

Abstract
We here report on our continued studies of ligands binding to the promising drug target angiotensin II type 2 receptor (AT2R). Two series of compounds were synthesized and investigated. The first series explored the effects of adding small substituents to the phenyl ring of the known selective nonpeptide AT2R antagonist C38, generating small but significant shifts in AT2R affinity. One compound in the first series was equipotent to C38 and showed similar kinetic solubility, and stability in both human and mouse liver microsomes. The second series was comprised of new bicyclic derivatives, amongst which one ligand exhibited a five‐fold improved affinity to AT2R as compared to C38. The majority of the compounds in the second series, including the most potent ligand, were inferior to C38 with regard to stability in both human and mouse microsomes. In contrast to our previously reported findings, ligands with shorter carbamate alkyl chains only demonstrated slightly improved stability in microsomes. Based on data presented herein, a more adequate, tentative model of the binding modes of ligand analogues to the prototype AT2R antagonist C38 is proposed, as deduced from docking redefined by molecular dynamic simulations.
Keywords: AT2 receptor, angiotensin II, medicinal chemistry, molecular docking, prototype antagonist
1. Introduction
The renin‐angiotensin‐aldosterone system (RAAS) is well‐known for its role in fluid‐electrolyte control and blood‐pressure regulation; there are several drugs on the market for the treatment of hypertension targeting proteins in RAAS. Although a series of bioactive components are formed in RAAS, the octapeptide angiotensin II (AngII) is considered to constitute the major effector peptide.1 Hence, inhibitors of the two proteases important for formation of AngII (angiotensin converting enzyme, ACE, and renin) or compounds acting as antagonists at its receptor (i. e. the sartans) are well established therapeutics. The angiotensin II type 1 receptor (AT1R) was long believed to be the only mediator of the effects elicited by the endogenous AngII. However, in the late 1980s the first evidence of a second protein binding AngII appeared in the literature, the angiotensin II type 2 receptor (AT2R).2,3 This receptor proved to be an enigmatic protein, which in recent years has emerged as a promising new drug target.4, 5, 6
AT2R is predominantly expressed in fetal tissue, indicating its important role in fetal development.7,8 In adults AT2R is mainly expressed in uterus, adrenal gland, smooth muscle, heart, and kidney.9,10 Notably, AT2R is strongly upregulated following tissue damage,11,12 such as vascular13 and neuronal injury,14 myocardial infarction15, 16, 17 and brain ischemia.18
There are currently two AT2R ligands in clinical trials, for different indications, again raising questions regarding the role(s) of this protein. The AT2R antagonist EMA401 (Figure 1), acquired by Novartis from Spinifex Pharmaceuticals Pty Ltd, Australia, is in phase II clinical trials for peripheral neuropathic pain.19,20 The AT2R agonist C21 (Figure 1), developed in our laboratory,21 is entering phase II clinical trials as a potential treatment for idiopathic pulmonary fibrosis. Recently published reviews detail the discovery of C21,22,23 and a large number of structurally related AT2R ligands were subsequently disclosed.24, 25, 26, 27, 28 In 2012 we reported that shifting the imidazole head group from the para position in the agonist C21 to meta position switches the pharmacological profile, resulting in the prototype antagonist C38 (Figure 1).29,30
Figure 1.
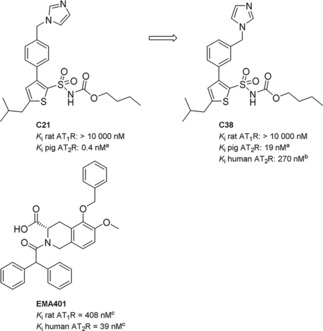
The first selective nonpeptide AT2R agonist C21, the structurally related AT2R antagonist C38, and the AT2R antagonist EMA401. [a] AT2R from pig uterus membrane assay. [b] HEK‐293 cells expressing human AT2R. [c] See Ref. [19].
The first crystal structure of AT1R binding antagonist ZD7155 was published in 2015 by Zhang et al., enabling further elucidation of the binding mode of AT1R ligands.31,32 The crystal structure allowed for comparison with our predicted inactive‐like homology model of both AT1R and AT2R, confirming the general topology and residue location in the binding cavity of our models. Minor structural changes resulting in an altered pharmacological profile. which could be rationalized in our AT2R homology model.33 Zhang et al. recently published the crystal structure of AT2R binding the selective AT2R antagonist L‐161,638, revealing a similar binding mode in comparison to ZD7155 from the previously published AT1R crystal structure.34 The published structures of both AT1R and AT2R provide insight in the structure‐function relationship and allows design of new selective ligands.
We herein report the impact on AT2R affinity of chemical modifications of the prototype AT2R antagonist C38, and consequently adjust our previously reported AT2 receptor‐antagonist model to the new SAR data through computational simulations using the newly published AT2R crystal structure.34
2. Results and Discussion
2.1. Chemistry
Two series of compounds were synthesized: the first series was motivated by our ambition to explore the impact of adding substituents to the central phenyl ring of the C38 core structure, as well as if replacing the phenyl for pyridine could improve solubility. In the second series we explored a new head group of C38 where the benzyl imidazole moiety was exchanged for bicyclic amides. Previous work from our group demonstrated that the affinity and selectivity of the ligands can be retained by replacing the imidazole with amides;29 with these new bicyclic amides we continue our exploration of the promising amide functionality by trying to ascertain the binding conformation. A majority of the bicyclic compounds retained a shortened sulfonyl carbamate chain, as we have recently found this to be beneficial for metabolic stability.35 The key building block for the synthesis of both series was MIDA boronate 3, that was synthesized according to the pathway previously published by our group.35 Microwave assisted Negishi coupling in a sealed vial of 5‐bromo‐N‐(tert‐butyl)thiophene‐2‐sulfonamide (1)] with in situ generated isobutylzinc chloride produced N‐(tert‐butyl)‐5‐isobutylthiophene‐2‐sulfonamide (2) in 51 % yield.36,37 Compound 2 was in turn converted to the MIDA boronate (3) in 76 % yield over 2 steps (Scheme 1). Substituted benzylimidazoles and pyridinemethyl imidazoles 16–27 were generated either via direct alkylation of 3‐halo benzylbromides (4–5, 10) with imidazole, or via chlorination or mesylation of the 3‐bromo benzylalcohols (6–9, 11–15) and subsequent alkylation with imidazole. Chlorination was initially tested for 3‐bromo benzylalcohols 6, 7, and 11, however generated no or low yields. Mesylation was instead explored and yielded the desired products 18, 19, and 23 in moderate to good yields (72 %, 43 %, and 66 % respectively). Once generated, the benzyl‐/pyridinemethyl imidazoles (16‐27) were coupled with the MIDA boronate 3 under Suzuki conditions in sealed vials either using conventional heating (16–18) or microwave assisted (19–27), delivering the tert‐butyl protected sulfonamides 28–39.36,37 A tert‐butyl deprotection followed by treatment with butyl chloroformate resulted in the target compounds 40–51, generated in low to fair yields over 3 steps (3‐28 %). The low yields obtained for some compounds (40, 42, 45, and 48 were isolated in 3 %, 3 %, 4 %, and 5 % respectively) may relate to electronic and/or steric properties effecting the Suzuki coupling efficiency.
Scheme 1.
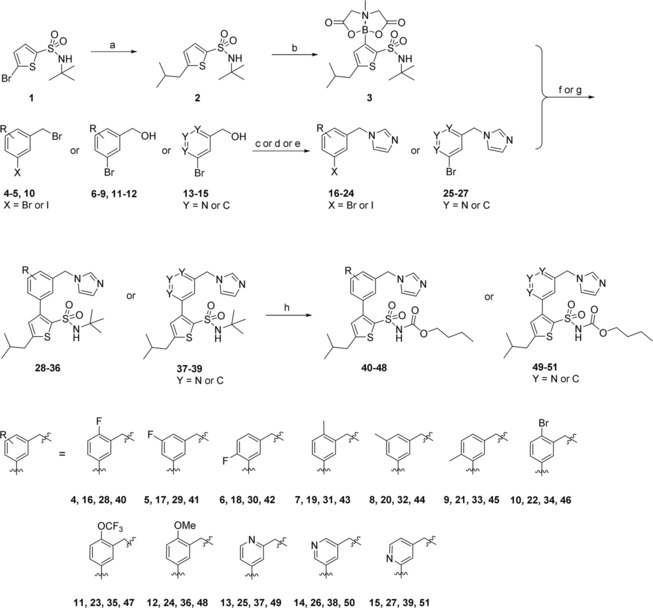
Synthesis of AT2R ligands 40–51. a) ZnCl2, isobutylmagnesium chloride, Pd(t‐Bu3P)2, toluene, THF; b) 1. n‐BuLi (4.5 eq.), triisopropyl borate, THF; 2. methyliminodiacetic acid, DMSO, toluene; c) imidazole, DCM (to 16–17) ; d) 1. TEA, MsCl, DCM; 2. imidazole, DMF (to 18–19, 22–23, 25–27); e) 1. thionyl chloride, DCM; 2. imidazole DMF (to 20–21, 24); f) PdCl2(dppf), K2CO3, DME, H2O (to 28–33, 35–39); g) Pd(PPh3)4, K2CO3, EtOH, H2O, THF (to 34); h) 1. TFA; 2. butyl chloroformate, Na2CO3, DCM, H2O.
The synthesis of the bicyclic compounds commenced with the acylation of isoindoline (52, 54) and tetrahydroisoquinoline (53, 55–56) yielding compounds 57–63 (Scheme 2). In addition, isoquinoline 55 was mesylated to form compounds 64. Bicyclic compounds 68–69 (Scheme 3) were generated by converting bromophenylacetic acid (65) into secondary amides 66–67 using thionyl chloride and methylamine or ethylamine. These were cyclized to the corresponding dihydroisoquinolinones 68–69 via a Pictet‐Spengler condensation/cyclization with paraformaldehyde, using Eaton's reagent (7.7 wt‐% P2O5 in MeSO3H) to replace the polyphosphoric acid traditionally used for this cyclization.38,39 The reaction was initiated with Eaton's reagent activating the aldehyde, followed by the N‐alkylated amide attacking the activated carbonyl carbon. Dehydration led to alkylideneacetamide formation and after an intramolecular electrophilic aromatic substitution, the dihydroisoquinolinones 68–69 were formed in 99 % and 94 % yield, respectively. Some of the bicyclic amide 69 obtained was dimethylated to yield compound 70. At this point the generated bicycles (57–64, 68–70) were coupled with MIDA boronate 3 under Suzuki conditions in sealed vials using conventional heating, to give compounds 71–75, 77–82 (Scheme 4). Isoindoline 54 was also coupled with MIDA boronate 3, generating compound 76. Subsequent deprotection and reaction with butyl or ethyl chloroformate produced the sulfonyl butyl carbamate products 83–86 and ethyl carbamate products 87–96 in moderate to good overall yield (15 %–82 %), with the exception of compound 94 and 96 that were isolated in 7 % and 5 % yield, respectively.
Scheme 2.
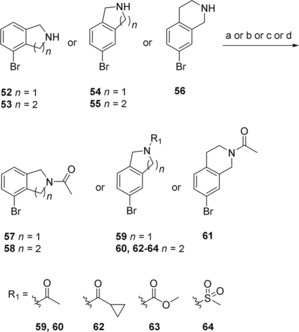
Synthesis of compounds 57–64. a) Acetic anhydride, K2CO3, MeCN (to 57–61); b) cyclopropanecarbonyl chloride, DIPEA, DCM (to 62); c) methyl chloroformate, DIPEA, DCM (to 63); d) MsCl, DIPEA, DCM (to 64).
Scheme 3.
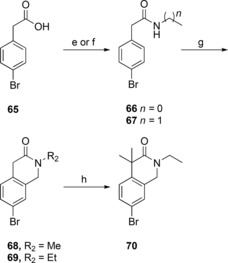
Synthesis for compounds 68–70. e) 1. Thionyl chloride, DMF, toluene; 2. methylamine, H2O (to 66); f) 1. thionyl chloride, DMF, toluene; 2. ethylamine, H2O (to 67); g) paraformaldehyde, Eaton's reagent; h) NaH, MeI, DME.
Scheme 4.
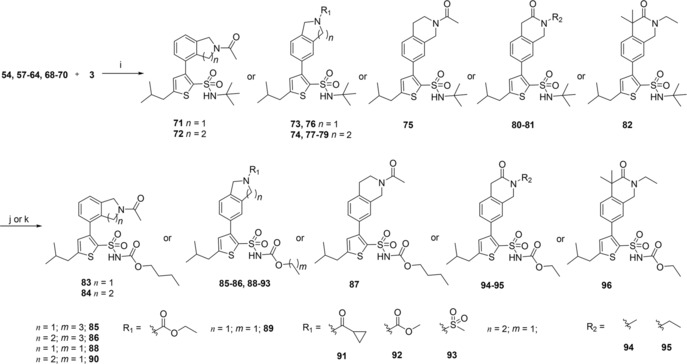
Synthesis of AT2R ligands 83–96. i) PdCl2(dppf), K2CO3, DME, H2O; j) 1. TFA; 2. butyl chloroformate, TEA, DCM (to 83–87); k) 1. TFA; 2. ethyl chloroformate, DMAP, DIPEA, DCM (to 88–96).
2.2. In Vitro Pharmacology
Selectivity for AT2R over AT1R was retained for all compounds in series 1, listed in Table 1. Introducing a fluoro substituent in ortho (40) or meta (41) position relative to the methylene imidazole group resulted in no change in affinity as compared to C38. Interestingly, compound 42 with a fluoro atom in the para position to the methylene imidazole exhibited a slight (3‐fold) reduction of affinity. This may relate to the altered electronic properties of the molecule or possibly a steric interaction. Replacing the fluoro substituent with methyl in the ortho position to the methylene imidazole was well tolerated (43). Adding a methyl to the meta or para position relative to the imidazole (44 and 45) did, however, result in a similar reduced affinity as was seen for compound 42, which further indicates a possible steric interaction. A bromide (46) or the larger trifluoromethoxy group (47) in the ortho position generated compounds with similar affinity as C38. Interestingly, a methoxy group (48) in the same position significantly decreased affinity. This may relate to the slightly lower lipophilicity of the methoxy as compared to the trifluoromethoxy moiety, or possibly the electron‐donating properties of the substituent. Replacing the phenyl ring for pyridine and placing the nitrogen of the pyridine in the ortho position relative to the methylene imidazole moiety (49) furnished a slightly reduced affinity while the equivalent meta and para pyridine analogues (50, 51) exhibit similar affinity for AT2R as C38.
Table 1.
Analogues of C38 with substituents on the central phenyl ring, synthesized via Scheme 1.
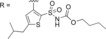
| |||
|---|---|---|---|
| Cmpd | Structure | K i hAT2R [nM][a] | [%] Inhibition of hAT1R at 10 μM[b] |
| C38 |
|
270 (19[c]) IC50=694 nM[d] | 7 IC50>10 000 nM[e] |
| 40 |
|
300 IC50=217 nM[d] | 8 IC50>10 000 nM[e] |
| 41 |
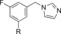
|
120 | 13 |
| 42 |
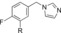
|
800 | 16 |
| 43 |
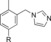
|
230 | 14 |
| 44 |
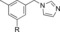
|
810 | 26 |
| 45 |
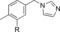
|
800 | 12 |
| 46 |
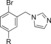
|
120 | 18 |
| 47 |
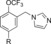
|
280 | 1.8 |
| 48 |
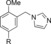
|
1300 | 23 |
| 49 |
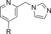
|
660 | 19 |
| 50 |
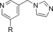
|
230 | 22 |
| 51 |
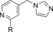
|
310 | 9.7 |
[a] Radioligand displacement from hAT2R in membranes from HEK‐293 cells overexpressing hAT2R (assay 1). N=6 for C38 and 40, N=2 for all other. [b] Inhibition of radioligand binding from hAT1R expressed in HEK‐293 cells (assay 1). [c] Radioligand displacement from AT2R in pig uterus membrane.29 [d] Radioligand displacement from hAT2R expressed in HEK‐293 cells (assay 2). [e] Radioligand displacement from hAT1R expressed in HEK‐293 cells, ligands were not active on hAT1R at the concentrations tested (assay 2). N=9.
As we have reported on previously, affinity to human AT2R expressed in HEK‐293 cells for C38 is reduced as compared to affinity for AT2R in pig uterus membrane (270 nM vs 19 nM).29,35 When evaluating affinity of the previously published, and very promising, amides C93, C97, and C102 (Table 2) for human AT2R in HEK‐293 cells we note a slight decrease in affinity for compound C93 (110 nM in human AT2R vs 29 nM in pig AT2R). For compound C97 the affinity did not change (110 nM in human AT2R vs 83 nM in pig AT2R), but notably for compound C102 the affinity dropped almost 200‐fold (420 nM in human AT2R vs 2.2 nM in pig AT2R). The affinity for human AT2R in HEK‐293 cells for the three tested amides were all in the same range as C38 in the same assay. Similar to the compounds in series 1, the bicyclic derivatives in series 2 (Table 2) also displayed a high selectivity for AT2R. Isoindoline 83 showed a significant reduction in affinity compared to C93 (>1500 nM vs 110 nM), suggesting a highly unfavorable orientation of the amide group. The isoquinoline 84, with similar amide orientation, also displayed reduced affinity. A more favorable amide orientation was obtained in isoindoline 85 and tetrahydroisoquinolines 86 and 87. Compound 85 and 87 displayed similar affinity as C38, while a 5‐fold improvement of affinity was seen for compound 86 compared to C38 (and a 2‐fold improvement compared to amides C93 and C97). Having identified the more favorable orientation of the amide, we explored compounds with a shorter and less lipophilic sulfonamide carbamate chain, as well as a few other moieties binding to the amide nitrogen. Interestingly the sulfonyl ethyl carbamate (88) resulted in a slightly decreased affinity as compared to the sulfonyl butyl carbamate (85).
Table 2.
Bicyclic amides with N‐ethoxycarbonyl or N‐butoxycarbonyl sulfonamide moieties synthesized according to Scheme 2–4.
| Cmpd | Structure | K i hAT2R [nM][a] | %‐Inhibition of hAT1R at 10 μM[b] | Cmpd | Structure | K i hAT2R [nM][a] | %‐Inhibition of hAT1R at 10 μM[b] |
|---|---|---|---|---|---|---|---|
|
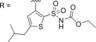
|
||||||
| C93 |
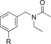
|
110 (29[c]) | 22 % | 88 |
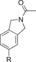
|
870 | 7.7 % |
| C97 |
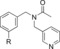
|
110 (83[c]) | 23 % | 89 |
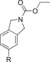
|
1300 | 26 % |
| C102 |
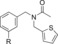
|
420 (2.2[c]) | 30 % | 90 |
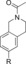
|
120 | NDI[g] |
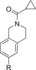
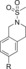
| 83 |
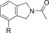
|
>1500[d] | 15 % | 91 |
|
120 | 28 % |
| 84 |
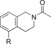
|
700 | 22 % | 92 |
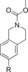
|
290 | 15 % |
| 85 |
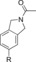
|
360 | 7.5 % | 93 |
|
760 | 19 % |
| 86 |
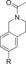
|
56 IC50=818 nM[e] | 34 % IC50 >10 000 nM[f] | 94 |
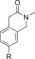
|
1300 | 1.5 % |
| 87 |
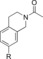
|
270 | 45 % | 95 |
|
850 | 11 % |
| – | – | – | – | 96 |
|
1100 | 15 % |
[a] Radioligand displacement from hAT2R in membranes from HEK‐293 cells overexpressing hAT2R (assay 1). N=2. [b] Inhibition of radioligand binding from hAT1R expressed in HEK‐293 cells (assay 1). [c] Radioligand displacement from AT2R in pig uterus membrane.29 [d] K i estimated to more than 1500 nM, IC50 was determined to be >3 000 nM. [e] Radioligand displacement from hAT2R expressed in HEK‐293 cells (assay 2). [f] Radioligand displacement from hAT1R expressed in HEK‐293 cells, ligands were not active on hAT1R at the concentrations tested (assay 2). N=9 [g] NDI=no detectable inhibition.
The affinity was further decreased when introducing a bicyclic N‐ethyl carbamate in combination with the sulfonamide ethyl carbamate (89), displaying a 20‐fold drop in affinity as compared to compound 86. Comparing compound 90 and compound 86, a slight decrease in affinity could also be detected with the shorter carbamate chain (120 nM vs 56 nM). This indicates the length of the sulfonyl carbamate chain may be more significant in the bicyclic scaffold than in the C38‐scaffold, where our previous studies showed a large tolerability in this moiety.35 The cyclopropane carboxamide 91 had a similar affinity as the amides C93/C97 and the bicyclic amide 90. Interestingly, the bicyclic methyl carbamate 92 was well tolerated in the binding cavity, in contrast to the ethyl carbamate 89 where an almost 5‐fold decrease in affinity was observed. The amide bioisosteric mesyl group was introduced in compound 93, which rendered a reduced affinity to AT2R. Lastly, we had synthesized a three compounds where the carbonyl of the amide function was incorporated as a lactam, locking the amide functionality in a different conformation compared to the previously synthesized bicycles, resulting in the lactam derivatives 94–96. These three compounds all demonstrated a significantly reduced affinity to AT2R.
To ensure quality we assessed three of the compounds and the AT2R agonist C21 in an orthogonal second assay using whole cells, performed in a different laboratory. The IC50 values at both hAT2R and hAT1R were examined for compounds C38, 40, and 86. The AT2R agonist C21 exhibited an IC50 of 1.47 nM at hAT2R in the orthogonal assay, correlating well with data obtained from the standard assay (K i=1.10 nM). Comparing C38 and 40, the IC50 at hAT2R was improved 3‐fold by introducing the para‐fluoro, resulting in an estimated 46‐fold selectivity for hAT2R over hAT1R (cf. 14‐fold hAT1R/hAT2R selectivity for C38). It is notable that C38 and 86 exhibited similar IC50 values in the orthogonal assay (C38; IC50=694 nM and 86; IC50=818 nM, respectively) although the affinities differed considerably in the standard assay applied herein (C38; Ki=270 nM and 86; Ki=56 nM, respectively).
2.3. Molecular Modelling
Using the published crystal structure of AT2R34, a comprehensive docking exploration with GLIDE revealed a common binding pose for the compounds in the first series (Table 1), which in each case was refined by MD equilibration.
Figure 2 depicts the binding mode for the most potent compound (41) in series 1, overlaid with the co‐crystalized AT2R antagonist L‐161,638. The sulfonyl carbamate is anchored via salt‐bridge interactions with R1824.64 and K2155.42 and a hydrogen bond of the carbonyl with T1253.33 (the Ballesteros‐Weinstein generic amino acid numbering scheme is indicated as superscript40). The phenyl ring is surrounded by W1002.60 and L1243.32, allowing the imidazole substituent to be accommodated within a hydrophobic cluster composed by residues Y511.39, Y1032.64, Y1042.65, Y1082.69, P3017.36 and I3047.39. The isobutyl group is placed in a deeper region of the transmembrane cavity, defined by residues L1243.32, M1273.35, W2696.48, F2726.51, and F3087.43. Finally, the ethyl substituent on the sulfonyl carbamate is located in the cavity between transmembrane helices TM3‐TM5 defined by the residues Pro1774.59, Met2145.41, Lys2155.42, and F1293.37.
Figure 2.
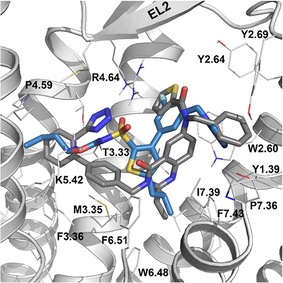
Compound 41 (blue) biding to the AT2 receptor (gray), overlaid with the co‐crystalized ligand L‐161,638 (PDB 5UNG, gray sticks).
The binding mode proposed explains to a big extent the SAR for the compounds in the first series (Figure 3), which are structurally related to the prototype antagonist C38. The experimental affinities (Table 1) show that introduction of a fluoro substituent in ortho position (40) relative to the methylene imidazole substituent is well tolerated, as is introducing a methyl group in the ortho position to the imidazole (43). This is consistent with the modelling of these substituents located in a cavity pointing towards the extracellular side (Figure 3a and Figure 3d). A meta‐fluoro substitution (41) slightly improves the affinity, probably due to favorable electrostatic interactions with Arg1824.64 (Figure 3b).
Figure 3.
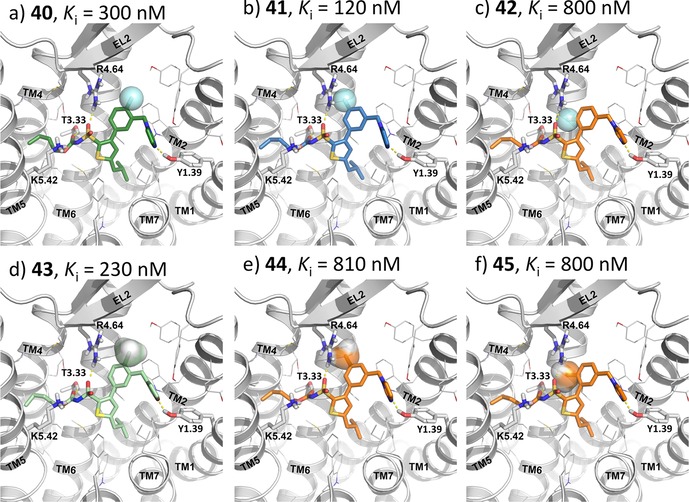
The docked ligands (40–45) in the most common pose on the modeled conformation of the AT2R. The ligands are color coded based on the binding affinities, blue – high, green – moderate, and orange – low binding affinity. The N‐terminal, EL3 and parts of TM6‐TM7 of the AT2R are not shown for better clarity.
Interestingly, compound 42 with a fluoro atom in the para position to the imidazole exhibited a slight (3‐fold) reduction of affinity. The interaction with Arg1824.64 cannot occur for compound 42 (Figure 3c), due to an electrostatic repulsion to the carbonyl of the sulfonyl carbamate. Adding a methyl to the meta or para position relative to the methylene imidazole (44 and 45) also result in a similar reduced affinity as was seen for compound 42, which correlates with a sub‐optimal fitting in the site between Arg1824.64 and Trp1002.61 as indicated in Figure 3e and 3f.
The pharmacological profile of the compounds in the second series has not been assessed due to lack of reliable functional biological models. The related amides reported by our group in 2012 displayed both agonistic and antagonist properties indicating a complex pharmacological relationship for ligands deviating from the imidazole head group29. Hence, tentative binding modes of the ligands in the second series were not studied in the antagonist binding model presented herein.
2.4. Stability in Liver Microsomes and Kinetic Solubility
Table 3 lists compounds that were evaluated for metabolic stability in human and mouse liver microsomes (HLM/MLM). For a selection of compounds the kinetic solubility was also determined. The previously reported AT2R ligands C38 and C93 were also, for comparison, evaluated in the same assays.29 The parent compound C38 exhibited a moderate stability in both human (12 min) and mouse (70 min) liver microsomes. All analogues related to the imidazole derivative C38 displayed a similar trend, with the compounds being more prone to undergo metabolism in human microsomes. Introduction of a fluoro atom onto phenyl rings is a well‐known strategy in medicinal chemistry to block phase I metabolism (oxidation), a potential problem suggested for C38.41,42 Adding a fluoro atom encouragingly displayed a retained affinity for AT2R (40, 41, Table 1). Unfortunately, the metabolic stability in human liver microsomes was not improved for any of the fluorinated compounds (40, 41, and 42). In mouse liver microsomes the metabolic stability was only retained for the ortho fluorinated compound (40) (Table 3). This implies that the phenyl ring is likely not the main site for oxidative metabolism of C38 in human liver microsomes. The solubility of fluoro‐analogue 40 was similar to C38 but was interestingly reduced for compound 41. Methylation of the phenyl ring produced slightly more lipophilic ligands (43‐46), for which the affinity could only be retained for compound 43. The metabolic stability was unsurprisingly reduced for these compounds, as they are likely better substrates for benzylic oxidation than C38. Methyl analogues 43–45 also displayed reduced solubility, likely related to the added lipophilicity.
Table 3.
Compounds evaluated for stability in mouse and human liver microsome assay. The kinetic solubility was also determined for a selection of compounds.
| Cmpd | HLM t [min][a] | MLM t [min][a] | Kinetic solubility [μM][b] |
|---|---|---|---|
| C38 | 12 | 70 | 90 |
| C93 | 15 | 5.8 | 72 |
| 40 | 12 | 88 | 81 |
| 41 | 7.3 | 41 | 52 |
| 42 | 7.8 | 48 | ND[c] |
| 43 | 11 | 39 | 37 |
| 44 | 7.0 | 35 | 24 |
| 45 | 5.2 | 11 | 46 |
| 46 | 5.1 | 99 | 21 |
| 47 | 10 | 103 | 52 |
| 48 | 5.3 | 31 | 32 |
| 49 | 7.6 | 6.5 | 50 |
| 50 | 17 | 18 | 58 |
| 51 | 7.6 | 13 | 44 |
| 83 | 15 | 6.9 | ND[c] |
| 84 | 6.4 | 3.8 | ND[c] |
| 85 | 2.4 | 1.8 | 30 |
| 86 | 8.7 | 2.9 | 45 |
| 87 | 5.4 | 2.2 | 31 |
| 88 | 7.8 | 21 | 35 |
| 89 | 2.3 | 2.1 | 58 |
| 90 | 32 | 14 | 76 |
| 91 | 13 | 7.4 | 47 |
| 92 | 13 | 12 | 60 |
| 93 | 22 | 4.6 | ND[c] |
| 94 | 7.8 | 23 | 61 |
| 96 | 25 | 3.9 | 78 |
[a] The metabolic stability was determined in 0.5 mg/mL human or mouse liver microsomes for compounds at a concentration of 1 μM in potassium phosphate buffer. [b] The kinetic solubility was determined at a final compound concentration of 100 μM in potassium phosphate buffer with 1 % DMSO. [c] ND=Not determined.
The bromo and trifluoromethoxy derivatives 46 and 47 displayed a retained stability profile in both mouse and human liver microsomes (Table 3). For methoxy compound 48 the metabolic stability was reduced. Introducing the bromo, trifluoromethoxy and methoxy resulted in reduced solubility compared to C38. Attempts to improve solubility of C38 by exchanging the phenyl ring for a pyridine gave unsatisfying results. Although exhibiting similar affinity as C38 the solubility was reduced for all three pyridine compounds 49–51. Moreover, the metabolic stability of the pyridines in mouse liver microsome assay was also reduced as compared to C38.
The solubility and stability in human liver microsomes of the amide C93 were similar to C38. Notably, in mouse liver microsomes the metabolic stability of C93 was significantly reduced as compared to C38. All of the bicyclic amides in the series demonstrated very low stability in mouse microsomes, with two exceptions (compound 88 and 94). For example, the bicyclic amides 85, 86, 87, and 89 rapidly decompose and are similarly unstable in human microsomes. The solubility was also reduced compared to the non‐cyclized amide C93. Neither the bicyclic acetyl amide 83 nor 84, regioisomers of 85 and 86, respectively, demonstrated any improved metabolic profile. Comparing the butyloxycarbonyl sulfonamide 85, one of the most metabolically unstable compounds in this report, with the corresponding ethoxycarbonyl compound 88 reveals only a slightly improved stability in mouse liver microsomes for the latter. A larger increase was expected in accordance with previously reported data.35 A larger impact on stability in human microsomes with a shortened carbamate alkyl chain can be noted when comparing 86 and with 90. The very low stability of the bicyclic amide 86 is unfortunate as compound 86 exhibits the highest affinity of all compounds assessed. Exchanging the acetyl of compound 90 gave the equipotent acetyl propyl 91 and carbamate 92, neither of which showed any improved solubility or metabolic stability. The bioisosteric sulfonamide 93 exhibit a metabolic stability of more than 20 min in human microsomes. The same was seen for compound 96, however both 93 and 96 are poor AT2R binders. The solubility was only comparable to C93 for compounds 90 and 96.
3. Conclusion
In summary, two series of new AT2R ligands were synthesized and evaluated. In the first series, small structural changes were introduced to the central phenyl ring of the known AT2R antagonist C38. These were well‐tolerated with half of the compounds synthesized exhibiting similar or slightly improved affinity to AT2R compared to C38. A common binding pose was identified for the compounds in the first series, a pose that could ascribe the reduced affinity for 3 out of 4 low‐affinity compounds to a sub‐optimal fit between Arg182 on helix TM4 and Trp100 on helix TM2. The highest affinity in the first series was displayed by the meta fluoro derivative 41 and the ortho bromo substituted derivative 46, of which only the latter displayed retained metabolic stability in mouse liver microsomes. In the second series where the imidazole heterocycle of C38 was replaced by bicyclic amides, the most favorable amide orientation was identified and explored. Compound 86 displayed the highest affinity to AT2R of all compounds assessed (K i=56 nM at AT2R). Unfortunately, all compounds in the second series exhibited a low metabolic stability both in human and mouse liver microsomes. The stability could be slightly improved by reducing the sulfonyl carbamate chain length (cf. compound 85 vs 88, and compound 86 vs 90).
Experimental Section
General Chemistry
All chemicals and solvents were purchased from Sigma Aldrich, Fisher Scientific, FluoroChem, and Enamine, and were used without further purification. Microwave heating was performed in a Biotage Initiator+ single‐mode microwave reactor. Automated flash column chromatography was performed on Biotage Isolera or Grace Reveleris instruments using commercial silica cartridges. Manual flash chromatography was performed on silica gel 60. Preparative reverse‐phase HPLC was performed using a C18 column with UV detection. Analytical HPLC/ESI‐MS was performed using electrospray ionization (ESI) and a C18 column. High resolution molecular masses (HRMS) were determined on a mass spectrometer equipped with an ESI source and 7‐T hybrid linear ion trap (LTQ). NMR spectra were recorded at 400 MHz for 1H and 101 MHz for 13C.
Synthesis
Butyl ((3‐(2‐acetyl‐1,2,3,4‐tetrahydroisoquinolin‐6‐yl)‐5‐isobutylthiophen‐2‐yl)sulfonyl)carbamate (86)
A 20 mL vial containing ZnCl2 (1.7 eq.) was dried in a vacuum oven at 120 °C overnight. The vial was capped and evacuated twice with vacuum/N2(g). After cooling to room temperature the ZnCl2 was dissolved in dry THF (5 mL). Isobutylmagnesium chloride (2 M in THF; 1.5 eq.) was added dropwise. After 10 min of stirring, a solution of 5‐bromo‐N‐(tert‐butyl)thiophene‐2‐sulfonamide (1, 7.3 mmol, 1.0 eq.) and Pd(t‐Bu3P)2 (0.015 eq.) in dry toluene (5 mL) was added. The mixture was microwave heated at 130 °C for 15 min after which it was partitioned between DCM and sat. aq. NH4Cl (3 : 2). The aqueous layer was extracted with DCM and the combined organic layers were washed with brine, dried with MgSO4 and concentrated under reduced pressure. The remaining residue was purified using silica gel flash chromatography (isohexanes with 10 % (v/v) EtOAc). N‐(tert‐Butyl)‐5‐isobutylthiophene‐2‐sulfonamide (2) was isolated in 51 % yield.35,43
N‐(tert‐Butyl)‐5‐isobutylthiophene‐2‐sulfonamide (2, 8.2 mmol, 1.0 eq.) was dissolved in dry THF (70 mL) and transferred to a dry three‐necked round bottom flask. The flask was cooled to −78 °C and evacuated thrice with vacuum/N2(g). To this was added n‐butyllithium (2.5 M in hexane; 4.5 eq.) dropwise after which the mixture was stirred for 1 h at −78 °C. The reaction was subsequently stirred at 0 °C for 1 h, after which it was again cooled to −78 °C and triisopropyl borate (2.5 eq.) was added. After 15 min the flask was again stirred at 0 °C for 3 h. The mixture was quenched with 2 M HCl (aq.) and partially evaporated before it was diluted with water and the product was extracted with DCM. The combined organic layers were dried with MgSO4 and the solvent was removed under reduced pressure. The crude residue was dissolved in DMSO (2 mL) and toluene (30 mL), methyliminodiacetic acid (1.3 eq.) was added and the mixture was refluxed for 3 h. The mixture was diluted with EtOAc and washed with 0.1 M HCl (aq.). The organic phase was dried with MgSO4 and the solvent was removed under reduced pressure. The residue obtained was dissolved in minimal amounts of acetone and equal amounts of diethyl ether after which hexane (100 mL) was added using a dropping funnel. The solid formed was filtered off and submitted to the same precipitation procedure once more. N‐(tert‐Butyl)‐5‐isobutyl‐3‐(6‐methyl‐4,8‐dioxo‐1,3,6,2‐dioxazaborocan‐2‐yl)thiophene‐2‐sulfonamide (3) was collected in 76 % yield.35
6‐Bromo‐1,2,3,4‐tetrahydroisoquinoline (55, 0.44 mmol, 1.0 eq.) and K2CO3 (2.3 eq.) were dissolved in MeCN (4 mL). Acetic anhydride (1.4 eq.) was added and the mixture was stirred overnight. The solvent was removed under reduced pressure and the product was purified by silica gel column chromatography (DCM with 5 % (v/v) MeOH). 1‐(6‐Bromo‐3,4‐dihydroisoquinolin‐2(1H)‐yl)ethan‐1‐one (60) was isolated in 97 % yield as 2 amide rotamers in 60 : 40 ratio at room temperature. 1H NMR (400 MHz, DMSO‐d 6, T=373 K) δ 7.45–7.28 (m, 2H), 7.21–7.09 (m, 1H), 4.57 (s, 2H), 3.65 (t, J=6.0 Hz, 2H), 2.84 (m, 2H), 2.07 (s, 3H). 13C NMR (101 MHz, DMSO‐d 6, T=353 K) δ 168.2, 130.5, 130.2, 130.1, 128.6, 128.0, 118.8, 42.7, 38.9, 28.9, 20.8.
MIDA‐boronate (3, 0.20 mmol, 1.0 eq.), K2CO3 (5.0 eq.), 1‐(6‐bromo‐3,4‐dihydroisoquinolin‐2(1H)‐yl)ethan‐1‐one (60) (1.0 eq.), and PdCl2(dppf) (0.05 eq.) were dissolved in DME (1 mL) and water (0.2 mL) in a 2–5 mL vial. The vial was flushed with N2 and the mixture was heated at 120 °C for 1 h. The reaction mixture was diluted with EtOAc and the layers were separated. The organic layer was purified by automated silica flash chromatography (isohexane with 50–100 % (v/v) EtOAc). The fractions containing product were collected and the solvent removed under reduced pressure. The crude 3‐(2‐acetyl‐1,2,3,4‐tetrahydroisoquinolin‐6‐yl)‐N‐(tert‐butyl)‐5‐isobutylthiophene‐2‐sulfonamide (74) was stirred in TFA (99.9 %; 65 eq.) at 40 °C overnight. The TFA was removed and the remaining crude material was dissolved in DCM (2 mL). To this was added triethylamine (2.1 eq.) and the mixture was stirred for 10 min at room temperature, after which butyl chloroformate (0.7 eq.) was added and the mixture was stirred at room temperature for 1 h. The reaction mixture was washed with 2 M HCl (aq.) and brine and dried with MgSO4. The solvent was evaporated and the product was purified by preparative RP‐HPLC (20‐100 % MeCN in water (0.05 % formic acid)). Butyl ((3‐(2‐acetyl‐1,2,3,4‐tetrahydroisoquinolin‐6‐yl)‐5‐isobutylthiophen‐2‐yl)sulfonyl)carbamate (86) was obtained in 28 % yield over 2 steps as a mixture of 2 amide rotamers in 60 : 40 ratio. 1H NMR (400 MHz, Chloroform‐d) δ 7.93 (overlapping; s, 2H), 7.35–7.28 (overlapping; m, 2H), 7.26–7.22 (overlapping; m, 2H), 7.19–7.12 (overlapping; m, 2H), 6.743 (minor; s, 1H), 6.737 (major; s, 1H), 4.74 (major; s, 2H), 4.65 (minor; s, 2H), 4.07 (minor; t, J=6.6, 2H), 4.06 (major; t, J=6.6, 2H), 3.81 (minor; t, J=5.9 Hz, 2H), 3.69 (major; t, J=5.9 Hz, 3H), 2.93 (major; t, J=5.9 Hz, 3H), 2.85 (minor; t, J=6.0 Hz, 2H), 2.70 (d, J=7.1 Hz, 4H), 2.18 (minor; s, 3H), 2.17 (major; s, 3H), 1.93 (overlapping; m, 2H), 1.52 (overlapping; m, 4H), 1.27 (overlapping; m, 4H), 0.99 (overlapping; d, J=6.6 Hz, 12H), 0.89 (overlapping; t, J=7.4 Hz, 6H). 13C NMR (101 MHz, Chloroform‐d) δ 169.8, 169.7, 151.7, 151.6, 150.4, 146.4, 146.3, 135.2, 134.2, 134.1, 133.2, 132.9, 132.6, 130.9, 129.64, 129.60, 129.5, 129.1, 127.3, 127.2, 126.7, 126.2, 67.0, 66.9, 48.1, 44.1, 44.0, 39.5, 30.7, 30.6, 29.5, 28.6, 22.4, 22.0, 21.7, 18.9, 13.7. MS (ESI): m/z calc'd for C24H32N2O5S2: 491.1674 [M−H]−; found: 491.1664
Further details on reaction conditions is available for all reactions in the supporting information. 1H NMR spectra were generated for all final compounds. Purity and elemental analyses were performed on all final compounds. 13C spectra were generated for a majority of the final compounds. All available spectral analysis is reported in the supplementary information.
Binding Assays
Assay 1 (K i Determination)
All synthesized ligands were evaluated in a radioligand assay by displacing [125I][Sar1Ile8]‐angiotensin II from human AT2R in HEK‐293 cells membrane preparations. [Sar1Ile8]‐angiotensin II (Sarile) acts as a nonselective AT2R agonist.44 The affinity was determined using a seven‐point dose‐response curve, each point performed in duplicates. All dose‐response curves are available in the Supplementary Information. Each new assay was validated using a selection of known ligands in accordance with Eurofins Cerep standard protocol. The compounds were also evaluated for inhibition of [125I][Sar1Ile8]‐angiotensin II binding to human AT1R in HEK‐293 cell membranes. For AT1R the percent inhibition was determined at 10 μM, in duplicates, with the endogenous ligand (angiotensin II) used as reference.
Assay 2 (IC50 Determination)
The IC50 values of C38, 40, 86 and C21 were assessed in whole cells assay using HEK293 cells expressing AT1R or AT2R as described previously45, 46, 47. Cells were grown to approximately 80 % confluence before being re‐plated into 48 well plates at 1×105 cells/well and grown for 48 h at 37 °C for a whole cell competition binding assay. [125I]‐Sar1Ile8Ang II at 50,000 cpm, incubated for 45 min at 37 °C, in the absence or presence of unlabeled ligands, prepared in binding buffer (DMEM, 0.1 % BSA), were used in the competition assays at concentrations ranging from 1 pM to 10 μM. For each experiment, each ligand concentration was tested in triplicate, and each experiment was repeated at least 3 separate times. Non‐specific binding (NSB) was defined in the presence of the unlabeled Ang II (10 μM). The ability of each ligand to inhibit specific binding of [125I]‐Sar1Ile8Ang II was measured on a gamma counter with all counts corrected for NSB. Non‐linear regression of the data using one‐site fit model was performed and IC50 values, representing the concentration at which each ligand displaced 50 % of [125I]‐Sar1Ile8Ang II binding, were calculated as affinity estimates for each ligand at AT1R and AT2R, using GraphPad Prism 6 (GraphPad Software Inc., San Diego, CA, USA).
Kinetic Solubility
The kinetic solubility was investigated for compounds 40–41, 43, 46–48, 50–51, 85–92, 94, 96. Kinetic solubility was measured at a final compound concentration of 100 μM and 1 % DMSO in 100 mM potassium phosphate buffer (pH 7.4) and incubated at 37 °C for at least 20 h. After incubation, the samples are centrifuged at 3000xg at 37 °C for 30 min to pellet insoluble material and an aliquot of the supernatant was taken for quantification of compound concentration by LC‐MS/MS analysis. The LC‐MS/MS system was an Acquity UPLC coupled to a triple quadrupole mass spectrometer (Waters), operating in multiple reaction monitoring (MRM) mode with positive or negative electrospray ionization. Mass spectrometric settings were optimized for each compound for one MRM transition. Chromatographic separation was typically done on a C18 Ethylene Bridged Hybrid (BEH) 1.7 μm column using a general gradient of 1 % to 90 % of mobile phase consisting of A, 5 % acetonitrile and 0.1 % formic acid in purified water, and B, 0.1 % formic acid in 100 % acetonitrile, over a total running time of 2 min. In a few cases, separation was done on a HSS T3 2×50 mm 2.1 μm column using a mobile phase consisting of A, 0.05 % heptafluorobutyric acid (HFBA) and 0.05 % propionic acid (PA) in water, and B, 0.05 % HFBA and 0.05 % PA in acetonitrile, with a total running time of 2 min. In both cases, the flow rate was set to 0.5 mL/min and 5 μL of the sample was injected.
Stability in Liver Microsomes
Human and mouse liver microsomes were used to assess the metabolic stability for all compounds (95 excluded). Metabolic stability was determined in 0.5 mg/mL human or mouse liver microsomes at a compound concentration of 1 μM in 100 mM potassium phosphate buffer (pH 7.4) in a total incubation volume of 500 μL. The reaction was initiated by addition of 1 mM NADPH. At various incubation times, i. e. at 0, 5, 10, 20, 40 and 60 min, a sample was withdrawn from the incubation and the reaction was terminated by addition of ice‐cold acetonitrile containing Warfarin as internal standard. The amount of parent compound remaining was analyzed by LC‐MS/MS as described above (Kinetic Solubility). In vitro half‐life (t1/2) and in vitro intrinsic clearance (Clint) were calculated using previously published models.48,49 Extraction ratio (E), i. e. the ratio of the hepatic clearance of a drug to the hepatic blood flow, can be generally classified as high (>0.7), intermediate (0.3–0.7) or low (<0.3), according to the fraction of drug removed during one pass through the liver. For human and mouse liver microsomes, E of 0.3 and 0.7 would correspond to a t1/2 of 126 min and 23 min, and 193 min and 35 min, respectively.
Molecular Modelling of the AT2 Receptor
The crystal structure of the human AT2R was retrieved from the Protein Data Bank (PDB code 5UNG with antagonist L‐161,638)31,34 and was subject to preparation and minor modifications with the Schrödinger suite (Schrödinger Release 2017–3, Schrödinger, LSS, New York, NY, 2017), including (i) deletion of the engineered B562RIL protein (fused to the truncated N‐terminus); (ii) addition of protons, assessment of the rotamers for Asn/Gln/His residues, and protonated state for titratable residues, resulting in all Asp, Gln, Lys, and Arg residues assigned to their default charged state and all His modelled as neutral with the proton on Nδ; (iii) addition of missing side chains, modelling the most probable conformer based on additional crystal structures of AT2 and the related AT1 receptor.
Ligand Docking
Ligands from Tables 1 were built and optimized their 3D conformation using the Maestro graphical interface and the LigPrep utility from the Schrödinger suite (Schrödinger Release 2017–3: Maestro, Schrödinger, LSS, New York, NY, 2017; Schrödinger Release 2017‐3: LigPrep, Schrödinger, LSS, New York, NY, 2017). This method also allowed determination of their most probable protonation state at physiological pH, with a net negative change localized on the sulfonylcarbamate group in all cases. Docking was performed with Glide SP using default settings (Schrödinger Release 2017–3: Glide, Schrödinger, LSS, New York, NY, 2017).50, 51, 52 The docking grid was placed taking as reference the coordinates of the co‐crystallized ligand L‐161,638, and expanding the cubic grid box to 30 Å on each dimensions. The selection of poses was done on the basis of a double criteria, combining the highest possible scoring while looking for the consensus among all ligands in the series.
Membrane Insertion and Molecular Dynamics Equilibration
Each ligand‐receptor complex obtained in the previous stage was subject to an MD equilibration following the PyMedDyn protocol, as implemented in a GPCR‐ModSim web server.53,54 Briefly, the receptor‐ligand complex was inserted in a pre‐equilibrated membrane consisting of 1‐palmitoyl‐2‐oleoyl phosphatidylcholine (POPC) lipids, with the transmembrane (TM) bundle aligned to its vertical axis. The simulation box was created with a hexagonal‐prism geometry, which was soaked with bulk water and energy‐minimized using the OPLS‐AA force field for proteins and ligands, combined with the Berger parameters for the lipids.53,55, 56, 57 It follows a molecular dynamics equilibration using periodic boundary conditions (PBC) and the NPT ensemble with the GROMACS simulation package.55
The first phase consists of 2.5 ns with a gradual release of harmonic restraints on protein (and ligand) heavy atoms. The second phase consists of free MD for another 2.5 ns, except for weak distance restraints between 24 pairs of interacting residues corresponding to conserved positions within the TM bundle of class‐A GPCRs with a structural role.54,58 The final snapshot was energy minimized and retained for analysis and figures.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the SciLifeLab Drug Discovery and Development Platform for support with compound synthesis and ADME evaluations, the Swedish National Infrastructure for Computing (SNIC) for synthetic and computational resources, the Kjell and Märta Beijer Foundation and the Swedish Research Council for financial support.
R. Isaksson, J. Lindman, J. Wannberg, J. Sallander, M. Backlund, D. Baraldi, R. Widdop, M. Hallberg, J. Åqvist, H. Gutierrez de Teran, J. Gising, M. Larhed, ChemistryOpen 2019, 8, 114.
References
- 1. Hallberg M., Med. Res. Rev. 2015, 35, 464–519. [DOI] [PubMed] [Google Scholar]
- 2. Whitebread S., Mele M., Kamber B., de Gasparo M., Biochem. Biophys. Res. Commun. 1989, 163, 284–291. [DOI] [PubMed] [Google Scholar]
- 3. Chiu A. T., Herblin W. F., McCall D. E., Ardecky R. J., Carini D. J., Duncia J. V., Pease L. J., Wong P. C., Wexler R. R., Johnson A. L., Biochem. Biophys. Res. Commun. 1989, 165, 196–203. [DOI] [PubMed] [Google Scholar]
- 4. Padia S. H., Carey R. M., Pflügers Arch. – Eur. J. Physiol. 2013, 465, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Foulquier S., Steckelings U. M., Unger T., Nature 2013, 493, S9. [DOI] [PubMed] [Google Scholar]
- 6. Unger T., Steckelings U. M., dos Santos R. A. S., The Protective Arm of the Renin Angiotensin System, Elsevier, 2015. [Google Scholar]
- 7. Grady E. F., Sechi L. A., Griffin C. A., Schambelan M., Kalinyak J. E., J. Clin. Invest. 1991, 88, 921–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bastien N. R., Ciuffo G. M., Saavedra J. M., Lambert C., Regul. Pept. 1996, 63, 9–16. [DOI] [PubMed] [Google Scholar]
- 9. Gasparo M. De, Catt K. J., Inagami T., Wright J. W., Unger T., Pharmacol. Rev. 2000, 52, 415–472. [PubMed] [Google Scholar]
- 10.“AGT2R,” can be found under https://www.proteinatlas.org/ENSG00000180772-AGTR2/tissue, 2018.
- 11. Steckelings U. M., Rompe F., Kaschina E., Namsolleck P., Grzesiak A., Funke-Kaiser H., Bader M., Unger T., J. Renin. Angiotensin. Aldosterone. Syst. 2010, 11, 67–73. [DOI] [PubMed] [Google Scholar]
- 12. Sumners C., Kloet A. D. De, Krause E. G., Unger T., Steckelings U. M., Curr. Opin. Pharmacol. 2015, 21, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nakajima M., Hutchinson H. G., Fujinaga M., Hayashida W., Morishita R., Zhang L., Horiuchi M., Pratf R. E., Dzau V. J., Berliner R. W., Proc. Mont. Acad. Sci. 1995, 92, 10663–10667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gallinat S., Yu M., Dorst A., Unger T., Herdegen T., Mol. Brain Res. 1998, 57, 111–122. [DOI] [PubMed] [Google Scholar]
- 15. Altarche-Xifro W., Curato C., Kaschina E., Grzesiak A., Slavic S., Dong J., Kappert K., Steckelings M., Imboden H., Unger T., Stem Cells 2009, 27, 2488–2497. [DOI] [PubMed] [Google Scholar]
- 16. Busche S., Gallinat S., Bohle R.-M., Reinecke A., Seebeck J. Rg, Franke F., Fink L., Zhu M., Sumners C., Unger T., Am. J. Pathol. 2000, 157, 605–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nio Y., Matsubara H., Murasawa S., Kanasaki M., Inada M., Clin. Invest. 1995, 95, 46–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li J., Culman J., Hörtnagl H., Zhao Y., Gerova N., Timm M., Blume A., Zimmermann M., Seidel K., Dirnagl U., FASEB J. 2005, 16, 617–619. [DOI] [PubMed] [Google Scholar]
- 19. Smith M. T., Wyse B. D., Edwards S. R., Pain Med. 2013, 14, 692–705. [DOI] [PubMed] [Google Scholar]
- 20. Rice A. S. C., Dworkin R. H., McCarthy T. D., Anand P., Bountra C., McCloud P. I., Hill J., Cutter G., Kitson G., Desem N., Lancet 2014, 383, 1637–1647. [DOI] [PubMed] [Google Scholar]
- 21. Wan Y., Wallinder C., Plouffe B., Beaudry H., Mahalingam A. K., Wu X., Johansson B., Holm M., Botros M., Karlén A., J. Med. Chem. 2004, 47, 5995–6008. [DOI] [PubMed] [Google Scholar]
- 22. Larhed M., Hallberg M., Hallberg A., Med. Chem. Rev. 2016, 51, 69–82. [Google Scholar]
- 23. Hallberg M., Sumners C., Steckelings U. M., Hallberg A., Med. Res. Rev. 2018, 38, 602–624. [DOI] [PubMed] [Google Scholar]
- 24. Wu X., Wan Y., Mahalingam a K., Murugaiah a M. S., Plouffe B., Botros M., Karlén A., Hallberg M., Gallo-Payet N., Alterman M., J. Med. Chem. 2006, 49, 7160–8. [DOI] [PubMed] [Google Scholar]
- 25. Murugaiah A. M. S., Wallinder C., Mahalingam A. K., Wu X., Wan Y., Plouffe B., Botros M., Karlén A., Hallberg M., Gallo-Payet N., Bioorg. Med. Chem. 2007, 15, 7166–83. [DOI] [PubMed] [Google Scholar]
- 26. Georgsson J., Sköld C., Botros M., Lindeberg G., Nyberg F., Karlén A., Hallberg A., Larhed M., J. Med. Chem. 2007, 50, 1711–1715. [DOI] [PubMed] [Google Scholar]
- 27. Wallinder C., Botros M., Rosenström U., Guimond M.-O., Beaudry H., Nyberg F., Gallo-Payet N., Hallberg A., Alterman M., Bioorg. Med. Chem. 2008, 16, 6841–9. [DOI] [PubMed] [Google Scholar]
- 28. Mahalingam A. K., Wan Y., Murugaiah A. M. S., Wallinder C., Wu X., Plouffe B., Botros M., Nyberg F., Hallberg A., Gallo-Payet N., Bioorg. Med. Chem. 2010, 18, 4570–90. [DOI] [PubMed] [Google Scholar]
- 29. Murugaiah A. M. S., Wu X., Wallinder C., Mahalingam A. K., Wan Y., Sköld C., Botros M., Guimond M.-O., Joshi A., Nyberg F., J. Med. Chem. 2012, 55, 2265–78. [DOI] [PubMed] [Google Scholar]
- 30. Guimond M.-O., Wallinder C., Alterman M., Hallberg A., Gallo-Payet N., Eur. J. Pharmacol. 2013, 699, 160–171. [DOI] [PubMed] [Google Scholar]
- 31. Zhang H., Unal H., Gati C., Han G. W., Liu W., Zatsepin N. A., James D., Wang D., Nelson G., Weierstall U., Cell 2015, 161, 833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang H., Unal H., Desnoyer R., Han G. Won, Patel N., Katritch V., Karnik S. S., Cherezov V., Stevens R. C., J. Biol. Chem. 2015, 290, 29127–29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sallander J., Wallinder C., Hallberg A., Åqvist J., Gutiérrez-De-Terán H., Bioorg. Med. Chem. Lett. 2016, 26, 1355–1359. [DOI] [PubMed] [Google Scholar]
- 34. Zhang H., Han G. W., Batyuk A., Ishchenko A., White K. L., Patel N., Sadybekov A., Zamlynny B., Rudd M. T., Hollenstein K., Nature 2017, 544, 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wannberg J., Isaksson R., Bremberg U., Backlund M., Sävmarker J., Hallberg M., Larhed M., Bioorg. Med. Chem. Lett. 2018, 28, 519–522. [DOI] [PubMed] [Google Scholar]
- 36. Nöteberg D., Schaal W., Hamelink E., Vrang L., Larhed M., J. Comb. Chem. 2003, 5, 456–464. [DOI] [PubMed] [Google Scholar]
- 37. Larhed M., Hallberg A., Drug Discovery Today 2001, 6, 406–416. [DOI] [PubMed] [Google Scholar]
- 38. Ulysse L. G., Yang Q., Mclaws M. D., Keefe D. K., Guzzo P. R., Haney B. P., Org. Process Res. Dev. 2010, 14, 225–228. [Google Scholar]
- 39. Eaton P. E., Carlson G. R., Lee J. T., J. Org. Chem. 1973, 38, 4071–4073. [Google Scholar]
- 40. Ballesteros J. A., Weinstein H., in Methods Neurosci. Academic Press, 1995, pp. 366–428. [Google Scholar]
- 41. Park B. K., Kitteringham N. R., O'neill P. M., Annu. Rev. Pharmacol. Toxicol. 2001, 41, 443–70. [DOI] [PubMed] [Google Scholar]
- 42. Müller K., Faeh C., Diederich F., Science 2007, 317, 1881–6. [DOI] [PubMed] [Google Scholar]
- 43. Kevin N. J., Rivero R. A., Greenlee W. J., Chang R. S. L., Chen T. B., Bioorg. Med. Chem. Lett. 1994, 4, 189–194. [Google Scholar]
- 44. Guimond M.-O., Hallberg M., Gallo-Payet N., Wallinder C., ACS Med. Chem. Lett. 2014, 5, 1129–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jones E. S., Del Borgo M. P., Kirsch J. F., Clayton D., Bosnyak S., Welungoda I., Hausler N., Unabia S., Perlmutter P., Thomas W. G., Hypertension 2011, 57, 570–576. [DOI] [PubMed] [Google Scholar]
- 46. Bosnyak S., Jones E. S., Christopoulos A., Aguilar M.-I., Thomas W. G., Widdop R. E., Clin. Sci. 2011, 121, 297–303. [DOI] [PubMed] [Google Scholar]
- 47. Borgo M. Del, Wang Y., Bosnyak S., Khan M., Walters P., Spizzo I., Perlmutter P., Hilliard L., Denton K., Aguilar M.-I., Clin. Sci. 2015, 129, 505–513. [DOI] [PubMed] [Google Scholar]
- 48. Houston J. B., Biochem. Pharmacol. 1994, 47, 1469–1479. [DOI] [PubMed] [Google Scholar]
- 49. Obach R. S., Drug Metab. Dispos. 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- 50. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 51. Halgren T. A., Murphy R. B., Friesner R. A., Beard H. S., Frye L. L., Pollard W. T., Banks J. L., J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- 52. Friesner R. A., Murphy R. B., Repasky M. P., Frye L. L., Greenwood J. R., Halgren T. A., Sanschagrin P. C., Mainz D. T., J. Med. Chem. 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- 53. Gutiérrez-de-Terán H., Bello X., Rodríguez D., Biochem. Soc. Trans. 2013, 41, 205–212. [DOI] [PubMed] [Google Scholar]
- 54. Esguerra M., Siretskiy A., Bello X., Sallander J., Gutiérrez-de-Terán H., Nucleic Acids Res. 2016, 44, W455–W462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hess B., Kutzner C., Spoel D. Van Der, Lindahl E., J. Chem. Theory Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- 56. Kaminski G. A., Friesner R. A., Tirado-Rives J., Jorgensen W. L., J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar]
- 57. Berger O., Edholm O., Jähnig F., Biophys. J. 1997, 72, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. Madan, Nature 2013, 494, 185–194. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary