLOGY 2017/18: Overview. Br J Pharmacol 174: S1–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Peters JA, Kelly E, Marrion NV, Faccenda E, Harding SD et al (2017d). The Concise Guide to PHARMACOLOGY 2017/18: Ligand‐gated ion channels. Br J Pharmacol 174: S130–S159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimakopoulou A, Panopoulos P, Chasapis CT, Coletta C, Zhou Z, Cirino G et al (2013). Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol 169: 922–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R (2017). Catalytic promiscuity and heme‐dependent redox regulation of H2S synthesis. Curr Opin Chem Biol 37: 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Zou CG (2005). Redox regulation and reaction mechanism of human cystathionine‐β‐synthase: a PLP‐dependent hemesensor protein. Arch Biochem Biophys 433: 144–156. [DOI] [PubMed] [Google Scholar]
- Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR et al (2015). Huntington disease. Nat Rev Dis Primers 1: 15005. [DOI] [PubMed] [Google Scholar]
- Chen X, Jhee KH, Kruger WD (2004). Production of the neuromodulator H2S by cystathionine β‐synthase via the condensation of cysteine and homocysteine. J Biol Chem 279: 52082–52086. [DOI] [PubMed] [Google Scholar]
- Cindrova‐Davies T, Herrera EA, Niu Y, Kingdom J, Giussani DA, Burton GJ (2013). Reduced cystathionine γ‐lyase and increased miR‐21 expression are associated with increased vascular resistance in growth‐restricted pregnancies: hydrogen sulfide as a placental vasodilator. Am J Pathol 182: 1448–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P et al (2012). Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium‐dependent vasorelaxation. Proc Natl Acad Sci U S A 109: 9161–9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d'Emmanuele di Villa Bianca R, Mitidieri E, Fusco F, Russo A, Pagliara V, Tramontano T et al (2016). Urothelium muscarinic activation phosphorylates CBS(Ser227) via cGMP/PKG pathway causing human bladder relaxation through H2S production. Sci Rep 6: 31491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwakar L, Ravindranath V (2007). Inhibition of cystathionine‐γ‐lyase leads to loss of glutathione and aggravation of mitochondrial dysfunction mediated by excitatory amino acid in the CNS. Neurochem Int 50: 418–426. [DOI] [PubMed] [Google Scholar]
- Doka E, Pader I, Biro A, Johansson K, Cheng Q, Ballago K et al (2016). A novel persulfide detection method reveals protein persulfide‐ and polysulfide‐reducing functions of thioredoxin and glutathione systems. Sci Adv 2: e1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enokido Y, Suzuki E, Iwasawa K, Namekata K, Okazawa H, Kimura H (2005). Cystathionine beta‐synthase, a key enzyme for homocysteine metabolism, is preferentially expressed in the radial glia/astrocyte lineage of developing mouse CNS. FASEB J 19: 1854–1856. [DOI] [PubMed] [Google Scholar]
- Ereno‐Orbea J, Majtan T, Oyenarte I, Kraus JP, Martinez‐Cruz LA (2014). Structural insight into the molecular mechanism of allosteric activation of human cystathionine β‐synthase by S‐adenosylmethionine. Proc Natl Acad Sci U S A 111: E3845–E3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic MR, Miljkovic J, Nauser T, Royzen M, Klos K, Shubina T et al (2012). Chemical characterization of the smallest S‐nitrosothiol, HSNO; cellular cross‐talk of H2S and S‐nitrosothiols. J Am Chem Soc 134: 12016–12027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipovic MR, Zivanovic J, Alvarez B, Banerjee R (2018). Chemical biology of H2S signaling through persulfidation. Chem Rev 118: 1253–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick NM, Bertho J, Patel KK, Petr GT, Bakradze E, Smith SB et al (2014). Dysregulation of system xc(‐) expression induced by mutant huntingtin in a striatal neuronal cell line and in R6/2 mice. Neurochem Int 76: 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Zhang W, Wu L, Yang G, Li H, Wang R (2012). Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci U S A 109: 2943–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco F, di Villa Bianca R, Mitidieri E, Cirino G, Sorrentino R, Mirone V (2012). Sildenafil effect on the human bladder involves the L‐cysteine/hydrogen sulfide pathway: a novel mechanism of action of phosphodiesterase type 5 inhibitors. Eur Urol 62: 1174–1180. [DOI] [PubMed] [Google Scholar]
- Gheibi S, Jeddi S, Kashfi K, Ghasemi A (2018). Regulation of vascular tone homeostasis by NO and H2S: implications in hypertension. Biochem Pharmacol 149: 42–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannakopoulou E, Konstantinou F, Ragia G, Tavridou A, Karaglani M, Chatzaki E et al (2017). Epigenetics‐by‐sex interaction for coronary artery disease risk conferred by the cystathionine γ‐lyase gene promoter methylation. OMICS 21: 741–748. [DOI] [PubMed] [Google Scholar]
- Group (1993). A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's disease collaborative research group. Cell 72: 971–983. [DOI] [PubMed] [Google Scholar]
- Guo C, Liang F, Shah Masood W, Yan X (2014). Hydrogen sulfide protected gastric epithelial cell from ischemia/reperfusion injury by Keap1 s‐sulfhydration, MAPK dependent anti‐apoptosis and NF‐kappaB dependent anti‐inflammation pathway. Eur J Pharmacol 725: 70–78. [DOI] [PubMed] [Google Scholar]
- Gupta S, Kruger WD (2011). Cystathionine β‐synthase deficiency causes fat loss in mice. PLoS One 6: e27598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y et al (2005). S‐nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol 7: 665–674. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M et al (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11: 619–633. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan MI, Boosen M, Schaefer L, Kozlowska J, Eisel F, von Knethen A et al (2012). Platelet‐derived growth factor‐BB induces cystathionine γ‐lyase expression in rat mesangial cells via a redox‐dependent mechanism. Br J Pharmacol 166: 2231–2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Harputlugil E, Zhang Y, Ruckenstuhl C, Lee BC, Brace L et al (2015). Endogenous hydrogen sulfide production is essential for dietary restriction benefits. Cell 160: 132–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine C, Kim HJ, Zhu Y, Harputlugil E, Longchamp A, Matos MS et al (2017). Hypothalamic‐pituitary axis regulates hydrogen sulfide production. Cell Metab 25: 1320–1333.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hourihan JM, Kenna JG, Hayes JD (2013). The gasotransmitter hydrogen sulfide induces Nrf2‐target genes by inactivating the Keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys‐226 and cys‐613. Antioxid Redox Signal 19: 465–481. [DOI] [PubMed] [Google Scholar]
- Ishii I, Akahoshi N, Yamada H, Nakano S, Izumi T, Suematsu M (2010). Cystathionine γ‐lyase‐deficient mice require dietary cysteine to protect against acute lethal myopathy and oxidative injury. J Biol Chem 285: 26358–26368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkowska H, Roman HB, Hirschberger LL, Sasakura K, Nagano T, Hanaoka K et al (2014). Primary hepatocytes from mice lacking cysteine dioxygenase show increased cysteine concentrations and higher rates of metabolism of cysteine to hydrogen sulfide and thiosulfate. Amino Acids 46: 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil H, Kabil O, Banerjee R, Harshman LG, Pletcher SD (2011a). Increased transsulfuration mediates longevity and dietary restriction in Drosophila. Proc Natl Acad Sci U S A 108: 16831–16836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Vitvitsky V, Xie P, Banerjee R (2011b). The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal 15: 363–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O, Yadav V, Banerjee R (2016). Heme‐dependent metabolite switching regulates H2S synthesis in response to endoplasmic reticulum (ER) stress. J Biol Chem 291: 16418–16423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamoun P, Belardinelli MC, Chabli A, Lallouchi K, Chadefaux‐Vekemans B (2003). Endogenous hydrogen sulfide overproduction in Down syndrome. Am J Med Genet A 116A: 310–311. [DOI] [PubMed] [Google Scholar]
- Kandil S, Brennan L, McBean GJ (2010). Glutathione depletion causes a JNK and p38MAPK‐mediated increase in expression of cystathionine‐γ‐lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem Int 56: 611–619. [DOI] [PubMed] [Google Scholar]
- Kery V, Poneleit L, Kraus JP (1998). Trypsin cleavage of human cystathionine β‐synthase into an evolutionarily conserved active core: structural and functional consequences. Arch Biochem Biophys 355: 222–232. [DOI] [PubMed] [Google Scholar]
- Kozich V, Sokolova J, Klatovska V, Krijt J, Janosik M, Jelinek K et al (2010). Cystathionine β‐synthase mutations: effect of mutation topology on folding and activity. Hum Mutat 31: 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK (2011). H2S‐induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal 4: ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger WD (2017). Cystathionine β‐synthase deficiency: of mice and men. Mol Genet Metab 121: 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P et al (2010). Aberrant Rab11‐dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington's disease. J Neurosci 30: 4552–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Kim WK, Choi YB, Kumar S, D'Emilia DM, Rayudu PV et al (1997). Neurotoxicity associated with dual actions of homocysteine at the N‐methyl‐D‐aspartate receptor. Proc Natl Acad Sci U S A 94: 5923–5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majtan T, Pey AL, Fernandez R, Fernandez JA, Martinez‐Cruz LA, Kraus JP (2014). Domain organization, catalysis and regulation of eukaryotic cystathionine β‐synthases. PLoS One 9: e105290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S, Yang G, Wang R (2011). A critical life‐supporting role for cystathionine γ‐lyase in the absence of dietary cysteine supply. Free Radic Biol Med 50: 1280–1287. [DOI] [PubMed] [Google Scholar]
- Martin JA, Pereda J, Martinez‐Lopez I, Escrig R, Miralles V, Pallardo FV et al (2007). Oxidative stress as a signal to up‐regulate γ‐cystathionase in the fetal‐to‐neonatal transition in rats. Cell Mol Biol (Noisy‐le‐Grand) 53 (Suppl): OL1010–OL1017. [PubMed] [Google Scholar]
- Martin JA, Sastre J, de la Asuncion JG, Pallardo FV, Vina J (2001). Hepatic γ‐cystathionase deficiency in patients with AIDS. JAMA 285: 1444–1445. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Shea TB (2003). Folate and homocysteine metabolism in neural plasticity and neurodegenerative disorders. Trends Neurosci 26: 137–146. [DOI] [PubMed] [Google Scholar]
- McBean GJ (2012). The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids 42: 199–205. [DOI] [PubMed] [Google Scholar]
- McBean GJ (2017). Cysteine, glutathione, and thiol redox balance in astrocytes. Antioxidants (Basel) 6: E62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBean GJ, Aslan M, Griffiths HR, Torrao RC (2015). Thiol redox homeostasis in neurodegenerative disease. Redox Biol 5: 186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier M, Janosik M, Kery V, Kraus JP, Burkhard P (2001). Structure of human cystathionine β‐synthase: a unique pyridoxal 5′‐phosphate‐dependent heme protein. EMBO J 20: 3910–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modis K, Coletta C, Erdelyi K, Papapetropoulos A, Szabo C (2013). Intramitochondrial hydrogen sulfide production by 3‐mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. FASEB J 27: 601–611. [DOI] [PubMed] [Google Scholar]
- Morikawa T, Kajimura M, Nakamura T, Hishiki T, Nakanishi T, Yukutake Y et al (2012). Hypoxic regulation of the cerebral microcirculation is mediated by a carbon monoxide‐sensitive hydrogen sulfide pathway. Proc Natl Acad Sci U S A 109: 1293–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosharov E, Cranford MR, Banerjee R (2000). The quantitatively important relationship between homocysteine metabolism and glutathione synthesis by the transsulfuration pathway and its regulation by redox changes. Biochemistry 39: 13005–13011. [DOI] [PubMed] [Google Scholar]
- Murano S (1995). Potential for pharmacological intervention in Werner syndrome. Drugs Aging 7: 449–458. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK et al (2009). H2S signals through protein S‐sulfhydration. Sci Signal 2: ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa AK, Sikka G, Gazi SK, Steppan J, Jung SM, Bhunia AK et al (2011). Hydrogen sulfide as endothelium‐derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res 109: 1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu WN, Yadav PK, Adamec J, Banerjee R (2015). S‐glutathionylation enhances human cystathionine β‐synthase activity under oxidative stress conditions. Antioxid Redox Signal 22: 350–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Sbodio JI, Snyder SH (2018). Cysteine metabolism in neuronal redox homeostasis. Trends Pharmacol Sci 39: 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Sbodio JI, Xu R, Vandiver MS, Cha JY, Snowman AM et al (2014). Cystathionine γ‐lyase deficiency mediates neurodegeneration in Huntington's disease. Nature 509: 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2012). H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol 13: 499–507. [DOI] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2014). Neurodegeneration in Huntington's disease involves loss of cystathionine γ‐lyase. Cell Cycle 13: 2491–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2015a). H2S: a novel gasotransmitter that signals by sulfhydration. Trends Biochem Sci 40: 687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2015b). Modes of physiologic H2S signaling in the brain and peripheral tissues. Antioxid Redox Signal 22: 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem Pharmacol 149: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietri R, Roman‐Morales E, Lopez‐Garriga J (2011). Hydrogen sulfide and hemeproteins: knowledge and mysteries. Antioxid Redox Signal 15: 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus DJ, Calvert JW, Butler J, Lefer DJ (2014). The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica (Cairo) 2014: 768607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudova A, Bauman Z, Braun A, Vitvitsky V, Lu SC, Banerjee R (2006). S‐adenosylmethionine stabilizes cystathionine β‐synthase and modulates redox capacity. Proc Natl Acad Sci U S A 103: 6489–6494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B, Cipriani S, Carino A, Simonetti M, Zampella A, Fiorucci S (2015). Reversal of endothelial dysfunction by GPBAR1 agonism in portal hypertension involves a AKT/FOXOA1 dependent regulation of H2S generation and endothelin‐1. PLoS One 10: e0141082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renga B, Mencarelli A, Migliorati M, Distrutti E, Fiorucci S (2009). Bile‐acid‐activated farnesoid X receptor regulates hydrogen sulfide production and hepatic microcirculation. World J Gastroenterol 15: 2097–2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose P, Moore PK, Zhu YZ (2017). H2S biosynthesis and catabolism: new insights from molecular studies. Cell Mol Life Sci 74: 1391–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salloum FN, Chau VQ, Hoke NN, Abbate A, Varma A, Ockaili RA et al (2009). Phosphodiesterase‐5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein‐kinase g‐dependent generation of hydrogen sulfide. Circulation 120: S31–S36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbodio JI, Snyder SH, Paul BD (2016). Transcriptional control of amino acid homeostasis is disrupted in Huntington's disease. Proc Natl Acad Sci U S A 113: 8843–8848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbodio JI, Snyder SH, Paul BD (2018). Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington's disease. Proc Natl Acad Sci U S A 115: 780–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer S, Kim HW (2018). Effects and mechanisms of taurine as a therapeutic agent. Biomol Ther (Seoul) 26: 225–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Paul BD, Gadalla MM, Mustafa AK, Sen T, Xu R et al (2012). Hydrogen sulfide‐linked sulfhydration of NF‐κB mediates its antiapoptotic actions. Mol Cell 45: 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibuya N, Tanaka M, Yoshida M, Ogasawara Y, Togawa T, Ishii K et al (2009). 3‐Mercaptopyruvate sulfurtransferase produces hydrogen sulfide and bound sulfane sulfur in the brain. Antioxid Redox Signal 11: 703–714. [DOI] [PubMed] [Google Scholar]
- Singh S, Madzelan P, Banerjee R (2007). Properties of an unusual heme cofactor in PLP‐dependent cystathionine β‐synthase. Nat Prod Rep 24: 631–639. [DOI] [PubMed] [Google Scholar]
- Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R (2009). Relative contributions of cystathionine β‐synthase and γ‐cystathionase to H2S biogenesis via alternative trans‐sulfuration reactions. J Biol Chem 284: 22457–22466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AD, Refsum H, Bottiglieri T, Fenech M, Hooshmand B, McCaddon A et al (2018). Homocysteine and dementia: an international consensus statement. J Alzheimers Dis 62: 561–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegborn C, Clausen T, Sondermann P, Jacob U, Worbs M, Marinkovic S et al (1999). Kinetics and inhibition of recombinant human cystathionine γ‐lyase. Toward the rational control of transsulfuration. J Biol Chem 274: 12675–12684. [DOI] [PubMed] [Google Scholar]
- Stipanuk MH, Ueki I (2011). Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J Inherit Metab Dis 34: 17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Collins R, Huang S, Holmberg‐Schiavone L, Anand GS, Tan CH et al (2009). Structural basis for the inhibition mechanism of human cystathionine γ‐lyase, an enzyme responsible for the production of H2S. J Biol Chem 284: 3076–3085. [DOI] [PubMed] [Google Scholar]
- Szabo C (2017). Hydrogen sulfide, an enhancer of vascular nitric oxide signaling: mechanisms and implications. Am J Physiol Cell Physiol 312: C3–C15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Ransy C, Modis K, Andriamihaja M, Murghes B, Coletta C et al (2014). Regulation of mitochondrial bioenergetic function by hydrogen sulfide. Part I. Biochemical and physiological mechanisms. Br J Pharmacol 171: 2099–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talaei F, van Praag VM, Henning RH (2013). Hydrogen sulfide restores a normal morphological phenotype in Werner syndrome fibroblasts, attenuates oxidative damage and modulates mTOR pathway. Pharmacol Res 74: 34–44. [DOI] [PubMed] [Google Scholar]
- Uthus EO, Brown‐Borg HM (2003). Altered methionine metabolism in long living Ames dwarf mice. Exp Gerontol 38: 491–498. [DOI] [PubMed] [Google Scholar]
- Vandiver MS, Paul BD, Xu R, Karuppagounder S, Rao F, Snowman AM et al (2013). Sulfhydration mediates neuroprotective actions of parkin. Nat Commun 4: 1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V, Garg SK, Banerjee R (2011). Taurine biosynthesis by neurons and astrocytes. J Biol Chem 286: 32002–32010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V, Thomas M, Ghorpade A, Gendelman HE, Banerjee R (2006). A functional transsulfuration pathway in the brain links to glutathione homeostasis. J Biol Chem 281: 35785–35793. [DOI] [PubMed] [Google Scholar]
- Wang R (2012). Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol Rev 92: 791–896. [DOI] [PubMed] [Google Scholar]
- Watanabe M, Osada J, Aratani Y, Kluckman K, Reddick R, Malinow MR et al (1995). Mice deficient in cystathionine β‐synthase: animal models for mild and severe homocyst(e)inemia. Proc Natl Acad Sci U S A 92: 1585–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman M, Li L, Kostetski I, Chu SH, Siau JL, Bhatia M et al (2006). Evidence for the formation of a novel nitrosothiol from the gaseous mediators nitric oxide and hydrogen sulphide. Biochem Biophys Res Commun 343: 303–310. [DOI] [PubMed] [Google Scholar]
- Wright DJ, Renoir T, Smith ZM, Frazier AE, Francis PS, Thorburn DR et al (2015). N‐acetylcysteine improves mitochondrial function and ameliorates behavioral deficits in the R6/1 mouse model of Huntington's disease. Transl Psychiatry 5: e492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pei Y, Teng H, Cao Q, Wang R (2011). Specificity protein‐1 as a critical regulator of human cystathionine γ‐lyase in smooth muscle cells. J Biol Chem 286: 26450–26460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K et al (2008). H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ‐lyase. Science 322: 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S et al (2013). Hydrogen sulfide protects against cellular senescence via S‐sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal 18: 1906–1919. [DOI] [PubMed] [Google Scholar]
- Zhang L, Yang G, Tang G, Wu L, Wang R (2011). Rat pancreatic level of cystathionine γ‐lyase is regulated by glucose level via specificity protein 1 (SP1) phosphorylation. Diabetologia 54: 2615–2625. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Martin E, Sharina I, Esposito I, Szabo C, Bucci M et al (2016). Regulation of soluble guanylyl cyclase redox state by hydrogen sulfide. Pharmacol Res 111: 556–562. [DOI] [PMC free article] [PubMed] [Google Scholar]