

Neisseria gonorrhoeae, the causative agent of gonorrhea, has evolved several mechanisms to subvert complement, including binding of the complement inhibitor factor H (FH). We previously reported FH binding to N. gonorrhoeae independently of lipooligosaccharide (LOS) sialylation.
KEYWORDS: factor H, Neisseria, NspA, complement
ABSTRACT
Neisseria gonorrhoeae, the causative agent of gonorrhea, has evolved several mechanisms to subvert complement, including binding of the complement inhibitor factor H (FH). We previously reported FH binding to N. gonorrhoeae independently of lipooligosaccharide (LOS) sialylation. Here we report that factor H-like protein 1 (FHL-1), which contains FH domains 1 through 7 and possesses complement-inhibitory activity, also binds to N. gonorrhoeae. The ligand for both FH and FHL-1 was identified as neisserial surface protein A (NspA), which has previously been identified as a ligand for these molecules on Neisseria meningitidis. As with N. meningitidis NspA (Nm-NspA), N. gonorrhoeae NspA (Ng-NspA) bound FH/FHL-1 through FH domains 6 and 7. Binding of FH/FHL-1 to NspA was human specific; the histidine (H) at position 337 of domain 6 contributed to human-specific FH binding to both Ng- and Nm-NspA. FH/FHL-1 bound Nm-NspA better than Ng-NspA; introducing Q at position 73 (loop 2, present in Ng-NspA) or replacing V and D at positions 112 and 113 in Nm-NspA loop 3 with A and H (Ng-NspA), respectively, reduced FH/FHL-1 binding. The converse Ng-NspA to Nm-NspA mutations increased FH/FHL-1 binding. Binding of FH/FHL-1 through domains 6 and 7 to N. gonorrhoeae increased with truncation of the heptose I (HepI) chain of LOS and decreased with LOS sialylation. Loss of NspA significantly decreased serum resistance of N. gonorrhoeae with either wild-type or truncated LOS. This report highlights the role for NspA in enabling N. gonorrhoeae to subvert complement despite LOS phase variation. Knowledge of FH-NspA interactions will inform the design of vaccines and immunotherapies against the global threat of multidrug-resistant gonorrhea.
INTRODUCTION
Neisseria gonorrhoeae is responsible for about 80 million cases of the sexually transmitted disease gonorrhea worldwide each year (1). There were 468,514 cases (145.8 gonorrhea cases per 100,000 population) reported in the United States in 2016, which represents a 46% increase from the number of reported cases in 2011 (2). The true incidence of disease is estimated to be about twice the reported incidence due to underreporting and because a number of infections go undiagnosed each year (3, 4). Undiagnosed infections further complicate effective control of this disease. N. gonorrhoeae infection of women can result in pelvic inflammatory disease, salpingitis, and loss of fertility. Effective vaccines to prevent the spread of N. gonorrhoeae are not available, and the bacterium is rapidly becoming untreatable with existing antibiotics (5–9). The growing reality of the threat of untreatable N. gonorrhoeae infection has ushered in an era where the discovery of new therapeutics and effective vaccines to thwart the unchecked spread of this disease is urgently needed (10).
Vaccines that protect against infection with Neisseria meningitidis are available. Several of these vaccines target meningococcal polysaccharide capsules and thus would not be effective against infection with N. gonorrhoeae. However, effective meningococcal vaccines that target the factor H binding protein (fHbp) and/or other surface proteins of N. meningitidis have also been developed (11–13); a recent study suggests that one such vaccine may also provide limited protection against gonococcal infection (14). Immunization with fHbp, for example, elicits serum bactericidal antibodies that protect against meningococcal infection. An added advantage of anti-fHbp vaccine antibodies may be their ability to block binding of the complement inhibitor human factor H (FH) to fHbp, thereby interrupting an important mechanism of immune evasion used by meningococci (15, 16).
FH regulates alternative complement pathway (AP) activation by acting as a cofactor for factor I-mediated cleavage of C3b to iC3b (cofactor activity) and also serves to irreversibly dissociate Bb from the AP C3 convertase C3b,Bb (decay-accelerating activity). FH is a 150-kDa glycoprotein that consists of 20 repetitive short consensus repeats (SCRs) that resemble beads on a string; each SCR is ∼60 amino acids and forms a globular structure (17, 18). Although similar in structure, the SCR domains exhibit different activities. Only SCRs 1 to 4 possess complement-inhibitory activities (19–21), while SCRs 7 and 20 bind polyanions such as heparin (22, 23). FH assists with discrimination of self from nonself by binding to host glycosaminoglycans (GAGs) and preventing unwanted complement attack (24).
FH-like protein 1 (FHL-1) is an alternatively spliced variant of FH that contains the seven N-terminal SCRs of FH with the distinction that the C-terminal SCR (SCR7) is differentially spliced and encodes four unique C-terminal amino acids (SFTL) (20, 25). FHL-1 retains all the regulatory functions of FH, which include cofactor activity for the factor I-mediated cleavage of C3b and accelerating the decay of alternative pathway C3/C5 convertases (26). Similar to FH (27), FHL-1 also binds select GAGs through SCR7 (28). In human blood, the concentration of FH is ∼500 µg/ml, and that of FHL-1 is ∼50 µg/ml, resulting in an FH/FHL-1 molar ratio of approximately 3:1 (28–30). The role of FHL-1 in AP regulation is often overlooked; however, FHL-1 has been identified recently as the main regulatory protein in the extracellular matrix underlying the human retina, called Bruch’s membrane (28).
Pathogenic bacteria, including N. meningitidis and N. gonorrhoeae, have evolved mechanisms to hijack host complement-regulatory molecules and protect themselves against unwanted complement activation (31). Both N. gonorrhoeae and N. meningitidis bind the complement regulator FH to evade killing by the innate immune system (32–39). Meningococcal fHbp and NspA proteins bind to FH SCR6 and -7 (32, 40). In addition, some N. meningitidis PorB types bind directly to FH SCR6 and -7 (34). Several other microbes also bind to FH SCR domains 6 and 7 (reviewed in reference 39). FHL-1, like FH, binds to the same microbial ligands that interact with FH SCR6 and -7. In contrast, N. gonorrhoeae strains bind FH SCRs 18 to 20 when they are grown in medium that enables lipooligosaccharide (LOS) sialylation (36, 37, 41); binding of FH to sialylated gonococci also requires N. gonorrhoeae porin (42). Sialic acid substituted on neisserial lacto-N-neotetraose (LNnT) LOS may function similarly to sialic acid on host cells and assist in subverting the AP by enhancing interactions with FH. N. gonorrhoeae has homologs of meningococcal fHbp and NspA. The gonococcal fHbp homolog, designated Ghfp (43), lacks a signal peptide, is not surface expressed, and does not contribute to FH binding (43). Gonococcal NspA (N. gonorrhoeae NspA [Ng-NspA]) shares 95% identity with N. meningitidis NspA (Nm-NspA), but the role of Ng-NspA in binding to FH and evasion of complement-mediated killing has not been investigated. We sought to determine the role of Ng-NspA in complement evasion and serum resistance and to compare the FH binding properties of Ng- and Nm-NspA.
Because of the importance of regulating the AP, the repertoire of FH ligands in Neisseria species, and the observation that some gonococcal strains bound FH in the absence of LOS sialylation, we sought to identify and characterize additional mechanisms of FH binding and AP regulation in gonococci. A prominent role for Ng-NspA in complement evasion is presented.
RESULTS
Gonococcal NspA mediates binding to FH and FHL-1 through FH SCRs 6 and 7 independently of LOS sialylation.
N. meningitidis binds FH SCRs 6 and 7 through interactions with PorB, fHbp, and NspA (32–34, 40). We used the unsialylated “serum-resistant” strain N. gonorrhoeae 15253, which has caused disseminated infection (44), expresses PorB.1A, and binds more FH than PorB.1B isolates but does not express LNnT LOS (expresses only lactose from both heptose I [HepI] [similar to the “2-Hex HepI LOS” expressed by ΔlgtA mutants described below] and HepII). N. gonorrhoeae 15253 binds FH as reported previously (37) and also binds FHL-1 (Fig. 1, left and middle panels, blue histograms). N. gonorrhoeae 15253 NspA and meningococcal NspA share ∼95% identity at the amino acid level (Fig. 5 shows sequence alignments). To determine if NspA was involved in FH/FHL-1 binding to wild-type N. gonorrhoeae 15253 (15253 wt), we examined the binding of FH and FHL-1 to an nspA deletion mutant (Fig. 1, left and middle panels, red histograms). Deletion of nspA decreased binding of FH and FHL-1 to N. gonorrhoeae 15253 ΔNspA by 2.6- and 3.1-fold, respectively, compared to N. gonorrhoeae 15253 wt. Similar to Nm-NspA, we determined that strain 15253 also bound FH and FHL-1 specifically through domains 6 and 7 by showing that a fusion protein comprising FH SCRs 6 and 7 fused to Fc (FH SCR6-7/Fc) (Fc was used as a target for detection) bound to N. gonorrhoeae 15253 wt but only minimally to N. gonorrhoeae 15253 ΔNspA (Fig. 1, right panel). Figure S1 in the supplemental material shows cumulative data from all experiments.
FIG 1.

Gonococcal NspA mediates binding to FH and FHL-1 through FH domains 6 and 7. Shown are data for binding of human FH (10 µg/ml) (left panel), recombinant human FHL-1 (10 µg/ml) (middle panel), and FH SCR6-7/Fc (5 µg/ml) (right panel) to gonococcal strain 15253 by flow cytometry. Binding to the wild-type strain is shown with a blue line, binding to the ΔNspA mutant is shown with a red line, and conjugate controls (bacteria plus fluorescent conjugate) with a gray shaded histogram. Numbers alongside the histograms represent the median fluorescence values of the entire bacterial population. The x axis shows fluorescence (log10 scale); the y axis shows counts. Figure S1 in the supplemental material shows cumulative data from all experiments.
N. gonorrhoeae has a homolog of meningococcal fHbp, named Ghfp, that lacks a signal peptide, is not expressed on the gonococcal surface, and does not bind FH (43). Inactivation of ghfp in strain 15253 did not affect the ability of 15253 to bind FH or to resist killing by normal human serum (NHS) (data not shown), and we concluded that Ghfp was not involved in binding to FH or serum resistance in strain 15253.
LOS glycans impact FH binding to NspA.
To extend the results observed with N. gonorrhoeae 15253 to other N. gonorrhoeae strains, we next examined N. gonorrhoeae strains UU1 and FA1090 for their ability to bind to FH and FHL-1. In contrast to 15253, wild-type unsialylated N. gonorrhoeae strains UU1 and FA1090 bound FH and FHL-1 only weakly (Fig. 2A, histograms depicted by broken green lines). Both UU1 and FA1090 express the extended LNnT LOS from HepI (“4-Hex HepI LOS”), as determined by binding of monoclonal antibody (mAb) 3F11 (data not shown) that recognizes the terminal lactosamine structure of LNnT (45). We previously showed that the length of HepI glycan extensions affects FH binding to NspA expressed on N. meningitidis; mutants that expressed lactose from HepI bound more FH through NspA than LNnT-expressing parent strains (33). Notably, N. gonorrhoeae 15253 harbors a natural defect in lgtB that results in the expression of lactose (2-Hex HepI) LOS, while both UU1 and FA1090 express the longer LNnT LOS (4-Hex HepI) species. We hypothesized that the shorter HepI glycan extension (lactose) from 15253 HepI would pose less hindrance for FH and FHL-1 binding to NspA than LNnT expressed from HepI of UU1 and FA1090. To determine if truncation of HepI LOS facilitated binding of FH and FHL-1 to NspA, we inactivated lgtA, which results in the shortening of 4-Hex HepI LOS (LNnT) to lactose extending from HepI of N. gonorrhoeae FA1090 and UU1, and assessed the role of FH and FHL-1 binding to NspA. Similar to 15253, binding of FH and FHL-1 to lgtA-null mutants of FA1090 and UU1 was readily detected by fluorescence-activated cell sorter (FACS) analysis (Fig. 2A). As described above for 15253 (Fig. 1B), this binding was also dependent upon the expression of NspA and involved binding to FH/FHL-1 SCRs 6 and 7 (Fig. 2B). These data provide evidence, for the first time, that gonococcal NspA plays a role in binding of FH and FHL-1 to N. gonorrhoeae in the absence of LOS sialylation. Figure S2 in the supplemental material shows cumulative data from all experiments.
FIG 2.

Truncation of heptose I (HepI) glycan extensions increases binding of FH and FHL-1 to N. gonorrhoeae. (A) Binding of human FH (10 µg/ml) and recombinant human FHL-1 (10 µg/ml) to gonococcal PorB.1A strain UU1 and PorB.1B strain FA1090 was measured by flow cytometry. Binding was compared in wild-type strains that express 4 hexose residues (LNnT) (“4 Hex HepI LOS”) from HepI LOS (green line), isogenic ΔlgtA mutant strains that express truncated LOS with only 2 hexose residues (Gal-Glc) from HepI (blue line) (“2 Hex HepI”), and isogenic mutant strains that lack both lgtA and NspA (red line) ΔlgtA ΔNspA). (B) Binding of FH domains 6 and 7 to ΔlgtA mutants occurs through NspA. Binding of FH SCR6-7/Fc (5 µg/ml) to ΔlgtA (blue line) (2 Hex Hep I LOS) and ΔlgtA ΔNspA (red line) isogenic mutants of N. gonorrhoeae UU1 and FA1090 was measured by flow cytometry. Numbers alongside histograms and axes are as described above for panel A. The result for one experiment that is representative of data from at least two independent experiments is shown. Figure S2 in the supplemental material shows cumulative data for all experiments.
LOS sialylation impedes binding of FH domains 6 and 7 to Ng-NspA.
We sought to determine whether sialylation of gonococcal LNnT LOS affected binding of FH domains 6 and 7 to Ng-NspA. Contrary to findings in N. meningitidis, where FH binding to Nm-NspA is enhanced by sialylation of LNnT LOS (33), sialylation of N. gonorrhoeae LNnT LOS impeded binding of FH SCR6-7/Fc to NspA (Fig. 3, top left graph). Deletion of nspA resulted in a loss of binding of FH SCR6-7/Fc to unsialylated strain FA1090 (Fig. 2B, bottom panel, and Fig. 3, bottom left panel). Sialylation of gonococcal LNnT LOS enhances binding of FH SCRs 18 to 20 to N. gonorrhoeae (41) and was used as a positive control (Fig. 3, top right graph). Deletion of NspA did not diminish binding of full-length FH to N. gonorrhoeae PorB in bacteria with sialylated LOS (data not shown) and in a corresponding fashion did not affect binding of FH SCR18-20/Fc (Fig. 3, bottom right graph). Deletion of NspA abrogated binding of FH, FHL-1 (Fig. 2A), and FH SCR6-7/Fc (Fig. 2B) to unsialylated FA1090 and binding of FH SCR6-7/Fc to sialylated FA1090 (Fig. 3, bottom left graph). These findings show that sialylation of gonococcal LNnT LOS enhances binding of FH SCRs 18 to 20 to N. gonorrhoeae (37, 42) but diminishes binding of FH SCRs 6 and 7 to Ng-NspA. Figure S3 in the supplemental material shows cumulative data from all experiments.
FIG 3.

Sialylation of lacto-N-neotetraose (LNnT) LOS decreases binding of FH SCR6-7/Fc to gonococci expressing NspA. Binding of FH SCR6-7/Fc (5 µg/ml) or FH SCR18-20/Fc (5 µg/ml) to wild-type gonococcal strain FA1090 (top panel) and its isogenic nspA deletion mutant (bottom panel) grown either with (shaded histograms) or without (solid lines) CMP-Neu5Ac (25 µg/ml) added to the growth medium was measured by flow cytometry. Negative-control reactions (no FH fusion protein added) are shown by shaded gray histograms. In all graphs, the x axis represents fluorescence intensity on a log10 scale, and the y axis represents the number of events; numbers are the median fluorescence values of FH SCR/Fc binding to the entire bacterial population. Data from one experiment that are representative of results from at least three independent experiments are shown. Figure S3 in the supplemental material shows cumulative data from two separate experiments.
Histidine residues in human SCR6 are important for FH binding to Ng-NspA.
Binding of FH to Ng- and to Nm-NspA (and also to N. meningitidis fHbp) is human specific (32, 33, 40, 46, 47). To determine if histidine (His) residues at positions 337, 360, and 371 in human FH SCR6, shown previously to be important in N. meningitidis fHbp binding (40, 47), were also important for binding of Ng-NspA to FH SCR6, we used a series of FH SCR5-8/Fc mutant proteins in which each of three His residues in human FH SCR6 were mutated to mirror the corresponding amino acid sequences present in the rhesus macaque counterpart of FH SCR6 (H337Y, H360P, and H371Y), either individually or in combination (40). Binding of FH to both Nm-NspA and Ng-NspA is human specific; rhesus FH5-8/Fc did not bind to NspA from either neisserial species (data not shown).
Flow cytometry was used to measure binding of FH SCR5-8/Fc and the seven His mutant derivatives of FH SCR5-8/Fc to N. gonorrhoeae strains 15253, UU1, and FA1090 and to an unencapsulated N. meningitidis mutant strain, A2594 ΔfHbp (32), devoid of fHbp expression (Fig. 4A). All three N. gonorrhoeae strains and the N. meningitidis strain bound FH in a manner analogous to that for fHbp protein variants 2 and 3 (subfamily A) (40), where mutation of H337 alone or in combination with either H360 or H371 had resulted in decreased binding of SCR5-8. Mutations in H360 or H371 alone and in combination increased binding compared to the unmutated FH SCR5-8/Fc protein (Fig. 4A). The specificity of these findings using intact N. gonorrhoeae were also determined by an enzyme-linked immunosorbent assay (ELISA) using Escherichia coli microvesicles (MVs) that expressed gonococcal NspA from strains 15253, UU1, and FA1090 (Fig. 4B). Similar to whole N. gonorrhoeae strains, mutations in H337 alone or in combination with either H360P or H371Y diminished binding of FH SCR5-8/Fc to all three Ng-NspA-containing vesicles. As reported previously using N. meningitidis that displayed variant 1 fHbp (40), mutating H337 in conjunction with H371 had the greatest effect. Together, these data highlight that Ng-NspA shares a binding mechanism with Nm-NspA and N. meningitidis fHbp; H337 and H371, two of the three histidine residues in SCR6 that are important for binding to polyanions, also participate in interactions between FH SCR6 and neisserial pathogens.
FIG 4.
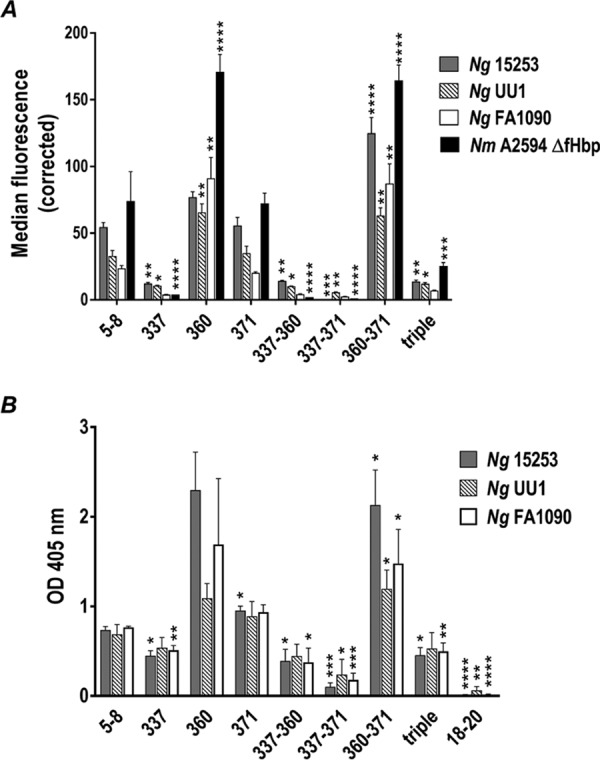
Role of human FH SCR6 histidine residues 337, 360, and 371 in binding of FH to gonococcal and meningococcal NspAs. (A) Binding of human FH SCR5-8/Fc with wild-type SCR6 (labeled “5-8”) and human FH SCR5-8/Fc with point mutations in histidine (His) residues 337, 360, and 371 in SCR6 (“triple” indicates that all 3 His residues have been changed, collectively referred to as FH/Fc below) to gonococcal strain 15253 (solid gray bars), the 2-Hex HepI LOS mutants of UU1 (striped bars) and FA1090 (white bars), and N. meningitidis strain A2594 ΔfHbp (solid black bars) measured by FACS analysis. The y axis represents the corrected median fluorescence of FH/Fc binding to the entire bacterial population, where binding of FH/Fc to isogenic mutant strains lacking NspA was subtracted from binding to the wild-type strain to represent net (corrected) NspA-specific binding. Each bar represents the mean (range) of data from two independent experiments. Comparison of binding of the different FH/Fc molecules to each strain was performed by one-way ANOVA (P < 0.0001 for all three strains), and pairwise comparisons of binding of each FH/Fc molecule with binding to FH SCR5-8/Fc on that strain were made using Dunnett’s multiple-comparison test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (B) Binding, measured by ELISAs, of FH SCR5-8/Fc and seven His mutants in SCR6 to E. coli membrane vesicles (MVs) expressing NspA from gonococcal strains 15253 (grey bars), UU1 (striped bars), and FA1090 (white bars). Negative controls included microtiter wells in which the FH/Fc fusion protein was omitted, a well that contained FH18-20/Fc (labeled “18-20”), and wells coated with MVs prepared from strains harboring the vector alone (vector control; no gonococcal NspA). Values from the “vector control” wells were subtracted as background binding. The y axis shows the absorbance (optical density [OD]) at 405 nm. Each bar represents the mean (range) of data from 3 independent experiments. Comparison of binding of the different FH/Fc molecules to each strain was performed by one-way ANOVA (P < 0.0001 for all three strains). Pairwise comparisons between the binding of each FH/Fc molecule with binding to FH SCR5-8/Fc on that strain were performed using the Holm-Sidak multiple-t-test method, and the adjusted P values are indicated (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Enhanced binding of FH and FHL-1 to Nm-NspA relative to Ng-NspA is mediated via V112 and D113 in surface loop 3.
Binding of full-length FH to E. coli MVs harboring Ng-NspA molecules from N. gonorrhoeae strains 15253, UU1, and FA1090 was compared to binding to MVs containing Nm-NspA. Dose-dependent binding of FH to Nm-NspA in MVs was readily detected, consistent with data from previous reports (33). Unexpectedly, full-length FH did not bind to Ng-NspA expressed in E. coli MVs (Fig. 5). This result was surprising because all three N. gonorrhoeae strains bound FH, FHL-1, FH SCR5-8/Fc, and FH SCR6-7/Fc in an NspA-dependent manner (Fig. 1 to 3), and E. coli MVs expressing Ng-NspA bound to FH SCR5-8/Fc (Fig. 4B). Ng-NspAs in MVs each showed equivalent amounts of expression compared to Nm-NspA in Coomassie blue-stained gels (data not shown); thus, different amounts of NspA did not explain the lack of binding. In the gonococcal background, binding of FH to NspA was observed best when the LOS HepI chain was truncated, and we surmised that the E. coli background was not optimal for Ng-NspA interactions with full-length FH. Binding of FH SCR5-8/Fc to Ng-NspA in E. coli MVs may have been facilitated by fewer steric constraints with the FH5-8 fragment than with intact FH. Consistent with this, we found that Ng-NspA expressed in a heterologous meningococcal background bound less FH than a heterologous Nm-NspA protein expressed in the same background (see Fig. S4 in the supplemental material).
FIG 5.

Comparison of FH binding to and sequences of meningococcal and gonococcal NspAs. (A) Binding of full-length human FH to recombinant meningococcal NspA 8047 (87) or gonococcal (strains UU1, 15253, and FA1090) NspAs expressed in E. coli MVs was compared by ELISA. Negative controls included ELISA measurements where FH was omitted and reactions that used MVs prepared from strains harboring the vector alone (vector control; no gonococcal NspA). Vector control optical density at 405 nm readings were considered the background and subtracted. FH concentrations are shown on the x axis, and absorbances at 405 nm are shown on the y axis. Data from a single experiment are shown, which are representative of results from at least 3 independent and reproducible experiments performed. (B) Comparison of the predicted amino acid sequences of NspAs from N. gonorrhoeae strains UU1, 15253, F62, 252, FA1090, and FA19. The predicted signal peptide (black line), surface-exposed loops (black boxes) (loops 1 to 4), and beta sheets (blue arrows) are indicated (48). Amino acids in the gonococcal NspAs that differed from meningococcal NspA are shaded, and the corresponding amino acid found in the meningococcal NspA is shown above the alignment in red font.
Based on the results in Fig. 5A, we speculated that Nm-NspA has a higher affinity for FH and FHL-1 than Ng-NspA. We compared the predicted amino acid sequences of NspAs from six gonococcal strains (15253, UU1, FA1090, 252, FA19, and F62) to those of meningococcal NspA sequences deposited in GenBank (Fig. 5B). Overall, NspA was highly conserved; gonococcal NspA proteins shared >97% identity with each other and at least 94% identity with meningococcal NspA (Fig. 5B). The NspA sequences from N. gonorrhoeae strains UU1 and 252 were identical, as were those from 15253 and F62.
The crystal structure of Nm-NspA indicates that the protein is an eight-stranded antiparallel beta-barrel with four short surface-exposed loops (48). Based on the crystal structure, we focused on divergent amino acids located in the surface-exposed loops of NspA (Fig. 5B) that were anticipated to interact with FH. The amino acid at position 157 of Nm-NspA was variable and predicted to be in loop 4; however, this residue was not further investigated because two of four of our gonococcal sequences (15253 and FA19) contained the same amino acid (T) as the one in meningococci; therefore, this residue was unlikely play a role in differential binding of FH. Three amino acids, two in loop 3 and one in loop 2, were targeted. Loop 3 was of particular interest because it is important for the binding of the protective anti-Nm-NspA mAbs AL12 and 14C7 (49). Ng-NspA was found to contain Ala (A) and His (H) exclusively at positions 112 and 113 in loop 3, while Nm-NspA encodes Val (V) and Asp (D) exclusively at positions 112 and 113. Position 73 in loop 2 of NspA from UU1, 15253, and FA1090 is occupied by a Gln (Q) residue that is absent from N. gonorrhoeae FA19 and all of the Nm-NspA sequences available in GenBank. Loop 2 may also play a role in the binding of protective mAbs, and the role of this Q residue in FH binding was also investigated.
Site-directed mutagenesis (SDM) was used to create Nm-NspA to Ng-NspA, and, conversely, Ng-NspA to Nm-NspA, exchanges at position 73 (loop 2) alone, positions 112 and 113 (loop 3) together, or positions 73, 112, and 113 simultaneously. Analysis of FH and FHL-1 binding to wild-type and SDM-mutated NspA proteins expressed in MVs indicated that V112 and D113 in surface loop 3 of Nm-NspA were important for enhanced binding of FH and FHL-1 to Nm-NspA relative to Ng-NspA; mutation of these Nm-NspA amino acids to their Ng-NspA counterparts (A112 and H113, respectively) decreased binding of both complement inhibitors (Fig. 6A). Introduction of Gln (Q) at position 73 of Nm-NspA decreased binding of FH but not FHL-1. The triple Nm-NspA to Ng-NspA mutant reduced binding of FH and FHL-1 to levels seen with Ng-NspA. Conversely, deletion of Q73 in Ng-NspA increased binding of FH and FHL-1 to levels that were ∼30% of those seen with Nm-NspA (Fig. 6B). N. gonorrhoeae to N. meningitidis mutations in NspA at positions 112 and 113 enhanced FH and FHL-1 binding to ∼60% and 100% of the levels seen with Nm-NspA. While the triple N. gonorrhoeae to N. meningitidis mutations in Ng-NspA restored binding of FH to levels seen with Nm-NspA, binding to FHL-1 was only 30% of that seen with Nm-NspA (Fig. 6B).
FIG 6.

Amino acids in loops 2 and 3 modulate binding of FH and FHL-1 to NspA. (A) Binding of FH (left graph) and FHL-1 (right graph) to E. coli MVs prepared from E. coli expressing recombinant wild-type meningococcal NspA (solid black squares) or meningococcal NspA bearing N. meningitidis-to-N. gonorrhoeae mutations, measured by ELISAs. The mutant meningococcal NspA molecules included (i) addition of Q at position 73 (+Q73) (solid red triangles); (ii) V-to-A and D-to-H mutations at positions 112 and 113, respectively (VD→AH) (inverted open blue triangles); or (iii) a combined +Q73,VD→AH mutation (filled green diamonds). E. coli MVs expressing Ng-NspA were included as a comparator (filled gray circles). x axis, concentration of FH or FHL-1; y axis, absorbance at 405 nm (optical density [OD] at 405 nm). (B) Binding of FH (left graph) and FHL-1 (right graph) to Ng-NspA expressed in E. coli MVs, measured by ELISAs. Gonococcal NspA was modified by (i) deleting Q at position 73 (−Q73) (open red triangles); (ii) making A-to-V and H-to-D mutations at positions 112 and 113, respectively (AH→VD) (solid blue inverted triangles); and (iii) a combined −Q73 and AH→VD triple mutation (open green diamonds). Nm-NspA-containing MVs (solid black squares) were included for comparison. Axes are as described above for panel A. Two-way ANOVA was used to compare binding of FH/FHL-1 to wild-type Nm-NspA with binding to Ng-NspA and the other mutant NspA molecules. In each instance, there was a statistically significant interaction between concentration and binding (P < 0.0001). Dunnett’s multiple-comparison test was used to compare binding of FH/FHL-1 to Nm-NspA with binding to each of the other NspA molecules; significance was set at a P value of 0.05. In the top left graph, Nm-NspA bound significantly more (P < 0.05) FH than each of the other molecules at all concentrations of ≥2.5 µg/ml. In the top right graph, Nm-NspA bound significantly more FHL-1 than each of the other NspA molecules at all concentrations tested (i.e., ≥0.125 µg/ml). In the bottom left graph, Nm-NspA bound significantly more FH than (i) Ng-NspA at all concentrations tested (i.e., ≥1.25 µg/ml), (ii) Ng-NspA −Q73 at a concentration of ≥2.5 µg/ml and Ng-NspA AH→VD, and (iii) Ng-NspA −Q73 AH→VD at 5 and 10 µg/ml. In the bottom right graph, Nm-NspA bound significantly more FHL-1 than all the tested NspA mutants, except Ng-NspA AH→VD, at all concentrations of ≥0.25 µg/ml.
Anti-Nm-NspA mAb Me-7 binds to loop 3 (49). MVs harboring mutated Nm-NspA encoding A113 and H114 also lost the ability to react with this mAb (data not shown). We previously showed that Me-7 blocked FH binding to N. meningitidis, which further supports the importance of loop 3 in interactions with FH.
Collectively, these data show that the amino acids at positions 73, 112, and 113 are important for FH and FHL-1 binding. FHL-1 binds better to NspA than FH, likely because of less steric hindrance for domains 6 and 7 in FHL-1 imposed by the 13 additional C-terminal domains of full-length FH. However, there are differences in how FH and FHL-1 interact with NspA; as an example, the triple N. gonorrhoeae to N. meningitidis NspA mutation fully restores binding of FH to levels seen with Nm-NspA but permits FHL-1 binding to only 30% of the levels seen with Nm-NspA.
Expression of Ng-NspA enhances serum resistance.
FH downregulates the alternative pathway of complement; therefore, bacteria that bind FH are predicted to be more resistant to complement-mediated serum killing than those that do not bind FH. The role of Ng-NspA in serum resistance was assessed with a serum bactericidal assay with NHS to compare serum resistance of N. gonorrhoeae strains UU1 and FA1090 and their isogenic NspA mutants (Fig. 7). Loss of NspA from N. gonorrhoeae strains UU1 and FA1090 and their isogenic lgtA mutants resulted in significantly increased sensitivity to killing by NHS (Fig. 7). FA1090 is intrinsically more serum resistant than UU1 by virtue of its ability to bind C4b binding protein (C4BP) (50); therefore, higher concentrations of NHS were required to achieve killing and reveal the role of NspA in serum resistance. Overall, these data support a role for NspA in gonococcal complement evasion.
FIG 7.
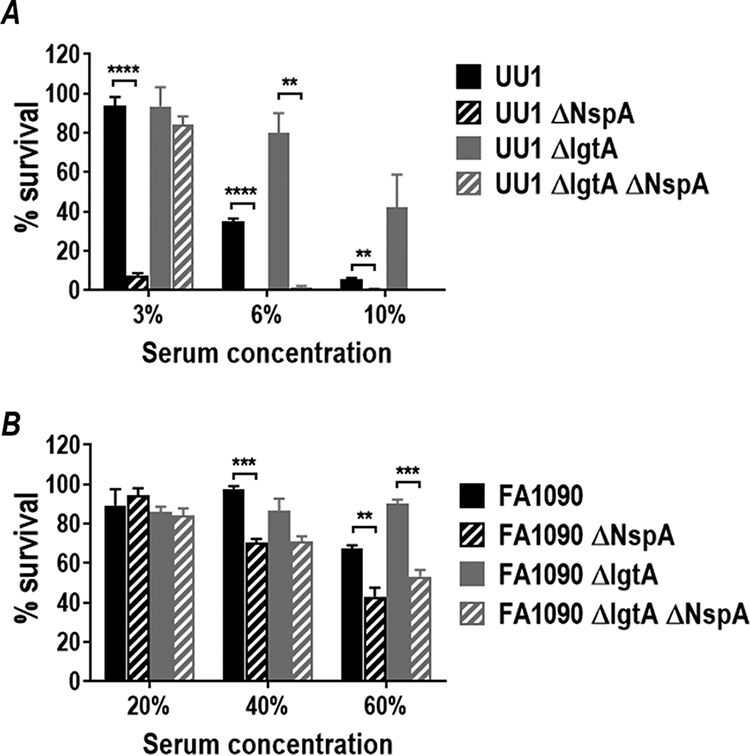
Expression of NspA enhances survival in normal human serum. Shown is survival in normal human serum (NHS) of gonococcal strains UU1 (A) and FA1090 (B) expressing either 4-Hex HepI LOS (LNnT) (wild-type strain, solid black bars) or 2-Hex HepI LOS (ΔlgtA mutant, solid gray bars) and their isogenic NspA mutants (black or gray striped bars, respectively). The percentage of NHS used is indicated on the x axis. The percent survival after incubation in serum at 30 min relative to CFU counts at time zero is plotted on the y axis (means [standard errors of the means {SEM}] of data from 3 independent experiments). Each bar represents the SEM of results from 3 independent observations. Pairwise comparisons were made using the t test. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001 (2-way ANOVA).
DISCUSSION
In this study, we have identified a key role for Ng-NspA in binding FH and complement evasion. As previously described for Nm-NspA (33), Ng-NspA binds FH and FHL-1 through SCR domains 6 and 7. We have identified NspA loops 2 and 3 as being important for interactions with FH and FHL-1 and elucidated differences in how FH interacts with Ng- and Nm-NspA. Similar to our findings with N. meningitidis (33), truncation of LOS glycan extensions from HepI increases binding of FH to Ng-NspA. As also noted previously with N. meningitidis (32), Ng-NspA functions to increase complement resistance on N. gonorrhoeae when HepI expresses LNnT LOS, despite low levels of FH binding detected by flow cytometry. Differences in the amounts of FH/FHL-1 bound by Ng- and Nm-NspA may be attributed to differences in amino acids at positions 72 (loop 2), 112 (loop 3), and 113 (loop 3); the presence of Q72, A112, and H113, as seen in most N. gonorrhoeae strains, decreases binding, while deletion (or absence) of Q at position 72 and the presence of V and D at positions 112 and 113, respectively, as seen in N. meningitidis, all increase FH/FHL-1 binding. In addition to the differences in affinities for FH, another noteworthy difference between the interactions of NspA on N. gonorrhoeae and N. meningitidis is the effect of LOS sialylation; while sialylation of N. meningitidis lacto-N-neotetraose (LNT) LOS enhances FH binding to NspA (33), the opposite is seen with N. gonorrhoeae. The reason for this difference is unclear, but a difference in the exposure of NspA in the presence of LOS sialylation is a possible explanation. However, the decrease in binding of FH domains 6 and 7 to Ng-NspA seen with N. gonorrhoeae LOS sialylation is offset by an increase in FH binding through domains 18 through 20. This dual mechanism of engaging human FH may permit the bacterium to evade complement while varying the LOS glycan composition, as discussed below.
The high incidence of invasive meningococcal disease and disseminated gonococcal infection (DGI) in persons deficient in terminal complement components (51–54) underscores the importance of complement in host defenses against these infections. Both N. gonorrhoeae and N. meningitidis have developed several mechanisms to escape killing by complement, including binding of complement inhibitors such as FH and C4b binding protein (C4BP) (35, 37, 38, 42, 50, 55, 56, 57, 59). Sialylation of LNT LOS of N. gonorrhoeae enhances binding of FH through its C terminus (domains 18 through 20) (36, 37, 41). LNT sialylation is important for establishing experimental urethral infection in men (60–62) and also in the mouse vaginal colonization model of gonorrhea (63, 64). The organism likely needs to finely balance the level of LOS sialylation. Excessive LOS sialylation may impede gonococcal colonization of men (61) because unsialylated LNT is required for binding to the asialoglycoprotein receptor (ASGP-R) to gain entry into male urethral epithelial cells (65). Phase variation of LOS may also enable the bacteria to evade anti-LOS immune responses (66). N. gonorrhoeae strains often express multiple LOS structures simultaneously (67, 68). A recent survey of 75 minimally passaged N. gonorrhoeae isolates obtained from men who attended a sexually transmitted diseases clinic in Nanjing, China, revealed that 100% of isolates expressed LNnT but also simultaneously expressed LOS with only lactose extensions from HepI, evidenced by reactivity with mAb 2-1-L8 (69–71). The ability to retain FH binding even in the absence of LNnT sialyation, in this instance by binding of FH SCR domains 6 and 7 by NspA, will enable complement evasion. It is worth emphasizing that while binding of FH and FHL-1 increases with HepI glycan truncation (i.e., when lgtA is phase varied “off”), NspA enhances serum resistance, even in wild-type isolates that also express LNnT LOS.
Meningococcal NspA was evaluated as a vaccine candidate, but those studies did not predict protection (72). Several reasons could have contributed to the predicted nonefficacy of the vaccine preparation. First, the recombinant protein that was produced in E. coli may not have folded properly and therefore may not have presented the same epitopes seen on native organisms. Second, binding of human FH to NspA may have reduced immunogenicity to the surface-exposed FH binding regions of NspA, which may also be targets for bactericidal antibodies such as Me-7. A similar phenomenon occurs when humans or mice that express human FH (human FH transgenic mice) are immunized with fHbp. Binding of human FH to N. meningitidis fHbp diverts the immune response to regions of the molecule that do not bind to FH (73–75). The elicited antibodies do not prevent FH from binding to bacteria, and the bacterium-bound FH dampens the bactericidal activity of vaccine antibodies. Amino acid mutations in fHbp that maintain the overall structure and immunogenicity, but which eliminate binding of human FH, elicit antibodies that divert FH away from the bacterial surface and are more bactericidal than antibodies that do not block FH binding (74). This study has identified the amino acids that modulate FH binding to NspA and could guide the rational design of neisserial NspA-based vaccines.
The worldwide emergence of multidrug-resistant gonococcal strains necessitates the development of novel treatments and immunotherapeutics against N. gonorrhoeae. An approach that our group has used to combat drug-resistant bacteria is to fuse the microbial binding domains of human FH to IgG Fc (39). The resulting fusion protein acts by blocking binding of FH to the microbial surface, while the Fc domain activates complement, which in turn can result in the insertion of lytic membrane attack complexes to kill Gram-negative bacteria or engage Fc receptors and complement receptors for C3 fragments on phagocytes. We have demonstrated the efficacy of a chimeric protein where FH domains 18 through 20 are fused to Fc against N. gonorrhoeae (39, 41, 76). Based on the finding that N. gonorrhoeae also binds FH domains 6 and 7, we have fused these two domains with human IgG1 Fc to create FH SCR6-7/Fc. Ongoing work has shown that FH SCR6-7/Fc is effective against N. gonorrhoeae both in vitro and in vivo (77, 78). We speculate that the use of FH SCR18-20/Fc and FH SCR6-7/Fc may provide additive or synergistic activity because these molecules bind to distinct targets on the bacterial membrane. Furthermore, their use in combination will also raise the barrier to the development of resistance.
In conclusion, we have defined NspA as a ligand for human FH and FHL-1 and an important factor that contributes to gonococcal complement resistance. Elucidating the molecular basis for the interaction between FH domains 6 and 7 and NspA will prove useful in informing the development of novel vaccines and therapeutics against multidrug-resistant gonorrhea that has emerged as a global threat to human health worldwide.
MATERIALS AND METHODS
Ethics statement.
This study was approved by the Committee for the Protection of Human Subjects in Research at the University of Massachusetts Medical School. All subjects who donated blood for this study provided written informed consent.
Bacterial strains, bacterial growth conditions, and mutagenesis.
N. gonorrhoeae PorB.1A strains UU1 (79, 80) and 15253 (81) and PorB.1B strain FA1090 (82) have all been described previously. N. meningitidis A2594 (also called WUE2594) is a serogroup A strain that expresses high levels of NspA (83); inactivation of mynB (to abrogate expression of capsule), fHbp, and nspA in A2594 was described previously (33). N. gonorrhoeae and N. meningitidis were routinely grown on chocolate agar plates supplemented with an IsoVitaleX equivalent at 37°C in an atmosphere enriched with 5% CO2. GC plates supplemented with the IsoVitaleX equivalent were used for antibiotic selection. DNA transformation of N. gonorrhoeae was performed as described previously (84). The following antibiotics were used when indicated; 100 µg/ml kanamycin, 50 µg/ml spectinomycin, 2 µg/ml erythromycin, and 10 mg/ml streptomycin. Escherichia coli strains (Invitrogen, Carlsbad, CA) were routinely cultured in Luria-Bertani (LB) broth or on LB agar. The following antibiotics were used as needed: 50 µg/ml kanamycin, 150 µg/ml ampicillin, 400 µg/ml erythromycin, and 100 µg/ml spectinomycin. Sialylation of LOS was achieved by growing bacteria in gonococcal liquid medium supplemented with cytidine-5′-monophospho-N-acetylneuraminic acid (CMP-Neu5Ac) (25 µg/ml).
Insertional inactivation of nspA in N. meningitidis has been described previously (33). A linearized plasmid harboring the meningococcal nspA gene with a 130-bp deletion and a spectinomycin resistance cassette was used to transform N. gonorrhoeae. NspA deletion mutants were similarly constructed in N. gonorrhoeae strains UU1, 15253, and FA1090. PCR was used to confirm the nspA::spc genotype, and Western blotting with anti-NspA mAb Me-7 (kindly provided by Dan Granoff, CHORI, Oakland, CA) was used to confirm the loss of NspA. Insertional inactivation of lgtA was accomplished as described previously (33, 85), using a plasmid harboring lgtA::kan. Insertional inactivation of lgtA was confirmed by PCR, and truncation of LOS was evidenced by migration on SDS-PAGE gels following staining with silver and by loss of reactivity with mAb 3F11 in Western blot assays.
DNA sequencing.
The nspA gene was amplified by PCR from N. gonorrhoeae strains UU1, 15253, FA1090, FA19, and F62, and the PCR product was directly sequenced. Sanger DNA sequencing of all PCR products and plasmid constructs was performed by GeneWiz (Cambridge, MA). Predicted amino acid sequences were compared using ClustalW (http://www.genome.jp/tools-bin/clustalw).
Sera.
Serum collected from healthy human volunteers without a history of neisserial disease was aliquoted and stored at −80°C until needed. Hemolytic activity of the serum was confirmed using a total hemolytic complement assay (Binding Site, Birmingham, UK). Complement activity in the sera was inactivated, when necessary, by heating at 56°C for 30 min. The sera used did not contain NspA-specific antibodies as revealed by Western blotting of whole bacterial lysates that were probed with serum.
Expression of NspA in E. coli and preparation of microvesicles.
E. coli BL21(DE3) (Invitrogen, Carlsbad, CA) harboring recombinant N. meningitidis nspA from N. meningitidis strain 8047 on plasmid pGMS 1.0 was described previously (49, 86). Gonococcal nspA genes were amplified from N. gonorrhoeae strains UU1, 15253, and FA1090 (primers NspABamH1_F and NspAHindIII_R [see Table S1 in the supplemental material]), cloned into pBluescript II SK+ (Stratagene, La Jolla, CA), digested with BamHI and HindIII, and transformed into E. coli BL21(DE3). The resulting clones, harboring plasmids pGMS_UU1, pGMS_15253, and pGMS_FA1090, were verified by DNA sequencing.
E. coli BL21(DE3) harboring recombinant nspA on plasmid pGMS 1.0, pGMS_UU1, pGMS_15253, or pGMS_FA1090 or E. coli harboring the pBluescript II SK+ vector alone was used to prepare microvesicles (MVs) as previously described (49, 86). MV preparations were quantified by a bicinchoninic acid (BCA) protein assay and visualized on 12% Bis-Tris polyacrylamide gels (Invitrogen, Carlsbad, CA) stained with Coomassie blue and by Western blotting with anti-NspA mAb Me-7.
Site-directed mutagenesis of nspA.
Site-directed mutagenesis (SDM) of nspA was used to alter specific amino acids in Nm- and Ng-NspA, and the wild-type and mutated NspA proteins were expressed in E. coli and isolated as MVs. Based on the sequence dissimilarity between Nm-NspA and Ng-NspA (Fig. 4), the following point mutations were constructed: insertion of Q73 (+Q73) in loop 2 and V112A and D113H (VD→AH) in loop 3 in Nm-NspA and deletion of Q73 (ΔQ73) and A112V and H113V (AH→VD) in Ng-NspA. Similarly, we created “double” mutants in both Nm-NspA (+Q73,VD→AH) and Ng-NspA (ΔQ73,AH→VD). Point mutations were created using the QuikChange II site-directed mutagenesis kit according to the manufacturer’s instructions (Stratagene, La Jolla, CA). Primers (see Table S1 in the supplemental material were designed using the QuikChange primer design program (Agilent Technologies). All constructs were validated by DNA sequencing.
Recombinant FH fragment/Fc fusion proteins.
FH/mouse IgG2a Fc fusion constructs that contain contiguous FH SCR domains (SCRs 5 to 8, SCRs 6 and 7, and SCRs 18 to 20) fused to the N terminus of the Fc fragment of murine IgG2a (FH/Fc fusion proteins) and human FH SCR5-8/Fc constructs with point mutations in His337, His360, and His 371 in SCR6 were described in detail previously (36, 40). These SCR 6×His residues are surface exposed and have been shown to be important for binding of FH to polyanions and to the meningococcal fHbp protein (27, 40). FH/Fc constructs were purified from culture supernatants collected from transiently transfected Chinese hamster ovary (CHO) cells using protein A/G-Sepharose (ThermoFisher, Waltham, MA) according to the manufacturer’s instructions. Purified FH/Fc constructs were concentrated by centrifugation using an Amicon Ultra-30 centrifugal filter unit, and the final protein concentration was determined by a BCA assay (Bio-Rad). Constructs were further assessed by both Coomassie blue staining of proteins separated by lithium dodecyl sulfate (LDS)-PAGE and Western blotting with both anti-FH polyclonal and anti-mouse IgG Fc antibodies. Goat anti-mouse IgG (Sigma-Aldrich, St. Louis, MO) was used to detect binding of recombinant FH/Fc fusion proteins to bacteria; the use of the IgG Fc region for detection permits equal detection of each fusion molecule.
Flow cytometry.
Flow cytometry to detect bound FH, recombinant FHL-1, or FH/Fc constructs was performed as described previously (40). Briefly, bacteria were grown overnight on chocolate agar plates, subcultured onto fresh chocolate agar, and grown at 37°C in 5% CO2. At 6 h, bacteria were washed with Hanks’ balanced salt solution (HBSS) containing 1 mM CaCl2 and 1 mM MgCl2 (HBSS++) and suspended to a final concentration of 3 × 108 cells/ml; 107 organisms were centrifuged and incubated with either FH purified from human plasma (Complement Technology, Inc., Tyler, TX) (concentration specified for each experiment), recombinant FHL-1 (purified from CHO cell supernatants using a polyclonal anti-FH affinity column as described above), or purified FH/Fc. Bound FH or FHL-1 was detected using either affinity-isolated sheep anti-human FH (Lifespan Biosciences, Seattle, WA) or anti-FH mAb 90X (catalog no. A254; Quidel, San Diego, CA). Fluorescein isothiocyanate (FITC)-conjugated anti-sheep IgG or anti-mouse IgG (Sigma-Aldrich, St. Louis, MO) was used as a secondary antibody. While the polyclonal anti-FH antibody provided higher sensitivity, relative differences in FH binding among strains using either of the two reagents were similar. Bound FH/Fc constructs were detected using goat anti-mouse IgG-FITC (Sigma-Aldrich, St. Louis, MO). All reactions were carried out in a solution containing HBSS++–1% bovine serum albumin (BSA). Flow cytometry was performed using a FACSCalibur instrument (Becton, Dickinson, Franklin Lakes, NJ), and data analysis was performed using the FlowJo data analysis software package (www.TreeStar.com). Reaction mixtures in which the primary ligand (FH, FHL-1, or FH/Fc) was omitted served as negative controls.
Assessment of binding of FH, FHL-1, and FH/Fc constructs to NspA-containing E. coli MVs using ELISAs.
ELISAs were used to detect binding of FH, FHL-1, and FH/Fc constructs to NspA-containing vesicles. Microtiter wells were coated with either NspA-containing or control MVs at a concentration of 10 µg/ml in phosphate-buffered saline (PBS) overnight at 22°C. Nonspecific binding was blocked with PBS–2.5% BSA for 2 h at 37°C. FH (concentrations ranging from 0 to 10 µg/ml), FHL-1 (concentrations ranging from 0 to 10 µg/ml), or FH/Fc (5 µg/ml) was added to wells for 1 h at 37°C, and bound ligand was detected using polyclonal sheep anti-human FH followed by anti-sheep IgG conjugated with alkaline phosphatase (for FH and FHL-1) or goat anti-mouse IgG conjugated with alkaline phosphatase (for FH/Fc constructs), each for 1 h at 37°C.
Serum bactericidal assays.
Bactericidal assays were used to compare serum resistances of wild-type N. gonorrhoeae and nspA deletion mutants. Serum bactericidal assays have been described previously (40, 87); the optimal concentration of serum was determined empirically for each strain. Bacteria from a culture grown overnight on chocolate agar plates were inoculated onto fresh chocolate agar and allowed to grow for 6 h at 37°C in 5% CO2. Briefly, 2,000 CFU of gonococci were incubated with serum (concentrations specified for each experiment) in a final reaction mixture volume of 150 µl. Aliquots of 25 µl were plated in duplicate at the start of the assay (time zero [t0]) and after incubating the reaction mixture at 37°C for 30 min (time 30 min [t30]). Survival was calculated as the number of viable colonies at t30 relative to baseline colony counts at t0. Each experiment was repeated at least three times. The average survival was calculated from at least three independent experiments, and error bars represent the standard deviations. A t test was used to determine significance.
Statistics.
Pairwise comparisons were made using the t test, and comparisons across groups were made using analysis of variance (ANOVA) (GraphPad Prism 7.0).
Supplementary Material
ACKNOWLEDGMENTS
We thank Mathew Carter, Bethany Chaves, Anthonia Beluchukwu, and Joseph Burbage for providing excellent technical support. We also thank Gregory R. Moe and Dan M. Granoff (Children’s Hospital Oakland Research Institute, Oakland, CA, USA) for providing plasmid pGMS 1.0 that harbors the meningococcal nspA gene, for plasmid pFP12, and for many informative discussions.
This work was supported by Public Health Service grants U01AI118161, R01AI114790, R01AI132296, and R01AI114710 from the National Institute of Allergy and Infectious Diseases (NIH).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00658-18.
REFERENCES
- 1.Newman L, Rowley J, Vander Hoorn S, Wijesooriya NS, Unemo M, Low N, Stevens G, Gottlieb S, Kiarie J, Temmerman M. 2015. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One 10:e0143304. doi: 10.1371/journal.pone.0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centers for Disease Control and Prevention, Division of STD Prevention, National Center for HIV/AIDS, Viral Hepatitis, STD, and TB Prevention. 2016. 2016 sexually transmitted diseases surveillance: gonorrhea. Centers for Disease Control and Prevention, Atlanta, GA: Accessed 26 September 2017 https://www.cdc.gov/std/stats16/default.htm. [Google Scholar]
- 3.Satterwhite CL, Torrone E, Meites E, Dunne EF, Mahajan R, Ocfemia MC, Su J, Xu F, Weinstock H. 2013. Sexually transmitted infections among US women and men: prevalence and incidence estimates, 2008. Sex Transm Dis 40:187–193. doi: 10.1097/OLQ.0b013e318286bb53. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention, Department of Health and Human Services. 2017. Gonorrhea—CDC fact sheet. Centers for Disease Control and Prevention, Atlanta, GA: Accessed 10 December 2018 https://www.cdc.gov/std/gonorrhea/stdfact-gonorrhea-detailed.htm. [Google Scholar]
- 5.Camara J, Serra J, Ayats J, Bastida T, Carnicer-Pont D, Andreu A, Ardanuy C. 2012. Molecular characterization of two high-level ceftriaxone-resistant Neisseria gonorrhoeae isolates detected in Catalonia, Spain. J Antimicrob Chemother 67:1858–1860. doi: 10.1093/jac/dks162. [DOI] [PubMed] [Google Scholar]
- 6.Gose S, Nguyen D, Lowenberg D, Samuel M, Bauer H, Pandori M. 2013. Neisseria gonorrhoeae and extended-spectrum cephalosporins in California: surveillance and molecular detection of mosaic penA. BMC Infect Dis 13:570. doi: 10.1186/1471-2334-13-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katz AR, Komeya AY, Kirkcaldy RD, Whelen AC, Soge OO, Papp JR, Kersh EN, Wasserman GM, O’Connor NP, O’Brien PS, Sato DT, Maningas EV, Kunimoto GY, Tomas JE. 2017. Cluster of Neisseria gonorrhoeae isolates with high-level azithromycin resistance and decreased ceftriaxone susceptibility, Hawaii, 2016. Clin Infect Dis 65:918–923. doi: 10.1093/cid/cix485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohnishi M, Golparian D, Shimuta K, Saika T, Hoshina S, Iwasaku K, Nakayama S, Kitawaki J, Unemo M. 2011. Is Neisseria gonorrhoeae initiating a future era of untreatable gonorrhea? Detailed characterization of the first strain with high-level resistance to ceftriaxone. Antimicrob Agents Chemother 55:3538–3545. doi: 10.1128/AAC.00325-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Unemo M, Shafer WM. 2014. Antimicrobial resistance in Neisseria gonorrhoeae in the 21st century: past, evolution, and future. Clin Microbiol Rev 27:587–613. doi: 10.1128/CMR.00010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alirol E, Wi TE, Bala M, Bazzo ML, Chen XS, Deal C, Dillon JR, Kularatne R, Heim J, Hooft van Huijsduijnen R, Hook EW, Lahra MM, Lewis DA, Ndowa F, Shafer WM, Tayler L, Workowski K, Unemo M, Balasegaram M. 2017. Multidrug-resistant gonorrhea: a research and development roadmap to discover new medicines. PLoS Med 14:e1002366. doi: 10.1371/journal.pmed.1002366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perez JL, Absalon J, Beeslaar J, Balmer P, Jansen KU, Jones TR, Harris S, York LJ, Jiang Q, Radley D, Anderson AS, Crowther G, Eiden JJ. 2018. From research to licensure and beyond: clinical development of MenB-FHbp, a broadly protective meningococcal B vaccine. Expert Rev Vaccines doi: 10.1080/14760584.2018.1483726:1-17. [DOI] [PubMed] [Google Scholar]
- 12.Gorringe AR, Pajon R. 2012. Bexsero: a multicomponent vaccine for prevention of meningococcal disease. Hum Vaccin Immunother 8:174–183. doi: 10.4161/hv.18500. [DOI] [PubMed] [Google Scholar]
- 13.O’Ryan M, Stoddard J, Toneatto D, Wassil J, Dull PM. 2014. A multi-component meningococcal serogroup B vaccine (4CMenB): the clinical development program. Drugs 74:15–30. doi: 10.1007/s40265-013-0155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petousis-Harris H, Paynter J, Morgan J, Saxton P, McArdle B, Goodyear-Smith F, Black S. 2017. Effectiveness of a group B outer membrane vesicle meningococcal vaccine against gonorrhoea in New Zealand: a retrospective case-control study. Lancet 390:1603–1610. doi: 10.1016/S0140-6736(17)31449-6. [DOI] [PubMed] [Google Scholar]
- 15.Konar M, Granoff DM, Beernink PT. 2013. Importance of inhibition of binding of complement factor H for serum bactericidal antibody responses to meningococcal factor H-binding protein vaccines. J Infect Dis 208:627–636. doi: 10.1093/infdis/jit239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Serruto D, Bottomley MJ, Ram S, Giuliani MM, Rappuoli R. 2012. The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: immunological, functional and structural characterization of the antigens. Vaccine 30:B87–B97. doi: 10.1016/j.vaccine.2012.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirkitadze MD, Barlow PN. 2001. Structure and flexibility of the multiple domain proteins that regulate complement activation. Immunol Rev 180:146–161. doi: 10.1034/j.1600-065X.2001.1800113.x. [DOI] [PubMed] [Google Scholar]
- 18.Ripoche J, Day AJ, Harris TJ, Sim RB. 1988. The complete amino acid sequence of human complement factor H. Biochem J 249:593–602. doi: 10.1042/bj2490593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. 1995. Identification of complement regulatory domains in human factor H. J Immunol 155:348–356. [PubMed] [Google Scholar]
- 20.Kuhn S, Skerka C, Zipfel PF. 1995. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol 155:5663–5670. [PubMed] [Google Scholar]
- 21.Kuhn S, Zipfel PF. 1996. Mapping of the domains required for decay acceleration activity of the human factor H-like protein 1 and factor H. Eur J Immunol 26:2383–2387. doi: 10.1002/eji.1830261017. [DOI] [PubMed] [Google Scholar]
- 22.Blackmore TK, Hellwage J, Sadlon TA, Higgs N, Zipfel PF, Ward HM, Gordon DL. 1998. Identification of the second heparin-binding domain in human complement factor H. J Immunol 160:3342–3348. [PubMed] [Google Scholar]
- 23.Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL. 1996. Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol 157:5422–5427. [PubMed] [Google Scholar]
- 24.Kajander T, Lehtinen MJ, Hyvarinen S, Bhattacharjee A, Leung E, Isenman DE, Meri S, Goldman A, Jokiranta TS. 2011. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci U S A 108:2897–2902. doi: 10.1073/pnas.1017087108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zipfel PF, Skerka C, Hellwage J, Jokiranta ST, Meri S, Brade V, Kraiczy P, Noris M, Remuzzi G. 2002. Factor H family proteins: on complement, microbes and human diseases. Biochem Soc Trans 30:971–978. doi: 10.1042/bst0300971. [DOI] [PubMed] [Google Scholar]
- 26.Zipfel PF, Jokiranta TS, Hellwage J, Koistinen V, Meri S. 1999. The factor H protein family. Immunopharmacology 42:53–60. doi: 10.1016/S0162-3109(99)00015-6. [DOI] [PubMed] [Google Scholar]
- 27.Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, Jowitt TA, Clark SJ, Tarelli E, Uhrin D, Barlow PN, Sim RB, Day AJ, Lea SM. 2007. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med 204:2277–2283. doi: 10.1084/jem.20071069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clark SJ, Schmidt CQ, White AM, Hakobyan S, Morgan BP, Bishop PN. 2014. Identification of factor H-like protein 1 as the predominant complement regulator in Bruch’s membrane: implications for age-related macular degeneration. J Immunol 193:4962–4970. doi: 10.4049/jimmunol.1401613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. 1987. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol 17:1485–1489. doi: 10.1002/eji.1830171015. [DOI] [PubMed] [Google Scholar]
- 30.Sofat R, Mangione PP, Gallimore JR, Hakobyan S, Hughes TR, Shah T, Goodship T, D’Aiuto F, Langenberg C, Wareham N, Morgan BP, Pepys MB, Hingorani AD. 2013. Distribution and determinants of circulating complement factor H concentration determined by a high-throughput immunonephelometric assay. J Immunol Methods 390:63–73. doi: 10.1016/j.jim.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 31.Kraiczy P, Wurzner R. 2006. Complement escape of human pathogenic bacteria by acquisition of complement regulators. Mol Immunol 43:31–44. doi: 10.1016/j.molimm.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 32.Lewis LA, Carter M, Ram S. 2012. The relative roles of factor H binding protein, neisserial surface protein A, and lipooligosaccharide sialylation in regulation of the alternative pathway of complement on meningococci. J Immunol 188:5063–5072. doi: 10.4049/jimmunol.1103748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lewis LA, Ngampasutadol J, Wallace R, Reid JE, Vogel U, Ram S. 2010. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PLoS Pathog 6:e1001027. doi: 10.1371/journal.ppat.1001027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis LA, Vu DM, Vasudhev S, Shaughnessy J, Granoff DM, Ram S. 2013. Factor H-dependent alternative pathway inhibition mediated by porin B contributes to virulence of Neisseria meningitidis. mBio 4:e00339-13. doi: 10.1128/mBio.00339-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madico G, Welsch JA, Lewis LA, McNaughton A, Perlman DH, Costello CE, Ngampasutadol J, Vogel U, Granoff DM, Ram S. 2006. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol 177:501–510. doi: 10.4049/jimmunol.177.1.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ngampasutadol J, Ram S, Gulati S, Agarwal S, Li C, Visintin A, Monks B, Madico G, Rice PA. 2008. Human factor H interacts selectively with Neisseria gonorrhoeae and results in species-specific complement evasion. J Immunol 180:3426–3435. doi: 10.4049/jimmunol.180.5.3426. [DOI] [PubMed] [Google Scholar]
- 37.Ram S, Sharma AK, Simpson SD, Gulati S, McQuillen DP, Pangburn MK, Rice PA. 1998. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med 187:743–752. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider MC, Exley RM, Chan H, Feavers I, Kang YH, Sim RB, Tang CM. 2006. Functional significance of factor H binding to Neisseria meningitidis. J Immunol 176:7566–7575. doi: 10.4049/jimmunol.176.12.7566. [DOI] [PubMed] [Google Scholar]
- 39.Ram S, Shaughnessy J, DeOliveira RB, Lewis LA, Gulati S, Rice PA. 2016. Utilizing complement evasion strategies to design complement-based antibacterial immunotherapeutics: lessons from the pathogenic Neisseriae. Immunobiology 221:1110–1123. doi: 10.1016/j.imbio.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shaughnessy J, Lewis LA, Jarva H, Ram S. 2009. Functional comparison of the binding of factor H short consensus repeat 6 (SCR 6) to factor H binding protein from Neisseria meningitidis and the binding of factor H SCR 18 to 20 to Neisseria gonorrhoeae porin. Infect Immun 77:2094–2103. doi: 10.1128/IAI.01561-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shaughnessy J, Gulati S, Agarwal S, Unemo M, Ohnishi M, Su XH, Monks BG, Visintin A, Madico G, Lewis LA, Golenbock DT, Reed GW, Rice PA, Ram S. 2016. A novel factor H-Fc chimeric immunotherapeutic molecule against Neisseria gonorrhoeae. J Immunol 196:1732–1740. doi: 10.4049/jimmunol.1500292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madico G, Ngampasutadol J, Gulati S, Vogel U, Rice PA, Ram S. 2007. Factor H binding and function in sialylated pathogenic neisseriae is influenced by gonococcal, but not meningococcal, porin. J Immunol 178:4489–4497. doi: 10.4049/jimmunol.178.7.4489. [DOI] [PubMed] [Google Scholar]
- 43.Jongerius I, Lavender H, Tan L, Ruivo N, Exley RM, Caesar JJ, Lea SM, Johnson S, Tang CM. 2013. Distinct binding and immunogenic properties of the gonococcal homologue of meningococcal factor H binding protein. PLoS Pathog 9:e1003528. doi: 10.1371/journal.ppat.1003528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Erwin AL, Haynes PA, Rice PA, Gotschlich EC. 1996. Conservation of the lipooligosaccharide synthesis locus lgt among strains of Neisseria gonorrhoeae: requirement for lgtE in synthesis of the 2C7 epitope and of the beta chain of strain 15253. J Exp Med 184:1233–1241. doi: 10.1084/jem.184.4.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Apicella MA, Mandrell RE, Shero M, Wilson ME, Griffiss JM, Brooks GF, Lammel C, Breen JF, Rice PA. 1990. Modification by sialic acid of Neisseria gonorrhoeae lipooligosaccharide epitope expression in human urethral exudates: an immunoelectron microscopic analysis. J Infect Dis 162:506–512. doi: 10.1093/infdis/162.2.506. [DOI] [PubMed] [Google Scholar]
- 46.Granoff DM, Welsch JA, Ram S. 2009. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect Immun 77:764–769. doi: 10.1128/IAI.01191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schneider MC, Prosser BE, Caesar JJ, Kugelberg E, Li S, Zhang Q, Quoraishi S, Lovett JE, Deane JE, Sim RB, Roversi P, Johnson S, Tang CM, Lea SM. 2009. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature 458:890–893. doi: 10.1038/nature07769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vandeputte-Rutten L, Bos MP, Tommassen J, Gros P. 2003. Crystal structure of neisserial surface protein A (NspA), a conserved outer membrane protein with vaccine potential. J Biol Chem 278:24825–24830. doi: 10.1074/jbc.M302803200. [DOI] [PubMed] [Google Scholar]
- 49.Hou VC, Moe GR, Raad Z, Wuorimaa T, Granoff DM. 2003. Conformational epitopes recognized by protective anti-neisserial surface protein A antibodies. Infect Immun 71:6844–6849. doi: 10.1128/IAI.71.12.6844-6849.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ram S, Cullinane M, Blom AM, Gulati S, McQuillen DP, Monks BG, O’Connell C, Boden R, Elkins C, Pangburn MK, Dahlback B, Rice PA. 2001. Binding of C4b-binding protein to porin: a molecular mechanism of serum resistance of Neisseria gonorrhoeae. J Exp Med 193:281–295. doi: 10.1084/jem.193.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Densen P. 1989. Interaction of complement with Neisseria meningitidis and Neisseria gonorrhoeae. Clin Microbiol Rev 2:S11–S17. doi: 10.1128/CMR.2.Suppl.S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Figueroa J, Andreoni J, Densen P. 1993. Complement deficiency states and meningococcal disease. Immunol Res 12:295–311. doi: 10.1007/BF02918259. [DOI] [PubMed] [Google Scholar]
- 53.Ross SC, Densen P. 1984. Complement deficiency states and infection: epidemiology, pathogenesis and consequences of neisserial and other infections in an immune deficiency. Medicine (Baltimore) 63:243–273. doi: 10.1097/00005792-198409000-00001. [DOI] [PubMed] [Google Scholar]
- 54.Ram S, Lewis LA, Rice PA. 2010. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev 23:740–780. doi: 10.1128/CMR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jarva H, Ram S, Vogel U, Blom AM, Meri S. 2005. Binding of the complement inhibitor C4BP to serogroup B Neisseria meningitidis. J Immunol 174:6299–6307. doi: 10.4049/jimmunol.174.10.6299. [DOI] [PubMed] [Google Scholar]
- 56.Ram S, McQuillen DP, Boden R, Gulati S, Pangburn MK, Rice PA. 1998. Neisseria gonorrhoeae regulate the classical pathway by directly binding C4-binding protein. Mol Immunol 35:398. doi: 10.1016/S0161-5890(98)90802-1. [DOI] [Google Scholar]
- 57.Ram S, McQuillen DP, Gulati S, Elkins C, Pangburn MK, Rice PA. 1998. Binding of complement factor H to loop 5 of porin protein 1A: a molecular mechanism of serum resistance of nonsialylated Neisseria gonorrhoeae. J Exp Med 188:671–680. doi: 10.1084/jem.188.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reference deleted.
- 59.Gulati S, Cox A, Lewis LA, Michael FS, Li J, Boden R, Ram S, Rice PA. 2005. Enhanced factor H binding to sialylated gonococci is restricted to the sialylated lacto-N-neotetraose lipooligosaccharide species: implications for serum resistance and evidence for a bifunctional lipooligosaccharide sialyltransferase in gonococci. Infect Immun 73:7390–7397. doi: 10.1128/IAI.73.11.7390-7397.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schneider H, Cross AS, Kuschner RA, Taylor DN, Sadoff JC, Boslego JW, Deal CD. 1995. Experimental human gonococcal urethritis: 250 Neisseria gonorrhoeae MS11mkC are infective. J Infect Dis 172:180–185. doi: 10.1093/infdis/172.1.180. [DOI] [PubMed] [Google Scholar]
- 61.Schneider H, Schmidt KA, Skillman DR, Van De Verg L, Warren RL, Wylie HJ, Sadoff JC, Deal CD, Cross AS. 1996. Sialylation lessens the infectivity of Neisseria gonorrhoeae MS11mkC. J Infect Dis 173:1422–1427. doi: 10.1093/infdis/173.6.1422. [DOI] [PubMed] [Google Scholar]
- 62.Hobbs MM, Sparling PF, Cohen MS, Shafer WM, Deal CD, Jerse AE. 2011. Experimental gonococcal infection in male volunteers: cumulative experience with Neisseria gonorrhoeae strains FA1090 and MS11mkC. Front Microbiol 2:123. doi: 10.3389/fmicb.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu H, Jerse AE. 2006. Alpha-2,3-sialyltransferase enhances Neisseria gonorrhoeae survival during experimental murine genital tract infection. Infect Immun 74:4094–4103. doi: 10.1128/IAI.00433-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lewis LA, Gulati S, Burrowes E, Zheng B, Ram S, Rice PA. 2015. Alpha-2,3-sialyltransferase expression level impacts the kinetics of lipooligosaccharide sialylation, complement resistance, and the ability of Neisseria gonorrhoeae to colonize the murine genital tract. mBio 6:e02465-14. doi: 10.1128/mBio.02465-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harvey HA, Ketterer MR, Preston A, Lubaroff D, Williams R, Apicella MA. 1997. Ultrastructural analysis of primary human urethral epithelial cell cultures infected with Neisseria gonorrhoeae. Infect Immun 65:2420–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang QL, Gotschlich EC. 1996. Variation of gonococcal lipooligosaccharide structure is due to alterations in poly-G tracts in lgt genes encoding glycosyl transferases. J Exp Med 183:323–327. doi: 10.1084/jem.183.1.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burch CL, Danaher RJ, Stein DC. 1997. Antigenic variation in Neisseria gonorrhoeae: production of multiple lipooligosaccharides. J Bacteriol 179:982–986. doi: 10.1128/jb.179.3.982-986.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mandrell R, Schneider H, Apicella M, Zollinger W, Rice PA, Griffiss JM. 1986. Antigenic and physical diversity of Neisseria gonorrhoeae lipooligosaccharides. Infect Immun 54:63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schneider H, Griffiss JM, Mandrell RE, Jarvis GA. 1985. Elaboration of a 3.6-kilodalton lipooligosaccharide, antibody against which is absent from human sera, is associated with serum resistance of Neisseria gonorrhoeae. Infect Immun 50:672–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.O’Connor ET, Swanson KV, Cheng H, Fluss K, Griffiss JM, Stein DC. 2008. Structural requirements for monoclonal antibody 2-1-L8 recognition of neisserial lipooligosaccharides. Hybridoma (Larchmt) 27:71–79. doi: 10.1089/hyb.2007.0552. [DOI] [PubMed] [Google Scholar]
- 71.Ram S, Gulati S, Lewis LA, Chakraborti S, Zheng B, DeOliveira RB, Reed GW, Cox AD, Li J, St Michael F, Stupak J, Su XH, Saha S, Landig CS, Varki A, Rice PA. 2018. A novel sialylation site on Neisseria gonorrhoeae lipooligosaccharide links heptose II lactose expression with pathogenicity. Infect Immun 86:e00285-18. doi: 10.1128/IAI.00285-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Halperin SA, Langley JM, Smith B, Wunderli P, Kaufman L, Kimura A, Martin D. 2007. Phase 1 first-in-human studies of the reactogenicity and immunogenicity of a recombinant meningococcal NspA vaccine in healthy adults. Vaccine 25:450–457. doi: 10.1016/j.vaccine.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 73.Beernink PT, Shaughnessy J, Pajon R, Braga EM, Ram S, Granoff DM. 2012. The effect of human factor H on immunogenicity of meningococcal native outer membrane vesicle vaccines with over-expressed factor H binding protein. PLoS Pathog 8:e1002688. doi: 10.1371/journal.ppat.1002688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beernink PT, Shaughnessy J, Braga EM, Liu Q, Rice PA, Ram S, Granoff DM. 2011. A meningococcal factor H binding protein mutant that eliminates factor H binding enhances protective antibody responses to vaccination. J Immunol 186:3606–3614. doi: 10.4049/jimmunol.1003470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Beernink PT, Shaughnessy J, Ram S, Granoff DM. 2010. Impaired immunogenicity of a meningococcal factor H-binding protein vaccine engineered to eliminate factor H binding. Clin Vaccine Immunol 17:1074–1078. doi: 10.1128/CVI.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ram S, Shaughnessy J, de Oliveira RB, Lewis LA, Gulati S, Rice PA. 2017. Gonococcal lipooligosaccharide sialylation: virulence factor and target for novel immunotherapeutics. Pathog Dis 75:ftx049. doi: 10.1093/femspd/ftx049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shaughnessy J, Vu DM, Punjabi R, Serra-Pladevall J, DeOliveira RB, Granoff DM, Ram S. 2014. Fusion protein comprising factor H domains 6 and 7 and human IgG1 Fc as an antibacterial immunotherapeutic. Clin Vaccine Immunol 21:1452–1459. doi: 10.1128/CVI.00444-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shaughnessy J, Lewis LA, Zheng B, Carr C, Bass I, Gulati S, DeOliveira RB, Gose S, Reed GW, Botto M, Rice PA, Ram S. 2018. Human factor H domains 6 and 7 fused to IgG1 Fc are immunotherapeutic against Neisseria gonorrhoeae. J Immunol 201:2700–2709. doi: 10.4049/jimmunol.1701666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wetzler LM, Blake MS, Gotschlich EC. 1988. Characterization and specificity of antibodies to protein I of Neisseria gonorrhoeae produced by injection with various protein I-adjuvant preparations. J Exp Med 168:1883–1897. doi: 10.1084/jem.168.5.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wetzler LM, Gotschlich EC, Blake MS, Koomey JM. 1989. The construction and characterization of Neisseria gonorrhoeae lacking protein III in its outer membrane. J Exp Med 169:2199–2209. doi: 10.1084/jem.169.6.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yamasaki R, Kerwood DE, Schneider H, Quinn KP, Griffiss JM, Mandrell RE. 1994. The structure of lipooligosaccharide produced by Neisseria gonorrhoeae, strain 15253, isolated from a patient with disseminated infection: evidence for a new glycosylation pathway of gonococcal lipooligosaccharide. J Biol Chem 269:30345–30351. [PubMed] [Google Scholar]
- 82.West SE, Clark VL. 1989. Genetic loci and linkage associations in Neisseria gonorrhoeae and Neisseria meningitidis. Clin Microbiol Rev 2:S92–S103. doi: 10.1128/CMR.2.Suppl.S92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, Brunelli B, Pieri A, Santini L, Savino S, Serruto D, Litt D, Kroll S, Welsch JA, Granoff DM, Rappuoli R, Pizza M. 2003. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med 197:789–799. doi: 10.1084/jem.20021911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lewis LA, Gipson M, Hartman K, Ownbey T, Vaughn J, Dyer DW. 1999. Phase variation of HpuAB and HmbR, two distinct haemoglobin receptors of Neisseria meningitidis DNM2. Mol Microbiol 32:977–989. doi: 10.1046/j.1365-2958.1999.01409.x. [DOI] [PubMed] [Google Scholar]
- 85.Ram S, Cox AD, Wright JC, Vogel U, Getzlaff S, Boden R, Li J, Plested JS, Meri S, Gulati S, Stein DC, Richards JC, Moxon ER, Rice PA. 2003. Neisserial lipooligosaccharide is a target for complement component C4b. Inner core phosphoethanolamine residues define C4b linkage specificity. J Biol Chem 278:50853–50862. doi: 10.1074/jbc.M308364200. [DOI] [PubMed] [Google Scholar]
- 86.Moe GR, Tan S, Granoff DM. 1999. Differences in surface expression of NspA among Neisseria meningitidis group B strains. Infect Immun 67:5664–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McQuillen DP, Gulati S, Rice PA. 1994. Complement-mediated bacterial killing assays. Methods Enzymol 236:137–147. doi: 10.1016/0076-6879(94)36013-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.