

Abstract
Non-Hodgkin lymphoma is one of the most common types of cancer. Relapsed and refractory diseases are still common and remain significant challenges as the majority of these patients eventually succumb to the disease. Herein, we report a translatable concurrent chemo-immuno-radiotherapy (CIRT) strategy that utilizes fully synthetic antibody mimic Selective High-Affinity Ligand (SHAL)-functionalized doxorubicin-encapsulated nanoparticles (Dox NPs) for the treatment of human leukocyte antigen-D related (HLA-DR) antigen-overexpressed tumors. We demonstrated that our tailor-made antibody mimic-functionalized NPs bound selectively to different HLA-DR-overexpressed human lymphoma cells, cross-linked the cell surface HLA-DR, and triggered the internalization of NPs. In addition to the direct cytotoxic effect by Dox, the internalized NPs then released the encapsulated Dox and upregulated the HLA-DR expression of the surviving cells, which further augmented immunogenic cell death (ICD). The released Dox not only promotes ICD but also sensitizes the cancer cells to irradiation by inducing cell cycle arrest and preventing the repair of DNA damage. In vivo biodistribution and toxicity studies confirm that the targeted NPs enhanced tumor uptake and reduced systemic toxicities of Dox. Our comprehensive in vivo anticancer efficacy studies using lymphoma xenograft tumor models show that the antibody-mimic functional NPs effectively inhibit tumor growth and sensitize the cancer cells for concurrent CIRT treatment without incurring significant side effects. With an appropriate treatment schedule, the SHAL-functionalized Dox NPs enhanced the cell killing efficiency of radiotherapy by more than 100% and eradicated more than 80% of the lymphoma tumors.
Short abstract
Antibody mimic Selective High-Affinity Ligand-functionalized doxorubicin-encapsulated nanoparticles have been engineered for concurrent chemo-immuno-radiotherapy of hematological cancer.
Introduction
The incidence of hematologic malignancies continues to rise in the United States (US).1 About half of the new cases (75 000 people) can be classified as non-Hodgkin Lymphoma (NHL)1 based on the presence of malignant lymphocytes. Approximately 1 in 46 people in the US will develop NHL during their lifetime.1 Unlike the cells in solid tumors, malignant lymphocytes commonly travel throughout the body to form systemic tumors. Standard NHL treatments include cytotoxic chemotherapy, immunotherapy, and radiotherapy (XRT).2−6 Clinical trials have demonstrated that concurrent chemo-radiotherapy (CRT) can be more effective than cytotoxic chemotherapy alone or sequential CRT in the treatment of some lymphoma types.7−9 However, concurrent CRT and concurrent chemo-immuno-radiotherapy (CIRT, the concurrent administration of chemotherapeutics, antibody-based immunotherapy, and XRT) also increases the side effects associated with different treatment modalities.8 In recent years, there has been growing interest in utilizing bio-nanotechnology to improve the therapeutic window by increasing therapeutic efficacy and reducing the side effects of traditional treatments.10,11 Liposomal doxorubicin (Dox) is the first in class to utilize nanoparticle formulation in delivering chemotherapeutic agents. However, liposomal Dox has been associated with only marginal improvement in efficacy and safety profiles compared to free Dox mainly because it lacks active targeting moieties to the tumor cell,12 and improvement of targeted drug delivery by conjugating tumor-specific ligands to the nanoparticle could potentially transform this approach. The next generation of nanoparticle-based drugs conjugated with tumor-specific ligands, such as antibodies, are currently being evaluated in various phases of clinical trials.
Cancer cell death can be triggered through various mechanisms. Immunogenic cell death (ICD) is a type of cancer cell death in which the dying cancer cells release antigens and trigger antigen-specific immune responses.13,14 The dying cancer cells often upregulate the expression of tumor-associated antigens. ICD can be induced by cytostatic ICD-inducing agents, such as Dox, or with radiation.13−18 Most recent studies in immuno-oncology have focused on the combination of the ICD effect and immune-checkpoint blockage for cancer immunotherapy.13,14 To the best of our knowledge, there have not been any published studies to investigate how ICD relates to the biodistribution and anticancer efficacy of targeted drug carriers and antibody-drug conjugates.
The rapid development of computational biology has facilitated the development of antibody mimics for new biomedical applications.19−22 These mimics can be broadly divided into protein-based antibody mimics (also known as non-antibody binding proteins, e.g., affirmers and affibodies) and fully synthetic antibody mimics.19 Fully synthetic antibody mimics are particularly attractive for biomedical applications because their structure can be tailored to increase binding affinities and selectivity, reduce immunogenicity, and lower the cost of producing antibodies.19 However, the absence of the Fc component in antibody mimics has hindered their therapeutic applications because many important cell killing mechanisms (e.g., the complement-dependent cytotoxicity and antibody-dependent cellular cytotoxicity mechanisms) require the Fc component to activate other immune cells (e.g., T cells).22 Selective High-Affinity Ligands (SHALs) belong to a family of fully synthetic antibody mimics that are designed in silico to bind to the Lym-1 epitope in the β-subunit of the human leukocyte antigen-D related (HLA-DR) antigen (Figures 1A and S1).21,23−26 This epitope is present in HLA-DR10 and some other closely related HLA-DRs27 that have been observed to be expressed by a subset of B-cell derived lymphomas, myelomas, and other cancers.28,29 Unlike traditional peptide-based fully synthetic antibody mimics, a SHAL is composed of two or three recognition ligands, each with low-moderate binding affinities to neighboring sites on the surface of the target protein, linked together through flexible linkers to allow specific, high-affinity binding with nanomolar to picomolar dissociation constants in vitro.21,23−26 Histopathological studies demonstrated that SHALs could be used to label HLA-DRs-overexpressed on human lymphoma and other cancer tissues.21,30 SHALs also showed very low in vivo toxicity in animal models.31,32 Preclinical and early phase studies using positron emission tomography (PET) confirmed that radionuclide-labeled SHAL could be used to detect HLA-DR-overexpressed lymphoma.33 Thus, SHALs may potentially overcome many challenges associated with chimeric HLA-DR antibodies for the treatment of hematologic and other malignancies.34
Figure 1.

Physicochemical properties of SHAL-functionalized Dox-encapsulated PEG-PLGA NPs. (A) Schematic of the binding of SHAL to the Lym-1 binding pocket in the β-subunit of HLA-DR antigen-overexpressed cells. The inset shows the chemical structure of the tridentate-based SHAL (SH7133) used in this study. SH7133 is composed of three recognition ligands that bind to neighboring sites on the surface of the target HLA-DR (highlighted in blue): 3-(2-([3-chloro-5-trifluoromethyl)-2-pyridinyl]oxy)-aniline-3-oxopropanionic acid) (Ct ligand), 4-[4-(4-chlorobenzyl)piperazino]-3-nitrobenzenecarboxylic acid (Cb ligand), and dansyl-l-valine (Dv ligand). (B) Schematic of the structure of SHAL-functionalized Dox NPs. (C) Representative TEM image of SHAL-functionalized Dox NPs. The mean number-average diameter (Dn) was found to be about 50 nm. (D) The plot of the intensity-average diameter (Dh) of SHAL-functionalized Dox NPs, as determined using the DLS method. The mean Dh of the Dox-encapsulated NPs was found to 82 ± 1 nm (polydispersity index = 0.205). (E) pH-dependent drug-release profiles of nontargeted and SHAL-functionalized Dox NPs in physiological conditions. (F) FACS binding assays for the Jurkat, Ramos, Daudi, and Raji cell lines: (i) biotin-functionalized SHAL (SH7129, labeled with PE-Cy5-labeled streptavidin) and (ii) rhodamine-labeled SHAL-functionalized PEG-PLGA NPs. (G) Representative CLSM images of Jurkat, Ramos, Daudi, and Raji cells: (i) unstained, (ii) stained with 200 nM of the biotin-functionalized SHAL SH7129 labeled with PE-Cy5-labeled streptavidin, and (iii) stained with rhodamine (Rhod)-labeled SHAL-functionalized PEG-PLGA NPs containing 200 nM of conjugated SHAL. The cells were stained at 4 °C for 30 min. (H) Representative time-dependent confocal images show the internalization of Raji cells pretreated with rhodamine-labeled SHAL-functionalized PEG-PLGA NPs after incubation at 37 °C for (i) 5 min, (ii) 30 min, and (iii) 60 min.
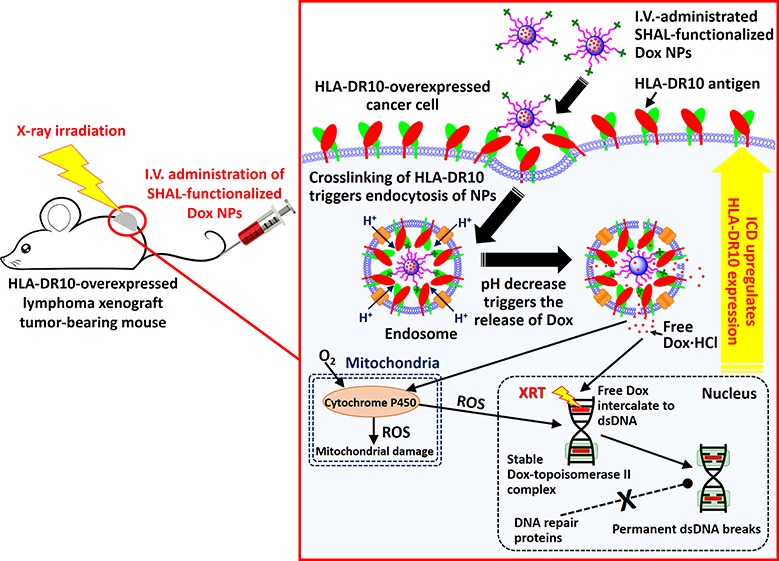
Herein, we report new treatment strategies involving chemo-immunotherapy and concurrent CIRT treatment strategies based on SHAL-functionalized Dox-encapsulated nanoparticles (Dox NPs) for the treatment of HLA-DR-overexpressed NHL. We hypothesized that improved HLA-DR-targeted therapy could be achieved by delivering high doses of an ICD-inducing agent and a radiosensitizer (a reagent that sensitizes cancer cells to XRT) directly into the cancer cells through a high-performance, biocompatible drug delivery system (Scheme 1). While HLA-DRs naturally travel back and forth from the surface of the cell to its interior35,36 carrying peptide fragments it presents to T cells, and this process can be hijacked to transport SHALs into cancer cells expressing HLA-DR,24,25 HLA-DR cannot internalize antibodies or nanoparticles unless it is cross-linked.37 SHAL-functionalized NPs under 100 nm in diameter can effectively cross-link the HLA-DR surface antigens and can trigger internalization of the NP through endocytosis (Scheme 1). The acidic endosomal environments trigger the release of the encapsulated drug. Dox was chosen for this study because, in addition to its direct cytotoxic effect, it is an effective radiosensitizer38 and ICD-inducing agent15 that sensitizes cells to radiation and upregulates the antigen expressions of dying cancer cells (Scheme 1). We also hypothesized that, with an appropriate treatment schedule, the ICD-triggered upregulation of HLA-DR antigen expression can enhance uptake of the SHAL-functionalized NPs and thus improve the overall treatment efficiency. To achieve these goals, we engineered tailor-made, SHAL-functionalized, Dox-encapsulated NPs. The physiochemical properties, in vitro binding affinities, and toxicities of these new antibody mimic-functionalized NPs, were investigated using four well-established NHL cell lines (Jurkat, Ramos, Daudi, and Raji cells) with varying degrees of expression of the specific HLA-DR target. We also studied the radiosensitizing mechanisms of the SHAL-functionalized Dox NPs. We investigated the ability to use these targeted Dox NPs to induce ICD and upregulate HLA-DR expression and evaluated the potential side effects associated with systemic administration of SHAL-functionalized Dox NPs compared to the free drug. Finally, we comprehensively evaluated the anticancer activities of HLA-DR antigen targeted NPs in two HLA-DR-overexpressed lymphoma mouse models.
Scheme 1. Mechanism of SHAL-functionalized Dox NPs for Chemo-Immunotherapy and Concurrent CIRT.

The systemically administered SHAL-functionalized Dox NPs selectively bind to the HLA-DR-overexpressed cancer cells, thus crosslinking the antigen and triggering the internalization of NPs. The acidic endosome protonates the hydrophobic Dox and triggers the release of free Dox from the endosomes, and the Dox subsequently enters the nucleus and mitochondria. The free Dox intercalates to the double-stranded DNA to form stable Dox-topoisomerase II complexes that prevent proteins from repairing DNA damage and that therefore cause ICD. The irreversible DNA damage becomes more significant when combined with X-ray irradiation, which effectively breaks the double-stranded DNA. Also, some of the released Dox enters the mitochondria, where it is metabolized by cytochrome P450 enzymes to form reactive oxygen species (ROS, e.g., hydroxyl radical), thus causing mitochondrial damage. Some ROS may enter the nucleus, where they break the double-stranded DNA and induce ICD. The inset illustrates that the multivalent SHAL-functionalized NPs works effectively in HLA-DR overexpressed cancer cells but not in healthy B cells, which contain one-tenth the amount of HLA-DR antigen.
Results
Fabrication and Characterization of SHAL-Functionalized Dox NPs
SHAL-functionalized Dox-encapsulated poly(ethylene glycerol)-block-poly(lactide-co-glycolide) (PEG-PLGA) NPs were prepared via nanoprecipitation in basic conditions (pH 9.0, Figures 1B and S2). Amine-functionalized SHAL (SH7133) was conjugated to poly(lactide)-block-poly(ethylene glycerol) N-hydroxysuccinimide ester (PLA(16K)-PEG(10K)-NHS) before preparing the NPs through the amine-NHS ester coupling reaction (Figures S3–S5). The number-average diameter and the intensity-average diameter of the targeted Dox NPs were 50 and 82 nm (Figures 1C,D and S6), as determined by transmission electron microscopy (TEM) and dynamic light scattering (DLS) techniques, respectively. By quantifying the number of NPs formed from each milligram of the polymer mixture using the nanoparticle tracking analysis (NTA) method (Figure S6C), we calculated that each NP contained approximately 70 conjugated SHAL molecules, which is an optimal number of targeting ligands for similar diameter NPs.39 Our target for Dox loading of the SHAL-functionalized NPs was 5% by weight, while the actual Dox loading was about 2.8% by weight (i.e., the encapsulation efficiency was about 57%; Figure S7). Nontargeted Dox NPs were prepared through the same method in the absence of PLA(16K)-PEG(10K)-SHAL. Drug-free rhodamine (Rhod)-labeled SHAL-functionalized PEG-PLGA NPs were prepared via the same nanoprecipitation method in the presence of 2.5% by weight of Rhod-labeled PLGA(20K) instead of Dox for in vitro binding and imaging studies. Both Dox-encapsulated NPs underwent pH-dependent controlled release in physiological conditions (Figures 1E and S7). Approximately 55%, 30%, and 15% of the encapsulated Dox was released at pH 5.5, 6.5, and 7.0 in the first 72 h, respectively. The faster Dox release in the acidic conditions was due to the protonation of the encapsulated Dox (pK1 of Dox = 8.2);40−42 This triggered the conversion of Dox from a hydrophobic form to a hydrophilic salt (Dox·HCl; Figure S8). The pH-dependent drug release ensures the majority of encapsulated Dox is released in the acid endosomes of the targeted cells after systemic administration, thus reducing the systemic side effects of Dox.
SHAL-Functionalized NPs Bind Selectively to HLA-DR-Overexpressed Lymphoma Cells
The binding affinities of unconjugated “free” SHAL (the biotin-functionalized tridentate SHAL (SH7129)) and SHAL-functionalized rhodamine-labeled SHAL NPs were quantified via a fluorescence-activated cell sorting (FACS) binding assay in four well-established human lymphoma cell lines with varying degrees of HLA-DR expression (Figure S9A). Both free SH7129 and SHAL functionalized NP bound selectively to the HLA-DR-overexpressed Ramos, Daudi, and Raji cells but not to the HLA-DR nonexpressing Jurkat cells (Figures 1F, S10, and S11). The binding affinities of SHAL-functionalized NPs were significantly higher than the free SH7129 in all three HLA-DR overexpressing cell lines due to the higher avidity of the SHAL-functionalized NPs. The macroscopic dissociation constant (Kd, Macro) of SHAL-functionalized NPs was calculated as 30 nM in the high HLA-DR10 expression Raji cell line, which is more than 3-fold lower than that of free SH7129 (Kd, Macro ≈ 100 nM, Figure 1F). The binding of free SHAL and SHAL-functionalized NPs was further confirmed by confocal laser scanning microscopy (CLSM) with a ring pattern of staining that can be observed in the CLSM images of HLA-DR overexpressed Ramos, Daudi, and Raji cells after staining with 200 nM of free SH7129 tagged with PE-Cy5-conjugated streptavidin (SH7129-SA) or SHAL functionalized NPs (Figure 1G). A further time-dependent CLSM study of the SHAL-functionalized NPs using NPs pretreated Raji cells confirmed the internalization of the SHAL-functionalized NPs in physiological conditions (37 °C), as a patchy staining pattern slowly replaced the sharp ring pattern, which was eventually sequestered in the cytoplasm (Figure 1H). Conversely, no significant internalization of the SH7129-SA complex was observed in the treated Raji cells (Figure S12). Our results were concordant with previous studies which showed similar cross-linking-induced endocytosis phenomena in HLA-DR-overexpressed epidermal cells after incubation with the cross-linked HLA-DR antibody.37 They also show the presence of the PE-Cy5-streptavidin conjugated to SH7129 prevents the internalization of free SHAL that has been observed to occur24,25 as the surface HLA-DR molecules move back into the cells for recycling.35,36
SHAL Functionalization Enhances the in Vitro Uptake of DOX and Improves Cytotoxic Effects in the HLA-DR-Overexpressed Lymphoma Cells
The in vitro uptake of free and encapsulated Dox in all three HLA-DR antigen overexpressed lymphoma cell lines was evaluated using the FACS method (Figure 2A). Cells from all three lines took up more Dox encapsulated in the SHAL-functionalized NPs than free Dox, and the uptake of the encapsulated Dox was directly proportional to the HLA-DR expression. The uptake of the Dox that was encapsulated in the nontargeted Dox NPs was much lower than that of the free Dox in all three lymphoma cell lines. Co-treatment with free SHAL or drug-free SHAL-functionalized NPs (SHAL/Dox = 1:2940, which is the molar ratio in SHAL-functionalized Dox NPs) did not significantly affect the uptake of free Dox or nontargeted Dox NPs (Figure 2A). Control study indicated free SHAL (SH7139, 200 nM) pretreatment significantly reduced the uptake of SHAL-functionalized Dox NPs (Figure S13) in the Daudi and Raji cell lines because free SHAL blocked the binding of the targeted Dox NPs. This validated the concept of HLA-DR targeting. A further CLSM study in the Raji cells confirmed the uptake of both free and encapsulated Dox. This study confirmed the release of encapsulated Dox from SHAL-functionalized Dox NPs; the entry of free Dox into the nucleus was further verified by the CLSM study (Figure 2B, including colocalization of Dox fluorescence with 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain).
Figure 2.

In vitro uptake and internalization of small-molecule (“free”) Dox and different Dox nanoformulations in HLA-DR-overexpressed lymphoma cell lines. (A) Representative flow cytometry histograms of (i) Ramos, (ii) Daudi, and (iii) Raji cells after 1 h of incubation with 1 μM of free Dox or different Dox-encapsulated nanoformulations (either nontargeted or SHAL-functionalized PEG–PLGA NPs), in the presence or absence of free or conjugated SHAL, and at physiological conditions (37 °C). (B) Representative CLSM images of Raji cells after incubation with free Dox or different Dox-encapsulated NPs (also in the presence or absence of free or conjugated SHAL) using the concentrations and conditions used in the flow cytometry experiment. The CLSM images confirm that Dox was released from the SHAL-functionalized NPs and entered the nucleus.
The in vitro toxicities of different forms of SHAL, Dox, and their combinations in all three HLA-DR10 overexpressed lymphoma cell lines using an MTS cell proliferation assay were determined to start 4 days after the initial treatment. Free Dox had the highest cytotoxicity in all three cell lines, with a half-maximal inhibitory concentration (IC50) of about 0.15 μM (Figure 3A). This was expected since the tumor cells were fully exposed to free DOX in the media in the in vitro setting. In comparison, free SHAL and drug-free SHAL-functionalized NPs demonstrated minimal cytotoxic effects (IC50 > 10 μM; Figure S14). The combination of free Dox and free SHAL (Dox/SHAL = 2940:1) did not significantly affect the cytotoxicity of free Dox in any of the three lymphoma cell lines (p = 0.1544 (Ramos), 0.0845 (Daudi), and 0.056 (Raji) vs the nontreatment control group, Figure 3A(i–iii)). Although nontargeted Dox NPs (and their combinations with SHAL-functionalized NPs) had significantly lower in vitro cytotoxicity than did free Dox (IC50 = 0.6–3.0 μM, p = 0.0242–0.0385 vs free Dox), the IC50 of SHAL-functionalized Dox NPs was comparable with that of free Dox combined with free SHAL in the Raji cells (IC50 = 0.15 ± 0.02 μM, p = 0.0341, Figure 3A(iii)) and was only slightly higher than that of free Dox combined with free SHAL in the Daudi cell line (Figure 3A(ii)). However, the cytotoxicity of SHAL-functionalized Dox NPs was significantly weaker than that of free Dox combined with free SHAL in the Ramos cells, which have a lower HLA-DR expression (p = 0.0416, Figure 3A(i)). The in vitro toxicity of the SHAL-functionalized Dox NPs was consistent with the cellular uptake of the targeted NPs and thus with the HLA-DR expression. The internalization of the targeted Dox NPs through endocytosis enhanced the cytotoxicity of the encapsulated Dox NPs, even though some of the encapsulated Dox was released inside the endosomes.
Figure 3.

In vitro toxicities and radiosensitizing properties of free Dox and different Dox nanoformulations for different HLA-DR-overexpressed lymphoma cells. (A) In vitro toxicities of free Dox and different Dox nanoformulations in the (i) Ramos, (ii) Daudi, and (iii) Raji cell lines. The cell viabilities were accessed via an MTS assay 4 days after the initial treatments (N.B., n = 8 per group; * denotes p < 0.05, i.e., statistically significant; n.s. denotes statistical insignificance). (B) Radiosensitizing responses of (i) Ramos, (ii) Daudi, and (iii) Raji cells after treatment with therapeutic doses of Dox (i.e., IC50 of free Dox) before 5 Gy X-ray irradiation. The numbers of live (AV–PI–), apoptotic (AV+PI–), and dead or necrotic (AV+PI+/AV–PI+) cells were quantified via a propidium iodide and A488-labeled annexin V live–dead cell apoptosis assay using the FACS method (n = 3; * denotes p < 0.05, i.e., statistically significant).
SHAL-Functionalized Dox NPs Sensitizes HLA-DR-Overexpressed Lymphoma Cells to Radiation in Vitro
The in vitro radiosensitizing properties of free and encapsulated Dox in all three HLA-DR-overexpressed lymphoma cell lines were evaluated using an annexin V (AV)-propidium iodide (PI) dead cell apoptosis assay. In all three lymphoma cell lines, less than half of the cells remained viable (AV–PI–) after treatment with therapeutic doses of Dox (i.e., IC50 of free Dox at 0.15 μM) for 96 h (Figure 3B). The population of necrotic and dead cells (AV+PI+/AV–PI+) varied from about 70% (Raji cells) to about 35% (Daudi cells) (Figure 3B). In general, SHAL-functionalized Dox showed higher toxicities than did nontargeted Dox NPs in all three types of lymphoma cell lines. The 5 Gy X-ray irradiation effectively led to 45–55% fewer AV–PI– cells than untreated cells. Dox pretreatment (24 h before irradiation) significantly decreased the number of viable cells after 5 Gy irradiation, with less than 5% of the Raji and Daudi cells remaining viable after treatment with free Dox or SHAL-functionalized Dox NPs, followed by radiation (Figure 3B). This finding indicates that Dox is an effective radiosensitizer as previously reported.38 Due to the poor cellular uptake of the nontargeted Dox NPs, pretreatment with nontargeted Dox NPs led to less significant radiosensitizing effects than treatment with free Dox or SHAL-functionalized Dox NPs. Co-administration of drug-free SHAL NPs with nontargeted Dox NPs (conjugated SHAL: encapsulated Dox = 1:2940) did not significantly affect the relative number of viable or apoptotic cells after irradiation, which indicates that SHAL alone did not sensitize radiation.
Cell cycle analyses were performed to investigate the radiosensitizing mechanism of free Dox and different Dox nanoformulations (Figure 4A). At 24 h after a therapeutic dose of free Dox or SHAL-functionalized Dox NPs (containing the IC50 of Dox), the percentage of cells in the G2/M phase significantly increased in all three lymphoma cell lines, indicating that the treatment induces cell cycle arrest in the radiosensitive G2/M phase (Figure 4A). Treatment with free Dox also reduced the population of cells in the more radioresistant S phase (Figure 4A). In contrast, treatment with the SHAL-functionalized Dox NPs did not significantly affect the percentage of cells in the S phase. This trend can be explained by slow drug release kinetics and an incomplete release of the encapsulated Dox, which could potentially lower the drug concentration. For the same reasons, in all three lymphoma cell lines, the nontargeted Dox NPs only slightly increased the population of the G2/M phase and did not significantly affect the percentage in the S phase (Figure 4A). Control studies revealed that cotreatment with free SHAL (SH7129) or drug-free SHAL NPs with either free Dox or nontargeted Dox NPs did not affect the cell cycle when compared to treatments without free or conjugated SHAL.
Figure 4.

Radiosensitization mechanisms of free Dox and different Dox nanoformulations. (A) Cell cycle analysis of (i) Ramos, (ii) Daudi, and (iii) Raji cells 24 h after treatment with therapeutic doses of free Dox or different Dox nanoformulations in the presence and absence of free SHAL SH7129 or SHAL-conjugated NPs. (B) 24-h postirradiation (5 Gy X-ray) and (C) 48-h post-treatment FACS histograms of anti-γH2AX-stained (i) Ramos, (ii) Daudi, and (iii) Raji cells. The fluorescence intensities of the stained cells are directly proportional to the number of double-stranded DNA breaks caused by X-ray irradiation (N.B., n = 3 per group; * denotes p < 0.05, i.e., statistically significant; n.s. denotes statistical insignificance).
DNA double-strand breaks induced by in vitro treatment with Dox and radiation for the three HLA-DR10-overexpressed lymphoma cell lines were quantified using a FACS-based γ-H2AX assay (Figure 4B). The γ-H2AX expressions of all three lymphoma cell lines slightly increased after treatment with therapeutic doses of Dox (i.e., IC50 of free Dox) because cytochrome P450 can metabolize Dox to generate hydroxide radicals, which diffuse into the nucleus and break double-stranded DNA. Also, Dox directly enters the nucleus and binds to double-stranded DNA to form a stable Dox-topoisomerase II complex that prevents proteins from repairing DNA damage (Scheme 1). In all three lymphoma cell lines, the increase in the γ-H2AX expressions induced by the SHAL-functionalized Dox NPs was comparable to that caused by free Dox, whereas treatment with nontargeted Dox NPs did not significantly affect γ-H2AX expression (Figure 4B). As in previous studies, 5 Gy X-ray irradiation effectively induced double-stranded breaks and increased the γ-H2AX expression in all three lymphoma cell lines. Pretreatment with therapeutic doses of free Dox or SHAL-functionalized Dox NPs (continued therapeutic doses of Dox) significantly increased the γ-H2AX expression relative to the treatment group that only received 5 Gy of X-ray irradiation because the Dox-topoisomerase II complex prevented proteins from repairing the DNA damage caused by the X-ray irradiation.
SHAL-Functionalized Dox NPs and X-ray Irradiation Induce Immunogenic Cell Death and Upregulate HLA-DR Antigen Expression in HLA-DR-Overexpressed Lymphoma Cells
Cytotoxic chemotherapy through XRT and an ICD-inducing agent such as Dox induces ICD, during which dying cancer cells may upregulate antigen expression in the cancer cells that survive. A time-dependent in vitro study was performed to investigate the calreticulin expression of Raji cells after treatment with a subtherapeutic dose of Dox and 5 Gy X-ray irradiation (Figures 5A and S15A), since the upregulation of calreticulin characterizes ICD.43,44 The calreticulin expression remained relatively constant 24 h after initial treatment in all treatment groups. The calreticulin expression of all treatment groups received 5 Gy XRT significantly increased 72 h after the irradiation (Figures 5A), but it returned to the background level 5 days after the treatment. The calreticulin expression of the Raji cells treated with SHAL-functionalized Dox NPs followed by 5 Gy XRT was nearly double 72 h after the irradiation but returned to background 2 days later. This confirmed the time-dependent nature of the ICD. Further time-dependent in vitro study was performed to investigate HLA-DR antigen expression in Raji cells after treatment with a subtherapeutic dose of Dox and 5 Gy X-ray irradiation (Figures 5B and S15B). Untreated Raji cells showed very stable HLA-DR expression (M.F.I. ≈ 4.4 × 105). In vitro treatment with a subtherapeutic dose of free Dox (i.e., IC25 of Raji = 80 nM) upregulated the HLA-DR expression 24 h after treatment. The HLA-DR expression reached its maximum (M.F.I. ≈ 6.8 × 105, about 55% higher than in the nontreatment group) 3 days after the initial treatment but dropped back to normal 5 days after treatment (Figures 5B and S15B). The combination of Dox and free SHAL (Dox/SHAL = 2940:1) did not significantly enhance HLA-DR expression. Conversely, in vitro treatment with SHAL-functionalized Dox NPs (continued IC25 of free Dox) showed much quicker and higher upregulation of HLA-DR expression (Figures 5 B and S15B). The HLA-DR expression of the survival fraction was higher than that of the nontreatment group cells 5 days after the initial treatment. Five Gy X-ray irradiation effectively upregulated HLA-DR expression (M.F.I. ≈ 5.7 × 105, 24 h after initial treatment, also about 55% higher than in the nontreatment group). As with free Dox, the HLA-DR expression of the surviving cells dropped back to the average level 5 days after the initial treatment (Figures 5B and S15B). The Dox pretreatment (with either free Dox or SHAL-functionalized Dox NPs) followed by the 5 Gy X-ray irradiation rapidly upregulated HLA-DR expression (45–66% higher than in the nontreatment group) 24 h after irradiation. The HLA-DR expression of the survival fractions of both treatment groups was 95–120% higher than those of the nontreatment group 3 days after irradiation but eventually dropped back to the average level 5 days after irradiation. This time-dependent study confirmed that Dox, X-ray irradiation, and their combination are all sufficient to upregulate HLA-DR expression in HLA-DR-overexpressed lymphoma cells but that the HLA-DR expression of the surviving cells eventually returns to average levels 5 days after treatment. Thus, with an appropriate schedule, the upregulation of HLA-DR antigen expression can be utilized to improve the uptake of SHAL-functionalized Dox NPs, both in vitro and in vivo.
Figure 5.

Calreticulin and HLA-DR expressions of Raji cells after treatment with a subtherapeutic dose (i.e., IC25) of free/encapsulated Dox with/without a therapeutic dose of X-ray irradiation. (A) The plot of mean fluorescence intensities (MFI) of unstained and α-calreticulin-stained Raji cells after treatment with IC25 of either free or encapsulated Dox, with or without 5 Gy X-ray irradiation (24 h after the initial drug treatment). (B) The plot of mean MFI of unstained and α-HLA-DR-stained propidium iodide-negative (PI–) variable Raji cells after treatment with IC25 of either free or encapsulated Dox, with or without 5 Gy X-ray irradiation (24 h after the initial drug treatment). Both antibodies were A488 stained (N.B., n = 3; * denotes p < 0.05, and hence statistically significant).
SHAL-Functionalized Dox NPs Improve the in Vivo Uptake and Reduce Systemic Side Effects of Dox
An ex vivo biodistribution study was performed to quantify the uptake of free Dox as well as Dox nanoformulations in Ramos, Daudi, and Raji tumor xenografts in athymic nude (Nu) mice. The Dox uptake was quantified via a well-established ex vivo fluorescence imaging technique at several time points after intravenous (i.v.) tail vein administration of different therapeutics (Figure S16). Dox fluorescence can be observed in all three different types of tumors when harvested 24 and 72 h postadministration of the therapeutics (Figures 6A, S17–S19). In the Raji tumor model, the tumor uptake of Dox delivered through SHAL-functionalized NPs was about 3.5 times higher than that of free Dox (p = 0.0143) at 24 h postadministration (Figures 6B and S17), but the uptake of Dox delivered through nontargeted NPs was similar to that of free Dox (p = 0.0539). The amount of Dox retained in the Raji tumor dropped significantly by 72 h postadministration (Figures 6B and S17), likely due to the cancer cells clearing the drug through circulation and metabolism. However, the amount of Dox retained in the tumor and delivered through the SHAL-functionalized NPs was still about 100% higher than that found in the group with free Dox. The Daudi tumor model had a very similar tumor uptake trend (Figures 6B and S18), but the Daudi tumor took up less of the Dox that was delivered through the SHAL-functionalized NPs than did the Raji tumor, presumably due to the lower HLA-DR expression of Daudi cells. However, in the Ramos tumor model, the tumor uptake of Dox was very similar, whether it was administered as a free drug or as a nanoformulation (p = 0.4341; Figures 6B and S19). The low HLA-DR expression could explain this effect in the Ramos cells. Nevertheless, our CLSM study on the harvested tumor sections confirmed the selective binding and uptake of the SHAL-functionalized Dox NPs (Figure 6C). A ring-stained pattern can be seen in the tumors that were preserved 24 h postadministration of the targeted Dox NPs. In contrast, a diffused pattern of Dox can be observed in tumor sections preserved 72 h postadministration of the targeted Dox NPs, which confirmed the release of the Dox from the NPs.
Figure 6.

Biodistributions of different forms of Dox in Ramos, Daudi, and Raji xenograft tumor-bearing mice. (A) Representative ex vivo fluorescent images of Ramos, Daudi, and Raji xenograft tumors harvested 24 or 72 h after i.v. tail vein administration of free Dox or different Dox nanoformulations (3.5 mg Dox/kg). The ex vivo fluorescence images were recorded using an IVIS Kinetic imaging system equipped with a DsRed emission filter (λem = 575–650 nm) with excitation at 465 ± 15 nm. (B) Biodistributions of free Dox and different Dox nanoformulations in Ramos, Daudi, and Raji xenograft tumor-bearing mice recorded 24 or 72 h after administration (n = 4 for the Ramos xenograft tumor model recorded 24 h after administration, n = 3 for the Ramos xenograft tumor model recorded 72 h after administration, n = 4 for the Daudi and Raji xenograft tumor models recorded 24 and 72 h after administration, respectively). (C) Representative CLSM images of (i) Ramos, (ii) Daudi, and (iii) Raji xenograft tumor sections preserved 24 or 72 h after the administration of free Dox or different Dox nanoformulations. The nuclei were stained with DAPI (N.B. * denotes p < 0.05, i.e., statistically significant).
A comprehensive in vivo toxicity study was performed to investigate the side effects of different forms of Dox at the maximum tolerated dose (M.T.D.) of Dox (10 mg/kg per week), both with free or conjugated SHAL (15 μg/kg) and without SHAL in healthy CD1 IGS mice. As shown in Table S1, the systemic administration of free Dox-induced significant hematological toxicities. In this study, the lymphocyte and reticulocyte counts were below the reference ranges for healthy mice (Figure 7A), likely due to the prolonged systemic exposure to active Dox. The coadministration of free or conjugated SHAL did not incur additional hematological toxicity. In contrast to free Dox, both Dox nanoformulations induced very low toxicities in healthy mice (Figure 7A). The postadministration lymphocyte and reticulocyte counts for both nanoformulations were also within the reference ranges for healthy mice. The reduction in side effects can be explained by the kinetics of prolonged Dox release at physiological pH (pH 7.0). The further histopathological study indicated that the administration of free Dox (or of free Dox plus free SHAL) induced significant myocardial toxicity. Substantial lesions and muscular fiber dissociation can be observed in both groups (Figure 7B) and likely result from prolonged exposure to free Dox after systemic administration. In contrast, the administration of neither of the Dox nanoformulations was associated with observable cardiotoxicity and hepatological toxicity because the Dox-encapsulated NPs could not effectively pass through the sinusoidal endothelium.45
Figure 7.

In vivo toxicities of small-molecule Dox and different Dox nanoformulations in healthy CD1 IGS mice. (A) (i) Reticulocyte (RET) and (ii) lymphocyte (LYMPH) counts of whole blood samples collected from healthy CD1 IGS mice 48 h after tail vein administration of M.T.D. of free Dox or different Dox nanoformulations (10 mg/kg) in the presence or absence of free or conjugated SHAL (15 μg/kg). The yellow highlighted regions show the normal ranges of RET and LYMPH counts (n = 5 per group). (B) Representative H&E-stained kidney, liver, spleen, lung, and heart histological sections harvested 48 h after tail vein i.v. administration of M.T.D. (i.e., 10 mg/kg) of free Dox or different Dox nanoformulations in the presence or absence of free or conjugated SHAL (15 μg/kg). The yellow arrows highlight the loosened myocardial fibers caused by the free Dox treatment.
Immunogenic Cell Death Enhances the in Vivo Anticancer Efficacy of SHAL-Functionalized Dox NPs for Concurrent Chemo-Immunotherapy and Concurrent CIRT
Comprehensive in vivo studies were performed in Daudi and Raji xenograft tumor models to investigate the anticancer efficiencies of SHAL-functionalized Dox NPs for chemo-immunotherapy and concurrent CIRT. The in vivo studies involved three short treatment cycles, with a rest period of 3 or 4 days between treatments to ensure that the second treatment started while the previous treatment was still upregulating the HLA-DR expression based on our previous in vitro data (Figure 8A). In the Daudi tumor model, treatment with a therapeutic dose of free Dox slowed down the tumor growth but only slightly increased the median survival time (M.S.T.) by 3 days versus the nontreatment group (Figure 8B). No significant tumor growth inhibition was observed after treatment with the nontargeted Dox NPs, presumably due to the poor uptake of nontargeted NPs. Similar to treatment with an anti-HLA-DR antibody, a single treatment or cotreatment with free SHAL (SH7129, p = 0.0352 vs the nontreatment group, Figure 8A) or drug-free SHAL-functionalized NPs (p = 0.0413 vs the nontreatment group, Figure 8B) did not significantly affect tumor growth in the immunocompromised mouse xenograft model. However, treatment with SHAL-functionalized Dox NPs (including a therapeutic dose of Dox) significantly slowed down tumor growth compared with the nontreatment control group, resulting in an absolute growth delay (A.G.D.) of about 7 days (Table S2). Although no complete response (CR) or long-term survival after treatment was observed with the SHAL-functionalized Dox NPs, it was calculated that the targeted Dox NPs enhanced the therapeutic efficiency of Dox by about 120%. In vivo fractionated XRT (3 × 5 Gy) significantly delayed the progression of cancer for 35 days. Concurrent administration of XRT with free SHAL or drug-free, SHAL-functionalized NPs did not improve the progression of the disease. Concurrent CRT with a therapeutic dose of free Dox prolonged the survival time (M.S.T. = 76 days versus 58 days for the XRT group), but the treatment only increased the regression time by an average of 6 days compared to the XRT treatment only group. Similar to chemotherapy, concurrent CIRT treatment with nontargeted Dox NPs only slightly slowed down the progression of the disease (M.S.T. = 68 days), and no treated mice achieved long-term survival. Concurrent CIRT with SHAL-functionalized Dox NPs effectively controlled the progression of tumor growth, with about 70% of the treated mice achieving a complete response and long-term survival (M.S.T. not reached at >80 days, CR = 70%); this method thus outperformed both concurrent CRT treatments with free Dox (M.S.T. = 76 days, CR = 13%, p = 0.0314) and free Dox plus free SHAL (M.S.T. = 72 days, CR = 14%, p = 0.0279). By comparing the average growth delays (A.G.D.) and normalized growth delays (N.G.D.) of the XRT treatment group and the chemotherapy group treated with SHAL-functionalized Dox NPs, it was calculated that the SHAL-functionalized Dox NPs enhanced the efficiency of XRT by more than 100% (Table S2). In addition to the superior treatment responses, concurrent CIRT with SHAL-functionalized Dox NPs did not induce any significant adverse effects (e.g., rapid weight loss; Figure S21), whereas significant weight loss after treatment was seen with free Dox plus free SHAL (either with or without further XRT), and about 30% of the treated mice were dead within 10 days of the final treatment.
Figure 8.

In vivo anticancer activities of free Dox and different Dox nanoformulations for chemo-immunotherapy and concurrent CIRT in the Daudi xenograft tumor model. (A) Treatment schedule and tumor growth curve of individual mice in the control and treatment groups. The treatments were three 3.5 mg/kg doses of free/encapsulated Dox and three 5 μg/kg doses of free/conjugated SHAL. The mice in the concurrent CIRT group received three 5 Gy XRT treatments 24 h after the i.v. administration of the therapeutics. (B) Kaplan–Meier survival curves for mice in the nontreatment group and in (i) the chemo-immunotherapy groups and (ii) the concurrent CIRT groups (n = 7 or 8 per group; * denotes p < 0.05, i.e., statistically significant).
The anticancer activities of SHAL-functionalized Dox NPs were further evaluated in the high HLA-DR antigen expressed and highly aggressive Raji xenograft tumor model. As in the Daudi tumor model, treatment with a therapeutic dose of free Dox or nontargeted Dox NPs only slightly delayed the progression of the tumor growth and increased the M.S.T. by only 10 and 7 days (Table S3; A.G.D. of the free Dox treatment group = 8 days; A.G.D. of nontargeted Dox NPs treatment group = 5 days), respectively. Overall, the Raji xenograft model was more resistant to chemotherapy with DOX, which only induced transient response followed by rapid tumor progression and death. This is consistent with the highly chemoresistant nature of Raji cells, which has overexpression of MDR1/P-glycoprotein (Figure S9B) and mutated p53 (Figure S9C).46 Therapy combining free Dox and free SHAL or combining nontargeted Dox NPs and drug-free SHAL NPs did not significantly affect the anticancer efficacy (Figures 9 and S22), but the combination of free Dox and free SHAL slightly reduced the M.S.T. due to the side effects associated with free drugs (about 25% of the mice were dead 10 days after the final treatment; Figure S23). In contrast, SHAL-functionalized Dox NPs effectively inhibited the progression of tumor growth, with a median progression time of 20 days. By comparing the A.G.D. with that of free Dox, it was calculated that the targeted NPs enhanced the anticancer efficiency of Dox by about 110% (Table S3). This is consistent with the higher average caspase 3 and HLS-DR expressions observed in the xenograft tumors treated with the SHAL-functionalized Dox NPs compared with that treated with free Dox plus free SHAL (Figures S25 and S26). Fractionated XRT (3 × 5 Gy) slowed the progression of the disease by an average of 20 days (Figure 9), but the concurrent administration of XRT with free SHAL or drug-free, SHAL-functionalized NPs did not further improve this efficacy. Concurrent CRT with free Dox only slightly slowed down the progression of the tumor (A.G.D. of the free Dox concurrent CRT group and the XRT group were 17 ± 2 days and 13 ± 3 days, respectively), indicating the Raji tumor was resistant not only to chemotherapy but also to radiation. In this model, the radiosensitizing effect of free Dox was limited (E.F. ≈ 1; Table S3), presumably due to the poor tumor uptake and rapid clearance of free Dox. For a similar reason, the nontargeted Dox NPs showed a very weak radiosensitizing effect (i.e., the mice survived an average of 3 days longer than in the XRT group). Co-administration of free SHAL or drug-free SHAL-functionalized NPs with either free Dox or nontargeted Dox NPs showed no further beneficial effects in concurrent CRT. Conversely, concurrent CIRT with the SHAL-functionalized Dox NPs significantly increased the length of the remission period even in this chemo-/radioresistant tumor model. At the study end point, 100% of the treated mice were alive with the follow-up of >80 days with 71% of them achieving a complete regression of the tumor. Histopathological study indicated the average caspase 3 expression of tumors treated with the SHAL-functionalized Dox NPs followed by 5 Gy XRT was 23% higher than that treated with free Dox plus free SHAL followed by 5 Gy XRT and 1.6 times higher than that without irradiation. Even in the mice with partial tumor regression, the treatment significantly increased their median remission time (∼ 55 days vs ∼15 days for the other treatment groups that received XRT). The improvement in survival rate was even more dramatic in this chemoresistant Raji tumor model when the group treated with CIRT with SHAL-functionalized Dox NPs (M.S.T. not reached at >80 days) was compared to other treatment groups, including CRT with free DOX (M.S.T. = 48 days, p = 0.0141) and free DOX plus free SHAL (M.S.T. = 52 days, p = 0.0323). Quantitatively, the antibody mimic-functionalized Dox NPs enhanced the efficiency of XRT by more than 100% (E.F. > 2.0). Taken together, concurrent therapy with SHAL-functionalized Dox NPs induced synthetic lethality even in tumors that are resistant to conventional chemotherapy and radiation.
Figure 9.

In vivo anticancer activities of free Dox and different Dox nanoformulations for chemo-immunotherapy and concurrent CIRT in the Raji xenograft tumor model. (A) Treatment schedule and tumor growth curve of individual mice in the control and treatment groups. The treatments were three 3.5 mg/kg doses of free/encapsulated Dox and three 5 μg/kg doses of free/conjugated SHAL. The mice in the concurrent CIRT group received three 5 Gy XRT treatments 24 h after the i.v. administration of the therapeutics. (B) Kaplan–Meier survival curves of mice in the nontreatment group and in (i) the chemo-immunotherapy groups and (ii) the concurrent CIRT groups (n = 6 or 7 per group; * denotes p < 0.05, i.e., statistically significant).
Additional in vivo studies were performed to investigate the significance of the upregulation of HLA-DR expression induced by ICD in the anticancer effects of SHAL-functionalized Dox NPs (Figure S24). A longer treatment cycle (with a six-day rest period between treatments) in this in vivo study using the Daudi xenograft model to allow the HLA-DR antigen expression of surviving cancer cells to return to the background level before subsequent treatment commenced. As in the shorter treatment cycle treatment schedule, the anticancer activities of the three weekly administrations of free SHAL, SHAL-functionalized Dox NPs, and nontargeted Dox NPs were comparable with those in the nontreatment control group. In contrast to the shorter treatment cycle protocol, the SHAL-functionalized Dox NPs were less effective at inhibiting tumor growth than was free Dox (M.S.T. SHAL-functionalized Dox NPs = 42 days vs M.S.T. of free Dox = 49 days; p = 0.0481; Figure S24). The survival probability of the mice treated with the SHAL-functionalized Dox NPs was similar to that of the nontreatment control group (p = 0.5112). This finding indicates that the ICD-induced upregulation of HLA-DR antigen expression directly enhanced the anticancer activity of the SHAL-functionalized Dox NPs.
Lastly, treatment sequence-dependent in vivo studies were performed to investigate how the chemo-immunotherapy and XRT treatment sequence affected the anticancer efficacy of SHAL-functionalized Dox NPs. As in the earlier efficacy study using the Raji tumor model, mice in the concurrent CIRT group received three treatments of 5 Gy XRT 24 h after the i.v. administration of SHAL-functionalized Dox NPs. In the sequential CIRT group, mice received three treatments of 5 Gy XRT, 3 days apart, starting 5 days after the final chemo-immunotherapy session (Figure S27). Although the sequential treatment schedule is more widely used than the concurrent schedule in certain clinical situations because of more toxicities associated with concurrent treatment, no significant side effects (e.g., weight loss) were observed in either treatment group in this study (Figure S27C). This absence of significant adverse events is likely because the SHAL-functionalized Dox NPs reduce any nonspecific uptake and systemic side effects associated with Dox, as justified by biodistribution and in vivo toxicity studies. However, the concurrent and sequential treatments showed drastically different antitumor effects. As in the earlier in vivo efficacy study, concurrent CIRT effectively inhibited the propagation of tumor growth, with a complete response rate of 78% (Figure S27A,D). In contrast, no mice in the sequential CIRT group achieved a complete response or long-term survival (Figure S27A,C). The sequential treatment only inhibited the tumor growth for approximately 4 weeks (from the date of initial treatment), and the M.S.T. for this group was only 9 days longer than that of the nontreatment control group. This is because systemic administration of SHAL-functionalized Dox NPs followed by 5 Gy XRT significantly increased the HLA-DR expression of cancer cells by about 45% compared without concurrent XRT treatment (Figure S28). The higher HLA-DR expression facilitates the uptake of the SHAL-functionalized NPs in subsequent treatment. These results indicate that the synergist effect of CIRT is maximized when they are given concurrently.
Discussion
Despite advances in the management of NHL, relapses and refractory diseases are still common and remain significant challenges with the majority of these patients eventually dying from the disease progression.47,48 Approximately 20 000 patients in the US will die from NHL each year.1 In particular, many NHL patients develop resistance to more than one type of treatment, including chemotherapy, radiation, and antibody therapy.49 In recent years, there has been increasing interest in the development of new treatment strategies to overcome these challenges. Because the HLA-DR antigen densities in malignant lymphocytes are at least 10 times higher than those in healthy lymphocytes,50 the development of an anti-HLA-DR antibody that targets the HLA-DRs overexpressed by cancer cells could allow for the use of a single antibody or synthetic antibody mimic to target a broad range of hematological cancers.21,23−26,30,50 In the past decade, several chimeric anti-HLA-DR antibodies and antibody-drug conjugates have been developed, but immunogenicity and poor anticancer efficacy limitations have hindered their clinical performance.51 The development of a fully synthetic anti-HLA-DR antibody mimic, such as the tridendate SHAL tested in this study, not only provides a unique opportunity to overcome these challenges but also facilitates the development of targeted drug delivery vehicles for high-performance chemo-immuno-radiotherapy.
In this study, we engineered a tailor-made SHAL-functionalized Dox NPs for the targeted and controlled delivery of Dox to HLA-DR10-overexpressed lymphocytes. An in vitro binding study confirmed that the SHAL-functionalized NPs bound selectively to three HLA-DR-overexpressed lymphoma cell lines with different levels of HLA-DR expression. A nanomolar dissociation constant for the high HLA-DR antigen expression of Raji cell line was determined, which is several times higher than that of unconjugated SHAL due to the high avidity of the antibody mimic-functionalized NPs. A time-dependent CLSM study revealed that the internalization of SHAL-functionalized NPs occurred in the Raji cell through HLA-DR, whereas no internalization was observed after incubation with free SHAL (SH7129) tagged with PE-Cy5-streptavidin. The endocytosis and internalization of NPs are likely triggered by cross-linking of HLA-DR antigens by the multivalent NPs. This phenomenon has been found in other HLA-DR overexpressed cells after incubation with cross-linked HLA-DR antibodies.37 This requirement for cross-linking is also supported by the lack of internalization observed with free SHAL tagged with PE-Cy5-streptavidin.
Comprehensive in vitro studies of HLA-DR-overexpressed lymphoma cells confirmed that SHAL-functionalized Dox NPs enhance the cellular uptake of Dox relative to nontargeted Dox NPs, as a significant amount of the released Dox accumulated in the nucleus. As a result, the in vitro cytotoxicity of SHAL-functionalized NPs was comparable to that of free Dox plus free SHAL. Further in vitro studies confirmed that therapeutic doses of SHAL-functionalized Dox NPs effectively killed most HLA-DR-overexpressed cells when combined with irradiation (5 Gy). Mechanistic studies confirmed that free Dox and SHAL-functionalized Dox NPs sensitized cancer cells to radiation by inducing cell cycle arrest in the radiosensitive G2 and M phases and increasing the number of unrepairable double-stranded DNA breaks due to the formation of a stable Dox-topoisomerase II complex, which prevents proteins from repairing radiation-induced DNA damage. Our study is the first time-dependent FACS study to reveal that the HLA-DR expression of malignant lymphocytes significantly increases shortly after in vitro treatment with Dox (either free Dox or SHAL-functionalized Dox NPs). The HLA-DR expression of surviving cells nearly doubled 3 days after treatment with either free Dox or SHAL-functionalized Dox NPs, followed by irradiation. The HLA-DR expression eventually dropped back to background levels 5 days after treatment, confirming our hypothesis that ICD, when induced by a cytostatic agent and radiation, can upregulate the HLA-DR expression of surviving cancer cells. Thus, by optimizing the treatment schedule, this phenomenon could be employed to improve the uptake of a targeted drug delivery system in subsequent treatments. The upregulation of HLA-DR expression induced by Dox may explain why IMMU-140—an SN38 (antineoplastic drug)-conjugated, anti-HLA-DR antibody-drug conjugate—improves in vivo anti-lymphoma efficacy after pretreatment with a subtherapeutic dose of Dox even when the same dose of the antibody–drug conjugate has very limited anticancer effects.51
Dox is a well-established cytostatic ICD-inducing agent15 and radiosensitizer,38 but its anticancer efficacy is often compromised by its cytotoxic side effects and poor cancer targeting capacity.52 Our comprehensive in vivo toxicity study of healthy CD1 IGS mice confirmed that SHAL-functional Dox NPs containing a maximum tolerated dose (MTD) of Dox significantly reduced the hematological toxicities and cardiotoxicity induced by free Dox. A biodistribution study in three different HLA-DR-overexpressed human lymphoma xenograft tumor models in mice indicated the SHAL-functionalized Dox NPs improved the uptake of Dox compared with free Dox. Due to the lack of enhanced permeability and also the retention effect in nonsolid tumors, an insignificant amount of nontargeted Dox NPs accumulated in xenograft tumors. In our Raji tumor model with a high HLA-DR antigen expression, the xenograft tumors contained about three times more Dox than free Dox at 24 h after the i.v. administration of therapeutics. This confirms that the SHAL-functionalized NPs overcome the limitations of poor tumor uptake and rapid clearance limitations found in conventional antibody-functionalized drug-encapsulated nanocarriers.53 These limitations are due to incorrect orientations of the conjugated antibodies and exposure of the Fc component. Our in vivo toxicity and biodistribution studies highlighted the key advantages of using a fully synthetic antibody mimic as a functionalized drug vehicle for high-performance targeted drug delivery.
Our comprehensive in vivo anticancer efficacy studies demonstrated that SHAL-functionalized Dox NPs are more effective than free Dox (or free Dox plus free SHAL) at inhibiting tumor growth in Daudi and Raji xenograft tumor models, which are known to be resistant to conventional chemotherapy and radiation through MDR1/G-glycoprotein overexpression and P53 mutation. In particular, concurrent CIRT with SHAL-functionalized Dox NPs and XRT effectively inhibited tumor growth, prolonging survival and eradicating more than 70% of HLA-DR-overexpressed tumors. Quantitatively, this concurrent treatment strategy enhanced the cell killing efficiency of XRT by more than 100% in both tumor models, confirming the potential for using such antibody mimic-functionalized Dox-encapsulated NPs in the treatment of hematological cancer. The radiosensitizing efficiency of free Dox with or without free SHAL was less than 10%, which is likely due to reduced uptake of free Dox in the two lymphoma tumor models.
One of the most notable findings to come out of these studies is that the addition of SHAL-functionalized Dox NPs overcame resistance to chemotherapy and radiation and resulted in significant improvements in response and survival rates with minimal toxicities in the xenograft models with known MDR1/G-glycoprotein overexpression. In contrast, treatment with free Dox with or without free SHAL, followed by XRT, induced significant side effects (e.g., weight loss and myocardial damages), and about 20% of the treated mice died within 10 days of the final treatment. Our time-dependent study indicated that the treatment schedule directly affected its anticancer efficacy, as well as the response and long-term survival rates. Although the sequential CIRT protocol is more common than the concurrent treatment protocol in the treatment of hematological cancers, a further treatment schedule-dependent study using the Raji xenograft tumor model indicated that the traditional sequential CIRT protocol did not cure cancer or significantly improve the long-term survival rate, although it conclusively delayed tumor growth and prolonged the survival time. Three underlining factors can explain the weaker antitumor efficacy of the sequential treatment protocol: the lower uptake of the SHAL-functionalized Dox NPs in the sequential CIRT due to weaker ICD-induced upregulation of HLA-DR antigen expression in the absence of XRT; the metabolic degradation or dissociation of intercalated Dox in the surviving cancer cells, which reduces the radiosensitizing efficiency in the sequential protocol; and the larger tumor size due to delayed radiation, which weakens the treatment efficiency of XRT.
Conclusion
In conclusion, we successfully engineered antibody mimic SHAL-functionalized Dox NPs for the targeted chemo-immuno-radiotherapy of HLA-DR-overexpressed hematological tumors. The antibody mimic-functionalized NPs effectively cross-linked HLA-DRs in HLA-DR-overexpressed lymphoma cells, triggering the internalization of NPs and inducing the release of encapsulated Dox in acidic endosomes. Our comprehensive in vitro and in vivo studies are the first to demonstrate that targeted Dox NPs effectively produce ICD and upregulate HLA-DR expression in surviving tumor cells. We have demonstrated that, with an appropriate treatment schedule, the ICD phenomenon can be used to improve the uptake of targeted NPs (and thus, the treatment efficiency), as justified in comprehensive in vivo studies of lymphoma xenograft tumors in mice. Also, our in vivo toxicity data show that SHAL-functionalized NPs containing MTD of Dox are not associated with any significant toxicities, while free Dox leads to substantial myocardial damages at the same dose. Lastly, our data demonstrate that concurrent therapy with SHAL-functionalized Dox NPs may overcome resistance to chemotherapy and radiation even in the presence of MDR1/G-glycoprotein overexpression and P53 mutation. The SHAL used in this study to functionalize Dox NPs has recently been shown to target a group of HLA-DRs, in addition to the HLA-DR10 expressed by Raji lymphoma cells, that are overexpressed by at least 15 non-hematological cancers.30 These results suggest the robust, targeted CIRT strategy described in the current study could translate to treatments for a broad range of HLA-DR overexpressed cancers.
Materials and Methods
Materials
Methoxy poly(ethylene glycol)-block-poly(d,l-lactic-co-glycolic) acid copolymer (mPEG(3K)-PLGA(30K), AK101; molecular weight ≈ (3 + 30) kDa ≈ 33 kDa), poly(d,l-lactide)-block-poly(ethylene glycol)-N-hydroxysuccinimide ester end-cap (PLA(16K)-PEG(10K)-NHS, AI068; molecular weight ≈ (16 + 10) kDa ≈ 26 kDa) and poly(lactide-co-glycolide) rhodamine B end-capped (PLGA-Rhod, AV027; molecular weight = 45–55 kDa) were purchased from Akina, Inc. (West Lafayette, IN). Primary amine-functionalized SHAL (SH7133), DOTA-functionalized SHAL (SH7139), and biotin-functionalized SHAL (SH7129) were provided by SHAL Technologies, Inc. (Livermore, CA). The synthesis, purification, and characterization of all SHALs were reported previously in refs (21, 22, 29, and 50). The SHALs used in this study were trifluoroacetate salts with the following purities as determined by LC/MS: SH7129, 96.2%; SH7133, 95.4%; SH7139, 95.0%. All SHALs were used without further purification. Doxorubicin hydrochloride salt (Dox·HCl, >99%) was purchased from LC Laboratories (Woburn, MA). Dimethyl sulfoxide (DMSO; anhydrous, ≥99.9%), triethylamine (TEA; ≥99.5%), methanol (HPLC grade, ≥99.9%), ethanol (200 proof, for molecular biology), dimethylformamide (anhydrous, ≥99.8%), diethyl ether (ACS reagent, ≥99.9%), acetonitrile (HPLC plus, ≥99.9%), deionized water (sterile-filtered, BioReagent), dichloromethane (anhydrous, ≥99.8%), propidium iodide solution (1 mg/mL in water), Triton X-100 (BioXtra), DNase-free RNase (from bovine pancreas), sodium azide (Laboratory grade), and bovine serum albumin (fraction V lyophilized powder) were purchased from Sigma (St. Louis, MO). Alexa Fluor 488-labeled antihuman HLA-DR antibody (clone L243), phycoerythrin-Cy5-labeled streptavidin, phycoerythrin (PE) anti-H2A.X phosphor (Ser139), antibody (clone 2F3) PE-labeled antihuman CD243 antibody (BioLegend, Clone: 4E3.16) and FITC-labeled antihuman p53 antibody (BioLegend, Clone DO-7) were purchased from BioLegend (San Diego, CA). Human BD Fc Block (antihuman CD16/CD32 antibody) was purchased from BD Bioscience (San Jose, CA). Alexa Fluor 488-labeled anti-calreticulin monoclonal antibody (clone: EPR3924) was purchased from Abcam (Cambridge, MA). Endogenous biotin-blocking kit and dead cell apoptosis kit (contain Alexa Fluor 488 Annexin V and propidium iodide solutions) were purchased from Fischer Scientific (Hampton, NH). All reagents, unless specified, were used without further purifications.
Methods
Synthesis of SHAL-Functionalized PEG-PLA
SHAL-functionalized PEG-PLA was prepared via a primary amine-NHS ester reaction between primary amine-functionalized SHAL (SH7133) and PLA-PEG-NHS ester. Briefly, amine-functionalized SHAL (SH7133, 4 mg, 2.06 μmol) was first dissolved in 0.8 mL of anhydrous DMSO before added to a DMF solution (0.5 mL) contained PLA-PEG-NHS (48 mg, 1.85 μmol) and triethylamine (1 μL, 7.2 μmol). The mixture was stirred at 20 °C in the dark for 18 h. The reaction was quenched by the addition of 1:1 v/v deionized water/methanol (10 μL). The SHAL-functionalized PEG-PLA was purified by precipitation into a large excess of cold 2:3 v/v of methanol/diethyl ether twice and cold diethyl ether 3 times. The precipitated polymers were collected by centrifugation (4000g, 15 min, 4 °C). After each precipitation step, the collected polymer pallet was dissolved in dichloromethane (1 mL) before reprecipitation. The purified polymer pallet was dry under nitrogen gas in the dark for 2 days. The dried polymer pallet was stored at −20 °C in the dark before further studies.
The number-average molecule (MN) of the unmodified PLA-PEG-NHS ester and PLGA-PEG-SHAL was 24 000 Da (P.D.I. = 1.36) and 28 600 Da (P.D.I. = 1.68), respectively, as determined by gel-permeation chromatography (GPC) used tetrahydrofuran as an elute and used different molecular weight standard polystyrenes (Agilent PS2) as a calibration standard. The GPC analysis was performed by Akina, Inc. (West Lafayette, IN).
The degree of functionalization of PLA-PEG with SHAL was quantified by UV–visible spectroscopy. Briefly, SHAL has a strong and characteristic visible absorption band centered at 452 nm (extinction coefficient at 452 nm, ε452nm = 21 500 M–1 cm–1 in DMSO). The degree of functionalized PLA-PEG (dissolved in a known amount of DMSO) was calculated from the extinction coefficient of SHAL at 452 nm.
Preparation of SHAL-Functionalized Dox-Encapsulated PEG-PLGA NPs and Nontargeted Dox-encapsulated PEG-PLGA NPs
Targeted and nontargeted Dox NPs were prepared via nanoprecipitation method. The target drug loading was 5 wt/wt Dox·HCl was converted to hydrophobic Dox in situ. Briefly, 1.5 mg of Dox·HCl was first dissolved in 30 μL of 1:1 v/v TEA/DMSO. The Dox solution was incubated in the dark for 30 min before the preparation of the NPs. For the preparation of 30 mg of SHAL-functionalized Dox-encapsulated NPs, 30 mg of mPEG(3K)-PLGA(30K) was first dissolved in 3 mL of acetonitrile before it was added to the Dox solution before the addition of 33.6 μL of SHAL-PEG-PLA solution (5 mg/mL, in anhydrous DMSO). The mixture was vortexed at 2000 rpm for 20 s before it was added slowly (1 mL/min) to 12 mL of deionized water under constant stirring (1000 r.p.m.). The pH of the mixture was about pH 9, as determined by pH paper. The mixture was stirred under reduced pressure in the dark at 20 °C for 2 h. The Dox-encapsulated NPs were washed 3 times with a 15 mL 30 000 nominal molecular weight cutoff Amicron Ultra ultrafiltration membrane filter (3000g for 15 min). After each wash, the NPs were resuspended in 3 mL of deionized water. At the final purification cycle, the NPs were first resuspended in 1.5 mL (final volume) of deionized water before mixed with 1.5 mL of 2× PBS to give a 10 mg/mL NP solution. Nontargeted Dox NPs were prepared via the same method except SHAL-PEG-PLA was not added to the mPEG(3K)-PLGA(30K) solution before the preparation of the NPs.
Preparation of Drug-free Rhodamine-Labeled SHAL-Functionalized PEG-PLGA NPs
Drug-free Rhod-labeled SHAL-functionalized NPs composed of 1 wt/wt% of PLGA-Rhod were prepared via a nanoprecipitation method. For the preparation of 30 mg of SHAL-functionalized Rhod-labeled NPs, 30 mg of mPEG(3K)-PLGA(30K) was first dissolved in 3 mL of acetonitrile contained 0.1 mg/mL of PLGA-Rhod before mixed with 33.6 μL of SHAL-PEG-PLA solution (5 mg/mL in anhydrous DMSO). The mixture was vortexed at 2000 rpm for 20 s before added slowly (1 mL/min) to 12 mL of deionized water under constant stirring (1000 rpm). The mixture was stirred under reduced pressure in the dark at 20 °C for 2 h. The NPs were washed 3 times with a 15 mL 30 000 nominal molecular weight cutoff Amicron Ultra ultrafiltration membrane filter (3000g for 15 min). After each wash, the NPs were resuspended in 3 mL of deionized water. At the final purification cycle, the NPs were first resuspended in 1.5 mL (final volume) of deionized water before being mixed with 1.5 mL of 2× PBS to give a 10 mg/mL NP solution.
Characterization of Nanoparticles
A transmission electron microscopy (TEM) image of different targeted and nontargeted NPs was recorded used a JEOL 1230 transmission electron microscope operated at 120 kV in the Microscopy Services Laboratory Core Facility at the UNC School of Medicine. Before TEM imaging, NPs samples were diluted to 10 μg/mL with deionized water before added to glow-discharged 400-mesh carbon-coated copper grids (10 μL per grid). After 5 min, extra water was removed from the grid via a filter paper before being stained with 4% uranyl acetate aqueous solution (10 μL per grid) for 20 s. The excess staining solution was removed by filter paper at the edge of the copper grid. The mean number-average diameter (Dn) and particle concentrations of different NP dispersions were determined by an NP-tracking analysis method recorded on a Nanosight NS500 instrument (Malvern, Inc.) in Microscopy Services Laboratory Core Facility at the UNC School of Medicine. All NP dispersions were diluted to 5 μg/mL before the NP tracking analysis. The average number of conjugated SHAL molecules per NP was calculated from the number of PLA-PEG-SHAL used per each mg of NPs and the number of NP per each mg of polymer used. Intensity-average diameter (Dh, also known as hydrodynamic diameter) and mean zeta potential (mean ζ) of different NP dispersions were determined by dynamic light scattering and an aqueous electrophoresis method using a Zetasizer Nano ZS Instrument (Malvern, Inc.). Before the measurements, NPs were diluted to 1 mg/mL with 1× PBS. All measurements were based on the average of three separate measurements.
Drug Loading and in Vitro Drug Release Study
The Dox loadings in the targeted and nontargeted NPs were quantified via the spectroscopic method as previously reported. UV–visible spectra of NP dispersions were recorded in a NanoDrop 1000 Microvolume spectrophotometer (Thermo Scientific). A molar extinction coefficient of 1 × 104 M–1 cm–1 for Dox at 495 nm (ε495nm) was used for the quantification. Drug-free targeted and nontargeted PEG-PLGA NPs (2 mg/mL) showed insignificant visible absorption at 495 nm. The pH-dependent in vitro drug release profile of targeted and nontargeted Dox NPs was recorded under conditions at pH 5.5, 6.5, or 7.0. Briefly, NP solutions at a concentration of 2 mg/mL were split into Slide-A-Lyzer MINI dialysis microtubes with a molecular cutoff of 10 kDa (Pierce, Rockford, IL) and subjected to dialysis against a large excess (2000 times) of 1× PBS at pH 5.5, 6.5, or 7.0 with gentle stirring at 37 °C in dark. The concentration of Dox retained in the NPs was quantified by the spectroscopic method through a NanoDrop 1000 Microvolume spectrophotometer. All measurements were performed in triplicate.
In Vitro Studies
Jurkat, Ramos, Daudi and Raji cells were obtained from the Tissue Culture Facility at UNC Lineberger Comprehensive Cancer Center that purchased the cancer cells from the American Type Culture Collection (ATCC). All lymphoblast cancer cell lines were cultured using RPMI-1640 medium (Gibco) supplemented with 10% (v/v) FBS and antibiotic–antimycotic (100 units/mL of penicillin, 100 μg/mL of streptomycin and 0.25 μg/mL of Gibco amphotericin B) in a 37 °C atmosphere supplemented with 5% CO2. The cell density was determined by a hemocytometer.
Flow Cytometry
Unless specified, viable cells were first washed three times with FACS buffer (0.1 M PBS with 5g/L of BSA, 1 g/L of sodium azide, and 2 mM of EDTA) via centrifugation (600g, 4 min)-redispersion method. The cell density was determined and adjusted to 10 × 106 cells/mL and blocked with human Fc blocker (antihuman CD16/32 antibody, 2 μg/million cells; BD) at 4 °C for 20 min before being stained with desired antibody/antibodies according to the manufacturer’s instructions. Stained cells were washed three times with FACS buffer before analysis on a Biosafety Level 2 (BSL2) Intellicyt iQue Screener PLUS flow cytometer in the UNC Flow Cytometry Core Facility at the UNC School of Medicine. All cells were analyzed within 2 h (at 4 °C) after staining and were analyzed without fixing. All collected FACS data were analyzed through a FlowJo V10.0.7 software pad.
Quantification of HLA-DR Expression
The HLA-DR expression of selected lymphoblast cancer cell lines were determined by FACS binding assay used A488-labeled antihuman HLA-DR antibody (clone L243) according to the manufacturer’s instructions.
Quantification of MDR-1 (CD243) and Intracellular p53 Expressions
The multidrug resistance protein 1 (MDR-1) antigen and intracellular p53 expressions of Raji, Daudi, and Ramos cells were determined by FACS binding assay used PE-labeled antihuman CD243 (clone 4E3.16) and FITC-labeled antihuman p53 antibody (clone DO-7) according to the manufacturer’s instructions. Briefly, the cells were first labeled with PE-labeled antihuman CD243 and fixed. The fixed cells were permeabilized with Intracellular Staining Perm Wash Buffer (BioLegend) before stained with the FITC-labeled antihuman p53 antibody (clone DO-7).
Quantification of Free SHAL Binding Affinity
The binding affinity biotin-functionalized free SHAL, SH7129, was quantified by FACS assay. Before the in vitro binding study, FACS buffer-washed cells were first blocked by endogenous biotin-blocking kit (Fisher) according to the manufacturer’s protocol. Blocked cells (1 × 106 cells/100 μL) were stained with different concentrations of SH7129 (0–200 nM) in FACS buffer at 20 °C for 30 min. After two washes (2000g, 3 min) with FACS buffer, membrane-bound SH7129 was labeled with phycoerythrin-Cy5-labeled streptavidin (BioLegend) according to the manufacturer’s protocol. The labeled cells were washed twice with FACS buffer before being analyzed on a BSL2 Intellicyt iQue Screener PLUS flow cytometer.
Cells stained with 200 nM of free SHAL followed by PE-Cy5 streptavidin were saved, fixed with 10% neutral buffered formalin at 20 °C for 15 min, and washed with PBS before mixed with equal volume of 4′,6-diamidino-2-phenylindole (DAPI)-containing ProLong Gold (Invitrogen) for confocal fluorescence imaging. Confocal fluorescence images were recorded using a Zeiss LSM 710 spectral confocal laser scanning microscope in the Microscopy Services Laboratory at the UNC School of Medicine.
Quantification of Drug-free Rhodamine-Labeled SHAL-Functionalized PEG–PLGA NPs Binding Affinity
FACS assay quantified the binding affinity rhodamine-labeled SHAL-functionalized PEG-PLGA NPs. Briefly, FACS buffer-washed cells (1 × 106 cells/100 μL) were stained with different concentrations of targeted NPs contained a known concentration of conjugated SHAL in the dark at 20 °C for 30 min. After two washes (2000g, 3 min) with FACS buffer, membrane-bound SH7129 was labeled by Alexa Fluor 610-R-phycoerythrin streptavidin. The labeled cells were washed twice with FACS buffer before analyzed on a BSL2 Intellicyt iQue Screener PLUS flow cytometer.
Cells stained with 200 nM of conjugated SHAL were saved, fixed with 10% neutral buffered formalin at 20 °C for 15 min, and washed with PBS before mixed with equal volume of DAPI-containing ProLong Gold (Invitrogen) for confocal fluorescence imaging. Confocal fluorescence images were recorded through a Zeiss LSM 710 spectral confocal laser scanning microscope in the Microscopy Services Laboratory at the UNC School of Medicine.
In Vitro Dox Uptake Study
The uptake of free Dox and different Dox-encapsulated NPs in Ramos, Daudi, and Raji cell lines was quantified by FACS and CLSM methods. Briefly, variable cells were washed twice with phenol red-free RPMI1640. The cell densities were adjusted to 10 × 106 cells/mL (in RPMI-1640), before being incubated with 10 μM (final concentration) of free and encapsulated Dox. After incubation at 37 °C for 1 h (in the dark), the treated cells were washed twice with cold FACS buffer (4 °C). Half of the cells were analyzed in a BSL2 Intellicyt iQue Screener PLUS flow cytometer in the UNC Flow Cytometry Core Facility at the UNC School of Medicine within 60 min. The Dox fluorescence was quantified in a PE-Texas Red channel (excitation at 561 nm, emission at 615–620 nm). The remaining cells were saved and fixed with 10% neutral buffered formalin at 20 °C for 15 min and washed with PBS before being mixed with an equal volume of DAPI-containing ProLong Gold (Invitrogen) for confocal fluorescence imaging. Confocal fluorescence images were recorded through a Zeiss LSM 710 spectral confocal laser scanning microscope in the Microscopy Services Laboratory at the UNC School of Medicine.
A control Dox uptake study was performed to validate the concept of HLA-DR targeting. In the control study, Daudi or Raji cells (10 × 106 cells/mL) were first treated with a saturated amount of free SHAL (SHAL7139, 200 nM) at 37 °C for 1 h to block all HLA-DR antigen, washed, before being further incubated with SHAL-functionalized Dox NPs contained 1 μM of encapsulated Dox at 37 °C for 1 h. The treated cells were washed twice with FACS buffer before being analyzed in a BSL2 Intellicyt iQue Screener PLUS flow cytometer.
In Vitro Toxicity
The in vitro toxicities of free Dox, different Dox nanoformulations and SHAL in Ramos, Daudi, and Raji cell lines were evaluated by a 3-(4,5-dimethylthiazol-2-yr)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) proliferation assay. In the in vitro toxicity study, cells were first washed twice with phenol red-free RPMI1640 before being resuspended in completed media (phenol red-free RPMI1640 supplemented with 10% FBS and antibiotic-antimycotic (100 units/mL of penicillin, 100 μg/mL of streptomycin and 0.25 μg/mL of Gibco amphotericin B). Cells were seeded at 10 × 103 cells per well in 96 well plates before being treated with desired concentrations of free Dox, different Dox nanoformulations, or free/conjugated SHAL at physiological conditions for 72 h. The proliferation of the lymphoma cells was quantified by MTS assay (Promega) according to the manufacturer’s protocol. Briefly, the drug-treated cells (100 μL/well) were incubated 20 μL of MTS/PMS solution in the dark at physiological conditions for 45 min (Raji cells) to 2.5 h (Daudi and Ramos cells). The cell viabilities were quantified via a plate reader by measuring the absorbance at 495 nm.
In Vitro Radiosensitizing Study
The radiosensitizing properties of free Dox and different Dox nanoformulations in the presence and absence of free SHAL SH7129 or conjugated SHAL were quantified using an Alexa Fluor 488 Annexin V (AV) and propidium iodide (PI)-based dead cell apoptosis kit via FACS. In the in vitro study, cells were first treated with therapeutic doses of free Dox or different Dox NPs (contained IC50 of free Dox) for 24 h before being subjected to 5 Gy X-ray irradiation through an X-RAD 320 X-ray irradiator (Precision X-ray Inc., CT) operated at 320 kVp and 12.5 mA. Cells were allowed to grow at physiological conditions for 3 days. Cells in the control groups were treated with different therapeutics at physiological conditions for 4 days. Treated cells were then washed twice with cold PBS (1×, 4 °C) before being resuspended in annexin-binding buffer (1×) at a cell density of about 1 × 106 cells/mL. A488-labeled Annexin V and PI were added to the cells and incubated in the dark for 15 min before being analyzed by a BSL2 Intellicyt iQue Screener PLUS flow cytometer. Viable cells were defined as AV–PI–, apoptotic cells were defined as AV+PI–, necrotic cells were defined as AV+PI+, and dead cells were defined as AV–PI+.
DNA Cell Cycle Analysis
The DNA contents of differently treated cells were quantified using a propidium iodide-based FACS assay. Cells were first treated with therapeutic doses of free Dox or different Dox NPs (contained IC50 of free Dox) for 24 h. Cells were then washed twice with cold PBS, and fixed in 70% ethanol at −20 °C for 24 h. Fixed cells were then washed once with PBS before being resuspended in 2 mL staining solution contained 0.1% Triton X-100, 0.4 mg of DNase-free RNase and 40 μL of 1 mg/mL PI solution. After being incubated at 20 °C for 30 min, stained cells were analyzed in a BSL2 Intellicyt iQue Screener PLUS flow cytometer.
Quantification of dsDNA Breaks
The dsDNA breaks (DBS) induced by Dox treatment, XRT, and their combinations were quantified by anti-H2AX-based FACS assay. Briefly, cells were first treated with therapeutic doses of free Dox or different Dox NPs (contained IC50 of free Dox) for 24 h, before being subjected to 5 Gy X-ray irradiation through an X-RAD 320 X-ray irradiator (Precision X-ray Inc., CT) operated at 320 kV and 12.5 mA. After another 24 h, cells were washed twice with cold PBS, fixed with 70% ethanol for 2 h, and washed twice with FACS buffer before being stained with PE-labeled anti-H2AX (BioLegend) for 30 min according to the manufacturer’s protocol. Stained cells were washed twice with FACS buffer before being analyzed in a BSL2 Intellicyt iQue Screener PLUS flow cytometer. Cells in the control groups only received Dox treatment and were fixed and analyzed 48 h after the initial treatments.
Quantification of Time-Dependent Calreticulin Expression after Different in Vitro Treatments
The change of calreticulin expression of Raji cells after treatment with Dox (with or without free SHAL) or SHAL-functionalized Dox NPs and X-ray irradiation wasquantified using an anti-HLA DR-based FACS assay. In the study, Raji cells were first treated with a subtherapeutic dose of free Dox (with/without free SHAL SH7129 at a molar ratio of SHAL/Dox = 1:2940) or SHAL-functionalized Dox NPs containing an IC25 of Dox for 24 h before being subjected to 5 Gy X-ray irradiation through an X-RAD 320 X-ray irradiator (Precision X-ray Inc., CT) operated at 320 kVp and 12.5 mA. Treated cells were then incubated at physiological conditions for 24, 72, and 120 h before being stained with the A488-labeled anti-calreticulin monocolonal antibody (Abcam, clone EPR3924) according to the manufacturer’s instructions. In the control groups (without X-ray irradiation), cells were treated with different therapeutics and incubated at physiological conditions for 48, 96, and 144 h before being stained for FACS analysis. Unstained cells were used as a control to demonstrate the background fluorescence from different Dox treatments would not interfere with the FACS study.
Quantification of Time-Dependent HLA-DR Expression after Different in Vitro Treatments
The change of HLA-DR expression of Raji cells after treatment with Dox (with or without free SHAL) or SHAL-functionalized Dox NPs and X-ray irradiation was quantified using an anti-HLA DR-based FACS assay. In the study, Raji cells were first treated with a subtherapeutic dose of free Dox (with/without free SHAL SH7129 at a molar ratio of SHAL/Dox = 1:2940) or SHAL-functionalized Dox NPs containing an IC25 of Dox for 24 h before subjected to 5 Gy X-ray irradiation through a X-RAD 320 X-ray irradiator (Precision X-ray Inc., CT) operated at 320 kVp and 12.5 mA. Treated cells were then incubated at physiological conditions for 24, 72, and 120 h before staining with the A488-labeled antihuman HLA-DR antibody (clone L243) according to the manufacturer’s instructions. In the control groups (without X-ray irradiation), cells were treated with different therapeutics and incubated at physiological conditions for 48, 96, and 144 h before being stained for FACS analysis. Unstained cells were used as a control to demonstrate the background fluorescence from different Dox treatments would not interfere with the FACS study.
In Vivo Studies
Animals were maintained in the Division of Comparative Medicine (an AAALAC-accredited experimental animal facility) under sterile environments at the University of North Carolina. All procedures involving experimental animals were performed in accordance with the protocols that the University of North Carolina Institutional Animal Care and Use Committee approved, and they conformed to the Guide for the Care and Use of Laboratory Animals (NIH publication No. 86-23, revised 1985). Athymic nude mice (Nu, also known as Nu/J) were obtained from UNC Animal Services Core (Chapel Hill, NC). The house breed Nu mice were originally obtained from the Jackson Lab. CD1 IGS mice were purchased from Charles River Laboratory (Durham, NC).
In Vivo Toxicity Study
In vivo toxicity of different small-molecule/conjugated SHAL and small-molecule/encapsulated Dox were evaluated in healthy tumor-free CD1 IGS mice (female, about 12 weeks old, 20–21 g). Mice in the control and treatment groups received the following treatments: (1) PBS (nontreatment control group); (2) small-molecules SHAL (SH7139, 300 ng/mouse); (3) drug-free SHAL-functionalized NPs (7.5 mg NPs per mouse, 300 ng conjugated SHAL per mouse); (4) small-molecule Dox (10 mg/kg, MTD of free Dox); (5) nontargeted Dox NPs (10 mg encapsulated Dox/kg); (6) SHAL-functionalized Dox NPs (10 mg encapsulated Dox/kg); (7) small-molecule SHAL (300 ng/mouse) plus small-molecule Dox (10 mg/kg); (8) SHAL NPs (contained 300 ng conjugated SHAL/mouse) plus nontargeted Dox NPs (10 mg encapsulated Dox/kg); and (9) drug-free mPEG–PLGA NPs (15 mg per mouse). 48 h after the i.v. Injection, mice were anesthetized via s.c. Injection of 100 μL of ketamine hydrochloride/xylazine hydrochloride solution (Sigma; St Louis, MO). Circulating blood was collected from the heart. 500 μL of each whole-blood sample was stored in an EDTA-coated tube and stores at 4 °C before blood toxicity study in the Animal Clinical Laboratory Core Facility at the UNC School of Medicine. Key organs (heart, lung, liver, spleen, and kidney) were preserved by 4% (v/v) neutral buffered formalin at 4 °C for 2 days and 40% ethanol at 4 °C for another 2 days before submitting to Animal Histopathology Core Facility at UNC School of Medicine for hematoxylin and eosin (H&E) stain. Representative H&E-stained tissue sections were imaged via an Olympus BX61 optical microscope in Microscopy Services Laboratory at UNC School of Medicine.
In Vivo Biodistribution Study
The biodistributions of Dox administered as a free drug or encapsulated in the nontargeted or SHAL-functionalized PEG–PLGA NPs were evaluated in Ramos, Daudi and Raji xenograft tumor-bearing Nu mice. Athymic Nu mice were chosen for this biodistribution study due to the absence of mature B cells in the body that would interfere with the in vivo study. Xenograft tumors were inoculated in the flank of male Nu mice via the subcutaneous injection of 2 × 106 Ramos, Daudi or Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI1640/Matrigel solution in the left flank. Each type xenograft tumor group contained 25–30 mice. Ten days after inoculation, mice in each group were randomized and divided into 7 subgroups. Mice in the 7 subgroups received the following treatments: (1) PBS (nontreatment control group); (2, 3) free Dox; (4,5) nontargeted Dox NPs; and (6,7) SHAL-functionalized Dox NPs. All Dox formulations were administered via a single tail-vein i.v. Injection of 3.5 mg/kg of free or encapsulated Dox. Mice in groups (1), (2), (4), and (6) were euthanized via s.c. injection of 100 μL of ketamine hydrochloride/xylazine hydrochloride solution 24 h after administration of therapeutics. Xenograft tumor, circulating blood and key organs (liver, kidney, lung, heart and spleen) were preserved. Mice in groups (3), (5) and (7) were euthanized 72 h after administration of therapeutics. Again, xenograft tumor, circulating blood and key organs (liver, kidney, lung, heart, and spleen) were preserved. Ex vivo images were recorded using an IVIS Kinetic imaging system equipped with a light source excited at 575–560 nm and a DsRed emission filter (λem = 575–650 nm). Preserved tumors were fixed in 4% (v/v) neutral buffered formalin at 4 °C for 2 days and 40% ethanol at 4 °C for another 2 days before being submitted to Animal Histopathology Core Facility at UNC School of Medicine for sectioning. Tumor sections were imaged via a Zeiss LSM710 Spectral Confocal Laser Scanning microscope in Microscopy Services Laboratory at UNC School of Medicine.
In Vivo Anticancer Efficacy Studies
The in vivo anticancer activities of free Dox, free SHAL SH7129, conjugated SHAL, nontargeted Dox NPs and SHAL-functionalized Dox NPs for chemo-immunotherapy and concurrent CIRT with a short treatment cycle (rest period between treatment = 3–4 days) were comprehensively evaluated in Duadi and Raji xenograft tumor models in Nu mice. Xenograft tumors were established via subcutaneous injection of 2 × 106 Duadi or Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI1640/Matrigel solution in the left flank. Each type of tumor model contained 120 female Nu mice (6–7 weeks old, 20–21 g). Four days (for Raji xenograft tumor model) or 7 days (for Daudi xenograft tumor model) postinoculation, mice were randomized and divided into 16 groups (6–7 mice per group) for different treatments. The control and treatment groups are (1) PBS (nontreatment group); (2) free SHAL SH7129; (3) drug-free SHAL-functionalized NPs; (4) free Dox; (5) nontargeted Dox NPs; (6) SHAL-functionalized Dox NPs; (7) free SHAL plus free Dox; (8) drug-free SHAL NPs plus nontargeted Dox NPs; (9) PBS (nontreatment group) followed by XRT; (10) free SHAL SH7129 followed by XRT; (11) drug-free SHAL-functionalized NPs followed by XRT; (12) free Dox followed by XRT; (13) nontargeted Dox NPs followed by XRT; (14) SHAL-functionalized Dox NPs followed by XRT; (15) free SHAL plus free Dox followed by XRT; and (16) drug-free SHAL NPs plus nontargeted Dox NPs followed by XRT. Mice in the treatment groups received 3 tail vein i.v. Injections of 3.5 mg/kg free/encapsulated Dox and 5 μg/kg of free SH7129 or conjugated SHAL at day 7, 11, and 14 (for Daudi tumor-bearing mice) or day 4, 8, and 11 (for Raji tumor-bearing mice) postinoculation. Mice in the concurrent CIRT groups received 5 Gy X-ray irradiation 24 h after administration of different therapeutics through a Precision X-RAD 320 (Precision X-ray, Inc.) machine operating at 320kVp and 12.5 mA. The source-subject distance of 70 cm and 50 cGy/min. Only the tumor regions (left flank) of the mice were irradiated, as the remaining parts of the body were lead-shielded. Tumor volume was measured every 3–4 days via a caliper. The bodyweight was measured every week. Tumor volumes were calculated by using the formula of volume (V) = 0.5 × a × b2, where, a and b are the larger and smaller diameters, respectively. Tumor growth for different treatment modalities was monitored until the volume increased to above 1,000 mm3 or loss more than 20% of the initial bodyweight at which point the animals were euthanized by an overdose of carbon dioxide.
The impact of upregulation of HLA-DR expression (induced by immunogenic cell death) on the in vivo anticancer efficacy of free Dox, free SHAL SH7129, conjugated SHAL, nontargeted Dox NPs and SHAL-functionalized Dox NPs for chemo-immunotherapy was evaluated in a Daudi xenograft tumor model in Nu mice. A longer treatment cycle (6 days rest day between treatments) was used to allow the HLA-DR expression of variable cancer cells return to background level before subsequent treatment. In the in vivo study, the Daudi xenograft tumors were established via subcutaneous injection of 2 × 106 Daudi cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI1640/Matrigel solution in the left flank in 48 female Nu mice (6–7 weeks old, 20–21 g). Seven days postinoculation, mice were randomized and divided into eight groups (six mice per group) for different treatments. The control and treatment groups are (1) PBS (nontreatment group); (2) free SHAL SH7129; (3) drug-free SHAL-functionalized NPs; (4) free Dox; (5) nontargeted Dox NPs; (6) SHAL-functionalized Dox NPs; (7) free SHAL plus free Dox; and (8) drug-free SHAL NPs plus nontargeted Dox NPs. Mice in the treatment groups received 3 tail vein i.v. Injections of 3.5 mg/kg free/encapsulated Dox and 5 μg/kg of free SH7129 or conjugated SHAL at day 7, 14, and 21 postinoculation. Each tumor volume was measured every 3–4 days via a caliper. The bodyweight was measured every week. Tumor volumes were calculated by using the formula of volume (V) = 0.5 × a × b2, where a and b are the larger and smaller diameters, respectively. Tumor growth for different treatment modalities was monitored until the volume increased to above 1000 mm3 or the animal lost more than 20% of its initial bodyweight at which point the animals were euthanized by an overdose of carbon dioxide.
A treatment sequence-dependent in vivo study was performed in a Raji xenograft tumor model in Nu mice to investigate how the radiotherapy schedule affects the treatment efficacy of the SHAL-functionalized Dox NPs. In the in vivo study, Raji xenograft tumors were established via subcutaneous injection of 2 × 106 Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI1640/Matrigel solution in the left flank in 26 male Nu mice (6–7 weeks old, 25–26 g). Five days postinoculation, mice were randomized and divided into 3 groups (8–9 mice per group) for different treatments. Mice in the treatment groups received 3 tail vein i.v. Injection SHAL-functionalized Dox NPs (contained 3.5 mg/kg encapsulated Dox and 5 μg/kg of conjugated SHAL) at day 5, 9, and 12 postinoculation. In the concurrent CIRT group, mice received 5 Gy X-ray irradiations 24 h after each i.v. administration of the therapeutics. Mice in the sequential CIRT group received three 5 Gy X-ray irradiations at day 17, 20, and 23 postinoculation. In vivo radiotherapy was performed using a Precision X-RAD 320 (Precision X-ray, Inc.) machine operating at 320 kV and 12.5 mA. The source–subject distance was 70 cm, and the dose was administered at a rate of 50 cGy/min. Only the tumor regions (left flank) of the mice were irradiated, as the remaining parts of the body were lead-shielded. Tumor volume was measured every 3–4 days via a caliper. The bodyweight was measured every week. Tumor volumes were calculated by using the formula of volume (V) = 0.5 × a × b2, where, a and b are the larger and smaller diameters, respectively. Tumor growth for different treatment modalities was monitored until the volume increased to above 1000 mm3 or there was a loss of more than 20% of the initial bodyweight at which point the animals were euthanized by an overdose of carbon dioxide.
Histopathological Study
Xenograft tumors were established via subcutaneous injection of 2 × 106 Raji cells in 200 μL of a 1:1 (v/v) mixture of a serum-free RPMI1640/Matrigel solution in the left flank. Four days postinoculation, mice were randomized and divided into 16 groups for different treatments. The control and treatment groups are (1) PBS (nontreatment group); (2) free SHAL SH7129; (3) drug-free SHAL-functionalized NPs; (4) free Dox; (5) nontargeted Dox NPs; (6) SHAL-functionalized Dox NPs; (7) free SHAL plus free Dox; (8) drug-free SHAL NPs plus nontargeted Dox NPs; (9) PBS followed by XRT; (10) free SHAL SH7129 followed by XRT; (11) drug-free SHAL-functionalized NPs followed by XRT; (12) free Dox followed by XRT; (13) nontargeted Dox NPs followed by XRT; (14) SHAL-functionalized Dox NPs followed by XRT; (15) free SHAL plus free Dox followed by XRT; and (16) drug-free SHAL NPs plus nontargeted Dox NPs followed by XRT. Mice in the treatment groups received a single tail vein i.v. injections of 3.5 mg/kg free/encapsulated Dox and 5 μg/kg of free SH7129 or conjugated SHAL at day 4 postinoculation. Mice in the concurrent CIRT groups received 5 Gy X-ray irradiation 24 h after administration of different therapeutics through a Precision X-RAD 320 (Precision X-ray, Inc.) machine operating at 320kVp and 12.5 mA. The source-subject distance of 70 cm and 50 cGy/min. Mice were euthanized 24 h to 5 days after the treatment. The tumors were collected and fixed in 4% neutral-buffered formalin for 24 h at 4 °C and then stored in 70% ethanol at 4 °C for 24 h before being submitted to the Animal Histopathology Core Facility at UNC Medical School for sectioning. Caspase 3, and HLA-DR immunohistochemistry stains were performed at the Translational Pathology Lab at the UNC Medical School. For quality control purposes, all staining was performed using a biological tissue automatic staining machine. All stained tumor sections were imaged on a Zeiss 710 Spectral CLSM confocal microscope in the Microscopy Services Laboratory Core Facility at the UNC School of Medicine.
Statistical Analysis
Quantitative data were expressed as mean ± SEM. The analysis of variance was completed using a one-way ANOVA in GraphPad Prism 6 software pack. The analysis of survival data was completed using a Log-rank (Mantel-Cox) test in GraphPad Prism 6 software pack. *p < 0.05 was considered statistically significant.
Safety Statement
No unexpected or unusually high safety hazards were encountered in this line of research.
Acknowledgments
We thank the Microscopy Service Laboratory Core, Animal Study Core, Small Animal Imaging Facility, Animal Clinical Laboratory, Animal Histopathology Core Facility, Translation Pathology Lab, UNC Flow Cytometry Core Facility, UNC Macromolecular Interactions Facility and UNC Michael Hooker Proteomics Centre in the School of Medicine, and Chapel Hill Analytical and Nanofabrication Laboratory (CHANL) at the University of North Carolina at Chapel Hill for their assistance with procedures in this manuscript. The UNC Flow Cytometry Core Facility is supported in part by P30CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. SHAL Technologies, Inc (Livermore, CA) is thanked for supplying SHALs and for permission to publish this work. R.B. and M.C.B. would like to thank the National Institutes of Health/National Cancer Institute for providing support to SHAL Technologies through SBIR grant R44CA159843. This work was supported by the University Cancer Research Fund from the University of North Carolina and R01CA178748 grant from the National Institutes of Health/National Cancer Institute. A.Z.W. was also supported by the National Institutes of Health Center for Nanotechnology Excellence Grant U54-CA151652.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00746.
Supporting figures and tables (PDF)
The authors declare the following competing financial interest(s): The University of North Carolina at Chapel Hill and SHAL Technologies, Inc. filed a patent to protect the IP for therapeutic applications of SHAL-functionalized nanoparticles.
Supplementary Material
References
- Siegel R. L.; Miller K. D.; Jemal A. Cancer statistics, 2018. Ca-Cancer J. Clin. 2018, 68 (1), 7. 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Coiffier B.; Lepage E.; Briere J.; Herbrecht R.; Tilly H.; Bouabdallah R.; Morel P.; Van Den Neste E.; Salles G.; Gaulard P.; et al. CHOP chemotherapy plus rituximab compared with CHOP alone in elderly patients with diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346 (4), 235. 10.1056/NEJMoa011795. [DOI] [PubMed] [Google Scholar]
- Feugier P.; Van Hoof A.; Sebban C.; Solal-Celigny P.; Bouabdallah R.; Ferme C.; Christian B.; Lepage E.; Tilly H.; Morschhauser F.; et al. Long-term results of the R-CHOP study in the treatment of elderly patients with diffuse large B-cell lymphoma: a study by the Groupe d’Etude des Lymphomes de l’Adulte. J. Clin. Oncol. 2005, 23 (18), 4117. 10.1200/JCO.2005.09.131. [DOI] [PubMed] [Google Scholar]
- DeVita V. T. Jr.; Chu E. A history of cancer chemotherapy. Cancer Res. 2008, 68 (21), 8643. 10.1158/0008-5472.CAN-07-6611. [DOI] [PubMed] [Google Scholar]
- Hennessy B. T.; Hanrahan E. O.; Daly P. A. Non-Hodgkin lymphoma: an update. Lancet Oncol. 2004, 5 (6), 341. 10.1016/S1470-2045(04)01490-1. [DOI] [PubMed] [Google Scholar]
- Lennard A. L.; Jackson G. H. Stem cell transplantation. BMJ. 2000, 321 (7258), 433. 10.1136/bmj.321.7258.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosset J. M. Chemoradiotherapy for localized non-Hodgkin’s lymphoma. N. Engl. J. Med. 1998, 339 (1), 44. 10.1056/NEJM199807023390110. [DOI] [PubMed] [Google Scholar]
- Basaran G.; Basaran M.; Onat H. Chemoradiotherapy for localized non-Hodgkin’s lymphoma: lessons from old studies. J. Clin. Oncol. 2005, 23 (13), 3161. 10.1200/JCO.2005.05.631. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M.; Tobinai K.; Oguchi M.; Ishizuka N.; Kobayashi Y.; Isobe Y.; Ishizawa K.; Maseki N.; Itoh K.; Usui N.; et al. Concurrent chemoradiotherapy for localized nasal natural killer/T-cell lymphoma: an updated analysis of the Japan clinical oncology group study JCOG0211. J. Clin. Oncol. 2012, 30 (32), 4044. 10.1200/JCO.2012.45.6541. [DOI] [PubMed] [Google Scholar]
- Chu T. W.; Yang J.; Zhang R.; Sima M.; Kopecek J. Cell surface self-assembly of hybrid nanoconjugates via oligonucleotide hybridization induces apoptosis. ACS Nano 2014, 8 (1), 719. 10.1021/nn4053827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au K. M.; Tripathy A.; Lin C. P.; Wagner K.; Hong S.; Wang A. Z.; Park S. I. Bespoke Pretargeted Nanoradioimmunotherapy for the Treatment of Non-Hodgkin Lymphoma. ACS Nano 2018, 12 (2), 1544. 10.1021/acsnano.7b08122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duggan S. T.; Keating G. M. Pegylated liposomal doxorubicin: a review of its use in metastatic breast cancer, ovarian cancer, multiple myeloma and AIDS-related Kaposi’s sarcoma. Drugs 2011, 71 (18), 2531. 10.2165/11207510-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Kroemer G.; Galluzzi L.; Kepp O.; Zitvogel L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51. 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- Galluzzi L.; Buque A.; Kepp O.; Zitvogel L.; Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17 (2), 97. 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- Casares N.; Pequignot M. O.; Tesniere A.; Ghiringhelli F.; Roux S.; Chaput N.; Schmitt E.; Hamai A.; Hervas-Stubbs S.; Obeid M.; et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202 (12), 1691. 10.1084/jem.20050915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesniere A.; Schlemmer F.; Boige V.; Kepp O.; Martins I.; Ghiringhelli F.; Aymeric L.; Michaud M.; Apetoh L.; Barault L.; et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene 2010, 29 (4), 482. 10.1038/onc.2009.356. [DOI] [PubMed] [Google Scholar]
- Golden E. B.; Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol 2015, 25 (1), 11. 10.1016/j.semradonc.2014.07.005. [DOI] [PubMed] [Google Scholar]
- Wang C.; Wang J.; Zhang X.; Yu S.; Wen D.; Hu Q.; Ye Y.; Bomba H.; Hu X.; Liu Z.; Dotti G.; Gu Z. In situ formed reactive oxygen species-responsive scaffold with gemcitabine and checkpoint inhibitor for combination therapy. Sci. Transl. Med. 2018, 10 (429), eaan3682. 10.1126/scitranslmed.aan3682. [DOI] [PubMed] [Google Scholar]
- Haussner C.; Lach J.; Eichler J. Synthetic antibody mimics for the inhibition of protein-ligand interactions. Curr. Opin. Chem. Biol. 2017, 40, 72. 10.1016/j.cbpa.2017.07.001. [DOI] [PubMed] [Google Scholar]
- Park B. W.; Zhang H. T.; Wu C.; Berezov A.; Zhang X.; Dua R.; Wang Q.; Kao G.; O’Rourke D. M.; Greene M. I.; et al. Rationally designed anti-HER2/neu peptide mimetic disables P185HER2/neu tyrosine kinases in vitro and in vivo. Nat. Biotechnol. 2000, 18 (2), 194. 10.1038/72651. [DOI] [PubMed] [Google Scholar]
- Balhorn R.; Hok S.; Burke P. A.; Lightstone F. C.; Cosman M.; Zemla A.; Mirick G.; Perkins J.; Natarajan A.; Corzett M.; et al. Selective high-affinity ligand antibody mimics for cancer diagnosis and therapy: initial application to lymphoma/leukemia. Clin. Cancer Res. 2007, 13 (18), 5621s. 10.1158/1078-0432.CCR-07-1128. [DOI] [PubMed] [Google Scholar]
- McEnaney P. J.; Fitzgerald K. J.; Zhang A. X.; Douglass E. F. Jr.; Shan W.; Balog A.; Kolesnikova M. D.; Spiegel D. A. Chemically synthesized molecules with the targeting and effector functions of antibodies. J. Am. Chem. Soc. 2014, 136 (52), 18034. 10.1021/ja509513c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hok S.; Natarajan A.; Balhorn R.; DeNardo S. J.; DeNardo G. L.; Perkins J. Synthesis and radiolabeling of selective high-affinity ligands designed to target non-Hodgkin’s lymphoma and leukemia. Bioconjugate Chem. 2007, 18 (3), 912. 10.1021/bc060305o. [DOI] [PubMed] [Google Scholar]
- DeNardo G. L.; Natarajan A.; Hok S.; Mirick G.; DeNardo S. J.; Corzett M.; Sysko V.; Lehmann J.; Beckett L.; Balhorn R. Nanomolecular HLA-DR10 antibody mimics: A potent system for molecular targeted therapy and imaging. Cancer Biother.Radiopharm. 2008, 23 (6), 783. 10.1089/cbr.2008.0589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhorn R.; Hok S.; DeNardo S.; Natarajan A.; Mirick G.; Corzett M.; Denardo G. Hexa-arginine enhanced uptake and residualization of selective high affinity ligands by Raji lymphoma cells. Mol. Cancer 2009, 8, 25. 10.1186/1476-4598-8-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhorn R. L.; Skorupski K. A.; Hok S.; Balhorn M. C.; Guerrero T.; Rebhun R. B. A selective high affinity ligand (SHAL) designed to bind to an over-expressed human antigen on non-Hodgkin’s lymphoma also binds to canine B-cell lymphomas. Vet. Immunol. Immunopathol. 2010, 137 (3–4), 235. 10.1016/j.vetimm.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Rose L. M.; Deng C. T.; Scott S. L.; Xiong C. Y.; Lamborn K. R.; Gumerlock P. H.; DeNardo G. L.; Meares C. F. Critical Lym-1 binding residues on polymorphic HLA-DR molecules. Mol. Immunol. 1999, 36 (11–12), 789. 10.1016/S0161-5890(99)00083-8. [DOI] [PubMed] [Google Scholar]
- Epstein A. L.; Marder R. J.; Winter J. N.; Stathopoulos E.; Chen F. M.; Parker J. W.; Taylor C. R. Two new monoclonal antibodies, Lym-1 and Lym-2, reactive with human B-lymphocytes and derived tumors, with immunodiagnostic and immunotherapeutic potential. Cancer Res. 1987, 47 (3), 830. [PubMed] [Google Scholar]
- Boegel S.; Lower M.; Bukur T.; Sahin U.; Castle J. C. A catalog of HLA type, HLA expression, and neo-epitope candidates in human cancer cell lines. Oncoimmunology 2014, 3 (8), e954893. 10.4161/21624011.2014.954893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balhorn M. C.; Balhorn R. Abstract 1171: Therapeutic applications of the selective high affinity ligand SH7139 may extend beyond NHL to many other types of solid tumors. Cancer Res. 2017, 77 (13 Supplement), 1171. 10.1158/1538-7445.AM2017-1171. [DOI] [Google Scholar]
- DeNardo G. L.; Mirick G. R.; Hok S.; DeNardo S. J.; Beckett L. A.; Adamson G. N.; Balhorn R. L. Molecular specific and cell selective cytotoxicity induced by a novel synthetic HLA-DR antibody mimic for lymphoma and leukemia. Int. J. Oncol. 2009, 34 (2), 511. 10.3892/ijo_00000176. [DOI] [PubMed] [Google Scholar]
- Balhorn R.; Balhorn M. C. Abstract 4079: Pre-clinical toxicology and safety of SH7139: The first of a new class of targeted therapeutics for non-Hodgkin’s lymphoma and other cancers. Cancer Res. 2017, 77 (13 Supplement), 4079. 10.1158/1538-7445.AM2017-4079. [DOI] [Google Scholar]
- DeNardo G. L.; Natarajan A.; Hok S.; Perkins J.; Cosman M.; DeNardo S. J.; Lightstone F. C.; Mirick G. R.; Miers L. A.; Balhorn R. L. Pharmacokinetic characterization in xenografted mice of a series of first-generation mimics for HLA-DR antibody, Lym-1, as carrier molecules to image and treat lymphoma. J. Nucl. Med. 2007, 48 (8), 1338. 10.2967/jnumed.107.041095. [DOI] [PubMed] [Google Scholar]
- Rech J.; Repp R.; Rech D.; Stockmeyer B.; Dechant M.; Niedobitek G.; Gramatzki M.; Valerius T. A humanized HLA-DR antibody (hu1D10, apolizumab) in combination with granulocyte colony-stimulating factor (filgrastim) for the treatment of non-Hodgkin’s lymphoma: a pilot study. Leuk. Lymphoma 2006, 47 (10), 2147. 10.1080/10428190600757944. [DOI] [PubMed] [Google Scholar]
- Roche P. A.; Teletski C. L.; Stang E.; Bakke O.; Long E. O. Cell surface HLA-DR-invariant chain complexes are targeted to endosomes by rapid internalization. Proc. Natl. Acad. Sci. U. S. A. 1993, 90 (18), 8581. 10.1073/pnas.90.18.8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinet V.; Vergelli M.; Martini R.; Bakke O.; Long E. O. Antigen presentation mediated by recycling of surface HLA-DR molecules. Nature 1995, 375 (6532), 603. 10.1038/375603a0. [DOI] [PubMed] [Google Scholar]
- Girolomoni G.; Cruz P. D. Jr.; Bergstresser P. R. Internalization and acidification of surface HLA-DR molecules by epidermal Langerhans cells: a paradigm for antigen processing. J. Invest. Dermatol. 1990, 94 (6), 753. 10.1111/1523-1747.ep12874611. [DOI] [PubMed] [Google Scholar]
- Bonner J. A.; Lawrence T. S. Doxorubicin decreases the repair of radiation-induced DNA damage. Int. J. Radiat. Biol. 1990, 57 (1), 55. 10.1080/09553009014550341. [DOI] [PubMed] [Google Scholar]
- Saha B.; Evers T. H.; Prins M. W. How antibody surface coverage on nanoparticles determines the activity and kinetics of antigen capturing for biosensing. Anal. Chem. 2014, 86 (16), 8158. 10.1021/ac501536z. [DOI] [PubMed] [Google Scholar]
- Yoo H. S.; Park T. G. Folate receptor targeted biodegradable polymeric doxorubicin micelles. J. Controlled Release 2004, 96 (2), 273. 10.1016/j.jconrel.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Fulop Z.; Gref R.; Loftsson T. A permeation method for detection of self-aggregation of doxorubicin in aqueous environment. Int. J. Pharm. 2013, 454 (1), 559. 10.1016/j.ijpharm.2013.06.058. [DOI] [PubMed] [Google Scholar]
- MacKay J. A.; Chen M.; McDaniel J. R.; Liu W.; Simnick A. J.; Chilkoti A. Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 2009, 8 (12), 993. 10.1038/nmat2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fucikova J.; Kralikova P.; Fialova A.; Brtnicky T.; Rob L.; Bartunkova J.; Spisek R. Human tumor cells killed by anthracyclines induce a tumor-specific immune response. Cancer Res. 2011, 71 (14), 4821. 10.1158/0008-5472.CAN-11-0950. [DOI] [PubMed] [Google Scholar]
- Kawano M.; Tanaka K.; Itonaga I.; Iwasaki T.; Miyazaki M.; Ikeda S.; Tsumura H. Dendritic cells combined with doxorubicin induces immunogenic cell death and exhibits antitumor effects for osteosarcoma. Oncol. Lett. 2016, 11 (3), 2169. 10.3892/ol.2016.4175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilmer S. N.; Cogger V. C.; Muller M.; Le Couteur D. G. The hepatic pharmacokinetics of doxorubicin and liposomal doxorubicin. Drug Metab. Dispos. 2004, 32 (8), 794. 10.1124/dmd.32.8.794. [DOI] [PubMed] [Google Scholar]
- Duthu A.; Debuire B.; Romano J.; Ehrhart J. C.; Fiscella M.; May E.; Appella E.; May P. p53 mutations in Raji cells: characterization and localization relative to other Burkitt’s lymphomas. Oncogene 1992, 7 (11), 2161. [PubMed] [Google Scholar]
- Hagemeister F. B. Treatment of relapsed aggressive lymphomas: regimens with and without high-dose therapy and stem cell rescue. Cancer Chemother. Pharmacol. 2002, 49 (Suppl 1), S13. 10.1007/s00280-002-0447-1. [DOI] [PubMed] [Google Scholar]
- Chao M. P. Treatment challenges in the management of relapsed or refractory non-Hodgkin’s lymphoma - novel and emerging therapies. Cancer Manage. Res. 2013, 5, 251. 10.2147/CMAR.S34273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. R. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene 2003, 22 (47), 7359. 10.1038/sj.onc.1206939. [DOI] [PubMed] [Google Scholar]
- DeNardo G. L.; Hok S.; Natarajan A.; Cosman M.; DeNardo S. J.; Lightstone F. C.; Mirick G. R.; Yuan A.; Perkins J.; Sysko V. V.; Lehmann J.; Balhorn R. Characteristics of dimeric (bis) bidentate selective high affinity ligands as HLA-DR10 beta antibody mimics targeting non-Hodgkin’s lymphoma. Int. J. Oncol. 2007, 31 (4), 729. 10.3892/ijo.31.4.729. [DOI] [PubMed] [Google Scholar]
- Cardillo T. M.; Govindan S. V.; Zalath M. B.; Rossi D. L.; Wang Y.; Chang C. H.; Goldenberg D. M. IMMU-140, a Novel SN-38 Antibody-Drug Conjugate Targeting HLA-DR, Mediates Dual Cytotoxic Effects in Hematologic Cancers and Malignant Melanoma. Mol. Cancer Ther. 2018, 17 (1), 150. 10.1158/1535-7163.MCT-17-0354. [DOI] [PubMed] [Google Scholar]
- Rodrigo M. R.; Rosenquist J. B.; Cheung L. K. Paracetamol and diflunisal for pain relief following third molar surgery in Hong Kong Chinese. Int. J. Oral Maxillofac Surg 1987, 16 (5), 566. 10.1016/S0901-5027(87)80107-8. [DOI] [PubMed] [Google Scholar]
- Cardoso M. M.; Peca I. N.; Roque A. C. Antibody-conjugated nanoparticles for therapeutic applications. Curr. Med. Chem. 2012, 19 (19), 3103. 10.2174/092986712800784667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.