

Abstract
Background and Purpose
The signalling associated with hydrogen sulfide (H2S) remains to be established, and recent studies have alluded to the possibility that H2S‐derived species play important roles. Of particular interest are hydropersulfides (RSSH) and related polysulfides (RSSnR, n > 1). This work elucidates the fundamental chemical relationship between these sulfur species as well as examines their biological effects.
Experimental Approach
Using standard analytical techniques (1H‐NMR and MS), the equilibrium reactions between H2S, disulfides (RSSR), RSSH, dialkyltrisulfides (RSSSR) and thiols (RSH) were examined. Their ability to protect cells from electrophilic and/or oxidative stress was also examined using cell culture.
Key Results
H2S, RSSR, RSSH, RSSSR and RSH are all in a dynamic equilibrium. In a biological system, these species can exist simultaneously, and thus, it is difficult to discern which species is (are) the biological effector(s). Treatment of cells with the dialkyl trisulfide cysteine trisulfide (Cys‐SSS‐Cys) resulted in high intracellular levels of hydropersulfides and protection from electrophilic stress.
Conclusions and Implications
In aqueous systems, the reaction between H2S and RSSR results in the formation of equilibria whereby H2S, RSH, RSSR, RSSH and RSSSR are present. In a biological system, any of these species can be responsible for the observed biological activity. These equilibrium species can also be generated via the reaction of RSH with RSSSR. Due to these equilibria, Cys‐SSS‐Cys can be a method for generating any of the other species. Importantly, HEK293T cells treated with Cys‐SSS‐Cys results in increased levels of hydropersulfides, allowing examination of the biological effects of RSSH.
Linked Articles
This article is part of a themed section on Chemical Biology of Reactive Sulfur Species. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.4/issuetoc
Abbreviations
- 2HED
2‐hydroxyethyl disulfide
- 2HET
2‐hydroxyethyl trisulfide
- 2MESSH
2‐ME hydropersulfide
- 2ME
2‐mercaptoethanol
- DSS
4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid
- CBS
cystathionine β‐synthase
- CSE
cystathionine γ‐lyase
- Cys‐SSS‐Cys
cysteine trisulfide
- Cys‐SH
cysteine
- Cys‐SS‐Cys
cystine
- GSH
glutathione
- GSSG
GSH disulfide
- GSSSG
GSH trisulfide
- IAM
iodoacetamide
- MCP‐SSH
MCPD hydropersulfide
- NAP
N‐acetyl penicillamine
- NEM
N‐ethylmaleimide
- MCPD
S‐methoxycarbonyl penicillamine disulfide
- HPE‐IAM
β‐(4‐hydroxyphenyl) ethyl iodoacetamide
Introduction
Hydrogen sulfide (H2S) is an endogenously generated small molecule bioregulator with a wide array of physiological functions (for recent reviews, see Kashfi and Olson, 2013; Polhemus and Lefer, 2014; Kimura, 2014). For example, H2S has been proposed to have a role in LTP, smooth muscle relaxation, inhibition of inflammation, activation of KATP channels and protection against ischaemia–reperfusion injury, just to name a few. However, it has also been hypothesized that some of the effects attributed to H2S may instead be due to the presence of hydropersulfides (RSSH) (e.g. Ida et al., 2014; Ono et al., 2014) since RSSH can be in equilibrium with H2S in the presence of an oxidized thiol (i.e. a disulfide, RSSR) via Reaction (1) (Cavallini et al., 1970; Francoleon et al., 2011; Vasas et al., 2015) and, possibly more importantly, the biosynthesis of H2S can occur via RSSH intermediacy (vide infra). (Note: the term RSSH will be used generally to describe both the neutral RSSH and the anionic RSS− forms, unless specifically noted.)
| (1) |
Although the forward and reverse reactions shown in Reaction (1) can be slow (at least in a purely chemical system) (Cuevasanta et al., 2015), it appears that H2S and RSSH are intimately linked and likely to be present simultaneously via Reaction (1) in a biological system. This makes the assignment of the compound responsible for the biological activity difficult and/or indicates that multiple effectors can be responsible for the observed effects. Furthermore, the chemistry represented by Reaction (1) is likely to be greatly oversimplified in that other reactions can potentially emanate from this initial equilibrium (e.g. see Bogdándi et al., 2019, this issue). For example, RSSH species are superior nucleophiles compared to the corresponding RSH species and can react further with other electrophiles in solution (i.e. RSSR) to give a dialkyltrisulfide (RSSSR, Reaction (2)) (e.g. Ono et al., 2014; Bogdándi et al., 2019, this issue).
| (2) |
Clearly, other chemical reactions/equilibria are possible (vide infra), making the simple reaction of H2S with RSSR potentially more complex than previously recognized.
It is important to note that the generation of RSSSR is not dependent on the presence of H2S in a biological system. For example, RSSH can be formed via the conversion of cystine (Cys‐SS‐Cys) to the corresponding cysteine hydropersulfide (Cys‐SSH) and pyruvate or serine via the actions of cystathionine γ‐lyase (CSE) or cystathionine β‐synthase (CBS) (Reaction (3)) (e.g. Yamanishi and Tuboi, 1981; Stipanuk, 1986; Ida et al., 2014).
| (3) |
In this system, subsequent reactions of the highly nucleophilic Cys‐SSH product with the electrophilic enzyme substrate Cys‐SS‐Cys would generate the corresponding cysteine trisulfide (or thiocystine, Cys‐SSS‐Cys, Reaction (2)) in the absence of H2S. Of course, H2S can eventually be formed in a biological system where thiols will undoubtedly be present via the simple reverse of Reaction (1), and the eventual formation of an equilibrium mixture of all species is likely. More recently, it has been reported that Cys‐SSH can also be generated from cysteine (Cys‐SH) via the actions of cysteine t‐RNA synthases, leading to translational incorporation of Cys‐SSH into proteins along with release of free Cys‐SSH (Akaike et al., 2017).
Numerous studies have utilized Reaction (1) to generate RSSH in situ, and there seems to be little doubt that RSSH can be generated in this way (e.g. Cavallini et al., 1970; Francoleon et al., 2011; Saund et al., 2015). However, the potential for Reaction (1) to lead to further chemistry (such as Reaction (2)) has not been adequately accounted for in these previous studies, and it is certain that other sulfur species besides those shown in the equilibrium reaction may be present. It is also possible that biologically generated RSSH participates in numerous other equilibria as well and a complete description of the chemical biology of hydropersulfides may require consideration of these other processes. Thus, this study examines Reaction (1) and evaluates the possibility of further chemistry occurring that may be relevant to biologically generated RSSH species. Importantly, a companion paper in this issue (Bogdándi et al., 2019) describes aspects of the equilibrium reactions mentioned above. The paper herein serves to examine the general scope of these equilibria with other RSH/RSSR as well as further investigates the generation and potential biological utility of an important species associated with this chemistry, specifically RSSSR.
Methods
Instrumentation
All NMR analyses were performed using an Agilent 400 MHz NMR spectrometer running on vnmrj software. Water suppression for all samples was achieved using a (1H) PreSat method in the vnmrj software. All NMR solvents were 10% D2O in 100 mM phosphate buffer (pH 7.4) containing 1 mM DSS as an internal standard.
Mass spectra of chemical studies were recorded using a Thermo Triple Quadrupole Electrospray Ionization Mass Spectrometer (ESI‐MS) in positive ion mode coupled with XCalibur 2.1 software. All samples were directly injected via a syringe pump at a flow rate of 5 μL·min−1 using N2 as a sheath gas and Ar as a collision gas. Ion optics were optimized using authentic samples of disulfides. The capillary temperature for all samples was 250°C and spray voltages were 3000 V for 2‐hydroxyethyl disulfide (2HED) samples and 2500 V for glutathione disulphide (GSSG) samples. Mass spectral analysis of cell lysates was obtained using a triple quadrupole (Q) mass spectrometer LCMS‐8050 (Shimadzu, Kyoto, Japan) coupled to the Nexera UHPLC system (Shimadzu, Kyoto, Japan).
Synthesis of cysteine trisulfide (Cys‐SSS‐Cys, thiocystine)
Cys‐SSS‐Cys was synthesized following the general method of Savige et al. (1964) as described briefly below.
Cystine‐S‐monoxide was synthesized by dissolving Cys‐SS‐Cys in 1 M H2SO4 (approximately 10 mL of H2SO4 solution.g‐1 cystine) in an ice bath. Two equivalents of peracetic acid were then added followed by stirring (on ice) for 2 h. After the sample had been stirred, the pH was adjusted to 4 using pyridine. Cystine‐S‐monoxide was then precipitated from solution via the addition of ethanol (approximately 3 times the volume of the initial H2SO4 solution). The precipitate was filtered and dried. The cystine‐S‐monoxide was then dissolved in 1M H2SO4 (approximately 10 mL of H2SO4 solution.g‐1 cystine‐S‐monoxide). Two equivalents of solid sodium hydrosulfide (NaHS) were then added to a separate flask and the cystine‐S‐monoxide solution added to the NaHS. After this addition, the flask was sealed with a septum and stirred at room temperature for 1 h. The pH was then adjusted to 3.5 using pyridine. A 50/50 mixture of ethanol/THF was then added (approximately 2–3 times the volume of the initial H2SO4 solution) to precipitate Cys‐SSS‐Cys. The precipitate was filtered, washed with 50/50 ethanol/THF and dried. Analysis was carried out using 1H‐NMR and ESI/MS. The typical purity of the resulting product was >90% (by NMR) with cystine as the only detectable impurity. Cys‐SSS‐Cys: 1H‐NMR (400 MHz, D2O) δ 4.015 (dd, J = 8.0, 4.0 Hz, 1H), δ 3.42 (dd, J = 15.2, 4.0 Hz, 1H), δ 3.23 (dd, J = 15.2, 8.0 Hz, 1H). Mass spectral analysis (ESI‐MS) m/z [M + H]+ calc. for C6H12N2O4S3, 273.37; found 272.98.
Synthesis of S‐methoxycarbonyl penicillamine disulfide (MCPD)
The hydropersulfide donor MCPD used previously by our lab (Millikin et al., 2016; Bianco et al., 2016) was synthesized via the method of Artaud and Galardon (2014).
NMR analysis of the NaHS/RSSR equilibrium
Stock solutions of 2HED (200 mM) and NaHS (50 and 100 mM) were prepared in 100 mM phosphate buffer (pH 7.4). DSS (internal standard) stock solutions (10 mM) were prepared in D2O. For all experiments, DSS was diluted in an NMR tube filled with buffer to a final concentration of 1 mM. For reactions between 2HED and NaHS, the stock solution of 2HED was added to the DSS‐containing NMR tube to a final concentration of 10 mM. NaHS was then added to the NMR tube to final concentrations of 5, 10 or 15 mM. All solutions were equilibrated at room temperature for 30 min, followed by 1H‐NMR analysis.
For cyanolysis reactions, stock solutions of 2HED and NaHS were diluted in an NMR tube containing buffer with 1 mM DSS to final concentrations of 20 and 10 mM respectively. The solution stood at room temperature for 30 min. At this time, a stock solution of sodium cyanide (100 mM in buffer) was diluted into the NMR tube to a final concentration of 10 mM. The cyanolysis reaction was allowed to proceed at room temperature for 1 h, followed by 1H‐NMR analysis.
ESI‐MS analysis of the NaHS/RSSR equilibrium
A stock solution of NaHS (50 mM) was prepared in 50 mM ammonium bicarbonate buffer (pH 7.4). Disulfide solutions were further diluted in the same buffer to final concentrations of 1 mM before ESI‐MS analysis. For the reaction of 2HED and NaHS, 2HED was diluted in buffer to a final concentration of 1 mM. NaHS was then added to a final concentration of 0.5 mM, and the mixture allowed to stand at room temperature for 30 min prior to ESI‐MS analysis.
NMR analysis for the competitive trapping of hydropersulfides
Stock solutions of 2HED, NaHS and DSS were prepared as stated above (NMR analysis of the NaHS/RSSR equilibrium). For reactions of 2HED, 10 mM NaHS was added to an NMR tube of 20 mM 2HED in 100 mM phosphate buffer (pH 7.4) with 1 mM DSS, and the mixture equilibrated for 30 min at room temperature. Iodoacetamide (IAM) or N‐ethylmaleimide (NEM; 1, 2, 3, 4 or 5 mM final concentration) was then added, and the reaction proceeded at room temperature for 1 h, followed by 1H‐NMR analysis.
ESI‐MS analysis for the competitive trapping of hydropersulfides
Stock solutions of 2HED and NaHS were prepared as above (ESI‐MS analysis of the NaHS/RSSR equilibrium). For reactions, 0.5 mM NaHS was added to a solution of 1 mM 2HED in 50 mM ammonium bicarbonate buffer (pH 7.4), and the mixture equilibrated for 30 min at room temperature. IAM (0.5 mM) was then added, and the reaction proceeded for 1 h at room temperature, followed by ESI‐MS analysis.
ESI‐MS analysis of the trisulfide equilibrium
Stock solutions of glutathione trisulfide (GSSSG; 25 mM) and N‐acetyl penicillamine (NAP; 25 mM) were prepared in 50 mM ammonium bicarbonate buffer (pH 7.4). For reactions, 250 μM NAP was added to a solution of 250 μM GSSSG in the same buffer, and the reaction proceeded at room temperature for 1 h, followed by ESI‐MS analysis.
ESI‐MS analysis for the reaction of RSSH and RSSR
A stock solution of GSSG (100 mM) was prepared in 50 mM ammonium bicarbonate buffer (pH 7.4). A stock solution of MCPD (20 mM) was prepared in deionized water. For the reaction, 200 μM MCPD was added to 1 mM GSSG, and the reaction proceeded at room temperature for 30 min followed by ESI‐MS analysis.
Cell culture and treatments for Cys‐SSS‐Cys uptake assay
HEK293T cells (CRL‐11268, American Type Culture Collection, Manassas, VA, USA) were cultured in DMEM (high‐glucose; Sigma‐Aldrich) that was supplemented with 10% FBS and 1% penicillin–streptomycin, under standard cell culture conditions (37°C, humidified, 5% CO2/95% air).
Analysis of intracellular hydropersulfides and other polysulfides with cultured HEK293T cells using LC‐ESI‐MS/MS analyses
Intracellular hydropersulfide levels in cultured HEK293T cells were quantified by LC‐MS/MS with HPE‐IAM as a trapping agent for hydropersulfides and polysulfides as reported recently (Numakura et al., 2017; Akaike et al., 2017). Briefly, the cultured cells were washed with PBS twice and homogenized in a cold methanol solution containing 1 mM HPE‐IAM, after which the cell lysates were incubated at 37°C for 20 min. After centrifugation, aliquots of the supernatants of the lysates were diluted 20 times with 0.1% formic acid containing known amounts of isotope‐labelled internal standards, which were then analysed via LC‐ESI‐MS/MS for per/polysulfide determination. A triple quadrupole (Q) mass spectrometer LCMS‐8050 (Shimadzu, Kyoto, Japan) coupled to the Nexera UHPLC system (Shimadzu, Kyoto, Japan) was used to perform LC‐ESI‐MS/MS. Per/polysulfide derivatives were separated by means of Nexera UHPLC with a YMC‐Triart C18 column (50 × 2.0 mm inner diameter) under the following elution conditions: mobile phases A (0.1% formic acid) with a linear gradient of mobile phases B (0.1% formic acid in methanol) from 5 to 90% for 15 min at a flow rate of 0.2 mL·min−1 at 40°C. MS spectra were obtained with each temperature of the ESI probe, desolvation line and heat block at 300, 250 and 400°C respectively, and the nebulizer, heating and drying nitrogen gas flows were set to 3, 10 and 10 L·min−1 respectively. Various per/polysulfide derivatives were identified and quantified by means of multiple reaction monitoring. Experiments were carried out 5 separate times (each time in triplicate). For each experiment, the values from the triplicates were averaged to give a single value. Experimental results are presented as the averages of the five individual experiments for an n = 5. Experimental protocols did not require sample blinding or randomization since sample analyses were not subjective and not susceptible to experimental bias.
Examination of Cys‐SSS‐Cys pretreatment on cell viability
HEK293T cells were plated at 3 × 104 cells per well in a 96‐well plate. The following day, media was removed and replaced with DMEM w/o Cys/Met (Gibco) alone or with 1 mM Cys‐SSS‐Cys. After 1.5 h, media were replaced and cells treated with H2O2 (Fisher Scientific, Waltham, MA, USA) or NEM (Sigma) for another 6 h in complete media. For WST8 assays (Cayman Chemical Co., Ann Arbor, MI, USA), the reagent was added for the final 2 h and then the absorbance read at 450 nm. For Sytox Green assays (Life Technologies, Carlsbad, CA, USA), the reagent was added after the 6 h and then the fluorescence (485/528 nm) measured. All treatments were performed in triplicate and samples read on a Synergy HT plate reader (BioTek Instruments, Winooski, VT, USA). Experiments were carried out 5 separate times. For each experiment, the values from the triplicates were averaged to give a single value. Experimental results are presented as the averages of the five individual experiments for an n = 5. Experimental protocols did not require sample blinding or randomization since sample analyses were not subjective and not susceptible to experimental bias.
Data and statistical analysis
Statistical analyses were performed with GraphPad Prism software ver. 6.0 (GraphPad Software, San Diego, CA, USA). Results are presented as means from five experiments run separately. For statistical comparisons, a two‐way ANOVA was utilized followed by Bonferroni post hoc tests, with significance set at P < 0.05. Post hoc tests were run only if F achieved P < 0.05, and there was no significant variance inhomogeneity. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2018).
Materials
Cystine (Cys‐SS‐Cys), cysteine (Cys‐SH), glutathione (GSH), GSH disulfide (GSSG), 2‐mercaptoethanol (2ME), 2‐hydroxyethyl disulfide (2HED), 4,4‐dimethyl‐4‐silapentane‐1‐sulfonic acid (DSS), iodoacetamide (IAM), N‐acetyl penicillamine (NAP), sulfasalazine and N‐ethylmaleimide (NEM) were purchased from Sigma‐Aldrich (St. Louis, MO, USA). Sodium hydrosulfide (NaHS) was purchased from Strem Chemicals, Inc. (Newburyport, MA, USA). S‐Benzyl‐L‐cysteine was purchased from Wako Chemicals (Osaka, Japan). β‐(4‐Hydroxyphenyl) ethyl iodoacetamide (HPE‐IAM) was purchased from Molecular BioSciences (Boulder, CO, USA). All other chemicals were purchased from commercial sources and were the highest purity available. GSH trisulfide (GSSSG) was a gift from Prof. Herbert Nagasawa and synthesized via previously published procedures (Savige et al., 1964).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
Results
Analysis of the NaHS/RSSR equilibrium
Bogdándi et al. (2019, this issue) have described the dynamic equilibrium that establishes between numerous sulfur and polysulfur‐containing species. For example, the reaction of GSSG with H2S results in the formation of GSSH, GSH and GSSSG as equilibrium partners with the initial reactants (Reactions (1) and (2)). To determine whether these reactions are specific to GSSG or are generally applicable to other disulfides, a series of studies analogous to those reported by Bogdándi et al. (2019, this issue) using a chemically simple disulfide have been carried out. Thus, the reaction of NaHS with 2‐hydroxyethyl disulfide (HOCH2CH2SSCH2CH2OH, 2HED), the disulfide of the thiol 2‐mercaptoethanol (HOCH2CH2SH, 2ME) was examined. Increasing additions of NaHS (5–15 mM) to 2HED (10 mM) caused subsequent concentration‐dependent increases in the formation of two new species after equilibration (Figure 1A). Comparison of the newly formed species with standard samples (data not shown) indicates these species are 2ME (δ = 2.67–2.70 and 3.68–3.72 ppm) and 2‐hydroxyethyl trisulfide (HOCH2CH2SSSCH2CH2OH, 2HET) (δ = 3.08–3.11 and 3.91–3.94 ppm). Of additional note is that the ratio of formed 2ME:2HET is 2:1, which would be expected for formation of 2HET via Reactions (1) and (2), as described below). Interestingly, addition of 10 mM NaCN to an equilibrated sample of 20 mM 2HED + 10 mM NaHS (Figure 1B, bottom spectrum) causes the disappearance of 2HET (Figure 1B, top spectrum), supporting the reaction of CN− with RSSSR.
Figure 1.
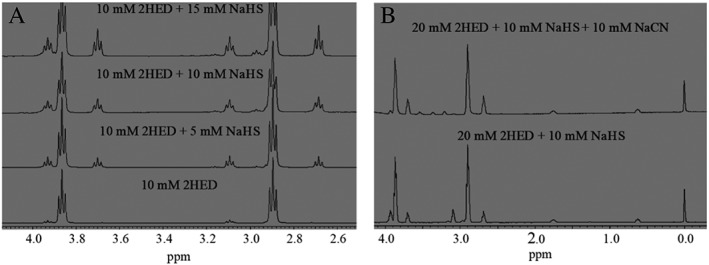
(A) Selected region for the stacked 1H‐NMR spectra of equilibrated samples (post 30 min) of 10 mM 2HED with increasing additions of NaHS (0, 5, 10 and 15 mM). The newly formed species correspond to 2HET (δ = 3.08–3.11 and 3.91–3.94 ppm) and 2ME (δ = 2.67–2.70 and 3.68–3.72 ppm) and increase in concentration with increasing additions of NaHS. (B) Stacked 1H‐NMR spectra of an equilibrated sample (post 30 min) of 20 mM 2HED + 10 mM NaHS (bottom spectrum) and addition of 10 mM NaCN (top spectrum) to this equilibrated sample. Addition of NaCN eliminates 2HET (δ = 3.08–3.11 and 3.91–3.94 ppm), making it absent in the top spectrum. All spectra were measured in 100 mM phosphate buffer (pH 7.4) with 10% D2O and 1 mM DSS as an internal reference.
Data collected from NMR analysis (Figure 1) indicate the addition of NaHS to 2HED results in the formation of RSH and RSSSR species after equilibration (consistent with the work of Bogdándi et al., 2019, this issue, with GSSG), indicating the general nature of this chemistry. ESI‐MS analysis of the 2HED/NaHS reaction also confirms the generation of the corresponding 2HET, which is consistent with the results of NMR analysis here and of Bogdandi and co‐workers for GSSG/NaHS (Bogdándi et al., 2019, this issue) (data not shown).
Analysis for the competitive trapping of hydropersulfides
The results above and those of Bogdándi et al. (2019, this issue) indicate that addition of NaHS to RSSR results in the formation of the corresponding RSH and RSSSR species as a general reaction. It has been widely accepted that the initial reaction of H2S with RSSR results in an equilibrated mixture with the corresponding RSH and RSSH species (Reaction (1)). However, lack of detection of RSSH in the equilibrated systems herein (described above) implies that under the conditions of these experiments, the RSSH species are subject to further reaction. This is also consistent with the detection of RSSSR (vide supra). It is therefore reasonable to consider that RSSH is fleeting and participates in a second equilibrium that ultimately results in RSSSR formation (Reaction (2)). If indeed a highly nucleophilic RSSH is fleeting due to subsequent reaction with electrophiles present in the reaction mixture (e.g. RSSR, Reaction (2)), it is expected that RSSSR formation could be avoided or lessened by the presence of an alternate electrophile capable of trapping the intermediate RSSH (Reaction (4)). In other words, trapping of RSSH by an exogenous electrophile (Reaction (4)) would be competitive with trapping of RSSH by RSSR (Reaction (2)) (as reported by Bogdándi et al., 2019, this issue, for GSSG), and thus by increasing the concentration of this exogenous electrophile, a leftward shift in the equilibrium shown in Reaction (2) would be expected, and RSSSR levels would decrease.
| (4) |
Indeed, increased addition of the electrophile IAM (0.5–2.5 mM) to an equilibrated sample of 20 mM 2HED + 10 mM NaHS caused subsequent decreases in the concentration 2HET formed (δ = 3.08–3.11 and 3.91–3.94 ppm) (Figure 2A). Additionally, increasing additions of the alternate electrophile NEM (0–5 mM) also caused subsequent decreases in the amount of 2HET formed (Figure 2B, black diamonds), albeit to a lesser extent compared to IAM (Figure 2B, black squares). Again, as was the case for GSSH (Bogdándi et al., 2019, this issue), it appears that the hydropersulfide of 2ME (2MESSH) preferentially reacts with 2HED in the presence of NEM, and this preference occurs to a lesser extent in the presence of IAM.
Figure 2.
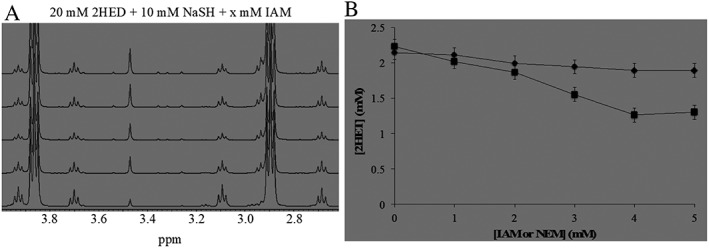
(A) Selected region for stacked 1H‐NMR spectra of 20 mM 2HED + 10 mM NaHS with increasing additions of IAM (1–5 mM, bottom to top spectrum respectively). Increasing additions of IAM causes subsequent decreases in the concentration of 2ME (δ = 2.67–2.70 and 3.68–3.72 ppm) and 2HET (δ = 3.08–3.11 and 3.91–3.94 ppm) because IAM competes with 2HED for the trapping of the intermediate 2ME‐SSH. (B) Plot for (2HET) as a function of increasing concentrations of exogenous electrophiles IAM (black squares) or NEM (black diamonds) (0–5 mM). Increasing addition of both electrophiles causes a subsequent decrease in the concentration of 2HET formed. All 1H‐NMR experiments were performed in 100 mM phosphate buffer (pH 7.4) with 10% D2O and 1 mM DSS as an internal reference.
ESI‐MS analysis for the competitive trapping of hydropersulfides
The above data indicate that exogenous addition of an electrophile competes with RSSR for the trapping of RSSH. To verify this further, the products formed after addition of 0.5 mM IAM to an equilibrated sample of 1 mM 2HED + 0.5 mM NaHS were analysed via ESI‐MS. The spectrum shown in Figure 3 indicates that exogenously added IAM to an equilibrated solution of 2HED + NaHS traps 2MESSH, resulting in the 2MESSH‐acetamide trapped adduct [(2MESS + AM+H)+, m/z = 168.05]. It should also be noted that 2HET is no longer present in the ESI‐MS spectrum of 2HED + NaHS treated with IAM, indicating that trapping of 2MESSH by IAM causes a leftward shift in Reaction (2) with subsequent loss of 2HET, consistent with the data presented in Figure 2.
Figure 3.
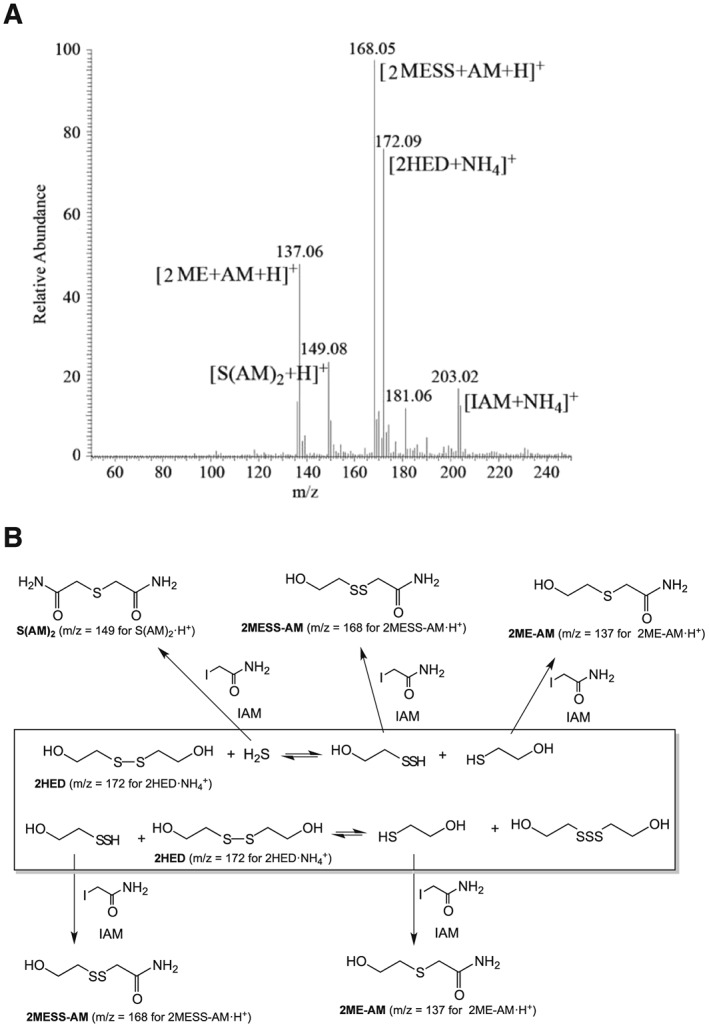
(A) ESI‐MS spectrum resulting from the addition of 0.5 mM IAM to an equilibrated solution (post 30 min) of 1 mM 2HED + 0.5 mM NaHS. Addition of IAM reacts with and traps HS− {[S (AM)2 + H]+}, 2ME [(2ME + AM+H)+] and 2MESSH, [(2MESS + AM+H)+]. (B) Scheme of equilibrium and trapped species.
ESI‐MS analysis of the trisulfide equilibrium
Summation of Reaction (1) and (2) leads to the overall equilibrium of Reaction (5).
| (5) |
Consistent with the relevance of Reaction (5), Figure 1A shows that treatment of 2HED with NaHS forms both RSSSR and RSH in an approximate 1:2 ratio respectively. Additionally, the equilibrium of Reaction (5) implies that reaction of RSH with RSSSR should lead to formation of the corresponding RSSR and H2S (the reverse of Reaction (5)). To test this, 250 μM GSSSG was reacted with 250 μM of the thiol N‐acetyl penicillamine (NAP) and analysed by ESI‐MS. Figure 4A shows that treatment of GSSSG with NAP (post 3 h) results in the mixed NAP‐SS‐G disulfide [(NAP+GS + H)+, m/z = 497.18] with no other alkyl polysulfur species formed, consistent with the reverse of Reaction (5).
Figure 4.
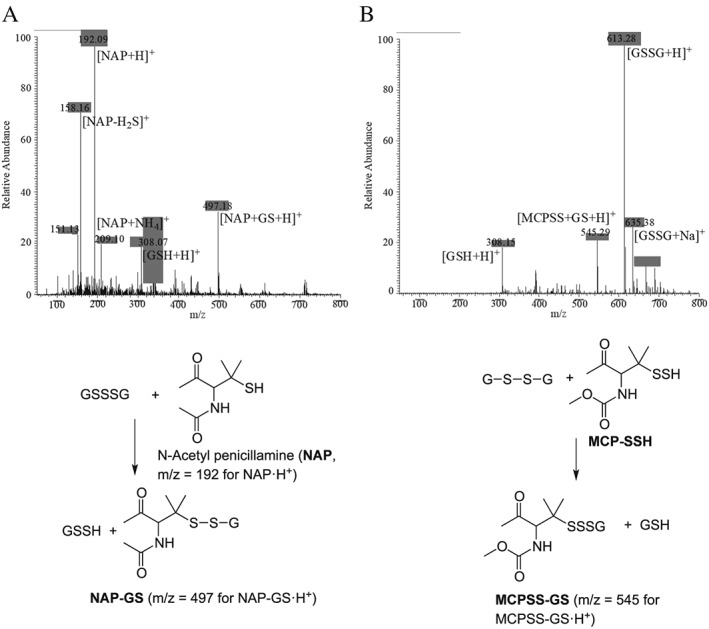
ESI‐MS spectrum for the reaction of (A) 250 μM NAP +250 μM GSSSG (post 3 h). The mixed disulfide [(NAP+GS + H)+, m/z = 497.18] is the only resulting species from this reaction. (B) Reaction of 200 μM MCP‐SSH + 1 mM GSSG. The mixed trisulfide [(MCPSS+GS + H)+, m/z = 545.29] and GSH [(GSH + H)+, m/z = 308.15] are the only resulting species from the reaction. All spectra were recorded in 50 mM ammonium bicarbonate buffer (pH 7.4). Relevant chemistry and mass assignments are shown directly below the spectra.
ESI‐MS analysis for the reaction of RSSH and RSSR
To this point, RSSH intermediates resulting from reaction of NaHS with RSSR have not been directly detected. As an indication of their presence, RSSH intermediates have been trapped via addition of the electrophiles IAM and NEM (Figures 2 and 3). To further confirm reaction of RSSH with RSSR, ultimately leading to formation of the corresponding RSSSR (Reaction (2)), a model H2S‐independent RSSH donor MCPD was used, as previously described by us (Millikin et al., 2016). The ESI‐MS spectrum corresponding to the decomposition of MCPD (releasing the MCP‐SSH hydropersulfide) in the presence of GSSG is shown in Figure 4B. As displayed in the spectrum, reaction of MCP‐SSH with GSSG leads to the mixed MCP‐SSS‐G trisulfide [Figure 4B, (MCPSS+GS + H)+, m/z = 545.29]. No other MCP‐SSH derived polysulfur species are detected, which is consistent with the primary fate of RSSH in the presence of RSSR being RSSSR formation (Reaction (2)). Interestingly, the persulfide donor MCPD is also a disulfide and yet no adduct associated with the persulfide reacting with the precursor donor disulfide species is detected. Thus, GSSG appears to outcompete this reaction since the only trisulfide that was readily detectable was the mixed trisulfide with GSSG (MCPSS+GS). It can also be envisioned that the persulfide formed in this experiment could have also reacted with a protonated hydropersulfide resulting in the corresponding trisulfide, RSSSR and HS− (RSS− + RSSH ➔ RSSSR + HS−). However, the pKa of RSSH has been reported to be 6.2–4.3 (Everett and Wardman, 1995; Cuevasanta et al., 2015). Thus, under the conditions of these experiments (pH 7.4), this reaction is unlikely to be competitive since the predominant species will be the nucleophilic anionic RSS− species with lesser amounts of the electrophilic protonated RSSH species present.
Dialkyltrisulfides as biological sources of hydropersulfides
Thus far, the results presented are consistent with the idea that RSSSR is in a dynamic equilibrium with RSSR, H2S, RSH and RSSH via Reactions (1) and (2). Moreover, RSSH levels are kept low in this system and higher order polysulfides are not readily generated (a rationale for this is given below). These results indicate that the equilibria set up via Reactions (1) and (2) (and summarized by Reaction (5)) can be approached starting from either RSSR/H2S or RSSSR/RSH (Reaction (5)). That is, RSSH can be generated starting with either equilibrium pair. In order to examine this in a biological system, cells were treated with cysteine trisulfide (Cys‐SSS‐Cys), with the hope that uptake would occur, thus establishing an intracellular RSSH equilibrium. Indeed, it was found that HEK293T cells treated with 0.2, 0.5 and 1 mM Cys‐SSS‐Cys for 1.5 h results in a concentration‐dependent and significant increase in intracellular levels of Cys‐SSH and also GSSH (Figure 5). Importantly, treatment with Cys‐SS‐Cys alone did not result in increased RSSH levels, indicating that the observed increases in intracellular RSSH were not due to CSE/CBS‐mediated conversion of Cys‐SS‐Cys to Cys‐SSH.
Figure 5.
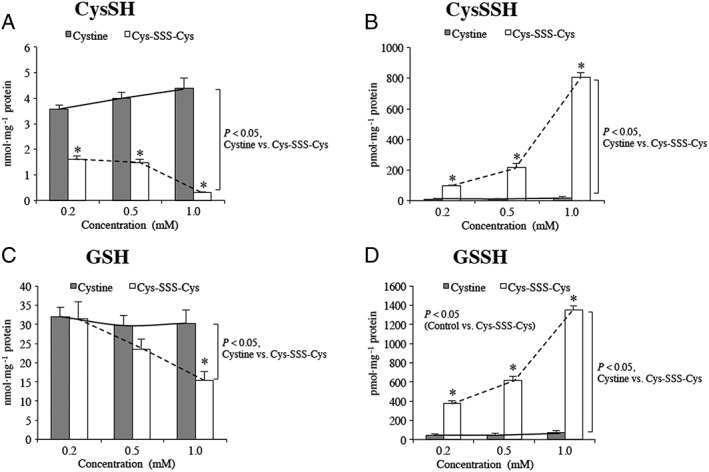
RSH and RSSH levels in HEK293T cells pretreated with either cystine (Cys‐SS‐Cys) or cysteine trisulfide (Cys‐SSS‐Cys). (A) Cysteine levels resulting from pretreatment with Cys‐SS‐Cys or Cys‐SSS‐Cys; (B) Cysteine hydropersulfide (Cys‐SSH) levels in cells pretreated with Cys‐SS‐Cys or Cys‐SSS‐Cys; (C) GSH levels in cells pretreated with Cys‐SS‐Cys or Cys‐SSS‐Cys and (D) GSSH levels in cells pretreated with Cys‐SS‐Cys or Cys‐SSS‐Cys. Each experiment was carried out 5 separate times (each time in triplicate). For each experiment, the values from the triplicates were averaged to give a single value. Data represent average ± SD (n = 5), * P < 0.05. Statistical analysis was performed as described in the Methods section.
Dialkyltrisulfide‐mediated cellular protection against oxidative and electrophilic stress
The results shown in Figure 5 indicate that HEK293T cells pretreated with Cys‐SSS‐Cys results in significant increases in intracellular Cys‐SSH and GSSH. It has been postulated that due to their unique chemistry (e.g. Ono et al., 2014; Millikin et al., 2016), a possible function of intracellular RSSH species is to protect cells from oxidative and/or electrophilic stress. Thus, Cys‐SSS‐Cys represents a novel method for examining the ability of increased intracellular RSSH levels in HEK293T cells to protect against electrophilic and/or oxidative stress. To test this, HEK293T cells were pretreated with 1 mM Cys‐SSS‐Cys for 1.5 h followed by exposure to either 0–1 mM H2O2 (oxidative/electrophilic stress) or 0–1 mM NEM (electrophilic stress). After 6 h of treatment with the stressor, cell viability was assessed using a WST8 assay (Ishiyama et al., 1997). The data indicate that pretreatment with Cys‐SSS‐Cys results in significant protection against NEM‐mediated toxicity (Figure 6B) but only marginal effects on H2O2 toxicity were observed (Figure 6A). Since viability measured by WST8 is actually an indicator of cellular reductive capacity (NADH/NADPH levels), another method for cell viability was used that interrogates another cell function in order to help confirm these results. Thus, the membrane impermeable nucleic acid dye, Sytox Green was also used as a measure of cytotoxicity (a measure of cell membrane integrity; Jones and Singer, 2001). Cells were again treated as before, and Sytox Green was added after 6 h (Figure 6C). As before, pretreatment with Cys‐SSS‐Cys protected cells against NEM‐mediated toxicity confirming the previous results using WST8.
Figure 6.
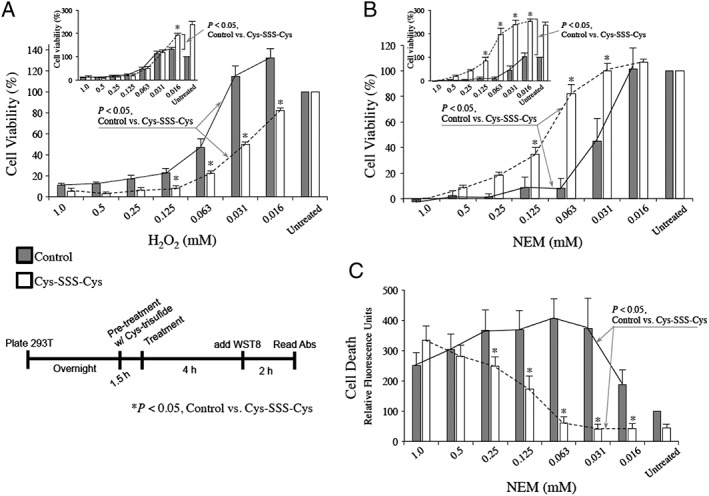
HEK293T cells were pre‐incubated with 1 mM cysteine trisulfide (Cys‐SSS‐Cys) for 1.5 h. Media was then replaced and cells treated with the indicated amount of H2O2 (A) or NEM (B, C) for 6 h. The cell viability indicator, WST8, was used for (A) and (B), while the cell death indicator Sytox Green was used in (C). For (A) and (B), graphs show data normalized to 100% viability for either untreated control or Cys‐SSS‐Cys only background, whereas insets show data normalized to 100% viability only in untreated controls. Each experiment was carried out 5 separate times (each time in triplicate). For each experiment, the values from the triplicates were averaged to give a single value. Experimental results are presented as the averages ± SEM of the five individual experiments, * P < 0.05. Statistical analysis was performed as described in the Methods section.
Discussion
Recent studies have alluded to the possible importance of RSSH species in biological signalling and in cellular protection against oxidative and/or electrophilic stress (e.g. Ono et al., 2014; Ida et al., 2014; Millikin et al., 2016; Saund et al., 2015). It is becoming increasingly apparent that H2S, RSSH and other polysulfides are all biologically prevalent and their chemistries intimately intertwined (e.g. Ida et al., 2014; Doka et al., 2016). As mentioned previously, numerous studies have utilized the reaction of H2S with RSSR (Reaction (1)) to generate RSSH for study in both chemical and biological systems. However, the work presented herein indicates this may be an oversimplified view of this reaction. It is clear that the RSSH product of Reaction (1) is very nucleophilic (most likely as the RSS− anion) and reacts further with the electrophilic RSSR to form RSSSR (Reaction (2)). It is important to note that RSSH is more acidic than the corresponding RSH by approximately 1–2 pKa units (and possibly more; Cuevasanta et al., 2015), indicating that a higher percentage of the nucleophilic anionic RSS− species will exist compared to the thiolate (RS−). It is also important to note that the higher levels of RSS− (compared to the protonated and electrophilic RSSH) will greatly decrease the rate of the reverse of Reaction (1) since an electrophilic RSSH is required in this reaction (Cuevasanta et al., 2015).
RSSSR generated from Reaction (2) is likely to be even more electrophilic than the corresponding RSSR (vide infra), indicating it may react further under these reaction conditions. In the case of a disulfide, RSSR, nucleophilic attack at either sulfur atom releases RSH/RS− as the leaving group (Reaction (6)). In the case of RSSSR, there are two types of sulfur atoms, a sulfane sulfur atom and non‐sulfane sulfur atoms (the bolded atoms of RSSSR). Nucleophilic attack at the sulfane sulfur atom results in an RS− leaving group (Reaction (7)) and attack at a non‐sulfane sulfur atoms results in expulsion of an RSS− leaving group (Reaction (8)).
| (6) |
| (7) |
| (8) |
Previous studies examining the nucleophilic cleavage of mixed disulfides (Reaction (6) where the R groups are different), found that the better leaving group (the weaker RS− base) governed the regiochemistry (Hiskey and Carroll, 1961). That is, the sulfur atom attacked was the one that had the better leaving group attached. Extrapolating this idea to the nucleophilic attack on RSSSR, it would be expected that attack at the non‐sulfane sulfur sites would predominate since RSS− is a significantly weaker base than RS− and thus a better leaving group (e.g. Nagy, 2013; Cuevasanta et al., 2015). Thus, in the reactions examined herein, the attack of a highly nucleophilic RSS− species on RSSSR is non‐productive since it gives back the same reactants (Reaction (9)).
| (9) |
(Note: prime notation given to follow reactants)
Of course other nucleophiles are present in solution that can also react with the electrophilic RSSSR species, namely, H2S and RSH. The reaction of HS− with RSSSR should also occur on the non‐sulfane sulfur atoms, leading to the generation of RSSH (Reaction (10)), and the reaction of RS− with RSSSR will lead to the formation of the corresponding RSSR and RSSH (Reaction (11)).
| (10) |
| (11) |
(Note: prime notation given to follow reactants)
In the case of the reaction of H2S with RSSR (Reaction (1)) and subsequent reactions (Reactions (2), (9), (10) and (11)), formation of the equilibrium partners RSSR, H2S, RSH, RSSSR and RSSH are expected without the generation of higher order polysulfides [i.e. RSSSnR or RSSnH (n > 1)]. Although chemical reactions can be envisioned that could lead to higher order polysulfides (Reactions (12) and (13)), based on the results and arguments presented here, these reactions appear unfavourable since the sulfane sulfur of RSSSR would have to be attacked.
| (12) |
| (13) |
Figure 7 schematically depicts the rationale explaining the lack of formation of higher order polysulfides (e.g. tetrasulfides, RSSSSR) from the reaction of RSS− with RSSSR. In all cases, the favoured reaction involves nucleophilic attack at the non‐sulfane sulfur atoms (Figure 7, pathway B).
Figure 7.
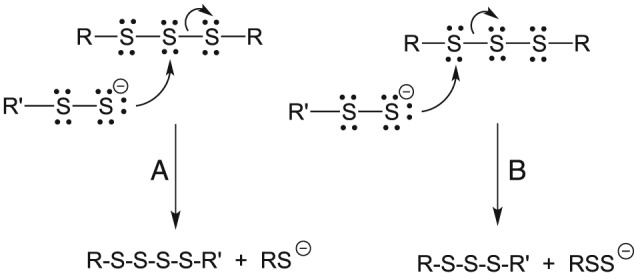
Reaction of a nucleophilic hydropersulfide with a trisulfide. (A) Nucleophilic attack at the sulfane sulfur atom and (B) nucleophilic attack at a non‐sulfane sulfur atom.
Another factor that may favour nucleophilic attack on the non‐sulfane sulfur atoms of RSSSR may involve electron pair repulsion between the attacking nucleophile and the lone pair electrons on the sulfur atoms of RSSSR. Nucleophilic attack on the central sulfane sulfur atom of RSSSR by RSS− (Figure 7, pathway A) would be inhibited by repulsion between the nucleophile and the lone pairs of electrons on both of the two adjacent sulfur atoms whereas nucleophilic attack on the non‐sulfane sulfur atoms (Figure 7, pathway B) involves repulsion from only one adjacent sulfur atom. Regardless of which effect predominates, both favour nucleophilic attack on the non‐sulfane sulfur sites of an RSSSR species in this purely chemical system. Scheme 1 illustrates the likely integrated chemistry that describes the results herein.
Scheme 1.
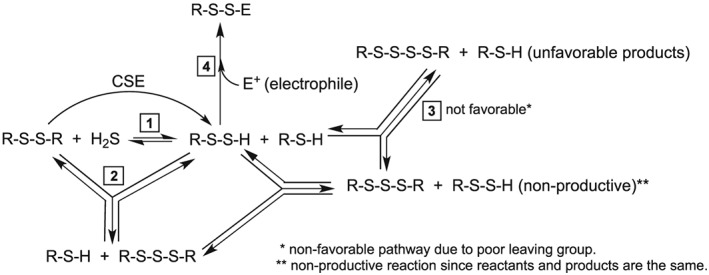
Integrated reaction pathways.
Support for the chemistry depicted in Scheme 1 is as follows:
Equilibrium 1 is a previously established reaction, which has been used by several labs to generate RSSH species in situ (Rao and Gorin, 1959; Cavallini et al., 1970; Francoleon et al., 2011).
Equilibrium 2 is evidenced by results presented in this study and the study of Bogdándi et al., 2019, this issue). Both NMR and mass spectral analysis indicate that the reaction of a disulfide (e.g. 2HED) with H2S results in detectable levels of thiol (2ME) and the corresponding trisulfide (2HET). Interestingly, RSSH species are not detected using either method of analysis, indicating that they may be at extremely low levels under the conditions of these experiments.
Reaction (3) (tetrasulfide, RSSSSR formation, Reaction (12), Figure 7A) appears unfavourable (vide supra) and, expectedly, RSSSSR species are not observed.
Reaction (4) the trapping of RSSH by an exogenous electrophile, occurs readily and serves as evidence for the proposed equilibria (although RSSH species are not directly detected).
It is important to note that care must be taken when extrapolating the chemical results described herein to a biological system. The kinetics of all the reactions examined (e.g. the forward and reverse reactions of Reactions (1) and (2)) are not firmly established and it is possible (if not likely) that competing reactions of the equilibrium species with other available biological molecules can occur. For example, RSSH species are highly redox active (Bianco et al., 2016; Francoleon et al., 2011) and can be intercepted by mild biological oxidants before it can participate in the equilibrium reactions described herein. Also, enzymatic conversion of, for example, RSSSR species via reduction processes to other species can occur, thus removing trisulfides from the equilibrium reactions. It is also worth noting that previous studies have examined the kinetics associated with the forward process of Reaction (1) (the reaction of H2S with a disulfide), and the rate constants are reported to be highly variable and dependent on the nature of the reactants (Vasas et al., 2015; Cuevasanta et al., 2015). For example, second order rate constants ranging from approximately 103 M−1·s−1 to 10−2 M−1·s−1 for the reaction of H2S with 5,5‐dithiobis‐(2‐nitrobenzoic acid) and 2HED, respectively, are reported. Since RSSH species are considered to be superior nucleophiles compared to thiols (and H2S), it is expected that they can react faster with electrophilic species compared to the corresponding thiol or H2S. Indeed, Cuevasanta et al. (2015) estimate that RSSH species can be as much as 20‐fold more reactive than the corresponding thiol at physiological pH.
Treatment of HEK293T cells with Cys‐SSS‐Cys led to a significant increase in the intracellular hydropersulfides Cys‐SSH and GSSH (Figure 5). It is established that the uptake of Cys‐SS‐Cys in mammalian cells occurs via the activity of the antiporter xc−, which exports glutamate while it takes up Cys‐SS‐Cys from extracellular solution (Conrad and Sato, 2012; Lewerenz et al., 2013). However, it is not known whether Cys‐SSS‐Cys is also transported via xc−. Regardless, Cys‐SSS‐Cys may represent a possible pharmacological/research tool for raising intracellular hydropersulfides (although more cell types need to be examined to determine if this is a general phenomenon). Importantly, trisulfides (e.g. Cys‐SSS‐Cys) are stable and can be stored and used similarly to disulfides (e.g. Cys‐SS‐Cys). In our hands, Cys‐SSS‐Cys is stable to acidic conditions, can be stored at room temperature, is stable to decomposition for months and is more soluble (approximately 10×) than Cys‐SS‐Cys in aqueous solution. On the other hand, hydropersulfides are inherently unstable to disproportionation and cannot be easily stored (Rao and Gorin, 1959).
Consistent with the idea that hydropersulfides can protect against specific stressors, HEK293T cells pretreated with Cys‐SSS‐Cys (which leads to increased intracellular levels of hydropersulfides, Figure 5) were significantly protected from the toxicity of the electrophile NEM (Figure 6B). However, a marginal effect (if any, depending on how samples are normalized) was observed with H2O2 treatment (Figure 6A). In the case of NEM, the increased levels of the highly nucleophilic hydropersulfides may be capable of intercepting/scavenging an otherwise toxic electrophile before it is able to modify critical cellular sites. Alternatively, hydropersulfide formation on critical protein thiols could be protected from irreversible electrophilic modification since modification will occur on the sacrificial sulfane sulfur atom, which can be reductively removed (Ono et al., 2014; Millikin et al., 2016). Importantly, persulfide‐mediated protection against thiophilic metal toxins has also been reported (Abiko et al., 2015). Although much more work remains to determine whether generation of per‐ and/or poly‐sulfides represents a protective mechanism against oxidative and/or electrophilic stress, the utility of Cys‐SSS‐Cys as a means of pharmacologically altering intracellular hydropersulfide levels may be an important tool in this regard.
Differences in the observed protection by Cys‐SSS‐Cys between H2O2 and NEM is intriguing. It is well established that NEM toxicity is primarily the result of thiol protein alkylation (e.g. Vimard et al., 2011). Thus, considering the high nucleophilicity of RSSH, it is not surprising that increased intracellular RSSH results in protection against NEM. Although H2O2‐mediated physiology (i.e. signalling) can either directly or indirectly involve oxidative thiol modification (e.g. Winterbourn, 2013; Jones, 2008), the toxicity of H2O2 is thought to primarily involve metal‐mediated generation of potent and indiscriminate oxidants (i.e. via the Fenton reaction) (Winterbourn, 1995). Importantly, the oxidants generated in this way are thought to be too reactive to be trapped or scavenged since they are immediately reactive and do not diffuse away from their site of generation. Thus, RSSH may have little effect on Fenton‐type oxidants.
It is important to note that intracellular increases in RSSH species may also lead to higher intracellular levels of RSSR, RSH and H2S via the equilibrium processes described above. Thus, it is naïve and even ill‐advised to conclude categorically that the observed protection is the sole result of increased intracellular RSSH levels. Since treatment of cells with Cys‐SSS‐Cys does not dramatically increase levels of RSH species (Figure 5), the observed protection is clearly not due to RSH. However, it remains possible that the protection results, at least partially, from increased intracellular H2S levels. Indeed, based on the results herein and those of Bogdándi et al. (2019, this issue), it is currently difficult to distinguish between the actions of RSSH from H2S in a biological system since they are equilibrium partners and should be present simultaneously. To be sure, there are chemical rationale for predicting protective functions associated with RSSH species (e.g. Ono et al., 2014; Millikin et al., 2016), but this remains to be unequivocally established.
Finally, it is interesting to note that treatment of cells with Cys‐SSS‐Cys alone leads to an increase in apparent “viability” (as measured by the WST8 assay) (Figure 6A, B) in control samples. That is, Cys‐SSS‐Cys treatment on its own appears to result in an increase in cellular reducing equivalents/reduction capacity (what WST8 ultimately measures; Ishiyama et al., 1997), which is construed as an increase in viability. It is noteworthy that hydropersulfides are considered to be good reductants (superior to, for example, thiols and akin to ascorbate and/or tocopherols) (e.g. Bianco et al., 2016; Chauvin et al., 2017). Thus, in spite of the fact that RSSH species are formally oxidized with respect of the corresponding RSH, they are significantly more reducing and may possibly be responsible for the greater reducing capacity observed. Interestingly, Akaike et al. (2017) reported that hydropersulfide generation in mitochondria is an important aspect of mitochondrial regulation and activity, and thus, it is intriguing to speculate that since NADH (one of the primary species assayed by WST8) is used by mitochondria as an electron source, the apparent increases in NADH levels in Cys‐SSS‐Cys pretreated cells may be the result of other reductants, possibly RSSH species, serving as alternative mitochondrial electron sources (proposed previously; Akaike et al., 2017) allowing NADH levels to rise.
Author contributions
Experimental work was carried out by C.L.B., T.I., C.F.H., R.N.G. and J.L. The manuscript was written by J.M.F. with significant assistance from J.L., T.A., P.N., V.B., J.P.T., Y.K. and C.L.B. The project was conceived by C.L.B., J.M.F., P.N., Y.K., J.L. and T.A.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Acknowledgements
The authors would like to acknowledge Dr. Michael Bartberger for helpful discussions regarding the chemical arguments presented. J.P.T. would like to acknowledge the National Science Foundation (NSF) (Division of Chemistry, CHE‐1566065) for support of this research. P.N. would like to acknowledge National Research, Development and Innovation Office (grants: KH17‐126766 and K 109843) for support.
Bianco, C. L. , Akaike, T. , Ida, T. , Nagy, P. , Bogdandi, V. , Toscano, J. P. , Kumagai, Y. , Henderson, C. F. , Goddu, R. N. , Lin, J. , and Fukuto, J. M. (2019) The reaction of hydrogen sulfide with disulfides: formation of a stable trisulfide and implications for biological systems. British Journal of Pharmacology, 176: 671–683. 10.1111/bph.14372.
Contributor Information
Joseph Lin, Email: linj@sonoma.edu.
Jon M Fukuto, Email: fukuto@sonoma.edu.
References
- Abiko Y, Yoshida E, Ishii I, Fukuto JM, Akaike T, Kumagai Y (2015). Involvement of reactive persulfides in biological bismethylmercury sulfide formation. Chem Res Toxicol 28: 1301–1306. [DOI] [PubMed] [Google Scholar]
- Akaike T, Ida T, Wei F‐W, Nishida M, Kumagai Y, Alam M et al (2017). Cysteinyl‐tRNA synthase governs cysteine polysulfidation and mitochondrial bioenergetics. Nature Comm 1177: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017). The Concise Guide To PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artaud I, Galardon E (2014). A persulfide analogue of the nitrosothiol SNAP: formation, characterization and reactivity. Chembiochem 15: 2361–2364. [DOI] [PubMed] [Google Scholar]
- Bianco CL, Chavez TA, Sosa V, Saund SS, Nguyen NN, Tantillo DJ et al (2016). The chemical biology of the persulfide (RSSH)/perthiyl (RSS) redox couple and possible role in biological redox signaling. Free Radic Biol Med 101: 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdándi V, Ida T, Sutton TR, Bianco C, Ditrói T, Koster G, et al (2019). Speciation of reactive sulfur species and their reactions with alkylating agents: Do we have any clue about what is present inside the cell? Br J Pharmacol 176: 646–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallini D, Federici G, Barboni E (1970). Interactions of proteins with sulfide. Eur J Biochem 14: 169–174. [DOI] [PubMed] [Google Scholar]
- Chauvin J‐PR, Griesser M, Pratt D (2017). Hydropersulfides: H‐atom transfer agents par excellence. J Am Chem Soc 139: 6484–6493. [DOI] [PubMed] [Google Scholar]
- Conrad M, Sato H (2012). The oxidative stress‐inducible cystine/glutamate antiporter, system xc −: cystine supplier and beyond. Amino Acids 42: 231–246. [DOI] [PubMed] [Google Scholar]
- Cuevasanta E, Lange M, Bonata J, Coitiño E, Ferrer‐Sueta G, Filipovic M et al (2015). Reaction of hydrogen sulfide with disulfide and sulfenic acid to form the strongly nucleophilic persulfide. J Biol Chem 290: 26,866–26,880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA et al (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol 175: 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doka E, Pader I, Biro A, Johansson K, Cheng Q, Ballago K et al (2016). A novel persulfide detection method reveals protein persulfide‐ and polysulfide‐reducing function of thioredoxin and glutathione systems. Sci Adv 2: e1500968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett SA, Wardman P (1995). Perthiols as antioxidants: radical‐scavenging and prooxidative mechanisms. Methods Enzymol 251: 55–69. [DOI] [PubMed] [Google Scholar]
- Francoleon NE, Carrington SJ, Fukuto JM (2011). The reaction of H2S with oxidized thiols: generation of persulfides and implications to H2S biology. Arch Biochem Biophys 516: 146–153. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiskey RG, Carroll FI (1961). Chemistry of aliphatic disulfides. II. Cyanide cleavage of unsymmetrical disulfides. J Am Chem Soc 83: 4644–4647. [Google Scholar]
- Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y et al (2014). Reactive cysteine persulfides and S‐polythiolation regulate oxidative stress and redox signaling. Proc Natl Acad Sci U S A 111: 7606–7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishiyama M, Miyazono Y, Sasamoto K, Ohkura Y, Ueno K (1997). A highly water‐soluble disulfonated tetrazolium salt as a chromogenic indicator for NADH as well as cell viability. Talanta 44: 1299–1305. [DOI] [PubMed] [Google Scholar]
- Jones DP (2008). Radical‐free biology of oxidative stress. Am J Physiol Cell Physiol 295: C849–C868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones lJ, Singer VL (2001). Fluorescence microplate‐based assay for tumor necrosis factor activity using SYTOX green stain. Anal Biochem 293: 8–15. [DOI] [PubMed] [Google Scholar]
- Kashfi K, Olson KR (2013). Biology and therapeutic potential of hydrogen sulfide and hydrogen sulfide‐releasing chimeras. Biochem Pharmacol 85: 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H (2014). The physiological role of hydrogen sulfide and beyond. Nitric Oxide 41: 4–10. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW et al (2013). The cystine/glutamate antiporter system xc − in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal 18: 522–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millikin R, Bianco CL, White C, Saund SS, Henriquez S, Sosa V et al (2016). The chemical biology of protein hydropersulfides: studies of a possible protective function of biological hydropersulfide generation. Free Radic Biol Med 97: 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P (2013). Kinetics and mechanism of thiol‐disulfide exchange convering direct substitution and thiol oxidation‐mediated pathways. Antioxid Redox Signal 18: 1623–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numakura T, Sugiura H, Akaike T, Ida T, Fujii S, Koarai A et al (2017). Production of reactive persulfide species in chronic obstructive pulmonary disease. Thorax 72: 1074–1083. 10.1136/thoraxjnl-2016-209359. [DOI] [PubMed] [Google Scholar]
- Ono K, Akaike T, Sawa T, Kumagai Y, Wink DA, Tantillo DJ et al (2014). The redox chemistry and chemical biology of H2S, hydropersulfides and derived species: implications of their possible biological activity and utility. Free Radic Biol Med 77: 82–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus DJ, Lefer DJ (2014). Emergence of hydrogen sulfide as an endogenous signaling molecule in cardiovascular disease. Circ Res 114: 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao GS, Gorin G (1959). Reaction of cystine with sodium sulfide in sodium hydroxide solution. J Org Chem 24: 749–753. [Google Scholar]
- Saund SS, Sosa V, Henriquez S, Nguyen QNN, Bianco CL, Soeda S et al (2015). The chemical biology of hydropersulfides (RSSH): chemical stability, reactivity and redox roles. Arch Biochem Biophys 588: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savige WE, Eager JA, Maclaren JA, Roxburgh CM (1964). The S‐monoxides of cystine, cystamine and homocystine. Tetrahedron Lett 5: 3289–3293. [Google Scholar]
- Stipanuk M (1986). Metabolism of sulfur‐containing amino acids. Annu Rev Nutr 6: 179–209. [DOI] [PubMed] [Google Scholar]
- Vasas A, Doka E, Fabian I, Nagy P (2015). Kinetic and thermodynamic studies on the disulfide‐bond reducing potential of hydrogen sulfide. Nitric Oxide 46: 93–101. [DOI] [PubMed] [Google Scholar]
- Vimard F, Saucet M, Nicole O, Feuilloley M, Duval D (2011). Toxicity induced by cumene hydroperoxide in PC12 cells: protective role of thiol donors. J Biochem Mol Toxicol 25: 205–215. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC (1995). Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett 82/83: 969–974. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC (2013). The biological chemistry of hydrogen peroxide. Methods Enzymol 528: 3–25. [DOI] [PubMed] [Google Scholar]
- Yamanishi T, Tuboi S (1981). The mechanism of the L‐cystine cleavage reaction catalyzed by rat liver γ‐cystathionase. J Biochem 89: 1913–1921. [DOI] [PubMed] [Google Scholar]