

Abstract
The intermembrane space (IMS) is a very small mitochondrial sub‐compartment with critical relevance for many cellular processes. IMS proteins fulfil important functions in transport of proteins, lipids, metabolites and metal ions, in signalling, in metabolism and in defining the mitochondrial ultrastructure. Our understanding of the IMS proteome has become increasingly refined although we still lack information on the identity and function of many of its proteins. One characteristic of many IMS proteins are conserved cysteines. Different post‐translational modifications of these cysteine residues can have critical roles in protein function, localization and/or stability. The close localization to different ROS‐producing enzyme systems, a dedicated machinery for oxidative protein folding, and a unique equipment with antioxidative systems, render the careful balancing of the redox and modification states of the cysteine residues, a major challenge in the IMS. In this review, we discuss different functions of human IMS proteins, the involvement of cysteine residues in these functions, the consequences of cysteine modifications and the consequences of cysteine mutations or defects in the machinery for disulfide bond formation in terms of human health.
Linked Articles
This article is part of a themed section on Chemical Biology of Reactive Sulfur Species. To view the other articles in this section visit http://onlinelibrary.wiley.com/doi/10.1111/bph.v176.4/issuetoc
Abbreviations
- CJ
cristae junctions
- CM
cristae membrane
- IBM
inner boundary membrane
- IMM
inner mitochondrial membrane
- IMS
intermembrane space
- MICOS
mitochondrial contact site and cristae organizing system
- OMM
outer mitochondrial membrane
The mitochondrial intermembrane space
Mitochondria are essential organelles with diverse functions in energy conversion, lipid and iron metabolism, heat production, signalling and cell death pathways. Their complex structure, which can be traced back to their endosymbiotic origin, consists of four distinct sub‐compartments: the matrix, the mitochondrial intermembrane space (IMS) and the outer (OMM) and inner (IMM) mitochondrial membranes. The IMS is enclosed by the OMM and IMM (Figure 1). It is further subdivided into the peripheral IMS, which is adjacent to the OMM and inner boundary membrane (IBM), and the cristae space, which is formed by invaginations of the IMM [cristae membrane (CM)]. Cristae junctions (CJ) separate the cristae space from the peripheral IMS. While shape and abundance of cristae differ strongly between organisms, tissues and various cellular conditions, such as stress and metabolism, the cristae junctions appear to be relatively uniform in size (Mannella, 2006). The OMM and IBM meet each other at contact sites. Recently identified distinct protein complexes define and constitute both, these contact sites and cristae junctions (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011; Kozjak‐Pavlovic, 2017).
Figure 1.
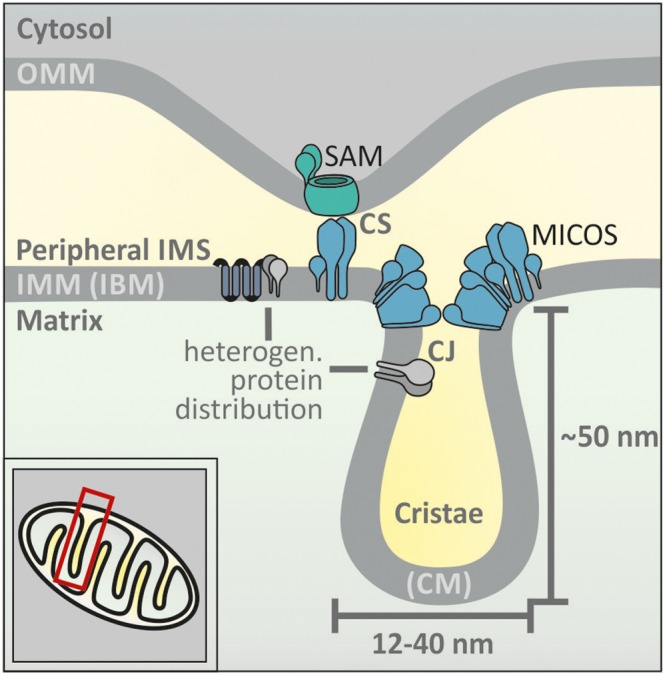
The layout of the mitochondrial intermembrane space. Mitochondria contain four sub‐compartments: the matrix, the OMM and IMM and the IMS. The IMS is subdivided into a peripheral IMS and cristae. The peripheral IMS is in contact with the OMM and the IBM. The cristae are enclosed by invaginations of the IMM and the CM. CJ segregate cristae and peripheral IMS. Contact sites (CS) formed by parts of the MICOS complex and the sorting and assembly machinery (SAM) complex mediate contacts between OMM and IBM. The OMM contains few proteins that have a high mobility in the membrane. Conversely, IMM proteins are more restricted in their movement. The structure of the IMM leads to heterogeneous protein distribution, for example, different protein sets between IBM and CM.
The subdivision of the IMS has functional consequences. For example, small molecules might be restricted in their diffusion by CJs resulting in chemical gradients within the IMS. This has been observed for protons that differ in their concentrations at different complexes of the respiratory chain (Rieger et al., 2014). Likewise, proteins appear to be actively sorted into either the IBM or the CM, as shown for membrane‐embedded subunits of the respiratory chain and the protein import machinery (Vogel et al., 2006; Appelhans and Busch, 2017; Stoldt et al., 2018). Such sorting has also been observed for soluble proteins such as cytochrome c (Scorrano et al., 2002), and it is likely that this is also found for other proteins.
The proteomes of the IMS in yeast and human cells and interactomes of central IMS proteins have been identified (Vogtle et al., 2012; Hung et al., 2014; Petrungaro et al., 2015; Morgenstern et al., 2017) (for a compiled list of human IMS proteins, see Supporting Information Table S1). While many of the proteins (approximately 150) so far identified, that either are soluble in the IMS or expose functional domains towards the IMS have been associated with specific functions, a significant share of proteins are so far uncharacterized and await further characterization in the future. Additionally, all approaches to determine the IMS proteome are hampered by the low amounts of many IMS proteins, by the very small size of many of these proteins and by the dual localization of many IMS proteins to the cytosol and the IMS that can change during different perturbations (e.g. oxidative stress). Thus, the IMS proteomes available as well as our knowledge on IMS processes should be considered incomplete, and it will be an exciting task in the future to expand our knowledge on IMS (protein) function.
Functions of IMS proteins
Most proteins of the IMS are of eukaryotic origin and developed during endosymbiosis to integrate mitochondrial activities into cellular functions. Enzymes of the IMS thus fulfil numerous functions in, for example, protein, lipid and metabolite transport, in protein folding, in signalling, in metabolism and in assembly and maintenance of the respiratory chain (Figure 2A).
Figure 2.
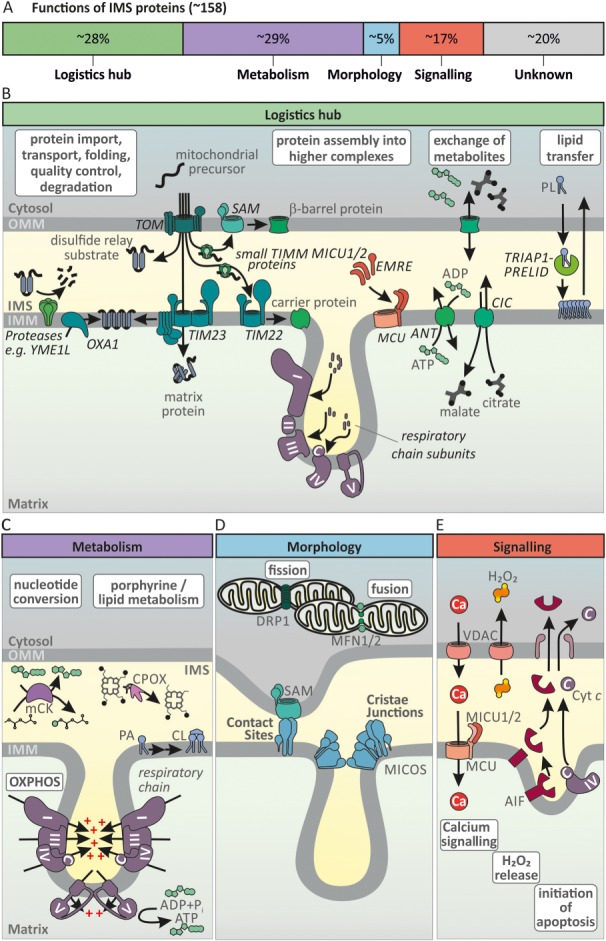
Functions of the mitochondrial intermembrane space. (A) A list of IMS proteins was compiled from the IMS proteomes of human cells, the interactome of CHCHD4 (Vogtle et al., 2012; Hung et al., 2014; Petrungaro et al., 2015; Morgenstern et al., 2017) and the bioinformatics analyses of twin CX9C‐like proteins (Cavallaro, 2010; Fischer et al., 2013) (Supporting Information Table S1). Some dually localized proteins were added manually to the list. Of the so far identified IMS proteins (about 150) approximately 20% are still of unknown function. According to published data or database entries, we assigned the remaining proteins to four different functional classes: Logistics hub, Metabolism, Morphology and Signalling. Proteins were assigned to the group ‘Logistics hub’ if they were annotated as being involved in transport of proteins/small molecules, protein degradation or assembly of protein complexes. Proteins of the ‘Metabolism’ group are integral subunits of respiratory chain complexes (e.g. ATP5I) or enzymes in metabolic pathways (e.g. CPOX). The ‘Morphology’ group contains proteins, which are involved in maintenance of mitochondrial structure (e.g. OPA1). Proteins were assigned to ‘Signalling’ if they were annotated as being involved in, for example, apoptotic, redox or calcium signalling. Assignment to multiple groups was undertaken where applicable (e.g. voltage‐dependent anion channels (VDAC1‐3) in ‘Logistics’, ‘Metabolism’ and ‘Signalling’). (B) Logistics hub: IMS proteins are involved in distributing mitochondrial proteins to their respective mitochondrial sub‐compartments. After entering through the TOM, proteins can become sorted into the OMM by the SAM complex. Chaperone complexes in the IMS support this process (small TIMM proteins). The translocase complex of the IMM, TIM23, either sorts proteins with mitochondrial targeting signals to the IMM or guides them to the matrix, while TIM22 handles members of the mitochondrial carrier family. OXA1 shuttles proteins into the IMM from the matrix side. Lastly, many soluble IMS proteins are folded by the mitochondrial disulfide relay. IMS proteins also contribute to folding, quality control and removal of proteins as well as their assembly into protein complexes such as the MCU or the respiratory chain. Additionally, the IMS plays an important role in transferring metabolites and lipids between cytosol and matrix and OMM and IMM respectively. PA, phosphatidic acid; CIC, mitochondrial citrate transporter; ANT, adenine nucleotide translocator; TIM, translocase of the inner membrane. (C) Metabolism: The cristae are functionally important for the formation of an electrochemical potential across the IMM. This electrochemical potential is utilized to generate ATP in the matrix (OXPHOS). The IMS also harbours enzymes that catalyse parts of metabolic pathways. For example, coproporphyrinogen oxidase (CPOX) catalyses the penultimate step in porphyrin biosynthesis. Nucleotide‐converting enzymes balance nucleotide pools and enable their efficient diffusion, membrane transport and enzyme function. They include adenylate kinase 2 and mitochondrial CK. In the IMM, membrane lipids are converted, for example, production of cardiolipin takes place. (D) Morphology: Proteins of the IMS and its adjacent membranes define mitochondrial morphology and dynamics. This not only includes formation of cristae junctions and contact sites by MICOS complex and SAM complex but also balancing membrane fission and fusion by DRP1 (also DNML1, dynamin‐1‐like protein) and MFN1/2 (mitofusin 1/2) respectively. (E) Signalling: The IMS is important for signalling processes. Regulators of mitochondrial calcium signalling such as MICU1/2 are resident in the IMS and modulate activity of the MCU. The IMS is an important production site for ROS and therefore is relevant in ROS (depicted as H2O2) release/signalling to the cytosol, for example, via VDACs in the OMM. Upon apoptotic stimuli and membrane remodelling IMS proteins, for example, cleaved AIF and cytochrome c can be released into the cytosol. For a detailed description, see text.
The IMS as a logistics hub
The IMS forms the interface between the cytosol and the mitochondrial matrix and thus takes an important function as a logistics hub to facilitate transport of proteins, lipids, metabolites and metal ions. It also serves to control protein levels and the spatial organization of protein complex assembly (Figure 2B).
Mitochondria are important hubs for the conversion of different lipid species (Scharwey et al., 2013; Tamura et al., 2014; Tatsuta and Langer, 2017). This includes the synthesis of cardiolipin, phosphatidylethanolamine and pregnenolone that takes place in the IMM. Thus, various precursors such as phospholipids, sphingolipids and cholesterol have to be imported into mitochondria and transferred via the IMS to the IMM. Lipid‐binding proteins and mitochondrial contact sites mediate this IMS transfer. Examples are the SLMO2–TRIAP1 or the PRELID1–TRIAP1 complexes, which shuttle phospholipids such as phosphatidic acid, phosphatidylserine and phosphatidylinositol across the IMS (Potting et al., 2010; Aaltonen et al., 2016). In steroidogenic cells, members of the StAR family control the supply of cholesterol to mitochondria and thereby steroidogenesis.
Like lipids, many other metabolites also have to find their way across the IMS (Figure 2B). They usually cross the OMM using VDAC channels (porins Por1 and Por2 in yeast) or other pores. The OMM often is considered a relatively non‐specific ‘sieve’ that is permeable for most molecules up to 5 kDa. However, recent findings indicate a higher degree of selectivity and regulation and potentially also the existence of additional OMM transporters (Kruger et al., 2017). In comparison, the IMM is impermeable and metabolites require dedicated transporters – many of which are coupled to the anti/symport of other metabolites or to the mitochondrial membrane potential. As there are not IMM transporters for all metabolites, some metabolites have to be converted to other metabolites for which transporters exist. For example, the malate–aspartate shuttle serves to export oxaloacetate from the matrix to the cytosol or to transfer NADH from the cytosol to the matrix. As there is no IMM transporter for oxaloacetate, it has to be converted to aspartate, which can be transported out of the matrix in an antiport with glutamate. In the cytosol/IMS [the aspartate aminotransferase GOT2 is found in the IMS proteome (Hung et al., 2014)], aspartate is converted back to oxaloacetate, which is reduced by NADH, yielding malate, which is transported into the matrix in an antiport with 2‐oxoglutarate.
The IMS also plays an important role in metal ion transport and homeostasis (Cobine et al., 2006). The copper chaperone for SOD1 (CCS1) and cytochrome c oxidase copper chaperone 17 (COX17) are both copper ion‐binding IMS proteins that shuttle copper ions into SOD1 or complex IV of the respiratory chain respectively. Through these two pathways, the IMS plays an important role in the control of cellular copper levels and distribution.
Most mitochondrial proteins that are destined for the different mitochondrial compartments have to pass the IMS (Figure 2B) (Hewitt et al., 2014; Schulz et al., 2015; Backes and Herrmann, 2017; Wiedemann and Pfanner, 2017). After passing the translocase of the OMM (TOM complex), matrix proteins utilize an ATP‐ and membrane potential‐dependent import route across the translocase of the IMM (TIM23 complex). These proteins are directed by N‐terminal presequences to these transport complexes. IMM proteins use a similar pathway if they are directed by so‐called bipartite presequences (an N‐terminal presequence followed by a hydrophobic segment) that result in their lateral release from the TIM23 complex. Alternatively, they use a pathway employing the TIM22 complex. To this end, IMM proteins rely on small heterooligomeric chaperones of the IMS (TIMM9/10, TIMM8/13) to transport them across the IMS to the TIM22 complex. Another pathway is mediated by the OXA (oxidase assembly factor) machinery, which mediates IMM insertion of a small subset of proteins including mitochondrial encoded proteins and few nuclear‐encoded proteins (Funes et al., 2011; Hennon et al., 2015). β‐Barrel proteins of the OMM also have to pass the IMS and likewise rely on these small IMS chaperones to transport them from the TOM complex to the OMM insertion machinery (also called the SAM complex). Lastly, IMS proteins can be imported in a TIM23‐dependent manner (bipartite presequences, e.g. Smac/Diablo or MICU1, in part with specific proteolysis to turn them into soluble IMS proteins). They can also follow pathways that do not require N‐terminal presequences but instead rely on folding and cofactor insertion or on conserved cysteine motifs (see The machinery for disulfide bond formation section).
IMS proteins are critical not only for the import of proteins but also for the assembly and maintenance of protein complexes such as the respiratory chain or the mitochondrial calcium uniplex (MCU) (Vahsen et al., 2004; Petrungaro et al., 2015; Friederich et al., 2017). Proteins such as COX17, COX11, SCO1 and SCO2 (synthesis of cytochrome c oxidase 1 and 2) contribute to copper ion insertion into complex IV. Small IMS proteins such as NDUFB7 and NDUFB10 (NADH:ubiquinone oxidoreductase subunits) are critical for the assembly, structural integrity and function of complex I. IMS proteases like ATP23, HTRA2, LACTB and YME1L1 monitor protein folding states, degrade proteins, prevent protein aggregation or specifically process IMS proteins for insertion into different complexes (Quiros et al., 2015). Taken together, controlled transport and handling of a wide array of molecules by IMS enzymes constitutes an important characteristic of this interface compartment.
IMS proteins in cellular metabolism
IMS proteins critically contribute to cellular metabolism (Figure 2C). For example, many IMS proteins contribute to the respiratory chain either by being part of respiratory chain complexes or by feeding electrons into the respiratory chain. The former group includes proteins such as the complex I subunits NDUFB7, NDUFB10, NDUFS5 and NDUFA8 that maintain complex I structure and activity (Zhu et al., 2016). Electrons are fed into the ubiquinone pool by the IMS localized glycerol‐3‐phosphate dehydrogenase (GPDH) that mediates oxidation of cytosolic NADH. Other proteins such as the augmenter of liver regeneration (ALR), a part of the mitochondrial disulfide relay (see The machinery for disulfide bond formation section) and sulfite oxidase (SUOX) feed electrons stemming from their respective activities into complex IV via cytochrome c (Wattiaux‐de Coninck and Wattiaux, 1971; Farrell and Thorpe, 2005). Cytochrome c mediates electron shuttling between complexes III and IV of the respiratory chain.
The IMS also contains enzymes that are important for energy homeostasis (Figure 2C) such as adenylate kinase 2 (AK2), mitochondrial creatine kinase (mCK) and the nucleoside–diphosphate kinase NME4/NDPK‐D. These enzymes balance and interchange nucleotide pools (e.g. AK2 converts two ADP to ATP and AMP) and thereby allow nucleotide exchange over the IMM, synthesis of nucleoside triphosphates other than ATP, efficient activity of nucleotide utilizing enzymes (e.g. NME4 increases GTP‐loading on the GTPase OPA1), AMP signalling and short‐term storage of chemical energy (Dzeja and Terzic, 2009).
The IMS also harbours individual enzymes, which are part of larger multistep metabolic pathways that take place in multiple compartments (Figure 2C). Coproporphyrinogen oxidase catalyses the penultimate step in porphyrin biosynthesis, a pathway, which also has reaction steps in the matrix and the cytosol (Hamza and Dailey, 2012). Another example is sulfite oxidase, which is an important enzyme in the oxidative degradation of the sulfur amino acids cysteine and methionine. Sulfite oxidase catalyses the oxidation of sulfite to sulfate and thereby shuttles electrons directly to cytochrome c (Kappler and Enemark, 2015; Schwarz, 2016). During de novo pyrimidine biosynthesis, dihydroorotate dehydrogenase (DHODH) performs the ubiquinone‐mediated oxidation of dihydroorotate to orotate (Chen and Jones, 1976). This is the only mitochondrial step in an otherwise cytosolic metabolic pathway.
IMS proteins in defining mitochondrial ultrastructure and biogenesis
Different IMS proteins are responsible for the maintenance of mitochondrial ultrastructure (Figure 2D). This includes the formation of cristae and CJs, the establishment of contact sites between the OMM and IMM and the dynamics of mitochondrial morphology. The recent identification of a large protein complex, the mitochondrial contact site and cristae organizing system (MICOS) (Harner et al., 2011; Hoppins et al., 2011; von der Malsburg et al., 2011), provided important insights into the determination of mitochondrial ultrastructure. Two important proteins of this complex are MIC10 and MIC60. MIC10 forms a complex that is important for the formation of CJs. It thereby oligomerizes and bends the IMM for CJ formation (Barbot et al., 2015; Bohnert et al., 2015). MIC60 contributes to a complex that is crucial for connecting IMM and OMM at contact sites. Proteins of the MICOS complex can become post‐translationally modified (Tsai et al., 2018), which might be important to control the structural plasticity of CJs.
Mitochondrial ultrastructure and biogenesis is also influenced by fusion and fission, two processes that are in part controlled by IMS proteins. For example, proteolytic processing of the GTPase OPA1 (in yeast: Mgm1) by the IMM proteases YME1L and OMA1 leads to a finely tuned balance between different forms of OPA1, which are required for fusion and fission (Anand et al., 2014).
The IMS as a signalling compartment
Its interface position also renders the IMS an important signalling compartment (Figure 2E). The IMS is closely integrated into different cell death networks. It harbours proapoptotic factors such as apoptosis inducing factor (AIF), cytochrome c and Smac/Diablo. Upon external and internal stimuli, OMM permeabilization takes place, and these proteins are released from the IMS to initiate cell death. This goes along with major structural rearrangements of the IMM and IMS (cristae membrane remodelling) (Scorrano et al., 2002; Cipolat et al., 2006; Frezza et al., 2006; Burke, 2017).
The IMS also serves important roles in other signalling pathways (Figure 2E). For example, release of hydrogen peroxide (H2O2) from mitochondria has been implied in hypoxia signalling. It is conceivable that not only H2O2‐consuming or ‐generating enzymes of the IMS [such as peroxiredoxins 3 and 4 (PRDX3/4)], GSH peroxidase 4 (GPx4) and SOD1) but also the transfer of H2O2 through pores in the OMM contribute to signal modulation and thus signalling (Riemer et al., 2015; Diebold and Chandel, 2016; Smith et al., 2017). Likewise, Ca2+ signalling is integrated in the IMS. The Ca2+ uniporter (MCU) in the IMM is the central entry gate for Ca2+ (coming from either the endoplasmic reticulum or outside the cell) (Kamer et al., 2014; Penna et al., 2018). The central regulatory subunits of MCU are MICU1, MICU2 and MICU3. These IMS proteins with EF hands (similar to other IMS proteins such as aspartate/glutamate exchangers or ATP‐Mg2+/Pi exchangers) modulate MCU activity, depending on the local Ca2+ levels (Del Arco et al., 2016).
Cysteine residues in IMS proteins
Of the approximately 150 IMS proteins that have so far been identified by different proteomic approaches, most contain conserved cysteines (Figure 3A and Supporting Information Table S1) (Vogtle et al., 2012; Hung et al., 2014; Petrungaro et al., 2015; Morgenstern et al., 2017). For many of these proteins, cysteine residues are important for protein folding and structure, activity and its regulation, as well as IMS localization (Figure 3B). Activity can thereby encompass roles in redox‐activity, metal ion binding, [Fe/S] cluster binding and metabolite coordination. For example, reduced cysteines play important roles in the copper chaperones SCO1, SCO2 and CCS1. In these proteins, they complex copper ions and allow their shuttling to complex IV and SOD1 respectively (Cobine et al., 2006; Leary, 2010; Nevitt et al., 2012). In IMS creatine kinase (mCK), a reduced cysteine in its thiolate state is important in complexing creatine (Bais and Edwards, 1982; Naor and Jensen, 2004). Cysteines undergoing cycles between the reduced and oxidized state are found for example in the oxidoreductase CHCHD4 (coiled‐coil‐helix‐coiled‐coil‐helix domain containing protein 4), in the thioredoxin TRX1 and the glutaredoxin GRX1 as well as in the peroxiredoxins PRDX3 and PRDX4 (Hung et al., 2014). Cysteines in structural disulfide bonds are important for the control of mitochondrial morphology (CHCHD3/MIC19 and CHCHD6/MIC25 as part of the MICOS complex), of Ca2+ signalling (MICU1, MICU2) and to facilitate mitochondrial protein import by the formation of chaperone interaction interfaces (TIMM proteins, CHCHD4) (Petrungaro et al., 2015; Modjtahedi et al., 2016). Additionally, the formation of disulfide bonds is for many proteins closely connected to mitochondrial import and retention in the IMS (see The machinery for disulfide bond formation section). Because the relevance (if any) of the conserved cysteine residues in many of the known IMS proteins remains unclear, it will be highly interesting to widen our understanding of their function in the future.
Figure 3.
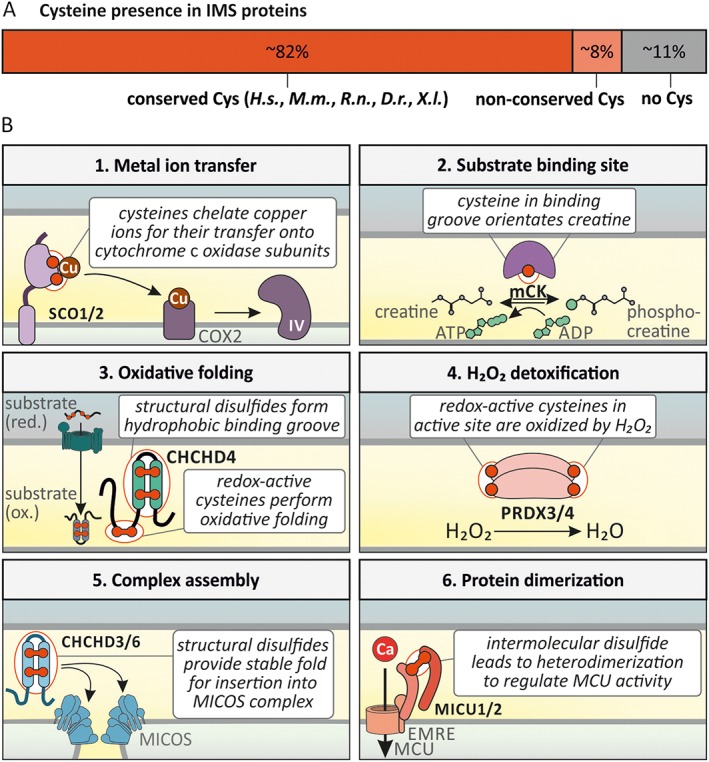
Conserved cysteines in mammalian IMS enzymes. (A) The list of IMS proteins (Supporting Information Table S1) was analysed for cysteine presence and their conservation. Many IMS proteins contain at least one conserved cysteine while only a few IMS proteins contain non‐conserved or no cysteines. Conservation was assessed in Homo sapiens (H.s.), Mus musculus (M.m.), Rattus norvegicus (R.n.), Danio rerio (D.r.) and Xenopus laevis (X.l.). (B) Examples for the function of conserved cysteine residues. (1) Reduced cysteines can act in metal ion transfer. Specifically, SCO1/2 can chelate copper and transfer it onto COX2, which assembles into complex IV of the respiratory chain. (2) Reduced cysteines can act in substrate binding sites. In CK, a reduced cysteine in its binding groove is involved in orientating creatine for phosphorylation. (3) Redox‐active cysteines are involved in oxidative folding while structural disulfides form a binding groove. In CHCHD4, the redox‐active cysteines C53 and C55 oxidize substrates during oxidative folding. The structural disulfides stabilize the hydrophobic binding groove necessary for substrate recognition. (4) Redox‐active cysteines are involved in the detoxification of H2O2. PRDX3/4 contain redox‐active cysteines, which are oxidized by H2O2, while H2O2 is reduced to H2O. (5) Structural disulfides are necessary for complex assembly. CHCHD3/6 are oxidatively folded via the disulfide relay. The stable fold conferred by two disulfides per protein allow for stable insertion into the MICOS complex. (6) Structural disulfides lead to protein dimerization. MICU1/2 form a disulfide‐linked heterodimer, which allows for calcium‐dependent regulation of the MCU. For a detailed description, see text.
Cysteine residue modifications in IMS proteins
Cysteine residues in IMS proteins can undergo diverse and often reversible post‐translational modifications. These modifications include disulfide bond formation and oxidation to, for example, sulfenic and sulfinic acid but also nitrosation and persulfidation (Figure 4A). Disulfide bonds are thereby formed intramolecularly and intermolecularly with other proteins or small molecules such as GSH (S‐glutathionylation). Oxidation of selected cysteine residues in proteins (usually peroxidases, for other proteins very unlikely) to sulfenic, sulfinic and sulfonic acid can be facilitated directly by H2O2 and depends on the reactivity of the targeted cysteine residues as well as the local H2O2 concentration.
Figure 4.
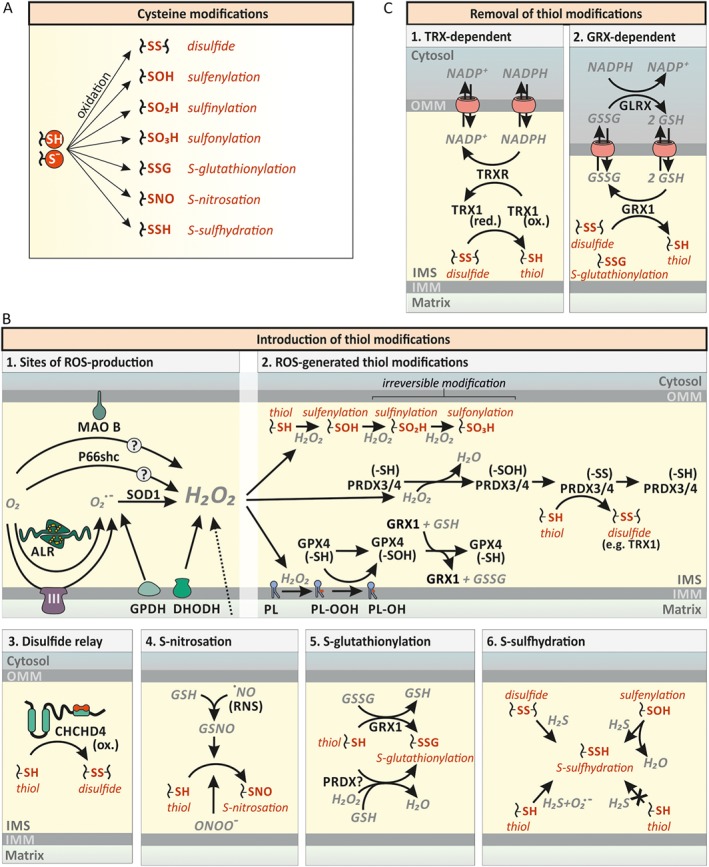
Cysteine modifications in the IMS. (A) Cysteine modifications. The reactive thiol group of cysteine residues can exist in the protonated state or as the deprotonated thiolate anion. This thiol group can be further modified as indicated. (B) Various thiol modifications are introduced into IMS proteins. (1) ROS‐generation in the IMS. Transfer of a single electron to O2 leads to formation of superoxide anion (O2°−). Superoxide generators of the IMS include complex III of the respiratory chain (CIII), the ALR, and GPDH. Superoxide is enzymically converted to H2O2 by SOD1. Dihydroorotate dehydrogenase (DHODH) can generate O2°− and H2O2. Likewise, MAO B and p66Shc have been reported to directly generate H2O2. (2) Cysteine modification by H2O2. H2O2 can result in the direct oxidation of target thiols (–SH) to sulfenic (–SOH), sulfinic (–SO2H) and sulfonic (–SO3H) acid depending on thiol reactivity and H2O2 availability. Most protein thiols exhibit low reactivity towards H2O2 and will thus be primarily metabolised by dedicated H2O2‐scavenging enzymes such as peroxiredoxins (PRDX). PRDX3/4 are located in the IMS and their oxidation by H2O2 can be reversed by thioredoxins (TRX), but might also be passed on to other proteins. H2O2 also mediates phospholipid (PL) peroxidation (PL–OOH). PL peroxidation can be removed by GSH peroxidase‐4 (GPX4). (3) The mitochondrial disulfide relay. During oxidative protein folding, reduced cysteine residues (–SH) are oxidized to disulfide bonds (–SS) by oxidized (ox.) CHCHD4. See Figure 5 for additional details. (4) S‐nitrosations of cysteine residues. Reduced GSH reacts with ·NO to S‐nitrosoglutathione (GSNO), inducing S‐nitrosations (–SNO). NO is also able to directly induce S‐nitrosations. (5) S‐glutathionylation of cysteine residues. Reduced glutaredoxin‐1 [GRX1 (–SH)] reacts with GSSG and becomes glutathionylated [GRX1 (–SSG)]. GRX1 subsequently mediates the S‐glutathionylation (–SSG). (6) S‐sulfhydration of cysteine residues. Disulfides and sulfenic acids (–SOH) can react with H2S, resulting in S‐sulfhydration (–SSH) of cysteines. Thiols on the other hand can only be sulfhydrated by H2S in the presence of superoxide anions. (C) Removal of thiol modifications. (1) Reduction of disulfide bonds by the thioredoxin system (TRX).Thioredoxin 1 (TRX1) is likely to mediate the reduction of disulfides and become oxidized itself. Thioredoxin reductase (TRXR) reduces TRX1 in an NADPH‐dependent manner. (2) Reduction of disulfide bonds and S‐glutathionylations by GRX1. GRX1 reduces disulfides and S‐glutathionylated thiols (–SSG) while oxidizing reduced GSH. GSH disulfide (GSSG) is reduced by GSH reductase (GLRX) in an NADPH‐dependent manner.
Introduction of disulfide bonds or S‐glutathionylation usually takes place in an enzyme‐mediated fashion (Figure 4B and for disulfide bond formation, see The machinery for disulfide bond formation section). S‐glutathionylation thereby could take place in a glutaredoxin‐ or GSH‐S‐transferase (GST)‐dependent fashion or by addition of GSH onto cysteine sulfenic acid residues. A glutaredoxin‐dependent process would require a local (at least temporary) accumulation of oxidized GSH (GSSG) as glutaredoxins predominantly catalyse deglutathionylation reactions at the reducing GSH redox potential (EGSH) that is present in the IMS (Gallogly and Mieyal, 2007; Kojer et al., 2012; Fischer et al., 2013; Deponte, 2017). Although such extreme GSSG accumulation might seem unlikely, GSH reductase has never been identified in the IMS in any screen and thus removal of GSSG through channels in the OMM might be too slow to efficiently remove GSSG under specific conditions allowing for protein S‐glutathionylation. Alternatively, S‐glutathionylation might proceed in a GST‐catalysed manner. However, so far, no GST has been identified in the IMS. Lastly, a H2O2‐mediated oxidation of reactive thiols (possibly catalysed by peroxiredoxins) and subsequent addition of GSH to the newly generated sulfenic acid might drive S‐glutathionylation.
Nitrosation and persulfidation depend on the availability of NO and H2S respectively (Bak and Weerapana, 2015; Wolhuter and Eaton, 2017; Zhang et al., 2017; Paul and Snyder, 2018). These cysteine modifications are normally tightly regulated and balanced, and their dysregulation can result in mitochondrial dysfunction and disease.
Sources of oxidizing power in the IMS
The IMS is adjacent to the respiratory chain, one of the main sources of ROS in the cell (Murphy, 2009; Wong et al., 2017) (Figure 4B). The proximal ROS produced at different sites of the respiratory chain are superoxide anions (O2°−). They can be released to both sides of the IMM depending on the generation site. Towards the IMS, O2°− are released from the Qo site of complex III, from the DQ site of DHODH and from the GQ site of GPDH. At these sites, electrons are transferred onto or from ubiquinone/ubiquinol (Wong et al., 2017). In the IMS, two molecules of O2°− are dismutated rapidly to H2O2 and O2 by SOD1 of which a fraction is localized to the IMS (Sturtz et al., 2001). H2O2 generated in the matrix can also traverse the IMM and contribute to the H2O2 load of the IMS. Further, ROS‐producing IMS enzymes include P66shc in combination with cytochrome c (Giorgio et al., 2005) and the flavoproteins ALR (see The machinery for disulfide bond formation section) (Bihlmaier et al., 2007; Daithankar et al., 2012), and MAO‐B, which in rat has been shown to face the IMS (Wang and Edmondson, 2011). The extent of ROS production by these enzyme systems remains however unclear.
H2O2 in the IMS can diffuse further to the cytosol, very likely in a process that is facilitated by pores in the OMM such as the VDACs. In addition, it can react with dedicated handling enzymes of the peroxiredoxin and GSH peroxidase family. Of these, mammalian PRDX3 and PRDX4 as well as GPX4 have been identified in the IMS (Hung et al., 2014). Data from yeast Gpx3 could indicate that even more redox enzymes might find their way into the IMS under specific conditions. Yeast Gpx3 is translated from an upstream start codon under conditions of oxidative stress, which results in a protein with mitochondrial targeting information that finds its way into the IMS (Gerashchenko et al., 2012; Kritsiligkou et al., 2017). After reaction with H2O2, both PRDX3 and PRDX4 contain a disulfide bond, which can be reduced by thioredoxins. At least in yeast, thioredoxin 1 (Trx1) and thioredoxin reductase 1 (Trr1) have also been found in the IMS (Vogtle et al., 2012). Additionally, PRDX3 and PRDX4 might transfer disulfide bonds directly onto specific target proteins for redox signalling. Although this has not been shown for IMS proteins, the concept has been demonstrated in yeast and mammalian cells for example for the Gpx3‐Yap1 and the PRX2‐STAT3 axis respectively (Delaunay et al., 2002; Sobotta et al., 2015). Importantly, the precise concentrations and activities of antioxidative enzymes but also the amounts of ROS (H2O2, O2°−) produced and present at any given time in the IMS of intact cells are not known.
Oxidation of cysteine residues in IMS proteins by mitochondrial ROS
Peroxiredoxins might contribute to cysteine residue oxidation in the IMS. However, a few cysteine residues could also become directly oxidized by H2O2 due to the potentially high local concentrations of H2O2 in the proximity of the respiratory chain (and potentially low activities of antioxidative systems in the IMS) (Figure 4B). Moreover, S‐glutathionylation in GS•‐, GSNO‐ or GSSG‐dependent and glutaredoxin‐catalysed reactions could contribute to oxidative cysteine modifications (Starke et al., 2003; Deponte, 2017). In most proteomic studies that focused on the identification of reversibly modified cysteine residues, approaches were employed that did not distinguish between these different modifications. Still some IMS proteins were identified to be differentially modified on their cysteine residues upon specific stimuli. For example, induction of ROS production by incubation of mitochondria, isolated from rat hearts, with the complex III inhibitor antimycin A, which results in O2°− release to the IMS, led to oxidation of cysteines in IMS proteins such as TIMM50, VDAC3, OPA1 and AK2 (Bleier et al., 2015). Oxidative modifications on VDAC3 but also VDAC2 were also reported by other studies (De Pinto et al., 2016; Reina et al., 2016a, 2016b). A different study characterizing IMS proteins as H2O2 targets in yeast mitochondria identified IMS proteins such as Pet191 and cytochrome b 2 (Topf et al., 2018). Neither study however addressed how these changes influence the activity of IMS enzymes and whether these modifications are reversible.
Reduction of oxidized cysteine residues in IMS proteins
Different reducing systems are present in the IMS (Figure 4C). In human cells, the dithiol‐glutaredoxin GRX1 (also GLRX) is localized to the IMS (Pai et al., 2007; Gallogly et al., 2008). It exhibits rate enhancements for deglutathionylation on the order of 104 for protein‐SSG substrates and thus likely represents the majority of deglutathionylase activity in the IMS (Chrestensen et al., 2000). Since k cat and K M values for glutathionylated proteins increase or decrease in parallel with the concentration of GSH, maximal rates of deglutathionylation depend on the GSH redox potential EGSH of the IMS. In yeast and mammalian tissue culture cells, IMS‐EGSH has been shown to be in the range of its cytosolic counterpart suggesting that GRX1 efficiently catalyses deglutathionylation (Kojer et al., 2012; Fischer et al., 2013). However, in contrast to the cytosol, short‐term fluctuations of EGSH might occur easily in the IMS caused, for example, by release of ROS. Given the fact that GSH reductase is absent from the IMS, fluctuations in EGSH might be more stable compared to the cytosol and could thus result in the short‐term accumulation of glutathionylated proteins in the IMS.
Notably, in yeast, it has been directly shown that glutaredoxins (in this case mainly Grx2) exhibit a lower activity in the IMS compared to the cytosol (Kojer et al., 2015). Similar data for mammalian cells are not yet available. Instead, the concentration of human GRX1 in the IMS has been estimated to be approximately 0.1 μM and, together with its enzyme kinetic parameters, it has been estimated that total deglutathionylation and GS• scavenging activities should be similar in the matrix and IMS (Gallogly et al., 2008). However, direct evidence for this activity in intact cells is lacking.
Besides glutaredoxins, thioredoxin and thioredoxin reductase have been reported to reside in the IMS of yeast and mammalian cells (Vogtle et al., 2012; Hung et al., 2014). Their role in counteracting cysteine residue modifications in IMS proteins has however not been addressed extensively.
Different reducing activities in the IMS, matrix and cytosol might have interesting implications for the kinetics of the reversal of oxidative modifications and thus the temporal control of signalling events. In this context, it is interesting to note that GRX1 levels in the IMS of ageing rats have been shown to be decreased by 50–60% (Gao et al., 2013). Thus, reduction–oxidation processes and repair of oxidative damage in the IMS might fluctuate over time.
Thiol nitrosation and persulfidation
Further cysteine modifications that have been reported to occur in the IMS are thiol nitrosation and persulfidation. These modifications are facilitated by the signalling molecules NO (NO through peroxynitrite (ONOO−) formed from NO and O2°− (Radi, 2013)) and H2S respectively. Persulfidation has been reported for example for VDAC1, the carnitine/acylcarnitine carrier (SLC25A20) and PRDX4 (Giangregorio et al., 2016; Longen et al., 2016). Nitrosation was for example detected on the IMS proteins VDAC1‐3, PRDX4 and mCK (Lam et al., 2010; Su et al., 2013; Chen et al., 2014). Also, for these modifications, the precise consequences for enzyme function remain unknown. However, it is interesting that some proteins might be affected by different modifications even at the same residue. It will thus be a major and exciting challenge in the future to unravel the consequences and the crosstalk of redox modifications of IMS proteins for mitochondrial function and cellular physiology.
Formation of structural disulfide bonds in the IMS
The machinery for disulfide bond formation
In addition to cysteine modifications by ROS and signalling molecules such as NO and H2S, the IMS also harbours the machinery for the introduction of structural disulfide bonds (Figure 5A) (Herrmann and Riemer, 2012; Stojanovski et al., 2012; Fischer and Riemer, 2013; Chatzi et al., 2016). Oxidative protein folding in the IMS is thereby conceptually very similar to the same processes in other oxidizing compartments such as the endoplasmic reticulum and bacterial periplasm (Depuydt et al., 2011; Herrmann and Riemer, 2014).
Figure 5.
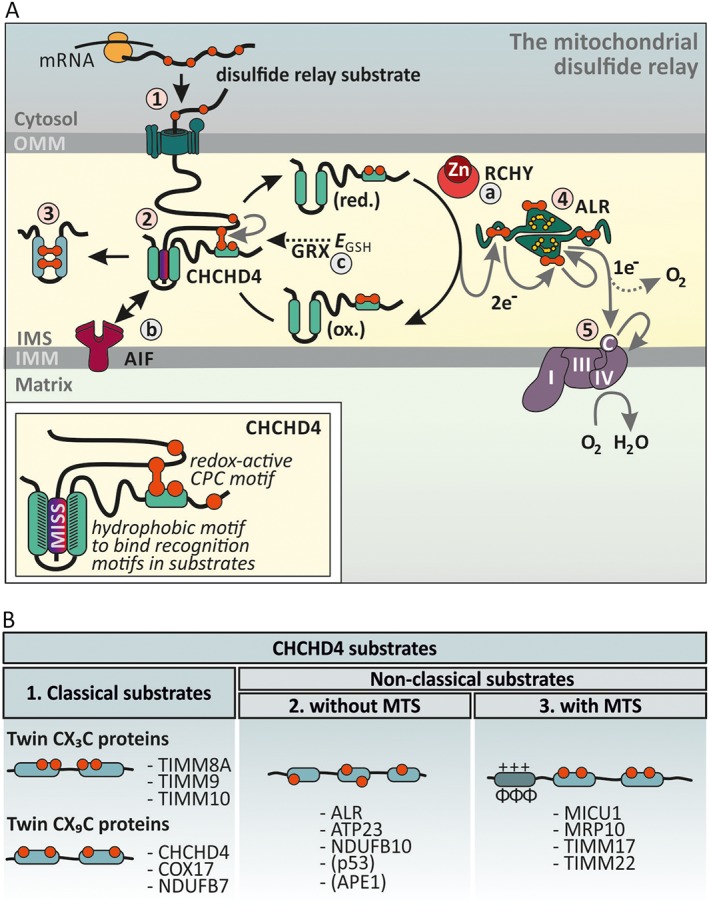
Enzyme‐catalysed disulfide formation in the IMS. (A) The mitochondrial disulfide relay. Disulfide relay substrates are synthesized on cytosolic ribosomes and are targeted to the TOM pore (step 1). During translocation, the substrates' mitochondrial IMS‐sorting signal (MISS, also ITS) binds the hydrophobic cleft of CHCHD4 and a mixed disulfide between the substrate and active site cysteines of CHCHD4 is formed (step 2). After releasing the oxidized and folded substrate (step 3), CHCHD4 is present in a reduced state. Electrons from reduced CHCHD4 are transferred to ALR (step 4). ALR shuffles electrons onto cytochrome c, which in turn transfers them to complex IV (step 5). The disulfide relay is supported by different auxiliary factors. RCHY/Hot13 complexes Zn2+ and thereby accelerates oxidation (step a). AIF facilitates CHCHD4 biogenesis and thus ensures the presence of this critical protein in the IMS (step b). The GSH redox buffer (EGSH) and glutaredoxins proofread wrongly formed or trapped disulfides. Glutaredoxins are present only in limiting amounts to allow disulfide formation (step c). (B) CHCHD4 substrates fall into three different structural classes. Proteins that lack classical mitochondrial targeting sequences and instead contain conserved cysteine residues form classes 1 and 2. The cysteines can either be ordered in twin CX3C of twin CX9C motifs (class 1) or not (class 2). Proteins from both classes acquire disulfide bonds during import, and it appears that this also is the prerequisite for mitochondrial accumulation and retention in the IMS. CHCHD4 also oxidizes substrates that rely on N‐terminal mitochondrial targeting sequences (MTS) for import (class 3).
In the human IMS, the so‐called disulfide relay machinery consists of two core components, the oxidoreductase CHCHD4 (in yeast Mia40) and the sulfhydryl oxidase ALR (in yeast Erv1). Most information on this system stems from work on the yeast system or from purified proteins. However, different primary sequences of Mia40 and CHCHD4 as well as Erv1 and ALR already indicate that there might be differences between the yeast and human disulfide relays. Indeed, recent experimental work supports that the mammalian system has an expanded substrate spectrum (e.g. MICU1, p53 and APE1), requires additional auxiliary factors (e.g. AIF) and has different physiological effects, for instance on the non‐conserved processes of hypoxia and Ca2+ signalling) (Yang et al., 2012; Hangen et al., 2015; Meyer et al., 2015; Petrungaro et al., 2015). The basic biochemical mechanisms of oxidative protein folding by the disulfide relay appear however to be conserved between yeast and human.
The oxidoreductase CHCHD4 serves a dual role as oxidizing enzyme and as an import receptor. In a first step, CHCHD4 recognizes its substrates, which are all synthesized in the cytosol (Figure 5A, step 1) right after they pass through the TOM pore (Figure 5A, step 2). In yeast, substrates do not rely on typical TOM receptor subunits such as Tom22 for recognition by the TOM complex (Gornicka et al., 2014). To interact with substrates, CHCHD4 is equipped with two functional parts, a redox‐active cysteine‐proline‐cysteine (CPC) motif in a flexible N‐terminal region and a hydrophobic patch in a central inflexible core domain (Banci et al., 2009; Banci et al., 2010; Weckbecker et al., 2012; Koch and Schmid, 2014a; Koch and Schmid, 2014b; Peleh et al., 2016). During its covalent (formation of a mixed disulfide bond between CHCHD4 and substrate) and non‐covalent (interaction of hydrophobic patches in substrate and CHCHD4) interactions with its substrates, CHCHD4 guides substrates into the IMS, oxidizes and folds them and possibly even helps to assemble them into larger complexes if required.
The release of an oxidized substrate leaves the CPC motif of CHCHD4 in a reduced state (Figure 5A, step 3). For another reaction with a new substrate, it needs to be reoxidized again. This reaction is performed by the sulfhydryl oxidase ALR (Figure 5A, step 4) (Banci et al., 2011a; Kojer et al., 2012; Fischer et al., 2013; Bien et al., 2010). To this end, each subunit of this homodimeric enzyme contains two redox‐active cysteine pairs. One cysteine pair is situated in a flexible arm and interacts with the CPC motif of CHCHD4. It then shuttles electrons to the other cysteine pair, which localizes to the core domain of ALR. This electron shuttle involves an intersubunit electron transfer. The core cysteine pair is in close proximity to the flavin cofactor of ALR. Finally, electrons are passed on from the flavin cofactor to terminal electron acceptors (Daithankar et al., 2010; Guo et al., 2012) (Figure 5A, step 5). Under most conditions, this is cytochrome c, which then passes the electrons on to complex IV of the respiratory chain leading to the formation of water (Farrell and Thorpe, 2005; Bihlmaier et al., 2007; Kojer et al., 2012). Under conditions of accumulation of reduced cytochrome c (or its absence), ALR can also utilize molecular oxygen as electron acceptor, however at the expense of producing H2O2 and with lower efficiency (Farrell and Thorpe, 2005; Bihlmaier et al., 2007). Alternatively, under hypoxic/anoxic conditions, the machinery can also utilize metabolic dehydrogenases at least in yeast (e.g. fumarate reductase Osm1) as electron acceptor (Neal et al., 2017).
Furthermore, CHCHD4 activity is supported by different ‘auxiliary’ factors (Figure 5A, steps a–c). The yeast homologue of RCHY, Hot13, abstracts zinc ions from the reduced cysteine residues of CHCHD4 substrates (and possibly also from the CPC motif of the oxidoreductase) and thus improves oxidation kinetics (Mesecke et al., 2008). A link to cellular NADH levels is provided by the interaction of CHCHD4 with the protein apoptosis inducing factor (AIF) (Figure 5A, step b) (Hangen et al., 2015; Meyer et al., 2015). AIF mediates the mitochondrial import of CHCHD4 by recognizing the N‐terminal 20–40 amino acids in CHCHD4. To this end, AIF has to dimerize which it does only upon binding of NADH. As CHCHD4 levels are limiting for substrate oxidation and import, this implies that oxidative folding kinetics and hence IMS protein import is controlled by the dynamics of the NADH/NAD+ ratio in the IMS. This specific role of AIF in the disulfide relay is not conserved to yeast, most likely because yeast Mia40 contains a bipartite mitochondrial targeting sequence that drives it into the mitochondrial IMS. Notably, AIF appears not to be essential for mitochondrial import of CHCHD4 as overexpression of CHCHD4 can result in the accumulation of CHCHD4 in mitochondria even upon concomitant AIF depletion (Meyer et al., 2015). Moreover, work on yeast cells with human CHCHD4 and ALR demonstrated that both proteins can enter the yeast IMS even in the absence of AIF (Chacinska et al., 2008; Sztolsztener et al., 2013).
CHCHD4 activity and oxidative protein folding are also linked to the GSH pool of the IMS (Figure 5A, step c). Like the cytosol, the IMS contains a highly reducing local GSH pool (Kojer et al., 2012; Fischer et al., 2013). In contrast to the cytosol, disulfide formation takes place for a wide variety of proteins in the IMS. In yeast, this is due to carefully balanced limiting amounts of glutaredoxin 2 (Grx2) in the IMS that directly influences the CHCHD4 redox state, the kinetics of oxidative folding and isomerization of substrates (Kojer et al., 2015) to an extent which still allows efficient Mia40 activity. The interplay with GSH also raised the question whether Mia40/CHCHD4 can serve as disulfide isomerase as many other oxidoreductases. In vitro results support such a function albeit at very low activity (Bien et al., 2010; Koch and Schmid, 2014a; Hudson and Thorpe, 2015).
Besides their roles in oxidative protein folding, both core components of the disulfide relay, ALR and CHCHD4, have also been implicated in other extra‐mitochondrial roles. This includes somewhat conflicting roles in iron sulfur cluster homeostasis and mitochondrial iron sulfur cluster export (Lange et al., 2001; Banci et al., 2011b; Spiller et al., 2013; Ozer et al., 2015; Haindrich et al., 2017). ALR has also been linked to the control of mitochondrial fission (Todd et al., 2010), and CHCHD4 levels influence the hypoxia response (Yang et al., 2012).
Substrates of the mitochondrial disulfide relay
Approximately 150 soluble proteins or proteins with soluble domains exposed to the IMS have so far been identified. More than a third of these proteins are targets of the mitochondrial disulfide relay and are thus likely to contain disulfide bonds (Petrungaro et al., 2015). The disulfide bonds and the involved cysteine residues of these substrates generally serve two purposes – they allow covalent interactions with the import receptor CHCHD4 and they provide the high stability of the mature protein fold. These substrate proteins fall into three groups (Figure 5B): (i) classical substrates that contain four conserved cysteine residues arranged in twin CX3C (small TIMM proteins) or twin CX9C motifs (or very similar motifs, e.g. proteins of the CHCHD family). Cysteines in these proteins are situated in antiparallel helices, which are brought together by disulfide formation. Notably, after oxidation, one of the two formed disulfide bonds is non‐consecutive. The proteins of this family lack classical targeting signals for the IMS and instead rely on oxidative folding and interaction with CHCHD4 for IMS import. Thus, if these proteins fail to become oxidized (e.g. because the machinery for oxidative folding is defective), they will accumulate in the cytosol (Fischer et al., 2013). (ii) Substrates with different conserved cysteine patterns. These proteins such as Atp23 in yeast (Weckbecker et al., 2012) or NDUFB10 in human cells (Friederich et al., 2017) contain, in part, very complex cysteine patterns and no mitochondrial targeting sequence. These proteins demonstrate the great versatility of CHCHD4 in handling proteins with diverse structures. Similar to twin CX9C and twin CX3C substrates, these proteins do not become imported into the IMS if disulfide formation fails. (iii) Substrates with conserved cysteines and mitochondrial targeting sequences. Representatives of this family are MICU1 (Patron et al., 2014; Petrungaro et al., 2015), Mrp10 (Longen et al., 2014), Tim22 (Wrobel et al., 2016) and Tim17 (Ramesh et al., 2016) (for the latter three proteins demonstrated in yeast). MICU1 is equipped with a mitochondrial targeting sequence and thus its mitochondrial import is independent of CHCHD4. It contains one conserved cysteine close to its C‐terminus that is oxidized by CHCHD4 and allows dimerization with its orthologue MICU2 (Petrungaro et al., 2015). Interestingly, this disulfide is required for the regulation of mitochondrial Ca2+ uptake. Absence of the disulphide results in increased mitochondrial Ca2+ uptake. In the presence of the disulfide bond, the MICU1‐MICU2 heterodimer binds to the Ca2+‐transporter MCU, depending on the IMS Ca2+ levels (Petrungaro et al., 2015). Mrp10 is a matrix protein and part of the mitochondrial ribosome. In yeast, it becomes oxidized by Mia40 during transit to the matrix that is driven by an unconventional proline‐rich matrix‐targeting sequence (Longen et al., 2014).
The disulfide relay appears also to be responsible for the mitochondrial accumulation of proteins that upon stress relocate from the cytosol to the mitochondria or are in general dually localized. These proteins include p53 (Zhuang et al., 2013) and APE1 (Vascotto et al., 2011; Barchiesi et al., 2015).
Strikingly, most if not all conserved cysteines, which interact with CHCHD4, are located in α‐helices in close proximity to hydrophobic amino acids. These hydrophobic areas have been shown to bind the hydrophobic patch of the core region of CHCHD4 and thereby serve as IMS‐targeting signals called MISS or ITS (Milenkovic et al., 2009; Sideris et al., 2009; Koch and Schmid, 2014b).
Cysteines in IMS proteins in disease
IMS proteins fulfil essential functions. Due to the wide substrate spectrum of the mitochondrial disulfide relay machinery and their important functions, missense mutations in the machinery for oxidative protein folding or in its substrates can result in severe disease phenotypes (Figure 6A). For example, mutations in COX6B1 sub‐unit result in mitochondrial complex IV deficiency and severe infantile encephalomyopathy (Massa et al., 2008). Another example are mutations in CHCHD10 that can lead to amyotrophic lateral sclerosis (ALS)/frontotemporal dementia or Charcot–Marie–Tooth disease (Auranen et al., 2015; Straub et al., 2018). Some of the mutations associated with the mitochondrial disulfide relay can be traced back directly to defects in redox processes and disulfide formation and will be discussed below.
Figure 6.
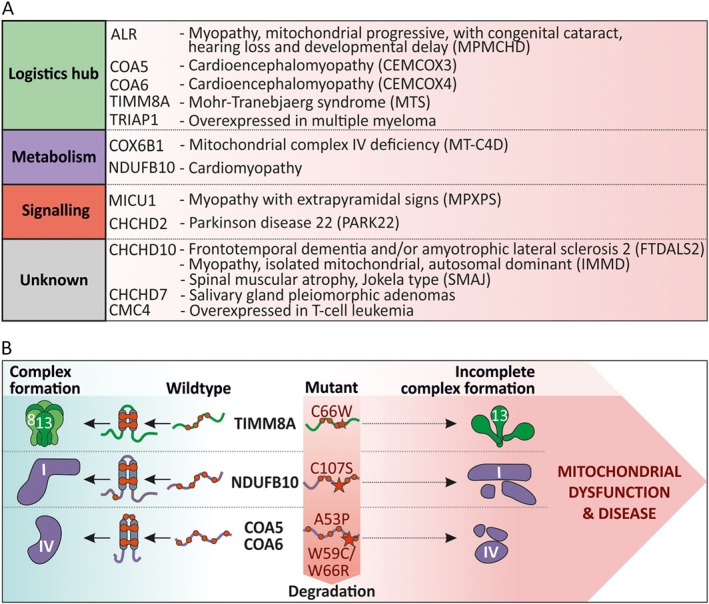
Cysteine‐linked defects in IMS enzymes associated with disease. (A) Substrates of the mitochondrial disulfide relay associated with disease phenotypes. Mutations in genes of diverse CHCHD4 substrates result in the indicated diseases. The CHCHD4 substrates are sorted according to their function as described in Figure 2 (also see Supporting Information Table S1). (B) Examples of CHCHD4 substrate mutations in which disulfide formation is affected. For TIMM8A (C66W) and NDUFB10 (C107S), cysteine mutations were reported to result in human disease. Specifically, these mutations affect disulfide formation, oxidative folding and mitochondrial import of TIMM8A and NDUFB10 that results in incomplete formation of the TIMM chaperone complex and complex I of the respiratory chain respectively. Disease‐associated mutations in COA5 (A53P) and COA6 (W59C/W66R) affect residues in proximity to cysteines which probably interferes with disulfide formation. For a detailed description, see text.
Disulfide relay components in disease
The core components of the mitochondrial disulfide relay, CHCHD4 and ALR as well as AIF, have all been implicated in different disease phenotypes. In human breast cancer and glioma, increased CHCHD4 expression correlates with increasing tumour grade and reduced patient survival (Yang et al., 2012). This is most likely because overexpression of CHCHD4 in tumour cells enhances HIF‐1α protein stabilization in hypoxic conditions and thus improves the hypoxia response, which is critical for tumour development. The molecular basis for this phenomenon is not yet understood.
Mutations in AIF are manifested as familial X‐linked diseases. Either AIF mutations cause severe paediatric mitochondriopathy linked to reduced expression and function of respiratory chain complexes or they cause the Cowchock syndrome (Ghezzi et al., 2010; Berger et al., 2011; Rinaldi et al., 2012). Recent data indicate that most of these phenotypes of AIF mutation might be due to the absence of CHCHD4, which depends on AIF for its mitochondrial import (Hangen et al., 2015).
For the ALR gene, there is one report of a mutation that results in a point mutation (R194H) in the ALR protein (Di Fonzo et al., 2009). This mutation causes an infantile mitochondrial disorder, which presents with progressive myopathy and partial combined respiratory‐chain deficiency, congenital cataract, sensorineural hearing loss and developmental delay. On a molecular level, respiratory chain complexes I, II and IV are reduced in their activity, substrates of the mitochondrial disulfide relay are reduced in their levels, mitochondria exhibited an abnormal ultrastructural morphology with an enlarged IMS and mitochondrial DNA deletions accumulated more quickly. Later studies revealed that the R194H mutation in ALR renders the protein less stable by decreasing its melting temperature, by increasing the rate of FAD dissociation from the holoenzyme and by enhancing the susceptibility to reduction of the intersubunit disulfide bonds in ALR by reduced GSH (Daithankar et al., 2010; Ceh‐Pavia et al., 2014). Taken together, these data point to a dysregulated (probably more reduced) CHCHD4 redox state caused by lowered amounts of active ALR.
Cysteine mutations and impairment in oxidative folding in disulfide relay substrates that lead to diseases
Loss‐of‐function mutations in the DDP1/TIMM8A gene result in the Mohr–Tranebjaerg syndrome, which is a progressive, neurodegenerative disorder (Hofmann et al., 2002; Roesch et al., 2002). A missense mutation known to cause this clinical phenotype is a cysteine to tryptophan exchange at position 66 (C66W, Figure 6B). Notably, the mutated human TIMM8/DDP1 is still efficiently imported into the IMS of isolated mitochondria. However, TIMM8/DDP1 with the C66W mutation loses its ability to assemble into a hetero‐hexameric 70‐kDa complex with its cognate partner protein human TIMM13 and thus fails to complement the function of its yeast homologue Tim8.
Compound heterozygous mutations in the NDUFB10 gene lead to a protein in which the conserved cysteine 107 is mutated to a serine (C107S, this cysteine is involved in a disulfide bond in NDUFB10) (Friederich et al., 2017) (Figure 6B). The affected infant died from fatal infantile lactic acidosis and cardiomyopathy. On a molecular level, NDUFB10 levels are strongly diminished in muscle and heart cells and less so in liver and fibroblasts. This in turn is due to failure of mitochondrial accumulation of NDUFB10. As a result, complex I biogenesis is perturbed at a late stage, and consequently, tissues from the infant had profoundly decreased activity of respiratory chain complex I in muscle, heart and liver. Interestingly, the phenotypes manifested to a very different extent in different tissues. While NDUFB10 and complex I activity were almost completely absent in muscle and heart, protein level and activity were almost normal in fibroblasts. This also implies that, despite the cysteine mutation, under certain conditions NDUFB10 can become imported, folded and assembled into complex I. The differences between the tissues might be explained by differences in redox environment, quality control and NDUFB10 turnover.
Mutations in the genes of COA5 and COA6 result in proteins with an A53P or a W66R mutation respectively (Huigsloot et al., 2011; Baertling et al., 2015) (Figure 6B). Although both mutations do not directly involve cysteine residues, they are close to the conserved cysteine residues in both proteins. Based on their position in the structure of both proteins, they are very likely to destroy the helix–loop–helix motif or the interaction with CHCHD4. Both mutations result in fatal infantile cardioencephalomyopathy due to cytochrome c oxidase deficiency and lactic acidosis.
Concluding remarks
The mitochondrial IMS is an important sub‐compartment at the interface of cytosol and mitochondria with a complex and only poorly explored redox biology. Many IMS proteins contain conserved cysteine residues, and for some, post‐translational modifications to disulfide bonds have been demonstrated. In spite of our lack of knowledge on the function of most conserved IMS cysteines, various diseases caused by their mutation underline their importance. It is however quite astonishing that distinct mutations result in highly organ‐specific phenotypes (e.g. NDUFB10 mutation which leads to defect in liver and heart and normal levels and function in fibroblasts), and it will thus in the future be necessary to explore the mitochondrial disulfide relay and its substrates in a tissue‐specific manner. Moreover, we do not have many pharmacological tools to interfere with the mitochondrial disulfide relay in vivo. One set of low MW inhibitors was discovered recently that will inhibit ALR oxidase activity (Dabir et al., 2013). With this inhibitor in hand, a role of ALR in human embryonic stem cell homeostasis was demonstrated. The future development of additional pharmacological tools might contribute to a further understanding of IMS redox biology and disulfide formation in vivo.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017a,b).
Conflict of interest
The authors declare no conflicts of interest.
Supporting information
Table S1 This table was generated by merging the datasets of the IMS‐proteome by Hung et al. (2014) (APEX), CHCHD4‐interactome by Petrungaro et al. (2015) and a bioinformatic analysis of Twin‐CX9C Motifs (some proteins were manually added e.g. dually localized like SOD1).
Acknowledgements
Research in the Riemer laboratory is supported by grants from the Deutsche Forschungsgemeinschaft (CRC1218 TP B02 and RI2150/1‐2). We thank Marcel Deponte for critical reading the manuscript.
Habich, M. , Salscheider, S. L. , and Riemer, J. (2019) Cysteine residues in mitochondrial intermembrane space proteins: more than just import. British Journal of Pharmacology, 176: 514–531. 10.1111/bph.14480.
References
- Aaltonen MJ, Friedman JR, Osman C, Salin B, di Rago JP, Nunnari J et al (2016). MICOS and phospholipid transfer by Ups2‐Mdm35 organize membrane lipid synthesis in mitochondria. J Cell Biol 213: 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Fabbro D, Kelly E, Marrion NV, Peters JA, Faccenda E et al (2017a). The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol 174: S272–S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD et al (2017b). The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol 174: S360–S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand R, Wai T, Baker MJ, Kladt N, Schauss AC, Rugarli E et al (2014. The i‐AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fission). J Cell Biol 204: 919–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appelhans T, Busch KB (2017). Dynamic imaging of mitochondrial membrane proteins in specific sub‐organelle membrane locations. Biophys Rev 9: 345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auranen M, Ylikallio E, Shcherbii M, Paetau A, Kiuru‐Enari S, Toppila JP et al (2015). CHCHD10 variant p.(Gly66Val) causes axonal Charcot–Marie–Tooth disease. Neurol Genet 1: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backes S, Herrmann JM (2017). Protein translocation into the intermembrane space and matrix of mitochondria: mechanisms and driving forces. Front Mol Biosci 4: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baertling F, A.M. van den Brand M, Hertecant JL, al‐Shamsi A, P. van den Heuvel L, Distelmaier F et al (2015). Mutations in COA6 cause cytochrome c oxidase deficiency and neonatal hypertrophic cardiomyopathy. Hum Mutat 36: 34–38. [DOI] [PubMed] [Google Scholar]
- Bais R, Edwards JB (1982). Creatine kinase. Crit Rev Clin Lab Sci 16: 291–335. [DOI] [PubMed] [Google Scholar]
- Bak DW, Weerapana E (2015). Cysteine‐mediated redox signalling in the mitochondria. Mol Biosyst 11: 678–697. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Calderone V, Cefaro C, Ciofi‐Baffoni S, Gallo A et al (2011a). Molecular recognition and substrate mimicry drive the electron‐transfer process between MIA40 and ALR. Proc Natl Acad Sci U S A 108: 4811–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cefaro C, Cenacchi L, Ciofi‐Baffoni S, Felli IC et al (2010). Molecular chaperone function of Mia40 triggers consecutive induced folding steps of the substrate in mitochondrial protein import. Proc Natl Acad Sci U S A 107: 20190–20195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, Bertini I, Cefaro C, Ciofi‐Baffoni S, Gallo A, Martinelli M et al (2009). MIA40 is an oxidoreductase that catalyzes oxidative protein folding in mitochondria. Nat Struct Mol Biol 16: 198–206. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Ciofi‐Baffoni S, Boscaro F, Chatzi A, Mikolajczyk M et al (2011b). Anamorsin is a [2Fe‐2S] cluster‐containing substrate of the Mia40‐dependent mitochondrial protein trapping machinery. Chem Biol 18: 794–804. [DOI] [PubMed] [Google Scholar]
- Barbot M, Jans DC, Schulz C, Denkert N, Kroppen B, Hoppert M et al (2015). Mic10 oligomerizes to bend mitochondrial inner membranes at cristae junctions. Cell Metab 21: 756–763. [DOI] [PubMed] [Google Scholar]
- Barchiesi A, Wasilewski M, Chacinska A, Tell G, Vascotto C (2015). Mitochondrial translocation of APE1 relies on the MIA pathway. Nucleic Acids Res 43: 5451–5464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger I, Ben‐Neriah Z, Dor‐Wolman T, Shaag A, Saada A, Zenvirt S et al (2011). Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol Genet Metab 104: 517–520. [DOI] [PubMed] [Google Scholar]
- Bien M, Longen S, Wagener N, Chwalla I, Herrmann JM, Riemer J (2010). Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol Cell 37: 516–528. [DOI] [PubMed] [Google Scholar]
- Bihlmaier K, Mesecke N, Terziyska N, Bien M, Hell K, Herrmann JM (2007). The disulfide relay system of mitochondria is connected to the respiratory chain. J Cell Biol 179: 389–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleier L, Wittig I, Heide H, Steger M, Brandt U, Drose S (2015). Generator‐specific targets of mitochondrial reactive oxygen species. Free Radic Biol Med 78: 1–10. [DOI] [PubMed] [Google Scholar]
- Bohnert M, Zerbes RM, Davies KM, Muhleip AW, Rampelt H, Horvath SE et al (2015). Central role of Mic10 in the mitochondrial contact site and cristae organizing system. Cell Metab 21: 747–755. [DOI] [PubMed] [Google Scholar]
- Burke PJ (2017). Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer 3: 857–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavallaro G (2010). Genome‐wide analysis of eukaryotic twin CX9C proteins. Mol Biosyst 6: 2459–2470. [DOI] [PubMed] [Google Scholar]
- Ceh‐Pavia E, Ang SK, Spiller MP, Lu H (2014). The disease‐associated mutation of the mitochondrial thiol oxidase Erv1 impairs cofactor binding during its catalytic reaction. Biochem J 464: 449–459. [DOI] [PubMed] [Google Scholar]
- Chacinska A, Guiard B, Muller JM, Schulze‐Specking A, Gabriel K, Kutik S et al (2008). Mitochondrial biogenesis, switching the sorting pathway of the intermembrane space receptor Mia40. J Biol Chem 283: 29723–29729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzi A, Manganas P, Tokatlidis K (2016). Oxidative folding in the mitochondrial intermembrane space: a regulated process important for cell physiology and disease. Biochim Biophys Acta 1863: 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JJ, Jones ME (1976). The cellular location of dihydroorotate dehydrogenase: relation to de novo biosynthesis of pyrimidines. Arch Biochem Biophys 176: 82–90. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Ching WC, Chen JS, Lee TY, Lu CT, Chou HC et al (2014). Decoding the s‐nitrosoproteomic atlas in individualized human colorectal cancer tissues using a label‐free quantitation strategy. J Proteome Res 13: 4942–4958. [DOI] [PubMed] [Google Scholar]
- Chrestensen CA, Starke DW, Mieyal JJ (2000). Acute cadmium exposure inactivates thioltransferase (Glutaredoxin), inhibits intracellular reduction of protein‐glutathionyl‐mixed disulfides, and initiates apoptosis. J Biol Chem 275: 26556–26565. [DOI] [PubMed] [Google Scholar]
- Cipolat S, Rudka T, Hartmann D, Costa V, Serneels L, Craessaerts K et al (2006). Mitochondrial rhomboid PARL regulates cytochrome c release during apoptosis via OPA1‐dependent cristae remodeling. Cell 126: 163–175. [DOI] [PubMed] [Google Scholar]
- Cobine PA, Pierrel F, Winge DR (2006). Copper trafficking to the mitochondrion and assembly of copper metalloenzymes. Biochim Biophys Acta 1763: 759–772. [DOI] [PubMed] [Google Scholar]
- Dabir DV, Hasson SA, Setoguchi K, Johnson ME, Wongkongkathep P, Douglas CJ et al (2013). A small molecule inhibitor of redox‐regulated protein translocation into mitochondria. Dev Cell 25: 81–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daithankar VN, Schaefer SA, Dong M, Bahnson BJ, Thorpe C (2010). Structure of the human sulfhydryl oxidase augmenter of liver regeneration and characterization of a human mutation causing an autosomal recessive myopathy. Biochemistry 49: 6737–6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daithankar VN, Wang W, Trujillo JR, Thorpe C (2012). Flavin‐linked Erv‐family sulfhydryl oxidases release superoxide anion during catalytic turnover. Biochemistry 51: 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pinto V, Reina S, Gupta A, Messina A, Mahalakshmi R (2016). Role of cysteines in mammalian VDAC isoforms' function. Biochim Biophys Acta 1857: 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Arco A, Contreras L, Pardo B, Satrustegui J (2016). Calcium regulation of mitochondrial carriers. Biochim Biophys Acta 1863: 2413–2421. [DOI] [PubMed] [Google Scholar]
- Delaunay A, Pflieger D, Barrault MB, Vinh J, Toledano MB (2002). A thiol peroxidase is an H2O2 receptor and redox‐transducer in gene activation. Cell 111: 471–481. [DOI] [PubMed] [Google Scholar]
- Deponte M (2017). The incomplete glutathione puzzle: just guessing at numbers and figures? Antioxid Redox Signal 27: 1130–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depuydt M, Messens J, Collet JF (2011). How proteins form disulfide bonds. Antioxid Redox Signal 15: 49–66. [DOI] [PubMed] [Google Scholar]
- Di Fonzo A, Ronchi D, Lodi T, Fassone E, Tigano M, Lamperti C et al (2009). The mitochondrial disulfide relay system protein GFER is mutated in autosomal‐recessive myopathy with cataract and combined respiratory‐chain deficiency. Am J Hum Genet 84: 594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold L, Chandel NS (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med 100: 86–93. [DOI] [PubMed] [Google Scholar]
- Dzeja P, Terzic A (2009). Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int J Mol Sci 10: 1729–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell SR, Thorpe C (2005). Augmenter of liver regeneration: a flavin‐dependent sulfhydryl oxidase with cytochrome c reductase activity. Biochemistry 44: 1532–1541. [DOI] [PubMed] [Google Scholar]
- Fischer M, Horn S, Belkacemi A, Kojer K, Petrungaro C, Habich M et al (2013). Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol Biol Cell 24: 2160–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M, Riemer J (2013). The mitochondrial disulfide relay system: roles in oxidative protein folding and beyond. Int J Cell Biol 2013: 742923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T et al (2006). OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell 126: 177–189. [DOI] [PubMed] [Google Scholar]
- Friederich MW, Erdogan AJ, Coughlin CR 2nd, Elos MT, Jiang H, O'Rourke CP et al (2017). Mutations in the accessory subunit NDUFB10 result in isolated complex I deficiency and illustrate the critical role of intermembrane space import for complex I holoenzyme assembly. Hum Mol Genet 26: 702–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funes S, Kauff F, van der Sluis EO, Ott M, Herrmann JM (2011). Evolution of YidC/Oxa1/Alb3 insertases: three independent gene duplications followed by functional specialization in bacteria, mitochondria and chloroplasts. Biol Chem 392: 13–19. [DOI] [PubMed] [Google Scholar]
- Gallogly MM, Mieyal JJ (2007). Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol 7: 381–391. [DOI] [PubMed] [Google Scholar]
- Gallogly MM, Starke DW, Leonberg AK, Ospina SM, Mieyal JJ (2008). Kinetic and mechanistic characterization and versatile catalytic properties of mammalian glutaredoxin 2: implications for intracellular roles. Biochemistry 47: 11144–11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XH, Qanungo S, Pai HV, Starke DW, Steller KM, Fujioka H et al (2013). Aging‐dependent changes in rat heart mitochondrial glutaredoxins – implications for redox regulation. Redox Biol 1: 586–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, Lobanov AV, Gladyshev VN (2012). Genome‐wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc Natl Acad Sci U S A 109: 17394–17399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi D, Sevrioukova I, Invernizzi F, Lamperti C, Mora M, D'Adamo P et al (2010). Severe X‐linked mitochondrial encephalomyopathy associated with a mutation in apoptosis‐inducing factor. Am J Hum Genet 86: 639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giangregorio N, Tonazzi A, Console L, Lorusso I, De Palma A, Indiveri C (2016). The mitochondrial carnitine/acylcarnitine carrier is regulated by hydrogen sulfide via interaction with C136 and C155. Biochim Biophys Acta 1860: 20–27. [DOI] [PubMed] [Google Scholar]
- Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C et al (2005). Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell 122: 221–233. [DOI] [PubMed] [Google Scholar]
- Gornicka A, Bragoszewski P, Chroscicki P, Wenz LS, Schulz C, Rehling P et al (2014). A discrete pathway for the transfer of intermembrane space proteins across the outer membrane of mitochondria. Mol Biol Cell 25: 3999–4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo PC, Ma JD, Jiang YL, Wang SJ, Bao ZZ, Yu XJ et al (2012). Structure of yeast sulfhydryl oxidase erv1 reveals electron transfer of the disulfide relay system in the mitochondrial intermembrane space. J Biol Chem 287: 34961–34969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haindrich AC, Boudova M, Vancova M, Diaz PP, Horakova E, Lukes J (2017). The intermembrane space protein Erv1 of Trypanosoma brucei is essential for mitochondrial Fe–S cluster assembly and operates alone. Mol Biochem Parasitol 214: 47–51. [DOI] [PubMed] [Google Scholar]
- Hamza I, Dailey HA (2012). One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim Biophys Acta 1823: 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangen E, Feraud O, Lachkar S, Mou H, Doti N, Fimia GM et al (2015). Interaction between AIF and CHCHD4 regulates respiratory chain biogenesis. Mol Cell 58: 1001–1014. [DOI] [PubMed] [Google Scholar]
- Harding SD, Sharman JL, Faccenda E, Southan C, Pawson AJ, Ireland S et al (2018). The IUPHAR/BPS guide to pharmacology in 2018: updates and expansion to encompass the new guide to immunopharmacology. Nucl Acids Res 46: D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harner M, Korner C, Walther D, Mokranjac D, Kaesmacher J, Welsch U et al (2011). The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J 30: 4356–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennon SW, Soman R, Zhu L, Dalbey RE (2015). YidC/Alb3/Oxa1 family of insertases. J Biol Chem 290: 14866–14874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Riemer J (2012). Mitochondrial disulfide relay: redox‐regulated protein import into the intermembrane space. J Biol Chem 287: 4426–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JM, Riemer J (2014). Three approaches to one problem: protein folding in the periplasm, the endoplasmic reticulum, and the intermembrane space. Antioxid Redox Signal 21: 438–456. [DOI] [PubMed] [Google Scholar]
- Hewitt VL, Gabriel K, Traven A (2014). The ins and outs of the intermembrane space: diverse mechanisms and evolutionary rewiring of mitochondrial protein import routes. Biochim Biophys Acta 1840: 1246–1253. [DOI] [PubMed] [Google Scholar]
- Hofmann S, Rothbauer U, Muhlenbein N, Neupert W, Gerbitz KD, Brunner M et al (2002). The C66W mutation in the deafness dystonia peptide 1 (DDP1) affects the formation of functional DDP1.TIM13 complexes in the mitochondrial intermembrane space. J Biol Chem 277: 23287–23293. [DOI] [PubMed] [Google Scholar]
- Hoppins S, Collins SR, Cassidy‐Stone A, Hummel E, Devay RM, Lackner LL et al (2011). A mitochondrial‐focused genetic interaction map reveals a scaffold‐like complex required for inner membrane organization in mitochondria. J Cell Biol 195: 323–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson DA, Thorpe C (2015). Mia40 is a facile oxidant of unfolded reduced proteins but shows minimal isomerase activity. Arch Biochem Biophys 579: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huigsloot M, Nijtmans LG, Szklarczyk R, Baars MJ, van den Brand MA, Hendriksfranssen MG et al (2011). A mutation in C2orf64 causes impaired cytochrome c oxidase assembly and mitochondrial cardiomyopathy. Am J Hum Genet 88: 488–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Zou P, Rhee HW, Udeshi ND, Cracan V, Svinkina T et al (2014). Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 55: 332–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, Sancak Y, Mootha VK (2014). The uniporter: from newly identified parts to function. Biochem Biophys Res Commun 449: 370–372. [DOI] [PubMed] [Google Scholar]
- Kappler U, Enemark JH (2015). Sulfite‐oxidizing enzymes. J Biol Inorg Chem 20: 253–264. [DOI] [PubMed] [Google Scholar]
- Koch JR, Schmid FX (2014a). Mia40 is optimized for function in mitochondrial oxidative protein folding and import. ACS Chem Biol 9: 2049–2057. [DOI] [PubMed] [Google Scholar]
- Koch JR, Schmid FX (2014b). Mia40 targets cysteines in a hydrophobic environment to direct oxidative protein folding in the mitochondria. Nat Commun 5: 3041. [DOI] [PubMed] [Google Scholar]
- Kojer K, Bien M, Gangel H, Morgan B, Dick TP, Riemer J (2012). Glutathione redox potential in the mitochondrial intermembrane space is linked to the cytosol and impacts the Mia40 redox state. EMBO J 31: 3169–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojer K, Peleh V, Calabrese G, Herrmann JM, Riemer J (2015). Kinetic control by limiting glutaredoxin amounts enables thiol oxidation in the reducing mitochondrial intermembrane space. Mol Biol Cell 26: 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozjak‐Pavlovic V (2017). The MICOS complex of human mitochondria. Cell Tissue Res 367: 83–93. [DOI] [PubMed] [Google Scholar]
- Kritsiligkou P, Chatzi A, Charalampous G, Mironov A Jr, Grant CM, Tokatlidis K (2017). Unconventional targeting of a thiol peroxidase to the mitochondrial intermembrane space facilitates oxidative protein folding. Cell Rep 18: 2729–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruger V, Becker T, Becker L, Montilla‐Martinez M, Ellenrieder L, Vogtle FN et al (2017). Identification of new channels by systematic analysis of the mitochondrial outer membrane. J Cell Biol 216: 3485–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YW, Yuan Y, Isaac J, Babu CV, Meller J, Ho SM (2010). Comprehensive identification and modified‐site mapping of S‐nitrosylated targets in prostate epithelial cells. PLoS One 5: e9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange H, Lisowsky T, Gerber J, Muhlenhoff U, Kispal G, Lill R (2001). An essential function of the mitochondrial sulfhydryl oxidase Erv1p/ALR in the maturation of cytosolic Fe/S proteins. EMBO Rep 2: 715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leary SC (2010). Redox regulation of SCO protein function: controlling copper at a mitochondrial crossroad. Antioxid Redox Signal 13: 1403–1416. [DOI] [PubMed] [Google Scholar]
- Longen S, Richter F, Kohler Y, Wittig I, Beck KF, Pfeilschifter J (2016). Quantitative persulfide site identification (qPerS‐SID) reveals protein targets of H2S releasing donors in mammalian cells. Sci Rep 6: 29808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longen S, Woellhaf MW, Petrungaro C, Riemer J, Herrmann JM (2014). The disulfide relay of the intermembrane space oxidizes the ribosomal subunit mrp10 on its transit into the mitochondrial matrix. Dev Cell 28: 30–42. [DOI] [PubMed] [Google Scholar]
- Mannella CA (2006). Structure and dynamics of the mitochondrial inner membrane cristae. Biochim Biophys Acta 1763: 542–548. [DOI] [PubMed] [Google Scholar]
- Massa V, Fernandez‐Vizarra E, Alshahwan S, Bakhsh E, Goffrini P, Ferrero I et al (2008). Severe infantile encephalomyopathy caused by a mutation in COX6B1, a nucleus‐encoded subunit of cytochrome c oxidase. Am J Hum Genet 82: 1281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesecke N, Bihlmaier K, Grumbt B, Longen S, Terziyska N, Hell K et al (2008). The zinc‐binding protein Hot13 promotes oxidation of the mitochondrial import receptor Mia40. EMBO Rep 9: 1107–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Buettner S, Ghezzi D, Zeviani M, Bano D, Nicotera P (2015). Loss of apoptosis‐inducing factor critically affects MIA40 function. Cell Death Dis 6: e1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic D, Ramming T, Muller JM, Wenz LS, Gebert N, Schulze‐Specking A et al (2009). Identification of the signal directing Tim9 and Tim10 into the intermembrane space of mitochondria. Mol Biol Cell 20: 2530–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modjtahedi N, Tokatlidis K, Dessen P, Kroemer G (2016). Mitochondrial proteins containing coiled‐coil‐helix‐coiled‐coil‐helix (CHCH) domains in health and disease. Trends Biochem Sci 41: 245–260. [DOI] [PubMed] [Google Scholar]
- Morgenstern M, Stiller SB, Lubbert P, Peikert CD, Dannenmaier S, Drepper F et al (2017). Definition of a high‐confidence mitochondrial proteome at quantitative scale. Cell Rep 19: 2836–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naor MM, Jensen JH (2004). Determinants of cysteine pKa values in creatine kinase and α1‐antitrypsin. Proteins 57: 799–803. [DOI] [PubMed] [Google Scholar]
- Neal SE, Dabir DV, Wijaya J, Boon C, Koehler CM (2017). Osm1 facilitates the transfer of electrons from Erv1 to fumarate in the redox‐regulated import pathway in the mitochondrial intermembrane space. Mol Biol Cell 28: 2773–2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevitt T, Ohrvik H, Thiele DJ (2012). Charting the travels of copper in eukaryotes from yeast to mammals. Biochim Biophys Acta 1823: 1580–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer HK, Dlouhy AC, Thornton JD, Hu J, Liu Y, Barycki JJ et al (2015). Cytosolic Fe–S cluster protein maturation and iron regulation are independent of the mitochondrial Erv1/Mia40 import system. J Biol Chem 290: 27829–27840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai HV, Starke DW, Lesnefsky EJ, Hoppel CL, Mieyal JJ (2007). What is the functional significance of the unique location of glutaredoxin 1 (GRx1) in the intermembrane space of mitochondria? Antioxid Redox Signal 9: 2027–2033. [DOI] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M et al (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53: 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul BD, Snyder SH (2018). Gasotransmitter hydrogen sulfide signaling in neuronal health and disease. Biochem Pharmacol 149: 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleh V, Cordat E, Herrmann JM (2016). Mia40 is a trans‐site receptor that drives protein import into the mitochondrial intermembrane space by hydrophobic substrate binding. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna E, Espino J, De Stefani D, Rizzuto R (2018). The MCU complex in cell death. Cell Calcium 69: 73–80. [DOI] [PubMed] [Google Scholar]
- Petrungaro C, Zimmermann KM, Kuttner V, Fischer M, Dengjel J, Bogeski I et al (2015). The Ca(2+)‐dependent release of the Mia40‐induced MICU1‐MICU2 dimer from MCU regulates mitochondrial Ca(2+) uptake. Cell Metab 22: 721–733. [DOI] [PubMed] [Google Scholar]
- Potting C, Wilmes C, Engmann T, Osman C, Langer T (2010). Regulation of mitochondrial phospholipids by Ups1/PRELI‐like proteins depends on proteolysis and Mdm35. EMBO J 29: 2888–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiros PM, Langer T, Lopez‐Otin C (2015). New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol 16: 345–359. [DOI] [PubMed] [Google Scholar]
- Radi R (2013). Peroxynitrite, a stealthy biological oxidant. J Biol Chem 288: 26464–26472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesh A, Peleh V, Martinez‐Caballero S, Wollweber F, Sommer F, van der Laan M et al (2016). A disulfide bond in the TIM23 complex is crucial for voltage gating and mitochondrial protein import. J Cell Biol 214: 417–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina S, Checchetto V, Saletti R, Gupta A, Chaturvedi D, Guardiani C et al (2016a). VDAC3 as a sensor of oxidative state of the intermembrane space of mitochondria: the putative role of cysteine residue modifications. Oncotarget 7: 2249–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina S, Guarino F, Magri A, De Pinto V (2016b). VDAC3 as a potential marker of mitochondrial status is involved in cancer and pathology. Front Oncol 6: 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieger B, Junge W, Busch KB (2014). Lateral pH gradient between OXPHOS complex IV and F(0)F(1) ATP‐synthase in folded mitochondrial membranes. Nat Commun 5: 3103. [DOI] [PubMed] [Google Scholar]
- Riemer J, Schwarzlander M, Conrad M, Herrmann JM (2015). Thiol switches in mitochondria: operation and physiological relevance. Biol Chem 396: 465–482. [DOI] [PubMed] [Google Scholar]
- Rinaldi C, Grunseich C, Sevrioukova IF, Schindler A, Horkayne‐Szakaly I, Lamperti C et al (2012). Cowchock syndrome is associated with a mutation in apoptosis‐inducing factor. Am J Hum Genet 91: 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roesch K, Curran SP, Tranebjaerg L, Koehler CM (2002). Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a‐TIMM13 complex. Hum Mol Genet 11: 477–486. [DOI] [PubMed] [Google Scholar]
- Scharwey M, Tatsuta T, Langer T (2013). Mitochondrial lipid transport at a glance. J Cell Sci 126: 5317–5323. [DOI] [PubMed] [Google Scholar]
- Schulz C, Schendzielorz A, Rehling P (2015). Unlocking the presequence import pathway. Trends Cell Biol 25: 265–275. [DOI] [PubMed] [Google Scholar]
- Schwarz G (2016). Molybdenum cofactor and human disease. Curr Opin Chem Biol 31: 179–187. [DOI] [PubMed] [Google Scholar]
- Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Mannella CA et al (2002). A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell 2: 55–67. [DOI] [PubMed] [Google Scholar]
- Sideris DP, Petrakis N, Katrakili N, Mikropoulou D, Gallo A, Ciofi‐Baffoni S et al (2009). A novel intermembrane space‐targeting signal docks cysteines onto Mia40 during mitochondrial oxidative folding. J Cell Biol 187: 1007–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KA, Waypa GB, Schumacker PT (2017). Redox signaling during hypoxia in mammalian cells. Redox Biol 13: 228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobotta MC, Liou W, Stocker S, Talwar D, Oehler M, Ruppert T et al (2015). Peroxiredoxin‐2 and STAT3 form a redox relay for H2O2 signaling. Nat Chem Biol 11: 64–70. [DOI] [PubMed] [Google Scholar]
- Spiller MP, Ang SK, Ceh‐Pavia E, Fisher K, Wang Q, Rigby SE et al (2013). Identification and characterization of mitochondrial Mia40 as an iron‐sulfur protein. Biochem J 455: 27–35. [DOI] [PubMed] [Google Scholar]
- Starke DW, Chock PB, Mieyal JJ (2003). Glutathione‐thiyl radical scavenging and transferase properties of human glutaredoxin (thioltransferase). Potential role in redox signal transduction. J Biol Chem 278: 14607–14613. [DOI] [PubMed] [Google Scholar]
- Stojanovski D, Bragoszewski P, Chacinska A (2012). The MIA pathway: a tight bond between protein transport and oxidative folding in mitochondria. Biochim Biophys Acta 1823: 1142–1150. [DOI] [PubMed] [Google Scholar]
- Stoldt S, Wenzel D, Kehrein K, Riedel D, Ott M, Jakobs S (2018). Spatial orchestration of mitochondrial translation and OXPHOS complex assembly. Nat Cell Biol 20: 528–534. [DOI] [PubMed] [Google Scholar]
- Straub IR, Janer A, Weraarpachai W, Zinman L, Robertson J, Rogaeva E et al (2018). Loss of CHCHD10‐CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum Mol Genet 27: 178–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC (2001). A fraction of yeast Cu,Zn‐superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem 276: 38084–38089. [DOI] [PubMed] [Google Scholar]
- Su D, Shukla AK, Chen B, Kim JS, Nakayasu E, Qu Y et al (2013). Quantitative site‐specific reactivity profiling of S‐nitrosylation in mouse skeletal muscle using cysteinyl peptide enrichment coupled with mass spectrometry. Free Radic Biol Med 57: 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sztolsztener ME, Brewinska A, Guiard B, Chacinska A (2013). Disulfide bond formation: sulfhydryl oxidase ALR controls mitochondrial biogenesis of human MIA40. Traffic 14: 309–320. [DOI] [PubMed] [Google Scholar]
- Tamura Y, Sesaki H, Endo T (2014). Phospholipid transport via mitochondria. Traffic 15: 933–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T, Langer T (2017). Intramitochondrial phospholipid trafficking. Biochim Biophys Acta 1862: 81–89. [DOI] [PubMed] [Google Scholar]
- Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U (2010). Growth factor erv1‐like modulates Drp1 to preserve mitochondrial dynamics and function in mouse embryonic stem cells. Mol Biol Cell 21: 1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topf U, Suppanz I, Samluk L, Wrobel L, Boser A, Sakowska P et al (2018). Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat Commun 9: 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PI, Lin CH, Hsieh CH, Papakyrikos AM, Kim MJ, Napolioni V et al (2018). PINK1 phosphorylates MIC60/mitofilin to control structural plasticity of mitochondrial Crista junctions. Mol Cell 69: 744–756 e746. [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N et al (2004). AIF deficiency compromises oxidative phosphorylation. EMBO J 23: 4679–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vascotto C, Bisetto E, Li M, Zeef LA, D'Ambrosio C, Domenis R et al (2011). Knock‐in reconstitution studies reveal an unexpected role of Cys‐65 in regulating APE1/Ref‐1 subcellular trafficking and function. Mol Biol Cell 22: 3887–3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel F, Bornhovd C, Neupert W, Reichert AS (2006). Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol 175: 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogtle FN, Burkhart JM, Rao S, Gerbeth C, Hinrichs J, Martinou JC et al (2012). Intermembrane space proteome of yeast mitochondria. Mol Cell Proteomics 11: 1840–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Malsburg K, Muller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T et al (2011). Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis. Dev Cell 21: 694–707. [DOI] [PubMed] [Google Scholar]
- Wang J, Edmondson DE (2011). Topological probes of monoamine oxidases A and B in rat liver mitochondria: inhibition by TEMPO‐substituted pargyline analogues and inactivation by proteolysis. Biochemistry 50: 2499–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattiaux‐de Coninck S, Wattiaux R (1971). Subcellular distribution of sulfite cytochrome c reductase in rat liver tissue. Eur J Biochem 19: 552–556. [DOI] [PubMed] [Google Scholar]
- Weckbecker D, Longen S, Riemer J, Herrmann JM (2012). Atp23 biogenesis reveals a chaperone‐like folding activity of Mia40 in the IMS of mitochondria. EMBO J 31: 4348–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann N, Pfanner N (2017). Mitochondrial machineries for protein import and assembly. Annu Rev Biochem 86: 685–714. [DOI] [PubMed] [Google Scholar]
- Wolhuter K, Eaton P (2017). How widespread is stable protein S‐nitrosylation as an end‐effector of protein regulation? Free Radic Biol Med 109: 156–166. [DOI] [PubMed] [Google Scholar]
- Wong HS, Dighe PA, Mezera V, Monternier PA, Brand MD (2017). Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem 292: 16804–16809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel L, Sokol AM, Chojnacka M, Chacinska A (2016). The presence of disulfide bonds reveals an evolutionarily conserved mechanism involved in mitochondrial protein translocase assembly. Sci Rep 6: 27484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Staples O, Thomas LW, Briston T, Robson M, Poon E et al (2012). Human CHCHD4 mitochondrial proteins regulate cellular oxygen consumption rate and metabolism and provide a critical role in hypoxia signaling and tumor progression. J Clin Invest 122: 600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Du J, Tang C, Huang Y, Jin H (2017). H2S‐induced sulfhydration: biological function and detection methodology. Front Pharmacol 8: 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Vinothkumar KR, Hirst J (2016). Structure of mammalian respiratory complex I. Nature 536: 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Wang PY, Huang X, Chen X, Kang JG, Hwang PM (2013). Mitochondrial disulfide relay mediates translocation of p53 and partitions its subcellular activity. Proc Natl Acad Sci U S A 110: 17356–17361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 This table was generated by merging the datasets of the IMS‐proteome by Hung et al. (2014) (APEX), CHCHD4‐interactome by Petrungaro et al. (2015) and a bioinformatic analysis of Twin‐CX9C Motifs (some proteins were manually added e.g. dually localized like SOD1).